

Abstract
A growing amount of evidence suggests that ubiquitination and deubiquitination of programmed death 1 (PD-1)/programmed death-ligand 1 (PD-L1) play crucial roles in the regulation of PD-1 and PD-L1 protein stabilization and dynamics. PD-1/PD-L1 is a major coinhibitory checkpoint pathway that modulates immune escape in cancer patients, and its engagement and inhibition has significantly reshaped the landscape of tumor clearance. The abnormal ubiquitination and deubiquitination of PD-1/PD-L1 influence PD-1/PD-L1-mediated immunosuppression. In this review, we describe the ubiquitination- and deubiquitination-mediated modulation of PD-1/PD-L1 signaling through a variety of E3 ligases and deubiquitinating enzymes (DUBs). Moreover, we briefly expound on the anticancer potential of some agents that target related E3 ligases, which further modulate the ubiquitination of PD-1/PD-L1 in cancers. Therefore, this review reveals the development of a highly promising therapeutic approach for cancer immunotherapy by targeting PD-1/PD-L1 ubiquitination.
Keywords: ubiquitination, deubiquitination, PD-1, PD-L1, immunotherapy
Graphical Abstract
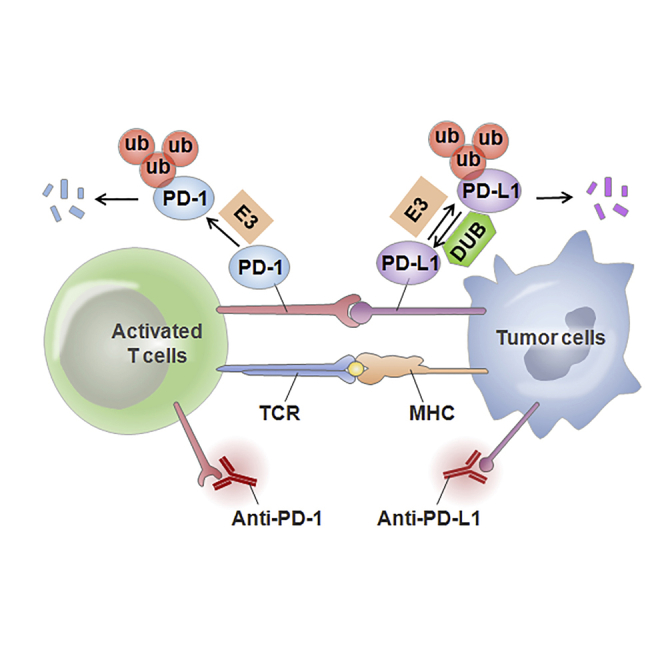
The abnormal ubiquitination and deubiquitination of PD-1/PD-L1 influence PD-1/PD-L1-mediated immunosuppression. Herein, we describe the ubiquitination- and deubiquitination-mediated modulation of PD-1/PD-L1 signaling through multiple E3 ligases and deubiquitination enzymes. This review suggests the development of a promising therapeutic strategy for cancer immunotherapy by targeting PD-1/PD-L1 ubiquitination.
Main Text
Human cancer is a complex disease that involves a variety of genetic and epigenetic alterations, which contribute to the production of tumor antigens that may lead to immune recognition, and even an immunological response. The immune system is a key modulator of tumor biology with the ability to promote or suppress tumorigenic potential.1 The association between immunity and carcinoma has been well established over the past several decades and was originally proposed by Rudolf Virchow in the 19th century.2 The multiple effects on cancer immunoediting comprise three major stages, that is, elimination, equilibrium, and escape, which result in tumor elimination, dormancy, and progression, respectively.3 Recently, the potential of tumors to avoid an immune response, in which T cell-mediated cytotoxicity plays a key role in killing cancer cells, has been identified as one of the most prominent cancer hallmarks.4 Through the selective identification and removal of pathogenic agents and abnormal cells such as cancer cells, T cell immunity is essential for preserving homeostasis.
Programmed death 1 (PD-1, also known as CD279), which was initially discovered in 1992 in a T cell hybridoma and a hematopoietic progenitor cell line in an apoptotic state,5 is a 55-kDa transmembrane protein in the B7-CD28 family. PD-1 is an important immune checkpoint receptor on activated T cells that negatively impacts the response to antigens.6 Accumulating evidence highlights the immunosuppressive function of PD-1 and its ligands programmed death-ligand 1 (PD-L1) (B7-H1) and PD-L2 (B7-DC) in the tumor microenvironment, which has dramatically reshaped the landscape of cancer therapy.7 Mechanistically, PD-L1 expressing on the surface of tumor cells binds to PD-1 receptors on activated T cells, resulting in the blockage of T lymphocyte proliferation, cytokine production, and the inhibition of the immune response.8 Additionally, PD-L1 is overexpressed in a variety of cancers, including colorectal cancer (CRC), gastric cancer, ovarian cancer, papillary thyroid cancer, and bladder cancer.9,10
Based on the above findings, inhibition of the PD-1/PD-L1 signaling pathway by antibodies can enhance T cell responses and improve preclinical anticancer effects. Currently, therapeutic antibodies against PD-1 (e.g., nivolumab, pembrolizumab, and cemiplimab) and PD-L1 (e.g., atezolizumab, avelumab, and durvalumab) have been approved by the US Food and Drug Administration (FDA) and have shown promising clinical outcomes in trials for a subset of malignancies.11 This treatment strategy has recently led to a 10%–40% increase in immunological responses among cancer patients.12,13 However, it is unclear why only PD-L1-positive cancers respond to PD-1/PD-L1 checkpoint inhibitors. A deeper exploration of the mechanisms that regulate PD-1/PD-L1 expression and stability may help increase the clinical effectiveness of PD-1/PD-L1 blockade.
The activity of PD-1/PD-L1 is complicated since it is modulated by multiple processes, including gene transcription, posttranscriptional modifications, posttranslational modifications (PTMs), and exosomal transport.14 PTMs (e.g., glycosylation, phosphorylation, ubiquitination, palmitoylation, SUMOylation, and acetylation) have been demonstrated to play a pivotal role in the modulation of protein stabilization and protein-protein interactions of the PD-1/PD-L1 axis.15 For example, Rho-associated protein kinase-dependent moesin phosphorylation stabilizes the PD-L1 protein level in breast cancer.16 Glycosylation of PD-L1, especially N-glycosylation, is important for modulating the immunosuppressive function and immune elimination in cancer. N-linked glycosylation of PD-1/PD-L1 proteins enhances their stability, which further improves the immune evasion ability of cancer cells.17,18 One recent study revealed that the acetylation-dependent modulation of PD-L1 inhibits its translocation and promotes the anti-cancer efficacy of PD-1/PD-L1 blockade.19 During the last decade, strong evidence has indicated that the expression of PD-1 and PD-L1 proteins is usually modulated by the ubiquitin (Ub)-mediated proteasome degradation pathway.20, 21, 22, 23 Ubiquitination is essential in the regulation of a subset of cellular processes, such as endogenous protein stabilization, receptor internalization, and immune responses.24 A more intensive exploration of the molecular mechanism of PD-1/PD-L1 protein expression and stability is essential for the improvement of immunotherapeutic strategies to treat human cancers. In this review, the crucial roles of ubiquitination and deubiquitination in the regulation of PD-1/PD-L1 in cancer and their therapeutic potential for targeting PD-1/PD-L1 are described.
Ubiquitin-Proteasome System (UPS)
The UPS serves as a major PTM mechanism that functions in protein degradation under physiological and pathological conditions.25 A well-accepted doctrine is that the UPS is composed of a wide range of important elements, including ubiquitin, ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), ubiquitin-protein enzymes (E3s), deubiquitinating enzymes (DUBs), and the 26S proteasome.26 Among them, ubiquitin is a highly conserved 76-aa protein in eukaryotes that serves as a posttranslational modifier that covalently binds to substrates through a series of enzyme-linked reactions mediated by E1, E2, and E3 ligases. Functionally, the E1-E2-E3 cascade consists of three steps. First, the carboxyl group (-COOH) of the C terminus of ubiquitin binds to an E1 cysteine residue along with ATP and is thus stimulated by a thioester link with E1. Second, the E2 ligase temporarily transfers ubiquitin moieties with a thioester linkage. Finally, activated ubiquitin is moved from E2 to the lysine residue on substrates by E3. Moreover, this enzyme-linked reaction changes ubiquitin polymerization into the polyubiquitin chain (Figure 1).27
Figure 1.

The Ubiquitination and Deubiquitination Processes Are Illustrated
The ubiquitin-proteasome system is composed of ubiquitin, ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), ubiquitin-protein enzymes (E3s), deubiquitinating enzymes (DUBs), and the 26S proteasome. Ubiquitination has a series of enzyme-linked reactions mediated by E1, E2, and E3 ligases. The carboxyl group (-COOH) of the C terminus of ubiquitin binds to an E1 cysteine residue along with ATP and is thus stimulated by a thioester link with E1. The E2 ligase temporarily transfers ubiquitin moieties with a thioester linkage. Activated ubiquitin is moved from E2 to the lysine residue on substrates by E3. Ubiquitination is controlled by E3 ligases, whose activities can be reversed by DUBs. Ubiquitination and deubiquitination play a crucial role in the regulation of PD-1/PD-L1 in cancer.
Ubiquitination is tightly controlled by E3 ligases, whose activities can be reversed by DUBs. It is well known that the substrate specificity of the UPS is modulated by E3s. E3 ligases, which are commonly divided into RING (really interesting new gene) E3s, HECT (homologous to E6AP C terminus) E3s, and RBR (RING-in-between-RING) E3s, are implicated in the modulation of various immune processes, including spontaneous lymphocyte activation and differentiation, induction of T cell-mediated tolerance, antigen presentation, and immune escape.28 Accumulating evidence indicates that insufficiency or mutations in many E3 enzymes such as casitas B cell lymphoma (Cbl)-b and ITCH, usually results in abnormal immunological responses in autoimmunity, carcinoma, and inflammation.29,30 Recent findings have demonstrated that ubiquitination and deubiquitination are extensively implicated in the modulation of the biological activities of the PD-1/PD-L1 pathway, which indicates that targeting E3s and DUBs is a novel strategy to improve anticancer immune responses.20,22,31,32
E3 Ligases in Regulation of PD-1/PD-L1
β-TrCP
Beta-transducin repeat-containing protein (β-TrCP) is well characterized as the substrate recognition subunit in the Skp1-Cullin 1-F-box (SCF)β-TrCP E3 ubiquitin ligase complex.33 The SCF E3 ubiquitin ligase complex can confer protein ubiquitination by inducing the specific recognition of substrates. Mechanistically, these SCFβ-TrCP complexes are implicated in the ubiquitination and degradation of a variety of proteins in a phosphorylation-dependent manner.34 SCFβ-TrCP plays a crucial role in the regulation of T cell function by triggering the activation of nuclear factor κB (NF-κB) cells by the ubiquitin-mediated proteasomal degradation of the inhibitory protein of IκB, which further leads to nuclear translocation and transcriptional activation.35
Glycogen synthase kinase 3β (GSK3β) is a serine-threonine kinase and one of the signaling regulators that serves as a major component of multiple pathways, especially the insulin and Wnt signaling pathways.36 GSK3β protein serves as a versatile switch by directly phosphorylating a broad spectrum of substrates, such as CRMP2, MCl-1, cyclin D1, c-Jun, c-myc, and Snail1.37,38 The phosphorylation-dependent regulation of GSK3β usually contributes to the recognition of the E3 ubiquitin ligase. For instance, β-catenin is usually targeted for ubiquitination-mediated proteolysis by the phosphorylation of GSK3β, followed by the incorporation of β-TrCP.39 Suppression of GSK3β induces the translocation of β-catenin into the nucleus, where it forms complexes with T cell factor/lymphoid enhancer factor, and thereby activates target gene expression.
A previous investigation demonstrated that GSK3β could lead to the phosphorylation-dependent degradation of PD-L1 by β-TrCP, when bound to nonglycosylated PD-L1. Conversely, inhibition of β-TrCP or a mutation in the GSK3β phosphorylation motif significantly blocks PD-L1 ubiquitination, which indicates that GSK3β or β-TrCP may modulate immune escape in cancer through PD-L1 ubiquitination and degradation.40 Thus, some special inhibitors that inactivate GSK3β could in turn suppress PD-L1 ubiquitination and promote its stability, thereby improving immunotherapy efficiency. For instance, a c-MET inhibitor,41 the PARP1 inhibitor olaparib,42 a tyrosine kinase inhibitor (TKI), and resveratrol40 have been shown to restrain GSK3β activity to further influence the interaction between PD-L1 and β-TrCP. A subsequent investigation found a previously undiscovered function of mammalian target of rapamycin (mTOR) complex 1 (mTORC1)/p70 S6 kinase (p70S6K) in the negative control of PD-L1 in cancer.43 That study indicated that inactivation of this signaling pathway by mTOR or p70S6K inhibitors significantly increased PD-L1 expression in lung carcinoma cells. Functionally, PD-L1 upregulation via suppression of the mTORC1/p70S6K pathway could be ascribed to inhibition of β-TrCP-mediated degradation of PD-L1.
CSN5
The constitutive photomorphogenesis 9 (COP9) signalosome (CSN) serves as a large multiprotein complex that is similar to the 19S lid of the 26S proteasome and plays an indispensable role in the modulation of cullin-RING ubiquitin E3 ligases (CRLs).44 CSN5 is identified as the fifth member of the CSN family and includes a conserved Jab1/Mpr1p and Pad1p N-terminal (MPN) domain metalloenzyme (JAMM) motif.45 JAMM has an important function in CSN-mediated deneddylation and subsequently modulates the activity of the SCF complex.46 The catalytic role of the CSNs, controlled by CSN5/Jab1, is centered on the deneddylation of the CRLs, which are the hydrolysates of the NEDD8 isopeptide bond.45 CSN acts a negative modulator of ubiquitin enzyme activity by deconjugating NEDD8 from cullin-NEDD8. Increasing numbers of studies have demonstrated that CSN5 participates in a subset of biological processes, including transcription factor specificity, deneddylation of NEDD8, and nuclear-to-cytoplasmic transportation of primary molecules.47 CSN5 has been demonstrated to be associated with cancer survival and is considered a poor prognostic biomarker in some tumors.48 Emerging evidence has shown that COP9 subunit CSN5 serves as an indispensable element of the innate immune system.49 For example, one group demonstrated that CSN5 is necessary for the promotion of the proinflammatory kinases p38 and extracellular signal-regulated kinase (ERK) and inhibition of the genes modulated by nuclear factor erythroid 2-related factor 2 (NRF2). Moreover, myeloid-specific CSN5-deficient mice with polymicrobial sepsis exhibit a lower mortality rate.49
A recent study reported a regulatory mechanism of immune surveillance by tumor cells involving the CSN5-mediated stability of PD-L1 by tumor necrosis factor (TNF)-α in breast cancer.20 In other words, inflammation-induced TNF-α enhances the activation of CSN5, whose expression is modulated by NF-κB p65, which further inhibits the ubiquitination and subsequent degradation of PD-L1 and results in immune system evasion. Particularly, repression of TNF-α-mediated PD-L1 stability in tumor cells enhances the tumor-infiltrating cytotoxic T cell immune response. Interestingly, as a DUB, CSN5 also has deubiquitination activity. For example, CSN5 deubiquitinates heat shock protein (HSP)70 and Snail to regulate the sorting of exosomal proteins50 and to stimulate tumor invasion and metastasis,51 respectively. Furthermore, CSN5 has been reported to deubiquitinate IκBa, Snail, and PD-L1, which enhances cancer progression and migration.20 These findings suggest that the deubiquitination function of the CSN5 protein plays an important role in the development and progression of cancer.
Tumor-associated macrophages (TAMs) are considered among the most efficient immune cell types in the tumor microenvironment of solid cancers, as they enhance the migratory and invasive abilities of cancer cells and lead to immune suppression and angiogenesis.52 Biologically, the inhibition of CD8+ T cell immunity by TAMs occurs through direct interaction with T cells or secretion of immunosuppressive molecules.53 CC chemokine receptor 5 (CCR5) is the receptor for C-C motif chemokine ligand 3 (CCL3), CCL4, and CCL5. Moreover, CCR5 plays an important role in the immune response through stimulation of diverse immune cells to migrate to damaged or infected sites.54 Emerging evidence has revealed the roles of CCR5 and its ligands, such as CCL5, in carcinogenesis and immunosuppression. In one study, macrophage-derived CCL5 attenuated T cell-mediated killing of CRC cells and improved immune escape through the stabilization of PD-L1.55 Functionally, CCL5 leads to the production of NF-κB p65/signal transducer and activator of transcription 3 (STAT3) complexes linked to the CSN5 promoter, further enhancing its activity. Furthermore, CSN5 modulates the deubiquitination and stabilization of PD-L1. CSN5 upregulation in CRC is correlated with poorer survival. Therefore, CSN5 plays an important role in PD-L1 regulation, and it may be a promising therapeutic target in cancer immunotherapy.
SPOP
Speckle-type POZ protein (SPOP) is a representative CRL3 adaptor protein that structurally consists of two conserved domains: an N-terminal meprin and a TRAF homology (MATH) domain, which recognizes substrates, and a C-terminal bric-a-brac and tramtrack and broad complex (BTB)/POZ domain that links to Cullin 3, which results in a functional multicomponent E3 ligase complex (Cullin 3/SPOP).56 CRLs are the major representative ubiquitin E3s in eukaryotes, including eight members of Cullin scaffold proteins (Cullin1, 2, 3, 4A, 4B, 5, 7, and 9).57 More than 240 E3 enzyme complexes are part of CRLs and are involved in a subset of key physiological processes. Similar to other CRL family members, CRL3 is composed of Cullin 3, the RING protein Rbx1, and a variable BTB domain adaptor protein, which is a substrate recognition element that recruits substrates into the complex for ubiquitination.58 The CRL3 subfamily is involved in diverse human diseases, including neurodegeneration and cancer.59 Recent studies have suggested that SPOP interacts with Cullin 3 to enhance ubiquitination-mediated degradation of target substrates. As a crucial E3 enzyme, SPOP has been reported to have dual effects on carcinogenesis. Previous studies found that SPOP plays a tumor suppressor role by facilitating the degradation of cancer promoters in certain tumors, including prostate, lung, colon, gastric, and liver carcinomas.60 However, evidence has also indicated the oncogenic effect of SPOP in kidney tumors, which suggests that the biological role of SPOP in cancer development may be context-dependent.
Cell cycle dysfunction is an important feature of human tumors, and targeting cyclin-dependent kinases (CDKs) to prevent cell proliferation is a promising antitumor treatment. CDK4 and CDK6 (CDK4/6) phosphorylate the retinoblastoma (Rb) tumor suppressor protein by interacting with D-type cyclins (CycDs) to further modulate the G1/S phase transition.61 Interruption of this CDK4/6-Rb axis is common in malignancies and usually arises as a result of CycD1 upregulation or depletion of the CDK4/6-specific suppressor p16INK4a, both of which promote CDK4/6 activity and result in unrestrained proliferation.62 New research has found that the upregulation of PD-L1 protein is modulated by cyclin D-CDK4 and Cullin 3SPOP through proteasome-mediated degradation. In other words, the cyclin D-CDK4 complex disrupts PD-L1 stability through Cullin 3SPOP, which was demonstrated to play a crucial role in PD-L1 ubiquitination. Moreover, suppression of CDK4/6 remarkably increases the expression of PD-L1 protein by blocking cyclin D-CDK4-mediated phosphorylation of SPOP, thus degrading SPOP by APC/CCdh1.63 Similarly, inactivating mutations in SPOP can perturb PD-L1 degradation by ubiquitination, which significantly upregulates PD-L1 expression and reduces the number of tumor-infiltrating lymphocytes (TILs) at the tumor site. Note that CDK4/6 suppressors combined with PD-L1 antibodies promote tumor elimination and clearly increase overall survival in vivo.63
STUB1
STIP1 homology and U-box containing protein 1 (STUB1), which encodes the E3 ubiquitin ligase carboxyl terminus of Hsp70-interacting protein (CHIP), contains a tetratricopeptide repeat and a U-box, which has been shown to enhance the ubiquitination of chaperone proteins.64 The STUB1 ubiquitin ligase is considered a negative co-chaperone for Hsp90/heat shock cognate protein 70 (Hsc70), and its expression is usually decreased or absent in various carcinomas, such as CRC.65 Several studies have shown that STUB1 acts as a tumor suppressor since it promotes the ubiquitination and degradation of some oncogenic proteins, such as YAP166 and MZF1.67 Moreover, STUB1 has been reported to play a vital role in the immune response. For instance, STUB1 negatively modulates the suppressive activity of regulatory T cells (Tregs) by promoting degradation of the transcription factor Foxp3.68 Similarly, another study found that STUB1 participates in the degradation of Foxp3, which is considered a negative modulator of PD-L1 activity, in Tregs in both haploid genetic screens.21 Inhibition of STUB1 significantly increases PD-L1 expression, which indirectly reveals STUB1 as an E3 enzyme that induces destabilization of PD-L1. Notably, STUB1 downregulation leads to a more significant upregulation of PD-L1 expression in CMTM6-insufficient cells compared with that in CMTM6-proficient cells, which suggests that STUB1 initiates the ubiquitination of PD-L1, either indirectly or through direct regulation of the lysine in the PD-L1 cytoplasmic domain.
CMTM6 is a widely expressed transmembrane protein that belongs to a family of eight MARVEL domain-containing proteins, and its function is still unclear. High expression of CMTM6 protein is strongly associated with various cancers, such as highly malignant gliomas,69 lung carcinoma,70 and head and neck squamous cell cancer.71 Additionally, CMTM6 is a key factor that modulates T cell activation and anticancer treatment. In 2017, two groups reported similar findings that CMTM6 acts as a positive modulator of PD-L1.21,32 Depletion of CMTM6 significantly suppresses PD-L1 protein expression in various human cancer cells and in primary dendritic cells (DCs). In addition to CMTM6, its closest family member, CMTM4, has been demonstrated to have a similar effect. One research team found that CMTM6, which is expressed on the cell surface, can interrupt PD-L1 ubiquitination and extend its half-life through the interaction of both proteins.21 In addition, CMTM6 significantly enhances the capacity of PD-L1-positive cancer cells to react to the immune response by upregulating the PD-L1 protein pool. Similarly, another group also demonstrated that CMTM6 is a common protein that binds to PD-L1 and maintains its level on the cell surface.32 CMTM6 is not essential for PD-L1 maturation, but it colocalizes with PD-L1 at the plasma membrane and in recycling endosomes, in which it prevents PD-L1 from lysosome-mediated degradation; this subsequently helps the cell escape immune surveillance. Moreover, CMTM6 has been identified as an important indicator that can predict the therapeutic effect of PD-1 inhibitors, in that cancer patients with high CMTM6 and PD-L1 expression have better overall survival after immune therapy.72 Cooperation between CMTM5 and PD-L1 may enhance immune response to PD-1 suppressors and further improve the survival of cancer patients.
FBXO38
F-box only protein 38 (FBXO38), a member of the F-box family that encodes some proteins with an F-box motif, is implicated in protein ubiquitination and degradation.73 F-box proteins can serve as subunits of the SCF E3 enzyme.74 One recent study uncovered a new mechanism of FBXO38 in PD-1 regulation and modulation of immune therapy and cancer inhibition.22 It is well known that the PD-1 protein is ubiquitinated after cell internalization and further degraded by the proteasome in activated T cells. FBXO38 upregulation leads to the enhancement of PD-1 ubiquitination, which is associated with decreased expression of PD-1 protein on the surface of activated T cells. Mechanistically, FBXO38 directly initiates Lys48-linked polyubiquitination of internalized PD-1 at Lys233, which results in the ubiquitination-mediated proteasomal degradation of PD-1. A similar result was observed in an animal model, indicating that FBXO38 depletion markedly increases the expression level of PD-1 on the surface of tumor-infiltrating T cells in vivo, which further promotes cancer development. Furthermore, this enhancement of tumor growth is reversed by anti-PD-1 treatment, which indicates that PD-1 is a major target of the FBXO38 ligase.
FBXO38 transcription is usually decreased in both human and mouse tumor-infiltrating CD8+PD-1+ T cells but can be upregulated by interleukin (IL)-2 administration, as this stimulates the transcription of FBXO38 through the STAT5 protein. In addition, treatment with IL-2 significantly downregulates the level of PD-1 protein, and wild-type (WT) mice exhibit a better antitumor outcome than do mice with T cell-specific deletion of FBXO38.22 The results above implicate FBXO38 as an important regulator of PD-1 degradation, which contributes to the maintenance of cancer immunotherapy. Targeting IL-2-mediated modulation of FBXO38 levels, which further promotes anticancer responses, may be a promising and novel therapeutic strategy to inhibit the PD-1 pathway in cancer.
DCUN1D1
Defective cullin neddylation 1 domain-containing 1/squamous cell carcinoma-related oncogene (DCUN1D1/SCCRO) serves as a RING finger domain-containing ubiquitin E3 enzyme that regulates the assembly and activity of CRLs by enhancing neddylation of proteins in the cullin family.75 DCUN1D1 is an oncogene, located on chromosome 3q26.3 and is commonly amplified in human squamous cell carcinoma (SCC).76 Emerging studies have indicated that DCUN1D1 is involved in a wide range of growth and metastasis processes in certain tumors, including glioma,77 prostate cancer,78 and colorectal carcinoma.79 A previous investigation indicated a high level of DCUN1D1 expression in CRC patients with a poorer clinical outcome.79 Moreover, overexpression of DCUN1D1 significantly promotes the migration and invasiveness of cervical cancer cells.80 Recently, one study showed DCUN1D1 exerts its oncogenic functions in non-small cell lung cancer (NSCLC) and serves as a poor prognostic marker.81 DCUN1D1 upregulation significantly enhances PD-L1 protein levels in lung cancer cell lines, indicating that DCUN1D1 may act as an endogenous promoter of PD-L1 protein expression in NSCLC. However, the molecular mechanism of the modulation of PD-L1 by the DCUN1D1 E3 ligase is still unclear.
Cbl-b and c-Cbl
The (Cbl) family, including the three isoforms, c-Cbl, Cbl-3, and Cbl-b, are RING finger E3 enzymes that can catalyze the transfer of ubiquitin from specific E2 enzymes to the target substrate.82 The Cbl-b gene was originally cloned and identified in 1995.83 Although c-Cbl is expressed exclusively in epithelial cells, studies have demonstrated that Cbl-b and c-Cbl are involved in many physiological processes by modulating multiple receptors and transcription signals. Emerging data have demonstrated that Cbl-b primarily plays a crucial tumor suppressive role in cancer. Functionally, Cbl-b dramatically suppresses tumor cell migration,84 the epithelial-to-mesenchymal transition (EMT), and metastasis,85 and it improves medical sensitivity to cancer therapy.86 Recent evidence has indicated that c-Cbl usually plays a suppressive role in angiogenesis and tumorigenesis by targeting proto-oncogenes, such as nuclear β-catenin87 and c-Src.88
Recently, various E3 ubiquitin enzymes have been shown to participate in the fine-tuning of immune responses. Among these, Cbl-b and c-Cbl serve as two of the most significant gatekeepers of immunological activation because of their role as nonredundant negative modulators of immune activation, especially T cell activation.28,89 Functionally, Cbl-b and c-Cbl increase the threshold for T cell activation through ubiquitylation and consequent interference with crucial T cell signaling members that are directly implicated in the T cell receptor pathway as well as in rearrangements of immune synapse.90 Cbl-b is well known to modulate T cell activity by degrading phosphatidylinositol 3-kinase (PI3K) downstream of the CD28 receptor. Cbl-b deficiency results in ineffective resistance and susceptibility to autoimmunity.91 Importantly, Cbl-b functions in the maintenance of self-tolerance by regulating the immunosuppressive roles of Tregs and transforming growth factor β (TGF-β) in T cells.92 One study showed that the Cbl-b−/− mutation remarkably inhibits the TGF-β receptor pathway in T cells, resulting in less susceptibility to immune suppression of T cells.93
Earlier studies have provided insight into Cbl-b and c-Cbl as promising targets for therapeutic manipulation in anti-PD1/PD-L1 cancer immunotherapy, as they can simultaneously regulate PD-1/PD-L1 signaling in T cells. One study demonstrated that T cells and natural killer (NK) cells in which Cbl-b is depleted become resistant to PD-1/PD-L1-mediated immunosuppression. In a mouse model of melanoma, where diverse liver metastases usually occur in WT mice in a PD-1 dependent manner, Cbl-b−/− mice develop significantly fewer liver metastases without treatment with a PD-1 inhibitor.31 Similarly, researchers found that cytotoxic T lymphocyte-associated protein 4 (CTLA4), but not PD-L1-based immunotherapy, selectively enhances the antitumor phenotype of Cbl-b-deficient mice. Consistently, in vitro evidence suggests that T cells in which Cbl-b is deleted are less susceptible to PD-L1-mediated inhibition of T cell growth and interferon (IFN)-γ secretion.29 One investigation has shown that Cbl-b accelerates ubiquitination of STAT5a and subsequently decreases PD-L1 levels in gastric cancer cells.94 Similarly, Cbl-b and c-Cbl enzymes suppress PD-L1 expression via inactivation of the STAT, AKT, and ERK signaling pathways in WT epidermal growth factor receptor (EGFR) lung cancer cells.95 Additionally, Cbl-b/c-Cbl levels are negatively associated with the PD-L1 level in NSCLC tissues. Emerging evidence indicates that c-Cbl destabilizes the PD-1 protein by ubiquitination-mediated degradation and reliance on the RING finger effect of c-Cbl. This result suggests that c-Cbl destabilizes and inhibits PD-1 activity through proteasomal degradation and further suppresses tumor development and immune infiltrates in CRC.96 Actually, during the T cell activation process, the PD-1/PD-L1 pathway promotes the accumulation of Cbl-b, and Cbl-b and c-Cbl antagonistically restrain PD-L1 expression, further alleviating immunosuppression in cancer. One study demonstrated that the binding between PD-L1 on DCs and PD-1 on CD8 T cells leads to the downregulation of ligand-induced TCR. This occurs through enhancement of the Cbl-b enzyme in CD8 T cells.97 However, more research is needed to determine the modulation mechanisms of PD-1/PD-L1 signaling by Cbl-b and c-Cbl E3 ligases in carcinomas.
HRD1
HMG-coenzyme A (CoA) reductase degradation protein 1 (HRD1) was initially known as an E3 ligase that regulates cholesterol accumulation by modulating the metabolism of the rate-limiting enzyme HMGCR in yeast.98 The cytoplasmic C terminus of HRD1 includes a RING domain that promotes ubiquitin transfer from E2s to targeted substrates. HRD1 is also identified as synoviolin because of its upregulation in synovial fibroblasts, which is commonly stimulated by proinflammatory cytokines, among rheumatoid arthritis patients.99 Further studies revealed that HRD1 plays a crucial role in endoplasmic reticulum (ER)-associated degradation (ERAD) of misfolded/unfolded proteins and prevents cells from ER stress-induced cell death.100 Additionally, HRD1 exerts its oncogenic activities in a variety of cancers through ubiquitination-mediated degradation of multiple proteins, such as sirtuin 2101 and PTEN.102
The E3 ligase HRD1 was recently implicated in immune modulation in the antigen-presenting function of DCs and in the sensitization of both T and B lymphocytes.103,104 For instance, one study demonstrated that deletion of the HRD1 gene significantly decreases T cell numbers, suppresses T cell clonal expansion and inhibits CD4+ T cell differentiation into T helper (Th)1, Th17 and Treg lineages. Mechanistically, p27Kip1 is considered a substrate of the HRD1 enzyme because HRD1 interacts with p27kip1 and then stimulates its degradation in T cells. Thus, HRD1 is identified as a positive modulator of T cell activity.103 Suppression of PD-L1/PD-1 signaling has been reported to be a novel treatment strategy in immunosuppressive therapy. One study found that AMP-activated protein kinase (AMPK) stimulated by metformin directly enhances S195 phosphorylation and leads to aberrant glycosylation of PD-L1. This then leads to the accumulation of ER and the enhancement of ERAD. Subsequently, metformin promotes CTL activity by inhibiting the stabilization and membrane localization of PD-L1. Functionally, a decrease in HRD1 remarkably suppresses the ubiquitination of endogenous PD-L1 stimulated by metformin and abolishes the major ubiquitination of the S195E mutant. Therefore, these data demonstrated that HRD1 acts as an E3 ligase during ERAD by targeting the PD-L1 protein with abnormal glycan constructs derived from S195 phosphorylation.105
DUB-Mediated Regulation of PD-L1
Emerging evidence highlights the importance of deubiquitination in the regulation of PD-L1 in cancer treatment. A previous study reported that RP-619, a broad-spectrum small molecule suppressor of DUBs, significantly decreases PD-L1 expression in 293T cells that stably express FLAG-PD-L1.106,107 This result suggests that DUBs may function as negative modulators of immune activity by deubiquitinating PD-L1. Ubiquitylation is a convertible process in which the deconjugation of ubiquitin is achieved by a series of enzymes called DUBs (also identified as deubiquitylases or deubiquitinases). DUBs can effectively eliminate ubiquitin from ubiquitinated proteins, which results in the stabilization of target substrates. Accumulating evidence suggests that DUBs have important functions in the modulation of diverse physiological and pathological processes, including embryonic development, immune homeostasis, carcinogenesis, and neurodegenerative disorders.108 For instance, a variety of DUBs have been shown to modulate the expression and activities of numerous cancer promoters and suppressors via their deubiquitylating functions.108 In most cases, DUBs regulate the overall level and activity of their substrates rather than in an “all-or-none” fashion.109 Recent studies have demonstrated that almost 99 DUBs are encoded by the human genome.110 It is well known that ubiquitin-specific proteases (USPs) comprise the largest family of cysteine proteases, which accurately modulate cellular processes via regulation of substrate stability.111
Recently, there has been growing interest in exploring the modulation and related mechanisms of the immune system by DUBs. One study has shown that ectopic expression of USP7 suppresses the polyubiquitination of the FOXP3 protein, which is an important transcription factor in the regulation of Treg differentiation, and further enhances its stabilization.112 Another report indicated that depletion of USP21 in Tregs significantly decreases the expression of FOXP3 and other Treg signature genes and inhibits their immunosuppressive effects.113 These data indicate that FOXP3 deletion in Tregs by targeting USP7 and USP21 is a promising strategy for immunosuppressive therapy in cancers. A recent study showed that inhibition of USP7 upregulates the level of PD-L1 protein in Lewis tumor cells. Moreover, the combination of P5091, an inhibitor of USP7, and anti-PD-1 exerts a synergistic anti-cancer effect.114 Emerging data support findings that USP22107 and USP9X115 function as modulators of T cell activity by inducing deubiquitination and stabilization of PD-L1.
USP22
Ubiquitin-specific protease 22 (USP22) serves as a component of the human Spt-Ada-Gcn5-acetyltransferase (SAGA) complex, which edits the histone code through the deubiquitination of H2A and H2B.116 USP22 also has some nonhistone substrates, such as FBP1, SIRT, and TRF1.117 Given the histone deubiquitinating role of USP22, its activity was originally associated with modulation of gene transcription as well as normal cell cycle progression.118 Recently, USP22 upregulation has been reported in various cancers by diverse research teams, which indicates its possible oncogenic function and potential as a novel therapeutic target in carcinomas.119,120 However, USP22 may also harbor tumor suppressive properties in some cancers. For example, USP22 deficiency inhibits myeloid differentiation and induces myeloid leukemia via oncogenic Kras.121 Moreover, USP22 plays an important role in cell cycle progression, apoptosis, tumorigenesis, and chemoresistance by deubiquitinating and stabilizing a variety of substrates, including Sirt1,122 FBP1,122 and EGFR.123
Emerging data have shown the association between USP22 and the anticancer immune response. For instance, one study showed that USP22 downregulation in pancreatic cancer cells decreases the penetration of myeloid cells and promotes the infiltration of T cells and NK cells, which improves immune-mediated tumor elimination in cancer immunotherapy.124 Additionally, USP22 depletion suppresses the c-MYC-mediated decrease in SIRT1 ubiquitination and leads to lower SIRT1 levels. Moreover, suppression of SIRT1 levels limits the proliferation of FLT3-ITD acute myeloid leukemia (AML) stem cells and remarkably promotes FLT3 TKI-mediated cell death.125 Therefore, UPS22 may play a crucial role in negatively regulating immune regulatory activity in tumors. A previous investigation revealed USP22 as a promising deubiquitylase of the PD-L1 protein via its interaction with the C terminus of PD-L1, which results in its deubiquitination and stability.107 This study indicated that USP22 downregulation remarkably decreases the level of PD-L1 and that USP22 inhibition could suppress tumor growth in H22 tumor-bearing mice. Furthermore, USP22 deletion not only promotes the treatment effect of PD-L1-targeted tumor immunological therapy but also enhances CDDP-based chemotherapy in vivo, which indicates the complex functions of USP22-PD-L1 signaling in relationship to the efficacy of cancer treatment.107 Similarly, a recent work also suggested that USP22 deubiquitinates PD-L1 and suppresses its degradation through the USP22-CSN5-PD-L1 axis. Moreover, USP22 depletion plays a tumor suppressor role due to increasing T cell cytotoxicity in NSCLC.126 In conclusion, targeting USP22 may be a novel and promising strategy to augment the evasion ability of PD-L1-positive tumor cells for immune-mediated elimination in immunotherapy.
USP9X
Ubiquitin-specific protease 9X (USP9X, also called FAM) is composed of an evolutionarily conserved sequence that contains homologous regions of the Drosophila fat facets gene.127 Biochemically, USP9X can cleave monoubiquitin from target substrates and diverse ubiquitin linkages consisting of K48-, K63-, and K29-linked ubiquitin chains. Emerging studies have demonstrated that USP9X can interact with nearly 35 proteins, some of which are substrates.128 The deubiquitinase USP9X participates in a variety of biological processes, such as cell polarity, death, and modulation of TGF signaling.129 In vivo experiments also showed that USP9X is involved in myriad diseases, including Parkinson’s disease and Alzheimer’s disease as well as autoimmune diseases.130 Additionally, USP9X has been described to have a context-dependent role in carcinogenesis and anticarcinoma.131,132 One study showed that USP9X levels are obviously increased in ERG-positive prostate cancer, and the USP9X inhibitor WP1130 can induce ERG degradation and further suppress tumor growth.131 Conversely, another study found that USP9X inhibits the tumorigenicity of pancreatic ductal adenocarcinoma (PDAC) by suppressing transformation and preventing anoikis in pancreatic cancer cells.133
USP9X has also been indicated to target several cytosolic proteins and to play an important role in immune regulation.23,115,134 One study indicated that in vivo downregulation of USP9X represses T cell growth and that USP9X depletion in T cells attenuates TCR signaling-mediated NF-κB activation. Additionally, naive CD4+ T cells from USP9X-silenced chimeric mice exhibit reduced cytokine production and Th cell differentiation. The abovementioned results implicated USP9X as an important positive modulator of the TCR pathway.134 Emerging data have demonstrated high USP9X expression in oral squamous cell carcinoma (OSCC) tissues. Moreover, USP9X suppresses cell proliferation via the deubiquitination-induced stabilization of PD-L1 and the subsequent accumulation of PD-L1 protein in OSCC cells.115 Therefore, targeting the PD-1/PD-L1 pathway via the inhibition of USP9X activity might be a promising anticancer therapeutic strategy. Another report showed that USP9X-insufficient T cells are hypoproliferative and could induce spontaneous lupus-like autoimmunity as well as lymphoproliferative lesions. However, USP9X knockdown cells remarkably enhance the expression of PD-1 mRNA. Consistently, mice with USP9X depletion have increasing numbers of PD-1+/high memory cells and lymph nodes.23 Therefore, further research is needed to elaborate on the physiological effect of USP9X in the regulation of the PD-1/PD-L1 pathway in carcinomas.
Targeting E3 and DUBs to Enhance PD-1/PD-L1 Therapy
Given the functions of ubiquitination and deubiquitination in the modulation of PD-1/PD-L1 expression and activity, the anticancer potential of targeting ubiquitin or DUBs implicated in PD-1/PD-L1 immunotherapy is being exploited. Emerging data suggest that suppression of CSN5 by curcumin reduces the PD-L1 level in cancer cells and improves the therapeutic efficacy of anti-CTLA4 therapy. In addition to the inhibition of ubiquitination, CSN5 could further directly induce the deubiquitination of PD-L1.20 Therefore, targeting PD-L1 stabilization through NF-κB/CSN5 inhibition is a potential strategy to treat cancer-related inflammation. Similarly, compound-15 (C-15) inhibits CSN5 and can bind to the active site of the enzyme, which leads to an interaction between the indazole moiety and the catalytic center of CSN5. Liu et al.55 found that C-15 obviously destabilizes PD-L1, which allows the cell to escape the immune response by inhibiting Cul1 deneddylation and CSN5 activity. Another study demonstrated that 2,5-dimethylcelecoxib (DMC), a targeted inhibitor of mPGES-1, could enhance ubiquitin-mediated degradation of HBx-induced PD-L1 protein in HCCs.135 Further evidence demonstrates that this process is mainly regulated by E3 ligase RBX1.135
Moreover, the small molecule WP1130, a selective deubiquitinase suppressor, attenuates the deubiquitinating potential of several DUBs, including USP5, USP9X, USP14, and USP37. WP1130 has been demonstrated to effectively inhibit cancers by triggering aggresome formation and promoting cancer cell apoptosis and chemosensitivity.136,137 An earlier study reported that WP1130 significantly decreases PD-L1 expression in OSCC cells through inhibition of the deubiquitination of PD-L1 by USP9X.115 These reports indicate that ubiquitin and DUBs, such as CNS5 and USP9X, might be attractive targets for the development of PD-1/PD-L1 blockade therapies. In addition, combination treatment with CDK4/6 inhibitors and PD-1/PD-L1 immune checkpoint blockade (ICB) leads to promoting therapeutic efficacy for cancers.63 USP22 was also identified as a potential target to improve the efficacy of cancer treatments based on ICB therapies.126,138 On-depth investigation is necessary to explore the role of targeting E3 ligases and DUBs as resistance mechanisms or synergistic effects to ICB therapies.
Conclusions
In conclusion, the ubiquitination- or deubiquitination-mediated regulation of the PD-1/PD-L1 pathway plays an important role in human cancer immunotherapy (Figure 2). Emerging evidence demonstrates that various agents targeting related E3 ligases could modulate PD-1/PD-L1 ubiquitination, while molecules targeting DUBs regulate the deubiquitination of PD-1/PD-L1; this in turn leads to the subsequent regulation of PD-1/PD-L1 activity and the modulation of immunosuppression and anticancer effects (Table 1). Therefore, this E3 ligase- or DUB-mediated modification of PD-1/PD-L1 provides a new concept for immunotherapy, especially in some PD-1/PD-L1-positive tumors.
Figure 2.

Ubiquitination-Mediated Regulation of PD-L1 Signaling Is Presented
The several E3 ligases, including SPOP, HRD1, Cbl-b, c-Cbl, STUB1, DCUND1, and β-TrCP, participate in PD-L1 ubiquitination. USP22, USP9X, and CSN5 are involved in PD-L1 deubiquitination in carcinoma.
Table 1.
Role of Ubiquitination and Deubiquitination in Regulation of PD-1/PD-L1 for Cancer Development and Immunotherapy
| Regulators | PTM | Mechanism | Regulation of PD-1/PD-L1 | Types of Cancer | Function in Immune or Tumor Cells | Related Molecules | References |
|---|---|---|---|---|---|---|---|
| β-TrCP | ubiquitination | regulates ubiquitination and degradation of PD-L1 | inhibits PD-L1 level | breast cancer | modulates immune escape | GSK3β, mTORC1/p70S6K | 40 |
| CSN5 | ubiquitination | suppresses the ubiquitination and degradation of PD-L1 | increases PD-L1 level | breast cancer | results in immune system evasion | TNF-α, NF-κB p65 | 20 |
| deubiquitination | promotes deubiquitination and stabilization of PD-L1 | upregulates PD-L1 expression | colorectal cancer | promotes cancer progression and migration | IκBa, Snail, CCL5, p65/STAT3 | 20,55 | |
| SPOP | ubiquitination | enhances ubiquitination-mediated degradation of PD-L1 | decreases PD-L1 level | prostate cancer | increases TILs in cancer site, inhibits tumor extinction | Cullin 3, cyclin D-CDK4 | 63 |
| STUB1 | ubiquitination | induces ubiquitination and destabilization of PD-L1 | downregulates level of PD-L1 | – | negatively modulates immune activity | – | 21 |
| FBXO38 | ubiquitination | enhances ubiquitination-mediated proteasomal degradation of PD-1 | decreases PD-1 expression | melanoma | enhances T cell antitumor activity and tumor regression | – | 22 |
| DCUN1D | ubiquitination | unknown | increases PD-L1 level | lung cancer | promotes the development of cancer | FAK pathway | 81 |
| Cbl-b/c-Cbl | ubiquitination | accelerates ubiquitination of STAT5a | decreases PD-L1 level | melanoma, gastric cancer, NSCLC | promotes immunosuppression | STAT5a, AKT, and ERK signaling pathways | 31,94,95 |
| c-Cbl | ubiquitination | enhances ubiquitination-mediated degradation of PD-1 | suppresses PD-1 activity | CRC | inhibits cancer development and immune infiltrates | – | 96 |
| HRD1 | ubiquitination | modulates ubiquitination of PD-L1 | downregulates PD-L1 level | – | improves the efficacy of immunosuppressive therapy | ERAD | 105 |
| USP22 | deubiquitination | promotes deubiquitination and stability of PD-L1 | increases expression of PD-L1 | H22 tumor, lung cancer | promotes tumor growth and inhibits immune elimination | CSN5 | 107,139 |
| USP9X | deubiquitination | modulates deubiquitination-induced stabilization of PD-L1 | increases PD-L1, inhibits PD-1 level | OSCC | suppresses cancer cell proliferation | – | 23,115 |
However, some important problems should be further investigated. For example, PD-1/PD-L1 has been validated to have multiple PTMs, including glycosylation, phosphorylation, ubiquitination, palmitoylation, SUMOylation, and acetylation.15 Is PD-1/PD-L1 ubiquitination more important than other PTMs to regulate the expression of PD-1/PD-L1? Are any of the other 600 E3 ligases involved in PD-1/PD-L1 regulation? In addition to USP22 and USP9X, do other DUBs govern the deubiquitination of PD-1/PD-L1 in cancer cells? Moreover, it is essential to develop suitable approaches for discovering new drugs that target these E3 ligases to inhibit PD-1/PD-1 activity and alleviate the inhibition of tumor-specific T cell activity. Additionally, more conventionally engineered mouse models in which E3 ligases or DUBs are targeted are needed to more extensively explore the roles and detailed mechanisms of ubiquitination and deubiquitination of the PD-1/PD-L1 pathway in cancer immunosuppression. We think that these explorations will provide new insight into the design of rational therapeutic strategies by targeting related E3 ligases and DUBs to regulate the PD-1/PD-L1 pathway in cancer immunotherapy.
Acknowledgments
This work was supported by grant from the Science and Technology Planning Project of Wenzhou City (no. Y20180082) and by the Research Fund for Lin He’s Academician Workstation of New Medicine and Clinical Translation.
Author Contributions
X.H., J.W., M.C., and Y.L. searched literature regarding to PD-1, PD-L1, and ubiquitination. X.H. and J.W. made the figures. X.H., X.Z., and Z.W. wrote the manuscript. All authors read and approved the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Contributor Information
Zhi-wei Wang, Email: zhiweichina@126.com.
Xueqiong Zhu, Email: zjwzzxq@163.com.
References
- 1.Gonzalez H., Hagerling C., Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32:1267–1284. doi: 10.1101/gad.314617.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grivennikov S.I., Greten F.R., Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 4.Gajewski T.F., Schreiber H., Fu Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishida Y., Agata Y., Shibahara K., Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akbay E.A., Koyama S., Carretero J., Altabef A., Tchaicha J.H., Christensen C.L., Mikse O.R., Cherniack A.D., Beauchamp E.M., Pugh T.J. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fife B.T., Bluestone J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008;224:166–182. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 8.Keir M.E., Butte M.J., Freeman G.J., Sharpe A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seo A.N., Kang B.W., Kwon O.K., Park K.B., Lee S.S., Chung H.Y., Yu W., Bae H.I., Jeon S.W., Kang H., Kim J.G. Intratumoural PD-L1 expression is associated with worse survival of patients with Epstein-Barr virus-associated gastric cancer. Br. J. Cancer. 2017;117:1753–1760. doi: 10.1038/bjc.2017.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X., Teng F., Kong L., Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. OncoTargets Ther. 2016;9:5023–5039. doi: 10.2147/OTT.S105862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohaegbulam K.C., Assal A., Lazar-Molnar E., Yao Y., Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015;21:24–33. doi: 10.1016/j.molmed.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topalian S.L., Hodi F.S., Brahmer J.R., Gettinger S.N., Smith D.C., McDermott D.F., Powderly J.D., Carvajal R.D., Sosman J.A., Atkins M.B. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brahmer J.R., Tykodi S.S., Chow L.Q., Hwu W.J., Topalian S.L., Hwu P., Drake C.G., Camacho L.H., Kauh J., Odunsi K. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun C., Mezzadra R., Schumacher T.N. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu J.M., Li C.W., Lai Y.J., Hung M.C. Posttranslational modifications of PD-L1 and their applications in cancer therapy. Cancer Res. 2018;78:6349–6353. doi: 10.1158/0008-5472.CAN-18-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng F., Su Y., Xu B. Rho-associated protein kinase-dependent moesin phosphorylation is required for PD-L1 stabilization in breast cancer. Mol. Oncol. 2020;14:2701–2712. doi: 10.1002/1878-0261.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailly C., Vergoten G. N-glycosylation and ubiquitinylation of PD-L1 do not restrict interaction with BMS-202: molecular modeling study. Comput. Biol. Chem. 2020;88:107362. doi: 10.1016/j.compbiolchem.2020.107362. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y.N., Lee H.H., Hsu J.L., Yu D., Hung M.C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J. Biomed. Sci. 2020;27:77. doi: 10.1186/s12929-020-00670-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao Y., Nihira N.T., Bu X., Chu C., Zhang J., Kolodziejczyk A., Fan Y., Chan N.T., Ma L., Liu J. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell Biol. 2020;22:1064–1075. doi: 10.1038/s41556-020-0562-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim S.O., Li C.W., Xia W., Cha J.H., Chan L.C., Wu Y., Chang S.S., Lin W.C., Hsu J.M., Hsu Y.H. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–939. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mezzadra R., Sun C., Jae L.T., Gomez-Eerland R., de Vries E., Wu W., Logtenberg M.E.W., Slagter M., Rozeman E.A., Hofland I. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. 2017;549:106–110. doi: 10.1038/nature23669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng X., Liu X., Guo X., Jiang S., Chen T., Hu Z., Liu H., Bai Y., Xue M., Hu R. FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature. 2018;564:130–135. doi: 10.1038/s41586-018-0756-0. [DOI] [PubMed] [Google Scholar]
- 23.Naik E., Webster J.D., DeVoss J., Liu J., Suriben R., Dixit V.M. Regulation of proximal T cell receptor signaling and tolerance induction by deubiquitinase Usp9X. J. Exp. Med. 2014;211:1947–1955. doi: 10.1084/jem.20140860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai J., Culley M.K., Zhao Y., Zhao J. The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell. 2018;9:754–769. doi: 10.1007/s13238-017-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pohl C., Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818–822. doi: 10.1126/science.aax3769. [DOI] [PubMed] [Google Scholar]
- 26.Puvar K., Luo Z.Q., Das C. Uncovering the structural basis of a new twist in protein ubiquitination. Trends Biochem. Sci. 2019;44:467–477. doi: 10.1016/j.tibs.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciechanover A. The unravelling of the ubiquitin system. Nat. Rev. Mol. Cell Biol. 2015;16:322–324. doi: 10.1038/nrm3982. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y.C. Ubiquitin ligases and the immune response. Annu. Rev. Immunol. 2004;22:81–127. doi: 10.1146/annurev.immunol.22.012703.104813. [DOI] [PubMed] [Google Scholar]
- 29.Peer S., Baier G., Gruber T. Cblb-deficient T cells are less susceptible to PD-L1-mediated inhibition. Oncotarget. 2017;8:41841–41853. doi: 10.18632/oncotarget.18360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lohr N.J., Molleston J.P., Strauss K.A., Torres-Martinez W., Sherman E.A., Squires R.H., Rider N.L., Chikwava K.R., Cummings O.W., Morton D.H., Puffenberger E.G. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am. J. Hum. Genet. 2010;86:447–453. doi: 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujiwara M., Anstadt E.J., Clark R.B. Cbl-b deficiency mediates resistance to programmed death-ligand 1/programmed death-1 regulation. Front. Immunol. 2017;8:42. doi: 10.3389/fimmu.2017.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burr M.L., Sparbier C.E., Chan Y.C., Williamson J.C., Woods K., Beavis P.A., Lam E.Y.N., Henderson M.A., Bell C.C., Stolzenburg S. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaik S., Nucera C., Inuzuka H., Gao D., Garnaas M., Frechette G., Harris L., Wan L., Fukushima H., Husain A. SCFβ-TRCP suppresses angiogenesis and thyroid cancer cell migration by promoting ubiquitination and destruction of VEGF receptor 2. J. Exp. Med. 2012;209:1289–1307. doi: 10.1084/jem.20112446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deshaies R.J. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 35.Kanarek N., Ben-Neriah Y. Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol. Rev. 2012;246:77–94. doi: 10.1111/j.1600-065X.2012.01098.x. [DOI] [PubMed] [Google Scholar]
- 36.Wu D., Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 2010;35:161–168. doi: 10.1016/j.tibs.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wakatsuki S., Saitoh F., Araki T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat. Cell Biol. 2011;13:1415–1423. doi: 10.1038/ncb2373. [DOI] [PubMed] [Google Scholar]
- 38.Wakatsuki S., Tokunaga S., Shibata M., Araki T. GSK3B-mediated phosphorylation of MCL1 regulates axonal autophagy to promote Wallerian degeneration. J. Cell Biol. 2017;216:477–493. doi: 10.1083/jcb.201606020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hart M., Concordet J.P., Lassot I., Albert I., del los Santos R., Durand H., Perret C., Rubinfeld B., Margottin F., Benarous R., Polakis P. The F-box protein β-TrCP associates with phosphorylated β-catenin and regulates its activity in the cell. Curr. Biol. 1999;9:207–210. doi: 10.1016/s0960-9822(99)80091-8. [DOI] [PubMed] [Google Scholar]
- 40.Li C.W., Lim S.O., Xia W., Lee H.H., Chan L.C., Kuo C.W., Khoo K.H., Chang S.S., Cha J.H., Kim T. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H., Li C.W., Li X., Ding Q., Guo L., Liu S., Liu C., Lai C.C., Hsu J.M., Dong Q. MET inhibitors promote liver tumor evasion of the immune response by stabilizing PDL1. Gastroenterology. 2019;156:1849–1861.e13. doi: 10.1053/j.gastro.2019.01.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiao S., Xia W., Yamaguchi H., Wei Y., Chen M.K., Hsu J.M., Hsu J.L., Yu W.H., Du Y., Lee H.H., Li C.W. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017;23:3711–3720. doi: 10.1158/1078-0432.CCR-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng L., Qian G., Zhang S., Zheng H., Fan S., Lesinski G.B., Owonikoko T.K., Ramalingam S.S., Sun S.Y. Inhibition of mTOR complex 1/p70 S6 kinase signaling elevates PD-L1 levels in human cancer cells through enhancing protein stabilization accompanied with enhanced β-TrCP degradation. Oncogene. 2019;38:6270–6282. doi: 10.1038/s41388-019-0877-4. [DOI] [PubMed] [Google Scholar]
- 44.Cavadini S., Fischer E.S., Bunker R.D., Potenza A., Lingaraju G.M., Goldie K.N., Mohamed W.I., Faty M., Petzold G., Beckwith R.E. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature. 2016;531:598–603. doi: 10.1038/nature17416. [DOI] [PubMed] [Google Scholar]
- 45.Echalier A., Pan Y., Birol M., Tavernier N., Pintard L., Hoh F., Ebel C., Galophe N., Claret F.X., Dumas C. Insights into the regulation of the human COP9 signalosome catalytic subunit, CSN5/Jab1. Proc. Natl. Acad. Sci. USA. 2013;110:1273–1278. doi: 10.1073/pnas.1209345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cope G.A., Suh G.S., Aravind L., Schwarz S.E., Zipursky S.L., Koonin E.V., Deshaies R.J. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science. 2002;298:608–611. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- 47.Shackleford T.J., Claret F.X. JAB1/CSN5: a new player in cell cycle control and cancer. Cell Div. 2010;5:26. doi: 10.1186/1747-1028-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adler A.S., Littlepage L.E., Lin M., Kawahara T.L., Wong D.J., Werb Z., Chang H.Y. CSN5 isopeptidase activity links COP9 signalosome activation to breast cancer progression. Cancer Res. 2008;68:506–515. doi: 10.1158/0008-5472.CAN-07-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng Z., Pardi R., Cheadle W., Xiang X., Zhang S., Shah S.V., Grizzle W., Miller D., Mountz J., Zhang H.G. Plant homologue constitutive photomorphogenesis 9 (COP9) signalosome subunit CSN5 regulates innate immune responses in macrophages. Blood. 2011;117:4796–4804. doi: 10.1182/blood-2010-10-314526. [DOI] [PubMed] [Google Scholar]
- 50.Liu Y., Shah S.V., Xiang X., Wang J., Deng Z.B., Liu C., Zhang L., Wu J., Edmonds T., Jambor C. COP9-associated CSN5 regulates exosomal protein deubiquitination and sorting. Am. J. Pathol. 2009;174:1415–1425. doi: 10.2353/ajpath.2009.080861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Y., Deng J., Rychahou P.G., Qiu S., Evers B.M., Zhou B.P. Stabilization of snail by NF-κB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X., Liu R., Su X., Pan Y., Han X., Shao C., Shi Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer. 2019;18:177. doi: 10.1186/s12943-019-1102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai J., Qi Q., Qian X., Han J., Zhu X., Zhang Q., Xia R. The role of PD-1/PD-L1 axis and macrophage in the progression and treatment of cancer. J. Cancer Res. Clin. Oncol. 2019;145:1377–1385. doi: 10.1007/s00432-019-02879-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zlotnik A., Yoshie O., Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7:243. doi: 10.1186/gb-2006-7-12-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu C., Yao Z., Wang J., Zhang W., Yang Y., Zhang Y., Qu X., Zhu Y., Zou J., Peng S. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ. 2020;27:1765–1781. doi: 10.1038/s41418-019-0460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhuang M., Calabrese M.F., Liu J., Waddell M.B., Nourse A., Hammel M., Miller D.J., Walden H., Duda D.M., Seyedin S.N. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell. 2009;36:39–50. doi: 10.1016/j.molcel.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deshaies R.J., Joazeiro C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 58.Dubiel W., Dubiel D., Wolf D.A., Naumann M. Cullin 3-based ubiquitin ligases as master regulators of mammalian cell differentiation. Trends Biochem. Sci. 2018;43:95–107. doi: 10.1016/j.tibs.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Genschik P., Sumara I., Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J. 2013;32:2307–2320. doi: 10.1038/emboj.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song Y., Xu Y., Pan C., Yan L., Wang Z.W., Zhu X. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol. Cancer. 2020;19:2. doi: 10.1186/s12943-019-1124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guiley K.Z., Stevenson J.W., Lou K., Barkovich K.J., Kumarasamy V., Wijeratne T.U., Bunch K.L., Tripathi S., Knudsen E.S., Witkiewicz A.K. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science. 2019;366:eaaw2106. doi: 10.1126/science.aaw2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Knudsen E.S., Witkiewicz A.K. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer. 2017;3:39–55. doi: 10.1016/j.trecan.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J., Bu X., Wang H., Zhu Y., Geng Y., Nihira N.T., Tan Y., Ci Y., Wu F., Dai X. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–95. doi: 10.1038/nature25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang J., Ballinger C.A., Wu Y., Dai Q., Cyr D.M., Höhfeld J., Patterson C. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 2001;276:42938–42944. doi: 10.1074/jbc.M101968200. [DOI] [PubMed] [Google Scholar]
- 65.Wang Y., Ren F., Wang Y., Feng Y., Wang D., Jia B., Qiu Y., Wang S., Yu J., Sung J.J. CHIP/Stub1 functions as a tumor suppressor and represses NF-κB-mediated signaling in colorectal cancer. Carcinogenesis. 2014;35:983–991. doi: 10.1093/carcin/bgt393. [DOI] [PubMed] [Google Scholar]
- 66.Tang D.E., Dai Y., Lin L.W., Xu Y., Liu D.Z., Hong X.P., Jiang H.W., Xu S.H. STUB1 suppresseses tumorigenesis and chemoresistance through antagonizing YAP1 signaling. Cancer Sci. 2019;110:3145–3156. doi: 10.1111/cas.14166. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Luan H., Mohapatra B., Bielecki T.A., Mushtaq I., Mirza S., Jennings T.A., Clubb R.J., An W., Ahmed D., El-Ansari R. Loss of the nuclear pool of ubiquitin ligase CHIP/STUB1 in breast cancer unleashes the MZF1-cathepsin pro-oncogenic program. Cancer Res. 2018;78:2524–2535. doi: 10.1158/0008-5472.CAN-16-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Z., Barbi J., Bu S., Yang H.Y., Li Z., Gao Y., Jinasena D., Fu J., Lin F., Chen C. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity. 2013;39:272–285. doi: 10.1016/j.immuni.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guan X., Zhang C., Zhao J., Sun G., Song Q., Jia W. CMTM6 overexpression is associated with molecular and clinical characteristics of malignancy and predicts poor prognosis in gliomas. EBioMedicine. 2018;35:233–243. doi: 10.1016/j.ebiom.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zugazagoitia J., Liu Y., Toki M., McGuire J., Ahmed F.S., Henick B.S., Gupta R., Gettinger S.N., Herbst R.S., Schalper K.A. Quantitative assessment of CMTM6 in the tumor microenvironment and association with response to PD-1 pathway blockade in advanced-stage non-small cell lung cancer. J. Thorac. Oncol. 2019 doi: 10.1016/j.jtho.2019.09.014. 14, 2084–2096.other. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen L., Yang Q.C., Li Y.C., Yang L.L., Liu J.F., Li H., Xiao Y., Bu L.L., Zhang W.F., Sun Z.J. Targeting CMTM6 suppresses stem cell-like properties and enhances antitumor immunity in head and neck squamous cell carcinoma. Cancer Immunol. Res. 2020;8:179–191. doi: 10.1158/2326-6066.CIR-19-0394. [DOI] [PubMed] [Google Scholar]
- 72.Koh Y.W., Han J.H., Haam S., Jung J., Lee H.W. Increased CMTM6 can predict the clinical response to PD-1 inhibitors in non-small cell lung cancer patients. OncoImmunology. 2019;8:e1629261. doi: 10.1080/2162402X.2019.1629261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin J., Cardozo T., Lovering R.C., Elledge S.J., Pagano M., Harper J.W. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–2580. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Skowyra D., Craig K.L., Tyers M., Elledge S.J., Harper J.W. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 75.Huang G., Kaufman A.J., Xu K., Manova K., Singh B. Squamous cell carcinoma-related oncogene (SCCRO) neddylates Cul3 protein to selectively promote midbody localization and activity of Cul3KLHL21 protein complex during abscission. J. Biol. Chem. 2017;292:15254–15265. doi: 10.1074/jbc.M117.778530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sarkaria I., O-charoenrat P., Talbot S.G., Reddy P.G., Ngai I., Maghami E., Patel K.N., Lee B., Yonekawa Y., Dudas M. Squamous cell carcinoma related oncogene/DCUN1D1 is highly conserved and activated by amplification in squamous cell carcinomas. Cancer Res. 2006;66:9437–9444. doi: 10.1158/0008-5472.CAN-06-2074. [DOI] [PubMed] [Google Scholar]
- 77.Broderick S.R., Golas B.J., Pham D., Towe C.W., Talbot S.G., Kaufman A., Bains S., Huryn L.A., Yonekawa Y., Carlson D. SCCRO promotes glioma formation and malignant progression in mice. Neoplasia. 2010;12:476–484. doi: 10.1593/neo.10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Z.H., Li J., Luo F., Wang Y.S. Clinical significance of SCCRO (DCUN1D1) in prostate cancer and its proliferation-inhibiting effect on Lncap cells. Eur. Rev. Med. Pharmacol. Sci. 2017;21:4283–4291. [PubMed] [Google Scholar]
- 79.Xiao J., Li G., Zhou J., Wang S., Liu D., Shu G., Zhou J., Ren F. MicroRNA-520b functions as a tumor suppressor in colorectal cancer by inhibiting defective in cullin neddylation 1 domain containing 1 (DCUN1D1) Oncol. Res. 2018;26:593–604. doi: 10.3727/096504017X14920318811712. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80.Jiang Z., Song Q., Zeng R., Li J., Li J., Lin X., Chen X., Zhang J., Zheng Y. MicroRNA-218 inhibits EMT, migration and invasion by targeting SFMBT1 and DCUN1D1 in cervical cancer. Oncotarget. 2016;7:45622–45636. doi: 10.18632/oncotarget.9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li J., Yu T., Yan M., Zhang X., Liao L., Zhu M., Lin H., Pan H., Yao M. DCUN1D1 facilitates tumor metastasis by activating FAK signaling and up-regulates PD-L1 in non-small-cell lung cancer. Exp. Cell Res. 2019;374:304–314. doi: 10.1016/j.yexcr.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 82.Weissman A.M., Shabek N., Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell Biol. 2011;12:605–620. doi: 10.1038/nrm3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Keane M.M., Rivero-Lezcano O.M., Mitchell J.A., Robbins K.C., Lipkowitz S. Cloning and characterization of cbl-b: a SH3 binding protein with homology to the c-cbl proto-oncogene. Oncogene. 1995;10:2367–2377. [PubMed] [Google Scholar]
- 84.Zhang L., Teng Y., Fan Y., Wang Y., Li W., Shi J., Ma Y., Li C., Shi X., Qu X., Liu Y. The E3 ubiquitin ligase Cbl-b improves the prognosis of RANK positive breast cancer patients by inhibiting RANKL-induced cell migration and metastasis. Oncotarget. 2015;6:22918–22933. doi: 10.18632/oncotarget.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H., Xu L., Li C., Zhao L., Ma Y., Zheng H., Li Z., Zhang Y., Wang R., Liu Y., Qu X. Ubiquitin ligase Cbl-b represses IGF-I-induced epithelial mesenchymal transition via ZEB2 and microRNA-200c regulation in gastric cancer cells. Mol. Cancer. 2014;13:136. doi: 10.1186/1476-4598-13-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Qu X., Zhang Y., Li Y., Hu X., Xu Y., Xu L., Hou K., Sada K., Liu Y. Ubiquitin ligase Cbl-b sensitizes leukemia and gastric cancer cells to anthracyclines by activating the mitochondrial pathway and modulating Akt and ERK survival signals. FEBS Lett. 2009;583:2255–2262. doi: 10.1016/j.febslet.2009.05.054. [DOI] [PubMed] [Google Scholar]
- 87.Lyle C.L., Belghasem M., Chitalia V.C. c-Cbl: an important regulator and a target in angiogenesis and tumorigenesis. Cells. 2019;8:498. doi: 10.3390/cells8050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee G.W., Park J.B., Park S.Y., Seo J., Shin S.H., Park J.W., Kim S.J., Watanabe M., Chun Y.S. The E3 ligase C-CBL inhibits cancer cell migration by neddylating the proto-oncogene c-Src. Oncogene. 2018;37:5552–5568. doi: 10.1038/s41388-018-0354-5. [DOI] [PubMed] [Google Scholar]
- 89.Thien C.B., Langdon W.Y. c-Cbl and Cbl-b ubiquitin ligases: substrate diversity and the negative regulation of signalling responses. Biochem. J. 2005;391:153–166. doi: 10.1042/BJ20050892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Paolino M., Penninger J.M. Cbl-b in T-cell activation. Semin. Immunopathol. 2010;32:137–148. doi: 10.1007/s00281-010-0197-9. [DOI] [PubMed] [Google Scholar]
- 91.Chiang Y.J., Kole H.K., Brown K., Naramura M., Fukuhara S., Hu R.J., Jang I.K., Gutkind J.S., Shevach E., Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 92.Gruber T., Hinterleitner R., Hermann-Kleiter N., Meisel M., Kleiter I., Wang C.M., Viola A., Pfeifhofer-Obermair C., Baier G. Cbl-b mediates TGFβ sensitivity by downregulating inhibitory SMAD7 in primary T cells. J. Mol. Cell Biol. 2013;5:358–368. doi: 10.1093/jmcb/mjt017. [DOI] [PubMed] [Google Scholar]
- 93.Adams C.O., Housley W.J., Bhowmick S., Cone R.E., Rajan T.V., Forouhar F., Clark R.B. Cbl-b−/− T cells demonstrate in vivo resistance to regulatory T cells but a context-dependent resistance to TGF-β. J. Immunol. 2010;185:2051–2058. doi: 10.4049/jimmunol.1001171. [DOI] [PubMed] [Google Scholar]
- 94.Fan Y., Che X., Hou K., Zhang M., Wen T., Qu X., Liu Y. miR-940 promotes the proliferation and migration of gastric cancer cells through up-regulation of programmed death ligand-1 expression. Exp. Cell Res. 2018;373:180–187. doi: 10.1016/j.yexcr.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 95.Wang S., Xu L., Che X., Li C., Xu L., Hou K., Fan Y., Wen T., Qu X., Liu Y. E3 ubiquitin ligases Cbl-b and c-Cbl downregulate PD-L1 in EGFR wild-type non-small cell lung cancer. FEBS Lett. 2018;592:621–630. doi: 10.1002/1873-3468.12985. [DOI] [PubMed] [Google Scholar]
- 96.Lyle C., Richards S., Yasuda K., Napoleon M.A., Walker J., Arinze N., Belghasem M., Vellard I., Yin W., Ravid J.D. c-Cbl targets PD-1 in immune cells for proteasomal degradation and modulates colorectal tumor growth. Sci. Rep. 2019;9:20257. doi: 10.1038/s41598-019-56208-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Karwacz K., Bricogne C., MacDonald D., Arce F., Bennett C.L., Collins M., Escors D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol. Med. 2011;3:581–592. doi: 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hampton R.Y., Rine J. Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J. Cell Biol. 1994;125:299–312. doi: 10.1083/jcb.125.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xu Y., Fang D. Endoplasmic reticulum-associated degradation and beyond: the multitasking roles for HRD1 in immune regulation and autoimmunity. J. Autoimmun. 2020;109:102423. doi: 10.1016/j.jaut.2020.102423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Baldridge R.D., Rapoport T.A. Autoubiquitination of the Hrd1 ligase triggers protein retrotranslocation in ERAD. Cell. 2016;166:394–407. doi: 10.1016/j.cell.2016.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu L., Yu L., Zeng C., Long H., Duan G., Yin G., Dai X., Lin Z. The E3 ubiquitin ligase HRD1 promotes lung tumorigenesis by promoting sirtuin 2 ubiquitination and degradation. Mol. Cell. Biol. 2020;40:e00257-19. doi: 10.1128/MCB.00257-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu L., Long H., Wu Y., Li H., Dong L., Zhong J.L., Liu Z., Yang X., Dai X., Shi L. HRD1-mediated PTEN degradation promotes cell proliferation and hepatocellular carcinoma progression. Cell. Signal. 2018;50:90–99. doi: 10.1016/j.cellsig.2018.06.011. [DOI] [PubMed] [Google Scholar]
- 103.Xu Y., Zhao F., Qiu Q., Chen K., Wei J., Kong Q., Gao B., Melo-Cardenas J., Zhang B., Zhang J. The ER membrane-anchored ubiquitin ligase Hrd1 is a positive regulator of T-cell immunity. Nat. Commun. 2016;7:12073. doi: 10.1038/ncomms12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kong S., Yang Y., Xu Y., Wang Y., Zhang Y., Melo-Cardenas J., Xu X., Gao B., Thorp E.B., Zhang D.D. Endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls B-cell immunity through degradation of the death receptor CD95/Fas. Proc. Natl. Acad. Sci. USA. 2016;113:10394–10399. doi: 10.1073/pnas.1606742113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cha J.H., Yang W.H., Xia W., Wei Y., Chan L.C., Lim S.O., Li C.W., Kim T., Chang S.S., Lee H.H. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol. Cell. 2018;71:606–620.e7. doi: 10.1016/j.molcel.2018.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Altun M., Kramer H.B., Willems L.I., McDermott J.L., Leach C.A., Goldenberg S.J., Kumar K.G., Konietzny R., Fischer R., Kogan E. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011;18:1401–1412. doi: 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 107.Huang X., Zhang Q., Lou Y., Wang J., Zhao X., Wang L., Zhang X., Li S., Zhao Y., Chen Q. USP22 deubiquitinates CD274 to suppress anticancer immunity. Cancer Immunol. Res. 2019;7:1580–1590. doi: 10.1158/2326-6066.CIR-18-0910. [DOI] [PubMed] [Google Scholar]
- 108.Galves M., Rathi R., Prag G., Ashkenazi A. Ubiquitin signaling and degradation of aggregate-prone proteins. Trends Biochem. Sci. 2019;44:872–884. doi: 10.1016/j.tibs.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 109.Harrigan J.A., Jacq X., Martin N.M., Jackson S.P. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat. Rev. Drug Discov. 2018;17:57–78. doi: 10.1038/nrd.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clague M.J., Urbé S., Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019;20:338–352. doi: 10.1038/s41580-019-0099-1. [DOI] [PubMed] [Google Scholar]
- 111.Hou P., Ma X., Zhang Q., Wu C.J., Liao W., Li J., Wang H., Zhao J., Zhou X., Guan C. USP21 deubiquitinase promotes pancreas cancer cell stemness via Wnt pathway activation. Genes Dev. 2019;33:1361–1366. doi: 10.1101/gad.326314.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.van Loosdregt J., Fleskens V., Fu J., Brenkman A.B., Bekker C.P., Pals C.E., Meerding J., Berkers C.R., Barbi J., Gröne A. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. 2013;39:259–271. doi: 10.1016/j.immuni.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li Y., Lu Y., Wang S., Han Z., Zhu F., Ni Y., Liang R., Zhang Y., Leng Q., Wei G. USP21 prevents the generation of T-helper-1-like Treg cells. Nat. Commun. 2016;7:13559. doi: 10.1038/ncomms13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dai X., Lu L., Deng S., Meng J., Wan C., Huang J., Sun Y., Hu Y., Wu B., Wu G. USP7 targeting modulates anti-tumor immune response by reprogramming tumor-associated macrophages in lung cancer. Theranostics. 2020;10:9332–9347. doi: 10.7150/thno.47137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jingjing W., Wenzheng G., Donghua W., Guangyu H., Aiping Z., Wenjuan W. Deubiquitination and stabilization of programmed cell death ligand 1 by ubiquitin-specific peptidase 9, X-linked in oral squamous cell carcinoma. Cancer Med. 2018;7:4004–4011. doi: 10.1002/cam4.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jeusset L.M., McManus K.J. Ubiquitin specific peptidase 22 regulates histone H2B mono-ubiquitination and exhibits both oncogenic and tumor suppressor roles in cancer. Cancers (Basel) 2017;9:167. doi: 10.3390/cancers9120167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Atanassov B.S., Dent S.Y. USP22 regulates cell proliferation by deubiquitinating the transcriptional regulator FBP1. EMBO Rep. 2011;12:924–930. doi: 10.1038/embor.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang X.Y., Varthi M., Sykes S.M., Phillips C., Warzecha C., Zhu W., Wyce A., Thorne A.W., Berger S.L., McMahon S.B. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell. 2008;29:102–111. doi: 10.1016/j.molcel.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liang J.X., Ning Z., Gao W., Ling J., Wang A.M., Luo H.F., Liang Y., Yan Q., Wang Z.Y. Ubiquitin-specific protease 22-induced autophagy is correlated with poor prognosis of pancreatic cancer. Oncol. Rep. 2014;32:2726–2734. doi: 10.3892/or.2014.3508. [DOI] [PubMed] [Google Scholar]
- 120.Ning Z., Wang A., Liang J., Xie Y., Liu J., Feng L., Yan Q., Wang Z. USP22 promotes the G1/S phase transition by upregulating FoxM1 expression via β-catenin nuclear localization and is associated with poor prognosis in stage II pancreatic ductal adenocarcinoma. Int. J. Oncol. 2014;45:1594–1608. doi: 10.3892/ijo.2014.2531. [DOI] [PubMed] [Google Scholar]
- 121.Melo-Cardenas J., Xu Y., Wei J., Tan C., Kong S., Gao B., Montauti E., Kirsammer G., Licht J.D., Yu J. USP22 deficiency leads to myeloid leukemia upon oncogenic Kras activation through a PU.1-dependent mechanism. Blood. 2018;132:423–434. doi: 10.1182/blood-2017-10-811760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lin Z., Yang H., Kong Q., Li J., Lee S.M., Gao B., Dong H., Wei J., Song J., Zhang D.D., Fang D. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell. 2012;46:484–494. doi: 10.1016/j.molcel.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 123.Zhang H., Han B., Lu H., Zhao Y., Chen X., Meng Q., Cao M., Cai L., Hu J. USP22 promotes resistance to EGFR-TKIs by preventing ubiquitination-mediated EGFR degradation in EGFR-mutant lung adenocarcinoma. Cancer Lett. 2018;433:186–198. doi: 10.1016/j.canlet.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 124.Li J., Yuan S., Norgard R.J., Yan F., Yamazoe T., Blanco A., Stanger B.Z. Tumor cell-intrinsic USP22 suppresses antitumor immunity in pancreatic cancer. Cancer Immunol. Res. 2020:282–291. doi: 10.1158/2326-6066.CIR-19-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li L., Osdal T., Ho Y., Chun S., McDonald T., Agarwal P., Lin A., Chu S., Qi J., Li L. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell. 2014;15:431–446. doi: 10.1016/j.stem.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Y., Sun Q., Mu N., Sun X., Wang Y., Fan S., Su L., Liu X. The deubiquitinase USP22 regulates PD-L1 degradation in human cancer cells. Cell Commun. Signal. 2020;18:112. doi: 10.1186/s12964-020-00612-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wood S.A., Pascoe W.S., Ru K., Yamada T., Hirchenhain J., Kemler R., Mattick J.S. Cloning and expression analysis of a novel mouse gene with sequence similarity to the Drosophila fat facets gene. Mech. Dev. 1997;63:29–38. doi: 10.1016/s0925-4773(97)00672-2. [DOI] [PubMed] [Google Scholar]
- 128.Murtaza M., Jolly L.A., Gecz J., Wood S.A. La FAM fatale: USP9X in development and disease. Cell. Mol. Life Sci. 2015;72:2075–2089. doi: 10.1007/s00018-015-1851-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grou C.P., Francisco T., Rodrigues T.A., Freitas M.O., Pinto M.P., Carvalho A.F., Domingues P., Wood S.A., Rodríguez-Borges J.E., Sá-Miranda C. Identification of ubiquitin-specific protease 9X (USP9X) as a deubiquitinase acting on ubiquitin-peroxin 5 (PEX5) thioester conjugate. J. Biol. Chem. 2012;287:12815–12827. doi: 10.1074/jbc.M112.340158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang X., Zhou J.Y., Chin M.H., Schepmoes A.A., Petyuk V.A., Weitz K.K., Petritis B.O., Monroe M.E., Camp D.G., Wood S.A. Region-specific protein abundance changes in the brain of MPTP-induced Parkinson’s disease mouse model. J. Proteome Res. 2010;9:1496–1509. doi: 10.1021/pr901024z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang S., Kollipara R.K., Srivastava N., Li R., Ravindranathan P., Hernandez E., Freeman E., Humphries C.G., Kapur P., Lotan Y. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc. Natl. Acad. Sci. USA. 2014;111:4251–4256. doi: 10.1073/pnas.1322198111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Khan O.M., Carvalho J., Spencer-Dene B., Mitter R., Frith D., Snijders A.P., Wood S.A., Behrens A. The deubiquitinase USP9X regulates FBW7 stability and suppresses colorectal cancer. J. Clin. Invest. 2018;128:1326–1337. doi: 10.1172/JCI97325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pérez-Mancera P.A., Rust A.G., van der Weyden L., Kristiansen G., Li A., Sarver A.L., Silverstein K.A., Grützmann R., Aust D., Rümmele P., Australian Pancreatic Cancer Genome Initiative The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012;486:266–270. doi: 10.1038/nature11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Park Y., Jin H.S., Liu Y.C. Regulation of T cell function by the ubiquitin-specific protease USP9X via modulating the Carma1-Bcl10-Malt1 complex. Proc. Natl. Acad. Sci. USA. 2013;110:9433–9438. doi: 10.1073/pnas.1221925110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen Z., Chen Y., Peng L., Wang X., Tang N. 2,5-Dimethylcelecoxib improves immune microenvironment of hepatocellular carcinoma by promoting ubiquitination of HBx-induced PD-L1. J. Immunother. Cancer. 2020;8:e001377. doi: 10.1136/jitc-2020-001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim S., Woo S.M., Min K.J., Seo S.U., Lee T.J., Kubatka P., Kim D.E., Kwon T.K. WP1130 enhances TRAIL-induced apoptosis through USP9X-dependent miR-708-mediated downregulation of c-FLIP. Cancers (Basel) 2019;11:344. doi: 10.3390/cancers11030344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu H., Chen W., Liang C., Chen B.W., Zhi X., Zhang S., Zheng X., Bai X., Liang T. WP1130 increases doxorubicin sensitivity in hepatocellular carcinoma cells through usp9x-dependent p53 degradation. Cancer Lett. 2015;361:218–225. doi: 10.1016/j.canlet.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 138.Li J., Yuan S., Norgard R.J., Yan F., Yamazoe T., Blanco A., Stanger B.Z. Tumor cell-intrinsic USP22 suppresses antitumor immunity in pancreatic cancer. Cancer Immunol. Res. 2020;8:282–291. doi: 10.1158/2326-6066.CIR-19-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huang X., Zhang X., Bai X., Liang T. Blocking PD-L1 for anti-liver cancer immunity: USP22 represents a critical cotarget. Cell. Mol. Immunol. 2020;17:677–679. doi: 10.1038/s41423-019-0348-4. [DOI] [PMC free article] [PubMed] [Google Scholar]