

Abstract
Historically poor clinical results of tumor vaccines have been attributed to weakly immunogenic antigen targets, limited specificity, and vaccine platforms that fail to induce high-quality polyfunctional T cells, central to mediating cellular immunity. We show here that the combination of antigen selection, construct design, and a robust vaccine platform based on the Synthetically Modified Alpha Replicon RNA Technology (SMARRT), a self-replicating RNA, leads to control of tumor growth in mice. Therapeutic immunization with SMARRT replicon-based vaccines expressing tumor-specific neoantigens or tumor-associated antigen were able to generate polyfunctional CD4+ and CD8+ T cell responses in mice. Additionally, checkpoint inhibitors, or co-administration of cytokine also expressed from the SMARRT platform, synergized to enhance responses further. Lastly, SMARRT-based immunization of non-human primates was able to elicit high-quality T cell responses, demonstrating translatability and clinical feasibility of synthetic replicon technology for therapeutic oncology vaccines.
Keywords: immuno-oncology, vaccine, RNA, replicon, self-replicating, tumor, T cell, neoantigen, synthetic
Graphical Abstract
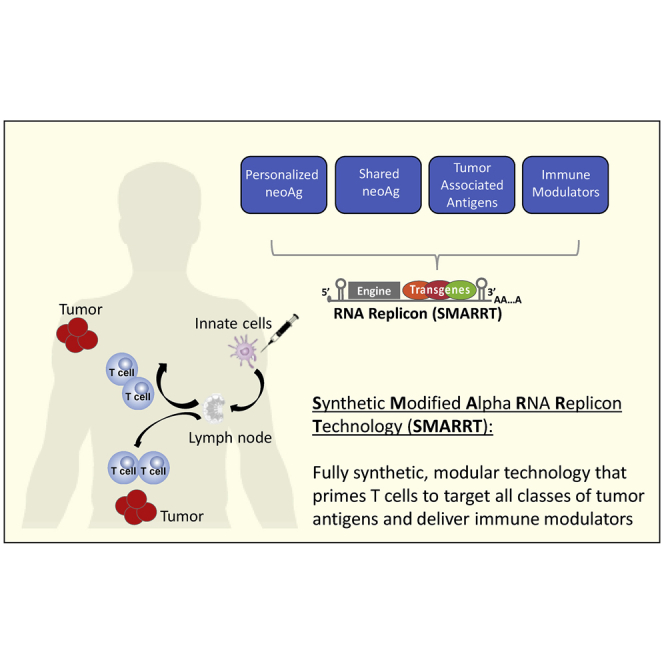
Maine and colleagues demonstrate the use of self-replicating RNA technology, SMARRT, to deliver combinatorial immunotherapies for use in oncology. Vaccination with this platform encoding neoantigens leads to tumor regression, mediated by polyfunctional T cell responses. Additionally, SMAART encoding a surrogate viral antigen demonstrates translatability of the platform to large animals.
Introduction
Despite advances in immunotherapeutic approaches for tumor therapy over the last decade, therapeutic cancer vaccines have demonstrated limited clinical success (reviewed in Hollingsworth and Jansen1). This is due, in part, to targets for tumor vaccination being poorly immunogenic as a result of being self-proteins. However, recent progress in next-generation sequencing technology has allowed us to take advantage of the non-self tumor mutanome as a source of epitopes, which have demonstrated superior immunogenicity compared to tumor-associated antigens.2 Numerous clinical trials are now focusing on neoantigens as vaccine targets allowing the development of both personalized and off-the-shelf vaccines.3 Two major factors contribute to the immunogenicity and subsequent effectiveness of neoantigen-based vaccine approaches. First, the quality of antigen encoded is critical for both inducing the desired phenotype of CD8+ and/or CD4+ T cells, as well as reducing the expansion of inhibitory regulatory T cells (Tregs).4 Second, the antigen must be presented in an inflammatory context to drive a robust, durable, and high-quality T cell response that is more resistant to peripheral tolerance (reviewed in Chen and Mellman5). To date, many class I and class II epitopes identified by prediction algorithms have failed to adequately correlate with immunogenicity. Similarly, traditional platforms such as peptides, viral particles, DNA, and mRNA have all been tested clinically and have disadvantages associated with poor T cell responses or bias toward undesirable T cell responses, and in some cases poor tolerability, capacity limitations of high cost of goods.6
Self-replicating RNAs, also termed replicons, have been used extensively as a vaccine and therapeutic modality for both infectious disease and oncology vaccines (reviewed in Lundstrom7). Replicon technology is commonly derived from alphavirus genomes. Replicons self-amplify once inside the host cell due to replicase machinery made of the viral non-structural gene products nsP1-4. These additional copies allow for higher and long-lasting expression of the gene of interest (i.e., the antigen) than conventional mRNA platforms. However, this amplification process can trigger innate immune pathways that, while desirable to add an adjuvant effect, can also lead to protein translational shutdown, such as RIG-I and PKR, which evolved to control viral spread during normal infection (reviewed in8) Here, we introduce a novel vaccine platform, termed Synthetically Modified Alpha Replicon RNA Technology (SMARRT), that has been engineered to drive more robust protein expression, while allowing for full triggering of innate immune pathways to maximize stimulation of the immune system.
Emerging data suggests that replicons could be an excellent vaccine platform for oncology indications. A single cycle viral replicon particle (VRP) version of the technology has had promising results in human phase I/IIa clinical trials targeting CEA, PSMA, or HER2, and for expression of therapeutic interleukin-12 (IL-12).9, 10, 11, 12 The platform has also shown efficacy across a wide range of tumor-associated antigens (TAAs) in preclinical small animal models.13, 14, 15, 16 Recently, clinical trials using a fully synthetic version of the technology have been used to encode neoantigens in a personalized medicine approach. This heterologous prime-boost vaccine regime relies on a chimp adenovirus prime followed by multiple synthetic replicon-based boosts (Gritstone Oncology, 2020, JP Morgan, conference). To date, no homologous prime-boost approach using replicons has been used clinically. Furthermore, while cytokines have been expressed from VRPs, there is only a single report showing expression from fully synthetic replicons.17 To date, fully synthetic replicons have not been used to express cytokines in combination with neoantigens. The ability to encode multiple molecules useful in combinatorial therapies and induce robust T cell responses, combined with relatively simple manufacturing and low cost of goods, suggests that synthetic replicons are potentially an ideal platform for use in oncology.
Here we show that therapeutic use of SMARRT results in significant induction of antigen specific T cell responses that control tumor growth in mice. For the identification of personalized neoantigens we have employed Ancer, an automated neo-epitope identification and characterization platform that integrates machine learning-based major histocompatibility complex (MHC) class I and MHC class II T cell epitope mapping tools along with specialized homology screenings to identify and remove potentially inhibitory (e.g., Treg) epitopes. To our knowledge, this is the first time that replicons alone can generate polyfunctional CD4 and CD8 T cell responses against a range of tumor antigen targets without requiring viral vector priming. We also show that appropriate design of the cassettes encoding multiple neoantigens is critical for inducing immune responses. Our data strongly support that Ancer-selected neoantigens delivered with SMARRT vaccines can inhibit tumor growth and that this technology can be used to target broadly shared neoantigens in an off-the-shelf approach. We further demonstrate that SMARRT can successfully be used to co-administer cytokines of interest along with antigens to provide an adjuvant effect that induces epitope spreading against weakly immunogenic targets. Finally, we show that SMARRT immunogenicity and safety in mice translates to non-human primates. Taken together, this study highlights the importance of antigen prediction and the context in which they are presented, and how combination therapies can be translated using novel gene delivery platforms.
Results
Ancer Prediction Algorithm Predicts Neoantigens in CT26 Cell Line
CT26 mutanomes and transcriptomes were retrieved from private and public sources.18 The 3,267 and 3,023 variants from the private and public mutanomes, respectively, were screened to extract 1,787 single nucleotide variants shared in both datasets. Of these, 1,002 mutations were contained in genes showing evidence of expression based on transcriptomic data. To further narrow the list of potential candidates, our analysis focused on 378 variants with at least 30X coverage in the tumor DNA. Mutated sequences were subsequently analyzed with the Ancer platform to identify BALB/c MHC class I (H2-Dd and H2-Kd) and MHC class II (I-Ad, I-Ed) restricted neo-epitopes. 135 of the 378 analyzed mutated sequences could be optimized to yield amino acid sequences that contained MHC class I- and MHC class II-restricted neo-epitopes displaying a low degree of self-similarity. These candidates were designed to maximize induction of effector responses while minimizing the risk to induce Tregs or to trigger immune-related adverse events. The 135 candidate sequences were then ranked and the highest ranking neoantigens were selected for in vivo studies (Table S1).
Following prediction and ranking of neoantigens from the CT26 cell line, we designed multiple epitope (polytope) constructs encoding top ranked neoantigens in a string-of-beads format. To exclude the hypothesis that neoantigen insert design has no effect on immunogenicity, we considered numerous factors when designing these cassettes, including the number of neoantigens, order of neoantigens (which can affect junctional immunogenicity, as well as polytope processing by peptidases), and addition of intracellular trafficking sequences to enhance MHC loading (Table 1; Figure 1A; Table S2).
Table 1.
Polytope Construct Design following Ancer Neoantigen Predictions
| Construct | No. of Neoantigens | Neoantigen Set | Junctional Immunogenicity | Trafficking Signal | Comments |
|---|---|---|---|---|---|
| C1 | 20 | top 20 | low | none | |
| C3 | 20 | top 31 | none | none | |
| C4 | 30 | top 50 | low | none | |
| C5 | 40 | top 50 | low | none | |
| C6 | 20 | top 31 | elevated | none | same neoantigens as C3 but ordered differently |
| C7 | 12 | top 20 | none | none | |
| C81 | 10 | top 20 | low | none | first half of C1 |
| C82 | 10 | top 20 | low | none | second half of C1 |
| C9 | 20 | top 20 | low | calreticulin | C1 with added trafficking signal |
| C10 | 20 | top 20 | low | class I secretion + MITD | C1 with added trafficking signal |
| C11 | 20 | top 20 | low | tissue plasminogen activator | C1 with added trafficking signal |
| C121 | 14 | top 20 | low | class I secretion + MITD | best class I neoantigens from the top 20 |
| C122 | 8 | top 20 | low | tissue plasminogen activator | best class II neoantigens from the top 20 |
Top Ancer predicted CT26 neoantigens were concatenated into several polytope designs to test the effect of neoantigen load (C1, C4, C5, C7), neoantigen ordering (C3, C6), splitting neoantigen load across different polytopes (C1, C81, C82, C121, C122), and inclusion of antigen presentation trafficking signals (C1, C9, C10, C11).
Figure 1.

Ancer-Designed Polytope Neoantigen Constructs Expressed in the SMARRT Platform Generate Polyfunctional CD4 and CD8 T Cell Responses upon Vaccination
(A) Schematic showing polytope design considerations following neoantigen identification. SMARRT replicons expressing neoantigen cassettes designed with Ancer were injected into BALB/c mice at a single dose of 10 μg, and spleens were removed 14 days later and restimulated with a peptide pool containing the top 50 Ancer predicted neoantigens from CT26. T cell function was measured by IFN-γ ELISpot (B), and intracellular cytokine staining is shown for selected replicons (C). Graphs show mean with standard deviation, n = 5 mice per group. Statistical testing was carried out with ordinary one-way ANOVA. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
SMARRT-Expressed Neoantigen Polytope Constructs Are Immunogenic In Vivo in Mice
Replicons have been known to trigger host immune responses that can result in protein translational shutdown (reviewed in Fros and Pijlman19). To circumvent this, we have generated a novel replicon platform that includes an RNA motif, placed upstream of the nonstructural proteins, that is naturally occurring in Old World alphaviruses called the downstream loop (DLP) that mediates resistance to host translational shutdown (Figure S1A). To test the efficacy of the DLP motif, we performed head-to-head comparison of SMARRT and a non-engineered replicon platform in vivo. Protein expression from each replicon platform was measured in the presence or absence of innate immune stimulation by pretreating some groups with poly(I:C), shown to induce an antiviral interferon response.20 As expected, pretreatment with poly(I:C) led to a significant decrease in protein expression from the unmodified replicon measuring over 3-fold less by day 3 (Figure S1B). In contrast, mice that were administered a single dose of SMARRT showed no decrease in protein expression. Thus, inclusion of the DLP motif in SMARRT technology allowed for enhanced protein expression, even in the presence of host innate immune responses.
To assess the feasibility of the SMARRT platform as a proof-of-concept cancer vaccine, we designed a polytope cassette encoding neoantigens from the CT26 mouse tumor cell line (Figure 1A; Table S2). The in silico-designed string-of-beads polytope constructs were introduced in the SMARRT platform for in vivo immunogenicity screening, which revealed differential T cell responses from surveyed constructs as measured by cytokine production. Of the 13 permutations of neoantigen cassettes tested, SMARRT encoding C1, C6, C8, C9, and C11 led to high interferon-γ (IFN-γ) responses in vivo after a single dose (Figure 1B). Notably, SMARRT encoding the same set of neoantigens in different orderings (C3 and C6) led to either strong (C6) or undetectable (C3) immune responses, emphasizing that cassette design is absolutely critical for proper antigen processing and presentation (Table 1; Figure 1).
Additional analysis of T cell responses revealed that SMARRT vaccination led to induction of antigen-specific CD4 and CD8 T cells, with a significant frequency of polyfunctional effector responders (Figure 1C). This is a critical feature of this technology, as many vaccine platforms fail to generate robust CD8 T cell responses.21, 22, 23 Thus, we have demonstrated that using the Ancer prediction pipeline and the SMARRT platform, we can successfully elicit T cell responses to neoantigens, inclusive of both CD4 and CD8 T cell subsets.
Prime and Boost Interval Greatly Influences Immunogenicity of SMARRT Vaccination in Mice
We further evaluated the T cell response following immunization of mice with an Ancer-neoantigen-expressing 20 epitope SMARRT replicon. Construct C9 was selected for follow-up studies and will be referred to as SMARRT.Ancer from this point onward. When varying prime/boost interval lengths. IFN-γ responses, as measured by ELISpot, were detectable at day 7 post-prime and increased at day 14. This response further increased with the addition of a boost, reaching significance with a 4-week interval between prime and boost (Figure S2A). A similar trend was observed using intracellular cytokine staining (ICS) to analyze CD8 T cell responses with detectable cytokine production at day 7, increasing at day 14. The addition of a boost dose reached significance at 4-week and 8-week intervals (Figures S2B and S2D). CD4 T cells exhibited altered kinetics compared to CD8, with cytokine production higher at 7 days post-prime compared to 14 days. The addition of a boost dose did not further increase cytokine production compared to day 7 but an 8-week interval significantly increased IFN-γ production compared to 14 days post-prime (Figures S2C and S2D). These data illustrate the importance of dose intervals when vaccinating with SMARRT.Ancer, highlighting the differential kinetics of the CD8 and CD4 T cell responses. Optimizing prime/boost intervals will be crucial when evaluating SMARRT vaccination in a clinical setting.
SMARRT.Ancer-Induced T Cell Responses Demonstrate Distinct Immunodominance to Select Neoantigens in Mice
To investigate immunodominance to polytope constructs expressed from the SMARRT replicon platform, we measured the functional T cell response to each of the 20 neoantigens encoded in SMARRT.Ancer. The production of IFN-γ by splenocytes revealed the dominance of 2 neoantigens across all time points regardless of prime/boost interval (Figures 2A and 2B). Intracellular cytokine staining revealed that the CD8 T cell response is dominated by neoantigen 20 across all time points and intervals analyzed (cytokine responses above mock stimulation displayed only). Furthermore, the CD8 T cell response to neoantigen 20 is polyfunctional as early as 14 days post-prime with significant increases in triple functional (IFN-γ+ tumor necrosis factor alpha [TNF-α+] IL-2+) CD8 T cells when the interval length reaches 4 weeks (Figure 2C). Neoantigen 4 is the dominant CD4 T cell neoantigen throughout all time points analyzed, however sub-dominant neoantigens are measurable throughout (neoantigens 5 and 8; Figure 2B). Neoantigen 4-specific CD4 T cells were mainly triple cytokine-positive throughout all prime/boost intervals (Figure 2D). Overall, these data suggest that, despite immunization with a 20-neoantigen SMARRT replicon, the CD4 and CD8 T cell response exhibits immunodominance to a minority of neoantigens. We do not discount that neoantigen immunodominance may be affected by the design of the specific polytope cassette expressed in SMARRT.Ancer. The varying IFN-γ responses observed across surveyed constructs suggest that alternative designs containing the same 20 neoantigens may lead to different immunogenicity patterns.
Figure 2.

SMARRT.Ancer Primes a Limited Number of Dominant T Cell Epitopes
SMARRT.Ancer (containing the top 20 CT26 neoantigens) was used to immunize BALB/c mice with varying prime/boost interval lengths. Splenocytes were analyzed by intracellular cytokine staining (on the indicated days post-final injection) by restimulating individually with all 20 neoantigens encoded in the cassette. (A) and (B) show the percentage of IFN-γ+ CD8 and CD4 T cells specific for peptides stimulating a response above mock (data not shown). (C) and (D) show the polyfunctionality of CD8 and CD4 T cells in response to the dominant neoantigen 20 and 4, respectively. Graphs show mean with standard deviation, n = 5 mice per group. Statistical analysis was performed using one-way ANOVA with Tukeys multiple comparison test. For (C) and (D), the triple functional subsets were compared. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001
Clinically Relevant, Single-Epitope SMARRT Vaccines Can Prime CD8 T Cell Responses In Vivo in Mice
To expand on the observation that a single dominant epitope can drive a polyfunctional CD8 T cell response, we evaluated the use of SMARRT replicon vaccines to immunize against a clinically relevant, shared neoantigen target. Immunization of HLA-A∗1101 transgenic mice with a SMARRT replicon encoding the G12V variant of KRAS, found in 5%–20% of lung, colorectal, and pancreatic cancer, leads to priming of polyfunctional CD8 T cells in 100% of mice (Figures 3A and 3B). No immune responses were detected against the wild-type (WT) version of KRAS, in both the groups of mice immunized with WT or KRAS G12V, indicating that the response is specific to the tumor variant. This result demonstrates how SMARRT vaccines may be used clinically to target single CD8 specific-tumor epitopes in the absence of CD4 T cell help.
Figure 3.

SMARRT Can Prime Polyfunctional T Cell Responses Specific to the Shared Neoantigen KRAS G12V
HLA-A∗1101 transgenic mice were vaccinated with SMARRT replicons encoding an epitope from either WT or G12V mutant KRAS and boosted on days 21 and 41. Spleens were analyzed 7 days post final boost by intracellular cytokine staining (A) and IFN-γ ELISpot (B) following restimulation ex vivo with the indicated peptides. Graphs show mean with standard deviation, n = 3 mice per group. Statistical analysis was performed using Mann-Whitney U Test, panel B compared double cytokine positive T cells. ∗p<0.05; ∗∗∗∗p<0.0001.
SMARRT.Ancer Vaccination in a Murine Tumor Model Can Inhibit Tumor Growth, Which Is Further Enhanced by Checkpoint Blockade Inhibition
To test in vivo efficacy of SMARRT.Ancer therapeutic vaccination, we immunized CT26 tumor-bearing mice. SMARRT.Ancer immunized mice significantly inhibited the growth of CT26 tumors, compared to irrelevant replicon (SMARRT.rFF) immunized mice or LNP treated controls (Figure 4A), leading to significantly increased survival (Figure 4B). The irrelevant SMARRT control was added to control for the fact that the SMARRT platform induces a type-I IFN response in the host cell. The addition of anti-PD-1 blocking antibody had an additive effect in combination with SMARRT.Ancer, resulting in further inhibition of tumor size (Figure 4C). Analysis of tumor infiltrating lymphocytes (TIL) at an intermediate time point revealed enhanced functional CD8 T cells in SMARRT.Ancer immunized animals compared to those treated with SMARRT.rFF and a further enhancement of polyfunctional CD8 T cells was observed when SMARRT.Ancer was combined with anti-PD-1 therapy (Figure 4D). Polyfunctional CD4 T cell infiltration into the tumor was enhanced when SMARRT.Ancer was combined with anti-PD-1 compared to SMARRT.Ancer alone (Figure 4E).
Figure 4.

Therapeutic Vaccination with SMARRT.Ancer Inhibits Tumor Growth and Enhances Survival
Mice were implanted with 3 × 105 CT26 cells s.c. at day 0. SMARRT was injected i.m. on days 3 and 17. Tumor growth (A) and overall survival (B) were measured (n = 18/group). In a separate study, mice (n = 6/group) were injected with CT26 and SMARRT constructs as described above; additionally, some groups received anti-PD-1 blocking mAbs on days 8, 11, 15, and 18. (C) Shows tumor growth until day 23. Tumors were excised on day 23 and T cells were analyzed by flow cytometry for cytokine production following ex vivo peptide re-stimulation. (D) Shows CD8 T cells and (E) shows CD4 T cells. Tumor growth curves show median tumor volume, and statistical testing was done with two-way ANOVA. Survival curve statistical testing was done using log-rank (Mantel Cox) test. Bar graphs show mean with standard deviation. ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
SMARRT Vaccine Platform Can Break Tolerance to Tumor-Associated Antigens, and Addition of Cytokine-Producing Replicons Results in Epitope Spreading in Mice
Combination therapies are becoming increasingly utilized in tumor therapy. We demonstrate above that SMARRT vaccines can be used in combination with checkpoint inhibition and we hypothesized that SMARRT vaccines would synergize with cytokines. We used the SMARRT platform to encode and express a series of cytokines that have been shown to enhance T cell priming, survival, and function. SMARRT successfully expressed mouse IL-7, IL-15, IL-7/15 fusion protein (mouse and human), IL-12, and GM-CSF in vitro (Figure 5A).
Figure 5.

SMARRT Can Be Used to Express Cytokines, Resulting in Epitope Spreading to Weakly Immunogenic Tumor-Associated Antigens
SMARRT expressing hIL-7/15 fusion, mIL-7/15 fusion, mIL-7, mIL-15, mGM-CSF, and IL-12 were used to transfect BHK cells. (A) Supernatants were harvested 24 h post-electroporation and cytokine concentrations were measured using ELISA. SMARRT.Trp2 was used to immunize C57BL/6 mice on days 0 and 14. Spleens were harvested on day 21 and restimulated with the dominant CD8 epitope TRP2180–188. (B and C) T cell function was measured by IFN-γ ELISpot (B) and intracellular cytokine staining (C). (D) Mice were immunized as described above with SMARRT.Trp2; in addition, mice were immunized with SMARRT.IL-12 at prime only, as indicated in the figure. The graph shows intracellular cytokine staining following stimulation with an overlapping peptide pool of TRP2 (Sub.) or TRP2180–188 (Dom.). Graphs in (B)–(D) show mean with standard deviation, n = 5 mice per group. Statistical testing for (B) and (C) was done using Mann-Whitney U test. Statistical testing in (D) was done using ordinary one-way ANOVA to compare the total IFN-γ+ CD8 T cells, ∗p < 0.05; ∗∗p < 0.01.
Previous attempts to break tolerance to TAAs have been reported with VRP platforms, but not fully synthetic replicons.13, 14, 15, 16 To test the ability of the SMARRT platform to successfully elicit immune responses to a TAA, we immunized animals with a SMARRT vaccine encoding tyrosinase-related protein 2 (Trp2), a melanoma TAA. Analysis of Trp2-specific T cell responses revealed significant effector CD8 T cells in 100% of the animals following two doses of the SMARRT vaccine (Figure 5B). In addition, single cell cytokine analysis demonstrated that ∼80% of antigen-specific CD8 T cells produced two or more inflammatory cytokines (i.e., polyfunctional), linked with high-quality effector responses (Figure 5C). Combination of SMARRT.TRP2 with SMARRT.IL-12 at prime showed a significant synergistic effect by enhancing the repertoire of Trp2-specific CD8 T cells, to a peptide pool containing subdominant epitopes (labeled Sub. in figure), in a dose-dependent manner (Figure 5D). IL-12 was chosen to combine with SMARRT.Trp2 based on previous findings that IL-12 expressed from VRPs led to enhancement of anti-tumor responses.12 This effect was also observed in vivo when a SMARRT vaccine, expressing the influenza antigen hemagglutinin (HA), was used in combination with cytokine-expressing SMARRT (Figure S3). In this case, we chose to co-administer the vaccine with SMARRT expressing IL-7 and/or IL-15, since both of these cytokines are crucial for enhancement of memory T cell responses, an important feature for the design of a prophylactic vaccine. Both CD8 and CD4 effector memory T cells were significantly expanded following the combination of SMARRT.HA and SMARRT expressing either IL-7 or IL-15. Thus, the SMARRT platform can be an effective vaccine to difficult immunological targets, such as TAAs and its potency can be further enhanced by addition of SMARRT-expressed cytokines, which broaden the T cell repertoire to subdominant epitopes.
SMARRT Vaccine Platform Immunogenicity Translates to Non-human Primates
Non-human primate studies are an important validation of the ability of a vaccine platform and formulation to translate from small animal mouse models into larger organisms. To this end, we immunized rhesus macaques with SMARRT encoding the influenza HA molecule as a surrogate antigen, as the ability of NHP to mount measurable immune responses to HA has been previously reported with other vaccine formats.24 To account for variable immunogenicity of different influenza strains25 vaccines to both A/California/07/2009 H1N1 and A/Vietnam/1203/2004 H5N1 were designed. Additionally, translatability and added synergy of the LNP formulation was also tested across a dose range. Detectable H1 and H5 specific T cells were present in 100% of SMARRT-immunized animals compared to saline administered groups (Figures 6A and 6B). Furthermore, both CD8 and CD4 T cell populations showed antigen-specific polyfunctional cytokine production at all 3 doses tested, including a group that received unformulated replicon (Figures 6C and 6D). Importantly, SMARRT was well-tolerated with no adverse events reported from clinical observations, inclusive of injection site reactogenicity, in any animal tested (data not shown). These data show that SMARRT immunogenicity translates to large animals at clinical feasible doses.
Figure 6.

SMARRT Primes Polyfunctional T Cell Responses in Non-human Primates
SMARRT replicons expressing HA from the H1N1 or H5N1 strains of influenza virus were used to immunize rhesus macaques on day 0 and 56 at the indicated doses. Saline immunization was used as a control. The animals were bled on day 77 and PBMCs were re-stimulated with peptide pools encoding either H1 or H5 HA sequences. (A) and (B) show activated (OX40+ CD25+) CD8 and CD4 T cells found in the PBMCs as measured by flow cytometry (solid circles, LNP formulated 100 μg; open circles, unformulated 100 μg; squares, formulated 10 μg; triangles, formulated 1 μg). (C) and (D) show cytokine production of CD8 and CD4 T cells measured by flow cytometry. Each bar/symbol corresponds to one animal. Statistical testing was carried out with one-tailed Mann-Whitney U test, ∗p < 0.05.
Discussion
Immunotherapeutic approaches to cancer therapy have revolutionized treatment options for patients across a wide range of malignancies. The generation of anti-tumor T cell responses has been clinically demonstrated using antibodies modulating immune pathways, cell therapies, and therapeutic vaccines with differing degrees of success. In this study we aimed to validate the use of self-replicating RNA for use in tumor vaccination using a range of antigen targets and cytokines. We showed that the SMARRT vaccine platform can prime polyfunctional CD4 and CD8 T cell responses in vivo, and that these can be further enhanced by combination therapies including SMARRT-expressed cytokines and traditional checkpoint inhibitors.
To date, cancer vaccines have failed to generate strong, durable clinical responses in patients. Vaccination approaches have suffered from the use of weakly immunogenic tumor targets and platforms. In recent years neoantigen prediction has improved clinical immunogenicity but vaccine platforms often fail to elicit both CD4 and CD8 T cell responses.3 We demonstrate in mice that SMARRT can be used in conjunction with both personalized and shared neoantigen vaccination strategies. Prediction of neoantigens with the Ancer platform from the mouse tumor cell line, CT26, was used to demonstrate a personalized neoantigen vaccine approach by encoding multiple MHC class I- and MHC class II-restricted epitopes in a bead-on-a-string format and expressing this construct in the SMARRT platform. Construct design of polytope inserts is highly complex, so numerous approaches were used to optimize epitope processing, presentation, and reduction of off-target junctional epitopes (Table 1; Table S2). Immunogenicity varied greatly when altering construct design and demonstrates an underappreciated fact, that optimization will require extensive assay development and in-depth studies before it can be used reliably in the clinic (Figure 1). It is likely that for each given set of antigens and platform, the optimal insert design will need to be determined empirically. Despite this, our data clearly show that one dose of SMARRT.Ancer can prime polyfunctional CD4 and CD8 T cell in vivo in mice and a boosting dose further enhances the magnitude and polyfunctionality of the CD8 response (Figure S2). Optimal prime/boost intervals will vary greatly between vaccine platforms and this study in mice analyzes this vaccination regimen in detail, describing for the first time, the kinetics of T cell function following replicon immunization.
SMARRT vaccination with a construct designed by the Ancer platform and expressing multiple neoantigens inhibited the growth of CT26 in vivo (Figures 4A and 4B), even at a prime-boost interval that is suboptimal. TIL analysis showed extensive infiltration of antigen-specific CD4 and CD8 T cells (Figures 4D and 4E). PD-1 blockade has shown efficacy in numerous human tumors and often synergizes with vaccines, likely because neoantigen specific T cells in the tumor express PD-1.26 Addition of anti-PD-1 blocking antibodies in combination with SMARRT.Ancer showed an additive effect, enhancing polyfunctional T cell infiltration into the tumor and further inhibiting tumor growth compared to the monotherapy in mice (Figure 4C). Interestingly the largest effect of anti-PD-1 blockade was observed on CD4 TILs in mice, which is in contrast to human clinical data showing PD-1 blockade mostly affects CD8 effector function.27 This result is encouraging for clinical translation of SMARRT as many cancer patients will be receiving checkpoint inhibitors. Recently, single cell sequencing of T cell clones following anti-PD-1 treatment in basal cell carcinoma patients revealed that, rather than reinvigorating pre-existing exhausted T cells, anti-PD-1 therapy can drive expansion of a distinct repertoire of tumor specific clones.28 Additionally, there is now a growing appreciation that, for some tumor types that are unresponsive to checkpoint blockade, e.g., ovarian cancer and microsatellite unstable colorectal cancer, the vast majority of tumor infiltrating T cells are bystander cells. Analysis of patient samples has revealed that very few PD-1+ CD8 T cells in these tumors (0%–30%) express a tumor reactive TCR.29 Our data indicate that SMARRT vaccination, in combination with checkpoint inhibition and careful selection of antigens, is a powerful therapeutic strategy that can enhance the frequency of tumor-reactive T cells that can infiltrate the tumor.
To further understand how SMARRT may be utilized in combination therapies, we successfully expressed a series of mouse and human cytokines from the replicon platform (Figure 5A). Previous attempts using VRPs had also successfully expressed IL-12 showing that it enhanced the anti-tumor activity of a TAA cancer vaccine in a mouse model.30 IL-12 VRP has also been used in a glioblastoma clinical trial as a monotherapy showing safety but no anti-tumor efficacy, possibly due to the fact that no antigen vaccine was used in combination.12 We show that the addition of SMARRT.IL-12 at prime in combination with SMARRT.TRP2 vaccination results in significantly enhanced CD8 T cell responses to a Trp2 overlapping peptide pool containing subdominant epitopes (Figure 5D). The T cell response to the dominant class I Trp2 epitope remained unchanged by the combination suggesting that SMARRT.IL-12 can induce epitope spreading. This effect is supported by a recent report describing how recombinant IL-12, in combination with tumor vaccines in mice, increased the clonality of the T cell repertoire.31 Our results demonstrate the utility of SMARRT outside of antigen expression.
Most neoantigen clinical trials immunize patients with a combination of peptide plus adjuvant. This approach has often struggled to generate potent anti-tumor responses, similar to mRNA vaccination, as both platforms often fail to generate robust CD8 T cell responses.20, 21, 22 Our data, however, indicate that SMARRT immunization results in priming of both tumor-specific CD8 and CD4 T cells. Recently a similar approach encoding multiple neoantigens on a single RNA molecule was reported in humans using traditional mRNA.26 Although 100% of patients had measurable T cell responses, this approach required up to 20 intra-nodal injections, at doses 5 times more than we anticipate being used in humans for SMARRT, and generated mainly class II restricted responses, which will limit this platform for widespread use. These studies demonstrate how platform technology can impact immunogenicity of tumor vaccines and that antigen selection is not the only important factor when designing effective vaccines.
Interestingly, SMARRT could inhibit tumor growth despite the T cell response being dominated by single epitopes in both CD8 and CD4 T cell compartments (Figures 2A and 2B). Optimization of construct design may expand the T cell pool to include subdominant epitopes, but we hypothesized that single epitope responses could be of benefit in creating off-the-shelf vaccines for shared neoantigen targets. Most neoantigen vaccine approaches have thus far concentrated on predicting personalized vaccines based on a snapshot of a patient’s tumor mutanome. This approach is limited due to expense of sequencing each patient’s tumor and the fact that the tumor is evolving rapidly and often loses passenger mutations that confer no growth advantage. Targeting shared driver mutations is a universal approach and can be used to mass-produce off-the-shelf vaccines that can be used across patients and tumor types. Another advantage of targeting broadly shared neoantigens is that construct design considerations are much simpler and will require less optimization than for complex personalized polytopes. One example of a shared neoantigen target is KRAS, a proto-oncogene that is frequently mutated at residue 12 in colon, lung, and pancreatic cancer. The KRAS G12V mutation is found in 5%–20% of patients with these malignancies and reactive T cells have been found to occur in patients bearing this mutation.32 We show that SMARRT expressing a single epitope derived from KRAS G12V can prime polyfunctional CD8 T cell responses in humanized mice with no cross-reactivity to WT KRAS (Figure 3). These data show that SMARRT expressing shared neoantigens is a viable clinical approach to develop universal cancer vaccines. As public tumor mutanome databases grow, the number of shared driver mutations will increase, allowing us to produce multiple SMARRT vaccines, which can be combined and tailored to the tumor mutations and HLA haplotype of each patient.
Mouse models often fail to predict outcome in clinical settings, especially in oncology.33 We demonstrate that SMARRT expressing an influenza antigen did translate from mice to NHP, with all immunized NHPs generating T cell responses after 2 doses (Figure 6). Furthermore, these animals generated both CD4 and CD8 T cell responses that were polyfunctional confirming translatability of data generated in mice. Also, the strength of the SMARRT platform in generating robust T cell responses in NHP is furthermore highlighted as a viral vector vaccine encoding equivalent HA antigens failed to produce effector T cell responses after two doses.24 Additionally, this NHP study also demonstrates that dosing of SMARRT in larger animals will be in the range of 1–100 μg, depending on the inherent immunogenicity of the antigen. Such a low dose combined with potentially fewer doses in total should provide important advantages to the SMARRT vaccine compared to mRNA, allowing development of a more cost-effective and globally utilized medicine compared to other nucleic acid platforms.
One caveat of using virally derived RNA vectors is the potential of protein translation inhibition by innate immune mechanisms, such as Toll-like receptors, anti-viral response proteins such as PKR and RIG-I, and type I IFNs.19 In the work presented here, we utilize the SMARRT platform, which includes an immune evasion motif that renders it resistant to these anti-viral mediators. This motif allows continued protein (antigen) expression from the SMARRT platform in the presence of innate immune responses (Figure S1). Thus, we expect that SMARRT vaccines would yield superior immune responses compared to traditional replicon platforms, especially in patients that may exhibit chronic inflammation.
This study shows that a fully synthetic, self-replicating RNA vaccine platform, SMARRT, can be used to generate anti-tumor T cell responses against personalized and shared neoantigens, as well as breaking tolerance to tumor-associated antigens in mice. SMARRT is a powerful platform that translates from mouse to NHP, synergizes with commonly used checkpoint inhibitors and should outperform traditional vaccine platforms despite using the same antigen targets. Future work will seek to clinically develop the SMARRT platform as an effective anti-tumor vaccination platform across multiple malignancies in combination with other immunotherapeutic modalities.
Materials and Methods
Mice
Female C57BL/6 and BALB/c mice were purchased from Charles River Labs. HLA-A∗1101 transgenic mice were purchased from Taconic. Mice were 6–8 weeks old at time study initiation.
On day of dosing, 10 μg (unless otherwise noted) of LNP-formulated SMARRT RNAs were administered intramuscularly (i.m.) in one or both quadricep muscles. Animals were monitored for body weight and other general observations throughout each study.
For cytokine studies, mice were immunized i.m. with LNP-formulated 1 μg of SMARRT encoding HA from influenza A/Vietnam/1203/2004 H5NA, in addition to human SMARRT-encoded IL-7, IL-15, or an IL-7/IL-15 hybrid.34
All procedures carried out in this experiment were conducted in compliance with all the laws, regulations, and guidelines of the National Institutes of Health (NIH) and with the approval of Explora Biolab’s Animal Care and Use Committee. Explora Biolabs is an AAALAC accredited facility.
In Vivo Protein Expression
Mice were administered a dose of 20 μg of poly(I:C) by hydrodynamic injection as previously described20 at 24 h before i.m. administration of 15 μg of replicon encoding red firefly luciferase. In vivo imaging of luciferase activity was done using IVIS at the indicated times.
Neoantigen Prediction and Polytope Construct Design
Pairs of mutated and normal matched WT 23-mer amino acid sequences were extracted for each of the CT26 cell line variants under study. Mutated sequences were extracted such that each mutation was surrounded by 11-mer flanks on both N- and C-termini. This length allows for the characterization of every 9- and 10-mer frames overlapping with the mutation while adding flanking residues for sequence design.
Each pair of mutated and normal peptides were uploaded to the Ancer platform, a machine learning-based neo-epitope screening and characterization tool. Ancer integrates EpiMatrix4 a commercial suite of MHC class I and MHC class II T cell epitope mapping tool, and JanusMatrix35 a specialized homology tool that compares epitopes to “self” sequences to avoid delivering epitopes capable of stimulating Tregs that hamper vaccine efficacy, unwanted cross-reactive immunity, or tolerated “dead end” epitopes that make no contribution to tumor control. Both EpiMatrix and JanusMatrix has been extensively validated in numerous prospective immunogenicity and efficacy studies.36, 37, 38, 39, 40, 41, 42
Each peptide was first parsed into overlapping 9- and 10-mer frames and each frame was evaluated with for its likelihood to bind to BALB/c MHC class I (H2-Dd and H2-Kd) and MHC class II (I-Ad and I-Ed) alleles. T cell epitopes predicted in mutated sequences were subsequently compared to normal matched sequences in order to identify neo-epitopes.
Additionally, sequences presenting a high likelihood of binding to MHC are screened using a customized homology search to remove epitopes containing combinations of TCR-facing residues that are commonly found in a reference proteome. This homology screen first considers all the predicted epitopes contained within a given protein sequence, divides each predicted epitope into its constituent agretope and epitope, and returns a homology score based on similarity with self-sequences.
T cells that recognize antigen-derived epitopes sharing TCR contacts with epitopes derived from self may be deleted or rendered anergic during thymic selection before they can be released to the periphery. As such, vaccine components targeting these T cells may be ineffective. On the other hand, vaccine-induced immune response targeting cross-reactive epitopes may induce unwanted autoimmune responses targeting the homologs of the cross-reactive epitopes identified by our homology search. As a result, vaccine safety may be reduced. A review of HLA class II-restricted T cell epitopes contained in the IEDB database indicates that there is a statistically significant relationship between high homology scores and observed production of the anti-inflammatory IL-10 cytokine and a statistically significant inverse relationship between high homology scores and observed production of the pro-inflammatory IL-4 cytokine.43
An optimization procedure is then run on each of the mutated sequences to design, if possible, candidate neoantigens. This optimization procedure has the effect of removing amino acid substrings containing putative Treg epitopes and other highly cross-conserved epitopes from mutated sequences. The resulting optimized sequences will solely contain MHC class I- and/or MHC class II-restricted neo-epitopes that exhibit low degree of similarity with self-sequences. Neoantigen sequences are then ranked according to immunogenicity-, sequencing-, and physicochemical-related features.
Neoantigen candidates were concatenated using the VaxCAD component of the Ancer platform to generate string-of-beads designs. VaxCAD is employed to evaluate the formation of junctional epitopes before merging two neoantigens.
All sequences of identified neoantigens and multi-epitope constructs are shown in Tables S1 and S2.
CT26 Tumor Model
The CT26 cell line was purchased from ATCC. Early cell passages were used for tumor experiments before using a new aliquot. For therapeutic vaccination modeling, 3 × 105 CT26 were inoculated subcutaneously (s.c.) into the flanks of BALB/c mice. Immunization with the SMARRT vaccine was administered on days 3 and 17 after tumor injection, unless indicated otherwise in the result section. Anti-PD-1 blocking antibody (clone RMP1-14 BioXcell) was administered on days 8, 11, 15, and 18, via intraperitoneal injection with 5 mg/kg/dose. Tumor sizes were measured twice a week, and mice were sacrificed when tumor diameter reached 2,000 mm3.
Tumor Infiltrating Lymphocyte Isolation
Tumors were excised, digested using Collagenase D (1 mg/mL, Roche) and DNase I (25 U/mL, New England Biolabs) in AIM V media, filtered, and lymphocyte single cell suspensions were prepared by lysing red blood cells per standard methods.
Non-human Primate Studies
Rhesus macaques were selected for influenza HA immunogenicity studies and enrolled based on weight and sex. Previous exposure to influenza was not measured at study initiation, but selected animals were confirmed to not have been used in prior influenza studies. Each group contained a male and female macaque. Animals were immunized i.m. with SMARRT encoding HA from either the A/California/07/2009 H1N1 or A/Vietnam/1203/2004 H5N1 strains of influenza on days 0 and 56. Multiple doses (1, 10, and 100 μg) of formulated SMARRT were used along with one group that received unformulated replicon at a dose of 100 μg. Saline immunized macaques were used as controls. Animals were bled weekly, and PBMCs were collected and used for downstream assays and monitored for any adverse effects, including standard clinical observations such as daily food intake, activity, and observation of dose site for signs of redness, swelling, and/or erythema. Weekly body weight measurements and complete blood count and differential were also completed. All work was carried out by New Iberia Research Center according to IACUC guidelines.
Generation of SMARRT Vaccines
Plasmid Construction
The TC-83 strain of Venezuelan Equine Encephalitis Virus (VEEV) genome sequence served as the base sequence used to construct the SMARRT replicon. This sequence was modified by placing the Downstream LooP (DLP) from Sindbis virus upstream of the non-structural protein 1 (nsP1) with the two joined by a 2A ribosome skipping element from porcine teschovirus-1. The first 193 nucleotides of nsP1 were duplicated downstream of the 5′ UTR and upstream of the DLP except for the start codon, which was mutated to TAG. This insured all regulatory and secondary structures necessary for replication were maintained but prevented translation of this partial nsp1 sequence. The structural genes were removed and EcoR V and Asc I restriction sites were placed downstream of the subgenomic promoter as a multiple cloning site (MCS) to facilitate insertion of any heterologous gene of interest. Each antigen was synthesized as a dsDNA fragment by IDT with 40 bp of homology to the MCS at their 5′ and 3′ ends. These were then cloned into the SMARRT replicon digested with EcoRV and AscI using NEB HiFi DNA assembly master mix (New England Biolabs). All constructs were sequenced verified.
RNA Transcription
Plasmids were purified using the Nucleobond xtra EF maxiprep kits (Machery-Nagel) followed by phenol/chloroform extraction and sodium acetate/ethanol precipitation. RNA was generated using the HiScribe T7 ARCA mRNA kit (New England Biolabs) and 1 μg of plasmid template linearized with Not I-HF (New England Biolabs). RNA was subsequently purified using RNeasy purification columns (QIAGEN) and eluted in water. RNA concentration was determined using a Qubit 4 fluorometer and Qubit RNA BR Assay Kit (Thermo Fisher).
Formulation for Lipid-Mediated Delivery of RNA
SMARRT replicon RNA was formulated in lipid nanoparticles by Arcturus Therapeutics using their LUNAR technology for in vivo delivery. Formulated SMARRT replicons were stored at −80°C until dosing day.
Potency
Potency of SMARRT vaccines was evaluated post-formulation using immunostaining for double-stranded RNA (dsRNA) intermediates and flow cytometry as a measure of replicon launch upon transfection into BHK-21 cells. In brief, RNA was extracted from LUNAR particles using Ambion kits and electroporated using a nucleofection (Lonza). Intracellular dsRNA was detected using mAb (J2, Scicons) and Cytox/Cytoperm intracellular staining kit (BD Biosciences). Frequency of dsRNA+ cells was quantified against a standard curve using a reference replicon lot.
ELISpot
IFN-γ ELISpot analysis was performed using the Mouse IFN-γ ELISpot PLUS Kit (HRP) (MabTech) as per manufacturer’s instructions. Briefly, splenocytes resuspended to a final concentration of 5 × 106 cells/mL in media containing peptides corresponding to the antigen(s), PMA/ionomycin as a positive control, or DMSO as mock stimulation.
Intracellular Cytokine Staining
Stimulation media containing peptides or PMA/ionomycin were added to a 96-well round-bottom plate (Corning) at 100 μL per well. 1 × 106 splenocytes were added to the well for a final volume of 200 μL per well. Samples were stimulated for 1 h at 37°C, 5% CO2. After 1 h, GolgiPlug protein transport inhibitor (BD Biosciences) was added to each well. Plates were incubated for an additional 5 h, prior to immunostaining.
Cells were stained with a predefined panel of surface markers and intracellular proteins shown below, as per standard methods and and analyzed on a LSRFortessa cell analyzer (BD Biosciences, USA). The acquired FCS files were analyzed using FlowJo software version 10.4.1
Mouse ICS Panel Antibodies
For mouse ICS panel antibodies, see Table 2.
Table 2.
Mouse ICS Panel Antibodies
| Channel | Specificity | Manufacturer | Clone | Dilution |
|---|---|---|---|---|
| FITC | CD8 | Invitrogen | 53-6.7 | 1:200 |
| APC-Cy7 | CD4 | Biolegend | GK1.5 | 1:200 |
| AF647 | IFN-γ | Biolegend | RPA-T8 | 1:200 |
| PE | IL-2 | Biolegend | JES6-5H4 | 1:200 |
| PE-TR | TNF-α | BD Biosciences | MP6-XT22 | 1:200 |
| PerCP Cy5.5 | B220 | Biolegend | B238128 | 1:200 |
| PerCP Cy5.5 | Gr-1 | Biolegend | RB6-8C5 | 1:200 |
| PerCP Cy5.5 | CD16/32 | Biolegend | M93 | 1:200 |
| PE-Cy7 | PD-1 | Biolegend | 29F.1A12 | 1:100 |
| BV421 | FoxP3 | Biolegend | MF-14 | 1:150 |
| BV785 | Tim-3 | Biolegend | RMT3-23 | 1:200 |
Ex Vivo Analysis of NHP PBMCs
Activation induced marker (AIM) assay44 was adapted to assess simultaneous induction of surface activation markers and intracellular cytokines by flow cytometry. Cryopreserved PBMCs were rested in AIM V media for 4 h prior to stimulation with 1 μg/mL of peptides pools and costimulatory antibodies anti-CD28 (clone CD28.2) and anti-CD49d (clone 9F10) overnight (see Table 3). GolgiPlug was added during the last 4 h of stimulation followed by immunostaining as described above.
Table 3.
NHP Panel Antibodies
| Channel | Specificity | Manufacturer | Clone | Dilution |
|---|---|---|---|---|
| APC/A647 | TNF-α | Biolegend | Mab11 | 1:200 |
| Ef780-APC | CD25 | eBioscience | BC96 | 1:200 |
| FITC | CD8 | eBioscience | RPA-T8 | 1:200 |
| PE | OX40 | BD Biosciences | L106 | 1:200 |
| PE-Dazzle | IFN-γ | Biolegend | B27 | 1:200 |
| Cy7-PE | CD3 | BD Biosciences | SP34-2 | 1:200 |
| PerCP Cy5.5 | CD4 | BD Biosciences | L-200 | 1:200 |
| BV421 | IL-2 | BD Biosciences | MQ1 | 1:200 |
Flow Cytometry for Memory T Cell Subsets
Single cell suspensions from spleens of immunized animals were immunostained as per standard protocols and analyzed on a LSRFortessa cell analyzer (BD Biosciences, USA). The acquired FCS files were analyzed using FlowJo software version 10.4.1.
Mouse T Cell Panel Antibodies
Please see Table 4 for information on mouse T cell panel antibodies.
Table 4.
Mouse T Cell Panel Antibodies
| Channel | Specificity | Manufacturer | Clone | Dilution |
|---|---|---|---|---|
| APC-Cy7 | CD44 | Invitrogen | IM7 | 1:100 |
| AF700 | CD4 | Biolegend | GK1.5 | 1:100 |
| APC | KLRG1 | Biolegend | 2F1 | 1:100 |
| FITC | CD127 | Biolegend | A7R34 | 1:50 |
| PE-Dazzle594 | CD62L | Biolegend | MEL-14 | 1:200 |
| PerCP Cy5.5 | B220 | Biolegend | RA3-6B2 | 1:200 |
| PerCP Cy5.5 | Gr-1 | Biolegend | RB6-8C5 | 1:200 |
| PerCP Cy5.5 | CD16/32 | Biolegend | 93 | 1:200 |
| PE-Cy7 | PD-1 | Biolegend | 9–12 | 1:100 |
| BV421 | CCR7 | Biolegend | 4B12 | 1:100 |
| BV785 | CD8 | Biolegend | 53-6.7 | 1:100 |
Peptides
For ex vivo restimulation of T cells, the following peptides were used. The top 50 neoantigens from CT26, predicted by the Ancer platform, were synthesized by New England Peptide and pooled together at 1 μg/mL per peptide. The top 20 neoantigen, predicted by Ancer, were used individually at 10 μg/mL for immunodominance studies (New England Peptide). Peptides surrounding the G12 residue of KRAS (WT and G12V versions of KRAS7–14, KRAS8–16, KRAS5–14, and KRAS7–16) were pooled and used to stimulate splenocytes at 10 μg/mL per peptide (New England Peptide). TRP180–188 was used at 10 μg/mL (Anaspec). Commercial PepMix Influenza A peptide pools (JPT) corresponding to H1N1 and H5N1 HA were used to stimulate PBMC from rhesus macaques.
ELISA
SMARRT replicons encoding mouse and human cytokines were used to transfect BHK cells; 24 h later, supernatant was collected and cytokine concentrations were determined by ELISA as per manufacturer’s instructions (Ready-Set-Go kits, Thermo Fisher Scientific).
Statistical Analysis
Statistical analysis was carried out using Graphpad Prism, and specific statistical tests are indicated in the figure legends.
Acknowledgments
We would like to thank Arcturus Therapeutics for LNP formulations and Jane Fontenot, Francois Villinger (New Iberia Research Center), and Philip J. Santangelo (Georgia Institute of Technology) for expertise with running non-human primate experiments.
Author Contributions
C.J.M., G.R., L.M., D.S.S., K.I.K., G.B., A.S.D., N.S.W., and P.A. designed experiments. J.S., R.P.S., O.G., D.S.S., M.C., J.M.C., A.A., M.D.O., S.J.M.-S., K.L., K.S., M.B., and J.L.D. performed experiments. C.J.M., G.R., J.S., L.M., R.P.S., O.G., D.S.S., M.C., J.M.C., A.A., M.D.O., S.J.M.-S., K.L., M.B., N.S.W., and P.A. analyzed and interpreted data.
Declaration of Interests
All authors affiliated with Synthetic Genomics Inc. declare no competing interests. A.D.G. is a senior officer and a majority shareholder, and L.M. is an employee of EpiVax, Inc., a privately owned immunoinformatics and vaccine design company. These authors acknowledge that there is a potential conflict of interest related to their relationship with EpiVax and attest that the work contained in this research report is free of any bias that might be associated with the commercial goals of the company. G.B. was previously a senior officer of EpiVax Therapeutics, Inc., and G.R. is currently an employee of EpiVax Therapeutics, Inc., a precision immunotherapy company and subsidiary of EpiVax, Inc. These authors acknowledge that there is a potential conflict of interest related to their relationship with EpiVax Therapeutics and attest that the work contained in this research report is free of any bias that might be associated with the commercial goals of the company.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.11.027.
Supplemental Information
References
- 1.Hollingsworth R.E., Jansen K. Turning the corner on therapeutic cancer vaccines. npj. Vaccines (Basel) 2019;4:1–10. doi: 10.1038/s41541-019-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castle J.C., Kreiter S., Diekmann J., Löwer M., van de Roemer N., de Graaf J., Selmi A., Diken M., Boegel S., Paret C. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72:1081–1091. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 3.Guo Y., Lei K., Tang L. Neoantigen Vaccine Delivery for Personalized Anticancer Immunotherapy. Front. Immunol. 2018;9:1499. doi: 10.3389/fimmu.2018.01499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Groot A.S., Moise L., Terry F., Gutierrez A.H., Hindocha P., Richard G., Hoft D.F., Ross T.M., Noe A.R., Takahashi Y. Better Epitope Discovery, Precision Immune Engineering, and Accelerated Vaccine Design Using Immunoinformatics Tools. Front. Immunol. 2020;11:442. doi: 10.3389/fimmu.2020.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen D.S., Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 6.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lundstrom K. Replicon RNA Viral Vectors as Vaccines. Vaccines (Basel) 2016;4:39. doi: 10.3390/vaccines4040039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawai T., Akira S. Innate immune recognition of viral infection. Nat. Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 9.Morse M.A., Hobeika A.C., Osada T., Berglund P., Hubby B., Negri S., Niedzwiecki D., Devi G.R., Burnett B.K., Clay T.M. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J. Clin. Invest. 2010;120:3234–3241. doi: 10.1172/JCI42672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slovin S.F., Kehoe M., Durso R., Fernandez C., Olson W., Gao J.P., Israel R., Scher H.I., Morris S. A phase I dose escalation trial of vaccine replicon particles (VRP) expressing prostate-specific membrane antigen (PSMA) in subjects with prostate cancer. Vaccine. 2013;31:943–949. doi: 10.1016/j.vaccine.2012.11.096. [DOI] [PubMed] [Google Scholar]
- 11.Crosby E.J., Gwin W., Blackwell K., Marcom P.K., Chang S., Maecker H.T., Broadwater G., Hyslop T., Kim S., Rogatko A. Vaccine-Induced Memory CD8+ T Cells Provide Clinical Benefit in HER2 Expressing Breast Cancer: A Mouse to Human Translational Study. Clin. Cancer Res. 2019;25:2725–2736. doi: 10.1158/1078-0432.CCR-18-3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren H., Boulikas T., Lundstrom K., Söling A., Warnke P.C., Rainov N.G. Immunogene therapy of recurrent glioblastoma multiforme with a liposomally encapsulated replication-incompetent Semliki forest virus vector carrying the human interleukin-12 gene--a phase I/II clinical protocol. J. Neurooncol. 2003;64:147–154. doi: 10.1007/BF02700029. [DOI] [PubMed] [Google Scholar]
- 13.Avogadri F., Merghoub T., Maughan M.F., Hirschhorn-Cymerman D., Morris J., Ritter E., Olmsted R., Houghton A.N., Wolchok J.D. Alphavirus replicon particles expressing TRP-2 provide potent therapeutic effect on melanoma through activation of humoral and cellular immunity. PLoS ONE. 2010;5:e12670. doi: 10.1371/journal.pone.0012670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avogadri F., Zappasodi R., Yang A., Budhu S., Malandro N., Hirschhorn-Cymerman D., Tiwari S., Maughan M.F., Olmsted R., Wolchok J.D., Merghoub T. Combination of alphavirus replicon particle-based vaccination with immunomodulatory antibodies: therapeutic activity in the B16 melanoma mouse model and immune correlates. Cancer Immunol. Res. 2014;2:448–458. doi: 10.1158/2326-6066.CIR-13-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durso R.J., Andjelic S., Gardner J.P., Margitich D.J., Donovan G.P., Arrigale R.R., Wang X., Maughan M.F., Talarico T.L., Olmsted R.A. A novel alphavirus vaccine encoding prostate-specific membrane antigen elicits potent cellular and humoral immune responses. Clin. Cancer Res. 2007;13:3999–4008. doi: 10.1158/1078-0432.CCR-06-2202. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Hernandez Mde.L., Gray A., Hubby B., Kast W.M. In vivo effects of vaccination with six-transmembrane epithelial antigen of the prostate: a candidate antigen for treating prostate cancer. Cancer Res. 2007;67:1344–1351. doi: 10.1158/0008-5472.CAN-06-2996. [DOI] [PubMed] [Google Scholar]
- 17.Manara C., Brazzoli M., Piccioli D., Taccone M., D’Oro U., Maione D., Frigimelica E. Co-administration of GM-CSF expressing RNA is a powerful tool to enhance potency of SAM-based vaccines. Vaccine. 2019;37:4204–4213. doi: 10.1016/j.vaccine.2019.04.028. [DOI] [PubMed] [Google Scholar]
- 18.Castle J.C., Loewer M., Boegel S., de Graaf J., Bender C., Tadmor A.D., Boisguerin V., Bukur T., Sorn P., Paret C. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genomics. 2014;15:190. doi: 10.1186/1471-2164-15-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fros J.J., Pijlman G.P. Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses. 2016;8:166. doi: 10.3390/v8060166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J., Huang S., Zhao X., Chen M., Lin Y., Xia Y., Sun C., Yang X., Wang J., Guo Y. Poly(I:C) treatment leads to interferon-dependent clearance of hepatitis B virus in a hydrodynamic injection mouse model. J. Virol. 2014;88:10421–10431. doi: 10.1128/JVI.00996-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baitsch L., Baumgaertner P., Devêvre E., Raghav S.K., Legat A., Barba L., Wieckowski S., Bouzourene H., Deplancke B., Romero P. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Appay V., Jandus C., Voelter V., Reynard S., Coupland S.E., Rimoldi D., Lienard D., Guillaume P., Krieg A.M., Cerottini J.-C. New generation vaccine induces effective melanoma-specific CD8+ T cells in the circulation but not in the tumor site. J. Immunol. 2006;177:1670–1678. doi: 10.4049/jimmunol.177.3.1670. [DOI] [PubMed] [Google Scholar]
- 23.Kreiter S., Vormehr M., van de Roemer N., Diken M., Löwer M., Diekmann J., Boegel S., Schrörs B., Vascotto F., Castle J.C. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Florek N.W., Weinfurter J.T., Jegaskanda S., Brewoo J.N., Powell T.D., Young G.R., Das S.C., Hatta M., Broman K.W., Hungnes O. Modified vaccinia virus Ankara encoding influenza virus hemagglutinin induces heterosubtypic immunity in macaques. J. Virol. 2014;88:13418–13428. doi: 10.1128/JVI.01219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hekele A., Bertholet S., Archer J., Gibson D.G., Palladino G., Brito L.A., Otten G.R., Brazzoli M., Buccato S., Bonci A. Rapidly produced SAM(®) vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013;2:e52. doi: 10.1038/emi.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sahin U., Derhovanessian E., Miller M., Kloke B.-P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 27.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J.M., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yost K.E., Satpathy A.T., Wells D.K., Qi Y., Wang C., Kageyama R., McNamara K.L., Granja J.M., Sarin K.Y., Brown R.A. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019;25:1251–1259. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scheper W., Kelderman S., Fanchi L.F., Linnemann C., Bendle G., de Rooij M.A.J., Hirt C., Mezzadra R., Slagter M., Dijkstra K. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019;25:89–94. doi: 10.1038/s41591-018-0266-5. [DOI] [PubMed] [Google Scholar]
- 30.Osada T., Berglund P., Morse M.A., Hubby B., Lewis W., Niedzwiecki D., Yang X.Y., Hobeika A., Burnett B., Devi G.R. Co-delivery of antigen and IL-12 by Venezuelan equine encephalitis virus replicon particles enhances antigen-specific immune responses and antitumor effects. Cancer Immunol. Immunother. 2012;61:1941–1951. doi: 10.1007/s00262-012-1248-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee K.L., Benz S.C., Hicks K.C., Nguyen A., Gameiro S.R., Palena C., Sanborn J.Z., Su Z., Ordentlich P., Rohlin L. Efficient Tumor Clearance and Diversified Immunity through Neoepitope Vaccines and Combinatorial Immunotherapy. Cancer Immunol. Res. 2019;7:1359–1370. doi: 10.1158/2326-6066.CIR-18-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran E., Robbins P.F., Lu Y.-C., Prickett T.D., Gartner J.J., Jia L., Pasetto A., Zheng Z., Ray S., Groh E.M. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mak I.W., Evaniew N., Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014;6:114–118. [PMC free article] [PubMed] [Google Scholar]
- 34.Song Y., Liu Y., Hu R., Su M., Rood D., Lai L. In Vivo Antitumor Activity of a Recombinant IL7/IL15 Hybrid Cytokine in Mice. Mol. Cancer Ther. 2016;15:2413–2421. doi: 10.1158/1535-7163.MCT-16-0111. [DOI] [PubMed] [Google Scholar]
- 35.Moise L., Gutierrez A.H., Bailey-Kellogg C., Terry F., Leng Q., Abdel Hady K.M., VerBerkmoes N.C., Sztein M.B., Losikoff P.T., Martin W.D. The two-faced T cell epitope: examining the host-microbe interface with JanusMatrix. Hum. Vaccin. Immunother. 2013;9:1577–1586. doi: 10.4161/hv.24615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Losikoff P.T., Mishra S., Terry F., Gutierrez A., Ardito M.T., Fast L., Nevola M., Martin W.D., Bailey-Kellogg C., De Groot A.S., Gregory S.H. HCV epitope, homologous to multiple human protein sequences, induces a regulatory T cell response in infected patients. J. Hepatol. 2015;62:48–55. doi: 10.1016/j.jhep.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 37.Wada Y., Nithichanon A., Nobusawa E., Moise L., Martin W.D., Yamamoto N., Terahara K., Hagiwara H., Odagiri T., Tashiro M. A humanized mouse model identifies key amino acids for low immunogenicity of H7N9 vaccines. Sci. Rep. 2017;7:1283. doi: 10.1038/s41598-017-01372-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu R., Moise L., Tassone R., Gutierrez A.H., Terry F.E., Sangare K., Ardito M.T., Martin W.D., De Groot A.S. H7N9 T-cell epitopes that mimic human sequences are less immunogenic and may induce Treg-mediated tolerance. Hum. Vaccin. Immunother. 2015;11:2241–2252. doi: 10.1080/21645515.2015.1052197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moise L., Buller R.M., Schriewer J., Lee J., Frey S.E., Weiner D.B., Martin W., De Groot A.S. VennVax, a DNA-prime, peptide-boost multi-T-cell epitope poxvirus vaccine, induces protective immunity against vaccinia infection by T cell response alone. Vaccine. 2011;29:501–511. doi: 10.1016/j.vaccine.2010.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moise L., Tassone R., Latimer H., Terry F., Levitz L., Haran J.P., Ross T.M., Boyle C.M., Martin W.D., De Groot A.S. Immunization with cross-conserved H1N1 influenza CD4+ T-cell epitopes lowers viral burden in HLA DR3 transgenic mice. Hum. Vaccin. Immunother. 2013;9:2060–2068. doi: 10.4161/hv.26511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffmann P.R., Hoffmann F.W., Premeaux T.A., Fujita T., Soprana E., Panigada M., Chew G.M., Richard G., Hindocha P., Menor M. Multi-antigen Vaccination With Simultaneous Engagement of the OX40 Receptor Delays Malignant Mesothelioma Growth and Increases Survival in Animal Models. Front. Oncol. 2019;9:720. doi: 10.3389/fonc.2019.00720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scholzen A., Richard G., Moise L., Baeten L.A., Reeves P.M., Martin W.D., Brauns T.A., Boyle C.M., Raju Paul S., Bucala R. Promiscuous Coxiella burnetii CD4 Epitope Clusters Associated With Human Recall Responses Are Candidates for a Novel T-Cell Targeted Multi-Epitope Q Fever Vaccine. Front. Immunol. 2019;10:207. doi: 10.3389/fimmu.2019.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moise L., Gutierrez A., Kibria F., Martin R., Tassone R., Liu R., Terry F., Martin B., De Groot A.S. iVAX: An integrated toolkit for the selection and optimization of antigens and the design of epitope-driven vaccines. Hum. Vaccin. Immunother. 2015;11:2312–2321. doi: 10.1080/21645515.2015.1061159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Havenar-Daughton C., Reiss S.M., Carnathan D.G., Wu J.E., Kendric K., Torrents de la Peña A., Kasturi S.P., Dan J.M., Bothwell M., Sanders R.W. Cytokine-Independent Detection of Antigen-Specific Germinal Center T Follicular Helper Cells in Immunized Nonhuman Primates Using a Live Cell Activation-Induced Marker Technique. J. Immunol. 2016;197:994–1002. doi: 10.4049/jimmunol.1600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.