

Abstract
The partial response of chronic hepatitis B virus (CHB) patients to interferon-α (IFN-α) therapy remains elusive, which requires a better understanding of the involved molecular mechanism. In our study, bioinformatics analysis was applied to relate IFN-α regulated candidate genes and RNA editing sites by RNA sequencing. Mitochondrial antiviral signaling protein (MAVS) antiviral effect was confirmed in HepG2.2.15 cells and in two mouse models. The associations between polymorphisms in MAVS gene and response to IFN-α therapy were confirmed in CHB patients. We found that IFN-α downregulates MAVS via RNA editing that was mediated by adenosine deaminase acting on RNA (ADAR1). ADAR1 inhibited MAVS expression via a human antigen R (HuR)-mediated post-transcriptional regulation. MAVS exerted an antiviral activity and reduced the level of hepatitis B virus (HBV) markers in vitro and in vivo. IFN-α antiviral effects were significantly enhanced by MAVS co-transfection. Hepatitis B core protein (HBc) interacted with SP1 to inhibit the promoter activity of MAVS that regulates its expression. CHB patients with a rs3746662A allele had higher MAVS expression and thus were more responsive to IFN-α treatment. In this work, we demonstrated that the decrease of MAVS expression is mediated by the IFN-α-ADAR1 axis. This study also highlighted the potential for the clinical application of MAVS in combination with IFN-α for the treatment of HBV infection.
Keywords: hepatitis B virus, RNA editing, adenosine deaminase acting on RNA, mitochondrial antiviral signaling protein, polymorphism
Graphical Abstract
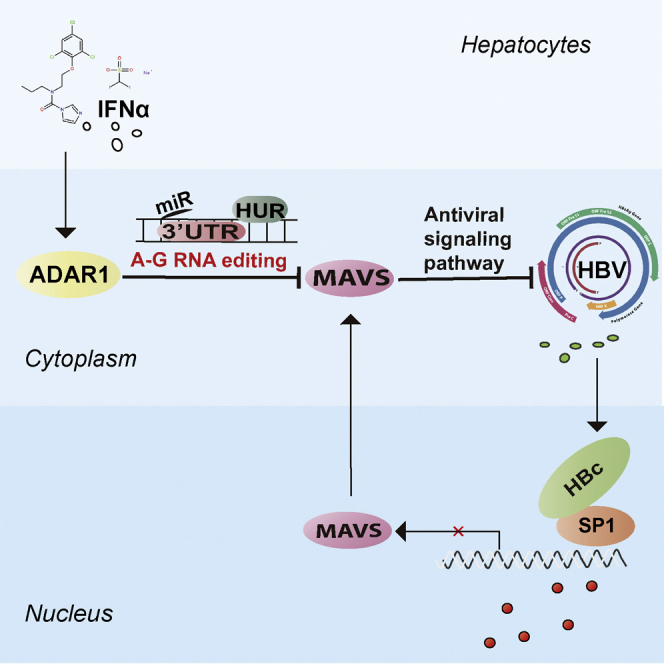
This study revealed the negative role of ADAR1 on expression of MAVS upon IFN-α stimulation. MAVS exerted antiviral activity to inhibit expression of HBV both in vitro and in vivo. The combination of MAVS with IFN-α provides an additional strategy for the treatment of HBV infection.
Introduction
Hepatitis B virus (HBV) is a hepatophilic DNA virus, which is considered a major cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma.1 The World Health Organization estimated that approximately 257 million persons, or 3.5% of the population, were living with chronic HBV (CHB) infection worldwide.2 Currently, interferon-α (IFN-α) and nucleoside analogs are the first-line therapies of CHB. Compared to nucleoside analogs, IFN-α is a promising therapy, given the limited and defined treatment course and the higher likelihood of hepatitis B surface antigen (HBsAg) clearance.3 IFN-α acts by inducing a cellular antiviral response through the activation of immune responses, the regulation of cytokine expression, and the subsequent activation of pathways that cause viral RNA breakdown.4 However, IFN-α therapeutic efficacy in CHB patients is still limited. It has been reported that only 30% of hepatitis B e antigen (HBeAg)-positive and 40% of HBeAg-negative cases have a sustained virological response at 6 months after completion of a 48-week course of Peg-IFN-α treatment.5,6 Hence, understanding the mechanisms of the various IFN therapeutic responses is crucial.
Adenosine deaminase acting on RNA (ADAR1) is an IFN-stimulated gene that catalyzes the covalent modification of highly structured RNA substrates by hydrolytic C-6 deamination of adenosine to yield inosine. A-to-I editing can cause changes in genetic decoding during translation or viral RNA replication because I base pairs as G with C, instead of A with U.7,8 ADAR1 long form (p150) is IFN inducible and localizes to the nucleus and the cytoplasm, whereas the short form (p110) is constitutively expressed and predominantly localizes in the nucleus.9 It has been reported that the IFN-inducible ADAR1 functions in the editing of viral RNA transcripts and cellular pre-mRNAs.10,11 Our previous study revealed that a single nucleotide polymorphism (SNP), rs4845384, in the ADAR1 gene was strongly associated with the outcome of IFN-α therapy in CHB patients.12 The SNP of rs4845384 contributes to HBsAg serum clearance in CHB patients13 through regulating ADAR1 expression.
Mitochondrial antiviral signaling protein (MAVS) acts as an adaptor protein and plays an important role in regulating the host’s innate immune signaling pathway. MAVS aggregates are formed after viral infection and are responsible for IRF3/IRF7 phosphorylation and IFN-β production.14,15 Cell pattern recognition receptors, such as Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), recognize invading pathogens and transmit signals to MAVS,16 which activate nuclear factor-κB (NF-κB) by stimulating downstream TBK1 and IKK complexes, respectively.17,18 Moreover, MAVS interacts with the stimulator of interferon genes (STING), which acts as a DNA sensor that is involved in antiviral cell response. This suggests a role of MAVS in cellular antiviral response,19,20 which is potentially involved in HBV regulation.
In this study, we aimed at discovering the mechanisms of the various responses to IFN-α therapy in CHB patients. Our previous studies have found that ADAR1 upregulates HBV expression through its RNA editing function (X.W., unpublished data); therefore, IFN-α treatment must result in editing ratio changes at the RNA editing sites that are mediated by ADAR1. Our goal is to identify genes and editing sites that are influenced by IFN-α using transcriptome sequencing analysis following IFN-α treatment.
Results
Downregulation of MAVS Expression by IFN-α in the HepG2.2.15 Cell Line
We first performed a transcriptome high-throughput RNA sequencing (RNA-seq) of the HepG2.2.15 cell line that can promote HBV replication and analyzed the genes and RNA editing sites that were changed by IFN-α treatment. A total of 23,576 nucleotide alterations were analyzed by RNA-seq. Then we screened 9,946 editing sites from A to G, or T to C in the transcriptome, and identified 1,492 statistically significant sites. After excluding SNPs, the remaining editing sites were 727 that were located in 292 genes, and among them, 20 were regulated by IFN-α (Figure 1A). Because our aim is to find out why IFN-α is not completely effective in the treatment of HBV infection, we mainly focused on genes that are related to viral immune response. Finally, we screened out 4 genes, including MAVS, C1R, APOL1, and HLA-A. The last 3 genes were antiviral and upregulated by IFN,21, 22, 23 which was consistent with our transcriptome results; only MAVS was abnormally downregulated by IFN-α. To analyze mismatches that are other than AG, we have also performed the exact same computational process with C-to-T and G-to-A mismatches. Among 23,576 positions with nucleic acid alteration acquired by RNA-seq, 7,681 positions underwent C-to-T or G-to-A alteration. We identified 849 statistically significant sites (p < 0.05) after IFN-α treatment, and 161 positions were non-SNP sites; IFN-α treatment increased T/C or A/G ratio of 90 positions located in 66 genes. Finally, HLA-C was determined as an IFN-α-regulated gene with IFN-α editing sites, which were excluded for its normal antiviral function and upregulation by IFN. Therefore, we focused on MAVS as a potential target in the present study. As the mRNA of ADAR1 p150 and p110 share almost the same sequence, we set qPCR primers on exon 8 and exon 10 and measured total mRNA expression of both p150 and p110. We verified that different IFN-α concentrations can influence the expression of ADAR1 and MAVS at the mRNA and protein levels (Figures 1B and 1C). We found that treatment with 500 U/mL IFN-α has the most obvious inhibitory effect on MAVS; therefore, this concentration was used for the following experiments. MAVS downregulation by IFN-α was further confirmed in HepG2, Hep3B, and mouse primary hepatocytes (Figure S1B)
Figure 1.

IFN-α Inhibited the Expression of MAVS in HepG2.2.15 Cells
(A) Twenty candidate genes for differential expression analysis and potential RNA editing screening of ADAR1 via sequence analysis of transcription group. (B) qPCR analysis of mRNA levels of ADAR1 and MAVS at different concentrations of IFN-α. (C) Western blot analysis of ADAR1 P150 and MAVS protein expression at different concentrations of IFN-α normalized to GAPDH. Right panel shows quantification from three replicates. (D) Western blot analysis and quantification of protein levels of ADAR1 and MAVS in ADAR1 overexpressing HepG2.2.15 cells at different concentrations of IFN-α. Data represent the mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We then explored whether IFN-α regulates MAVS expression via ADAR1. After ADAR1 (P150) overexpression, IFN-α could no longer inhibit MAVS expression, suggesting that IFN-α inhibits MAVS through ADAR1 (Figure 1D). In conclusion, IFN-α can stimulate ADAR1 to enhance its RNA editing activity, leading to a lower MAVS expression.
MAVS Is Downregulated by ADAR1 through RNA Editing
To prove that the change of MAVS is truly related to ADAR1, ADAR1 and sh-ADAR1 plasmids were overexpressed in HepG2.2.15 cells, and the MAVS 3′UTR chr20:3870562 editing ratio was measured by Sanger sequencing (Sanger-seq) (Figure 2A). The results showed that ADAR1 editing ability for MAVS 3′UTR A to G is enhanced and that interfering with ADAR1 expression reduces its editing intensity to MAVS 3′UTR. ADAR1 affected MAVS, as MAVS was significantly inhibited following ADAR1 overexpression (Figure 2B left), and remarkably increased following ADAR1 knockdown (Figure 2B, right). These results were confirmed in HepG2, Hep3B, and primary hepatocytes (Figure S2A).
Figure 2.

ADAR1 Inhibited MAVS Expression by RNA Editing
(A) ADAR1-P150, FLAG-NC, sh-ADAR1, and sh-NC were overexpressed in HepG2.2.15 cells, Sanger sequencing was used to detect the chr20:3870562 site of MAVS 3′UTR. (B) HepG2.2.15 cells were transfected with ADAR1-overexpressing or knockdown (sh-ADAR1) plasmids. qPCR analysis of mRNA levels of MAVS. (C) Western blot analysis and quantification of MAVS protein expression. (D) HepG2.2.15 cells were transfected with NC-FLAG, ADAR1, ADAR1 EAA, and ADAR1 E912A plasmid. Western blot analysis and quantification of ADAR1 and MAVS. (E) Pyrosequencing results of RNA editing rates in ADAR1 overexpression, NC-FLAG, sh-ADAR1, and sh-NC groups. (F) HepG2.2.15 cells were transfected with ADAR1 or NC-FLAG plasmid (left) or treatment with IFN-α or PBS negative control (right) and normal A allele or edited G allele reporter plasmid, respectively. NC, non-specific control. Analyzed for relative luciferase activity. Data represent the mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To determine whether ADAR1 RNA editing activity regulates MAVS expression, ADAR1-E912A and ADAR1-EAA,24 two mutants without RNA editing activity (ADAR1-E912A) or RNA binding activity (ADAR1-EAA; i.e., K554E-K555A-K558A, K665E-K666A-K669A, and K777E-K778A-K781A), were used. MAVS was expressed at a low level in ADAR1-expressing cells; however, no difference in MAVS expression was observed in the E912A, EAA, and NC groups, demonstrating that an ADAR1 RNA binding-domain mutant (EAA), such as E912A mutant, failed to suppress MAVS expression (Figure 2D). To further verify its RNA editing activity toward MAVS, we performed pyrosequencing to confirm that MAVS-3′UTR chr20:3870562 is the editing site and that the proportion of the editing A to G allele negatively correlates with sh-ADAR1 expression. There was a 50% elevation in the normal A allele ratio when ADAR1 was knocked down (Figures 2E and S2B). Meanwhile, AZIN1, which is known to be modified (T to C) by ADAR1, was included as a control to ensure the ADAR1 RNA editing activity (Figure S2C). Furthermore, to confirm whether the change in A/G allele frequency at the editing site (chr20:3870562) influences MAVS expression, we conducted luciferase assay using two reporter plasmids, chr20:3870562-A and -G. The relative luciferase activity of the edited G allele was significantly lower than that of the normal A allele in HepG2.2.15 and Hep3B cells (Figure 2F, left, and S2D). The relative luciferase activity significantly decreased when ADAR1 was overexpressed in the chr20:3870562-A group but not in the chr20:3870562-G group (Figure 2F, left). The same trend was found in non-HBV-positive SMMC-7721 cells (Figure S2E). Additionally, when incubated with IFN-α for 48 h, the relative luciferase activity of chr20:3870562-G was comparable between the NC and IFN-α groups, while the activity of chr20:3870562-A decreased after treatment with IFN-α (Figure 2F, right). Furthermore, the ADAR1 effect on the A allele activity was stronger than that of IFN-α (Figure 2F, right). These data suggest that ADAR1 suppresses MAVS expression through MAVS 3′UTR editing.
ADAR1 Inhibits MAVS Expression via Human Antigen R (HuR)-Mediated Post-transcriptional Regulation
The decay rates of relative luciferase activity of the chr20:3870562-A and -G luciferase reporter plasmids were measured in a time-course assay and after actinomycin D mediated inhibition of transcription. The relative activity of the G group decayed more significantly than that of the A group over a 48 h period, indicating that the G allele protein instability was higher than that of the A allele (Figure 3A, left). To determine whether the degradation of mRNA in MAVS is affected by the expression of ADAR1, we performed an mRNA decay assay, and the result suggested that the MAVS mRNA degradation rate is accelerated after ADAR1 overexpression (Figure 3A, right). These data demonstrate that the 3′UTR editing changed MAVS mRNA stability.
Figure 3.

ADAR1 Inhibits MAVS Expression by HuR-Mediated Post-transcriptional Regulation
(A) The decay rate of relative luciferase activity of normal A allele or edited G allele after using actinomycin D (1 μg/mL) in HepG2.2.15 cells. HepG2.2.15 cells were transfected with NC-FLAG and ADAR1 plasmid. The decay rate of mRNA was detected at prescribed time points after using actinomycin D (1 μg/mL) is shown. (B) Luciferase assay of A or G alleles after transfected miRNAs in HepG2.2.15 cells. (C) The interaction of HuR with the MAVS 3′UTR by RNA immunoprecipitation (RIP) in HepG2.2.15 cells. Western blot analysis for HuR protein following RIP with a HuR-specific antibody or an IgG control and qPCR analysis of MAVS 3′UTR mRNA in HepG2.2.15 cells are shown. (D and E) RIP analysis after transiently transfecting with 3870562-A and 3870562-G luciferase reporter plasmids via gel electrophoresis and qPCR. Data represent the mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To explore the downstream mechanism of ADAR1 3′UTR editing activity in regulating MAVS expression, we hypothesized on the potential ways that affect mRNA stability through 3′UTR. Frequently, microRNAs (miRNAs) target the 3′UTR and reduce the stability of mRNAs.25 By matching the miRNA database http://microrna.org/,26 we identified 2 miRNAs that may target the 3′UTR around the chr20:3870562 site. The two miRNAs were expressed via miRNA mimics, and no difference in luciferase activity was observed among the 3 groups: miR-1260, miR-1260b, and miR-NC (Figure 3B). Next, we analyzed other potential ways that may regulate the stability of mRNAs and found that the sequence contained several adenosine- and uridine-rich elements (AREs) around the MAVS-3′UTR editing site (Figure S3). Previous studies have shown that mammals can code a protein, HuR (also named ELAVL1), that binds to almost all types of AREs and that impairs mRNA stability to control gene expression by enabling HuR-mediated post-transcriptional regulation.27,28 To determine whether ADAR1-mediated RNA editing in MAVS 3′UTR would influence HuR recruitment and, consequently, affect MAVS mRNA stability, we first detected HuR potential binding to MAVS-3′UTR by immunoprecipitation using extracts from HepG2.2.15 cells (Figure 3C). When transiently transfecting chr20:3870562-A and -G luciferase reporter plasmids followed by HuR-RIP, significantly higher amounts of A sequence were immunoprecipitated compared with that of G, as detected by gel electrophoresis and qPCR (Figures 3D and 3E). This result indicated that chr20:3870562-A and -G affinities to HuR are different. Altogether, these data showed that ADAR1-mediated RNA editing can regulate the MAVS mRNA stability at the post-transcriptional level.
MAVS Has an Anti-HBV Effect, and the Combined Use with IFN-α Could Enhance the Antiviral Effect of IFN-α in HBV-Positive Cell Lines
To assess the impact of the MAVS signaling pathway on the clearance of HBV infection in HBV-positive cell lines, plasmids that overexpress or knock down MAVS were transiently transfected into HepG2.2.15 cells, and MAVS protein level was examined by western blot analysis (Figure 4A, left). Compared with the control group, cell-attached slides were stained with hepatitis B virus X (HBx), which confirmed that MAVS overexpression could significantly decrease HBx expression in HepG2.215 cells (Figure 4A, right). Moreover, when measuring HBsAg concentrations, we found that MAVS reduced HBV replication, HBeAg, and HBV-DNA expression in the supernatant (Figures 4B, upper panels, and S4A). The levels of intracellular HBV total RNA, pre-genomic RNA (pgRNA), and covalently closed circular DNA (cccDNA), which reflect HBV expression efficiency, were negatively correlated with MAVS expression level (Figure 4B, upper panels, and S4A), while high HBV expression activities were noticed in MAVS knocked down cells (Figure 4B, lower panels, and S4B). Furthermore, MAVS overexpression was associated with the attenuation of HBc protein expression compared with that in the control group (Figure 4D). The MAVS signaling pathway can activate the expression of type I IFN and mainly secrete IFN-β to inhibit virus replication in hepatocytes. ELISA of IFN-β also supported this result and showed that the expression of MAVS enhances IFN-β production (Figure 4C). In addition, MAVS activates IRF3/IRF7 by upregulating the phosphorylation of IRF3 and IRF7 (Figure S4C). Because HepG2.2.15 cells weakly expressed HBV, we co-transfected 1.3∗HBV genotype B isolate S64 on Puc18 plasmid and MAVS plasmid in HepG2 cells to confirm the effect of MAVS on HBV expression. The supernatant HBsAg and HBV-DNA levels were significantly reduced by 50% compared with the control group. Meanwhile, the intracellular expression of cccDNA and pgRNA in the MAVS overexpression group were significantly lower than those in the control group (Figures 4E and S4D). These data demonstrated that MAVS expression inhibits HBV replication at the DNA replication and transcription, protein expression, and viral antigen packaging levels.
Figure 4.

MAVS Inhibits the Expression Levels of HBV Markers In Vitro
(A) HepG2.2.15 cells were transfected with MAVS-overexpressing or knockdown (sh-MAVS) plasmid. Western blot analysis of MAVS protein expression normalized to GAPDH, which served as a protein loading control and IHC of cell-attached slides assay detected the HBx protein expression level are shown. (B) ELISA analysis of HBsAg in HepG2.2.15 cell supernatants and qRT-PCR determination of HBV cccDNA levels. Cells were transfected with the MAVS overexpression or knockdown plasmid. (C) ELISA analysis of IFN-β expression in HepG2.2.15 cells in MAVS overexpression, NC-FLAG, sh-MAVS, and sh-NC groups. (D) Western blot analysis and quantification of HBc expression in HepG2.2.15 cells in MAVS overexpression, NC-FLAG, sh-MAVS, and sh-NC groups. (E) ELISA analysis of HBsAg in HepG2.2.15 cell supernatants and qRT-PCR determination of HBV DNA, cccDNA, and pgRNA levels. Cells were co-transfected with the HBV and MAVS overexpression or NC plasmid. (F) HepG2.2.15 cells were transfected with MAVS-overexpressing plasmid or the empty FLAG vector and treated with or without IFN-α. ELISA analysis of HBsAg in HepG2.2.15 cell supernatants and qRT-PCR determination of HBV cccDNA and HBV pgRNA levels are shown. Data represent the mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
IFN-α is a conventional medication for treating CHB patients, but our data demonstrated that it could reduce MAVS expression, which might weaken the MAVS antiviral signal pathway. On the contrary, when MAVS was overexpressed in HepG2.2.15 cells, the effect of ADAR1 on MAVS was masked (Figure S4E). Therefore, we hypothesized that MAVS expression could enhance the antiviral effects of IFN-α. IFN-α notably decreased the level of HBV markers compared with NC, while this effect was strikingly enhanced when combined with MAVS overexpression (Figures 4F and S4F). These results supported the conclusion that the antiviral effects of IFN-α can be enhanced by using compensatory MAVS.
The Combined Application of MAVS and IFN-α Has a Better Anti-HBV Response in Two HBV Transgenic Mouse Models
To assess the in vivo antiviral effects of MAVS and IFN-α combined treatment, similar studies were performed in C57BL/6J-Tg (Alb1HBV)44Bri/J mice expressing both HBsAg and HBx proteins. To simulate the effect of MAVS, hydrodynamic transfection of mice with the MAVS plasmid was performed, and we investigated its effect 10 days after the injection (Figure S5A). Compared with the NC group, HBsAg expression in sera was decreased in IFN-α + NC group by day 5, and the decrease in IFN-α + MAVS group was more noticeable (Figure 5A). Moreover, the expression of serum HBsAg in the IFN-α + MAVS group decreased by close to 2/3 at 10 days post-injection. When compared with the IFN-α + NC group, this downregulation was decreased by nearly half 10 days post-injection (Figure 5A). A similar trend for the HBsAg particle was observed in hepatic parenchymal cells using immunohistochemistry on the tenth day after primary DNA injection. We also observed lower expression and smaller numbers of hepatocytes positive for HBx in the IFN-α + MAVS group compared with other groups (Figure 5B).
Figure 5.

MAVS Inhibits HBV Replication in Two HBV Tg Mouse Strains
(A) ELISA analysis of HBsAg in serum after hydrodynamic injection and treatment of IFN-α (105 U/kg) and MAVS DNA 20 μg (n = 6). (B) IHC assay of HBsAg and HBx in the liver of HBV mouse (n = 6). Scale bar, 50 μm. (C and D) Continuous ELISA analysis of HBsAg and qPCR analysis of HBV DNA level in serum after AAV injection (n = 8). (E) IHC assay of HBs, HBc, NLRX1, and CASP1 in the liver of HBV mouse. (F) Analysis of HBsAg and HBV DNA levels in serum of the full HBV Tg mouse after AAV-NC or AAV-MAVS injection and treated with or without IFN-α (n = 4). Analysis of the full HBV Tg mouse liver Ifnb1 mRNA level after AAV-NC or AAV-MAVS injection and treated with or without IFN-α (n = 4) is shown. Scale bar, 50 μm. Data represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The HBV replication ability is mainly controlled by the transcriptional state of HBV DNA and the yield of pgRNA. However, HBV transgenic B6 mice only expressed HBs and HBx proteins. More importantly, to further assess the effect of MAVS on the HBV genome, full-HBV-Tg mice were infected with AAV-MAVS, an adeno-associated virus that specifically acts on the liver for a long term and stably expresses MAVS, to verify its impact on HBV DNA, pgRNA, and other HBV replication markers. To verify the effectiveness of MAVS expression, the spatial expression profile showed that the MAVS gene was specifically expressed in liver and had an obvious inhibitory effect on HBc in the full-HBV-Tg mice liver (Figure S5B). After 3, 5, and 7 weeks of AAV infection, we continuously monitored the changes in HBsAg and HBV DNA content in the mice serum. ELISA for HBsAg indicated that the expression levels accumulated over time in the mice, but MAVS inhibitory effect on HBsAg also enhanced with time (Figure 5C). Similar results were obtained with a serum HBV-DNA level that was assessed by qPCR (Figure 5D). After 7 weeks of infection, the hepatocyte content in HBV pgRNA and total RNA in the AAV-MAVS group was lower than that in the control group, indicating that the replication of HBV might be repressed by AAV-MAVS (Figures S5C and S5D). Immunohistochemistry (IHC) analysis yielded similar results and confirmed that the frequencies of HBsAg and HBcAg-positive cells in the AAV-MAVS group were significantly lower than those in the AAV-NC group. In addition, the positive rate of the inflammatory factors NLRX1 and CASP1, and the glutamic-pyruvic transaminase/glutamic-oxalacetic transaminase (ALT/AST) liver damage, were all attenuated compared to those in the AAV-NC group (Figures 5E and S5E). Altogether, these data indicated that MAVS could suppress HBV expression, inflammatory reaction, and liver damage.
Next, mice were divided into four groups: the BALB/c-wild-type (WT) group, the BALB/c-full-HBV-Tg NC group, the BALB/c-full-HBV-Tg mice treated with IFN-α group, and the BALB/c-full-HBV-Tg mice treated with IFN-α + MAVS group. Similar experimental methods were used to monitor the expression of HBV markers, respectively. The IFN-α group notably decreased the HBsAg, HBV-DNA, and pgRNA levels compared with the HBV NC group, while this effect was strikingly enhanced when combined with MAVS overexpression (Figures 5F and S5F). In addition, the level of IFNb1 in the full-HBV-Tg mice livers was lower than that in the BALB/c-WT mice. Compared with the IFN-α group, the expression of IFNb1was significantly higher in the IFN-α + MAVS group, suggesting that MAVS might inhibit HBV replication via stimulating IFNb1 (Figure 5F). In summary, MAVS efficiency could enhance the anti-HBV effects of IFN-α.
HBc Inhibits MAVS Promoter Activity through Its Interaction with SP1
Besides the IFN-α inhibitory effect of MAVS, we found that MAVS expression in the HepG2.2.15 cell line was lower than that in HepG2 cell line (Figure S6A). Therefore, we hypothesized that HBV could inhibit the expression of MAVS. First, we overexpressed the HBV plasmid in HepG2 cells and found that MAVS mRNA expression in HepG2 cells was significantly lower than that in the control group (Figure S6B). To verify if HBV influences MAVS in natural infection models, sodium taurocholate cotransporting polypeptide (NTCP)-HepG2 cells were incubated in the presence or absence of HBV virus for 16 h. Western blot assays revealed that MAVS protein level in the HBV-infected NTCP-HepG2 cells was clearly downregulated (Figure S6C).
Next, considering the studies that found that HBx, HBc, and the viral small molecular proteins were frequently involved in the immune regulation, we hypothesized that HBx or HBc protein may acts on MAVS. HBx and HBc expression plasmids were constructed, and the results showed that HBc, but not HBx, suppresses MAVS mRNA expression level (Figure 6A). Moreover, HBc overexpression elicited a robust repression of the MAVS protein level in HepG2.2.15 cells (Figure 6B).
Figure 6.

SP1 Regulates the Expression of MAVS by Interacting with HBc
(A) HepG2 cells were transfected with FLAG-NC, HBx, or HBc plasmid. qPCR analysis of MAVS mRNA expression is shown. (B) HepG2.2.15 cells were transfected with FLAG-NC or HBc plasmid. Western blot analysis and quantification of MAVS protein expression is shown. (C) MAVS mRNA level in liver tissues of the full HBV Tg mouse was detected by qPCR. (D) Detection of the activity of MAVS promoter fragment by dual luciferase reporter gene assay. (E) The relative luciferase activity of promoter-full and promoter-3 was detected after overexpression of HBc and FLAG. (F) After overexpression of HBc and FLAG, the level of MAVS promoter-3 enriched by SP1 was detected. (G) After the overexpression of HBc and SP1 in the HepG2.2.15 cells, they were detected by coIP. Data represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Then, we compared MAVS expression between the full-length HBV Tg and BALB/c-WT mice. The full-length HBV Tg mice had a significantly lower MAVS mRNA expression level in the mouse liver (Figure 6C). Because HBc could repress MAVS expression at the transcriptional level, we constructed a luciferase reporter plasmid containing 672 bp (−657/14, 0 means the transcriptional start site) of the MAVS promoter (named promoter-full) to verify that HBc has an effect on its promoter. We also constructed 3 additional luciferase reporter plasmids containing the early, middle, and late segments of the full MAVS promoter (promoter-1, -2, and -3, respectively). The luciferase assays showed that only promoter-3 (−211/14) had apparent promoter activity after segmentation (Figure 6D). The activity of the full MAVS promoter and promoter-3 could be significantly inhibited by HBc overexpression (Figure 6E). These results indicated that HBc mainly inhibits the activity of the MAVS promoter through its effect on the region 226 bp (−211/14), which is the closest to the transcriptional start site.
Furthermore, we elucidated the mechanism by which HBc inhibits the MAVS promoter. Given that HBc can interact with a transcription factor to regulate the promoter activity of the host gene,29 we predicted the transcription factor binding sites in the promoter region −211/14, using the online promoter analysis tool, the JASPAR database (http://jaspar.genereg.net/). We found that there were several predicted SP1 transcription factor binding sites on the sequence of this promoter (Figure S6D). The luciferase reporter assays also showed that SP1 overexpression could increase the promoter activity of MAVS promoter-3 (Figure S6E) and that SP1 could directly interact with MAVS promoter as revealed by electrophoretic mobility shift assay (EMSA) (Figure S6F). Chromatin immunoprecipitation (ChIP) assays revealed that SP1 was able to bind to the MAVS promoter and that HBc could decrease the interaction between SP1 and the MAVS promoter (Figure 6F). We also investigated whether HBc affects the expression of SP1, and the results showed that HBc had no effect on the expression of SP1 at mRNA and protein levels (Figure S6G). Therefore, we speculated on whether there is a protein interaction between HBc and SP1. The co-immunoprecipitation (coIP) assays further demonstrated that HBc interacts with SP1 in HepG2.2.15 cells (Figure 6G). Overall, we suggest that HBc decreases the SP1 binding to the MAVS promoter by interacting with SP1, thus inhibiting MAVS expression.
Polymorphisms in the MAVS Gene Affect the Response to IFN Therapy of Chronic Hepatitis B in Chinese Han
We performed pharmacogenetic assays to analyze if MAVS polymorphisms are associated with the response to IFN-α therapy. Six polymorphisms, rs17857295, rs2326369, rs3746660, rs3746661, rs3746662, and rs3899452, were genotyped in 240 CHB patients. Patients were evaluated for the efficacy of IFN-α by the combined assessment criteria. The overall sustained virologic response (SVR) rate among CHB patients was 30.8% (74/240). There was no significant difference in age and gender distributions or in mean levels of ALT and AST between SVR and no response (NR) patients (Table S1). The genotype distributions of the studied polymorphisms were in Hardy-Weinberg equilibrium. In the single locus analysis, the frequency of the C allele of rs3746662 was significantly lower in the SVR group than in the NR group (4.7% versus 13.4%; p = 0.005). The genotype distributions of this polymorphism also pointedly contrasted between the SVR and NR groups (p = 0.02) (Table S2). Patients with an AA genotype had a significantly higher SVR rate than that in AC or CC carriers (35.1% versus 14.9%; p = 0.007) (Figure 7A). We were also interested in determining whether this polymorphism had a biological function. Rs3746662 is located at 531bp downstream of the MAVS transcription terminator. Interestingly, this position is also in the MAVS-3′UTR area, the same area as the ADAR1 RNA editing site. Therefore, a MAVS-rs3746662(A/C) luciferase plasmid was constructed to compare the relative luciferase activities. The data confirmed that the rs3746662A allele had a potently higher luciferase activity than that of the rs3746662C allele, indicating that the rs3746662A allele might have a higher protein production (Figure 7B). Furthermore, MAVS IHC analysis in 64 HCC patients confirmed that patients with a rs3746662AA genotype had a higher MAVS expression compared to that of the AC and CC genotypes in hepatocarcinoma paratumors (Figure 7C). Altogether, these data show that CHB patients who carry the rs3746662A allele have a better MAVS expression and, thus, have potentially a better response to the IFN-α treatment of HBV.
Figure 7.

Polymorphisms in the MAVS Gene Affect Response to IFN Therapy for Chronic Hepatitis B in Han Chinese
(A) SVR rate between AG (or GG) and AA genotypes. (B) The luciferase assay of rs3746662C allele and rs3746662A allele. (C) IHC assay of MAVS in human liver tissues. Scale bar, 50 μm. Data represent the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
In this study, we report that IFN-α increases the expression of ADAR1 P150 in HepG2.2.15 cells, and that ADAR1 can edit the schr20:3870562 site in the 3′UTR of the MAVS mRNA, resulting in changes of its stability via a HuR-mediated post-transcriptional regulation. Preceding studies also showed that IFN-α could rouse the IFN-stimulated response element (ISRE) in the ADAR1 promoter region to specifically induce the production of ADAR1 P150.30,31 In addition, almost 1% of cellular transcripts are extensively edited in the IFN-inducible cytoplasmic isoform ADAR1 P150, which is more than the constitutively expressed nuclear isoform ADAR1 P110.32 In addition, there were reports that showed that RNA editing enables the recruitment of the stabilizing RNA-binding protein HuR to the 3′UTR of the CTSS transcript, thereby controlling CTSS mRNA stability and expression.28 Although the edited CTSS is more stable, this is not the case for the edited MAVS, which may be related to the different binding affinity between the binding site and the protein HuR. HuR is protective in our results, while in the Xu et al.33 report, HuR could destabilize the mRNA and maintains MAVS protein at low levels, which is possibly due to the HepG2.2.15 cell line that was used in this study compared to HeLa cells in their report. Moreover, this can also be explained by the assays used that were mostly performed in vitro in the Xu et al.33 study. It has also been proved that HuR plays a protective role in the regulation of CTSS stability.
In this study, the editing site at MAVS is located within the Alu repeats. Editing of Alu within the UTR is common and exists in thousands of genes. Hence, the regulation mechanism may be involved in a huge portion of the human transcriptome. However, the Alu sequence is prevalent in human genomes, not in other non-primate mammals.34 We assumed this mechanism was not conserved across mammalian genomes, where there could be more different mechanisms participating in the regulation of MAVS and antiviral defense.
Recent studies showed that the induction of ADAR1 P150 isoform by IFN specifically regulates the MDA5-MAVS dependent autoimmunity pathway, where ADAR1 acts as a positive regulator of the MDA5-MAVS antiviral response.35 The Adar1 knockout mutation is lethal by embryonic day E12.5 in mice; however, this ADAR1-induced lethality changed in Adar1 and Mavs double-knockout mice, suggesting that Adar1ko leads to an uncontrolled MAVS pathway.36 The MAVS-mediated IFN response to measles virus (MeV) infection, which is effective at later stages of antiviral response, is also regulated by ADAR1.32 In addition, the ADAR1 knockout leads to the enhancement of MAVS-mediated immune infiltration of tumor cells. These reports are consistent with our results that showed an inhibition of MAVS expression by ADAR1.37 However, the role of ADAR1 in the RLRs-MAVS signaling pathway has been reported by Pujantell et al.,38 which showed that ADAR1 knockdown increases the expression of MDA5, RIG-I, IRF7, and phsopho-STAT1, but not MAVS. This might be due to the different efficacy of ADAR1 knockdown between macrophages in Pujantellet al.38 and HepG2.2.15 cells in our study. Bajad et al.39 argued that AdarΔ7–9 mice are editing deficient and die at E12.5 showing heightened immune response. While most AdarΔ7–9 Mavs−/− mice lived until ∼15 days after birth, a few mice survived for several months, which revealed that ADAR1 negatively regulates MAVS and is consistent with our results.39 Furthermore, our study also indicates that ADAR1 inhibitory effects can be attributed to the RNA editing in the chr20:3870562 site of the MAVS 3′UTR (Figure 2D).
It is known that MAVS is essential in driving antiviral innate immunity in response to viral infection.40,41 The MAVS antiviral signal is initiated when RIG-I and MDA5 sense viral dsRNA. This signal is then propagated through the assembly of a MAVS complex, containing TRAF3, TRAF6, TANK, and TRADD, which induces IRF3/7 phosphorylation and the production of IFN type I and pro-inflammatory cytokines.42,43 However, its direct role in DNA viral HBV infection remains to be elucidated. In this study, we confirmed that MAVS overexpression could restrict HBV replication in HBV-positive cell lines and HBV transgenic mice. Whether HBV stimulates the MAVS signaling pathway remains to be further studied.
IFN-α is one of the first-line treatments for CHB patients; however, approximately 30% of patients respond to it. The rs7574865 site of STAT4 is a steady forecaster of IFN-α treatment response in HBeAg-positive CHB patients.44 In this study, we found that the MAVS higher expression in the rs3746662AA allele has a better response to IFN-α therapy in CHB patients. This allele is also located in the MAVS 3′UTR region, suggesting that the 3′UTR of MAVS mRNA has an association with the response to IFN-α.
Our data demonstrated that the decrease of MAVS expression attenuates its antiviral effect through the IFN-α -ADAR1 axis. While suppressed, the MAVS antiviral signal pathway leads to the inhibition of the expression of the endogenous type I IFNs.45 However, IFN-α is only produced in leukocytes; therefore, HepG2.2.15 cells only produce IFN-β, not IFN-α. At this point, further studies are still required to confirm whether IFN-α can inhibit the expression of endogenous IFN-α through MAVS in CHB, which may destroy the homeostasis of natural antiviral immunity.
In addition, HBX interacted with MAVS and promoted the degradation of MAVS through the ubiquitination of its Lys136, thus preventing IFN-β induction.46 MAVS may also be relevant, as HBV replicates through an RNA intermediate.47 These strongly indicate that enhancing the MAVS antiviral pathway would be a promising strategy in promoting the production of endogenous type I IFNs and to further improve IFN response in CHB patients.
In addition, the stimulation of IFN production in the murine liver through the administration of synthetic RNA decreases the viral infection in an adenovirus-HBV model, demonstrating that IFN possesses an anti-HBV activity in the liver. However, hepatocytes do not produce IFN type I in response to foreign DNA or HBV infection in humans and mice.48 This suggests that it is a good therapeutic strategy to inhibit HBV replication through the RNA-activated MAVS-mediated IFN pathway.
In conclusion, our study demonstrated that IFN-α inhibits the expression of MAVS via ADAR1 through RNA editing of the MAVS 3′UTR and HuR-mediated post-transcriptional regulation (Figure 8). The IFN-α antiviral effects are also enhanced by a combined treatment with MAVS in vivo and in vitro. These imply that the therapy based on MAVS could be a promising strategy to improve the effect of IFN-α treatment in patients with HBV infection.
Figure 8.

Schematic Representation of Present Study
Materials and Methods
Additional materials and methods can be found in the Supplemental Materials and Methods.
Cell Cultures
HepG2.2.15 cells were purchased from China Center for Type Culture Collection (Wuhan, Hubei, P.R. China), and HepG2 and Hep3B cells were purchased from the Chinese Academy of Medical Sciences Cell Culture Center (Beijing, P.R. China). NTCP-HepG2 cells were kind gifts from professor Wenhui Li (National Institute of Biological Sciences, Beijing, P.R. China). The cells were cultured in Minimum Essential Medium and Dulbecco’s Modified Eagle’s Medium (Gibco; Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, USA). The mouse primary hepatocytes were isolated from the livers of female full-HBV-Tg mice and cultured in RPMI-1640 containing 10% FBS, 100 units/mL penicillin, and 0.1 mg/mL streptomycin. The cell lines were maintained at 37°C in a moist atmosphere with 5% CO2.
Human Subjects
A total of 324 IFN-α treatment-naive and HBeAg-positive CHB patients were recruited from the Beijing Youan Hospital between November 2005 and May 2008. The inclusion and exclusion criteria, and the therapeutic methods, were previously described.44 Detailed instructions are provided in the Supplemental Materials and Methods.
Quantification of the Viral RNA and DNA
Following the isolation of total RNA, serum HBV DNA, and cccDNA, their amounts were measured by qRT-PCR using appropriate primer pairs as previously described.49,50
Statistical Analysis
Student’s t test was used for comparison between two independent groups. Hardy-Weinberg equilibrium tests of the genotyped SNPs in patients were performed using the χ2 test. The data are presented as the mean ± SD of three independent experiments. The comparisons for continuous variables, between different groups of subjects, were conducted using a t test (for mean comparison) or the Mann-Whitney U test (for median comparison). p <0.05 was considered statistically significant, and all statistical tests were two-sided. Statistical analyses were performed using Statistical Product and Service Solutions (SPSS) v.20.0.
Acknowledgments
We are thankful to all subjects who participated in this study. This work was supported by grants from the CAMS Innovation Fund for Medical Sciences of China (2018-I2M-1-004 and 2016-I2M-1-004).
Author Contributions
X.W. and Y.L. designed the experiments; T.L., X.Y., J.S., and X.Z. conducted the experiments and wrote the paper; and W.L. and Z.L. collected human subjects enrolled in this study.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.11.031.
Contributor Information
Xiaopan Wu, Email: wuxiaopanpumc@163.com.
Ying Liu, Email: liuyingpumc@163.com.
Supplemental Information
References
- 1.Ganem D., Prince A.M. Hepatitis B virus infection--natural history and clinical consequences. N. Engl. J. Med. 2004;350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization . 2017. Global Hepatitis Report, 2017.https://extranet.who.int/iris/restricted/handle/10665/255016 [Google Scholar]
- 3.Ghany M.G. Current treatment guidelines of chronic hepatitis B: The role of nucleos(t)ide analogues and peginterferon. Best Pract. Res. Clin. Gastroenterol. 2017;31:299–309. doi: 10.1016/j.bpg.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Rang A., Bruns M., Heise T., Will H. Antiviral activity of interferon-alpha against hepatitis B virus can be studied in non-hepatic cells and Is independent of MxA. J. Biol. Chem. 2002;277:7645–7647. doi: 10.1074/jbc.C100729200. [DOI] [PubMed] [Google Scholar]
- 5.Perrillo R. Benefits and risks of interferon therapy for hepatitis B. Hepatology. 2009;49(5, Suppl):S103–S111. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- 6.Kim V., Abreu R.M., Nakagawa D.M., Baldassare R.M., Carrilho F.J., Ono S.K. Pegylated interferon alfa for chronic hepatitis B: systematic review and meta-analysis. J. Viral Hepat. 2016;23:154–169. doi: 10.1111/jvh.12418. [DOI] [PubMed] [Google Scholar]
- 7.George C.X., Gan Z., Liu Y., Samuel C.E. Adenosine deaminases acting on RNA, RNA editing, and interferon action. J. Interferon Cytokine Res. 2011;31:99–117. doi: 10.1089/jir.2010.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hogg M., Paro S., Keegan L.P., O’Connell M.A. RNA editing by mammalian ADARs. Adv. Genet. 2011;73:87–120. doi: 10.1016/B978-0-12-380860-8.00003-3. [DOI] [PubMed] [Google Scholar]
- 9.Patterson J.B., Samuel C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol. Cell. Biol. 1995;15:5376–5388. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bass B.L. RNA editing and hypermutation by adenosine deamination. Trends Biochem. Sci. 1997;22:157–162. doi: 10.1016/s0968-0004(97)01035-9. [DOI] [PubMed] [Google Scholar]
- 11.Bass B.L., Nishikura K., Keller W., Seeburg P.H., Emeson R.B., O’Connell M.A., Samuel C.E., Herbert A. A standardized nomenclature for adenosine deaminases that act on RNA. RNA. 1997;3:947–949. [PMC free article] [PubMed] [Google Scholar]
- 12.Wu X., Xin Z., Zhu X., Pan L., Li Z., Li H., Liu Y. Polymorphisms in ADAR1 gene affect response to interferon alpha based therapy for chronic hepatitis B in Han Chinese. Antiviral Res. 2012;94:272–275. doi: 10.1016/j.antiviral.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Wu X., Shi W., Wu J., Zhu X., Chen K., Zheng S., Li Z., Duan Z., Li H., Liu Y. A functional polymorphism in ADAR1 gene affects HBsAg seroclearance both spontaneously and interferon induced. Liver Int. 2014;34:1560–1565. doi: 10.1111/liv.12444. [DOI] [PubMed] [Google Scholar]
- 14.Liu B., Gao C. Regulation of MAVS activation through post-translational modifications. Curr. Opin. Immunol. 2018;50:75–81. doi: 10.1016/j.coi.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K.J., Takeuchi O., Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 16.Jacobs J.L., Coyne C.B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 2013;425:5009–5019. doi: 10.1016/j.jmb.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seth R.B., Sun L., Ea C.K., Chen Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 18.Hou F., Sun L., Zheng H., Skaug B., Jiang Q.X., Chen Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishikawa H., Ma Z., Barber G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crux N.B., Elahi S. Human Leukocyte Antigen (HLA) and Immune Regulation: How Do Classical and Non-Classical HLA Alleles Modulate Immune Response to Human Immunodeficiency Virus and Hepatitis C Virus Infections? Front. Immunol. 2017;8:832. doi: 10.3389/fimmu.2017.00832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong X., Chen M., Ding M., Zhong M., Li B., Wang Y., Fu S., Yin X., Guo Z., Ye J. C1r and C1s from Nile tilapia (Oreochromis niloticus): Molecular characterization, transcriptional profiling upon bacterial and IFN-γ inductions and potential role in response to bacterial infection. Fish Shellfish Immunol. 2017;70:240–251. doi: 10.1016/j.fsi.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 23.Nichols B., Jog P., Lee J.H., Blackler D., Wilmot M., D’Agati V., Markowitz G., Kopp J.B., Alper S.L., Pollak M.R., Friedman D.J. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2015;87:332–342. doi: 10.1038/ki.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen T., Xiang J.F., Zhu S., Chen S., Yin Q.F., Zhang X.O., Zhang J., Feng H., Dong R., Li X.J. ADAR1 is required for differentiation and neural induction by regulating microRNA processing in a catalytically independent manner. Cell Res. 2015;25:459–476. doi: 10.1038/cr.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vasuri F., Visani M., Acquaviva G., Brand T., Fiorentino M., Pession A., Tallini G., D’Errico A., de Biase D. Role of microRNAs in the main molecular pathways of hepatocellular carcinoma. World J. Gastroenterol. 2018;24:2647–2660. doi: 10.3748/wjg.v24.i25.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Betel D., Wilson M., Gabow A., Marks D.S., Sander C. The microRNA.org resource: Targets and expression. Nucleic. Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barreau C., Paillard L., Osborne H.B. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2006;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stellos K., Gatsiou A., Stamatelopoulos K., Perisic Matic L., John D., Lunella F.F., Jaé N., Rossbach O., Amrhein C., Sigala F. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat. Med. 2016;22:1140–1150. doi: 10.1038/nm.4172. [DOI] [PubMed] [Google Scholar]
- 29.Lee S.G., Rho H.M. Transcriptional repression of the human p53 gene by hepatitis B viral X protein. Oncogene. 2000;19:468–471. doi: 10.1038/sj.onc.1203312. [DOI] [PubMed] [Google Scholar]
- 30.Samuel C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.George C.X., John L., Samuel C.E. An RNA editor, adenosine deaminase acting on double-stranded RNA (ADAR1) J. Interferon Cytokine Res. 2014;34:437–446. doi: 10.1089/jir.2014.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfaller C.K., Donohue R.C., Nersisyan S., Brodsky L., Cattaneo R. Extensive editing of cellular and viral double-stranded RNA structures accounts for innate immunity suppression and the proviral activity of ADAR1p150. PLoS Biol. 2018;16:e2006577. doi: 10.1371/journal.pbio.2006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu L., Peng L., Gu T., Yu D., Yao Y.G. The 3'UTR of human MAVS mRNA contains multiple regulatory elements for the control of protein expression and subcellular localization. Biochim. Biophys. Acta. Gene Regul. Mech. 2019;1862:47–57. doi: 10.1016/j.bbagrm.2018.10.017. [DOI] [PubMed] [Google Scholar]
- 34.Tsirigos A., Rigoutsos I. Alu and b1 repeats have been selectively retained in the upstream and intronic regions of genes of specific functional classes. PLoS Comput. Biol. 2009;5:e1000610. doi: 10.1371/journal.pcbi.1000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pestal K., Funk C.C., Snyder J.M., Price N.D., Treuting P.M., Stetson D.B. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity. 2015;43:933–944. doi: 10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mannion N.M., Greenwood S.M., Young R., Cox S., Brindle J., Read D., Nellåker C., Vesely C., Ponting C.P., McLaughlin P.J. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014;9:1482–1494. doi: 10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H., Golji J., Brodeur L.K., Chung F.S., Chen J.T., deBeaumont R.S., Bullock C.P., Jones M.D., Kerr G., Li L. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med. 2019;25:95–102. doi: 10.1038/s41591-018-0302-5. [DOI] [PubMed] [Google Scholar]
- 38.Pujantell M., Riveira-Muñoz E., Badia R., Castellví M., Garcia-Vidal E., Sirera G., Puig T., Ramirez C., Clotet B., Esté J.A., Ballana E. RNA editing by ADAR1 regulates innate and antiviral immune functions in primary macrophages. Sci. Rep. 2017;7:13339. doi: 10.1038/s41598-017-13580-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bajad P., Ebner F., Amman F., Szabó B., Kapoor U., Manjali G., Hildebrandt A., Janisiw M.P., Jantsch M.F. An internal deletion of ADAR rescued by MAVS deficiency leads to a minute phenotype. Nucleic Acids Res. 2020;48:3286–3303. doi: 10.1093/nar/gkaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar H., Kawai T., Kato H., Sato S., Takahashi K., Coban C., Yamamoto M., Uematsu S., Ishii K.J., Takeuchi O., Akira S. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006;203:1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ning X., Wang Y., Jing M., Sha M., Lv M., Gao P., Zhang R., Huang X., Feng J.M., Jiang Z. Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol. Cell. 2019;74:19–31.e7. doi: 10.1016/j.molcel.2019.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Michallet M.C., Meylan E., Ermolaeva M.A., Vazquez J., Rebsamen M., Curran J., Poeck H., Bscheider M., Hartmann G., König M. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity. 2008;28:651–661. doi: 10.1016/j.immuni.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 43.Oganesyan G., Saha S.K., Guo B., He J.Q., Shahangian A., Zarnegar B., Perry A., Cheng G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 44.Jiang D.K., Wu X., Qian J., Ma X.P., Yang J., Li Z., Wang R., Sun L., Liu F., Zhang P. Genetic variation in STAT4 predicts response to interferon-α therapy for hepatitis B e antigen-positive chronic hepatitis B. Hepatology. 2016;63:1102–1111. doi: 10.1002/hep.28423. [DOI] [PubMed] [Google Scholar]
- 45.Zevini A., Olagnier D., Hiscott J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017;38:194–205. doi: 10.1016/j.it.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei C., Ni C., Song T., Liu Y., Yang X., Zheng Z., Jia Y., Yuan Y., Guan K., Xu Y. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010;185:1158–1168. doi: 10.4049/jimmunol.0903874. [DOI] [PubMed] [Google Scholar]
- 47.Joo S.S., Won T.J., Kim M.J., Hwang K.W., Lee D.I. Interferon signal transduction of biphenyl dimethyl dicarboxylate/amantadine and anti-HBV activity in HepG2 2.2.15. Arch. Pharm. Res. 2006;29:405–411. doi: 10.1007/BF02968591. [DOI] [PubMed] [Google Scholar]
- 48.Thomsen M.K., Nandakumar R., Stadler D., Malo A., Valls R.M., Wang F., Reinert L.S., Dagnaes-Hansen F., Hollensen A.K., Mikkelsen J.G. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology. 2016;64:746–759. doi: 10.1002/hep.28685. [DOI] [PubMed] [Google Scholar]
- 49.Okuyama-Dobashi K., Kasai H., Tanaka T., Yamashita A., Yasumoto J., Chen W., Okamoto T., Maekawa S., Watashi K., Wakita T. Hepatitis B virus efficiently infects non-adherent hepatoma cells via human sodium taurocholate cotransporting polypeptide. Sci. Rep. 2015;5:17047. doi: 10.1038/srep17047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.