

Abstract
Tissue-resident macrophages (TRMs) are sentinel cells for maintaining tissue homeostasis and organ function. In this study, we discovered that lipopolysaccharide (LPS) administration dramatically reduced TRM populations and suppressed their self-renewal capacities in multiple organs. Using loss- and gain-of-function approaches, we define Sectm1a as a novel regulator of TRM self-renewal. Specifically, at the earlier stage of endotoxemia, Sectm1a deficiency exaggerated acute inflammation-induced reduction of TRM numbers in multiple organs by suppressing their proliferation, which was associated with more infiltrations of inflammatory monocytes/neutrophils and more serious organ damage. By contrast, administration of recombinant Sectm1a enhanced TRM populations and improved animal survival upon endotoxin challenge. Mechanistically, we identified that Sectm1a-induced upregulation in the self-renewal capacity of TRM is dependent on GITR-activated T helper cell expansion and cytokine production. Meanwhile, we found that TRMs may play an important role in protecting local vascular integrity during endotoxemia. Our study demonstrates that Sectm1a contributes to stabling TRM populations through maintaining their self-renewal capacities, which benefits the host immune response to acute inflammation. Therefore, Sectm1a may serve as a new therapeutic agent for the treatment of inflammatory diseases.
Keywords: tissue-resident macrophage, proliferation, Sectm1a, acute inflammation, LPS, T helper cell, IL-4
Graphical Abstract
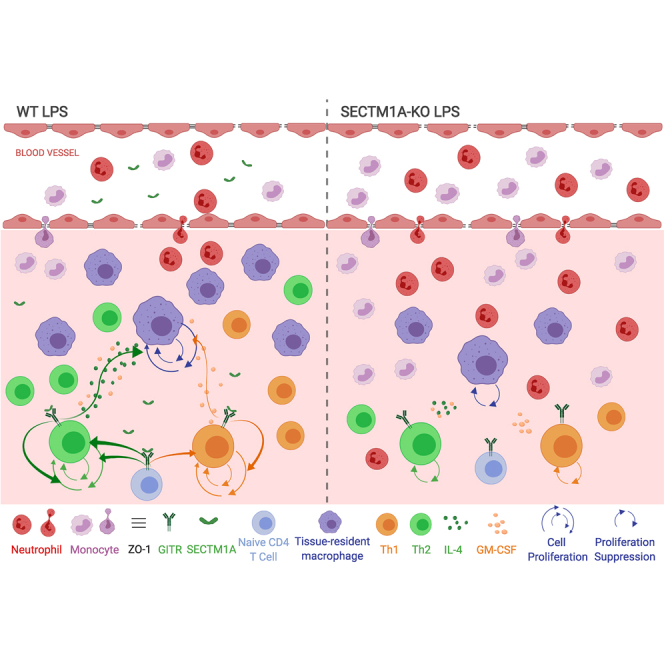
In this article, Mu et al. identified a novel regulatory mechanism of innate immune response: governing the self-renewal capacity of tissue-resident macrophages (TRMs) might greatly benefit the control of inflammation. They also revealed that Sectm1a contributed to maintaining the self-renewal capacity of TRMs through boosting T helper cells during endotoxemia.
Introduction
Sepsis, as a systemic inflammatory disease, remains a major challenge for worldwide healthcare systems, with 31.5 million cases of sepsis and 19.4 million cases of severe sepsis occurring annually in high-income countries.1,2 Notably, sepsis accounts for more than 50% of hospital deaths and has a considerable economic impact (13% of total US hospital costs).3,4 Endotoxin (lipopolysaccharide [LPS]) is recognized as the most potent microbial mediator implicated in the pathogenesis of septic shock.5,6
As a fundamental component of innate immunity, macrophages are the most abundant immune cells in many tissues and the first responders to local damage or inflammation, and they play essential roles throughout all phases of inflammation disease.7,8 The long-held belief that all macrophages are terminally differentiated immune cells derived from circulating monocyte infiltration has been challenged by recent evidence showing that most TRM (tissue-resident macrophage) populations originate from embryonic progenitors, persist into adulthood, and self-maintain by local proliferation rather than monocyte recruitment.9, 10, 11, 12 In mammals, TRMs are found in all organs with similar functions (i.e., scavenging of necrotic or apoptotic cells, invading pathogens, and maintaining homeostasis) and specific functions (i.e., participating in iron recycling, metabolism, synaptic information transfer, or cardiac conduction) depending on their anatomical location.12,13
During acute inflammation, recruitment of circulating monocytes triggered by LPS is a double-edged sword, which plays important roles in antimicrobial immune defense and tissue healing, but also contributes to the pathogenesis of inflammatory and degenerative diseases through promoting the inflammation and hindering the resolution.14 However, unlike bone marrow-derived monocytes/macrophages (BMDMs), TRMs are able to conduct silent clearance of apoptotic cells with low expression of Toll-like receptor 9 (TLR9) and less TLR responsiveness to nucleic acids, which does not promote the inflammatory response.15 Notably, recent evidence proved that TRMs prevented neutrophil influx, neutrophil-mediated monocyte infiltration, and inflammatory damage through prostrating their membranes to cloaking the minor tissue damage.16 Furthermore, TRMs are found to successively lose self-renewal capacity (the capability of proliferation) with age, leading to the reduction of their population in organs.17, 18, 19, 20 Although, as the first defense line, TRMs sit in the tissue and work silently without detrimental effects, there is still a heavy influx of inflammatory monocytes and neutrophils during acute inflammation. Herein, we hypothesized that there was a reduction in TRMs under acute inflammation, which generated demand for the recruitment of macrophages and triggered the infiltration of neutrophils and inflammatory monocytes. Therefore, upregulating the local proliferation of TRMs may contribute to stabling the TRM populations in tissues, preventing the detrimental infiltration of monocytes/neutrophils and favoring homeostasis/healing over further tissue damage caused by acute inflammation.
Mouse Sectm1a (secreted and transmembrane 1A) shares the greatest homology to human Sectm121, 22, 23 and has been characterized as an alternative CD7 ligand that is able to co-stimulate T cell proliferation and interleukin-2 (IL-2) release.24,25 In mouse, Sectm1a also acted as a co-stimulator of T cells to enhance T cell proliferation and IL-2 production, but this action was implicated to work through binding to GITR (glucocorticoid-induced tumor necrosis factor receptor-related protein) rather than CD7.23 Recently, Tsalik et al.26 reported that expression levels of Sectm1 were higher in peripheral blood of sepsis survivors compared with non-survivors. However, to our knowledge, the functional role of Sectm1a has never been investigated in TRM biology.
In this study, we discovered that Sectm1a, as a novel GITR ligand, contributed to maintaining the self-renewal capacity of TRMs through boosting Th2 effector cells during endotoxemia. Furthermore, the administration of recombinant Sectm1a (rSectm1a) significantly attenuated LPS-induced reduction of TRMs and detrimental influx of inflammatory monocytes/neutrophils and improved the mouse survival following LPS injection. Hence elevation of Sectm1 levels could be a new therapeutic approach for inflammatory disease.
Results
Loss of Sectm1a Aggravates Endotoxin-Induced Multi-organ Damage in Mice
To determine whether Sectm1a levels are altered in mice after LPS injection, we collected different organs at indicated time points. We found that LPS administration significantly downregulated Sectm1a expression levels in spleens (2.2-fold repression, p = 0.004), lungs (3.5-fold decrease, p < 0.001), and hearts (2.8-fold reduction, p = 0.027) at 6 h post-LPS injection (Figures 1A–1C). Sectm1a remained to be downregulated only in the spleen as late as 36 h post-LPS injection, whereas in the lung, Sectm1a had a 1.7-fold increase at 36 h post-LPS treatment compared with vehicle controls (p = 0.012). In addition, our immunofluorescence stain results also confirmed the downregulation of Sectm1a in the spleen (p < 0.001) and heart (p < 0.001) of LPS-treated mice (Figures S1A–S1D). Similarly, we found that sepsis patients had significantly lower levels of Sectm1 in the plasma compared with healthy donors (35.7 versus 132.2 ng/mL; p = 0.033) (Figure S1E). Taken together, these data suggest that the reduction of Sectm1a may contribute to the pathogenesis of acute inflammation. Given that Sectm1a is a secreted transmembrane protein, we therefore utilized a global Sectm1a-knockout (KO) mouse model to investigate its possible role in endotoxin-induced organ damages (Figure S2A). The loss of Sectm1a in KO mouse was confirmed in the spleen (Figure S2B) and heart (Figure S2C) by using the immunofluorescence staining technique. As shown in Figures S2D and S2E, there were no morphological abnormalities in KO mouse spleen and lung, compared with wild type (WT), at different ages. In addition, our complete blood count data did not present any significant difference between WT and KO mice at different ages (Table S1).
Figure 1.

Loss of Sectm1a Aggravates Endotoxin-Induced Multi-organ Damage in Mice
(A–C) After the administration of LPS (10 μg/g BW) to WT mice, the spleen (A), lung (B), and heart (C) were collected at indicated time points. Gene expression of Sectm1a in these tissues was determined by qRT-PCR (n = 4). (D–F) Sectm1a-KO mouse was generated as shown in Figure S2. At 24 h post-LPS (10 μg/g) or PBS injection, the spleen-to-body weight ratio (D) in WT and Sectm1a-KO mice was quantified, and the percentage increase (E) induced by LPS was calculated (n = 17–21). The ratio of spleen wet weight to dry weight (F) was also analyzed at the same time point post-LPS treatment (n = 6–9). (G) LPS-induced pulmonary vascular leakages were assessed by the extravasation of EB at 24 h post-LPS (10 μg/g) treatment. EB accumulation in different groups was normalized to control (n = 6–9). (H) Following 24 h of LPS (10 μg/g) treatment, ratios of lung wet weight to dry weight in WT and Sectm1a-KO mice were compared (n = 8–9). (I and J) Representative H&E staining images of lung sections at 24 h after PBS or LPS injection (I) are shown at ×200 original magnification. Scale bars, 50 μm. Lung injury scores (J) were assessed (n = 5–6). (K and L) Cardiovascular permeability was assessed by quantifying the extravasation of EB in the heart at 24 h post-treatment of PBS or LPS (10 μg/g). Blue represents DAPI, and red represents EB. Scale bars, 50 μm. n = 5–6. Data in all panels were pooled from at least two independent experiments. All results are presented as mean ± SEM and analyzed by Student’s t test, one-way ANOVA, or two-way ANOVA.
Spleen, the largest second lymphoid organ in human, plays important roles in immunity.27 Loss of function of the spleen, either by splenectomy or infiltrative disease, is known to prominently increase the risk for severe infection and sepsis (five to six times higher).28 Considering that Sectm1a is highly expressed in the spleen,23 we then first determined whether loss of Sectm1a augmented splenomegaly or spleen edema in mice during acute inflammation. Interestingly, under basal PBS conditions, the ratio of spleen weight (SW) to body weight (BW) was significantly lower in Sectm1a-KO mice than that in WT mice (p = 0.002) (Figure 1D). However, the administration of LPS increased the ratio of SW/BW to a similar degree in both WT and KO mice at 24 h post-injection (p = 0.395) (Figure 1D). Hence further analysis showed that LPS actually caused higher increase of SW in KO mice than that in WT mice (p = 0.001) (Figure 1E). Accordingly, KO spleens possessed a remarkably higher ratio of wet weight/dry weight (p = 0.02) (Figure 1F) and more serious damage (evidenced by more enlargement of white pulp zone and larger presence of germinal centers) (p < 0.001) (Figures S2F and S2G) than WT spleens after LPS treatment, indicating that the loss of Sectm1a aggravated LPS-induced spleen edema and damage.
Next, we assessed lung injury characterized by pulmonary vascular hyperpermeability and neutrophilic inflammation in WT and Sectm1a-KO mice at 24 h post-LPS injection. As shown in Figure 1G, the Evans blue (EB) dye leakage in lung tissue was significantly increased in both WT and KO mice upon LPS treatment compared with vehicle controls, but such increase was more pronounced in KO lungs than WT controls (p = 0.025). Accordingly, similar effects and trends were observed in the wet weight/dry weight ratio of the lung (p = 0.028) (Figure 1H), indicating that LPS injection leads to more severe lung edema in KO mice than WT mice. At the same time point, we examined lung histology and found that both WT lungs and KO lungs exhibited normal structure under basal conditions. However, lung tissues collected from LPS-treated WT mice had extensive morphological damage and pathological abnormalities compared with PBS-WTs, including infiltration of neutrophils into the interstitium and alveolar spaces, increased hyaline membranes, increased alveolar wall thickness, and alveolar collapse (Figure 1I). Notably, the endotoxin-induced histopathological damage was aggravated in the Sectm1a-KO group (injury score, 0.66) compared with WT controls (injury score, 0.54) (p = 0.033) (Figure 1J). Altogether, these data indicate that loss of Sectm1a significantly exacerbates endotoxin-induced pathological lesions of lung.
Lastly, it is well recognized that a sepsis-induced systemic inflammatory response can directly induce cardiac vascular leakage and impair cardiac function;29,30 thus, we assessed cardiovascular permeability in the heart of WT and KO mice with EB at 24 h post-LPS injection. As shown in Figures 1K and 1L, the red fluorescence intensity of EB extravasation was elevated by 2-fold in LPS-treated WT hearts, but this EB extravasation was further aggravated in LPS-treated KO hearts (p = 0.019). These results demonstrated that Sectm1a deficiency could exaggerate cardiovascular leakage in response to acute inflammation induced by endotoxin.
Ablation of Sectm1a Further Reduces the TRM Population in Mouse Spleen and Lung after LPS Injection
In the spleen, there are four sub-populations of macrophages, including red pulp macrophages (RPMs), marginal zone macrophages, marginal zone metallophilic macrophages, and tingible body macrophages, defined by anatomical compartments of red and white pulp regions. However, only RPMs are recognized as TRM, because they are seeded prenatally and maintained independently on substantial monocytic input.31,32 In addition, as the largest distinct organ of the lymphatic system, the spleen also acts as a reservoir of inflammatory monocytes that rapidly deploy to damaged heart or lung during acute inflammation.33,34 Once splenic monocytes localized at these sites, they are able to release inflammatory cytokines and promote the recruitment of neutrophils, which, in turn, exaggerate tissue damage in the heart and lung.34, 35, 36, 37 Therefore, in this study, we first utilized flow cytometry to analyze the population change of RPMs and splenic monocytes in both WT and Sectm1a-KO mice in response to acute inflammation. According to previous reports,34,38 we characterized RPMs as CD45.2+ Lin− CD11c− F4/802+ CD11blow/− cells and splenic monocytes as CD45.2+ Lin− CD11c− F4/80+ CD11b2+ cells, and the gating strategy was shown in Figure S3A. Our analysis results showed that LPS (10 μg/g) treatment resulted in a loss of half of the RPM (blue gate) population in WT mice at 18 h post-injection (Figures 2A and 2B), and this reduction was dose dependent as shown in Figure 2C (p = 0.007). Interestingly, LPS exposure led to a further reduction (70.8%) of RPMs in Sectm1a-KO mice compared with WT mice (50.6%) (p = 0.013) (Figures 2A and 2B). As for splenic monocytes, fewer monocytes remained in KO spleens (0.67% of live cells) compared with that in WT spleens (0.87% of live cells) following LPS injection (p = 0.04) (Figure 2D). This means that the spleen in KO mice may release more monocytes into the circulating system compared with WT mice (p = 0.02) (Figure 2E).
Figure 2.

Sectm1a Deficiency Further Diminishes Tissue-Resident Macrophages in the Spleen and Lung after LPS Injection
The whole spleen and lung tissues were collected from WT and Sectm1a-KO mice at 18 h post-PBS or LPS (10 or 20 μg/g) injection and subjected to flow cytometry analyses. (A) Representative flow cytometry plots of red pulp macrophages (CD45.2+ Lin− CD11c− F4/802+ CD11blow/− cells, blue gate) and splenic monocyte (CD45.2+ Lin− CD11c− F4/80+ CD11b2+ cells, red gate) following PBS or LPS (10 μg/g) treatment. Gating strategy is shown in Figure S3A. (B) Percentages of RPM population in WT and KO spleen were summarized following PBS or LPS treatment (n = 6). (C) The percentage change of the RPM population in the spleen of WT mice in response to different doses (10 μg/g, 20 μg/g) of LPS (n = 8–10). (D) Calculation of the percentage of splenic monocytes in WT and Sectm1a-KO mice after PBS or LPS (10 μg/g) treatment (n = 7–11). (E) Quantification percentages of released monocytes in WT and KO mice with the formula: Percentage of released monocytes = [(percentage of monocytes in PBS group) − (percentage of monocytes remaining in LPS group)]/(percentage of monocytes in PBS group) (n = 11). (F–H) Representative flow cytometry plots of AMs (F) (CD45.2+ Ly-6G− CD3− CD19− MerTK+ CD64+ CD11c+ Siglec-F+ CD11b−) in the lung of WT and Sectm1a-KO mice after PBS or LPS injection. Gating strategy was shown in Figure S3B. Percentages of AMs in total live cells (G) and percentages of AMs in live cells excluding neutrophils (H) were quantified (n = 8). (I and J) Representative flow cytometry plots (I) of inflammatory monocytes (CD45.2+ Ly-6G− CD3− CD19− MerTK− Siglec-F− CD64+ CD11b+ Ly-6C+) in the lung following PBS or LPS administration. Percentages of infiltrated inflammatory monocytes in total live cells (J) are shown (n = 5). (K) Flow cytometry showing the infiltration of neutrophil (CD45.2+ Ly-6G+ CD11b+ Ly-6C+) in the lung after LPS injection. (L) Percentages of infiltrated neutrophils in total live cells (n = 8).
In the lung, alveolar macrophages (AMs) are well recognized as resident macrophages and play a critical role in surfactant homeostasis and immune surveillance.39 To investigate the population change of AMs at the earlier stage of acute inflammation, we collected all lung samples from mice at 18 h post-LPS treatment (10 μg/g) for flow cytometry analysis. The gating strategies for AMs (CD45.2+ Ly-6G− CD3− CD19− MerTK+ CD64+ CD11c+ Siglec-F+ CD11b−), infiltrated monocytes (CD45.2+ Ly-6G− CD3− CD19− MerTK− Siglec-F− CD64+ CD11b+ Ly-6C+), and neutrophils (CD45.2+ Ly-6G+ CD11b+ Ly-6C+) were shown in Figure S3B. We observed that the AM population (of live cells) dropped from 8.35% (PBS control) to 2.5% (LPS-treated) in WT mice (Figures 2F and 2G), However, LPS-triggered reduction of AMs was more pronounced in Sectm1a-KO lungs, which was 1.4-fold less than that in WT lungs (p = 0.002) (Figures 2F and 2G). Given that LPS-triggered recruitment of a high amount of neutrophils also contributes to the drop of AM percentage in live cells, we then excluded neutrophils from live cells and re-calculated the percentage of AMs (of live cells). As shown in Figure 2H, our re-analysis results also confirmed that Sectm1a deficiency resulted in further decline of the AM population in response to acute inflammation (p < 0.001). In parallel, we found that the recruitment of inflammatory monocytes and neutrophils was further increased by 40% (p = 0.04) (Figures 2I and 2J) and 20% (p = 0.003) (Figures 2K and 2L), respectively, in LPS-treated KO lungs compared with those in LPS-treated WTs.
Loss of Sectm1a Exacerbates LPS-Induced Reduction of Cardiac-Resident Macrophages (CRMs) in the Heart
In the heart, TRMs were recently characterized as CD45.2+ Ly-6G− F4/80+ CD64+ CD11b+ Ly-6C− and play important roles in promoting coronary development, promoting cardiac regeneration, facilitating electrical conduction, and protecting cardiac functions.40, 41, 42 However, whether the CRM population is affected by endotoxin remains unknown. Herein, we determined the CRM population change following LPS injection and found that the percentage of CRMs (gating strategy as shown in Figure S3C) in the total macrophage/monocyte population was decreased by more than half in WT mice at 18 h post-LPS injection (Figures 3A and 3B). Notably, LPS caused further reduction of CRMs in Sectm1a-KO mice (p < 0.001) (Figures 3A and 3B). Similar to what we observed in the lung, the percentages of cardiac inflammatory monocytes (CD45.2+ Ly-6G− F4/80+ CD64+ CD11b+ Ly-6C+ MHCII−) were respectively increased to 56.8% in WT mice and 63.7% in KO mice after LPS challenge (p < 0.001) (Figures 3C and 3D). Further analysis revealed that the ratio of inflammatory monocytes to CRMs was much higher in LPS-KO mice than LPS-WTs, indicating more influx of inflammatory monocytes to KO hearts (p < 0.001) (Figure 3E). Similar to human hearts, mouse CRMs can also be partitioned into two distinct subsets depending on the expression of CCR2. In this study, our flow cytometry results revealed that LPS treatment significantly increased the ratio of CCR2+ to CCR2− in both WT and KO hearts, but there was no difference between the two mouse groups in this ratio before or after LPS treatment (Figures 3F and 3G). However, Sectm1a depletion significantly exaggerated LPS-induced infiltration of neutrophils into the heart, compared with WTs (p = 0.008) (Figures 3H and 3I).
Figure 3.

Loss of Sectm1a Exacerbates LPS-Induced Reduction of Cardiac-Resident Macrophages (CRMs) in the Heart
The whole hearts were collected from WT and Sectm1a-KO mice at 18 h after PBS or LPS (10 μg/g) injection and were subjected to flow cytometry analyses. (A and B) Representative flow cytometry plots of CRM (CD45.2+ Ly-6G− F4/80+ CD64+ CD11b+ Ly-6C−) (green gate) in WT and Sectm1a-KO mice in response to endotoxin (A). Gating strategy is shown in Figure S3C. Percentages of CRM (B) in the total macrophage and monocyte population (n = 8). (C–E) Representative flow cytometry plots of inflammatory monocytes (CD45.2+ Ly-6G− F4/80+ CD64+ CD11b+ Ly-6C+ MHCII−) in the heart following LPS administration (C). Percentages of infiltrated inflammatory monocytes (D) in the total macrophage and monocyte population (n = 8). Comparative analysis of the ratio of inflammatory monocytes to CRM (E) in WT and Sectm1a-KO mice after LPS injection (n = 8). (F and G) Representative flow cytometry plots (F) and ratios of CCR2+ to CCR2− CRMs (G) in WT and Sectm1a-KO mice after PBS or LPS injection (n = 7). Gating strategy is shown in Figure S3C. (H and I) Flow cytometry showing the infiltration of neutrophils (CD45.2+ CD11b+ Ly-6G+ Ly-6C+) in the heart (H) after PBS or LPS injection. Percentages of infiltrated neutrophils in total leukocytes of heart (n = 8) (I). Data in all panels were pooled from at least two independent experiments. All results are presented as mean ± SEM and analyzed by Student’s t test or one-way ANOVA.
Sectm1a Deficiency Exacerbates Endotoxin-Induced Proliferation Suppression in TRMs
To understand why the loss of Sectm1a augments LPS-induced reduction of TRMs, we first examined apoptosis/necrosis in mouse RPMs and AMs at 16 h post-LPS injection. Our flow cytometry results revealed that there were not any differences in the degree of apoptosis/necrosis between WT-RPMs and KO-RPMs in response to endotoxin treatment (Figures S4A–S4C). A similar degree of apoptosis/necrosis was also observed between WT-AMs and KO-AMs after endotoxin challenge (Figures S4D–S4F). These results exclude the contribution of cell mortality to the further reduction of TRMs in KO mice during acute inflammation compared with WT mice. Therefore, we speculated that such Sectm1a deficiency-increased reduction of TRMs may be ascribed to the decreased local proliferation. Using intracellular staining flow cytometry, we analyzed the percentage of Ki-67+ (a widely used cellular marker for proliferation) cells in TRMs before and after LPS treatment. As shown in Figures 4A and 4B, proportions of Ki-67+ RPMs were respectively dropped by 1.6-fold and 2.4-fold in WT mice and KO mice at 12 h post-LPS injection, and the proportion of Ki-67+ RPMs in KO mice was 2-fold less than that in WT mice after LPS treatment (p < 0.001). In addition, our results also revealed that Ki-67+ RPMs were reduced significantly in the Sectm1a-KO mice compared with WT mice under PBS treatment (Figure 4B). This reduction may be ascribed to the abundance of Sectm1a and T cells in the mouse spleen. Next, these flow cytometry results were further confirmed by co-immunofluorescence staining of spleens with antibodies for F4/80 and Ki-67 (Figure 4C).
Figure 4.

Sectm1a Deficiency Exacerbates Endotoxin-Induced Proliferation Suppression in Tissue-Resident Macrophages
The spleen, lung, and heart in WT and Sectm1a-KO mice were collected and subjected to flow cytometry analysis at 16 h after PBS or LPS (10 μg/g) administration. (A, D, and G) Representative flow cytometry plots showing Ki-67+ RPMs (A), AMs (D), and CRMs (G), respectively. (B, E, and H) Percentages of Ki-67+ RPMs (B), AMs (E), and CRMs (H) after PBS or LPS injection (n = 5–10). (C) Immunofluorescent staining of spleen sections with F4/80 (blue) and Ki-67 (magenta) at 16 h after PBS or LPS treatment. Scale bar: 50 μm. (F and I) Quantification of mean fluorescence intensity (MFI) in Ki-67+ AMs (F) and CRMs (I) (n = 8–10). (J and K) Percentages of Ki-67+ cell population in CCR2− (J) and CCR2+ (K) CRMs following PBS or LPS injection (n = 7–8). Data in all panels were pooled from at least two independent experiments. All results are presented as mean ± SEM and analyzed by Student’s t test or one-way ANOVA.
Similarly, we found that the proliferation of lung-resident macrophages (AMs) was also significantly inhibited in both WT and KO mice upon LPS challenge, but KO mice presented a larger degree of reduction (p = 0.017), as evidenced by a 2-fold decrease of Ki-67+ AMs in WT mice versus a 3-fold decrease in KO mice at 12 h post-LPS treatment (Figures 4D and 4E). Meanwhile, the mean fluorescence intensity (MFI) of Ki-67 was significantly lower in Ki-67+ AMs from LPS-treated KO mice (p = 0.013) (Figure 4F). With regard to CRMs, the fractions of Ki-67+ CRMs (p = 0.001) and their MFI (p = 0.01) were also decreased to a significantly lower degree in Sectm1a-KO mice compared with that in WT mice after LPS stimulation (Figures 4G–4I). In addition, our further analysis revealed that although the local proliferations of CCR2− (p < 0.001) and CCR2+ (p = 0.023) CRMs were both significantly downregulated by LPS administration, more reductions were observed in Sectm1a-KO mice compared with WTs (Figures 4J and 4K). Interestingly, Ki-67+ CCR2− CRMs were decreased more than Ki-67+ CCR2+ CRMs after LPS injection in both WT and KO mice, which may explain the increased ratio of CCR2+ to CCR2− in the CRM population. Together, these data suggest that Sectm1a could be essential for local proliferation of TRMs and contribute to the maintenance of their population in multiple organs upon endotoxin stress.
rSectm1a Promotes the Proliferation of TRMs through Boosting the Expansion of T Helper (Th) Cells
To further determine the effect of Sectm1a on TRM proliferation, we isolated RPMs from spleens of WT or KO mice as described in our recent publication.43 Then RPMs were labeled with CellTracker fluorescence reagent and grown for 48 h to monitor cell proliferation. Unexpectedly, the fluorescence intensity showed no difference between WT-RPMs and Sectm1a-deficient RPMs (Figure S5A), indicating that ablation of Sectm1a in RPMs has no effect on their proliferation. In addition, treatment of WT-RPMs with rSectm1a did not affect RPM proliferation (Figure S5B). However, in our positive control group (using IL-4 as a stimulator of macrophage growth),44,45 there was remarkably higher fluorescence intensity compared with the PBS-treated group (p < 0.05) (Figure S5B). These results were further confirmed by the staining of RPMs with 5-ethynyl-2′-deoxyuridine (EdU) (cell proliferation marker), as evidenced by a similar proliferation rate in RPMs isolated from either WT or Sectm1a KO mice, regardless of the addition of rSectm1a (Figures S5C and S5D). Taken together, all these data indicate that Sectm1a could not directly regulate TRM proliferation. Hence we speculated that Sectm1a might affect other cell types, which, in turn, crosstalk with TRMs and impact their proliferation.
Given that Sectm1 and Sectm1a were both reported to be able to co-stimulate T cell proliferation and IL-2 release,23, 24, 25 we thus hypothesized that Sectm1a might also promote the differentiation of Th2 and the release of IL-4, which, in turn, stimulated the proliferation of TRMs. To this end, we co-cultured naive CD4 T cells with RPMs. As shown in Figure 5A, RPMs were seeded on the coverslip of the lower chamber at the same density, and naive CD4 T cells were isolated from mice and cultured on the upper insert, which was coated with rSectm1a (2 μg/mL) or IgG2a (1.25 μg/mL). At the same time, Dynabeads covalently coupled to anti-CD3 and anti-CD28 antibodies were added to stimulate CD4 naive T cells. After 4 days of stimulation, half of the cells were treated with 200 ng/mL LPS for 24 h. Then supernatants were collected to measure cytokine levels, and RPMs were stained with F4/80 (red) and EdU (green). Meanwhile, to assess the effect of rSectm1a on the differentiation of naive CD4 T cells, we carried out intracellular cytokine assays for interferon (IFN)-γ and IL-4 following PMA and ionomycin stimulation. We observed that RPM populations in the rSectm1a-treated group were significantly increased by 2.3-fold (p < 0.001) in the absence of LPS and 2.5-fold (p < 0.001) in the presence of LPS (Figures 5B and 5C), respectively, compared with their isotype controls. It is important to note here that the application of LPS led to 50% reduction in the RPM population, which is consistent with our in vivo experiment results (Figures 2A–2C). Accordingly, the percentage of EdU+ RPMs in the rSectm1a-treated group was around two times higher than that in isotype control under LPS exposure (p < 0.001) (Figures 5D and 5E), indicating a higher proliferating rate induced by rSectm1a treatment. Meanwhile, our flow cytometry results showed that rSectm1a dramatically elevated the percentage of IL-4-producing CD4 T cells by 18-fold in the absence of LPS and 15-fold in the presence of LPS in vitro compared with isotype control groups (Figure 5F). In line with previous reports about rSectm1a-mediated Th1 polarization, we also observed that the number of IFN-γ-producing CD4 T cells was significantly increased by 10-fold in the absence of LPS and 2-fold in the presence of LPS in vitro after rSectm1a treatment (Figure 5F). Next, we compared the differentiation of Th1 and Th2 cells in vivo between WT and KO mice at 12 h post-LPS treatment. As shown in Figures 5G–5I, Sectm1a depletion significantly reduced Th2 (IL-4+ CD4 cells) (p = 0.007) and Th1 (IFN-γ+ CD4 cells) (p < 0.001) populations in the spleen of LPS-treated mice, coinciding with our in vitro results. As for Th2-associated cytokines (i.e., IL-4 and IL-5), we found that both IL-4 (p = 0.04) and IL-5 (p = 0.049) levels were significantly higher in the co-culture supernatants collected from the rSectm1a-treated group than those collected from isotype controls (Figures 5J and 5K). Furthermore, levels of GM-CSF (granulocyte-macrophage colony stimulating factor), another stimulator of TRM proliferation, were also remarkably increased by rSectm1a treatment (p < 0.001) (Figure 5L). Consistent with these in vitro findings, serum levels of IL-4 (p = 0.031), GM-CSF (p = 0.025), and macrophage colony-stimulating factor (M-CSF; p = 0.008) were significantly lower in Sectm1a-KO mice than WT mice following LPS treatment (Figures 5M–5O). Taken together, our results demonstrate that Sectm1a positively promotes RPM proliferation through driving Th1 and Th2 polarization and relevant cytokine release.
Figure 5.

rSectm1a Promotes the Proliferation of Tissue-Resident Macrophages through Boosting the Expansion of T Helper Cells
(A) RPM and naive CD4 T cell co-culture experiment design. (B) Representative bright-field image and immunofluorescence stain image of RPMs after co-culturing with IgG2a-treated or rSectm1a-treated naive CD4 T cells in the absence or presence of LPS (red represents F4/80; blue represents DAPI). Scale bars, 100 μm. (C) The proliferation of RPMs was assessed by counting cell numbers per microscopic field. All determinations were performed for at least three independent experiments (n = 8). (D and E) Representative immunofluorescence stain images (D) and quantification (E) of EdU-positive RPMs under co-culture condition following different treatments. Results are shown as means ± SEM of four independent experiments, and 200–300 cells were analyzed per experiment (n = 4). Scale bars, 100 μm. (F) Percentages of CD4+ IFN-γ+ T cell (Th1) and CD4+ IL-4+ T cell (Th2) after co-culture with RPMs in the absence or presence of LPS were shown in representative dot plots. (G–I) The intracellular cytokine assays for IFN-γ and IL-4 in CD4 T cell isolated from the spleen of WT or KO mice after LPS injection (G). (H and I) Percentages of CD4+ IL-4+ T cell (Th2) (H) and CD4+ IFN-γ+ T cell (Th1) (I) in vivo in response to endotoxin treatment (n = 4). (J–L) After co-culture, cytokine levels of IL-4 (J), IL-5 (K), and GM-CSF (L) in cell culture supernatants were measured by using Luminex technology (n = 3–4). (M–O) At 12 h post-LPS (10 μg/g) injection, cytokine levels of IL-4 (M), GM-CSF (N), and M-CSF (O) in sera were determined in WT and Sectm1a KO mice by using Luminex. (n = 5–8). All results are presented as mean ± SEM and analyzed by Student’s t test or one-way ANOVA.
GITR Is Critical for the Sectm1a-Mediated Regulatory Effect on the Activation of Th Cells
Although Howie et al.23 identified GITR as a Sectm1a receptor on T cells in vitro, it remains unclear whether Sectm1a-mediated effects on primary Th cells are dependent on GITR in vivo. Therefore, we conducted the following experiments with GITR-KO mice. First, we isolated naive CD4 T cells from GITR-KO mice and co-cultured them with RPMs from WT mice following the same protocol used above (Figure 5A). After 6 days of co-culture, we found that rSectm1a treatment did not significantly increase the RPM population (Figures 6A and 6B). As expected, using naive CD4 T cells isolated from GITR-KO mice, rSectm1a treatment also failed to stimulate the differentiation of either Th1 or Th2 (Figures 6C–6E). In addition, the administration of rSectm1a to GITR-KO mice displayed no effects on LPS-induced animal mortality (Figure 6F). Accordingly, rSectm1a treatment did not attenuate the decrease of either RPMs or splenic monocytes in GITR-KO mice during acute inflammation (Figures 6G–6I). Similarly, rSectm1a injection also failed to increase the AMs population (Figures 6J and 6K) and reduce the infiltration of inflammatory monocytes (Figures 6L and 6M) and neutrophils (Figures 6N and 6O) in the lung of LPS-treated GITR-KO mice. However, the administration of rSectm1a to WT mice significantly improved the animal survival in response to endotoxin challenge (Figure 6P). In addition, rSectm1a treatment significantly increased the RPM population (p = 0.01) and attenuated the reduction of splenic monocytes (p = 0.018) in LPS-treated WT mice (Figures 6Q–6S). As expected, the injection of rSectm1a also significantly enhanced the AM population (p = 0.01) (Figures 6T and 6U) and reduced the infiltration of neutrophils (p = 0.019) (Figures 6V and 6W) in the lung of WT mice during acute inflammation. In addition, the administration of rSectm1a did not cause any morphological change in mouse spleen and lung according to our H&E staining results (Figure S6A). Our complete blood count data did not present any significant difference after the administration of rSectm1a (Table S2). Furthermore, we also tried to determine the cytokine (TNF-α and IL-6) levels in the serum of IgG2a-treated and rSectm1a-treated mice at 24 h post-treatment. As expected, both of them were undetectable. These results indicate that the administration of rSectm1a is not toxic for mice. Taken together, both our in vitro and in vivo results suggest that GITR is pivotal for the Sectm1a-mediated impact on the expansion of Th cells and TRMs.
Figure 6.

GITR Is Critical for a Sectm1a-Mediated Regulatory Effect on the Activation of T Helper Cells
(A–E) Following the same co-culture design in Figure 5A, RPMs isolated from WT mice were co-cultured with naive CD4 T cells isolated from GITR-KO mice. (A) Representative immunofluorescence stain image of RPMs after treatment (red represents F4/80, and blue represents DAPI). Scale bars, 50 μm. (B) The proliferation of RPMs was assessed by counting cell numbers per microscopic field. All determinations were performed for at least three independent experiments (n = 4). (C) Representative flow cytometry dot plots of CD4+ IFN-γ+ T cells (GITR−/−) and CD4+ IL-4+ T cells (GITR−/−) after co-culture with RPMs in the absence or presence of rSectm1a. (D and E) Percentages of CD4+ IL-4+ T cells (GITR−/−) (D) and CD4+ IFN-γ+ T cells (GITR−/−) (E) after co-culture (n = 4). (F–O) After intraperitoneal (i.p.) injection with LPS (10 μg/g), GITR-KO mice were immediately injected intravenously (i.v.) with IgG2a (125 μg/kg) (isotype) or rSectm1a (200 μg/kg). (F) Mouse survival was monitored for up to 96 h post-LPS treatment (n = 8–9). (G–O) At 18 h after last treatments, spleen and lung tissues were collected and subjected to flow cytometry experiments. (G–I) Representative flow cytometry plots (G) of RPMs (blue gate) and splenic monocytes (red gate) in GITR-KO mice with rSectm1a treatment or IgG2a treatment. The percentage of RPMs (H) and monocytes (I) in the live cells were calculated (n = 5–6). (J and K) Representative flow cytometry plots (J) and quantification (K) of AMs in the lung after different treatments. (L and M) Representative flow cytometry plots (L) and quantification (M) of inflammatory monocytes in the lung. (N and O) Representative flow cytometry plots (N) and quantification (O) of neutrophils in the lung. (P) After i.p. injection with LPS (15 μg/g), WT mice were immediately injected i.v. with IgG2a (125 μg/kg) (isotype) or rSectm1a (200 μg/kg) and monitored for survival up to 96 h post-LPS treatment (n = 10–16). (Q–W) WT mice were immediately injected i.v. with IgG2a (125 μg/kg) (isotype) or rSectm1a (200 μg/kg) following LPS (10 μg/g) i.p. injection. At 18 h after last treatments, the spleen and lung tissues were collected and subjected to flow cytometry experiments. (Q–S) Representative flow cytometry plots (Q) of RPMs (blue gate) and splenic monocyte (red gate) in WT mice with rSectm1a (or isotype) treatments and quantification of RPMs (R) and splenic monocytes (S) (n = 5). (T and U) Representative flow cytometry plots (T) and quantification of AMs (U) in the lung after different treatments (n = 5–6). (V and W) Representative flow cytometry plots (V) and quantification of neutrophils (W) in the lung (n = 5–6). Data in all panels were pooled from at least two independent experiments. All results are presented as mean ± SEM and analyzed by Mann-Whitney test, Student’s t test, or one-way ANOVA.
TRMs Are Essential for Maintaining Local Vascular Integrity during Acute Inflammation
Lastly, to understand why the reduction of TRMs was associated with the recruitment of inflammatory leukocytes into multiple organs during endotoxemia, we pre-depleted TRMs with clodronate liposome and determined its consequential effect on the LPS-induced vascular leakage, a major contributor to sepsis-caused organ failure. After 3-day application of clodronate liposome, our flow cytometric analysis confirmed that TRMs in spleens and lungs were successfully diminished (Figures 7A and 7B). At 24 h post-LPS injection, we observed that the ratio of spleen wet weight/dry weight in macrophage pre-depleted mice was significantly higher than that in control mice (p = 0.013) (Figure 7C). In addition, LPS administration also resulted in more EB leakage (p = 0.038) and morphological damage (p = 0.026) in lungs of clodronate-treated groups compared with controls (Figures 7D–7F). Similarly, pre-depletion of macrophages also significantly increased cardiovascular permeability in response to endotoxin (p = 0.032) (Figures 7G and 7H). In line with our in vivo findings, our in vitro results showed that the presence of RPMs significantly attenuated the thrombin-induced endothelial barrier disruption (p = 0.003) (Figure 7I). Mechanistically, we found that the RPM conditioned medium significantly upregulated mRNA levels of ZO-1 (tight-junction proteins) (p = 0.012), VE-cadherin (adherens-junction protein) (p = 0.01), and occludin (p = 0.003) in endothelial cells (ECs) (Figures 7J–7L), and our western blot results also confirmed the upregulation of ZO-1 and VE-cadherin (Figure S6B). Moreover, our confocal microscopy examination clearly presented a more dense distribution of tight-junction protein between ECs in the presence of RPM medium (Figure 7M). Accordingly, under thrombin or PMA stimulation, less fragmentation and depletion of ZO-1 were observed between ECs cultured in RPM-conditioned medium compared with that cultured in normal medium (Figure 7M), suggesting that RPM-conditioned medium helped to maintain ZO-1 dense distribution and linear pattern between ECs. Taken together, our results indicate the important role of TRMs in stabilizing the local vascular integrity.
Figure 7.

Tissue-Resident Macrophages Are Essential for Maintaining Local Vascular Integrity during Endotoxemia
(A) Graphic illustration of mouse treatment scheme. To pre-deplete tissue-resident macrophages, we injected (i.v.) WT mice with clodronate-liposome (200 μL, clodronate 5 mg/mL). The control mice received vehicle-liposome (200 μL, no clodronate). After 3 days, mice were injected (i.p.) with LPS (5 μg/g). At 24 h after LPS injection, the spleen, lung, and heart were collected for the following experiments. (B) Representative flow cytometry plots showing the reduction of RPMs in the spleen (upper plot) and AMs in the lung (lower plot) on day 3 after clodronate liposome treatment. (C) The ratio of spleen wet weight to dry weight in control and macrophage pre-depleted mice was measured at 24 h after LPS treatment (n = 6). (D) Meanwhile, LPS-induced pulmonary vascular leakages were assessed by the extravasation of EB (n = 6). (E) Representative H&E staining images of lungs at 24 h after LPS injection were shown at ×200 original magnification; scale bars, 50 μm. (F) The lung injury scores were calculated (n = 5–6). (G and H) Cardiovascular permeability was assessed by quantifying the extravasation of EB in the heart at 24 h after LPS treatment. Frozen heart sections (G) were observed under a confocal LSM 710 microscope. Blue represents DAPI, and red represents EB; scale bars, 50 μm. The relative intensity of red fluorescence emitted by EB was quantified (H) with ImageJ software (n = 4–5). (I) Mouse cardiac endothelial cells (MCECs) (on the insert of transwell) were cultured in the presence or absence of RPMs on the bottom chamber for 4 days until the endothelial monolayer was established. Then fluorescein isothiocyanate (FITC)-dextran particle was added into the upper insert, and thrombin (3 U/mL) was used to stimulate MCECs for 1 h. The degree of EC monolayer leakage was determined by measuring the intensity of FITC-dextran fluorescence in the basolateral chamber (n = 4). (J–L) MCECs were cultured in the control medium (C-M) or RPM conditioned medium (RPM-M) for 1 day. qRT-PCR was used to determine mRNA levels of ZO-1 (J), VE-cadherin (K), and occludin (L), in the presence or absence of RPM conditioned medium (n = 4). (M) MCECs were cultured with control medium or RPM conditioned medium for 3 days to reach the monolayer on the coverslip. Representative images of immunofluorescent staining for ZO-1 (green) at 1 h after thrombin (3 U/mL) or PMA (40 ng/mL) treatment. White arrows indicate the integrity linear pattern of ZO-1. Yellow arrows present the fragmentations and depletions of ZO-1. Scale bars, 20 μm. All results are presented as mean ± SEM and analyzed by Student’s t test or one-way ANOVA.
Discussion
In this study, we demonstrated that LPS administration dramatically reduced the TRM population along with the recruitment of inflammatory monocytes and neutrophils in multiple organs. Notably, Sectm1a null mice had a smaller TRM population and more inflammatory monocytes and neutrophils in organs, which were associated with more than severe organ damage compared with WT mice upon LPS insult. Both our in vitro and in vivo data showed that Sectm1a was able to promote TRM proliferation through boosting Th cells and driving the release of cytokines (i.e., IL-4, GM-CSF, and M-CSF). Importantly, we identified that GITR, as a receptor of Sectm1a, was essential for Th cell-mediated boosting effects on macrophage self-renewal. Furthermore, we found that TRMs played a critical role in maintaining local vascular integrity. Collectively, our study uncovers that Sectm1a could positively regulate local proliferation of TRMs in the context of acute inflammation.
Recently, TRMs have been shown to play important roles in maintaining organ homeostasis, maintaining immune defense, and promoting tissue healing during inflammatory diseases, and maintaining an appropriate size of TRM pool has the potential to preserve and restore tissue homeostasis upon acute inflammation challenge.46 In line with our study, Shults et al.47 reported that AMs were diminished by 50% in the bronchoalveolar lavage (BAL) at 24 h after burn injury and binge ethanol intoxication. In addition, the reduction of CRM was also observed in numerous mouse models of cardiac disease, including permanent myocardial infarction, reperfused myocardial infarction, and diphtheria toxin cardiomyocyte ablation.48,49 These reductions have mainly been attributed to increased cell death.50 However, the self-renewal capacity, as the most important trait of TRMs, has been overlooked, and it remains unclear how to modulate the self-renewal capacity of TRMs in inflammatory diseases.46 In the present study, using a global KO mouse model, we observed that loss of Sectm1a impaired self-renewal capacity of residential macrophages in the spleen, lung, and heart of mice during acute inflammation. By contrast, administration of mice with rSectm1a protein attenuated LPS-induced reduction of TRMs and, in turn, improved mouse survival.
Our mechanistic study reveals that rSectm1a-mediated beneficial effects rely on the crosstalk between Th cells and macrophages in a GITR-dependent manner. GITR serves as a receptor of Sectm1a on T cells,23 and such interaction of Sectm1a/GITR could promote the expansion of Th cells and the release of cytokine, leading to positive regulation on the proliferation of TRMs. Although GITR was previously thought to be a marker of Treg (regulatery T cell) precursor cells, recent studies have demonstrated that GITR is highly expressed in the activated CD4+ T cells.51, 52, 53, 54 Functionally, in addition to stimulating the proliferation and differentiation of CD4+ naive T cells, GITR is also engaged in promoting the cytokine release from fully polarized Th2,55, 56, 57 leading to a positive feedback on Th2 differentiation and cytokine production. Consistent with these reports, using co-culture of naive CD4 T cells with macrophages, we observed that rSectm1a treatment increased the Th2 population to a greater degree than the Th1 population regardless of LPS stimulation, indicating a skewing effect on Th2 polarization. Furthermore, it was reported that the activation of GITR increased T cell survival and abrogated the suppressive action of Treg cells.58,59 Regarding the factors/signals that stimulate self-renewal of TRMs, several cytokines (i.e., IL-4, GM-CSF, M-CSF, and IL-34) have been validated thus far.9,60 Nonetheless, among these stimulators, GM-CSF, M-CSF, and IL-34 are all context and tissue specific in promoting TRM proliferation,9 and only IL-4 is able to induce rapid local proliferation in diverse tissues.44,45,61 Therefore, our present work, together with these prior studies, supports that Sectm1a, as a novel GITR ligand, promotes the expansion of Th cells, especially Th2, and increases secretion of Th2 cytokines, which, in turn, stimulate the local proliferation of TRMs and improve animal survival following endotoxin treatment. Consistent with our findings, Scumpia et al.62 observed that treatment of mice with DTA-1, a GITR ligand, restored CD4+ T cell proliferation, increased Th2 cytokine production, and improved survival in mice after injection of bacteria. In addition, after analyzing >150 million human patients in the Truven MarketScan insurance claim database, Krishack et al.63 found that patients with activation of type 2 allergic immune response (characterized by the expansion of Th2 and production of Th2 cytokines) possess a significantly lower incidence of sepsis and/or sepsis-related mortality, suggesting that pre-existing Th2 activation could protect against the development of sepsis.
Although TRMs are known to promote angiogenesis in the context of tumor growth,64 whether TRMs influence vascular integrity during sepsis remains unclear. Under sepsis condition, loss of endothelial barrier accounts for the vascular leakage and contributes to infiltration of leukocytes, tissue edema, and organ damage.65,66 In the present study, using in vitro splenic RPMs as a model of TRMs, we observed that RPMs helped to maintain EC monolayer integrity by upregulation of tight-junction-associated proteins (i.e., ZO-1, Occludin, and VE-Cadherin). Furthermore, pre-depletion of TRM in vivo augmented LPS-induced vascular leakage in multiple organs of mice. Together, both in vitro and in vivo data suggest that TRMs (especially those located near vessels) may work as a “gatekeeper” and play a pivotal role in maintaining the vascular integrity. Therefore, the elevation of TRMs may protect against sepsis-induced vascular hyper-permeability and organ failure.
Currently, it is well recognized that there is a special monocyte reservoir in the spleen (splenic reservoir monocytes). As a major source of monocytes, splenic monocytes egress from the spleen, migrate to sites of inflammation, and promote chronic inflammation and organ damages in response to myocardial infarct, stroke, and atherosclerosis.34, 35, 36, 37 Nevertheless, how to regulate such monocyte migration from spleen to other organs remains obscure. In the present study, we observed that global depletion of Sectm1a in mice caused more release of splenic monocytes, contributing to the further infiltration of inflammatory monocytes into the lung and the heart compared with WT mice following endotoxin treatment. By contrast, administration of mice with rSectm1a protein exhibited the opposite effects. These findings suggest that Sectm1a may be a critical factor to coordinate crosstalk between spleen and other organs during sepsis.
Some limitations of our study should be acknowledged. Although our data show that TRMs are essential for maintaining local vascular integrity during acute inflammation, the further molecular mechanism behind this needs to be explored in the future. In addition to causing pulmonary failure and cardiovascular dysfunction, severe sepsis also resulted in other organ dysfunctions, such as kidney dysfunction (reduced urine volume) and liver dysfunction (jaundice and cholestasis). Thus, in the future, the population change of TRM at the earlier stage of acute inflammation should also be determined in other organs. Furthermore, although the data in this study focused on Sectm1a’s effects during early inflammatory insults, we did not investigate its effects at a later stage, the inflammation resolution phase. During this phase, TRMs would undergo a transient and intense proliferative burst in situ to repopulate the tissue and facilitate tissue repair processes.46 Interestingly, our results showed that the gene expression levels of Sectm1a in the lung and heart were significantly increased at a later time point after LPS treatment, implying a positive regulatory effect of Sectm1a on the self-renewal capacity of TRMs during the inflammation resolution phase. Besides, given that the successive loss in self-renewal capacity with age may help to explain the dramatic increases of sepsis incidence and mortality rates among aged patients (≥65 years) compared with younger patients,17, 18, 19,46 it would be very interesting and important to decipher the role of Sectm1a in mediating the local proliferation of TRM in aged mice, which might have a great biomedical potential. Together, future new discoveries in the field of TRMs would be very helpful in the development of novel drugs that treat a wide range of acute inflammatory diseases.
Materials and Methods
Mice
All mice were maintained and bred under specific pathogen-free conditions in the Division of Laboratory Animal Resources at the University of Cincinnati Medical Center. All animal experiments conformed to the Guidelines for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences, published by the National Institutes of Health, and approved by the University of Cincinnati Animal Care and Use Committee. The mouse models used in this study were described in the electronic supplementary material.
Cell Culture
We isolated naïve CD4+ T cells and CD4+ T cell from mouse spleen by using EasySep Mouse Naïve CD4+ T Cell Isolation Kit (STEMCELL) and EasySep™ Mouse CD4+ T Cell Isolation Kit (STEMCELL), respectively. The detailed cell preparations and cell culture conditions of CD4+ T Cell, RPMs, and mouse cardiac endothelial cells (MCECs) were briefly described in the electronic supplementary material.
Flow Cytometry Analysis
Single-cell suspensions were prepared according to previous studies, which are described in the Supplemental Materials and Methods. Cells were stained with LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen) for the detection of live/dead cells before staining of the cell surface. A CD16/32 antibody (clone 93) (eBioscience, 1:100 dilution) was used to block the non-specific binding to Fc receptors before surface staining. All antibodies used in this study are listed in Table S3. The detailed method descriptions for the surface marker staining and intracellular Ki-67 staining are available in the Supplemental Materials and Methods.
Co-culture of RPMs with Naïve CD4 T Cells
RPMs were seeded on the coverslip at the same density (1 × 105) and cultured on the lower chamber of Transwell (Corning Life Sciences). Before co-culture, the insert was coated with IgG2a (1.25 μg/mL) or rSectm1a (2 μg/mL) at 4°C for 24 h. Naïve CD4+ T cells were isolated from spleen by using EasySep Mouse Naïve CD4+ T Cell Isolation Kit (STEMCELL) and cultured on the pre-coated insert (1.5 × 106/insert). At the same time, Dynabeads covalently coupled to anti-CD3, and anti-CD28 antibodies were added to stimulate naïve CD4 T cells. After 4 days stimulation, half of the Transwells were treated with 200 ng/mL LPS for 2 days. Then, supernatants were collected to determine the cytokine level. RPMs were stained with EdU (green) and F4/80 (red). Meanwhile, intracellular cytokine assays for IFN-γ and IL-4 were carried out following PMA (20 ng/mL) and ionomycin (1 μg/mL) stimulation to determine the Th1 and Th2 differentiation.
EdU Incorporation Assay and Immunofluorescence Stain
EdU (Click-iT EdU Cell Proliferation Imaging Kit, Invitrogen) is a nucleoside analog of thymidine. It is incorporated into DNA during active DNA synthesis. Cell proliferation was determined by the EdU incorporation assay according to the manufacturer’s instruction. After co-culture, RPMs were incubated with an EdU-labeling solution for 6 h at 37°C and then fixed with 4% cold formaldehyde for 30 min at room temperature. After permeabilization with 1% Triton X-100, cells were reacted with Click-iT reaction cocktails (Invitrogen) for 30 min. After washing, cells were incubated with PE-conjugated anti-mouse F4/80 for 1 h at room temperature. Subsequently, the DNA contents of the cells were stained with Hoechst 33342 for 30 min. Finally, cells were mounted with ProLong Diamond Antifade Mounting medium with DAPI (Invitrogen). Images were captured with Zeiss LSM710 LIVE Duo Confocal Microscope (Live Microscopy Core, University of Cincinnati). The detailed immunofluorescence stain method descriptions are available in the Supplemental Materials and Methods.
Measurement of Vascular Leakage in Multiple Organs
The pulmonary microvascular permeability was determined in each experimental group by measuring the EB accumulation in lung tissue and lung wet to dry weight ratio. The detailed methods are described in the Supplemental Materials and Methods. For the measurement of cardiac vascular permeability, EB accumulation in the heart was determined as described previously, which is also briefly described in the Supplemental Materials and Methods.
Spleen/Lung Histology and Injury Score
The spleen and lung tissues were collected from mice at 24 h post-LPS injection. The detailed histology and injury score method descriptions are available in the Supplemental Materials and Methods.
In Vitro Endothelial Permeability Assay
MCECs (8 × 104 cells/well) were seeded on the insert of 12-well Transwell (Corning) and cultured in the presence or absence of RPMs for 3 days to form a monolayer. Just before stimulating ECs with thrombin (Sigma) (5 U/mL), the fluorescein isothiocyanate (FITC)-dextran (10 kDa) (Sigma) stock solution was added to the medium on the insert to achieve a final concentration of 1 mg/mL. After 30 min, 50 μL samples of the medium were taken in triplicate from the lower chamber and placed in a 96-well cluster plate to measure the fluorescent intensity (excitation at 485 nm and emission at 535 nm).
RNA Isolation and Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from cultured cells or tissues by using the RNeasy Kit (Qiagen) following the manufacturer’s manual. cDNA was synthesized from 0.5–1.0 μg RNA by using Superscript II Reverse Transcriptase (Invitrogen). Then, qRT-PCR was performed in triplicate with the ABI PRISM 7900HT Sequence Detection System (ABI) using SYBR green (Genecopoeia). Relative mRNA levels were calculated and normalized to 18S rRNA. The primers were as follows: ZO-1 forward 5′-GCTAAGAGCACAGCAATGGA-3′, reverse 5′GCATGTTCAACGTTATCCAT; VE-cadherin forward 5′-TCTTGCCAGCAAACTCTCCT-3′, reverse 5′-TTGGAATCAAATGCACATCG-3′; Occludin forward 5′-GGACCCTGACCACTATGAAACAGACTAC-3′, reverse 5′-ATAGGTGGATATTCCCTGACCCAGTC-3′; Sectm1a forward 5′-ATGATGACCTGCCCTTCAGTGC-3′, reverse 5′-CTGGGTGTCTTTGATCACAAGC-3′; and 18S rRNA forward 5′-GCAATTATTCCCCATGAACG-3′, reverse 5′-GGCCTCACTAAACCATCCAA-3′.
Measurement of Cytokines
Blood was collected 12 h after LPS injection, and plasma samples were prepared by centrifugation at 3000 × g for 15 min at 4°C. Cell culture supernatants from RPM and naïve CD4 T cell co-culture experiments were collected and centrifugation at 800 × g for 15 min at 4°C to remove cell debris. All plasmas and cell culture supernatants were put into -80 °C until use. The concentration of cytokines was measured using Luminex Multiplex Immunoassay (Invitrogen) according to the manufacturer’s protocol.
Western Blot Analysis
The total protein was extracted from MCECs cultured with control medium or RPM-conditioned medium. Equal amounts of protein were subjected to SDS-PAGE and gel electrophoresis as described in a previous study.1 The following antibodies were used: rabbit anti-ZO-1 (61-7300) (1:250; ThermoFisher); rabbit anti-VE-Cadherin (ab33168) (1:500; Abcam); and rabbit anti-GAPDH (1:1000; Cell Signaling Technology).
Statistical Analysis
All data were tested for normality with Shapiro-Wilks test before statistical analysis. Statistical calculations were performed with GraphPad Prism 5.0 and all data were presented as mean ± SEM. Data were analyzed using two tailed Student’s t test (normally distributed data) or Mann-Whitney test (non-normally distributed data) to determine the significance between population means when two groups were compared. For comparison of more than two groups, one-way ANOVA or two-way ANOVA were used to determine the significance. The survival rates were constructed using the Kaplan-Meier method, and differences in mortality were compared using log-rank (Mantel-Cox) test. A p <0.05 was considered statistically significant.
Acknowledgments
This study was partially supported by the National Institutes of Health (NIH) (grants R01 GM-126061 and GM-132149) and American Heart Association (AHA) Established Investigator Award (17EIA33400063 to G.-C.F.).
Author Contributions
X.M. conceived the project, designed and performed experiments, and wrote the manuscript. H.F. measured the plasma level of Sectm1 in sepsis patients and healthy donors. P.W., K.E., Y.L., and Q.L. helped to sacrifice mice and collect samples. P.W. and K.D. helped to conduct flow cytometry experiments. Y.L. and X.W. reviewed/edited the manuscript. J.C. and T.P helped to analyze data. G.-C.F. analyzed results, reviewed/edited the manuscript, provided financial and administrative support, and gave final approval of the manuscript.
DECLARATION of Interest
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.001.
Contributor Information
Xingjiang Mu, Email: muxingjiang@163.com.
Guo-Chang Fan, Email: fangg@ucmail.uc.edu.
Supplemental Information
References
- 1.Fleischmann C., Scherag A., Adhikari N.K., Hartog C.S., Tsaganos T., Schlattmann P., Angus D.C., Reinhart K., International Forum of Acute Care Trialists Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am. J. Respir. Crit. Care Med. 2016;193:259–272. doi: 10.1164/rccm.201504-0781OC. [DOI] [PubMed] [Google Scholar]
- 2.Paoli C.J., Reynolds M.A., Sinha M., Gitlin M., Crouser E. Epidemiology and Costs of Sepsis in the United States-An Analysis Based on Timing of Diagnosis and Severity Level. Crit. Care Med. 2018;46:1889–1897. doi: 10.1097/CCM.0000000000003342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaieski D.F., Edwards J.M., Kallan M.J., Carr B.G. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit. Care Med. 2013;41:1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 4.Hershey T.B., Kahn J.M. State Sepsis Mandates – A New Era for Regulation of Hospital Quality. N. Engl. J. Med. 2017;376:2311–2313. doi: 10.1056/NEJMp1611928. [DOI] [PubMed] [Google Scholar]
- 5.Dunn D.L. Prevention and treatment of multiple organ dysfunction syndrome: lessons learned and future prospects. Surg. Infect. (Larchmt.) 2000;1:227–236. doi: 10.1089/109629600750018150. discussion 236–237. [DOI] [PubMed] [Google Scholar]
- 6.Opal S.M. Endotoxins and other sepsis triggers. In: Ronco C., editor. Endotoxemia and Endotoxin Shock. Karger Publishers; 2010. pp. 14–24. [Google Scholar]
- 7.Hotchkiss R.S., Moldawer L.L., Opal S.M., Reinhart K., Turnbull I.R., Vincent J.L. Sepsis and septic shock. Nat. Rev. Dis. Primers. 2016;2:16045. doi: 10.1038/nrdp.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar V. Targeting macrophage immunometabolism: Dawn in the darkness of sepsis. Int. Immunopharmacol. 2018;58:173–185. doi: 10.1016/j.intimp.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 9.Sieweke M.H., Allen J.E. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013;342:1242974. doi: 10.1126/science.1242974. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto D., Chow A., Noizat C., Teo P., Beasley M.B., Leboeuf M., Becker C.D., See P., Price J., Lucas D. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheng J., Ruedl C., Karjalainen K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity. 2015;43:382–393. doi: 10.1016/j.immuni.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 12.Ginhoux F., Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44:439–449. doi: 10.1016/j.immuni.2016.02.024. [DOI] [PubMed] [Google Scholar]
- 13.Epelman S., Lavine K.J., Randolph G.J. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi C., Pamer E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts A.W., Lee B.L., Deguine J., John S., Shlomchik M.J., Barton G.M. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity. 2017;47:913–927.e6. doi: 10.1016/j.immuni.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uderhardt S., Martins A.J., Tsang J.S., Lämmermann T., Germain R.N. Resident macrophages cloak tissue microlesions to prevent neutrophil-driven inflammatory damage. Cell. 2019;177:541–555.e17. doi: 10.1016/j.cell.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molawi K., Wolf Y., Kandalla P.K., Favret J., Hagemeyer N., Frenzel K., Pinto A.R., Klapproth K., Henri S., Malissen B. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014;211:2151–2158. doi: 10.1084/jem.20140639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gentek R., Molawi K., Sieweke M.H. Tissue macrophage identity and self-renewal. Immunol. Rev. 2014;262:56–73. doi: 10.1111/imr.12224. [DOI] [PubMed] [Google Scholar]
- 19.Hoeffel G., Ginhoux F. Ontogeny of tissue-resident macrophages. Front. Immunol. 2015;6:486. doi: 10.3389/fimmu.2015.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chmielewski S., Olejnik A., Sikorski K., Pelisek J., Błaszczyk K., Aoqui C., Nowicka H., Zernecke A., Heemann U., Wesoly J. STAT1-dependent signal integration between IFNγ and TLR4 in vascular cells reflect pro-atherogenic responses in human atherosclerosis. PLoS ONE. 2014;9:e113318. doi: 10.1371/journal.pone.0113318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y., Lua I., French S.W., Asahina K. Role of TGF-β signaling in differentiation of mesothelial cells to vitamin A-poor hepatic stellate cells in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016;310:G262–G272. doi: 10.1152/ajpgi.00257.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamata H., Yamamoto K., Wasserman G.A., Zabinski M.C., Yuen C.K., Lung W.Y., Gower A.C., Belkina A.C., Ramirez M.I., Deng J.C. Epithelial cell–derived secreted and transmembrane 1a signals to activated neutrophils during pneumococcal pneumonia. Am. J. Respir. Cell Mol. Biol. 2016;55:407–418. doi: 10.1165/rcmb.2015-0261OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howie D., Garcia Rueda H., Brown M.H., Waldmann H. Secreted and transmembrane 1A is a novel co-stimulatory ligand. PLoS ONE. 2013;8:e73610. doi: 10.1371/journal.pone.0073610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lyman S.D., Escobar S., Rousseau A.M., Armstrong A., Fanslow W.C. Identification of CD7 as a cognate of the human K12 (SECTM1) protein. J. Biol. Chem. 2000;275:3431–3437. doi: 10.1074/jbc.275.5.3431. [DOI] [PubMed] [Google Scholar]
- 25.Wang T., Huang C., Lopez-Coral A., Slentz-Kesler K.A., Xiao M., Wherry E.J., Kaufman R.E. K12/SECTM1, an interferon-γ regulated molecule, synergizes with CD28 to costimulate human T cell proliferation. J. Leukoc. Biol. 2012;91:449–459. doi: 10.1189/jlb.1011498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsalik E.L., Langley R.J., Dinwiddie D.L., Miller N.A., Yoo B., van Velkinburgh J.C., Smith L.D., Thiffault I., Jaehne A.K., Valente A.M. An integrated transcriptome and expressed variant analysis of sepsis survival and death. Genome Med. 2014;6:111. doi: 10.1186/s13073-014-0111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bronte V., Pittet M.J. The spleen in local and systemic regulation of immunity. Immunity. 2013;39:806–818. doi: 10.1016/j.immuni.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgren G., Almqvist R., Hartman M., Utter G.H. Splenectomy and the risk of sepsis: a population-based cohort study. Ann. Surg. 2014;260:1081–1087. doi: 10.1097/SLA.0000000000000439. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y.C., Yu M.M., Shou S.T., Chai Y.F. Sepsis-induced cardiomyopathy: mechanisms and treatments. Front. Immunol. 2017;8:1021. doi: 10.3389/fimmu.2017.01021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mu X., Wang X., Huang W., Wang R.T., Essandoh K., Li Y., Pugh A.M., Peng J., Deng S., Wang Y. Circulating Exosomes Isolated from Septic Mice Induce Cardiovascular Hyperpermeability Through Promoting Podosome Cluster Formation. Shock. 2018;49:429–441. doi: 10.1097/SHK.0000000000000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yona S., Kim K.W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies L.C., Jenkins S.J., Allen J.E., Taylor P.R. Tissue-resident macrophages. Nat. Immunol. 2013;14:986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borges da Silva H., Fonseca R., Pereira R.M., Cassado Ados.A., Álvarez J.M., D’Império Lima M.R. Splenic macrophage subsets and their function during blood-borne infections. Front. Immunol. 2015;6:480. doi: 10.3389/fimmu.2015.00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swirski F.K., Nahrendorf M., Etzrodt M., Wildgruber M., Cortez-Retamozo V., Panizzi P., Figueiredo J.L., Kohler R.H., Chudnovskiy A., Waterman P. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Francis M., Sun R., Cervelli J.A., Choi H., Mandal M., Abramova E.V., Gow A.J., Laskin J.D., Laskin D.L. Editor’s highlight: Role of spleen-derived macrophages in ozone-induced lung inflammation and injury. Toxicol. Sci. 2017;155:182–195. doi: 10.1093/toxsci/kfw192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leuschner F., Panizzi P., Chico-Calero I., Lee W.W., Ueno T., Cortez-Retamozo V., Waterman P., Gorbatov R., Marinelli B., Iwamoto Y. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ. Res. 2010;107:1364–1373. doi: 10.1161/CIRCRESAHA.110.227454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsiao H.M., Fernandez R., Tanaka S., Li W., Spahn J.H., Chiu S., Akbarpour M., Ruiz-Perez D., Wu Q., Turam C. Spleen-derived classical monocytes mediate lung ischemia-reperfusion injury through IL-1β. J. Clin. Invest. 2018;128:2833–2847. doi: 10.1172/JCI98436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose S., Misharin A., Perlman H. A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytometry A. 2012;81:343–350. doi: 10.1002/cyto.a.22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hussell T., Bell T.J. Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014;14:81–93. doi: 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- 40.Frantz S., Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc. Res. 2014;102:240–248. doi: 10.1093/cvr/cvu025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aurora A.B., Porrello E.R., Tan W., Mahmoud A.I., Hill J.A., Bassel-Duby R., Sadek H.A., Olson E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Invest. 2014;124:1382–1392. doi: 10.1172/JCI72181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hulsmans M., Clauss S., Xiao L., Aguirre A.D., King K.R., Hanley A., Hucker W.J., Wülfers E.M., Seemann G., Courties G. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169:510–522.e20. doi: 10.1016/j.cell.2017.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mu X., Wang P., Wang X., Li Y., Zhao H., Li Q., Essandoh K., Deng S., Peng T., Fan G.C. Identification of a Novel Antisepsis Pathway: Sectm1a Enhances Macrophage Phagocytosis of Bacteria through Activating GITR. J. Immunol. 2020;205:1633–1643. doi: 10.4049/jimmunol.2000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jenkins S.J., Ruckerl D., Cook P.C., Jones L.H., Finkelman F.D., van Rooijen N., MacDonald A.S., Allen J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jenkins S.J., Ruckerl D., Thomas G.D., Hewitson J.P., Duncan S., Brombacher F., Maizels R.M., Hume D.A., Allen J.E. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J. Exp. Med. 2013;210:2477–2491. doi: 10.1084/jem.20121999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mu X., Li Y., Fan G.C. Tissue-Resident Macrophages in the Control of Infection and Resolution of Inflammation. Shock. 2021;55:14–23. doi: 10.1097/SHK.0000000000001601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shults J.A., Curtis B.J., Boe D.M., Ramirez L., Kovacs E.J. Ethanol intoxication prolongs post-burn pulmonary inflammation: role of alveolar macrophages. J. Leukoc. Biol. 2016;100:1037–1045. doi: 10.1189/jlb.3MA0316-111R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bajpai G., Bredemeyer A., Li W., Zaitsev K., Koenig A.L., Lokshina I., Mohan J., Ivey B., Hsiao H.M., Weinheimer C. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 2019;124:263–278. doi: 10.1161/CIRCRESAHA.118.314028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honold L., Nahrendorf M. Resident and monocyte-derived macrophages in cardiovascular disease. Circ. Res. 2018;122:113–127. doi: 10.1161/CIRCRESAHA.117.311071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barth M.W., Hendrzak J.A., Melnicoff M.J., Morahan P.S. Review of the macrophage disappearance reaction. J. Leukoc. Biol. 1995;57:361–367. doi: 10.1002/jlb.57.3.361. [DOI] [PubMed] [Google Scholar]
- 51.Kanamaru F., Youngnak P., Hashiguchi M., Nishioka T., Takahashi T., Sakaguchi S., Ishikawa I., Azuma M. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J. Immunol. 2004;172:7306–7314. doi: 10.4049/jimmunol.172.12.7306. [DOI] [PubMed] [Google Scholar]
- 52.Nocentini G., Giunchi L., Ronchetti S., Krausz L.T., Bartoli A., Moraca R., Migliorati G., Riccardi C. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA. 1997;94:6216–6221. doi: 10.1073/pnas.94.12.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kwon B., Yu K.Y., Ni J., Yu G.L., Jang I.K., Kim Y.J., Xing L., Liu D., Wang S.X., Kwon B.S. Identification of a novel activation-inducible protein of the tumor necrosis factor receptor superfamily and its ligand. J. Biol. Chem. 1999;274:6056–6061. doi: 10.1074/jbc.274.10.6056. [DOI] [PubMed] [Google Scholar]
- 54.Clouthier D.L., Watts T.H. Cell-specific and context-dependent effects of GITR in cancer, autoimmunity, and infection. Cytokine Growth Factor Rev. 2014;25:91–106. doi: 10.1016/j.cytogfr.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 55.Motta A.C., Vissers J.L., Gras R., Van Esch B.C., Van Oosterhout A.J., Nawijn M.C. GITR signaling potentiates airway hyperresponsiveness by enhancing Th2 cell activity in a mouse model of asthma. Respir. Res. 2009;10:93. doi: 10.1186/1465-9921-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van der Werf N., Redpath S.A., Phythian-Adams A.T., Azuma M., Allen J.E., Maizels R.M., Macdonald A.S., Taylor M.D. Th2 responses to helminth parasites can be therapeutically enhanced by, but are not dependent upon, GITR-GITR ligand costimulation in vivo. J. Immunol. 2011;187:1411–1420. doi: 10.4049/jimmunol.1100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ward-Kavanagh L.K., Lin W.W., Šedý J.R., Ware C.F. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity. 2016;44:1005–1019. doi: 10.1016/j.immuni.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shevach E.M., Stephens G.L. The GITR-GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat. Rev. Immunol. 2006;6:613–618. doi: 10.1038/nri1867. [DOI] [PubMed] [Google Scholar]
- 59.Esparza E.M., Arch R.H. Glucocorticoid-induced TNF receptor functions as a costimulatory receptor that promotes survival in early phases of T cell activation. J. Immunol. 2005;174:7869–7874. doi: 10.4049/jimmunol.174.12.7869. [DOI] [PubMed] [Google Scholar]
- 60.Röszer T. Understanding the biology of self-renewing macrophages. Cells. 2018;7:103. doi: 10.3390/cells7080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Minutti C.M., Jackson-Jones L.H., García-Fojeda B., Knipper J.A., Sutherland T.E., Logan N., Ringqvist E., Guillamat-Prats R., Ferenbach D.A., Artigas A. Local amplifiers of IL-4Rα-mediated macrophage activation promote repair in lung and liver. Science. 2017;356:1076–1080. doi: 10.1126/science.aaj2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scumpia P.O., Delano M.J., Kelly-Scumpia K.M., Weinstein J.S., Wynn J.L., Winfield R.D., Xia C., Chung C.S., Ayala A., Atkinson M.A. Treatment with GITR agonistic antibody corrects adaptive immune dysfunction in sepsis. Blood. 2007;110:3673–3681. doi: 10.1182/blood-2007-04-087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krishack P.A., Wang K., Rzhetsky A., Solway J., Sperling A.I., Verhoef P.A. Preexisting type 2 immune activation protects against the development of sepsis. Am. J. Respir. Cell Mol. Biol. 2017;57:628–630. doi: 10.1165/rcmb.2017-0277LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poh A.R., Ernst M. Targeting macrophages in cancer: from bench to bedside. Front. Oncol. 2018;8:49. doi: 10.3389/fonc.2018.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kadl A., Leitinger N. The role of endothelial cells in the resolution of acute inflammation. Antioxid. Redox Signal. 2005;7:1744–1754. doi: 10.1089/ars.2005.7.1744. [DOI] [PubMed] [Google Scholar]
- 66.Leenders G.J., Smeets M.B., van den Boomen M., Berben M., Nabben M., van Strijp D., Strijkers G.J., Prompers J.J., Arslan F., Nicolay K., Vandoorne K. Statins promote cardiac infarct healing by modulating endothelial barrier function revealed by contrast-enhanced magnetic resonance imaging. Arterioscler. Thromb. Vasc. Biol. 2018;38:186–194. doi: 10.1161/ATVBAHA.117.310339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.