

Abstract
In this work, we pioneered a combination of ultralow flow (ULF) high-efficiency ultranarrow bore monolithic LC columns coupled to MS via a high-field asymmetric waveform ion mobility spectrometry (FAIMS) interface to evaluate the potential applicability for high sensitivity, robust, and reproducible proteomic profiling of low nano-gram-level complex biological samples. As a result, ULF LC-FAIMS-MS brought unprecedented sensitivity levels and high reproducibility in bottom-up proteomic profiling. In addition, FAIMS improved the dynamic range, signal-to-noise ratios, and detection limits in ULF LC–MS-based measurements by significantly reducing chemical noise in comparison to the conventional nanoESI interface used with the same ULF LC–MS setup. Two, three, or four compensation voltages separated by at least 15 V were tested within a single LC–MS run using the FAIMS interface. The optimized ULF LC-ESI-FAIMS-MS/MS conditions resulted in identification of 2,348 ± 42 protein groups, 10,062 ± 285 peptide groups, and 15,734 ± 350 peptide-spectrum matches for 1 ng of a HeLa digest, using a 1 h gradient at the flow rate of 12 nL/min, which represents an increase by 38%, 91%, and 131% in respective identifications, as compared to the control experiment (without FAIMS). To evaluate the practical utility of the ULF LC-ESI-FAIMS-MS platform in proteomic profiling of limited samples, approximately 100, 1,000, and 10,000 U937 myeloid leukemia cells were processed, and a one-tenth of each sample was analyzed. Using the optimized conditions, we were able to reliably identify 251 ± 54, 1,135 ± 80, and 2,234 ± 25 protein groups from injected aliquots corresponding to ~10, 100, and 1,000 processed cells.
Graphical Abstract
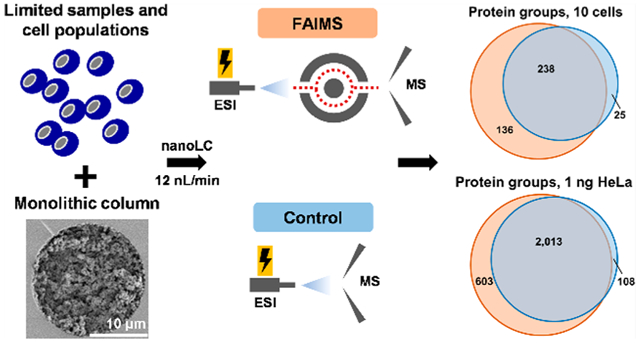
Deep proteomic profiling (i.e., at the level of thousands of characterized proteins) of scarce biological and clinical samples is still a major challenge. The ability to qualitatively and quantitatively characterize numerous proteins and their post-translational modifications (PTM) present in limited samples (e.g., microneedle biopsies, microsampled liquid biopsies, populations of rare cells, and even individual single cells) is highly important for acquiring new knowledge in fundamental biology research and enabling novel diagnostic and prognostic studies.1–4 In bottom-up proteomic approaches, proteins of highly complex samples are enzymatically cleaved into numerous peptides of vastly different structures, concentration levels (i.e., the wide dynamic range), and physicochemical properties, including peptide length, polarity, hydrophobicity, ionization efficiency, and chemical stability, which collectively pose a significant challenge for in-depth LC–MS-based proteomic profiling. Therefore, high-efficiency separation, e.g., capillary electrophoresis1,5–7 or LC,8 is needed to increase the detection sensitivity by ESI-MS. In the past decades, significant efforts were devoted to improving the detection sensitivity, separation performance, and throughput of LC–MS analysis by decreasing the flow rate, downscaling the column diameter, and increasing the column length.9–13 With an increased need for the improved separation efficiency and, therefore, the depth of profiling sensitivity in LC–MS-based proteomics,14,15 the development of nanoLC columns has been rapid, and sub-2-μm bead-packed,16,17 monolithic2,11,18–20 and open-tubular2,21–25 capillary columns, as well as pillar array columns,26 have been developed, as overviewed in several recent reviews.27–30
Currently, conventional nanoLC separations are carried out in packed fused-silica capillary columns with internal diameters (IDs) between 50 and 100 μm, typically operated at a flow rate between 100 and 400 nL/min, as the process of high quality packing in nanoLC columns with IDs below 50 μm is reportedly challenging.17,29,31 Monolithic stationary phases have been an attractive alternative for high complexity samples in proteomics applications as a result of their high permeability, low backpressure, ease of preparation, diverse surface chemistries, significant control on the porosity and structure of the stationary phase, high separation efficiency, and broad selectivity.11,18,27,28,32–34 While in the past, monolithic columns posed issues with inconsistent reproducibility, more recent reports show reasonable reproducibility of column preparation and performance; moreover, there are examples of monolithic columns that were commercialized.11,29,35 Rapid and highly efficient separation of complex peptide, protein, metabolite, oligonucleotide, and other small or large molecule mixtures can be routinely achieved using monolithic columns.28,32,36 When connected to MS, high-efficiency ultranarrow bore monolithic LC columns (≤20 μm ID) operated at ultralow flow (ULF) rates (≤20 nL/min) significantly increase the sensitivity of nanoESI-MS, resulting in an improved depth of qualitative and quantitative molecular profiling of limited clinical and biological samples.11,18 Smaller eluent droplets generated by the nanoESI emitter with ULF LC–MS result in higher ionization efficiency of analyte species, lower ion suppression, and a miniaturized nanoESI plume, leading to higher ion transfer efficiency and improved MS sensitivity. The performance of ULF columns has been effectively demonstrated in high sensitivity LC–MS-based bottom-up and top-down proteomics applications and has also established the potential of ULF LC–MS for reaching the sensitivity levels required for proteomic profiling of individual cells.2,22,31,37 Despite the reported attractive advantages, the current implementations of the ULF LC–MS technology still lack the sufficient robustness and reproducibility required to become commercialized and widespread.
During the last 15 years, mass spectrometric technologies have undergone a substantial evolution, as evident from the improvements in the speed of data acquisition, detection limits, mass range, mass resolution, and mass accuracy.14,38,39 Several key advances were made to improve ion sampling in sources of mass spectrometers and interfacing of liquid-phase separations with MS. One example of such advances is high-field asymmetric waveform ion mobility spectrometry (FAIMS), which is an atmospheric pressure ion mobility technology that separates gas-phase ions by their characteristic differences in mobility in an asymmetric electric field.40,41 Over the past several years, high-resolution FAIMS technologies were developed.42 The benefits of the FAIMS interface were demonstrated for diverse qualitative and quantitative proteomics applications, including top-down,43,44 middle-down,45 and bottom-up proteomics39,46–48 and lipidomics.49,50 Comprehensive overviews of the state-of-the-art FAIMS MS-based proteomics techniques can be found in several recent reviews.51–53 FAIMS has shown significant advantages by focusing and separating multiply charged peptide ions from interfering singly charged background ions, resulting in improved dynamic range and detection limits in MS-based measurements.47,54,55 However, it was reported that a fraction of multiply charged ions are lost in the interface, and therefore, FAIMS can decrease the ion transmission efficiency and the MS signal.47,55–57 The total depth of molecular profiling may be therefore compromised if the ion transmission efficiency is decreased by FAIMS, especially when minute amounts of starting material are used (i.e., low ng-level). On the basis of our knowledge, there are no reports showing a practical utility of the FAIMS technology on proteomic analysis on such low amounts of complex biomedical samples.
In this work, for the first time, we demonstrate the combination of high-efficiency ULF separation, using ultranarrow bore monolithic nanoLC capillary columns interfaced with MS using the FAIMS technology. To the best of our knowledge, this is the first report showing the increased depth of proteomic profiling for low-ng proteomic samples and LC columns operated at flow rates below 250 nL/min.45 In-house polymerized polystyrene-divinylbenzene (PS-DVB) monolithic columns, that were operated at a flow rate of 12 nL/min, and the FAIMS Pro technology interfaced with an Orbitrap Fusion Lumos significantly increased the depth of proteomic profiling for low-ng standard HeLa digest sample as well as limited samples containing approximately 10, 100, and 1,000 U937 myeloid leukemia cells per LC injection compared to the control experiments (without FAIMS).
MATERIALS AND METHODS
Nano-Liquid Chromatography.
The monolithic column preparation procedure followed a previously published protocol with minor changes.2,11,18 In brief, a fused silica capillary was pretreated with 1 M NaOH and the inner surface was silanized. The polymerization of the polymerization mixture was initiated using elevated temperature. More experimental details are provided in the Supporting Information. An Ultimate 3000 nanoLC system (Thermo Fisher Scientific, Waltham, MA) was used for nanoLC separations. Several system adjustments to plumbing and connections were required to generate flow rates below 50 nL/min (see schematic in Figure 1). The monolithic column was connected to a column switching valve via a NanoViper LC Column to MS Tubing (20 μm × 360 μm × 1 m, P/N 6041.5293, Thermo) to a microtee (P/N P888, IDEX Health & Science, Lake Forest, IL). The end of the monolithic column was connected butt-to-butt to the ESI emitter (FS360-10-5-D-20, NewObjective, Woburn, MA) via a Teflon union (P/N 161030, Thermo) to achieve a zero dead volume connection. The ESI voltage was applied to a distal coated emitter, and a Nanospray Flex Ion Source (Thermo) was used to generate stable nanoESI of the eluate. A 15 cm × 10 μm ID capillary connected to the tee union served as a restrictor to generate the ULF at 12 nL/min (Figure 1b). During the sample loading step, the splitting capillary was connected to a plug through a switching valve; otherwise, the restrictor was connected to a waste line to generate the desired flow rate. The sample was loaded at 50 nL/min for 1 h using 1% of solvent B. Peptides were eluted at 12 nL/min from the column with a 1 h linear gradient from 1% B to 15% B, where solvent A consisted of 0.1% formic acid (FA) in water and solvent B of 0.1% FA in acetonitrile (ACN). The solvent composition was switched from 15% B to 80% B over 2 min and held constant for 3 min. Finally, the mobile phase composition was changed from 80% B to 1% B over 0.1 min and then held constant at 1% B for 45 min. The HeLa protein digest standard (P/N 88328, Thermo) was resuspended in 1% FA in water (to 1, 10, 50, and 100 ng/μL) and analyzed in triplicate.
Figure 1.
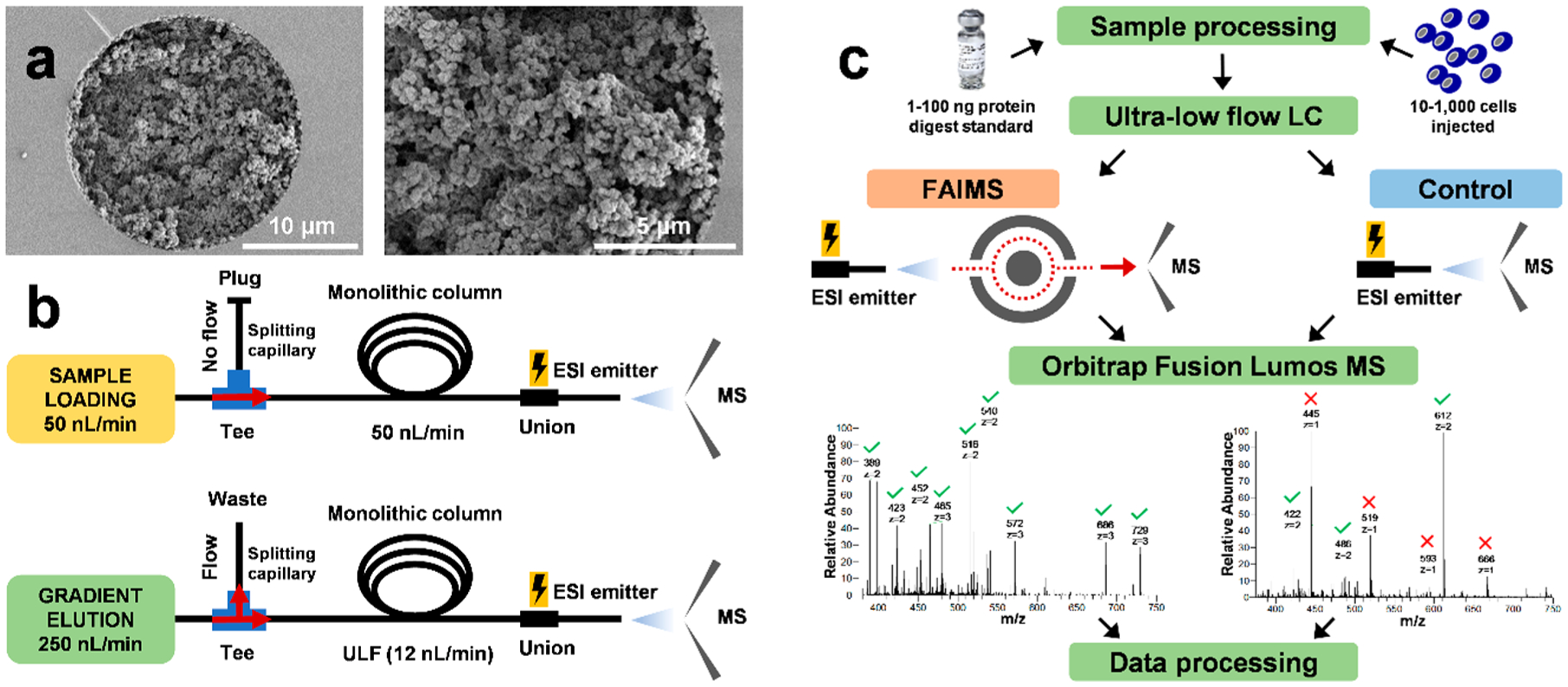
Overview of the ULF LC-ESI-FAIMS-MS/MS experimental design and workflow. (a) SEM images of the 20 μm ID PS-DVB-based monolithic column show the bed structure, column porosity, and the attachment of the stationary phase to the capillary wall. (b) Schematics of plumbing and configuration of the ULF LC–MS system during sample loading at 50 nL/min and gradient elution at 12 nL/min. (c) The experimental workflows with and without the FAIMS Pro interface for in-depth proteomic analysis of HeLa protein digest standard (1–100 ng injected) and samples derived from 10 to 1,000 cells. Green checkmarks and red × in representative MS1 spectra indicate doubly charged and singly charged ions, respectively.
Mass Spectrometry and FAIMS.
MS experiments were performed using an Orbitrap Fusion Lumos equipped with an Ultimate 3000 nanoLC system (both Thermo). LC–MS-based profiling experiments were conducted with or without a FAIMS Pro interface (Thermo) mounted to the MS to assess the impact on the results of proteomic profiling. These experiments were labeled in the text and figures as “FAIMS” or “control,” respectively. The ion transfer tube temperature was set to 275 °C. The ESI voltage was set to 1.5 kV using a Nanospray Flex Ion Source.
The ULF LC-FAIMS-MS/MS experiments were performed using the following experimental settings: the temperatures of the FAIMS inner electrode, outer electrode 1, and outer electrode 2 were set to 100 °C (Standard Resolution Mode) and the FAIMS user set gas flow of 0 L/min was used. Multiple compensation voltages (CVs) were evaluated within each single stage MS data acquisition method (“internal CV stepping” type of experiment, see Table S1) followed by tandem MS data acquisition as described below. An example of the MS method where 4 CVs were used within 6 s total MS cycle time is in Figure S1.
Data Analysis.
All raw files were analyzed using Proteome Discoverer (v. 2.3, Thermo) and MaxQuant (v. 1.6.14.0)58 software using matched parameters wherever possible, and the UniProtKB/Swiss-Prot human database (Release 2020_01, containing 20,302 reviewed sequences) was used. A nonredundant contaminant database was created combining the MaxQuant and the common Repository of Adventitious Proteins (cRAP) contaminant fasta databases (containing 276 sequences) to filter out common contaminant proteins.59
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE60 partner repository with the data set identifier PXD020669. Additional experimental details about materials, reagents, and methods, including U937 cell culture, on-micro SPE tip-based sample processing, preparation of monolithic column, mass spectrometry, and data analysis are provided in the Supporting Information.
RESULTS AND DISCUSSION
Coupling ULF liquid phase separations to MS proved to be promising for increasing the sensitivity and depth of proteomic profiling of limited samples (low-ng), as compared to conventional nanoflow techniques.10,13 The FAIMS interface was successfully applied in nano- and microflow LC–MS proteomic applications using the flow rate of ~250 nL/min to 400 μL/min,45,61 and the majority of sample amounts were ranging from microgram to milligrams levels of complex proteome-level samples.39,45,47,54,62–65 Examples of the analysis of ng-level samples, 5 ng39 and 100 ng47 of tryptic peptides, were recently shown. However, the applicability of FAIMS in analysis of limited samples (i.e., low ng-level) and in combination with ULF separation techniques, where the ion transmission efficiency and the general compatibility of the FAIMS interface with both very low influx of ions and ULF of the eluent (sub-20 nL/min) are critical, has not yet been reported, to the best of our knowledge.
In order to demonstrate how ULF LC, using polymeric ultranarrow bore (20 μm ID) monolithic columns coupled with MS via the FAIMS Pro interface, can enhance the proteomic profiling of limited samples, we first analyzed a commercially available digested HeLa cell lysate standard as a representative high-complexity sample. Sample aliquots were diluted to desired concentration levels corresponding to total amounts of digested protein ranging from 1 to 100 ng, which approximately corresponds to 5–500 HeLa cells, respectively.31 The specimens were analyzed with and without the FAIMS Pro interface placed between the nanoESI emitter of the column outlet and the inlet of the mass spectrometer (Figure 1c).
Experimental Setup.
An in-house prepared PS-DVB-based monolithic column (20 μm ID × 35 cm) with incorporated C18 moieties to increase the column hydro-phobicity was used in this study (Figure 1a). The zoomed-in SEM image reveals a homogeneous and continuous bed structure attached to the capillary wall without any gaps. A conventional nanoLC system was used to load the sample at 50 nL/min and to apply the gradient elution at 12 nL/min using split flow (Figure 1b). To each FAIMS experiments, replicate control experiments (without FAIMS Pro interface incorporated into the workflow) were conducted to assess the change in the profiling sensitivity (Figure 1c).
Background Noise Reduction in ULF LC–MS of Low ng Samples Using FAIMS Pro Interface.
We assessed if the application of the FAIMS technology to our ULF LC–MS workflow in the analysis of low ng samples would benefit from the same reduction in noise level, as previously reported, and if the reduced noise level is one of the reasons for the improved profiling sensitivity.39,46 We compared LC–MS base peak intensity chromatograms (BICs) for 1 ng of the HeLa standard analyzed with and without the FAIMS Pro interface (Figure 2a–b). To plot BIC traces for the FAIMS experiments, we combined the LC–MS BIC for all CVs. The initial examination of the BICs expectedly showed similar patterns in peak distribution throughout the elution profile and comparable signal intensity in the FAIMS experiments for the same sample amount. Background ion signal was estimated by observing the BIC signal during the “empty” retention time (RT) ranges of the chromatograms (before or after the elution of sample peaks and during column equilibration). When the control experiments are compared to the FAIMS experiments, the background chemical noise reduction observed is 3 orders of magnitude lower in the FAIMS experiments (Figure 2c–d, Figure S2a–g). The background noise level in the FAIMS experiments is in the range of 1.0E3–1.0E4, while in the control experiments, the noise level was at much higher values of 1.0E6–1.0E7. Most of the background noise signal in the control experiments corresponds to singly charged polysiloxane ions that are partially or completely removed by the FAIMS interface depending on the used CV. In fact, the highest intensity polysiloxane (m/z 445.12) was ~100% removed with all evaluated CVs (Figure 2e–f). These results, combined with comparative peptide signal measurements, confirm that the FAIMS technology helped substantially decrease the level of chemical noise without a corresponding drastic reduction in the signal intensity for peptides (Figure 1c, Figure 2a–b, and Figure S3), which collectively led to, on average, improved signal-to-noise (S/N) ratios and detection limits in nanoLC- MS-based proteomic profiling. To assess the effect of FAIMS on S/N ratios, we randomly selected several (n = 12) multiply charged high abundance peptide peaks across the whole gradient elution profile. The representative data show an ~1.6-fold improvement in the S/N ratio for most of the selected high abundance peaks (Figure S3).
Figure 2.
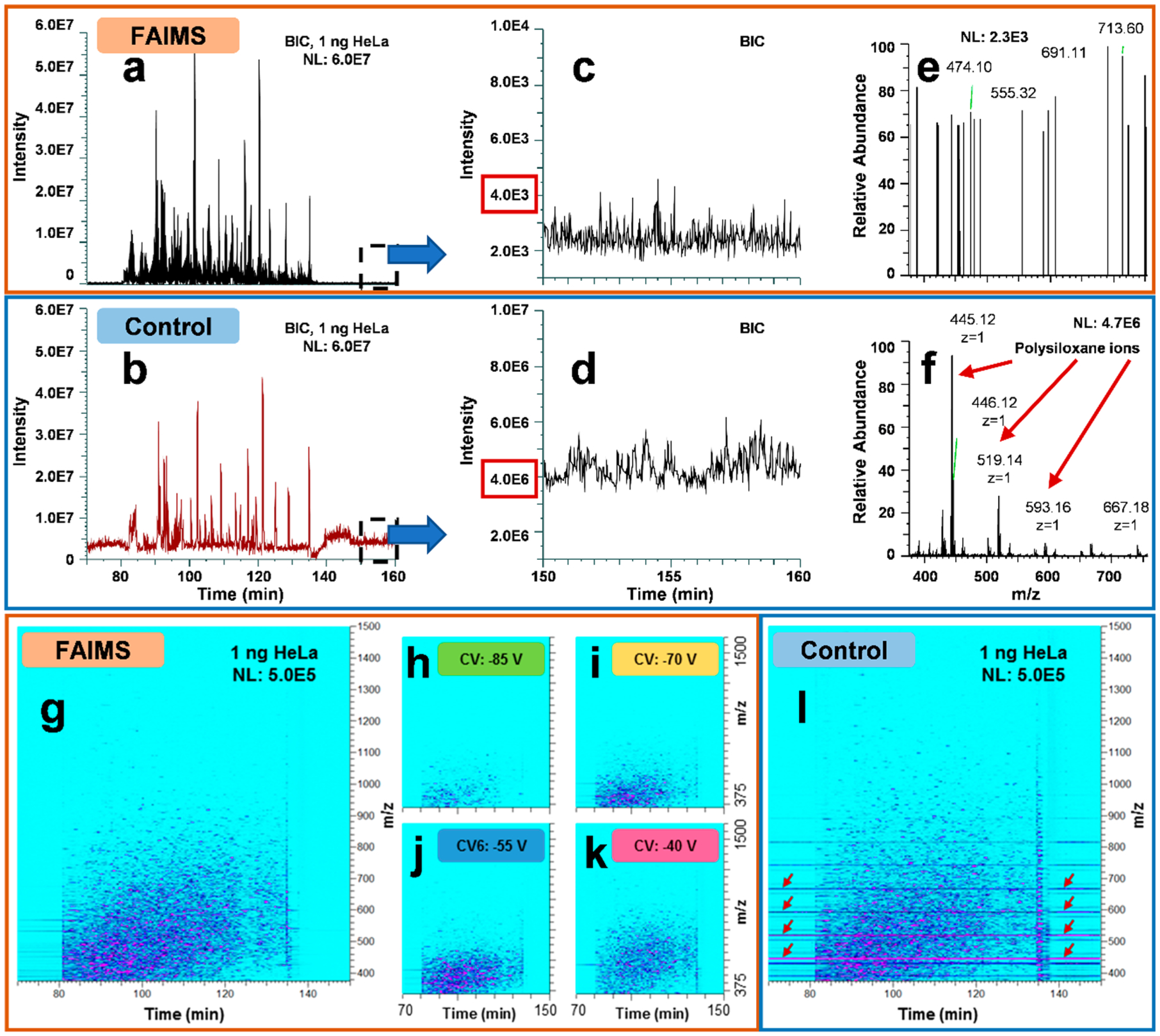
Background noise reduction in ULF LC–MS of 1 ng HeLa protein digest standard samples using the FAIMS Pro interface. BICs for both (a) FAIMS and (b) control experiments. The FAIMS plot is a result of a combination of 4 CVs (−85, −70, −55, and −40 V) used within one internal CV stepping method. BIC background intensity levels within the “peptide-free” portion (column equilibration) of the chromatogram is plotted for (c) FAIMS (at CV −55 V) and (d) control experiments. (e and f) The representative FAIMS and control MS1 scans for the RT range acquired at ~154.7 min. Polysiloxane background ions typical for control experiments are indicated by red arrows. Ion density maps for the (g–k) FAIMS and (l) control ULF LC–MS experiments at signal intensities are shown normalized to 1.0E6. For the FAIMS experiments, (h–k) ion density maps for individual CVs and (g) the total signal integrated for all CVs are shown. Red arrows highlight high-intensity streaking lines corresponding to polysiloxanes (m/z 445.12, 519.14, 593.16, and 667.18).
Similar observations regarding the reduction of the background chemical noise, signal suppression, and ion interference due to the effective removal of singly charged ions by the FAIMS interface versus control experiments, using identical data acquisition parameters (e.g., automatic gain control, ion accumulation time), were made after examining the generated ULF LC–MS ion density maps for analyses of ng-level HeLa samples (1 and 100 ng, see Figure 2g–l and Figure S4a–f, and for 10 and 50 ng, data not shown). For example, in a representative LC–MS ion density map for the analysis of 1 ng of the HeLa standard, multiple singly charged contaminant ions (e.g., siloxanes, polymers) were detected across the wide range of RTs in the control experiments and significantly removed or decreased in their intensity levels in the FAIMS experiments (Figure 2g–l). Such ion species with m/z values of 445.12, 462.15, 519.14, 536.17, etc. that were predominant in the control experiments were substantially diminished in intensity (often to the level below the LOD, i.e., completely removed from the experimental data) in the FAIMS experiments, mainly correspond to polysiloxanes, common contaminants in LC–MS.66 Thus, there are no detected peaks corresponding to polysiloxanes in the described “empty” RT ranges (Figure 2g–k) that dramatically reduced overall noise. Additionally, numerous singly charged ions at the end of the gradient elution (at around 136 min), which most likely correspond to polymeric and other hydrophobic contaminants, were significantly reduced in the FAIMS experiments (Figure 2g–l). A few low-intensity streaking peaks (e.g., with m/z of 450.77 and 567.78) that are observed between the retention times of 70 and 80 min (Figure 2g–k) correspond to highly hydrophilic peptides, that are poorly retained on the monolithic column and elute in broad tailing peaks. Ion density maps plotted for different CVs show distinct peptide patterns (Figure 2h–k). Similarly, unique peptide detection patterns and the decreased noise level were observed in LC–MS ion density maps, when larger amounts of the HeLa standard were analyzed (Figure S4a–f).
Next, we examined BICs for individual CVs, using a normalized intensity scale for 1 ng of the standard HeLa digest analyzed at CV steps of −85, −70, −55, and −40 V (Figure 3a). The lowest CV (−85 V) showed slightly lower BIC intensity compared to the three remaining CV steps, but in general, comparable base peak intensities and numbers of detectable peaks were observed. The distribution of peptide-spectrum matches (PSMs) counts for each CV value was nonuniform and showed a significant number of PSMs detected across all CV steps (Figure 3b). A level of redundancy in PSMs between different CV steps is shown in the Venn diagram (Figure 3c). From the total number of 47,003 PSMs identified in the FAIMS experiment for 1 ng of the HeLa standard (vs 20,381 in the control experiment under optimized conditions), approximately 8%, 14%, 24%, and 33% are unique for −85, −70, −55, and −40 V, respectively. These results support the expectations that MS data acquisition using different CVs will result in complementary sets of peptide identifications in ULF LC–MS.
Figure 3.
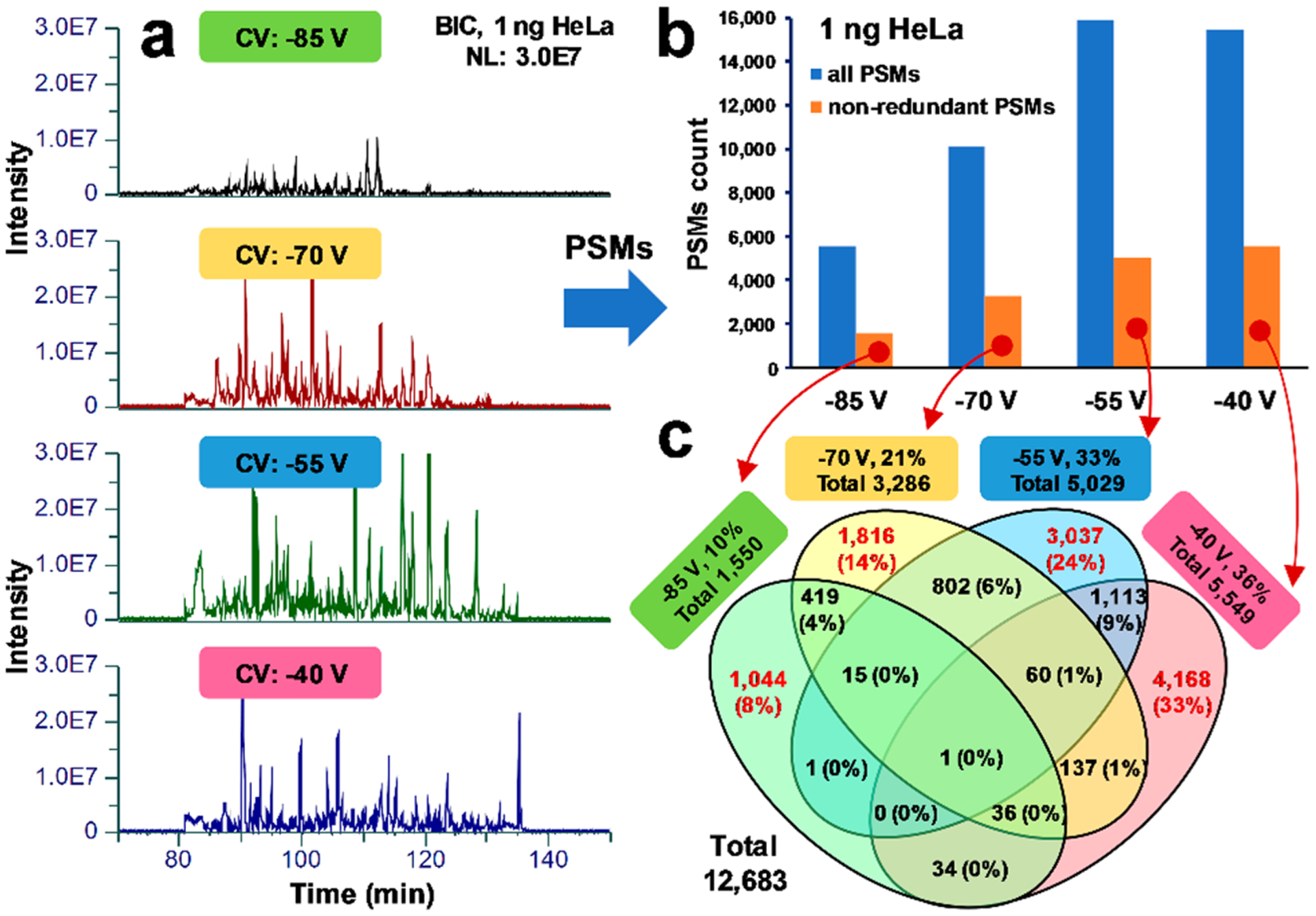
Assessment of redundancy in proteomic identification results between CV steps in ULF LC-FAIMS-MS experiments. (a) BICs for individual CVs, using a normalized intensity scale (3.0E7) for 1 ng of the standard HeLa digest analyzed at CV steps of −85 V, −70 V, −55 V and −40 V. (b) PSM count for all three technical replicates plotted for each CV value. Blue and orange bars represent all and nonredundant PSMs, respectively, identified in each CV step and within three technical replicates. (c) Venn diagram shows overlaps in identifications and unique PSMs between −85, −70, −55, and −40 V CV steps.
Optimization of FAIMS Parameters and Total MS Cycle Time.
Traditionally, there are two approaches in conducting ESI-based experiments using a FAIMS technology: (i) the external stepping method, when each analysis is conducted using a single CV, and (ii) the internal stepping method, when multiple CVs are used within a single analysis.47,62 The internal stepping method was used in our study. We initiated our ULF LC-FAIMS-MS method optimization efforts in the analysis of the HeLa digest standard using the reported conditions for a 1 h gradient with minor modifications (single CV, −60 V).47 However, in our hands, these pilot experiments did not result in an improved depth of proteomic profiling in comparison to the control experiments for low-ng HeLa samples (data not shown). Next, we evaluated two, three, and four CVs keeping the same total MS data-dependent acquisition (DDA) cycle time or “cycle time,” as referred below, of 3 s (e.g., two CVs: −75 V and −55 V, 1.5 s/CV, see Table S1). It is worth noting that the total number of CVs or total cycle time is not limited. To increase the selectivity of the FAIMS Pro interface in the transmission efficiency for various peptide ion species depending on their charge and structural features and to decrease an overlap between precursor ion selection for MS/MS fragmentation, the used CVs were spaced at least 10–15 V apart.39,47 In the control experiments, the same total cycle time of 3 s was used (see Table S1 for additional experimental details).
The control experiments (total cycle time 3 s) for 1 ng of the HeLa standard resulted in the identification of ~1,700 protein groups and ~5,300 peptide groups (Figure 4a, Table S2). The initial experiment, using two CVs, did not outperform the 3 s cycle time control experiment. Therefore, we evaluated the impact of the increased number of CVs. Three and four CVs were used in the following experiments. Figure 4 shows the positive impact of the increased number of CVs, using the same total MS cycle time, on the protein identification results. On the basis of this evaluation, four CVs (−85, −70, −55, and −40 V) were selected for all subsequent experiments.
Figure 4.
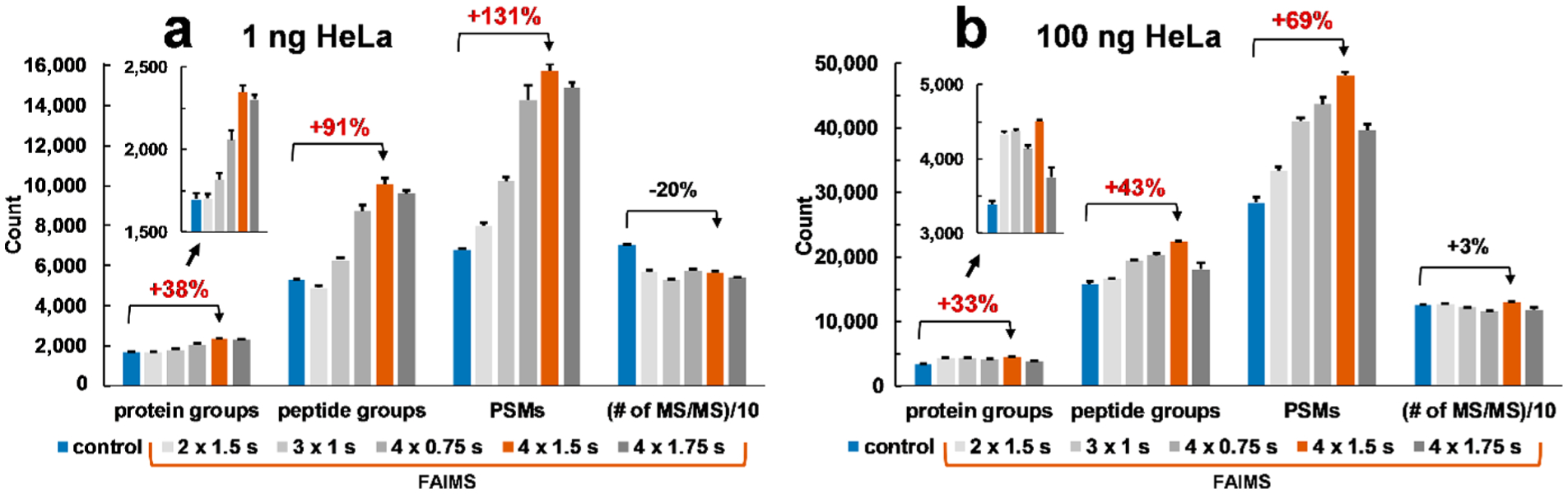
Protein identification results for FAIMS and control ULF LC–MS analyses of (a) 1 ng and (b) 100 ng HeLa protein digest standard at 1% FDR. Bar charts show average identification results and error bars correspond to a standard deviation from 3 technical replicates. The control experiments were conducted using 3 s total MS cycle time. Insets are zoomed-in bar charts corresponding to the number of protein groups. The blue bars show identification results for the control experiments conducted under optimized conditions (3 s); red bars show the results of the FAIMS experiments under optimized conditions. Data labels, e.g., “2 × 1.5 s,” represent conditions of a FAIMS experiment, which in this example correspond to two CVs and 1.5 s total MS cycle time/CV. See Table S1, Table S2, and Figure S1 for more details.
As the next step in our method optimization, the total MS cycle time was optimized to allow more MS2 scans to be acquired and to further increase the depth of proteomic profiling. The time allowed for one CV was increased from 0.75 s/CV up to 1.75 s/CV (Figure 4, Table S1, and Table S2). In Figure 4, values between 0.75 and 1.5 s/CV were not included for clarity purposes but can be seen in Table S2. These experiments demonstrated an increasing trend in the number of identification attributes from 0.75 to 1.5 s/CV followed by a drop at 1.75 s/CV, indicating that the highest values in protein/peptide identification attributes were reached at 1.5 s/CV using the evaluated experimental conditions that included 4 CVs in the internal stepping method. The control experiments were conducted using the total cycle time varied from 3 s up to 7 s (in 1 s increments), with a maximal protein/peptide identification results in most of the evaluated sample loads acquired at or around the cycle time of 3 s (Table S2). In the figures presenting the HeLa digest experiments, we show the results for 3 s cycle time control experiments compared to the results of the 1.5 s/CV, 4 CVs FAIMS experiments (Figure 1–3, Figure 5, and Figure S2–S5). The optimized ULF LC-ESI-FAIMS-MS/MS conditions resulted in the identification of 2,348 ± 42 protein groups, 10,062 ± 285 peptide groups, and 15,734 ± 350 PSMs from 1 ng of the HeLa digest standard as compared to the control experiment, which resulted in 1,697 ± 41 protein groups, 5,271 ± 32 peptide groups, and 6,809 ± 77 PSMs), using a 1 h gradient at the flow rate of 12 nL/min (Figure 4a, Table S2). This represents an increase of 38%, 91%, and 131% in protein groups, peptide groups, and PSMs, respectively. It is worth noting that the number of MS/MS scans remained approximately the same across all experiments. The detailed protein, peptide, and PSM identification results for all sample loads (1, 10, 50, and 100 ng of the standard HeLa sample), and all MS methods (7 FAIMS methods, 5 control methods) are summarized in Table S2.
Figure 5.
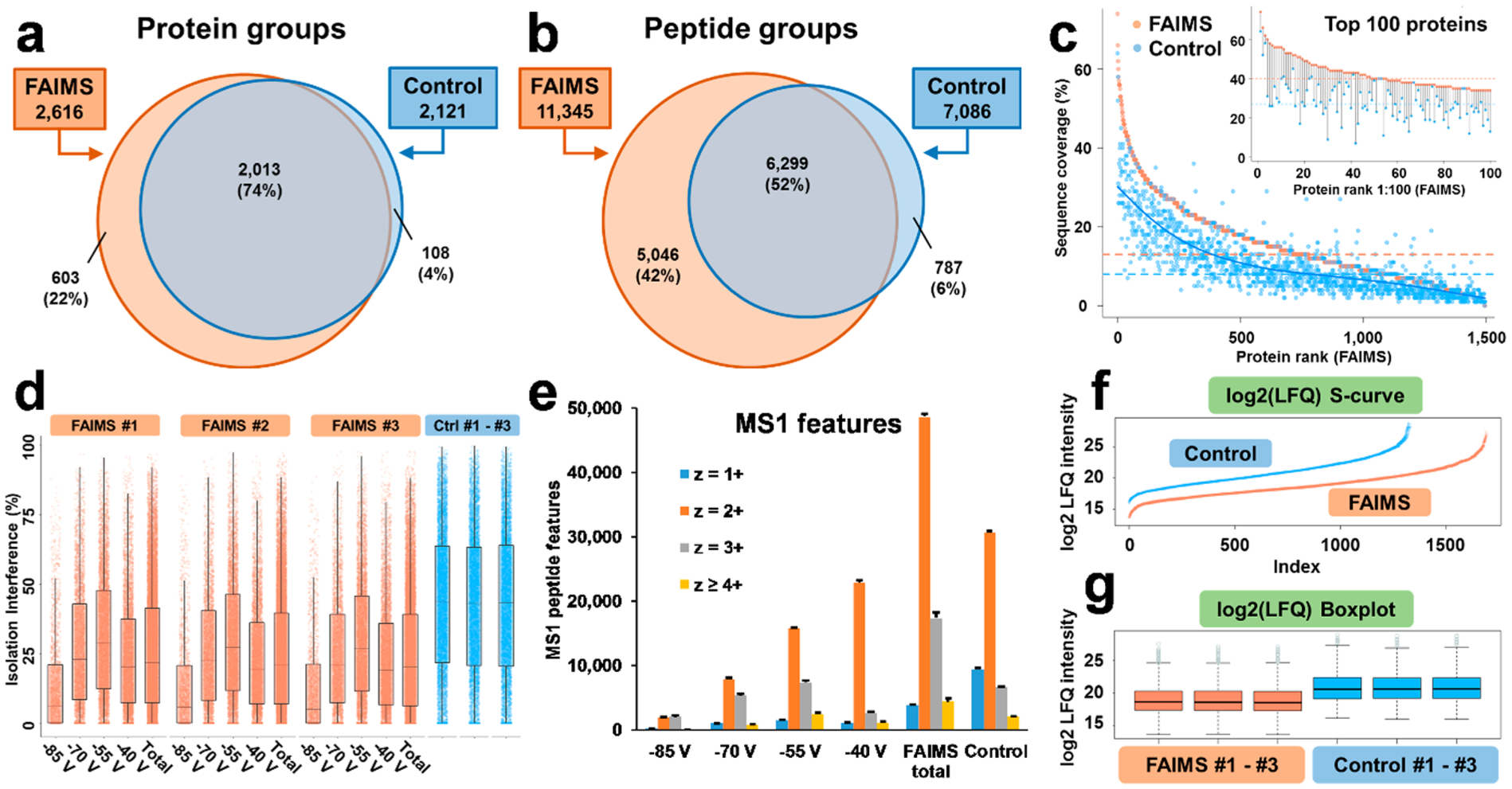
Comparative assessment of the depth of proteomic profiling with and without FAIMS for 1 ng HeLa protein digest standard. (a–b) Venn diagrams illustrating common and unique identifications for nonredundant protein and peptide groups with FAIMS compared to the control experiments. (c) The comparison of protein SeqCov. The highest abundance 1,500 proteins identified using FAIMS are plotted from high to low SeqCov, and the matched SeqCov values for the same proteins identified in the control experiments are plotted against FAIMS proteins’ values. Matched protein pairs that were identified in at least two out of three replicates and which were detected in both FAIMS and control samples were considered. Of the multiple replicate coverage values, the maximum value was chosen for each protein for FAIMS and control. Orange and blue horizontal lines represent the median SeqCov values. A spline polynomial function was used to fit a smoothed trendline for the control data (blue curve). The inset represents the lollipop plot for the top abundance 100 proteins based on the FAIMS analyses and the corresponding values from the control experiments. (d) Ion isolation interference in the selection of precursor ion species for MS2 fragmentation is shown for 3 technical replicates for the FAIMS (4 individual CVs and combined) and control experiments. Box = inner 50%, line = median, whiskers = 10% and 90%. (e) Distribution of precursor charge states for the individual FAIMS CVs, combined FAIMS CVs (total), and control experiments. The results of LFQ are shown as (f) S-curves and (g) Box-plot for log2 LFQ intensities. Box = inner 50%, line = median, whiskers = Q1−1.5*IQR and Q3 + 1.5*IQR, respectively. Dots = outliers. Q1 = first quartile, Q2 = second quartile, and IQR (interquartile range) = Q3-Q1.
Our results represent a significant increase in protein identifications in comparison to the recent reports using state-of-the-art techniques. For example, in a recent study, the sample was loaded onto 20 μm ID bead packed nanoLC column that was operated at ~20 nL/min using a 120 min long gradient and was coupled to an Orbitrap Fusion Lumos MS and ~1,650 proteins and ~9,100 peptides were identified using 2 ng of the HeLa digest standard.31 In this study, we used a shorter gradient (1 h) and 1 ng of the HeLa digest standard. Using the optimized ULF LC-ESI-FAIMS-MS/MS conditions, we were able to identify +42% and 11% more protein groups and peptide groups, respectively. We expect that using a longer gradient, the depth of proteomic profiling may be further improved with our approach.16
On the basis of our observations, using both monolithic and bead-packed column (data not shown), the optimal total cycle time (time per each CV) and the number of CVs are highly dependent on the column performance, in particular, the chromatographic peak widths of the peptides being separated. The FAIMS settings were optimized for the described ULF LC–MS/MS conditions, and additional optimization might be required for modified conditions. For the monolithic columns we used in this study, the average peak widths at 10% and 50% peak height were 30.4 and 13.4 s, respectively, and the peak capacity was ~750 (data for 1 ng standard HeLa sample).
FAIMS Increases Protein Sequence and Proteome Coverage in ULF LC–MS-Based Profiling.
The total number of protein groups identified in the analyses of 1 ng of the HeLa standard using the optimized experimental ULF LC–MS conditions for both, the FAIMS (4 CVs, 6 s total cycle time), and control (3 s total cycle time) experimental setups, was 2,724 (100%), with 603 (22%) unique to FAIMS, 108 (4%) unique to the control experiment, and 2,013 (74%) were observed in both experiments (Figure 5a–b). A similar trend was observed for peptide groups and PSMs. In addition, 28% and 71% more of unique protein and peptide groups were detected using FAIMS that were not detected in the control experiments.
The FAIMS Pro interface not only increased the number of identified proteins and peptides compared to the control ULF LC–MS experiments but also improved the protein sequence coverage (SeqCov) (Figure 5c) as the median SeqCov increased from 8% to 13%. These results demonstrate potential improvements to quantitation, PTM analysis, protein characterization, and other applications in proteomics, where both the SeqCov and the depth of profiling are critical.
FAIMS Improves Isolation Interference in ULF LC–MS-Based Proteomic Profiling.
In a typical bottom-up DDA proteomics workflow, the precursor ion detected in the MS1 full scan mode is selected for fragmentation with a user-defined ion isolation m/z window. This selection step can reduce the chemical noise level, and in the ideal case scenario, only one precursor ion species is fragmented in a single MS2 scan. In reality, however, other closely eluting precursor ions (singly- or multiply charged) with similar m/z values (or their naturally occurring isotopes) are often coisolated and fragmented together within the precursor isolation window.67,68 Such coisolation may compromise the reliability and sensitivity of peptide/protein identification, as well as the accuracy of MS2-based quantitation. In our ULF LC–MS experiments, the FAIMS Pro interface significantly reduced the isolation interference for individual CVs and for the combined results (Figure 5d), resulting in superior spectral quality and lower quality q-values for peptide identifications (arithmetic mean: 0.00206 for FAIMS and 0.00248 for control; median: 0.00091 for FAIMS and 0.00169 for control), which, when combined, can contribute to the improved sensitivity and depth of the proteomic profiling. The reduced coisolation is also expected to improve the accuracy of quantification (e.g., iTRAQ or TMT).68
FAIMS’ Selectivity for Charge States of Peptides Improves the Profiling Sensitivity of ULF LC–MS.
Next, we evaluated the charge state distributions for MS1 features detected in the FAIMS and control experiments for 1 ng of the HeLa standard (Figure 5e) during the time range of the effectual elution gradient (70–150 min, see Figure 2, Figure 3, and Figure S4). In our DDA ULF LC–MS/MS experiments, singly charged ions were not subjected to MS2 fragmentation. However, the presence of high abundance singly charged ions could lead to detrimental effects decreasing the overall depth and sensitivity of LC–MS-based proteomic profiling (e.g., the fill time with a set AGC target, S/N levels for multiply charged ions in the same scan, space charging, overlaps in isotope envelopes of multiply charged ions leading to an increased isolation interference). Therefore, singly charged ions fill the Orbitrap more quickly without FAIMS than with FAIMS applied and some of the multiply charged ions might either not be detected in MS1 scans, detected at the levels below the threshold for triggering MS/MS acquisition, or may not be selected by the DDA method for MS/MS during their elution due to the presence of multiple higher intensity singly charged precursor ions. Using a FAIMS Pro interface, the number of detected singly charged ion features was significantly reduced in the FAIMS experiments in comparison to the control experiments (by ~59%, 3,885 for FAIMS, 9,460 for control, Figure 5e). For specific CVs in the FAIMS experiments, the number of doubly charged ions increased with the voltage changing from −85 to −40 V (Figure 5e). At the same time, the number of triply charged ions increased by 364% with the change of CV values from −85 to −55 V and then decreased by 64% with the change from −55 to −40 V. The total number of each type of multiply charged ions detected using FAIMS (2+, 3+, and ≥4+) exceeded the total number of these ions 1.6–2.6-fold compared to the control experiment (Figure 5e).
Protein LFQ in FAIMS and Control ULF LC–MS Experiments.
Next, we evaluated protein LFQ in FAIMS and control experiments (Figure 5f–g). The determined dynamic range of the proteome coverage based on LFQ values were approximately the same and slightly shifted down in the FAIMS compared to control experiments (FAIMS 1.3E4–1.5E8, log2:13.7–27.2 and control 6.8E4–4.5E8, log2:16.1–28.7), while in the FAIMS experiment, we quantified 364 additional proteins on average (Figure 5f). In addition, we observed high reproducibility and high level of correlation in log2 protein intensities between technical replicates (n = 3) in both, FAIMS and control experiments, with R-squared of R2 = 0.97 and R2 = 0.98 (Figure 5g, Figure S5).
Deep Proteomic Profiling of U937 Myeloid Leukemia Cells at the Level of 10–1,000 Cells.
To evaluate the potential of the ULF LC-ESI-FAIMS-MS/MS platform in the bottom-up proteomic analysis of limited samples, approximately 100, 1,000, and 10,000 U937 myeloid leukemia cells were processed using in-house prepared microSPE columns.2,69 According to our estimates based on the published reports, an individual U937 myeloid leukemia cell contains ~200–500 pg of protein per cell.4 One-tenth of each microSPE-processed sample was analyzed in each technical replicate. Figure S6a–b shows an ion density map corresponding to an ~10 cell-equivalent from ~100 processed U937 myeloid leukemia cells. Similarly to the HeLa experiments described before, the background noise level is significantly decreased, and singly charged polysiloxanes ions are getting effectively removed when the FAIMS technology is applied compared to the control experiments. The high-intensity peaks, at 94 min (m/z 523.29) and 99 min (m/z 421.76, correspond to trypsin autocleavage peptides. Singly charged trypsin autocleavage peptides (e.g., m/z 842.51 at 98 min) were effectively removed by the FAIMS Pro interface (average intensity values from 3 technical replicates: FAIMS: 5.6E4 vs control: 9.3E6). No high-intensity (>5.0E6) Lys-C autocleavage peptides were observed. The overlap in protein identification results and the corresponding Venn diagrams generated for protein and peptide groups (identified using the optimized experimental ULF LC–MS conditions for the FAIMS (4 CVs, 6 s total cycle time) and control (3 s total cycle time) experiments are shown in Figure 6a–b. The total number of protein groups identified in both experimental setups was 399 (100%), with 136 (34%) unique to FAIMS, 25 (6%) unique to the control experiment, and 238 (60%) were observed in both experiments. The protein identification results for all sample loads and blanks are shown in Figure 6c–d and Table S3. The optimized ULF LC-ESI-FAIMS-MS/MS conditions resulted in the identification of 251 ± 54 protein groups, 578 ± 199 peptide groups, and 931 ± 250 PSMs from the injected sample equivalent to ~10 cells from ~100 processed U937 myeloid leukemia cells, using a 1 h gradient at the flow rate of 12 nL/min. The control experiment resulted in 176 ± 26 protein groups, 381 ± 89 peptide groups, and 448 ± 104 PSMs (Figure 6c–d, Table S3). This comparison represents an average increase of 43%, 52%, and 108% in protein groups, peptide groups, and PSMs, respectively. The gain in protein identification results is less pronounced for larger sample amounts, e.g., for a 1,000 cell sample equivalent, the gain in reflected in an increase of 14%, 46% and 67% in protein groups, peptide groups, and PSMs, respectively (Table S3).
Figure 6.
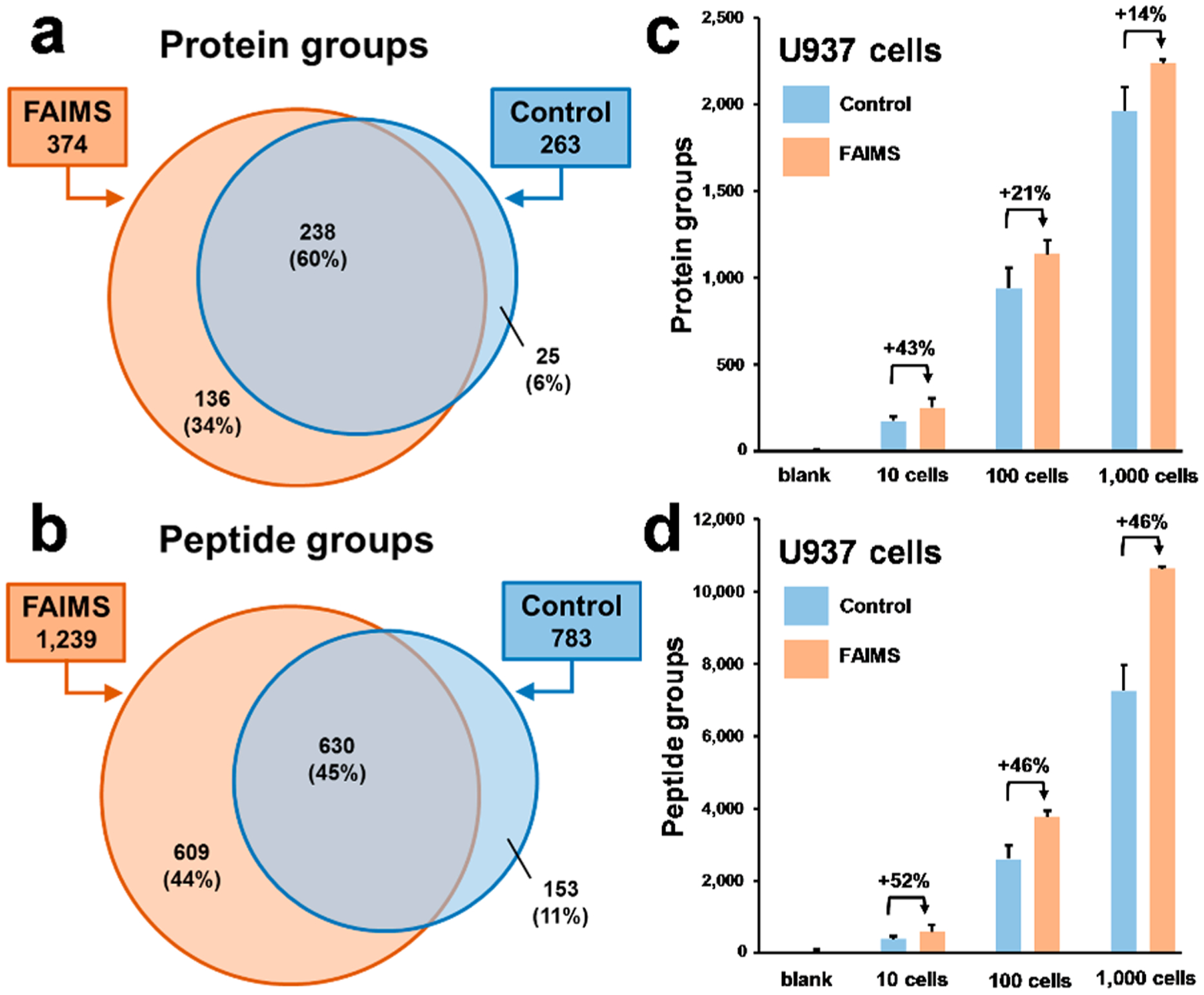
Protein identification results for U937 myeloid leukemia cells at the level of 10–1,000 cells. (a–b) Venn diagrams illustrate unique and common nonredundant protein and peptide groups identified in FAIMS compared to the control experiments. (c–d) Protein identification results for ~10, 100, and 1,000 cell equivalents resulted from ~100, 1,000, and 10,000 processed U937 myeloid leukemia cells. Bar charts show the average identification results and error bars correspond to a standard deviation from 3 replicates. The control experiments were conducted using a 3 s total MS cycle time.
CONCLUSIONS
In this work, for the first time, we have demonstrated how ULF LC coupled with tandem MS via a FAIMS interface can improve the depth and sensitivity of proteomic profiling of limited samples. Using commercially available instrumentation and an in-house prepared high-efficiency polymeric monolithic column operated at 12 nL/min, we showed improvements in proteomic characterization of low-ng samples (HeLa protein digest standard and the digested total lysate of U937 myeloid leukemia cells). These advances show the potential applicability of the described approach to proteomic analysis of single cells. However, further improvements in the efficiency of sample processing and the recovery of the processed samples are needed to effectively cope with the deep profiling of individual cells.
In this study, we showed that the FAIMS Pro interface decreased the noise level in ULF LC–MS experiments by 3 orders of magnitude, and comparable peptide signal intensities were observed in the FAIMS and control experiments. The number of CVs, their values, and the total MS cycle time was optimized to improve protein identification results for applied chromatographic parameters. The optimized ULF LC-ESI-FAIMS-MS/MS resulted in the identification of ~2,348 protein groups and ~10,062 peptide groups for 1 ng of a HeLa digest, and ~215 protein groups and ~578 peptide groups were identified from the analysis of ~10 cell equivalents from ~100 processed U937 myeloid leukemia cells, both using a 1 h gradient. These identification results show at least 38% improvement over the control experiments.
The field of nanoflow-LC coupled to MS has made incredible advances during the past decade. Multiple vendors made a variety of robust nanoLC instrumentation and columns commercially available. We envision that ULF columns of the type we described in this study together with appropriate pumping and sample delivery systems will be commercialized during this decade to enable various novel, exciting biomedical applications requiring detection sensitivity levels that the conventional nanoLC-based approaches cannot attain. Coupling these ULF LC–MS technologies to commercially available orthogonal techniques like FAIMS can further enhance the performance in solving challenging biological problems.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr. Slavov for kindly providing aliquots of U937 myeloid leukemia cells. This work was supported by the National Institutes of Health under Awards R01GM120272 (ARI), R01CA218500 (ARI), and R35GM136421 (ARI). We acknowledge the team of Thermo Fisher Scientific for its support through a technology alliance partnership program.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03262.
Detailed descriptions of methods and experimental results, including an example of a FAIMS-MS method with 6 s total MS cycle time, background noise intensity levels in FAIMS experiments, extracted ion chromatograms, peak intensities, and signal-to-noise ratios, ion density maps acquired in the ULF LC–MS analysis of 100 ng HeLa protein digest standard, comparative assessment of protein abundance estimated using label-free quantitation, key settings, including CVs and total MS cycle time for FAIMS and control, protein identification results for 1, 10, 50, and 100 ng HeLa protein digest standard, ion density maps and protein identification results for U937 samples (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c03262
The authors declare no competing financial interest.
REFERENCES
- (1).Lombard-Banek C; Moody SA; Manzini MC; Nemes P Anal. Chem 2019, 91 (7), 4797–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Li S; Plouffe BD; Belov AM; Ray S; Wang X; Murthy SK; Karger BL; Ivanov AR Mol. Cell. Proteomics 2015, 14 (6), 1672–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian WJ; Kelly RT Nat. Commun 2018, 9 (1), 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Huffman RG; Chen A; Specht H; Slavov N J. Proteome Res 2019, 18 (6), 2493–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Amenson-Lamar EA; Sun L; Zhang Z; Bohn PW; Dovichi NJ Talanta 2019, 204, 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhang Z; Hebert AS; Westphall MS; Qu Y; Coon JJ; Dovichi NJ Anal. Chem 2018, 90 (20), 12090–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Krenkova J; Kleparnik K; Luksch J; Foret F Electrophoresis 2019, 40 (18–19), 2263–2270. [DOI] [PubMed] [Google Scholar]
- (8).Sorensen MJ; Miller KE; Jorgenson JW; Kennedy RT J. Chromatogr A 2020, 1611, 460575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Eghbali H; Sandra K; Detobel F; Lynen F; Nakanishi K; Sandra P; Desmet G J. Chromatogr A 2011, 1218 (21), 3360–6. [DOI] [PubMed] [Google Scholar]
- (10).Zhu Y; Zhao R; Piehowski PD; Moore RJ; Lim S; Orphan VJ; Pasa-Tolic L; Qian WJ; Smith RD; Kelly RT Int. J. Mass Spectrom 2018, 427, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ivanov AR; Zang L; Karger BL Anal. Chem 2003, 75 (20), 5306–16. [DOI] [PubMed] [Google Scholar]
- (12).Valaskovic GA; Kelleher NL; Little DP; Aaserud DJ; McLafferty FW Anal. Chem 1995, 67 (20), 3802–5. [DOI] [PubMed] [Google Scholar]
- (13).Shen Y; Zhao R; Berger SJ; Anderson GA; Rodriguez N; Smith RD Anal. Chem 2002, 74 (16), 4235–49. [DOI] [PubMed] [Google Scholar]
- (14).Shishkova E; Hebert AS; Coon JJ Cell Syst 2016, 3 (4), 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Specht H; Slavov N J. Proteome Res 2018, 17 (8), 2565–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kawashima Y; Ohara O Anal. Chem 2018, 90 (21), 12334–12338. [DOI] [PubMed] [Google Scholar]
- (17).Kovalchuk SI; Jensen ON; Rogowska-Wrzesinska A Mol. Cell. Proteomics 2019, 18 (2), 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang J; Wu SL; Kim J; Karger BL J. Chromatogr A 2007, 1154 (1–2), 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Vaast A; Terryn H; Svec F; Eeltink SJ Chromatogr A 2014, 1374, 171–179. [DOI] [PubMed] [Google Scholar]
- (20).Dores-Sousa JL; Fernandez-Pumarega A; De Vos J; Lammerhofer M; Desmet G; Eeltink SJ Sep Sci. 2019, 42 (2), 522–533. [DOI] [PubMed] [Google Scholar]
- (21).Yue G; Luo Q; Zhang J; Wu SL; Karger BL Anal. Chem 2007, 79 (3), 938–46. [DOI] [PubMed] [Google Scholar]
- (22).Vehus T; Roberg-Larsen H; Waaler J; Aslaksen S; Krauss S; Wilson SR; Lundanes E Sci. Rep 2016, 6, 37507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Xiang P; Zhu Y; Yang Y; Zhao Z; Williams SM; Moore RJ; Kelly RT; Smith RD; Liu S Anal. Chem 2020, 92 (7), 4711–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hara T; Izumi Y; Nakao M; Hata K; Baron GV; Bamba T; Desmet G J. Chromatogr A 2018, 1580, 63–71. [DOI] [PubMed] [Google Scholar]
- (25).Collins DA; Nesterenko EP; Brabazon D; Paull B Anal. Chem 2012, 84 (7), 3465–72. [DOI] [PubMed] [Google Scholar]
- (26).De Malsche W; De Bruyne S; Op De Beek J; Sandra P; Gardeniers H; Desmet G; Lynen F J. Chromatogr A 2012, 1230, 41–7. [DOI] [PubMed] [Google Scholar]
- (27).Moravcova D; Rantamaki AH; Dusa F; Wiedmer SK Electrophoresis 2016, 37 (7–8), 880–912. [DOI] [PubMed] [Google Scholar]
- (28).Eeltink S; Wouters S; Dores-Sousa JL; Svec F J. Chromatogr A 2017, 1498, 8–21. [DOI] [PubMed] [Google Scholar]
- (29).Wilson SR; Vehus T; Berg HS; Lundanes E Bioanalysis 2015, 7 (14), 1799–815. [DOI] [PubMed] [Google Scholar]
- (30).Lam SC; Sanz Rodriguez E; Haddad PR; Paull B Analyst 2019, 144 (11), 3464–3482. [DOI] [PubMed] [Google Scholar]
- (31).Cong Y; Liang Y; Motamedchaboki K; Huguet R; Truong T; Zhao R; Shen Y; Lopez-Ferrer D; Zhu Y; Kelly RT Anal. Chem 2020, 92 (3), 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ivanov AR; Horvath C; Karger BL Electrophoresis 2003, 24 (21), 3663–73. [DOI] [PubMed] [Google Scholar]
- (33).Gharbharan D; Britsch D; Soto G; Weed AM; Svec F; Zajickova Z J. Chromatogr A 2015, 1408, 101–7. [DOI] [PubMed] [Google Scholar]
- (34).Xie C; Ye M; Jiang X; Jin W; Zou H Mol. Cell. Proteomics 2006, 5 (3), 454–61. [DOI] [PubMed] [Google Scholar]
- (35).Hara T; Izumi Y; Hata K; Baron GV; Bamba T; Desmet GJ Chromatogr A 2020, 1616, 460804. [DOI] [PubMed] [Google Scholar]
- (36).Chocholouskova M; Komendova M; Urban J J. Chromatogr A 2017, 1488, 85–92. [DOI] [PubMed] [Google Scholar]
- (37).Cifani P; Kentsis A Mol. Cell. Proteomics 2017, 16 (11), 2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Meier F; Geyer PE; Virreira Winter S; Cox J; Mann M Nat. Methods 2018, 15 (6), 440–448. [DOI] [PubMed] [Google Scholar]
- (39).Bekker-Jensen DB; Martinez-Val A; Steigerwald S; Ruther P; Fort KL; Arrey TN; Harder A; Makarov A; Olsen JV Mol. Cell. Proteomics 2020, 19 (4), 716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Guevremont R J. Chromatogr A 2004, 1058 (1–2), 3–19. [PubMed] [Google Scholar]
- (41).Purves RW; Guevremont R; Day S; Pipich CW; Matyjaszczyk MS Rev. Sci. Instrum 1998, 69 (12), 4094–4105. [Google Scholar]
- (42).Shvartsburg AA; Li F; Tang K; Smith RD Anal. Chem 2006, 78 (11), 3706–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sarsby J; Griffiths RL; Race AM; Bunch J; Randall EC; Creese AJ; Cooper HJ Anal. Chem 2015, 87 (13), 6794–800. [DOI] [PubMed] [Google Scholar]
- (44).Shvartsburg AA; Smith RD Anal. Chem 2013, 85 (14), 6967–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Shliaha PV; Gorshkov V; Kovalchuk SI; Schwammle V; Baird MA; Shvartsburg AA; Jensen ON Anal. Chem 2020, 92 (3), 2364–2368. [DOI] [PubMed] [Google Scholar]
- (46).Pfammatter S; Bonneil E; McManus FP; Thibault P J. Proteome Res 2019, 18 (5), 2129–2138. [DOI] [PubMed] [Google Scholar]
- (47).Hebert AS; Prasad S; Belford MW; Bailey DJ; McAlister GC; Abbatiello SE; Huguet R; Wouters ER; Dunyach JJ; Brademan DR; Westphall MS; Coon JJ Anal. Chem 2018, 90 (15), 9529–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Rosting C; Yu J; Cooper HJ J. Proteome Res 2018, 17 (6), 1997–2004. [DOI] [PubMed] [Google Scholar]
- (49).Contrepois K; Mahmoudi S; Ubhi BK; Papsdorf K; Hornburg D; Brunet A; Snyder M Sci. Rep 2018, 8 (1), 17747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hanson AJ; Banks WA; Bettcher LF; Pepin R; Raftery D; Craft S Metabolomics 2020, 16 (1), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Swearingen KE; Moritz RL Expert Rev. Proteomics 2012, 9 (5), 505–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Cooper HJ J. Am. Soc. Mass Spectrom 2016, 27 (4), 566–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Bonneil E; Pfammatter S; Thibault P J. Mass Spectrom 2015, 50 (11), 1181–95. [DOI] [PubMed] [Google Scholar]
- (54).Pfammatter S; Bonneil E; McManus FP; Prasad S; Bailey DJ; Belford M; Dunyach JJ; Thibault P Mol. Cell. Proteomics 2018, 17 (10), 2051–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Canterbury JD; Yi X; Hoopmann MR; MacCoss MJ Anal. Chem 2008, 80 (18), 6888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Prasad S; Belford MW; Dunyach JJ; Purves RW J. Am. Soc. Mass Spectrom 2014, 25 (12), 2143–53. [DOI] [PubMed] [Google Scholar]
- (57).Purves RW; Prasad S; Belford M; Vandenberg A; Dunyach JJ J. Am. Soc. Mass Spectrom 2017, 28 (3), 525–538. [DOI] [PubMed] [Google Scholar]
- (58).Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Mol. Cell. Proteomics 2014, 13 (9), 2513–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Mellacheruvu D; Wright Z; Couzens AL; Lambert JP; St-Denis NA; Li T; Miteva YV; Hauri S; Sardiu ME; Low TY; Halim VA; Bagshaw RD; Hubner NC; Al-Hakim A; Bouchard A; Faubert D; Fermin D; Dunham WH; Goudreault M; Lin ZY; Badillo BG; Pawson T; Durocher D; Coulombe B; Aebersold R; Superti-Furga G; Colinge J; Heck AJ; Choi H; Gstaiger M; Mohammed S; Cristea IM; Bennett KL; Washburn MP; Raught B; Ewing RM; Gingras AC; Nesvizhskii AI Nat. Methods 2013, 10 (8), 730–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Perez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaino JA Nucleic Acids Res. 2019, 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Bailly-Chouriberry L; Cormant F; Garcia P; Lonnberg M; Szwandt S; Bondesson U; Popot MA; Bonnaire Y Analyst 2012, 137 (10), 2445–53. [DOI] [PubMed] [Google Scholar]
- (62).Creese AJ; Shimwell NJ; Larkins KP; Heath JK; Cooper HJ J. Am. Soc. Mass Spectrom 2013, 24 (3), 431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Bridon G; Bonneil E; Muratore-Schroeder T; Caron-Lizotte O; Thibault P J. Proteome Res 2012, 11 (2), 927–40. [DOI] [PubMed] [Google Scholar]
- (64).Zhao H; Cunningham DL; Creese AJ; Heath JK; Cooper HJ J. Proteome Res 2015, 14 (12), 5077–87. [DOI] [PubMed] [Google Scholar]
- (65).Pfammatter S; Bonneil E; Thibault PJ Proteome Res. 2016, 15 (12), 4653–4665. [DOI] [PubMed] [Google Scholar]
- (66).Schlosser A; Volkmer-Engert R J. Mass Spectrom 2003, 38 (5), 523–5. [DOI] [PubMed] [Google Scholar]
- (67).Houel S; Abernathy R; Renganathan K; Meyer-Arendt K; Ahn NG; Old WM J. Proteome Res 2010, 9 (8), 4152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Sandberg A; Branca RM; Lehtio J; Forshed J J. Proteomics 2014, 96, 133–44. [DOI] [PubMed] [Google Scholar]
- (69).Rappsilber J; Mann M; Ishihama Y Nat. Protoc 2007, 2 (8), 1896–906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.