

Abstract
The complexity of neurodegeneration restricts the ability to understand and treat the neurological disorders affecting millions of people worldwide. Therefore, there is an unmet need to develop new and more effective therapeutic strategies to combat these devastating conditions and that will only be achieved with a better understanding of the biological mechanism associated with disease conditions. Recent studies highlight the role of DNA damage, particularly, DNA double-strand breaks (DSBs), in the progression of neuronal loss in a broad spectrum of human neurodegenerative diseases. This is not unexpected because neurons are prone to DNA damage due to their non-proliferative nature and high metabolic activity. However, it is not clear if DSBs is a primary driver of neuronal loss in disease condition or simply occurs concomitant with disease progression. Here, we provide evidence that supports a critical role of DSBs in the pathogenesis of the neurodegenerative diseases. Among different kinds of DNA damages, DSBs are the most harmful and perilous type of DNA damage and can lead to cell death if left unrepaired or repaired with error. In this review, we explore the current state of knowledge regarding the role of DSBs repair mechanisms in preserving neuronal function and survival and describe how DSBs could drive the molecular mechanisms resulting in neuronal death in neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. We also discuss the potential implications of DSBs as a novel therapeutic target and prognostic marker in patients with neurodegenerative conditions.
Keywords: DNA damage, Genomic instability, Neurodegeneration, Alzheimer’s disease, Parkinson’s disease, DNA repair
Introduction
Neurodegenerative diseases are an incurable and debilitating condition, characterized by the gradual and progressive deterioration of brain function, with a consequential decrease in the quality of life for patients and their family members. Alzheimer’s disease and Parkinson’s disease are the most common types, with approximately 44 million people suffering from Alzheimer’s disease and 10 million people living with Parkinson’s disease globally (Dos Santos Picanco et al. 2018; Kiebish and Narain 2019). Neurodegenerative diseases occur when nerve cells in the central or peripheral nervous system begin to deteriorate over time and ultimately die. The onset of neurodegenerative diseases mainly occurs in mid-to late-life, so the prevalence is expected to increase as the population ages (Rulten and Caldecott 2013). Knowledge of the precise molecular mechanisms underlying neurodegeneration remains incomplete, and these gaps in our knowledge about fundamental neurobiology are a major barrier to therapeutic discovery. Thus, there is a critical and urgent need to improve our understanding of the molecular mechanisms underlying the neurodegenerative process and to identify therapeutic targets for treatment and prevention. DNA damage, particularly DNA double-strand-breaks (DSBs), has been recently implicated in neurodegenerative diseases, but it remains unknown whether elevated DNA damage is simply a bystander effect or primary driver of disease progression. Emerging findings suggest that an imbalance between DNA damage and repair in the brain contributes to neuronal damage and has been documented in several neurodegenerative disorders (Rulten and Caldecott 2013; Madabhushi et al. 2014; Merlo et al. 2016; White and Vijg 2016; Mitra et al. 2019; Shanbhag et al. 2019)
In this review, recent studies that offer evidence supporting a role for the deterioration of DNA integrity as a central contributing factor in the pathogenesis of neurodegenerative diseases will be discussed. DNA, the central store of genetic information, is constantly subjected to damage from myriad sources including intra-cellular metabolism, physiological neuronal activity, transcription, replication, and exposure to genotoxic agents (Suberbielle et al. 2013; Madabhushi et al. 2015; Schwer et al. 2016). Of note, accumulation of DNA damage is a pervasive phenomenon in aged brains, and it is elevated in brains of AD, PD, and ALS patients (Myung et al. 2008; Suberbielle et al. 2015; Milanese et al. 2018; Mitra et al. 2019). It has been estimated that each mammalian cell endures as many as 10,000 single-strand and 10 to 50 double-strand DNA breaks per day (Madabhushi et al. 2014). Among possible DNA damage, DSBs are the most harmful and dangerous type of DNA damage and can lead to cell death if left unrepaired or repaired with errors. In proliferating cells, unrepaired DSBs can lead to cell cycle arrest and cell death (Zhou and Elledge 2000; Ambrosio et al. 2016), while the misrepaired DSBs can lead to mutations, deletions, or chromosomal translocations (van Gent et al. 2001; Cannan and Pederson 2016). Unlike proliferating cells, which can resort sister chromatids to repair DSBs in an error-free manner by homologous recombination (HR), postmitotic neurons utilize error-prone mechanisms of DSBs repair by non-homologous end joining (NHEJ) (Madabhushi et al. 2014; Kannan et al. 2018). Thus, DSBs could be particularly deleterious to neuronal function and survival. Each neuronal cell is equipped with complex networks of DNA repair mechanisms that preserve genomic integrity (Merlo et al. 2016). Homologous recombination and the NHEJ pathways are the two major pathways to repair DSBs (McKinnon and Caldecott 2007; Merlo et al. 2016; Chang et al. 2017). All of these observations suggest that efficient DNA repair capacity is critical for adequate neuronal survival and functions. Consistent with this concept, a substantial body of work links mutations in genes that encode DNA repair proteins to several neurodegenerative diseases (Sheng et al. 1998; Jacobsen et al. 2004; Suberbielle et al. 2015; White and Vijg 2016; Chang et al. 2017; Wang and Hegde 2019).
There is burgeoning interest in investigating the regulatory mechanisms underlying DSBs and the development of strategies to target this pathway since there is substantial evidence for the participation of DSBs in several neural and extra-neural diseases (Sheng et al. 1998; Jacobsen et al. 2004; McKinnon and Caldecott 2007; Suberbielle et al. 2013, 2015; Nakanishi et al. 2015; Merlo et al. 2016; Chang et al. 2017). Furthermore, the basis of current knowledge and the concept of DSBs are derived mainly from proliferative cells or nonneuronal cells and role of DSBs in normal mature neurons remains largely unexplored. Therefore, a significant gap of knowledge exists regarding the role of DSBs in neuronal cells during pathological conditions. We demonstrate that investigation of DSBs holds promise to reveal novel therapeutic targets for neurological diseases including Alzheimer’s and Parkinson’s diseases in human. This review highlights recent progress towards the understanding of the role of the DNA damage and repair in neurodegenerative diseases. Specifically, we focus on the possible implication of DSBs and repair mechanisms in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, and the potential use as prognostic markers and a novel therapeutic target for these clinical conditions.
DNA damage response: an overview
DNA is a multifaceted molecule and precisely regulated chemical sequence that harbors all the information required for a cell to survive and function. In cells, DNA is under continuous attack by several endogenous stresses, such as metabolic processes, DNA replication and transcription, or by exogenous stresses such as ultraviolet radiation, chemicals, and pollution that can ultimately lead to DNA damage (El-Khamisy et al. 2005; Valko et al. 2006). Whereas errors in other biological molecules are relatively short-lived, when DNA acquires an error, it could result in a long-term irreversible outcome leading to a change within the final RNA and/or protein product. While proliferating (nonneuronal) cells endure this alteration for a short time before they are replaced, in contrast, neurons are retained for life and therefore require the means to contend with a lifetime of damage (Madabhushi et al. 2014; Merlo et al. 2016). There are several types of damage to DNA, such as base modifications, strand breaks (single or double), crosslinks, and mismatches, and they can affect both the nuclear and the mitochondrial genome.
Neurons are constantly exposed to high levels of DNA damage including DSBs from both physiological and pathological sources (Suberbielle et al. 2013; Madabhushi et al. 2015; Shanbhag et al. 2019). In the brain, neocortical and cerebellar neurogenesis involves phases of rapid proliferation, differentiation, and migration which yield higher incidences of DNA breaks originating from elevated levels of DNA transcription, replication, or naturally occurring modifications due to oxidation (Rulten and Caldecott 2013; Chang et al. 2017; Yu et al. 2018). The rich abundance of easily peroxidizable polyunsaturated fatty acids, high level of iron, a potent reactive oxygen species (ROS) catalyst, and the relative paucity of antioxidants and related enzymes in brain make it an easily susceptible region for cellular damage (Cadet and Davies 2017). Neurodegenerative states are associated with the high levels of DNA damage resulting from elevated transcription and/or replication (Massey and Jones 2018). Unrepaired or erroneously repaired DSBs accumulation in neurons results in loss of genome integrity, and the subsequent increased risk of errors in the RNA and protein synthesis may lead to neuronal dysfunction. To counter this threat, neurons activate a highly evolved and complex DNA damage response (DDR) pathway that not only senses and repairs damaged DNA, but also coordinates DNA repair with several other cellular processes such as chromatin remodeling, transcription, and inflammation, that mediate either apoptosis or survival (Madabhushi et al. 2015; Fielder et al. 2017). The response to DSBs formation involves the rapid post-translational modification and recruitment of several proteins to broken DNA ends and the surrounding chromatin. An extremely well-studied histone modification in the DDR is the phosphorylation of the histone H2A.X, which represents a sub-family of histone H2A (Crowe et al. 2006; Madabhushi et al. 2014). Phosphorylation of H2A.X spreads rapidly over megabases of genomic DNA flanking the DSBs and plays a crucial role in imbedding DDR proteins to the damaged sites (Barzilai et al. 2008). Adequate function of DDR is important during both neuronal development and in the mature nervous system. In the brain, DSBs are repaired using one of two main pathways: NHEJ, which involves direct ligation of the broken DNA ends and is error prone, or HR, that uses the undamaged sister chromatid as a template to guide repair (Merlo et al. 2016; Chang et al. 2017; Massey and Jones 2018). HR, relatively less error-prone repair pathway, is important for DNA repair in neural progenitors and nonneuronal cells in the brain while NHEJ is the primary pathway of DSB repair in mature neurons. It is important to note, while an efficient repair of DSBs is crucial for preserving genomic integrity in dividing neural progenitors during development, the postmitotic neurons lack DNA replication-dependent DNA repair processes. Failure to preserve the DNA repair function and initiate repair is most likely a key mechanism for early neuronal damage in neurodegenerative diseases. For instance, complete loss of either Xrcc2 or Lig4, which are essential for DSBs repair through HR and NHEJ, respectively, results in embryonic lethality that is also associated with extensive neural apoptosis (Orii et al. 2006). Therefore, regulated balance of DSBs accumulation and DDR pathways is required to preserve genome stability and adequate neuronal function. The efficiency of the DNA repair system appears to decrease with advancing age, leading to the accumulation of DNA damage in the brain (Rulten and Caldecott 2013; Merlo et al. 2016; Massey and Jones 2018). The association of DNA repair defects with both elevated predisposition to cancer and increased rates of neurodegeneration and aging, sometimes in the same genetic disease, is particularly intriguing, because cancer is a disease of excessive cell growth and survival, whereas neurodegeneration is a disease of excessive cell dysfunction and death. Opposite cellular end-points can thus arise from defects in common or related processes (Gorman 2008). Here, we highlight the recent progress in the signaling of DSBs, and new potential approaches to design effective therapeutic strategies for the neurodegenerative diseases.
DNA double-strand breaks and repair mechanisms
DNA DSBs are a type of DNA damage that causes fractures in both strands of the DNA helix. Such an occurrence is rare among the many types of DNA damage that a cell suffers at any time (Suberbielle et al. 2013; Merlo et al. 2016). Of the many different types of damage, DSBs are the most significant and lethal since they do not leave an intact complementary strand to be used as a template for DNA repair. If left unrepaired or misrepaired, they can ultimately lead to chromosome breakage and genome instability, immune system activation, and neurodegeneration (Sheng et al. 1998; Gorman 2008; Suberbielle et al. 2013). The frequency of DSBs is estimated to be 10 to 50 events per cell per day and a single DSBs can lead to the death of a cell or can muddle its genomic integrity (Jackson and Bartek 2009). In neurons, DSBs are generated in response to several exogenous and endogenous stresses. Intriguingly, a study by Suberbielle and coworkers demonstrated that physiologic brain activity causes neuronal DSBs and suppression of aberrant neuronal activity in mice reduces DSBs accumulation (Suberbielle et al. 2013). Emerging evidence suggests that impact of DSBs and repair is not restricted to cellular stress and pathological conditions as previously thought, but instead it is also involved in fundamental physiological functions of neurons. The interesting results from Madabhushi and coworkers support an imminent view that neuronal activity triggers DSBs formation on promoters of a subset of early-response genes, which are crucial for experience-driven changes to synapses, learning, and memory (Madabhushi et al. 2015). They further suggested that activity-dependent DSBs formation is likely mediated by the type II topoisomerase, topoisomerase IIβ (Topo IIβ), and silencing of Topo IIβ extenuates both DSBs formation and early-response gene expression. In addition, recent studies demonstrated that robust induction of transcriptional programs by a wide array of nuclear hormone receptors and other transcription factors demands the recruitment of several factors involved in DNA damage and repair (Ju et al. 2006; Haffner et al. 2011; Schwer et al. 2016). In this regard, several studies documented increased and sustained DSBs in human brains with neurodegenerative condition (Gorman 2008; Federico et al. 2012; Merlo et al. 2016).
DNA repair is essential for neuronal survival and maintenance of tissue homeostasis. In response to DSBs, cells initiate the DNA damage response, a complex network of interconnected pathways that muster and regulate arsenals of repair enzymes, modify the chromatin structure, act as a scaffold for repair and signaling factors, regulate chromosome mobility, cell cycle, and transcription, thus providing time for damaged cells to repair their DNA (McKinnon 2009; Ceccaldi et al. 2016; Yu et al. 2018). The phosphatidylinositol-3 kinase-related kinases (PIKK) play an important role in different stages of DSBs signaling through their ability to phosphorylate several substrates leading to the broadcasting of DSBs signaling. Three members of the PIKK family, such as DNA-dependent protein kinase catalytic subunit (DNA-PKcs), ataxia telangiectasia mutated (ATM), and ataxia telangiectasia, and Rad3 related (ATR) are essential for the response to DSBs. ATM and DNA-PKcs are critical for the signaling of DSBs while ATR is mainly involved in response to DNA single-strand breaks and stalled replication fork (Shibata et al. 2011; Herrup et al. 2013; Maréchal and Zou 2013; Merlo et al. 2016).
The MRN complex, a protein complex consisting of MRE11, RAD50 and NBS1, and the Ku70/Ku80 complex are important for sensing DSBs and activation of ATM and DNA-PKcs kinase activity in neurons. The MRN complex summons ATM, while Ku70/80 summons DNA-PKcs to the sites of DNA damage. As a first responder to DSBs, MRN promotes appropriate repair by either NHEJ or HR pathways, playing essential roles via its 3′−5′ exonuclease and single-stranded and DNA hairpin endonuclease activities (Shibata et al. 2014). The primary role of MRN in HR is DSB sensing. Similar to MRN, a unique set of proteins are employed for detecting, synapsing, and processing DSBs in NHEJ. It is presently unclear how MRN out-competes the NHEJ machinery for its place at DSBs termini during the S and G2 phases of the cell cycle. Initial analyses of the importance of MRN to NHEJ produced conflicting results but emerging data have now firmly established roles for MRN in both C- and A-NHEJ (Lamarche et al. 2010). ATM is a crucial protein kinase that functions at the peak of a signaling pathway that yields cell cycle arrest after DNA damage to stop proliferation. p53, checkpoint kinase 2, breast cancer associated 1 (BRCA1), structural maintenance of chromosomes 1A (SMC1A), and NBS1 are found to be the major cell cycle checkpoints activated by ATM-dependent phosphorylation (Shibata et al. 2014; Nakanishi et al. 2015; Suberbielle et al. 2015). DSBs also induce rapid phosphorylation of histone H2AX, facilitating the retention of numerous proteins that assemble at the break, including NBS1, mediator of DNA damage checkpoint 1, and p53-binding protein 1 (53BP1) (McKinnon 2009). H2A.X is a key factor in the repair process of DSBs and often used as an indicator of the DSBs. H2A.X is rapidly phosphorylated at serine 139 by three PI-3 kinases, ATM, ATR, and DNA-PK, resulting in discrete γ-H2A.X (phosphorylated H2A.X) foci at the sites of DNA damage (Burma et al. 2001; Myung et al. 2008). H2A.X is a member of the H2A histone family and part of nucleosomes and in response to DSBs, H2A.X localizes to sites of DNA damage at subnuclear foci. Phosphorylation of H2A.X may be a factor involved in the structural change in chromatin observed following induction of apoptotic cell death. Many studies of γH2A.X (Ser139) have focused on the relationship between severe DSBs and the activation of neuronal death pathways. In proliferating cells, its activation is typically associated with a pause in cell cycle progression. In cultures of differentiated neurons, γH2A.X (Ser139) increases transiently after stimulation with N-methyl-D-aspartate, suggesting a potential link between neuronal activity and DSBs (Myung et al. 2008; Herrup et al. 2013; Suberbielle et al. 2013). Similarly, increased and early accumulation of γH2A.X (Ser139) are observed in the brains of patients with neurodegenerative diseases, suggesting early accumulation of DSBs may contribute to neuronal dysfunction and neurodegeneration (Shanbhag et al. 2019; Mitra et al. 2019).
Two major repair pathways for DSBs exist in mammalian cells: the homologous recombination and the NHEJ (McKinnon and Caldecott 2007; Ceccaldi et al. 2016; Chang et al. 2017). The HR pathway is error-free but requires a sister chromatin to act as a template for accurate repair and is confined to late S and G2 phases of cell cycle, whereas NHEJ involves direct ligation of the broken DNA and is active throughout the entire cell cycle but most prevalent in G1 and early S phases (Lieber 2010; Bee et al. 2013; Su et al. 2015). The choice of HR or NHEJ pathway for repairing DSBs is dependent on the phases of the cell cycle (Shibata et al. 2014; Ceccaldi et al. 2016). Since neurons are postmitotic cells, NHEJ is the primary pathway of DSBs repair, although HR is probably important for DNA repair in neural progenitors and nonneuronal cells in the brain (Su et al. 2015). In addition, alternative error-prone DSBs repair pathways, namely alternative end joining and single-strand annealing have been recently shown to operate in many different conditions and to contribute to genome rearrangements and oncogenic transformation (Ceccaldi et al. 2016). The alternative end joining pathways involve different proteins; they can be initiated by poly (ADP-ribose) polymerase 1 (PARP1) and can also involve factors that function in HR, such as BRCA1 (Yu et al. 2018; Seol et al. 2018). The choice of pathway used for repair of individual breaks is crucial for ensuring faithful restoration of genome integrity and elaborate regulatory mechanisms underlie this decision (Ceccaldi et al. 2016; Schwertman et al. 2016).
Non-homologous end joining (NHEJ)
Non-homologous end joining is the preeminent DSBs repair pathway in postmitotic cells such as neurons throughout the cell cycle, including during S and G2 phase (Chang et al. 2017). NHEJ involves initial recognition of a DSBs by the Ku70-Ku80 heterodimer, followed by the assembly of additional factors including the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), Artemis, X-ray cross-complementing protein 4 (XRCC4), and XRCC4-like factor (XLF; also called Cernunnos), with XRCC4 playing a prime role in recruiting DNA ligase IV to carry out the DSBs-joining reaction (Fig. 1) (Jacobsen et al. 2004; Jette and Lees-Miller 2015; Chang et al. 2017). The Ku70/80 heterodimer was discovered as an autoimmune antigen with the unusual property of binding with high affinity to ends of dsDNA in a DNA sequence independent manner (Jette and Lees-Miller 2015). It is now established that the Ku70/80 heterodimer binds ends of dsDNA and recruits DNA-PKcs to DSBs in vitro and in vivo (Kanungo 2013; Jette and Lees-Miller 2015). Highly purified DNA-PKcs has weak protein kinase activity that, in the presence of dsDNA, is stimulated 5–10 fold in the presence of Ku (Jette and Lees-Miller 2015).
Fig. 1.
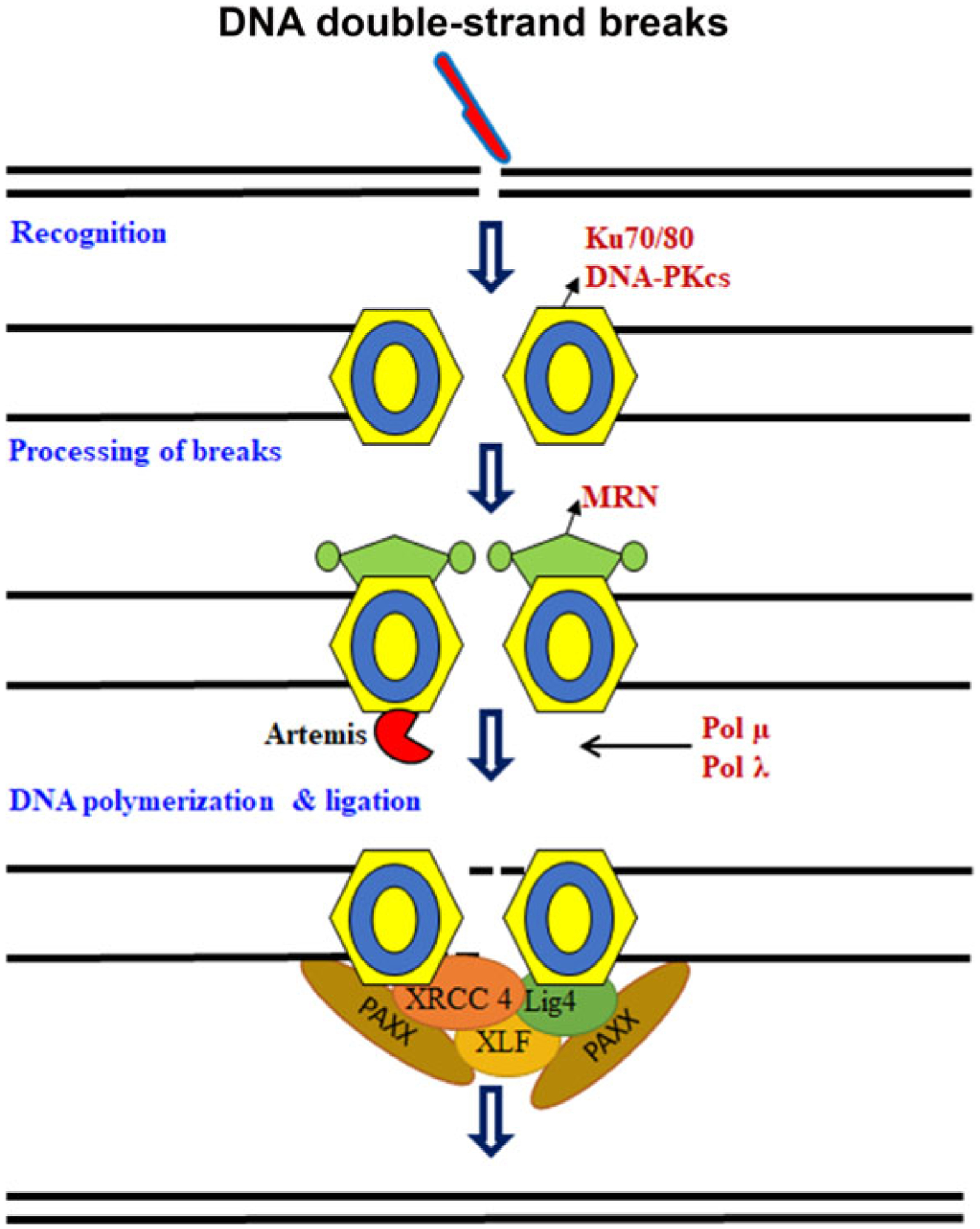
Schematic representation of non-homologous end joining. DNA double-strand break (DSBs) formation is detected by the Ku70/80–DNA-PKcs (DNA-dependent protein kinase). The ends are processed by MRN complex (MRE11, meiotic recombination11; NBS1, Nijmegen breakage syndrome 1). The gaps are filled by polymerase μ and λ and ligated by DNA ligase IV (LIG4). Artemis, X-ray cross-complementing protein 4 (XRCC4), and XRCC4-like factor (XLF; also called Cernunnos) playing a prime role in recruiting LIG4 to carry out the DSBs-joining reaction.
Moreover, mice expressing a kinase-dead DNA-PKcs, and targeted disruption of the genes encoding ligase IV or XRCC4 are embryonic lethal and exhibit neuronal apoptosis, suggesting that the components of NHEJ play an important role in developing and maintaining neurons (Ochi et al. 2015; Chang et al. 2017). XRCC4 and XLF, together with spindle-assembly abnormal protein 6, comprise a homologous superfamily of structurally related proteins. Recently, PAXX (also known as XLS) has been identified as a new NHEJ component protein. PAXX, a paralog of XRCC1 and XLF, interacts with a key repair pathway protein, Ku, and helps promote ligation of the broken DNA (Ochi et al. 2015).
Current models in mammalian cells suggest that the abundant Ku70/Ku80 heterodimer rapidly binds to all two-ended DSBs, allowing NHEJ to make the first attempt at DSBs rejoining (Shibata et al. 2014). Thus, even in G2 where HR functions, NHEJ rejoins most DSBs but subsequently repair switches to HR, necessitating resection (Fang et al. 2019). Resection of two-ended DSBs is a critical step that initiates and potentially commits to repair by HR when NHEJ stalls. MRE11 nuclease activities promote resection, but their roles are unclear; furthermore, MRE11 exonuclease has the wrong polarity to drive resection (Stracker and Petrini 2011; Shibata et al. 2014).
Homologous recombination (HR)
Homologous recombination predominates in the mid-S and mid-G2 cell cycle phases, when DNA replication is ongoing or has ocuured and the sister template is available (Ceccaldi et al. 2016; Wright et al. 2018). HR requires the activity of several proteins including BRCA1, BRCA2, XRCC2, XRCC3, and RAD51. HR is initiated by nucleolytic resection of 5′-termini at DSBs in a CtIP/MRN-dependent dependent manner, after which RAD51 facilitates the invasion of the single-stranded 3′-tail into the homologous sister chromatid, thereby enabling accurate repair of the DSBs. The process is typically error-free even though completion of HR often requires error-prone polymerases (Merlo et al. 2016; Ceccaldi et al. 2016; Wright et al. 2018). HR contributes to distinct processes, including meiotic recombination, replication fork stabilization, and one-ended DSBs repair, and overlaps with NHEJ to repair two-ended DSBs in late S/G2 phase (Ranjha et al. 2018; Wright et al. 2018). Recent studies revealed that HR is involved in HR-mediated fork rescue mechanisms during replication fork stalling, before or after a DSBs occurred. Due to its error-prone nature, it will be desirable for genome integrity that HR-mediated fork rescue mechanisms are postponed as last-resort options. HR-dependent fork reactivation mechanisms are liable to generate genetic instability by instigating microhomology-driven strand invasion or by establishing a replication fork whose progression is error-prone (Branzei and Szakal 2016).
HR (but not NHEJ) functions during meiosis. Meiotic DSBs are introduced by Spo11, a topoisomerase II-like protein, which bridges DNA ends; DSBs opening and Spo11 removal require Mre11 nuclease activity (Garcia et al. 2011). In yeast, DSBs processing creates a ssDNA nick up to 300 bp from the DSBs end followed by bidirectional resection. Mre11 3′−5′ exonuclease activity digests toward the DSBs end, and exonuclease 1 (Exo1) generates ssDNA moving 5′−3′. Current data suggest that Mre11 endonuclease activity makes the initial ss nick, with the combined activities promoting the removal of covalently end-bound Spo11. For HR in mitotic cells, Sae2/MRX (CtIP/MRN) initiates DSBs resection, enabling 5′−3′ resection by Exo1/Sgs1 (EXO1/BLM), although further details are unclear (Fig. 2). Mre11 mutations impact either its exonuclease activity, endonuclease activity or both or disturb Mre11 interactions with interfacing Rad50 or NBS1. The role of MRE11 nuclease activities during resection would require the ability to specifically ablate endo- or exonuclease activity, which in turn depends on structural insight into regions on MRE11 (Shibata et al. 2014).
Fig. 2.
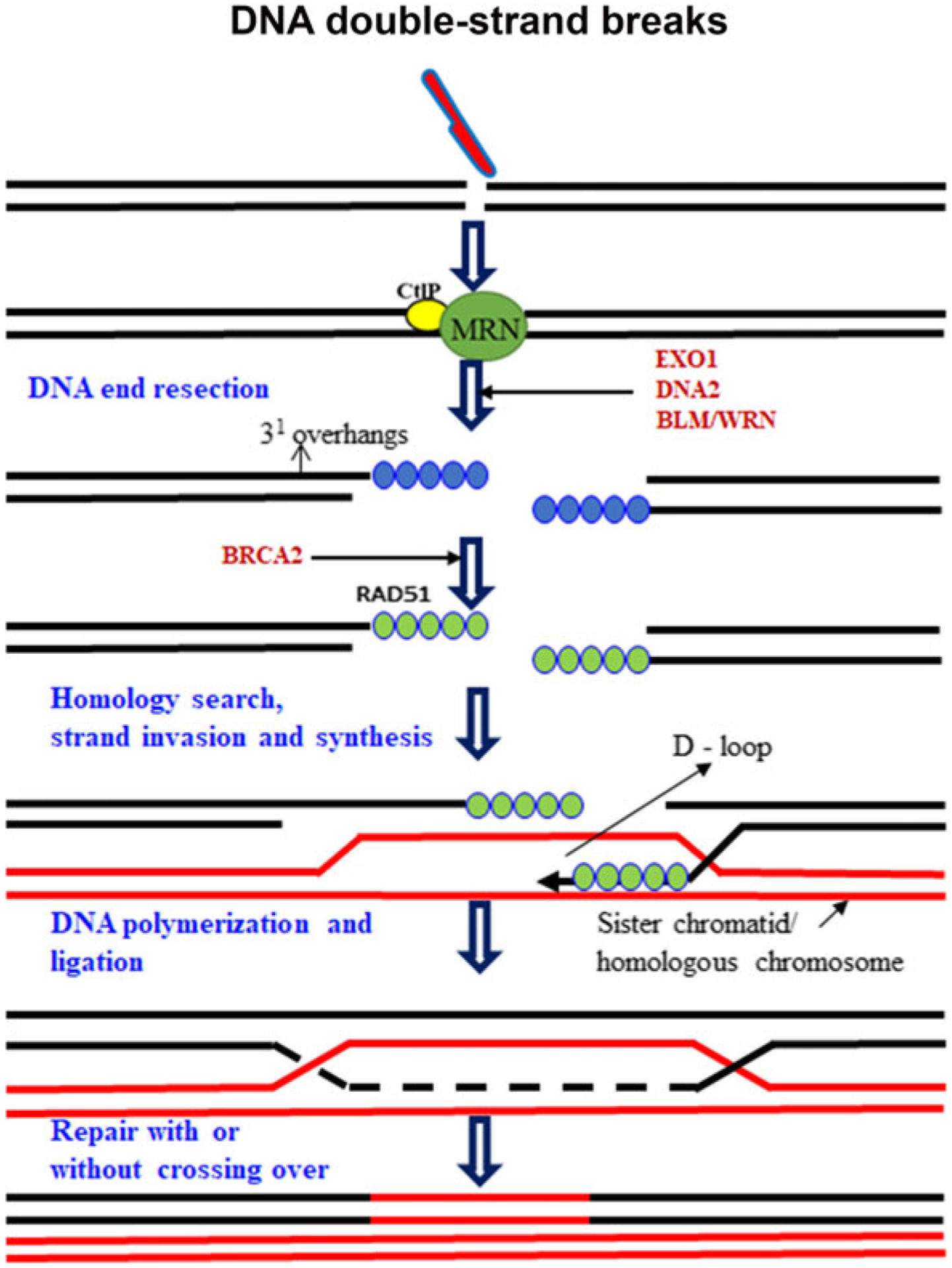
Schematic representation of homologous recombination. HR involves repair of DSBs in the S and G2 phases using the genetic information coded in the sister chromatid. DSBs are recognized by MRE11-RAD50-NBS1 (MRN) complex and CtIP/MRN initiates DSBs resection, enabling 5′−3′ resection by Exo1/Sgs1 (EXO1/BLM/WRN/DNA2). RAD51 facilitates invasion of the single-stranded 3′-tail into the homologous sister chromatid and forms D-loop; this ensure the highly specific error-free repair of damaged DNA
Alternative NHEJ (A-NHEJ)
In recent years, the notion of an alternative end joining pathway in addition to C-NHEJ has become more evident. The use of alternative end joining for DSBs repair has been described in various cellular contexts, but the mechanistic details of this pathway remain unclear (Ceccaldi et al. 2016; Chang et al. 2017; Seol et al. 2018). Alternative NHEJ (A-NHEJ), which includes microhomology-mediated end joining (MMEJ), repairs DNA DSBs by annealing 2–20-bp stretches of overlapping bases flanking the DSB. Not all A-NHEJ produces repair junctions with MH; therefore, MMEJ likely only corresponds to a subset of A-NHEJ (Seol et al. 2018). Genetically, MMEJ does not require Ku70/Ku80, RAD51, BRCA2, or LIG4, but both processes require the MRE11-RAD50-NBS1 complex [MRN complex; Mre11-Rad50-Xrs2 (MRX) complex in yeast], DNA polymerase theta (Polθ) or the B and X family polymerases in yeast (Polδ and Pol4), CtBP-interacting protein (CtIP; Sae2 in yeast), poly(ADP-ribose) polymerase 1 (PARP1), ataxia telangiectasia mutated (ATM; Tel1 in yeast), and flap endonuclease 1 (Fig. 3) (Chang et al. 2017; Sallmyr and Tomkinson 2018). Despite the high mutation burden associated with MMEJ, it acts as a back up repair pathway and provides valuable physiological functions. Recent findings illustrate the emerging possibility of MMEJ as an attractive anti-cancer and neurodegenerative disease drug target (Seol et al. 2018; Sallmyr and Tomkinson 2018).
Fig. 3.
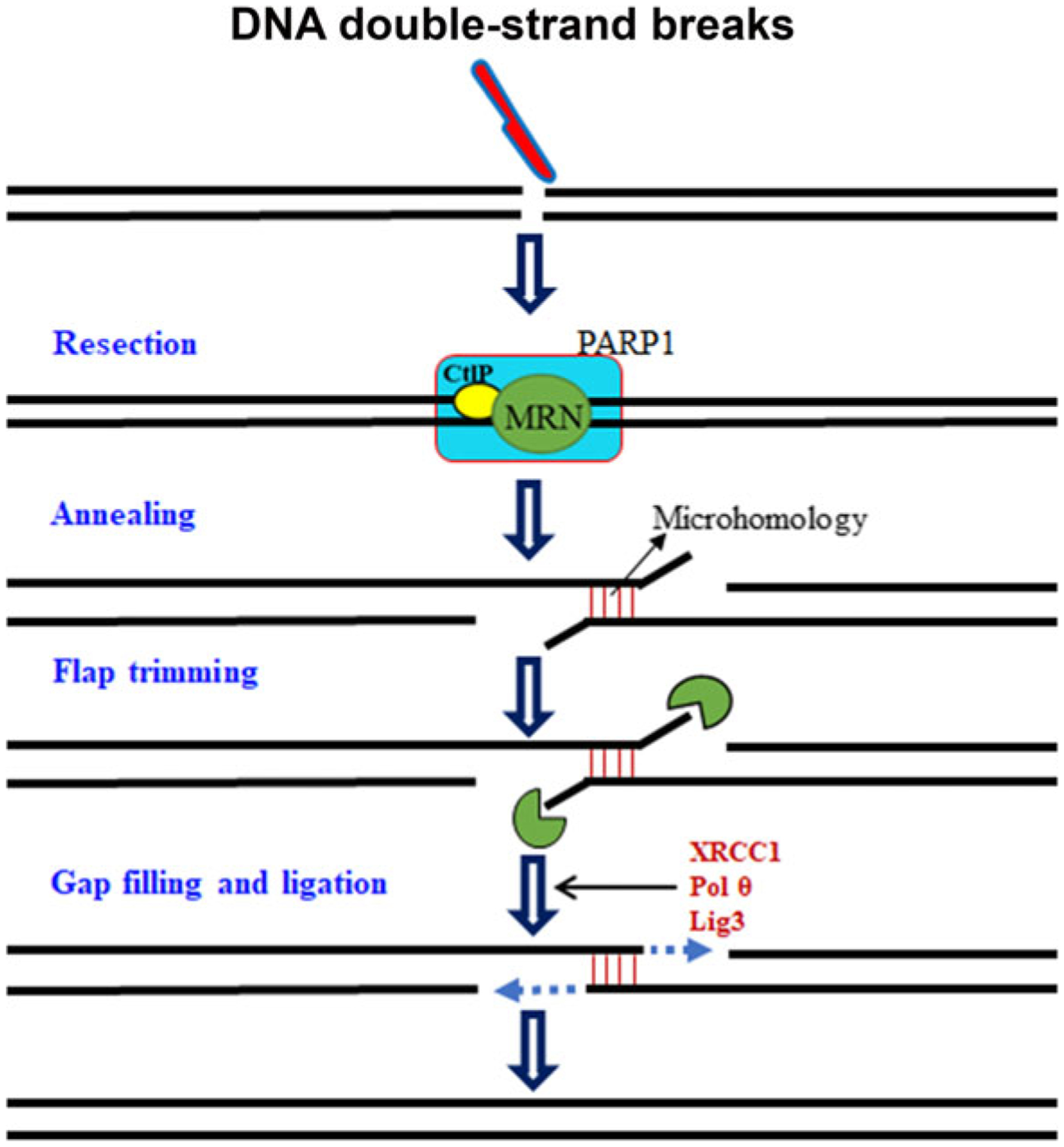
Schematic representation of alternative/microhomology-mediated end joining. Alternative/microhomology-mediated end joining acts as a back up pathway for DSBs repair, by annealing 2–20-bp stretches of overlapping bases flanking the DSBs (all alternative pathways does not require microhomology). MRE11-RAD50-XRS2? complex [MRN complex; MRE11-RAD50-XRS2], CtBP-interacting protein (CtIP), and poly(ADP-ribose) polymerase 1 (PARP1) resect the DNA ends and gaps are filled by DNA polymerase theta (Polθ), ligated by Lig3, and mediated by XRCC1
DNA double-strand breaks and neurodegenerative diseases
Preserving genomic integrity is a critical function of each cell. Each cell is equipped with arsenals of unique and efficient DNA repair proteins that preserve genome stability and ensure the integrity of DNA. When DNA repair proteins are deficient or defective, DNA breaks are not repaired or incorrectly repaired, resulting in progressive and permanent neuronal damage and impairing cognitive and motor functions (Madabhushi et al. 2014; Alt and Schwer 2018; Khan et al. 2018). DSBs are detrimental for neurons as they remarkably impact on genome integrity, transcriptional activity, cellular proteostasis, and energy starvation (Ferrer et al. 1993; Casafont et al. 2011; Pan et al. 2014; Madabhushi et al. 2015; Mata-Garrido et al. 2018). Studies suggested that DSBs accumulation causes alteration in transcription from gene promoters located near break sites, which results in altered neuronal and synaptic function (Kruhlak et al. 2007; Shanbhag et al. 2010). Emerging findings suggest that an imbalance between DSBs accumulation and repair in brain contributes to neuronal damage, impaired learning and memory, and has been documented in the pathogenesis of a broad spectrum of human neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (Rulten and Caldecott 2013; Madabhushi et al. 2014; Merlo et al. 2016; White and Vijg 2016; Yu et al. 2018; Milanese et al. 2018; Wang and Hegde 2019).
DNA double-strand breaks in Alzheimer’s disease:
Alzheimer disease (AD) is a progressive neurodegenerative disease and accounts for most dementia cases. The disease is characterized by progressive neuronal death associated with neuropathological findings of neurofibrillary tangles and senile plaques. These characteristics are further accompanied by synaptic dysfunction, memory loss, and cognitive decline. Despite extensive research efforts, the molecular mechanisms underlying the development and progression of AD in aged populations remains poorly understood (Khan et al. 2018; Yu et al. 2018).
DNA damage, particularly DSBs, has recently emerged as a driving force behind memory loss and neuronal degeneration in AD or AD-related dementia (Myung et al. 2008; Kanungo 2013; Suberbielle et al. 2015; Hou et al. 2017). Several studies suggest that increased DSBs accumulation or decreased levels of DNA repair proteins occur in brains of AD patients and DNA damage may precede tangle formation (Su et al. 1997; Sheng et al. 1998). A study by Shanbhag and colleagues suggest that DSBs accumulate in neurons and astrocytes at early stages and during the progression of AD, and may contribute to neuronal dysfunction and degeneration (Shanbhag et al. 2019). Furthermore, sustained and increased DSBs have also been reported in mouse models of AD (Shen et al. 2016; Yu et al. 2018). For instance, 3xTg-AD/Polβ mice exhibit more extensive DSBs, and neuronal loss in the hippocampus than 3xTg-AD mice, suggesting that impairment in DNA repair machinery can render the brain more vulnerable to AD-related pathologies (Hou et al. 2017). Similarly, a study by Suberbielle and colleagues reported that APP transgenic mice, which overexpress the amyloid precursor protein, had more severe and prolonged neuronal DNA strand breaks compared to control mice (Suberbielle et al. 2013). DSBs occurred in multiple brain regions, and were most abundant in the dentate gyrus, which is involved in learning and memory (Suberbielle et al. 2013). A related study by Yu and coworkers reported that APP/PSEN1 mice showed increased and sustained DSBs and apoptosis in hippocampal neurons, along with diminished expression of RAD51, indicating reduced DSBs repair capacity (Yu et al. 2018). The breast and ovarian cancer susceptibility protein 1 protein plays an important role in DSBs repair and its level is reduced in the brains of AD patients (Suberbielle et al. 2013). Depletion of BRCA1 in the hippocampus of mice increases neuronal DSBs, alters neuronal structure and function, reduces neuronal size, increases neuronal excitability, and impairs learning and memory (Suberbielle et al. 2015). Thus, BRCA1 may serve as a potential therapeutic target for treating AD or AD-related dementias. CDKN1A-interacting zinc finger protein 1 (CIZ1) plays a critical role in DNA replication, DNA repair, and cell cycle progression at the G1/S checkpoint (Coverley et al. 2005; Xiao et al. 2016; Khan et al. 2018). Recent study suggests that deficiency of CIZ1 causes increased neuronal DSBs which contribute to neuronal loss and cognitive decline in aged mouse, indicating a role of CIZ1 in DSBs repair (Khan et al. 2018). Although a direct association between CIZ1 and AD progression has yet not been established, genome-wide association studies have consistently identified variants in cell cycle genes as susceptibility factors for mild cognitive impairment and dementia (Scacchi et al. 2013), and two polymorphisms in CDKN1A are associated with increased risk and earlier age-of-onset of AD (Yates et al. 2015). A better understanding of the molecular mechanisms by which CIZ1 regulates DNA damage and memory loss may uncover novel therapeutic targets for AD-related dementia. Ataxia telangiectasia mutated is a key cellular protein involved in a cell cycle checkpoint control during the repair of DNA damage (Herrup et al. 2013; Maréchal and Zou 2013). In response to DSBs, the ATM kinase phosphorylates several downstream targets associated with DNA damage repair, cell cycle arrest, and apoptosis (Maréchal and Zou 2013). A recent study by Shen et al reported that cultured Atm+/− cortical neurons as well as neurons in the brains of Atm+/− mice demonstrate cellular abnormalities similar to those seen in the AD brain (Shen et al. 2016). They further reported that loss of ATM is highly correlated with neuronal cell death in human AD brain, indicating ATM signaling is a key part of the neurodegeneration mechanism during AD pathogenesis (Burma et al. 2001; Herrup et al. 2013; Shen et al. 2016). Therefore, targeting ATM-associated mechanisms may hold therapeutic promise against neurodegenerative diseases. These findings strongly suggest that alterations in DSBs repair proteins and/or accumulation of DSBs make an important early step in the pathway toward neural damage and degeneration in AD (Fig. 4). Interestingly, a study by Lee at el. suggests that neuronal DSBs formation could also offer entry points for the retro-insertion of genomic complementary DNAs and induce mosaic mutations of the APP gene which encodes the amyloid precursor protein in AD (Lee et al. 2018). Somatic gene recombination in AD is dysregulated, producing many more numbers and forms of APP genomic complementary DNA, including 11 single-nucleotide variations that are considered pathogenic APP mutations when they appear in families. Therefore, reducing disease-related somatic gene recombination using reverse transcriptase inhibitors could represents a possible therapeutic option for AD.
Fig. 4.
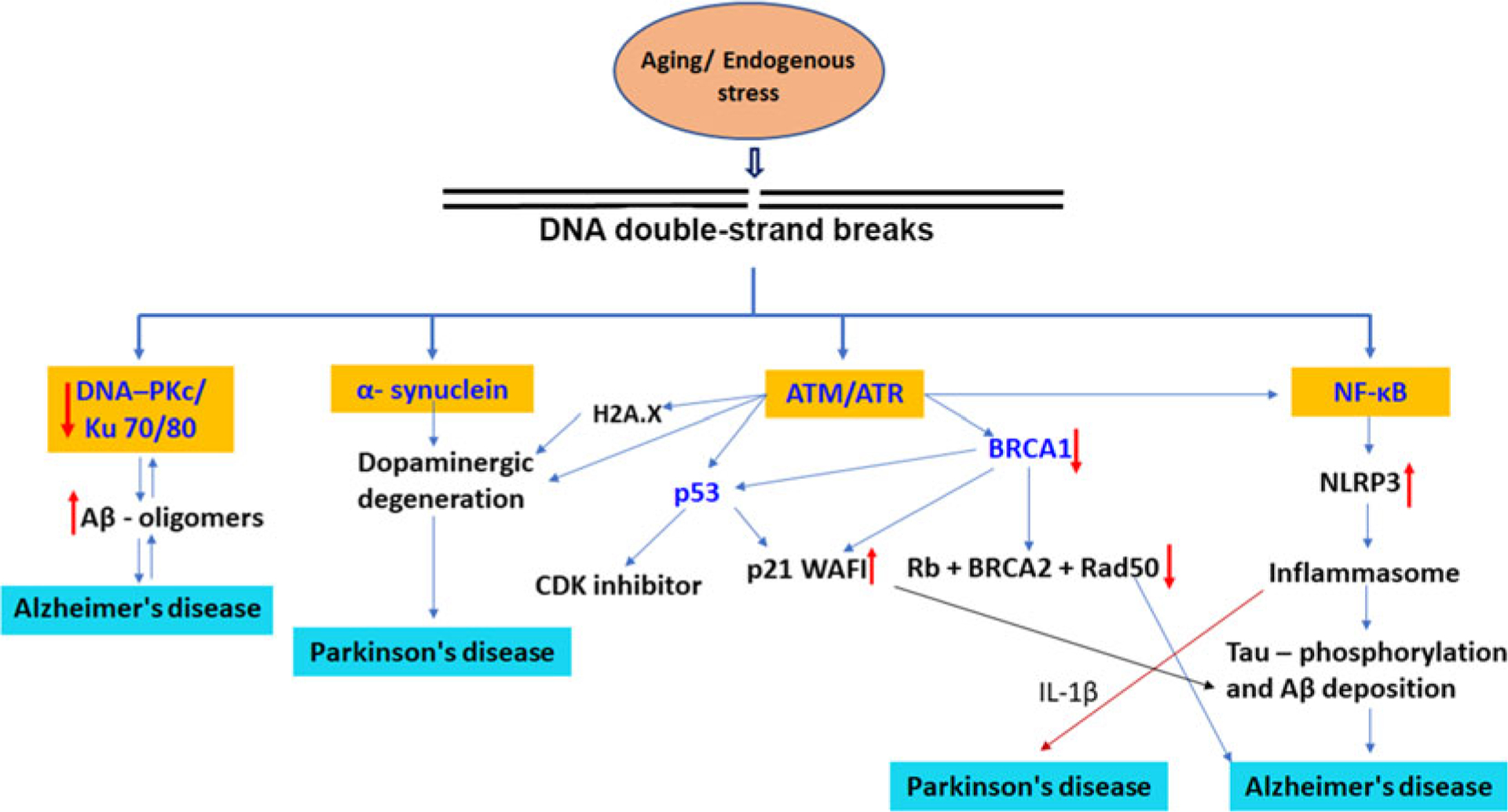
Proposed mechanisms for the involvement of DNA double-strand breaks in onset and progression of neurodegenerative diseases. DNA double-strand breaks signaling pathways are intertwined through a variety of mechanisms including, ATM activation, ROS generation, and NF-κB activation. The elevated levels of DSBs induce a reduction in the levels of DSBs repair proteins such as DNA-PKcs/Ku70/80 complex and MRN complex proteins, and contributes to progression of AD. ATM/ATR operates through different pathways by p53, BRCA1 proteins, and dopaminergic degeneration. BRCA1 interacts with BRCA2, retinoblastoma protein (Rb), RAD50, RAD51. BRCA1 stimulates the CDK inhibitor p21 WAF1 and p53 tumor suppressor protein. DSBs directly activate NF-κB in a membrane receptor-independent manner or activated through ATM. NF-κB upregulation triggers NLRP3 activation and secretion of proinflammatory cytokines in Alzheimer disease and Parkinson’s disease.
Of the two main pathways for DSBs repair, NHEJ is the predominant dsDNA repair pathway in mammalian cells. This pathway requires DNA-dependent protein kinase (DNA-PK), composed of a catalytic subunit, DNA-PKcs, and a regulatory component, Ku70/Ku80 heterodimer. Mice deficient in NHEJ components such as DNA Ligase IV, XRCC4, Ku70, and Ku 80 exhibit increased apoptosis in postmitotic neurons (Kanungo 2013). Furthermore, mice with defective NHEJ exhibit accelerated aging. Similarly, a study by Davydov et al. demonstrated the DNA-PK complex, DNA-PKcs and Ku70/80, was reduced in AD midfrontal cortex brain tissues, suggesting the impairment of this DNA damage repair protein complex in AD progression (Davydov et al. 2003). A study by Kanungo reported reduced NHEJ activity in AD brains along with reduced levels of DNA-PKcs and the Ku proteins, indicating a potential link between AD and DSBs accumulation (Kanungo 2013). Mre11 complex proteins play a critical role in cellular responses to DSBs and were substantially reduced in the cortical neurons of AD patients compared to non-AD cohorts (Jacobsen et al. 2004). It is plausible that the lack of Mre11 complex could lead to an inability to sense and fix DNA damage (Garcia et al. 2011; Shibata et al. 2014). These findings point to a possible connection between the impairment of the DSBs repair protein complex and the disease progression of AD patients.
Postmitotic neurons that are terminally differentiated, when triggered to re-enter cell cycle following chronic or acute insults inducing DNA damage and/or oxidative stress, undergo apoptosis (Kruman et al. 2004). Neurons re-entering cell cycle are prone to accrue DNA damage (Yang et al. 2001). Therefore, it is possible that DNA replication is a consequence of cell cycle re-entry that precedes neurodegeneration in AD brain. In addition, reactive oxygen/nitrogen species can cause misdirected and inefficient DNA replication, called “replication stress” during AD pathogenesis can lead to genomic instability, thus facilitating Aβ accumulation and deregulation of cell cycles (Yurov et al. 2011). In postmitotic neurons, these adverse events can be further amplified with the existence of defective DNA repair systems leading to accumulation of additional DNA damage and genomic instability.
Neuronal death in response to DSBs proceeds through signaling pathways that that are intertwined through a variety of mechanisms, including ATM activation, ROS generation, and NF-κB activation. Several studies have offered evidence that DSBs directly activate NF-κB in a membrane receptor-independent manner, which involves a retrograde signaling cascade from the nucleus to the cytoplasm (Huang et al. 2000; Janssens and Tschopp 2006; McCool and Miyamoto 2012). However, other studies suggested that ATM is essential for NF-κB activation in response to DSBs (Piret et al. 1999; Li et al. 2001). A study by Tilstra and colleagues demonstrated that accumulation of DNA damage drives the aging process through upregulation of NF-κB activation and pharmacological inhibition of NF-κB activation leads to attenuation of age-related pathologies (Tilstra et al. 2012).
The tumor suppressor protein p53 is considered the guardian of the genome that regulates the cellular response to various stress signals, including DNA damage. The role of p53 has been considered as a double-edged sword depending on the severity of DNA damage. Early in the DSBs response, p53 relays a wide range of prosurvival signals that allow the cells to repair the damage. If DSBs continue to accumulate, p53 switches gears and promotes apoptosis or senescence (Nakanishi et al. 2015; Suberbielle et al. 2015). Evidence of the oxidative DNA damage profile in AD generates speculation that BRCA1 may play an important role in AD pathogenesis. BRCA1 has binding properties for BRCA2, Rb, RAD50, and RAD51, in order to activate the cell cycle checkpoints (Shibata et al. 2014; Broustas and Lieberman 2014). BRCA1 stimulates proteins that control cell cycle check points such as the CDK inhibitor p21 WAF1 and p53. BRCA1 is colocalized with DNA recombinase Rad51 and may be involved in DSBs repair. Recent work has reported that BRCA1 functions in telomere maintenance, a distinctive feature of degenerating neurons in the AD brain (Nakanishi et al. 2015)
However, the exact mechanism by which accumulated DSBs in the brain contribute to neurodegeneration and cognitive deficits in AD remains unsolved. Although recent studies have increased regarding the association between DSBs accumulation and AD and simultaneously provided insight into potential new targets and novel strategies for the treatment of AD, continued research is necessary to elucidate the molecular and cellular mechanisms connecting accumulation of DSBs to its impact on AD onset and progression.
DNA double-strand breaks in Parkinson’s disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that affects 10 million people worldwide. PD is characterized by degeneration of dopaminergic neurons in substantia nigra of the midbrain and loss of both motor and non-motor features (Milanese et al. 2018). Several hypotheses have been proposed to explain the cause of PD, including loss of dopaminergic neurons, mitochondrial dysfunction, oxidative stress, and α-synuclein deposition (Lewy bodies), but the exact causes of PD are still unclear. More recent studies have highlighted the role of DNA damage, particularly, DSBs in the progression of neuronal loss in PD (Camins et al. 2010; Kirshner et al. 2012; Brochier and Langley 2013; Wang et al. 2016; Milanese et al. 2018).
Elevated levels of DSBs were detected in the dopaminergic neurons of the substantia nigra in PD patients (Hegde et al. 2006; Camins et al. 2010) and in mouse models of PD (Camins et al. 2010; Sepe et al. 2016; Wang et al. 2016; Milanese et al. 2018). A study by Wang et al reported that DSBs accumulation preceded onset of motor phenotype and dopaminergic degeneration in α-synuclein (A53T) overexpressing transgenic mice (Wang et al. 2016). Similarly, Sepe and coworkers reported that specific deletion of DNA excision repair protein Ercc1 in dopaminergic neurons induces loss of tyrosine hydroxylase immunoreactivity in the mouse brain, suggesting defects in DNA repair impact the dopaminergic system and are associated with human PD pathology (Sepe et al. 2016). In fact, a strong correlation has been reported between the DSBs and severity of PD (Migliore et al. 2001; Hegde et al. 2006; Milanese et al. 2018). For instance, a study by Hegde and colleagues examined the topology and DNA damage in PD brains and age-matched non-PD controls (Hegde et al. 2006). In this study, they reported the structural integrity and topology of genomic DNA is altered predominantly in the substantia nigra of PD patients, but also in other regions of the brain, such as the caudate putamen, hippocampus, and thalamus. The changes included DSBs accumulation, single-strand DNA damage, and DNA instability. Similarly, Milanese et al. demonstrated a significantly increased immunoreactivity of γH2AX and 53BP1 foci, and phospho-ATM in dopaminergic neurons in synucleinopathy models of PD, which collectively indicate the role of accumulation of DSBs and inefficient DNA repair capacity in PD (Milanese et al. 2018). Moreover, mutations in several genes that encode proteins, critically involved in DNA repair, have been shown to contribute PD progression (Camins et al. 2010; Kirshner et al. 2012; Ogino et al. 2016). For example, mice deficient for the DNA repair protein ATM exhibit locomotor abnormalities similar to a PD, accompanied with loss of dopaminergic neurons and striatal degeneration (Eilam et al. 1998, 2003). Considering the role of ATM in PD pathogenesis, a study by Shackelford et al has stated that iron chelation therapy had a protective effect on dopaminergic neurons, which was attributed to ATM activation and subsequent ATM-mediated upregulation in DNA repair and antioxidant pathways (Shackelford et al. 2006). Related to this finding, Camins and coworkers presented evidence that MPP+ treatment induces DSBs with rapid activation of ATM and constitutes an early signal in the apoptotic route of this neurotoxin (Camins et al. 2010). In this study, they also reported increased levels of p-γH2AX (Ser139), pATM, and p53 (Ser15) in the brains of PD patients, suggesting the potential role of DSBs in PD progression. These findings provided proof-of-concept evidence that accumulation of unrepaired or erroneously repaired DSBs can render the brain more vulnerable to PD-related pathologies. Thus, targeting the DSBs-associated neuronal death pathway might offer a novel approach to prevent or mitigate PD progression. Further studies and a better understanding of DSBs in PD are needed to shed new insights into the possible mechanism and new therapeutic approaches for the prevention and treatment of PD.
DNA double-strand breaks in amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects neurons in the motor cortex, brainstem, and upper and lower spinal cord, slowly inducing muscle atrophy, denervation, and severe motor dysfunction (Mitra et al. 2019). Despite extensive research, there is no effective treatment to halt, or reverse, the progression of the disease. More than 90% of cases are sporadic, while 5 to 10% of the cases have a familial origin. The most prevalent genetic causes of familial ALS are mutations in the superoxide dismutase 1 (SOD 1), transactivation response DNA-binding protein (TDP-43), fused in sarcoma (FUS) genes, and chromosome 9 open reading frame 72 (C9ORF72).
There is growing evidence that ALS-related mutations affect several genes specifically involved in the maintenance of genomic stability and DNA repair function. For instance, mutations in FUS (Qiu et al. 2014; Wang et al. 2018; Wang and Hegde 2019), TDP-43 (Hill et al. 2016; Mitra et al. 2019; Guerrero et al. 2019), and C9orf72 (Walker et al. 2017; Konopka and Atkin 2018; Lopez-Gonzalez et al. 2019) are associated with ALS and have been demonstrated to play an important role in DDR pathways. A study by Walker et al demonstrated that C9orf72 repeat expansions cause impaired DNA repair function and increased R-loop formation as assessed by decreased H2A ubiquitylation, impaired 53BP1, altered ATM phosphorylation, and accumulation of HDAC4 (Walker et al. 2017). Increased DDR activation in C9orf72-ALS patients has been corroborated by recent findings (Farg et al. 2017; Walker et al. 2017), which demonstrated elevated DNA damage in postmortem brain and spinal cord tissues of C9ORF72 patients. Similarly, a study by Lopez-Gonzalez and coworkers showed elevated levels of DNA damage markers γH2AX, ATR, GADD45, and p53 in motor neurons differentiated from iPSC lines from C9orf72 ALS patients, suggesting the role of DSBs in ALS patients (Lopez-Gonzalez et al. 2016).
Recent studies have shown a correlation between FUS aggregation and impaired DNA repair function (Hill et al. 2016; Wang et al. 2018; Wang and Hegde 2019). Earlier studies showed that FUS is rapidly recruited at the sites of DNA damage in a poly(ADP-ribose) polymerase (PARP)-dependent manner. In response to DSBs-inducing stimuli, FUS is phosphorylated by DNA damage sensor protein kinases ATM and DNA-dependent protein kinase, both of which are involved in DSBs repair pathway (Deng et al. 2014; Wang et al. 2018). FUS interacts with histone deacetylase 1 (HDAC1), suggesting the role of FUS in DSBs repair pathways (Wang et al. 2018). Interestingly, loss of FUS abrogated both HR and NHEJ efficiency in exogenomic vector-based assays (Wang et al. 2013). Similarly, depletion of FUS reduced DSBs repair pathways indicating FUS as an upstream participant in major DSBs repair pathways (Mastrocola et al. 2013). Transgenic mice expressing ALS-associated mutant FUS-R521C exhibited DNA damage in cortical neurons and spinal motor neurons (Qiu et al. 2014). Similarly, increased expression of γH2AX foci has been detected in ALS patients carrying FUS-R521C or FUS-P525L mutations (Wang et al. 2013). A study by Naumann et al demonstrated that FUS mutations in the nuclear localization sequence cause impairment of PARP-dependent DDR, which leads to neurodegeneration and the formation of FUS aggregates (Naumann et al. 2018). Wang and colleagues reported that FUS mutations cause defects in DNA nick ligation and oxidative damage repair in ALS patients (Wang et al. 2018).
TDP-43, an RNA/DNA-binding protein, plays an important role in the regulation of RNA transcription, splicing, and transport. A significant accumulation of DSBs is observed in TDP-43-linked diseases (Hill et al. 2016) and identification of DNA repair protein Ku in the TDP-43 immunocomplex from human cells indicates the possibility of TDP-43’s involvement in DSBs damage repair (Freibaum et al. 2010). In healthy neurons, TDP-43 facilitates the optimal recruitment of XRCC4/DNA ligase IV complex at DSBs sites and regulates DSBs ligation and repair (Guerrero et al. 2019). In ALS, nuclear loss of function of TDP-43 causes deficient repair of DSBs, leading to DSB accumulation, and persistent DDR activation, contributing to neurodegeneration. A recent study by Mitra et al. found out that conditional depletion of TDP-43 markedly increases accumulation of DSBs by impairing NHEJ repair function, and thereby, sensitizing neurons to DSBs stress. The study also reveals that the strong correlation of TDP-43 pathology with DSBs repair defects, and damage accumulation in the neuronal genomes of sporadic ALS patients and in Caenorhabditis elegans mutant with TDP-1 loss of function (Mitra et al. 2019).
These finding strongly endorse the possibility that an inadequate DNA repair function and/or accumulation of DSBs significantly contribute to neurodegenerative phenotype in ALS and targeting DSBs signaling could lead to novel therapeutic routes for attenuating ALS. Further studies are required to dissect the mechanisms describing how DSBs accumulation or impaired DNA repair function contribute to neurodegeneration and motor deficits in ALS.
DNA double-strand breaks: a target for the treatment of neurodegenerative diseases
DNA DSBs are the most deleterious type of DNA damage and a feature of many age-related neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Thus, understanding how DSBs promote neuronal loss in the brain may allow for the development of effective therapies for neurodegenerative diseases. Based on the available data in cellular and animal models and studies involving postmortem brain tissue from patients with neurodegenerative diseases, several potential candidates of the DSBs machinery have been exposed as possible drug targets for the treatment of neurodegenerative diseases. For instance, an intriguing report by Suberbielle and colleagues reported that BRCA1 suppression in the mouse dentate gyrus increased DSBs accumulation and led to memory loss (Suberbielle et al. 2015). In this study, they also observed reduced levels of BRCA1 in the brains of AD patients and human amyloid precursor protein transgenic mice, suggesting BRCA1 could serve as a therapeutic target for AD and AD-related dementia. Similarly, the protein kinase ATM is employed in chromatin remodeling and in epigenetic alterations that are required for DSBs repair. Studies have shown that reduced levels of ATM are positively correlated with neuronal loss in AD brains, indicating ATM signaling is a key part of the neurodegeneration mechanism (Herrup et al. 2013; Shen et al. 2016). Similarly, Eilam and coworkers reported that Atm-deficient mice exhibit dopaminergic neuronal loss and motor abnormalities (Eilam et al. 2003). In this regard, Edwin Shackelford and colleagues suggested that iron chelating flavonoid quercetin directly activates ATM and protects neuronal cells from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridin-induced toxicity (Edwin Shackelford et al. 2005). Given the role of ATM in AD and PD progression, pharmacological activator of ATM may have significant therapeutic potential in the treatment of these diseases.
Studies reported that mice harboring mutations/deficiency in genes involved in NHEJ show signs and symptoms of premature aging and neurodegeneration (Espejel et al. 2004; White and Vijg 2016). Since NHEJ are decreased in neurodegenerative diseases, application of an NHEJ stimulator may be particularly beneficial in these disorders. Although it is pharmacologically easier to target a single enzymatic step, a strategy could be advanced to enhance the whole NHEJ process by up-regulating all enzymatic steps at the same time.
MRN complex, comprising the Mre11, Rad50, and NBS1 proteins senses early process of DSBs. A recent study by Tuxworth and coworkers reported that Mirin, a Mre11 inhibitor, prevented synapsin loss in primary hippocampal neurons exposed to Aβ1–42 oligomers (Tuxworth et al. 2019). Similarly, Kirshner et al reported that inactivation of the Nbs1 gene in mouse brain leads to loss of dopaminergic neurons in substantia nigra and a reduction of dopamine transporter in the striatum (Kirshner et al. 2012). These findings imply that pharmacological agents that enrich the DSBs repair capacity to boost neuronal survival and function might be effective to recuperate brain health and function in patients with neurodegenerative diseases.
Sirt1 deacetylates and stimulates the enzymatic activity of HDAC1, which was necessary for DSBs repair through the NHEJ. Neurons lacking either SIRT1 or HDAC1 are more susceptible to DSB-inducing agents, suggesting SIRT1 or HDAC1 activation offers an important therapeutic avenue in several neurodegenerative diseases (Dobbin et al. 2013). Castillo et al reported that galantamine treatment protects against beta amyloid peptide-induced DNA damage in SH-SY5Y human neuroblastoma cell (Castillo et al. 2016). They further suggested that antigenotoxic effects of galantamine may be related to the activation of DNA repair function which may favor neuronal restoration. A study by Yang et al showed that inhibition of GSK3β accelerates DSBs repair function through enhanced end joining of DSBs in irradiated mouse hippocampal neurons (Yang et al. 2011).
Interestingly, antisense oligonucleotides (ASOs) targeting the C9orf72 repeat expansion are currently showing promise for ALS (Konopka and Atkin 2018). Although the exact molecular mechanisms by which C9orf72 repeat expansions account for neurodegeneration have not yet been decoded, some potential therapeutics, such as ASOs targeting hexanucleotide GGGGCC repeats in mRNA, were successful in pre-clinical trials and are awaiting phase 1 clinical trials (Babić Leko et al. 2019). Furthermore, a study by Gendron et al documented that ASO treatment targeting poly GP reduced both repeat-containing RNA foci and poly GP concentrations in C9orf72 ALS iPSC-derived neurons (Gendron et al. 2017). Similarly, ASOs targeting C9orf72 RNA and small molecules (KPT-276, or TMPyP4) treatments inhibited nuclear import impairment by the C9orf72 repeat expansion in fly models, as well as in C9orf72 iPSC-derived neurons, and reduced neurodegeneration (Zhang et al. 2015). A recent study by Lopez-Gonzales and coworkers reported that partial suppression of Ku80, a DNA repair protein, in C9ORF72 iPSC-derived patient neurons through CRISPR/Cas9-mediated ablation or small RNAs-mediated knockdown reduced the apoptotic pathway, suggesting inhibition of the hyperactivated Ku80-dependent DNA repair pathway is a promising therapeutic approach in C9ORF72-ALS (Lopez-Gonzalez et al. 2019). Becker et al. reported that ASOs targeting ataxin 2 administered directly to the central nervous system of TDP-43 mice, markedly extended survival and reduced pathology in TDP-43 mice, suggesting targeting ataxin 2 could represent a broadly effective therapeutic strategy for ALS (Becker et al. 2017). McGurk and colleagues demonstrated that small-molecule inhibition of tankyrase-1/2 reduces TDP-43 accumulation in the cytoplasm and greatly lessens TDP-43-mediated neurodegeneration (McGurk et al. 2018). In a recent study, Fang el al. identified several classes of small molecules that prevented stress granule (SG) localization of TDP-43 and inhibited accumulation of TDP-43 into persistent cytoplasmic puncta in human-induced pluripotent stem cell-derived motor neurons. This study suggests that small-molecule disruption of RNA-RNA-binding proteins interactions in SGs is a potential therapeutic strategy for ALS (Fang et al. 2019). These findings suggest that ASOs/small molecules targeting DNA damage response pathways in CNS represent a feasible treatment for ALS. However, the major challenge in using ASOs in neurodegenerative disorders is the poor bioavailability, susceptible nuclease degradation nature, and limited blood-brain barrier permeability. Considering the advances in the field of the nanotechnologies, solutions have been proposed to facilitate the CNS targeted distribution of ASOs.
Conclusion
DNA double-strand breaks are the most lethal and dangerous type of lesions with severe consequences for neuronal survival, function, and the preservation of neuronal genome. To protect the neuronal genome, cells are equipped with a myriad of DNA repair proteins and unique pathways, commonly referred to as the DNA damage response. DNA damage response is a tightly regulated signaling cascade that senses DNA damage, recruits DNA repair proteins to the damaged sites, and executes DNA repair. If DSBs repair fails or DNA is misrepaired, the damaged DNA triggers myriad of cellular responses leading to activation of downstream cell death signaling pathways. However, the precise molecular mechanisms by which damaged DNA leads to cell death remains enigmatic. During the past few years, significant progress has been made toward better understanding of the relationship between DSBs responses and neurodegenerative diseases. Recent studies in the human postmortem brain samples and mice brains revealed several molecular mechanisms underlying the DSBs and their links to neurodegenerative diseases; however, many important issues must still be resolved. In this review, we provided a comprehensive summary of the DSBs and DSBs repair mechanisms in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis.
From a therapeutic perspective, it will be of great importance to understand how the DSBs repair pathways can be pharmacologically manipulated to switch from a prodeath to a prosurvival pathway. Understanding the crosstalk among the DSBs as well as how these signaling pathways are intertwined with initiation of inflammatory and apoptotic cascade will provide new treatment options for several neurodegenerative diseases including Alzheimer’s and Parkinson’s disease. As we achieve a better understanding of DSBs repair mechanisms and the regulation of pathways, it is most likely that basic mechanistic insights will translate into clinical benefits. Moreover, several DSBs repair proteins including the BRCA1, DNA-PKc, FUS, PARP1, HDAC1, ATM, and TDP-43 represent a rich set of potential targets to be utilized in the development of more effective therapeutic strategies in neurodegenerative diseases. These targets may also be useful as biomarkers of neuronal genome instability to detect disease condition in its earliest stages. Future studies are warranted to identify of how DSBs accumulation can lead to neuronal death in disease condition. Such studies will build a solid foundation for future work on the DSBs-associated mechanisms in human neurodegenerative diseases.
Funding information
Our research on DNA damage is supported by Department of Defense grant W81XWH-17-1-0062; William and Ella Owens Medical Research Foundation, NIH R21 GM118962, R03 NS101485, Neuroscience Institute, and the Division of Rehabilitation Sciences, College of Health Professions, University of Tennessee Health Science Center.
Abbreviations:
- AD
Alzheimer’s disease
- ALS
Amyotrophic lateral sclerosis
- ASOs
Antisense oligonucleotides
- A-NHEJ
Alternative NHEJ
- ATM
Ataxia telangiectasia mutated
- ATR
Ataxia telangiectasia and Rad3 related
- BER
Base excision repair
- BRCA1
Breast and ovarian cancer susceptibility protein 1
- C9ORF72
Chromosome 9 open reading frame 72
- CtIP
CtBP-interacting protein
- CIZ1
CDKN1A-interacting zinc finger protein 1
- DDR
DNA damage response
- DNA
Deoxyribonucleic acid
- DSBs
DNA double-strand-breaks
- DNA-
DNA-dependent protein kinase catalytic
- PKcs
subunit
- DNA-PK
DNA-dependent protein kinase
- Exo1
Exonuclease 1
- FUS
Fused in sarcoma
- HDAC1
Histone deacetylase 1
- H2AX
H2A histone family member X
- HR
Homologous recombination
- LIG4
DNA ligase IV
- MMR
Mismatch repair
- MMEJ
Microhomology-mediated end joining
- MRE11
Meiotic recombination11
- NBS1
Nijmegen breakage syndrome 1
- NER
Nucleotide excision repair
- NHEJ
Non-homologous end joining
- PD
Parkinson’s disease
- PIKK
Phosphatidylinositol-3 kinase-related kinases
- PARP1
Poly (ADP-ribose) polymerase 1
- SMC1A
Structural maintenance of chromosomes 1A
- SOD 1
Superoxide dismutase 1
- TDP-43
Transactivation response DNA-binding protein
- Topo Iiβ
Topoisomerase Iiβ
- XRCC4
X-ray cross-complementing protein 4
- XLF
XRCC4-like factor
- 53BP1
p53-binding protein 1
Footnotes
Conflict of interest The authors declare that they have no conflicts of interest.
Contributor Information
Nidheesh Thadathil, Department of Neurology, College of Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, USA.
Roderick Hori, Department of Microbiology, Immunology and Biochemistry, University of Tennessee Health Science Center, Memphis, TN, USA.
Jianfeng Xiao, Department of Neurology, College of Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, USA.
Mohammad Moshahid Khan, Department of Neurology, College of Medicine, University of Tennessee Health Science Center, Memphis, TN 38163, USA; Division of Rehabilitation Sciences and Department of Physical Therapy, College of Health Professions, University of Tennessee Health Science Center, Memphis, TN, USA; Department of Neurology, Neuroscience Institute, University of Tennessee Health Science Center, 855 Monroe Avenue, 432 Link Building, Memphis, TN 38163, USA.
References
- Alt FW, Schwer B (2018) DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair 71:158–163. 10.1016/j.dnarep.2018.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosio S, Di Palo G, Napolitano G et al. (2016) Cell cycle-dependent resolution of DNA double-strand breaks. Oncotarget 7:4949–4960. 10.18632/oncotarget.6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babić Leko M, Župunski V, Kirincich J et al. (2019) Molecular Mechanisms of neurodegeneration related to C9orf72 hexanucleotide repeat expansion. Behav Neurol 2019: 2909168. 10.1155/2019/2909168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai A, Biton S, Shiloh Y (2008) The role of the DNA damage response in neuronal development, organization and maintenance. DNA Repair 7:1010–1027. 10.1016/j.dnarep.2008.03.005 [DOI] [PubMed] [Google Scholar]
- Becker LA, Huang B, Bieri G et al. (2017) Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544:367–371. 10.1038/nature22038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bee L, Fabris S, Cherubini R et al. (2013) The efficiency of homologous recombination and non-homologous end joining systems in repairing double-strand breaks during cell cycle progression. PloS One 8:e69061. 10.1371/journal.pone.0069061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Szakal B (2016) DNA damage tolerance by recombination: molecular pathways and DNA structures. DNA Repair 44:68–75. 10.1016/j.dnarep.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier C, Langley B (2013) Chromatin modifications associated with DNA double-strand breaks repair as potential targets for neurological diseases. Neurotherapeutics 10:817–830. 10.1007/s13311-013-0210-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broustas CG, Lieberman HB (2014) DNA damage response genes and the development of cancer metastasis. Radiat Res 181: 111–130. 10.1667/RR13515.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M et al. (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276:42462–42467. 10.1074/jbc.C100466200 [DOI] [PubMed] [Google Scholar]
- Cadet J, Davies KJA (2017) Oxidative DNA damage & repair: an introduction. Free Radic Biol Med 107:2–12. 10.1016/j.freeradbiomed.2017.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camins A, Pizarro JG, Alvira D et al. (2010) Activation of ataxia telangiectasia muted under experimental models and human Parkinson’s disease. Cell Mol Life Sci CMLS 67:3865–3882. 10.1007/s00018-010-0408-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannan WJ, Pederson DS (2016) Mechanisms and consequences of double-strand DNA break formation in chromatin. J Cell Physiol 231:3–14. 10.1002/jcp.25048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casafont I, Palanca A, Lafarga V et al. (2011) Effect of ionizing radiation in sensory ganglion neurons: organization and dynamics of nuclear compartments of DNA damage/repair and their relationship with transcription and cell cycle. Acta Neuropathol (Berl) 122:481–493. 10.1007/s00401-011-0869-0 [DOI] [PubMed] [Google Scholar]
- Castillo WO, Aristizabal-Pachon AF, de Lima Montaldi AP et al. (2016) Galanthamine decreases genotoxicity and cell death induced by β-amyloid peptide in SH-SY5Y cell line. Neurotoxicology 57:291–297. 10.1016/j.neuro.2016.10.013 [DOI] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, D’Andrea AD (2016) Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 26:52–64. 10.1016/j.tcb.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR (2017) Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18:495–506. 10.1038/nrm.2017.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov V, Hansen LA, Shackelford DA (2003) Is DNA repair compromised in Alzheimer’s disease? Neurobiol Aging 24: 953–968 [DOI] [PubMed] [Google Scholar]
- Deng Q, Holler CJ, Taylor G et al. (2014) FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J Neurosci Off J Soc Neurosci 34:7802–7813. 10.1523/JNEUROSCI.0172-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbin MM, Madabhushi R, Pan L et al. (2013) SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci 16:1008–1015. 10.1038/nn.3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos Picanco LC, Ozela PF, de Fatima de Brito Brito M et al. (2018) Alzheimer’s disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr Med Chem 25:3141–3159. 10.2174/0929867323666161213101126 [DOI] [PubMed] [Google Scholar]
- Edwin Shackelford R, Manuszak RP, Heard SC et al. (2005) Pharmacological manipulation of ataxia-telangiectasia kinase activity as a treatment for Parkinson’s disease. Med Hypotheses 64:736–741. 10.1016/j.mehy.2004.08.029 [DOI] [PubMed] [Google Scholar]
- Eilam R, Peter Y, Elson A et al. (1998) Selective loss of dopaminergic nigro-striatal neurons in brains of Atm-deficient mice. Proc Natl Acad Sci U S A 95:12653–12656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilam R, Peter Y, Groner Y, Segal M (2003) Late degeneration of nigro-striatal neurons in ATM−/− mice. Neuroscience 121:83–98 [DOI] [PubMed] [Google Scholar]
- El-Khamisy SF, Saifi GM, Weinfeld M et al. (2005) Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 434:108. [DOI] [PubMed] [Google Scholar]
- Fang MY, Markmiller S, Vu AQ et al. (2019) Small-Molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 103:802–819.e11. 10.1016/j.neuron.2019.05.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farg MA, Konopka A, Soo KY et al. (2017) The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet 26:2882–2896. 10.1093/hmg/ddx170 [DOI] [PubMed] [Google Scholar]
- Federico A, Cardaioli E, Da Pozzo P et al. (2012) Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci 322: 254–262 [DOI] [PubMed] [Google Scholar]
- Ferrer I, Serrano T, Alcantara S et al. (1993) X-ray-induced cell death in the developing hippocampal complex involves neurons and requires protein synthesis. J Neuropathol Exp Neurol 52:370–378. 10.1097/00005072-199307000-00004 [DOI] [PubMed] [Google Scholar]
- Freibaum BD, Chitta RK, High AA, Taylor JP (2010) Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 9:1104–1120. 10.1021/pr901076y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Phelps SEL, Gray S, Neale MJ (2011) Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479:241–244. 10.1038/nature10515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Chew J, Stankowski JN et al. (2017) Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med 9. 10.1126/scitranslmed.aai7866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman AM (2008) Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J Cell Mol Med 12:2263–2280. 10.1111/j.1582-4934.2008.00402.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero EN, Mitra J, Wang H et al. (2019) Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum Mol Genet. 10.1093/hmg/ddz062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner MC, De Marzo AM, Meeker AK et al. (2011) Transcription-induced DNA double strand breaks: both oncogenic force and potential therapeutic target? Clin Cancer Res Off J Am Assoc Cancer Res 17:3858–3864. 10.1158/1078-0432.CCR-10-2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Gupta VB, Anitha M et al. (2006) Studies on genomic DNA topology and stability in brain regions of Parkinson’s disease. Arch Biochem Biophys 449:143–156. 10.1016/j.abb.2006.02.018 [DOI] [PubMed] [Google Scholar]
- Herrup K, Li J, Chen J (2013) The role of ATM and DNA damage in neurons: upstream and downstream connections. DNA Repair 12:600–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Song H, Croteau DL et al. (2017) Genome instability in Alzheimer disease. Mech Ageing Dev 161:83–94. 10.1016/j.mad.2016.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Wuerzberger-Davis SM, Seufzer BJ et al. (2000) NF-kappaB activation by camptothecin. A linkage between nuclear DNA damage and cytoplasmic signaling events. J Biol Chem 275:9501–9509 [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461:1071–1078. 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen E, Beach T, Shen Y et al. (2004) Deficiency of the Mre11 DNA repair complex in Alzheimer’s disease brains. Brain Res Mol Brain Res 128:1–7. 10.1016/j.molbrainres.2004.05.023 [DOI] [PubMed] [Google Scholar]
- Janssens S, Tschopp J (2006) Signals from within: the DNA-damage-induced NF-kappaB response. Cell Death Differ 13:773–784. 10.1038/sj.cdd.4401843 [DOI] [PubMed] [Google Scholar]
- Jette N, Lees-Miller SP (2015) The DNA-dependent protein kinase: a multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol 117:194–205. 10.1016/j.pbiomolbio.2014.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju B-G, Lunyak VV, Perissi V et al. (2006) A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 312:1798–1802. 10.1126/science.1127196 [DOI] [PubMed] [Google Scholar]
- Kannan A, Bhatia K, Branzei D, Gangwani L (2018) Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res 46:8326–8346. 10.1093/nar/gky641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanungo J (2013) DNA-dependent protein kinase and DNA repair: relevance to Alzheimer’s disease. Alzheimers Res Ther 5:13. 10.1186/alzrt167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MM, Xiao J, Patel D, LeDoux MS (2018) DNA damage and neurodegenerative phenotypes in aged Ciz1 null mice. Neurobiol Aging 62:180–190. 10.1016/j.neurobiolaging.2017.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebish MA, Narain NR (2019) Enabling biomarker discovery in Parkinson’s disease using multiomics: challenges, promise and the future. Pers Med 16:5–7. 10.2217/pme-2018-0115 [DOI] [PubMed] [Google Scholar]
- Kirshner M, Galron R, Frenkel D et al. (2012) Malfunctioning DNA damage response (DDR) leads to the degeneration of nigro-striatal pathway in mouse brain. J Mol Neurosci MN 46:554–568. 10.1007/s12031-011-9643-y [DOI] [PubMed] [Google Scholar]
- Konopka A, Atkin JD (2018) The emerging role of dna damage in the pathogenesis of the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Int J Mol Sci 19. 10.3390/ijms19103137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak M, Crouch EE, Orlov M et al. (2007) The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 447:730–734. 10.1038/nature05842 [DOI] [PubMed] [Google Scholar]
- Kruman II, Wersto RP, Cardozo-Pelaez F et al. (2004) Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41:549–561 [DOI] [PubMed] [Google Scholar]
- Lamarche BJ, Orazio NI, Weitzman MD (2010) The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett 584:3682–3695. 10.1016/j.febslet.2010.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-H, Siddoway B, Kaeser GE et al. (2018) Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 563:639–645. 10.1038/s41586-018-0718-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Banin S, Ouyang H et al. (2001) ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. J Biol Chem 276:8898–8903. 10.1074/jbc.M009809200 [DOI] [PubMed] [Google Scholar]
- Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79:181–211. 10.1146/annurev.biochem.052308.093131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gonzalez R, Lu Y, Gendron TF et al. (2016) Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA Damage in iPSC-derived motor neurons. Neuron 92:383–391. 10.1016/j.neuron.2016.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gonzalez R, Yang D, Pribadi M et al. (2019) Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD. Proc Natl Acad Sci U S A 116:9628–9633. 10.1073/pnas.1901313116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, Tsai L-H (2014) DNA damage and its links to neurodegeneration. Neuron 83:266–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR et al. (2015) Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161:1592–1605. 10.1016/j.cell.2015.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal A, Zou L (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5. 10.1101/cshperspect.a012716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey TH, Jones L (2018) The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech 11:dmm031930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola AS, Kim SH, Trinh AT et al. (2013) The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem 288:24731–24741. 10.1074/jbc.M113.497974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata-Garrido J, Tapia O, Casafont I et al. (2018) Persistent accumulation of unrepaired DNA damage in rat cortical neurons: nuclear organization and ChIP-seq analysis of damaged DNA. Acta Neuropathol Commun 6:68. 10.1186/s40478-018-0573-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool KW, Miyamoto S (2012) DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol Rev 246:311–326. 10.1111/j.1600-065X.2012.01101.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk L, Gomes E, Guo L et al. (2018) Poly(ADP-Ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol Cell 71:703–717.e9. 10.1016/j.molcel.2018.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ (2009) DNA repair deficiency and neurological disease. Nat Rev Neurosci 10:100–112. 10.1038/nrn2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ, Caldecott KW (2007) DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet 8:37–55. 10.1146/annurev.genom.7.080505.115648 [DOI] [PubMed] [Google Scholar]
- Merlo D, Mollinari C, Racaniello M et al. (2016) DNA double strand breaks: a common theme in neurodegenerative diseases. Curr Alzheimer Res 13:1208–1218 [DOI] [PubMed] [Google Scholar]
- Migliore L, Scarpato R, Coppede F et al. (2001) Chromosome and oxidative damage biomarkers in lymphocytes of Parkinson’s disease patients. Int J Hyg Environ Health 204:61–66 [DOI] [PubMed] [Google Scholar]
- Milanese C, Cerri S, Ulusoy A et al. (2018) Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis 9:818. 10.1038/s41419-018-0848-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra J, Guerrero EN, Hegde PM et al. (2019) Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A. 10.1073/pnas.1818415116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung N-H, Zhu X, Kruman II et al. (2008) Evidence of DNA damage in Alzheimer disease: phosphorylation of histone H2AX in astrocytes. Age Dordr Neth 30:209–215. 10.1007/s11357-008-9050-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi A, Minami A, Kitagishi Y et al. (2015) BRCA1 and p53 tumor suppressor molecules in Alzheimer’s disease. Int J Mol Sci 16:2879–2892. 10.3390/ijms16022879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann M, Pal A, Goswami A et al. (2018) Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun 9:335. 10.1038/s41467-017-02299- [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi T, Blackford AN, Coates J et al. (2015) PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 347:185–188. 10.1126/science.1261971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino M, Ichimura M, Nakano N et al. (2016) Roles of PTEN with DNA repair in Parkinson’s disease. Int J Mol Sci 17. 10.3390/ijms17060954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orii KE, Lee Y, Kondo N, McKinnon PJ (2006) Selective utilization of nonhomologous end-joining and homologous recombination DNA repair pathways during nervous system development. Proc Natl Acad Sci U S A 103:10017–10022. 10.1073/pnas.0602436103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Penney J, Tsai L-H (2014) Chromatin regulation of DNA damage repair and genome integrity in the central nervous system. J Mol Biol 426:3376–3388. 10.1016/j.jmb.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret B, Schoonbroodt S, Piette J (1999) The ATM protein is required for sustained activation of NF-kappaB following DNA damage. Oncogene 18:2261–2271. 10.1038/sj.onc.1202541 [DOI] [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y et al. (2014) ALS-associated mutation FUS R521C causes DNA damage and RNA splicing defects. J Clin Invest 124:981–999. 10.1172/JCI72723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjha L, Howard SM, Cejka P (2018) Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma 127:187–214. 10.1007/s00412-017-0658-1 [DOI] [PubMed] [Google Scholar]
- Rulten SL, Caldecott KW (2013) DNA strand break repair and neurodegeneration. DNA Repair 12:558–567 [DOI] [PubMed] [Google Scholar]
- Sallmyr A, Tomkinson AE (2018) Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J Biol Chem 293:10536–10546. 10.1074/jbc.TM117.000375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scacchi R, Gambina G, Moretto G, Corbo RM (2013) P21 gene variation and late-onset Alzheimer’s disease in the Italian population. Dement Geriatr Cogn Disord 35:51–57. 10.1159/000345788 [DOI] [PubMed] [Google Scholar]
- Schwer B, Wei P-C, Chang AN et al. (2016) Transcription-associated processes cause DNA double-strand breaks and translocations in neural stem/progenitor cells. Proc Natl Acad Sci U S A 113:2258–2263. 10.1073/pnas.1525564113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwertman P, Bekker-Jensen S, Mailand N (2016) Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat Rev Mol Cell Biol 17:379–394. 10.1038/nrm.2016.58 [DOI] [PubMed] [Google Scholar]
- Seol J-H, Shim EY, Lee SE (2018) Microhomology-mediated end joining: good, bad and ugly. Mutat Res Mol Mech Mutagen 809:81–87. 10.1016/j.mrfmmm.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepe S, Milanese C, Gabriels S et al. (2016) Inefficient DNA repair is an aging-related modifier of Parkinson’s disease. Cell Rep 15:1866–1875. 10.1016/j.celrep.2016.04.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford RE, Fu Y, Manuszak RP et al. (2006) Iron chelators reduce chromosomal breaks in ataxia-telangiectasia cells. DNA Repair 5:1327–1336. 10.1016/j.dnarep.2006.05.041 [DOI] [PubMed] [Google Scholar]
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C et al. (2010) ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141:970–981. 10.1016/j.cell.2010.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag NM, Evans MD, Mao W et al. (2019) Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol Commun. 7:77. 10.1186/s40478-019-0723-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Chen J, Li J et al. (2016) Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro:3. 10.1523/ENEURO.0124-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WS (1998) Progressive neuronal DNA damage associated with neurofibrillary tangle formation in Alzheimer disease. J Neuropathol Exp Neurol 57: 323–328 [DOI] [PubMed] [Google Scholar]
- Shibata A, Conrad S, Birraux J et al. (2011) Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 30:1079–1092. 10.1038/emboj.2011.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS et al. (2014) DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53:7–18. 10.1016/j.molcel.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Petrini JHJ (2011) The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 12:90–103. 10.1038/nrm3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su JH, Deng G, Cotman CW (1997) Neuronal DNA damage precedes tangle formation and is associated with upregulation of nitrotyrosine in Alzheimer’s disease brain. Brain Res 774:193–199 [DOI] [PubMed] [Google Scholar]
- Su Y, Ming G, Song H (2015) DNA damage and repair regulate neuronal gene expression. Cell Res 25:993–994. 10.1038/cr.2015.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E, Sanchez PE, Kravitz AV et al. (2013) Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat Neurosci 16:613–621. 10.1038/nn.3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E, Djukic B, Evans M et al. (2015) DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat Commun 6:8897. 10.1038/ncomms9897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilstra JS, Robinson AR, Wang J et al. (2012) NF-κB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest 122:2601–2612. 10.1172/JCI45785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuxworth RI, Taylor MJ, Martin Anduaga A et al. (2019) Attenuating the DNA damage response to double-strand breaks restores function in models of CNS neurodegeneration. Brain Commun 1. 10.1093/braincomms/fcz005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Rhodes CJ, Moncol J et al. (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 160:1–40. 10.1016/j.cbi.2005.12.009 [DOI] [PubMed] [Google Scholar]
- van Gent DC, Hoeijmakers JH, Kanaar R (2001) Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2:196–206. 10.1038/35056049 [DOI] [PubMed] [Google Scholar]
- Walker C, Herranz-Martin S, Karyka E et al. (2017) C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat Neurosci 20:1225–1235. 10.1038/nn.4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hegde ML (2019) New mechanisms of DNA repair defects in fused in sarcoma-associated neurodegeneration: stage set for dna repair-based therapeutics? J Exp Neurosci 13:1179069519856358. 10.1177/1179069519856358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Yu T, Liu Y et al. (2016) DNA damage preceding dopamine neuron degeneration in A53T human α-synuclein transgenic mice. Biochem Biophys Res Commun 481:104–110. 10.1016/j.bbrc.2016.11.008 [DOI] [PubMed] [Google Scholar]
- Wang H, Guo W, Mitra J et al. (2018) Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat Commun 9:3683. 10.1038/s41467-018-06111-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RR, Vijg J (2016) Do DNA double-strand breaks drive aging? Mol Cell 63:729–738. 10.1016/j.molcel.2016.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WD, Shah SS, Heyer W-D (2018) Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem 293:10524–10535. 10.1074/jbc.TM118.000372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Vemula SR, Xue Y et al. (2016) Motor phenotypes and molecular networks associated with germline deficiency of Ciz1. Exp Neurol 283:110–120. 10.1016/j.expneurol.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci Off J Soc Neurosci 21:2661–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ES, Nowsheen S, Wang T et al. (2011) Glycogen synthase kinase 3beta inhibition enhances repair of DNA double-strand breaks in irradiated hippocampal neurons. Neuro-Oncol 13:459–470. 10.1093/neuonc/nor016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates SC, Zafar A, Rabai EM et al. (2015) The effects of two polymorphisms on p21cip1 function and their association with Alzheimer’s disease in a population of European descent. PloS One 10:e0114050. 10.1371/journal.pone.0114050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Harrison FE, Xia F (2018) Altered DNA repair; an early pathogenic pathway in Alzheimer’s disease and obesity. Sci Rep 8:5600. 10.1038/s41598-018-23644-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurov YB, Vorsanova SG, Iourov IY (2011) The DNA replication stress hypothesis of Alzheimer’s disease. ScientificWorldJournal 11:2602–2612. 10.1100/2011/625690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR et al. (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. 10.1038/nature14973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408:433–439. 10.1038/35044005 [DOI] [PubMed] [Google Scholar]