

Summary
Background
Isocitrate dehydrogenase-1 (IDH1) is mutated in up to 25% of cholangiocarcinomas, especially intrahepatic cholangiocarcinoma. Ivosidenib is an oral, targeted inhibitor of mutant IDH1 (mIDH1) approved in the USA for the treatment of mIDH1 acute myeloid leukaemia in newly diagnosed patients ineligible for intensive chemotherapy and patients with relapsed or refractory disease. Ivosidenib is under clinical evaluation in a phase 1 study that aims to assess its safety and tolerability in patients with mIDH1 solid tumours. Here we report data for the mIDH1-cholangiocarcinoma cohort.
Methods
We did a phase 1 dose-escalation and expansion study of ivosidenib monotherapy in mIDH1 solid tumours at 12 clinical sites in the USA and one in France. The primary outcomes were safety, tolerability, maximum tolerated dose, and recommended phase 2 dose. Eligible patients had a documented mIDH1 tumour based on local testing, an Eastern Cooperative Oncology Group performance status of 0 or 1, one or more previous lines of therapy, and evaluable disease by Response Evaluation Criteria in Solid Tumors version 1.1. During dose escalation, ivosidenib was administered orally at 200–1200 mg daily in 28-day cycles in a standard 3 + 3 design; during expansion, patients received the selected dose on the basis of pharmacodynamic, pharmacokinetic, safety, and activity data from dose escalation. Safety and clinical activity analyses were reported for all patients with mIDH1-cholangiocarcinoma who were enrolled and received at least one dose of study treatment. Enrolment is complete, and the study is ongoing. This trial is registered at ClinicalTrials.gov, number NCT02073994.
Findings
Between March 14, 2014 and May 12, 2017, 73 patients with mIDH1-cholangiocarcinoma were enrolled and received ivosidenib. No dose-limiting toxicities were reported and maximum tolerated dose was not reached; 500 mg daily was selected for expansion. Common (≥20%) adverse events, regardless of cause, were fatigue (31 [42%]; two [3%] grade ≥3), nausea (25 [34%]; one [1%] grade ≥3), diarrhoea (23 [32%]), abdominal pain (20 [27%]; two [3%] grade ≥3), decreased appetite (20 [27%]; one [1%] grade ≥3), and vomiting (17 [23%]). Common grade 3 or worse adverse events were ascites (four [5%]) and anaemia (three [4%]); the only treatment-related grade 3 or worse adverse event in more than one patient was fatigue (two [3%]). Two (3%) patients had serious adverse events leading to on-treatment death (Clostridioides difficile infection and procedural haemorrhage); neither was assessed by the investigator as related to treatment. 46 (63%) patients had adverse events deemed related to ivosidenib, of which four (5%) were grade 3 or higher (two [3%] for fatigue; one [1%] each for decreased blood phosphorus and increased blood alkaline phosphatase). One serious adverse event was considered possibly related to treatment (grade 2 supraventricular extrasystoles). Four (5%; 95% CI 1·5-13·4) patients had a partial response. Median progression-free survival was 3·8 months (95% CI 3·6-7·3), 6-month progression-free survival was 40·1% (28·4-51·6), and 12-month progression-free survival was 21·8% (12·3-33·0). Median overall survival was 13·8 months (95% CI 11·1-29·3); however, data were censored for 48 patients (66%).
Interpretation
Ivosidenib might offer a well tolerated option for patients with mIDH1-cholangiocarcinoma. An ongoing, global phase 3 study is evaluating ivosidenib versus placebo in patients with previously treated nonresectable or metastatic mIDH1-cholangiocarcinoma.
Funding
Agios Pharmaceuticals, Inc.
Introduction
Cholangiocarcinoma is a rare, genetically diverse, and aggressive malignancy arising from the intrahepatic, perihilar, or extrahepatic biliary epithelium.1,2 Standard-of-care treatment for patients with inoperable disease is a chemotherapy regimen of gemcitabine and cisplatin.3 No standard second-line therapies exist. With a median survival of less than 24 months in advanced cases and 5-year survival of 5–10%,1,4 there is an unmet need for effective treatments for patients with cholangiocarcinoma.
Gain-of-function mutations within the isocitrate dehydrogenase (IDH)-1 enzyme are among the most common driver genetic alterations in cholangiocarcinoma, particularly in intrahepatic cholangiocarcinoma, where they have been reported to occur in up to 25% of patients.5–7 These mutations result in the excessive production of the oncometabolite D-2-hydroxyglutarate (2-HG),8 reduction of the endogenous intermediary metabolite α-ketoglutarate,9 and consequent stimulation of multiple oncogenic processes, including aberrant metabolism and widespread epigenetic dysregulation.8,10–12 Thus, mutant IDH1 (mIDH1) represents a therapeutic target in cholangiocarcinoma. Preclinical work showed that treatment of in-vitro IDH1-mutant mouse hepatoblasts with a mIDH1 inhibitor resulted in reduction of 2-HG production and restoration of cellular differentiation, providing a rationale for the clinical use of mIDH1 inhibitors.13
Ivosidenib (Agios Pharmaceuticals, Inc, Cambridge, MA, USA) is an oral, potent inhibitor of mIDH1, approved in the USA for the treatment of acute myeloid leukaemia with a susceptible IDH1 mutation, as detected by a US Food & Drug Administration-approved test, in newly diagnosed adults aged 75 years or older or who have comorbidities that preclude use of intensive induction chemotherapy and adults with relapsed or refractory disease.14 In this Article, we report clinical and translational data from the cohort of patients with mIDH1-cholangiocarcinoma enrolled in an ongoing, phase 1 trial of ivosidenib that aimed to assess its safety and activity in patients with advanced mIDH1 solid tumours.
Methods
Study design and participants
We did a phase 1, multicentre, open-label, dose-escalation and dose-expansion study of ivosidenib monotherapy in adults with mIDH1 solid tumours across 12 teaching hospitals and cancer institutes in the USA and one in France (appendix p 14). This Article focuses solely on the patients with mIDH1-cholangiocarcinoma who were treated with ivosidenib in this study (other cohorts will be reported elsewhere).
Patients aged 18 years or older with an Eastern Cooperative Oncology Group performance status of 0 or 1 and an advanced solid tumour that had recurred or progressed following standard therapy with a documented IDH1 mutation by local testing were eligible for escalation. For expansion, patients with cholangiocarcinoma were required to have histologically confirmed, unresectable stage II-IV (intrahepatic, extrahepatic, or perihilar) radiographically measurable disease by Response Evaluation Criteria in Solid Tumors version 1.1, and progression following a gemcitabine-based regimen. Additional eligibility criteria were adequate bone marrow function (absolute neutrophil count ≥1·5 × 109 cells per L, haemoglobin ≥9 g/dL, and platelets ≥75 × 109 per L), adequate hepatic function (total bilirubin ≤1·5 times the upper limit of normal, except for patients with Gilbert’s syndrome, and aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase ≤2·5 times the upper limit of normal), adequate renal function (serum creatinine ≤2 times the upper limit of normal and creatinine clearance >40 mL/min), recovery from the toxic manifestations of previous treatments, and a minimum expected survival of at least 3 months. Patients were excluded if they had a heart rate-corrected QT interval of more than 450 ms, were pregnant or breastfeeding, or had an active severe infection, known hypersensitivity to any component of ivosidenib, a history of cardiovascular disease, a history of (or risk factors for) prolonged QT interval syndrome, known infection with HIV or active hepatitis B or C, or known conditions that reduce the ingestion or absorption of orally administered drugs. Exclusion criteria also included systemic anticancer therapy or radiotherapy within 3 weeks of study start, receipt of an investigational agent within 2 weeks of study start, and use of certain CYP3A4 or P-glycoprotein transporter-sensitive substrate medications.
The study was done in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. The protocol was approved by the appropriate review board at each site. All patients provided written informed consent before undergoing baseline screening evaluations, which occurred within 28 days before day 1 of the study. Data were collected at participating study centres (appendix p 2).
Procedures
For dose escalation, a standard design of three to six patients per dose was used and was to continue until two or more patients had dose-limiting toxicities. The starting dose of ivosidenib was 100 mg twice daily, following which dosing proceeded once daily, on the basis of the favourable pharmacokinetic profile, at 300 mg, 400 mg, 500 mg, 800 mg, and 1200 mg once daily. Dose-limiting toxicities were evaluated during cycle 1 of the dose-escalation phase and defined as any grade 3 or higher event reported to be related or possibly related to ivosidenib (appendix p 3). In expansion, treatment was continued until disease progression, unacceptable toxicity, confirmed pregnancy, death, withdrawal of consent, or loss to follow-up. Daily dosing began on day 1 and continued in consecutive 28-day cycles. Ivosidenib dose reduction to a dose approved by the medical monitor or interruption of dosing was permitted in the event of grade 1 or 2 toxicities that were assessed as possibly or probably related to treatment, following discussion with the medical monitor. In the escalation part of the study, the dose could be reduced in multiples of 50 mg, and the dose could be reduced to 250 mg in the expansion part. Reduction back to the starting dose or an intermediate dose was permitted with medical monitor approval. Patients who had toxicity of grade 3 or higher that was assessed as possibly or probably related to treatment who were in the opinion of the investigator benefiting from treatment could continue treatment with medical monitor approval. If the time required for recovery from toxicity (ie, a return to at least baseline levels) was more than 28 days, the risks and benefits ofthe patient’s continuation in the study was discussed with the medical monitor.
A single dose of ivosidenib was administered orally three days before starting daily dosing (day −3), and was followed by 3 days of pharmacokinetic sampling. Details of pharmacokinetic and pharmacodynamic assessments, including plasma and tumour 2-HG evaluation at baseline and after treatment, are provided in the appendix (p 4).
All patients had CT or MRI evaluations to obtain tumour measurements at screening and approximately every 56 days (± 3 days) thereafter while on treatment, independent of dose delays or dose interruptions, or at any time when progression of disease was suspected.
Exploratory assessments on archived and fresh-frozen tumour samples included confirmation of baseline mIDH1 status and identification of co-occurring mutations by next-generation sequencing and Ki-67 proliferation marker by immunohistochemistry (appendix p 4–5).
Outcomes
Primary outcomes were safety, tolerability, maximum tolerated dose of ivosidenib, and recommended dose for further phase 2 evaluation. Key secondary outcomes were dose-limiting toxicity, pharmacokinetic and pharmacodynamic effects, and clinical activity.
Toxicity was evaluated by assessing adverse events. All toxicities were graded and documented according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. ECG QT prolongation is an identified risk associated with ivosidenib15 and is considered an adverse event of special interest; thus, it was closely monitored and managed with electrolyte repletion and ivosidenib dose modification as needed.
Tumour responses were evaluated with serial CT or MRI using Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, and classified as complete response, partial response, stable disease, or progressive disease. Endpoints of clinical activity included objective response (proportion of patients with a complete response or partial response), progression-free survival, defined as time from first dose to disease progression or death, and time to response, defined as time from first dose to the first documentation of response (further details are provided in the appendix p 4). Overall survival (time from first does to death due to any cause) was added as a secondary endpoint as part of a protocol amendment (July 27, 2016). Per protocol, participants who had disease progression by RECIST assessments who were, in the opinion of the investigator, benefiting from treatment could continue the study drug with approval of the medical monitor.
Statistical analysis
On the basis of the planned dose escalation scheme, it was estimated that approximately 170 patients would be enrolled in the study overall (approximately 45 in the dose escalation phase and 125 in the expansion phase). On the basis of 50 patients in the cholangiocarcinoma expansion cohort, the chance of observing at least one adverse event would be 99·5%, with a true underlying event rate of 10%, and 92·3% with a true underlying event rate of 5%. On the basis of 25 patients in the expansion cohort, the chance of observing at least one adverse event would be 92·8%, with a true underlying event rate of 10%, and 72·3%, with a true underlying event rate of 5%. Additionally, for the secondary endpoint of preliminary anti-tumour activity, on the basis of about 50 patients in the cholangiocarcinoma expansion cohort and an exact binomial distribution, the maximum width of the 95% CI around the proportion of patients achieving an objective response would be 0·289.
Safety data are reported for the safety analysis set, comprising all patients with cholangiocarcinoma who were enrolled and received at least one dose of ivosidenib in the dose-escalation and dose-expansion cohorts, classified according to the actual treatment received. All other analyses are reported for the full analysis set, comprising all patients who were enrolled and received at least one dose of study treatment, classified according to the assigned dose. Descriptive statistics are reported for safety outcomes and other clinical, pharmacokinetic, and progressive disease parameters. Time-to-event endpoints (progression-free survival and overall survival) were estimated using Kaplan-Meier methods, and the median with associated 95% CI produced.
Statistical analyses were done with SAS software version 9.3 or higher. This study is registered with ClinicalTrials.gov, number NCT02073994.
Role of the funding source
This study was designed by the sponsor in close collaboration with the investigators. Data were collected by investigators and their research staff and analysed by the sponsor. Statistical analyses were done by a contract research organisation, overseen by qualified statisticians employed by the sponsor. All authors had access to the raw data on request. The paper was drafted by the first and last authors in collaboration with the study sponsor and was revised in collaboration with all authors. The corresponding author had full access to the data in the study and had final responsibility for the decision to submit for publication.
Results
This study was started on March 14, 2014, and included 168 patients with a variety of mIDH1 solid tumours (73 [43%] cholangiocarcinoma, 66 [39%] glioma, 21 [13%] chondrosarcoma, and eight [5%] other). At the analysis cutoff date (May 12, 2017), enrolment was complete, and the study was ongoing.
Of the 73 patients with mIDH1-cholangiocarcinoma enrolled, 24 (33%) received ivosidenib in dose escalation and 49 (67%) in dose expansion. The majority had intrahepatic cholangiocarcinoma (65 [89%]) and had received a median of two (range 1–5) previous lines of therapy (table 1). In dose expansion, ivosidenib was administered at doses of 200–1200 mg daily. There were no dose-limiting toxicities to establish the maximum tolerated dose. The dose of 500 mg once daily was selected for dose expansion (appendix p 14) on the basis of pharmacodynamic (2-HG inhibition), pharmacokinetic, safety, and activity data from the dose escalation phase. Combined data from the dose escalation and expansion portions confirmed that the dose of 500 mg once daily ivosidenib was appropriate.
Table 1:
Baseline characteristics
| Patients with cholangiocarcinoma (N=73) | |
|---|---|
| Sex | |
| Female | 49 (67%) |
| Male | 24 (33%) |
| Age, years | 60 (32–81) |
| ECOG performance status | |
| 0 | 24 (33%) |
| 1 | 48 (66%) |
| 2 | 1 (1%) |
| Cholangiocarcinoma subtype | |
| Intrahepatic | 65 (89%) |
| Extrahepatic | 8 (11%) |
| TNM stage at diagnosis | |
| Grade I | 0 |
| Grade II | 16 (22%) |
| Grade III | 4 (5%) |
| Grade IV | 45 (62%) |
| Unknown | 8 (11%) |
| Previous systemic therapies | 2 (1–5) |
| Gemcitabine-based | 72 (99%) |
| Fluorouracil-based | 37 (51%) |
| Duration of last line of previous systemic therapy, months | 3·2 (0·2–26·2) |
| Mutant IDH1 allele | |
| R132C* | 56 (77%) |
| R132L† | 8 (11%) |
| R132G | 5 (7%) |
| R132H | 2 (3%) |
| R132S | 2 (3%) |
Data are n, n (%), or median (range). ECOG=Eastern Cooperative Oncology Group.
Seven of eight patients with extrahepatic cholangiocarcinoma had an R132C allele.
One of eight patients with extrahepatic cholangiocarcinoma had an R132L allele.
Six (8%) patients received less than 500 mg once daily (two 100 mg twice daily, three 300 mg once daily, and one 400 mg once daily), 62 (85%) received 500 mg once daily (13 in escalation and 49 in expansion), and five (7%) received more than 500 mg once daily (two 800 mg once daily and three 1200 mg once daily). Five (7%) patients required dose reductions.
At the analysis cutoff date, 12 (16%) patients remained on treatment (appendix p 15). Overall median treatment duration was 3·7 months (range 0·6–23·4). Reasons for on-study treatment discontinuation were radiographic (50 [68%]) or clinical progression of disease (seven [10%]), withdrawal of consent (two [3%]), adverse event (one [1%]), and death (one [1%]).
In patients with mIDH1-cholangiocarcinoma, following a single oral dose on day −3, ivosidenib was rapidly absorbed, with a median half-life of 56 h (range 26–112). Steady state was achieved within 14 days of dosing. Plasma exposure increased less than proportionally with increasing doses (300–1200 mg once daily).
Maximal 2-HG inhibition in plasma (up to 98·4%) relative to baseline was observed during cycle 1 in all patients treated with 500 mg once daily, with no additional inhibition observed beyond 28 days or at higher doses (800 or 1200 mg once daily). After multiple doses of ivosidenib, plasma 2-HG concentrations were substantially reduced at all doses tested. At 500 mg once daily, mean plasma 2-HG concentrations decreased by 88% (median, IQR 71–92) to concentrations seen in healthy volunteers, with no additional plasma 2-HG reduction observed at doses greater than 500 mg once daily, further supporting selection of the 500 mg once daily regimen.
In dose escalation, ivosidenib was administered at doses up to 1200 mg once daily. All 73 (100%) patients with cholangiocarcinoma across both study phases had an adverse event. ECG QT prolongation was reported in eight patients (11%; grade 3 in one, grade 1 or 2 in the remainder; table 2; appendix pp 6–7); all events were non-serious and managed with appropriate guidance. The most common grade 3 or higher adverse events, irrespective of cause, were ascites (four [5%]) and anaemia (three [4%]; table 2; appendix pp 6–7). 20 (27%) patients had serious adverse events. Two patients (3%) had serious adverse events leading to on-treatment deaths (Clostridioides difficile infection, procedural haemorrhage); neither were assessed by the investigator as related to study drug (appendix p 8). 46 (63%) patients had adverse events deemed related to ivosidenib (appendix p 9). Of these, four (5%) were grade 3 or higher: fatigue (two [3%]) and one (1%) each for decreased blood phosphorus and increased blood alkaline phosphatase. One serious adverse event that was deemed possibly related to treatment (grade 2 supraventricular extrasystoles) occurred.
Table 2:
Treatment-emergent adverse events occurring in more than 10%* of patients with cholangiocarcinoma
| 500 mg once daily (N=62) |
Overall (N=73) |
|||||
|---|---|---|---|---|---|---|
| Grade 1 or 2 | Grade 3 | Grade 4 | Grade 1 or 2 | Grade 3 | Grade 4 | |
| Fatigue | 28 (45%) | 1 (2%) | 0 | 29 (40%) | 2 (3%) | 0 |
| Nausea | 21 (34%) | 1 (2%) | 0 | 24 (33%) | 1 (1%) | 0 |
| Diarrhoea | 19 (31%) | 0 | 0 | 23 (32%) | 0 | 0 |
| Abdominal pain | 16 (26%) | 2 (3%) | 0 | 18 (25%) | 2 (3%) | 0 |
| Decreased appetite | 19 (31%) | 0 | 0 | 19 (26%) | 1 (1%) | 0 |
| Vomiting | 15 (24%) | 0 | 0 | 17 (23%) | 0 | 0 |
| Ascites | 7 (11%) | 3 (5%) | 0 | 9 (12%) | 4 (5%) | 0 |
| Peripheral oedema | 11 (18%) | 0 | 0 | 13 (18%) | 0 | 0 |
| Pyrexia | 11 (18%) | 0 | 0 | 12 (16%) | 0 | 0 |
| Cough | 10 (16%) | 0 | 0 | 10 (14%) | 1 (1%) | 0 |
| Abdominal distension | 6 (10%) | 2 (3%) | 0 | 8 (11%) | 2 (3%) | 0 |
| Back pain | 10 (16%) | 0 | 0 | 10 (14%) | 0 | 0 |
| Musculoskeletal pain | 9 (15%) | 0 | 0 | 10 (14%) | 0 | 0 |
| Anaemia | 5 (8%) | 2 (3%) | 0 | 6 (8%) | 3 (4%) | 0 |
| Abdominal pain upper | 6 (10%) | 0 | 0 | 8 (11%) | 0 | 0 |
| ECG QT prolonged | 7 (11%) | 1 (2%) | 0 | 7 (10%) | 1 (1%) | 0 |
| Hypokalaemia | 6 (10%) | 0 | 1 (2%) | 7 (10%) | 0 | 1 (1%) |
Data are n (%).
Based on the overall population of 73 patients.
17 patients (23%) had adverse events leading to ivosidenib being withheld. Three (4%) patients required dose reductions for adverse events. One (1%) patient discontinued treatment owing to cystitis and hyponatraemia deemed unrelated to ivosidenib. Further information on adverse events is provided in the appendix (pp 6–9).
All-cause mortality within 30 days of first dose was 1% (one of 73 patients) and within 60 days was 4% (three of 73 patients). No deaths on study were deemed treatment-related.
Four (5%, 95% CI 1·5–13·4) patients achieved an objective response, with four partial responses (all at ≤500 mg once daily; figure 1A and B; appendix pp 10–11). 41 (56%) patients had stable disease. Median progression-free survival was 3·8 months (95% CI 3·6–7·3), 6-month progression-free survival was 40·1% (28·4–51·6), and 12-month progression-free survival was 21·8% (12·3–33·0; figure 1C; appendix p 12). Median overall survival was 13·8 months (95% CI 11·1-29·3); however, data were censored for 48 patients (66%). Time to response data are in the appendix (p 10).
Figure 1: Duration on treatment, best percentage change in target lesion size, and progression-free survival.
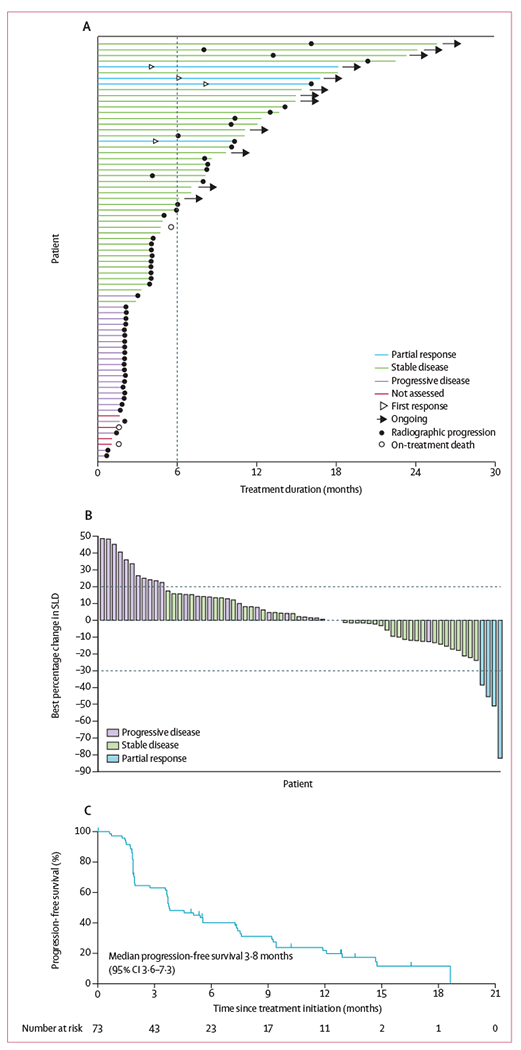
(A) Swim plot of duration on ivosidenib treatment. The bar lengths represent treatment duration as of May 12, 2017, for each patient. The vertical dashed line shows the 6-month time point. (B) Best percentage change from baseline in the sum of target lesion diameter for the 68 patients for whom post-baseline changes in target lesions were calculable. The dashed line at −30% denotes the minimum change necessary for partial response, and the dashed line at 20% denotes the minimum change necessary for progressive disease, according to Response Evaluation Criteria in Solid Tumors v1.1. (C) Kaplan-Meier estimate of progression-free survival. Tick marks indicate censored observations.
In the majority of patients (n=69), even those with progressive disease, plasma 2-HG decreased substantially and persistently and remained at low concentrations, approximating the range seen in healthy volunteers (appendix p 16).
Of 73 treated patients with cholangiocarcinoma, 63 (86%) had either archival formalin-fixed paraffin-embedded or pretreatment frozen tissue samples of sufficient quality for DNA sequencing analysis. Of these, mIDH1 status was confirmed in 55 (87%) patients (figure 2). Of the eight (13%) patients without confirmed mIDH1, six had baseline plasma 2-HG concentrations outside the range seen in healthy volunteers (appendix p 13), suggesting that low tumour content might have contributed to the inability to confirm mIDH1 status centrally.
Figure 2: Baseline co-occurring mutations.
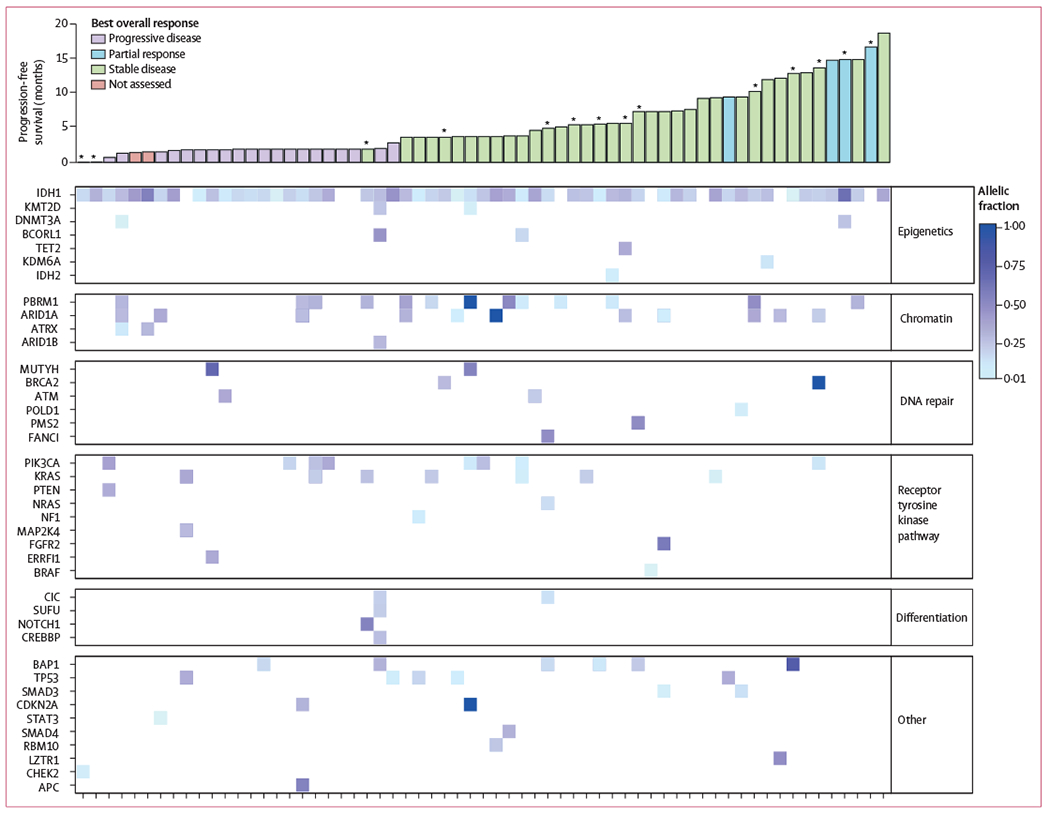
Baseline mutation profiles and their association with progression-free survival and best response in the 63 patients with samples available. *Progression-free survival value was censored.
In the 63 patients with baseline genetic profiling data, a median of two (range 0–8) additional mutations were detected; the most common known or likely oncogenic comutations were in PBRM1 (n=13; 21%), ARID1A (n=11; 17%), PIK3CA (n=8; 13%), and KRAS (n=7; 11%). However, no association emerged between progression-free survival or best response and comutations in any single gene or gene groups defined by selected biological pathways (figure 2). 37 (59%) patients had paired tissue available from pretreatment and at least one post-treatment time point for next-generation sequencing. New known or likely oncogenic mutations emerged at an allele frequency of 5% or more during treatment in six of these patients, spanning seven genes from multiple functional pathways (table 3). One patient developed an IDH2-R172V and one an IDH1-R132F mutation at disease progression, and comutations in TP53, ARID1A, POLE, PIK3R1, and TBX3 emerged in four other patients (table 3).
Table 3:
Emerging known or likely oncogenic variants in paired pretreatment and post-treatment tumour biopsies with VAF >5%
| Best response | End of treatment relative day | IDH1 mutation |
Emerging known or likely mutations (not present at baseline) |
Plasma 2-hydroxyglutarate |
Tumour 2-hydroxyglutarate |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline (VAF) | After dose (VAF) | After dose (VAF) | Relative day after dose (visit) | Response at visit | Baseline | After dose | Relative day after dose (visit) | Tumour concentration | Relative day | |||
| 1 | Partial response | 448 | R132C (18%) | R132C (30%) | IDH2 R172V (10%) | 447 (Cycle 17, day 1) | Progressive disease | 5990 ng/mL | 103 ng/mL | 449 (Cycle 17, day 1) | 1080 ng/mg | 462 |
| 2 | Stable disease | 625 | R132C (34%) | R132C not detected (converted to IDH1 R132F) | IDH1 R132F (37%) | 567 (Cycle 19, day 1) | Progressive disease | 5220 ng/mL | 569 ng/mL | 567 (Cycle 19, day 1) | 11000 ng/mg | 568 |
| 3 | Stable disease | 196 | R132G (24%) | R132G (24%) | PIK3R1 G376R (10%) | 168 (Cycle 7, day 1) | Stable disease | 444 ng/mL | No data | NA | 3·25 ng/mg | 168 |
| 4 | Stable disease | 168 | R132C (35%) | R132C (39%) | ARID1A splice site (10%) | 55 (Cycle 3, day 1) | Stable disease | 993 ng/mL | 107 ng/mL | 57 (Cycle 3, day 1) | 149 ng/mg | 53 |
| 4 | Stable disease | 168 | R132C (35%) | R132C (35%) | TBX3 E175* (20%) | 165 (Cycle 7, day 1) | Progressive disease | .. | No data | NA | 23·2 ng/mg | 165 |
| 5 | Stable disease | 343 | R132C (17%) | R132C (49%) | TP53 D281H (55%) | 58 (Cycle 3, day 1) | Stable disease | 485 ng/mL | 66·8 ng/mL | 60 (Cycle 3, day 1) | Below quantification limit | 59 |
| 6 | Progressive disease | 83 | R132C (45%) | R132C (34%) | POLEY726* (6%) | 38 (Cycle 3, day 1) | Unknown | 3610 ng/mL | 232 ng/mL | 28 (Cycle 2, day 1) | 368 ng/mg | 58 |
No emerging variants with VAF greater than 5% were observed in other patients (n=31). IDH=isocitrate dehydrogenase. VAF=variant allele frequency. NA=not applicable.
Denotes nonsense mutations.
13 patients (nine stable disease, two progressive disease, and two partial responses) had baseline and post-treatment tumour samples available for assessment of Ki-67 proliferation index (appendix pp 17–18). At cycle 3 day 1, nine patients had a reduction in Ki-67-positive cells (including six with stable disease); the median reduction across all 13 patients was −22·6% (range −80·7 to 186·7).
Discussion
In patients with advanced, unresectable mIDH1-cholangiocarcinoma, ivosidenib was well tolerated and without dose-limiting toxicities as defined by the trial protocol. The maximum tolerated dose was not reached. Most adverse events reported were consistent with those expected from the underlying disease as well as with previously reported data on ivosidenib in mIDH solid tumours.16–18 A low frequency of grade 3 or higher treatment-related adverse events occurred (four patients; 5%). The adverse event of interest, ECG QT prolongation, was reported in eight patients (11%; one grade ≥3). These events were managed with electrolyte repletion and ivosidenib dose modification.
Cholangiocarcinoma is an aggressive malignancy. First-line gemcitabine and cisplatin treatment in a randomised, phase 2 study3 of patients with biliary tract cancers resulted in a median overall survival of 11·7 months. Published data on patients with biliary tree carcinomas, including cholangiocarcinoma with unknown mIDH1 status, have generally shown small proportions of patients achieving a response following second-line chemotherapy regimens and beyond, with average median progression-free survival of 2–3 months and overall survival of 7–11 months.19–21 In a phase 2 study22 of monotherapy with the targeted multikinase inhibitor regorafinib in patients with advanced, pretreated biliary tract cancers, median progression-free survival was 3·9 months and median overall survival was 8 months.
In this study, ivosidenib therapy was administered to a heavily pretreated cohort of patients with mIDH1-cholangiocarcinoma, who had received a median of two (range 1–5) previous systemic therapies, with the last line of therapy continuing for a median of 3·2 months (range 0·2–26·2). In these patients, ivosidenib resulted in a median progression-free survival of 3·8 months (95% CI 3·6–7·3), 6-month progression-free survival of 40·1% (28·4–51·6) and 12-month progression-free survival of 21·8% (12·3–33·0), and a median overall survival of 13·8 months (11·1–29·3). Stable disease was noted in 56% of patients, which is clinically relevant given that it is comparable to the proportion of patients with stable disease receiving front-line gemcitabine-cisplatin therapy3 and that this is a refractory population. Of note, four patients have continued treatment for more than 1·5 years, one of whom had been receiving treatment for more than 2 years, and multiple patients continued on the trial for many months after documented progression. Considering the cytostatic mechanism of action of ivosidenib, the signal of stable disease as the primary RECIST-defined assessment is unsurprising and is consistent with that observed with other targeted non-cytotoxic drugs. In comparison to historical data, which has shown relatively short progression-free survival and overall survival in patients with heavily pretreated biliary tract cancer, we observed durable disease control associated with ivosidenib. Although no prospective studies have been done evaluating survival outcomes among patients with cholangiocarcinoma receiving primarily third-line chemotherapy, overall survival with chemotherapy in the second-line setting for biliary tract cancers has been reported in the range of 7–11 months.19–21 These studies, however, included other biliary tract cancers, thus they have limitations as direct historical comparators. Nonetheless, a median overall survival of 13·8 months in this heavily pretreated population is promising. Therefore, the data suggest that ivosidenib therapy offers a well-tolerated option in addition to cytotoxic chemotherapies of unproven efficacy that are known to be associated with both acute and chronic toxicities. The evidence to date suggests that mIDH1 is not prognostic or predictive of more favourable outcomes in patients with cholangiocarcinoma in relation to available treatments,23–28 implying that the clinical benefits seen in this study reflect the therapeutic effects of ivosidenib rather than mIDH1 status conferring a better prognosis. This is further supported by the observation that about 30% of patients progressed early within the first 2 months. Targeted therapies that have a mechanism of action distinct from typical cytotoxic agents might not elicit large proportions of patients achieving a response but might still be associated with prolonged disease control and a clinically meaningful cytostatic effect. The results of an ongoing phase 3 trial (NCT02989857) of ivosidenib in a similar population are awaited to confirm these phase 1 non-randomised activity data.
Although analyses of baseline comutations did not identify any genes or biological pathways to be predictive of disease control with ivosidenib, we identified six patients who acquired mutations after treatment. Of note, emergence of a secondary IDH1 or IDH2 mutation in two patients with mIDH1-cholangiocarcinoma with previously prolonged stable disease was observed (one of which has been previously reported by Harding and colleagues29). The biological and clinical significance of these and other acquired mutations (TP53, ARID1A, POLE, PIK3R1, TBX3) warrants further investigation. We observed a reduction in Ki-67 proliferation index in nine of 13 samples tested, which has been previously correlated with better prognosis in other cancer types.30,31 mIDH1 might block normal hepatocyte differentiation and increase the pool of hepatic progenitor cells, promoting susceptibility or vulnerability to cholangiocarcinoma development. Additionally, mIDH1-cholangiocarcinomas are characterised by upregulation of a hepatic progenitor cell transcriptional signature.13 On the basis of these observations, we hypothesise that prolonged stable disease in some patients with mIDH1-cholangiocarcinoma in this study might be due to an ivosidenib-induced differentiation effect on cholangiocarcinoma cells. Additional analyses of tumour morphology and gene expression in patient samples are ongoing.
This study has several limitations, including its non-randomised design and enrolment of a molecularly defined patient population, for which little historical reference data for progression-free survival and overall survival exist. Although we obtained multiple paired biopsies, the small number of samples for patients with different treatment outcomes (ie, longer vs shorter progression-free survival) precludes the identification of distinct molecular markers of primary and acquired treatment resistance. Additionally, information on disease factors, including cirrhosis, chronic inflammatory biliary disease, and viral hepatitis, was not collected comprehensively, therefore associations with specific epidemiological risk factors cannot be made. Most patients had IDH1-R132C mutations, therefore conclusions regarding varying responses between different alleles cannot be drawn.
In this study, ivosidenib was associated with low toxicity, objective responses, and durable disease control in heavily pretreated patients with advanced mIDH1-cholangiocarcinoma. Moreover, preliminary assessments suggest that ivosidenib treatment was associated with molecular changes consistent with therapeutic efficacy independent of traditional RECIST-defined radiographic responses. On the basis of the observations in this study, ivosidenib is being assessed in a global, randomised, placebo-controlled, phase 3 trial (NCT02989857) in previously treated patients with mIDH1-cholangiocarcinoma.
Supplementary Material
Research in context.
Evidence before this study
Cholangiocarcinoma is a rare, aggressive malignancy. Gemcitabine and cisplatin combination chemotherapy remains the sole standard first-line treatment for inoperable disease.
No standard second-line therapies exist. Outcomes remain poor. Mutations in the metabolic enzyme isocitrate dehydrogenase 1 (IDH1) are reported in up to 25% of patients with intrahepatic cholangiocarcinoma and result in overproduction of the oncometabolite D-2-hydroxyglutarate (2-HG). Preclinical work showed that treatment of IDH1-mutant mouse hepatoblasts with a mutant IDH1 (mIDH1) inhibitor in vitro resulted in reduction of 2-HG production and restoration of cellular differentiation, providing a rationale for the clinical use of mIDH1 inhibitors.
Added value of this study
To our knowledge, this is the first clinical report of mIDH1 inhibitor treatment in cholangiocarcinoma. In this ongoing, phase 1 trial of ivosidenib in patients with advanced mIDH1 solid tumours, ivosidenib had a favourable safety profile and low toxicity in the cohort of patients with heavily pretreated advanced cholangiocarcinoma. Ivosidenib treatment resulted in objective responses and durable disease control, with median progression-free survival and overall survival that compare favourably with best supportive care. Additionally, preliminary data suggest that ivosidenib treatment was associated with molecular changes consistent with therapeutic activity, including a reduction in Ki-67 proliferation index.
Implications of all the available evidence
No standard second-line treatment options exist for advanced cholangiocarcinoma. The identification of specific, relevant genetic mutations justifies a targeted therapy approach (eg, mIDH1). Our data suggest that ivosidenib is a well tolerated option for the treatment of mIDH1 advanced cholangiocarcinoma, and might offer patients some clinical benefit in this molecularly defined population. On the basis of these findings, ivosidenib is being assessed in a global, phase 3, randomised, placebo-controlled trial in previously treated patients with mIDH1 cholangiocarcinoma (NCT02989857).
Acknowledgments
This study was funded by Agios Pharmaceuticals, Inc. Writing assistance was provided by Yvonna Fisher-Jeffes (Agios Pharmaceuticals, Inc), with support from Helen Varley (Excel Medical Affairs, Horsham, UK). The authors thank Ania Tassinari (Agios Pharmaceuticals, Inc) for next-generation sequencing support and Julia Auer (Agios Pharmaceuticals, Inc) for operational contributions and Vikram Deshpande (Massachusetts General Hospital) for pathology support.
Declaration of interests
MAL is on advisory boards for Agios, Celgene, Pfizer, and Roche. HAB reports a grant from Agios during the conduct of the study, and grants from Acerta Pharma, Agios, Amplimmune, Array BioPharma, AstraZeneca, BIND Therapeutics, BioAtla, BioMed Valley Discoveries Inc, Bristol-Myers Squibb, Celgene, CicloMed, Clovis Oncology, Cytomx, eFFECTOR Therapeutics, Eli Lilly, Exelixis, Genentech, Gilead, GlaxoSmithKline, Harpoon Therapeutics, Hengrui Therapeutics, Immunocore, Intellikine, Janssen, Jounce Therapeutics, Loxo Oncology, MacroGenics, MedImmune, Medivation, Merck, Mirna Therapeutics, Moderna, Novartis, Pfizer, Revolution Medicines, Seattle Genetics, Stem CentRx, Takeda, Tesaro, TG Therapeutics, Verastem, and Vertex Pharmaceuticals, non-financial support from AstraZeneca, Eli Lilly, Hoffman LaRoche, Millennium Pharmaceuticals, Merck, Novartis, and Tesaro, and payment to their institution for consulting or expert witness services from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Eisai, Forma, Hoffman-La Roche, Janssen, MedImmune, Novartis, and Tolero, outside the conduct of this study. FJ has received research funding from Agios, Asana, Astellas, Bayer, BioMed Valley Discoveries, Bristol-Myers Squibb, Deciphera, FujiFilm Pharma, Genentech, Novartis, Piqur, Plexxikon, Proximagen, and Symphogen, has consulted for Deciphera, Guardant Health, Grail, IFM Therapeutics, Immunomet, Illumina, Novartis, PureTech Health, Sotio, Synlogic, Trovagene, and Valeant, has ownership interests in Trovagene, and has been loaned laboratory equipment at no cost by Bio-Rad and Biocartis. RTS has received grants from Agios, Celgene, Eli Lilly, Halozyme, and Pieris, is an advisory board member for Codiak Biosciences, Debiopharm, Exelixis, Merck, and Seattle Genetics, and is a consultant for QED Therapeutics. JMC has received research funding from Merck and Tesaro, is a consultant to Bristol-Myers Squibb, has received honorarium from Agios, and has received travel funding from Agios, Bristol-Myers Squibb, and Roche. NSA reports no competing interests. LGoy is on scientific advisory boards of Agios, Debiopharm, and Pieris Pharmaceuticals, is a consultant for H3 Biomedicine, and has received travel funding from Taiho Pharma. EAM has received research funding from Agios. LGor is on advisory boards and has consulted for Amgen, Celgene, OnKure, and Roche/Genentech and holds stock in Amgen, Celgene, Clovis Oncology, and Sanofi. AH was principal investigator for Agios, has received personal fees from Amgen, Eisai, Gritstone Oncology, and Merck Serono, is on the advisory board for Debiopharm, and has received non-financial support from Amgen, Lilly, and Servier. MB reports institutional research funding from Agios and personal fees from Genentech. JCT is on advisory boards for Agios, Blueprint, Deciphera, Epizyme, Janssen, Daiichi Sankyo, Lilly, and Novartis and has received research funding (clinical trial support) from Agios, Blueprint, Deciphera, Plexxicon, Lilly, and Novartis. LJ, BF, EA-F, CG, and SSP are employees of, and hold stock in, Agios Pharmaceuticals, Inc. SC and BW are employees of, hold stock in, and hold patents, royalties, or other intellectual property with Agios Pharmaceuticals, Inc. SVA was an employee of and held stock in Agios Pharmaceuticals, Inc at the time of the study. AXZ has acted as advisor or consultant to AstraZeneca, Bayer, Bristol-Myers Squibb, Eisai, Exelixis, Lilly, Merck, Novartis, and Roche. GKA-A has received research support (to institution) from ActaBiologica, Agios, Array, AstraZeneca, Bayer, Beigene, Bristol-Myers Squibb, Casi, Celgene, Exelixis, Genentech, Halozyme, Incyte, Lilly, Mabvax, Novartis, OncoQuest, Polaris Puma, QED, and Roche, and is a consultant for 3DMedcare, Agios, Alignmed, Amgen, Antengene, Aptus, Aslan, Astellas, AstraZeneca, Bayer, Beigene, Bioline, Bristol-Myers Squibb, Boston Scientifc, Bridgebio, Carsgen, Celgene, Casi, Cipla, CytomX, Daiichi, Debio, Delcath, Eisai, Exelixis, Genoscience, Halozyme, Hengrui, Incyte, Inovio, Ipsen, Jazz, Janssen, Kyowa Kirin, LAM, Lilly, Loxo, Merck, Mina, Novella, Onxeo, PCI Biotech, Pfizer, Pieris, QED, Redhill, Sanofi, Servier, Silenseed, Sillajen, Sobi, Targovax, Tekmira, Twoxar, Vicus, Yakult, and Yiviva.
Footnotes
Data sharing
The data collected for the study will not be made available to others. We encourage investigators interested in data sharing and collaboration to contact the corresponding author.
See Online for appendix
References
- 1.Blechacz B, Gores GJ. Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment. Hepatology 2008; 48: 308–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brivio S, Cadamuro M, Fabris L, Strazzabosco M. Epithelial-to-mesenchymal transition and cancer invasiveness: what can we learn from cholangiocarcinoma? J Clin Med 2015;4: 2028–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010; 362: 1273–81. [DOI] [PubMed] [Google Scholar]
- 4.Ramírez-Merino N, Aix SP, Cortés-Funes H. Chemotherapy for cholangiocarcinoma: an update. World J Gastrointest Oncol 2013; 5: 171–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012; 17: 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borger DR, Goyal L, Yau T, et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin Cancer Res 2014; 20: 1884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goyal L, Govindan A, Sheth RA, et al. Prognosis and clinicopathologic features of patients with advanced stage isocitrate dehydrogenase (IDH) mutant and IDH wild-type intrahepatic cholangiocarcinoma. Oncologist 2015; 20: 1019–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462: 739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009; 324: 261–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dang L, Su SM. Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem 2017; 86: 305–31. [DOI] [PubMed] [Google Scholar]
- 11.Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol 2016; 27: 599–608. [DOI] [PubMed] [Google Scholar]
- 12.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012; 483: 474–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saha SK, Parachoniak CA, Ghanta KS, et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014; 513: 110–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agios Pharmaceuticals. TIBSOVO (ivosidenib tablets), for oral use prescribing information. Agios Pharmaceuticals, 2018. https://www.tibsovo.com/pdf/prescribinginformation.pdf (accessed Feb 7, 2019).
- 15.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 2018; 378: 2386–98. [DOI] [PubMed] [Google Scholar]
- 16.Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. Proc Am Soc Clin Oncol 2017; 35 (suppl): 4015 (abstr). [Google Scholar]
- 17.Tap W, Villalobos VM, Cote GM, et al. A phase 1 study of AG-120, an IDH1 mutant enzyme inhibitor: results from the chondrosarcoma dose escalation and expansion cohorts. Connective Tissue Oncology Society Annual Meeting; Lisbon, Portugal; Nov 9–12, 2016. 138 (poster). [Google Scholar]
- 18.Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol 2017;(suppl 6): vi10–11. [Google Scholar]
- 19.Brieau B, Dahan L, De Rycke Y, et al. Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: a large multicenter study by the Association des Gastro-Enterologues Oncologues. Cancer 2015; 121: 3290–97. [DOI] [PubMed] [Google Scholar]
- 20.Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014; 25: 2328–38. [DOI] [PubMed] [Google Scholar]
- 21.Lowery MA, Williams Goff L, Jordan E, et al. Second-line chemotherapy (CTx) outcomes in advanced biliary cancers (ABC): a retrospective multicenter analysis. Proc Am Soc Clin Oncol 2016; 34 (suppl): 437 (abstr). [Google Scholar]
- 22.Sun W, Patel A, Normolle D, et al. A phase 2 trial of regorafenib as a single agent in patients with chemotherapy-refractory, advanced, and metastatic biliary tract adenocarcinoma. Cancer 2019; 125: 902–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Churi CR, Shroff R, Wang Y, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One 2014; 9: e115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Javle M, Bekaii-Saab T, Jain A, et al. Biliary cancer: utility of next-generation sequencing for clinical management. Cancer 2016; 122: 3838–47 [DOI] [PubMed] [Google Scholar]
- 25.Ruzzenente A, Fassan M, Conci S, et al. Cholangiocarcinoma heterogeneity revealed by multigene mutational profiling: clinical and prognostic relevance in surgically resected patients. Ann Surg Oncol 2016; 23: 1699–707 [DOI] [PubMed] [Google Scholar]
- 26.Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013; 32: 3091–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu AX, Borger DR, Kim Y, et al. Genomic profiling of intrahepatic cholangiocarcinoma: refining prognosis and identifying therapeutic targets. Ann Surg Oncol 2014; 21: 3827–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boscoe AN, Rolland C, Kelley RK. Frequency and prognostic significance of IDH1 mutations in cholangiocarcinoma: a systematic literature review. J Gastrointest Oncol 2019; published online March 18. DOI: 10.21037/jgo.2019.03.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harding JJ, Lowery MA, Shih AH, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov 2018; 8: 1540–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol 2000; 182: 311–22. [DOI] [PubMed] [Google Scholar]
- 31.de Azambuja E, Cardoso F, de Castro G Jr, et al. Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12 155 patients. Br J Cancer 2007; 96: 1504–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.