

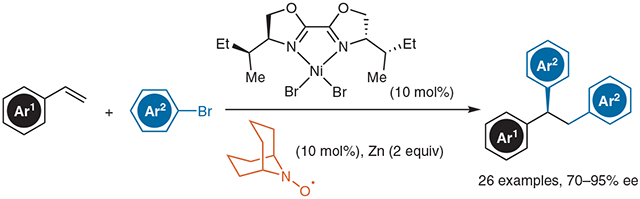
Abstract
Alkenes are an appealing functional group that can be transformed into a variety of structures. Transition-metal catalyzed dicarbofunctionalization of alkenes can efficiently afford products with complex substitution patterns from simple substrates. Under reductive conditions, this transformation can be achieved while avoiding stoichiometric organometallic reagents. Asymmetric difunctionalization of alkenes has been underdeveloped, in spite of its potential synthetic utility. Herein, we present a summary of our efforts to control enantioselectivity for alkene diarylation with a nickel catalyst. This reaction is useful for preparing triarylethanes. The selectivity is enhanced by an N-oxyl radical additive.
Keywords: asymmetric catalysis, nickel catalysis, alkene difunctionalization, reductive coupling, triarylethanes
Graphical Abstract
Introduction
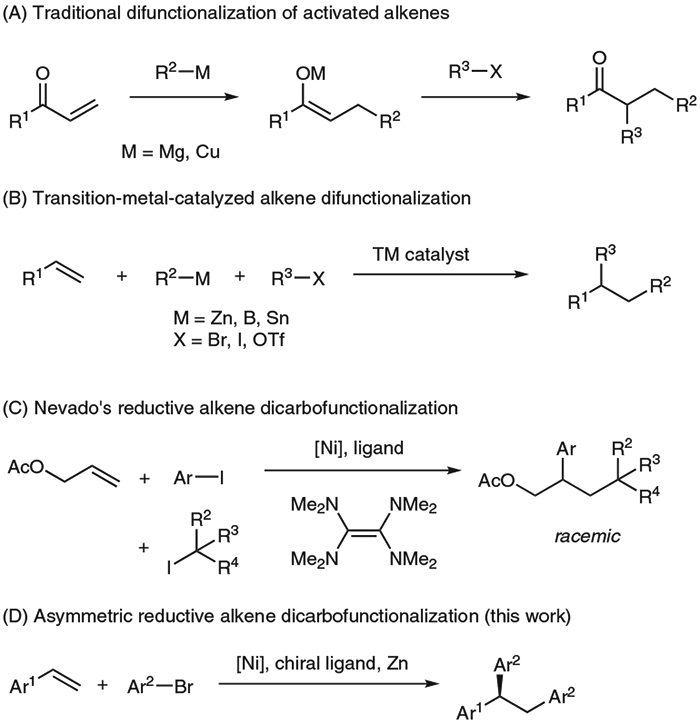
Dicarbofunctionalization of alkenes represents a compelling approach to efficiently access substituted, saturated molecular scaffolds from simple, abundant starting materials.1 Traditional methods to difunctionalize α,β-unsaturated carbonyl compounds have been achieved through a two-step process, in which a nucleophile undergoes conjugate addition with an activated alkene and the resulting enolate is then trapped with an electrophile (Scheme 1A).2 This strategy is limited to activated alkenes in conjugation with a carbonyl group, and functional groups must be compatible with the highly reactive organometallic nucleophiles.
Scheme 1.
Strategies for difunctionalization of alkenes
Transition metal catalysis has allowed for one-step difunctionalization of alkenes using a nucleophile and an electrophile (Scheme 1B).1a This redox-neutral strategy has employed palladium catalysts and aryl or vinyl nucleophiles and electrophiles to achieve the regioselective difunctionalization of vinylarenes,3 dienes,4 and, using a directing group, unactivated alkenes.5 A few asymmetric variants have been reported, but the scope has been restricted to diene substrates6 or intramolecular reactions.7 Nickel catalysts have expanded the scope of nucleophiles and electrophiles to include alkyl groups,8 but asymmetric reactions have been restricted to intramolecular examples.9
Reductive coupling reactions have recently emerged to eschew stoichiometric nucleophiles and thus expand the substrate scope to include moieties sensitive to organometallic nucleophiles.10 Nevado developed a Ni-catalyzed reductive dicarbofunctionalization of alkenes using alkyl and aryl iodides in combination with tetrakis(dimethylamino)ethylene (TDAE) as a reductant (Scheme 1C).11 In this reaction, Ni mediates the formation of an alkyl radical, which is added to the alkene. Combination of the radical with Ni forms a Ni-alkyl intermediate, which undergoes reductive elimination to afford the product. While the C(sp3) electrophile is restricted to tertiary alkyl iodides, this method is notable for its mild conditions.
This example highlights the radical reactivity of nickel compared to its Group 10 congener palladium, which favors two-electron pathways.12 The access to radical intermediates by nickel catalysts provides new opportunities for controlling enantioselectivity. In particular, for palladium-catalyzed arylation of alkenes, the enantioselectivity is dictated by either the alkene coordination or migratory insertion step (Scheme 2, steps i and ii),13 while for a nickel-mediated pathway involving single-electron redox chemistry, radical capture by nickel (v) or reductive elimination (vi) could be enantio-determining.14 We applied the radical properties of nickel catalysts to develop an intermolecular asymmetric difunctionalization of alkenes (Scheme 1D).
Scheme 2.
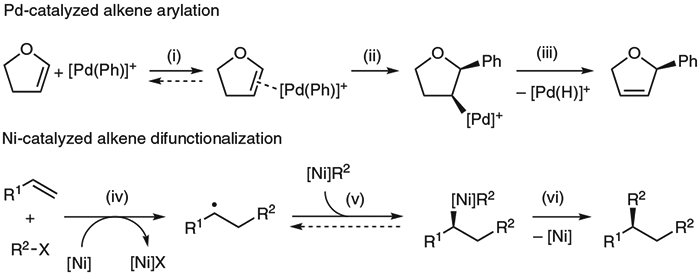
Mechanisms of palladium- and nickel-catalyzed asymmetric alkene functionalization
Reaction Development
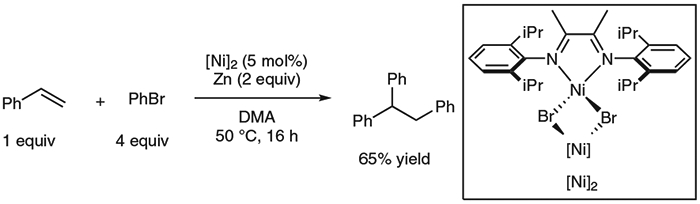
We began our investigation by developing conditions for racemic diarylation of styrene using bromobenzene. A dimeric nickel(I) catalyst has been previously shown to enable the carbofunctionalization of dienes15, and we were able to utilize this catalyst to conduct diarylation on styrene to generate 1,1,2-triphenylethane in good yield, so these conditions were used as a basis from which an asymmetric reaction could be evolved (Equation 1). Polar, aprotic solvents such as N,N-dimethylacetamide (DMA) or dimethylpropyleneurea (DMPU) were necessary in order to achieve high yields of the desired difunctionalization products. Heterogeneous reductants such as manganese and magnesium offered some diarylation product, while zinc powder gave the highest yield. Among various classes of chiral ligands, bioxazoline (biOx) ligands, commonly used in nickel catalysis,16 proved to be the most effective for achieving high yield and enantioselectivity.
 |
Equation 1 |
Racemic diarylation of styrene with bromobenzene
We observed a marked decrease in enantioselectivity from 38% to only 2% when the styrene substrate was distilled prior to use rather than used directly from its commercial bottle (Table 1, entries 1 and 2). Due to their propensity for auto-polymerization, styrenes are typically stabilized with radical inhibitors such as 2,6-di-tert-butyl-4-methylphenol (BHT). We speculated that the presence of such an additive, or the stabilized O-radical resultant from quenching of a radical by BHT, could be responsible for this discrepancy. While the use of BHT did not result in higher enantioselectivity (entry 3), the addition of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) had a dramatic effect, raising the enantiomeric excess to 49% (entry 4). Furthermore, equimolar TEMPO loading with respect to nickel was paramount in order to achieve optimal results, with lower loading engendering worse enantioselectivity (entry 5), while higher loading shut down the reaction (entry 6).
Table 1.
Optimization of Reaction Conditionsa
 | |||
|---|---|---|---|
| Entry | Additive (mol%) | Yield (%)b | ee (%)c |
| 1d | none | 73 | 38 |
| 2 | none | 61 | 2 |
| 3 | BHT (10%) | 62 | 14 |
| 4 | TEMPO (10%) | 44 | 49 |
| 5 | TEMPO (5%) | 72 | 26 |
| 6 | TEMPO (20%) | 1 | – |
| 7 | ABNO (10 mol%) | 57 | 76 |
| 8e | ABNO (8 mol%) | 90 | 91 |
Reaction conditions: 4-acetoxystyrene (0.2 mmol), bromobenzene (0.8 mmol), (dme)NiBr2 (10 mol%), iPr-biOx (13 mol%), Zn (0.4 mmol), DMPU (0.2 mL). Reaction was performed for 16 h, then ester was hydrolyzed with aqueous NaOH prior to purification.
Yields were determined by 1H NMR analysis with mesitylene as internal standard.
Enantiomeric excess was determined by HPLC with a chiral column after column chromatography purification.
4-Acetoxystyrene was used directly from commercial bottle without distillation.
Reaction performed with (dme)NiBr2 (10 mol%), sec-Bu-biOx (20 mol%), and DMPU/THF (0.1 mL each) at 10 °C.
These observations led us to evaluate various accessible N-oxyl radicals. The enantioselectivity exhibits a strong correlation with the percent buried volume (%Vbur), a computationally derived steric descriptor,17 of the N-oxyl radical. Less bulky N-oxyl additives gave higher enantioselectivities (Figure 1). 9-Azabicyclo[3.3.1]nonane N-oxyl (AB-NO) delivered the difunctionalized product with the highest enantiomeric excess (Table 1, entry 7). Enantioselectivity was further improved by modifying the substituent on the biOx ligand, lowering the reaction temperature, and using a mixture of DMPU and tetrahydrofuran (THF) as the solvent (entry 8).
Figure 1.

Steric effect of N-oxyl additives on enantioselectivity
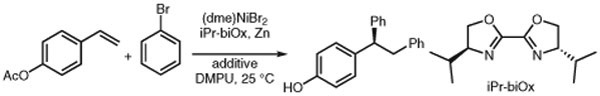
With optimized conditions in hand, we explored the substrate scope of this reaction (Scheme 3).18 Electron-donating and -withdrawing substituents had little effect on the enantioselectivity, although higher catalyst loadings were required to obtain good yields for electron-poor vinylarene substrates. Disubstituted alkenes were not well-tolerated, with 1,1-disubstituted vinylarenes showing no reactivity, although diarylation of indene gave a 17% yield of the trans-difunctionalized product.
Scheme 3.
Selected asymmetric diarylation substrate scope. Reagents and conditions: alkene (0.2 mmol), aryl bromide (0.8 mmol), (dme)Ni-Br2 (10 mol%), sec-Bu-biOx (20 mol%), ABNO (8 mol%), Zn (0.4 mmol), DMPU/THF (0.1 mL each). Reactions were performed for 16 h. Yields were measured by 1H NMR analysis with an internal standard. a Product was hydrolyzed with aqueous NaOH prior to isolation. b 20 mol% (dme)NiBr2, 40 mol% sec-Bu-biOx, and 16 mol% ABNO were used.
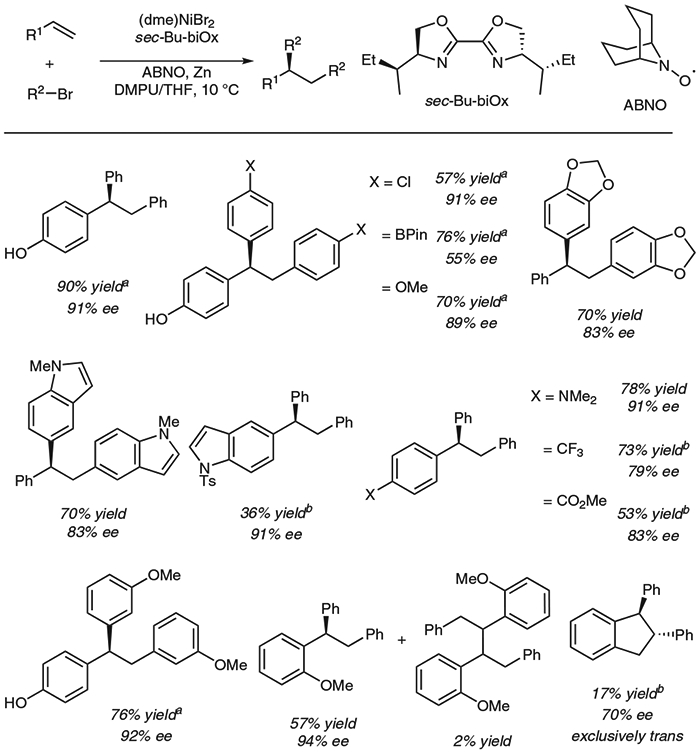
Mechanistic Investigation
We gained insight into the mechanism by considering several observations. First, formation of a dimer at the benzylic position suggests the presence of a benzylic radical (Scheme 4A). Second, the diarylation of indene resulted solely in the formation of the trans-diarylation product (Scheme 4B). One would expect a syn-addition from a typical migratory insertion, so the observed product diastereomer suggests the formation of a radical that combines with nickel on its less-hindered face (Scheme 4C). Third, the presence of excess N-oxyl radical with respect to nickel inhibited the reaction, mostly likely via interference of radical intermediates. Finally, no evident linear correlation was found between the observed enantiomeric ratio and the electronic effect of vinylarenes, suggesting that the enantio-determining step could involve a radical species.
Scheme 4.
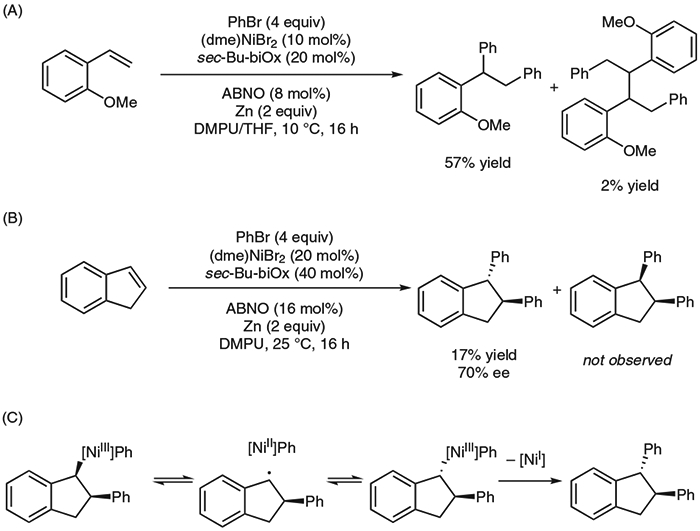
Mechanistic experiments and proposed origin of trans-diastereoselectivity
We performed control experiments to investigate the possible in situ generation of an organozinc reagent, which could afford the difunctionalized alkene as in redox-neutral strategies. When phenylzinc chloride was used in place of bromobenzene in the absence of zinc powder, no diarylation product was observed. This experiment suggests that direct oxidative addition of zinc to the aryl bromide does not enable the reaction.
A mechanistic hypothesis consistent with our findings is presented in Scheme 5. Oxidative addition and reduction of a nickel(I) bromide species to the aryl bromide electrophile results in an arylnickel(I) species.19 This intermediate undergoes migratory insertion with the alkene substrate. Oxidative addition of another molecule of aryl bromide to the arylnickel(I) makes a nickel(III) species. This open-shell intermediate can eject a stabilized benzylic radical, which accounts for our observations of both the benzylic dimer byproduct and the trans-diastereoselectivity with indene. This reversible radical ejection is consistent with computational work performed by Kozlowski and co-workers.14 Recombination of this radical with the chiral nickel catalyst followed by reductive elimination furnishes the diarylated product and regenerates the nickel(I) bromide.
Scheme 5.
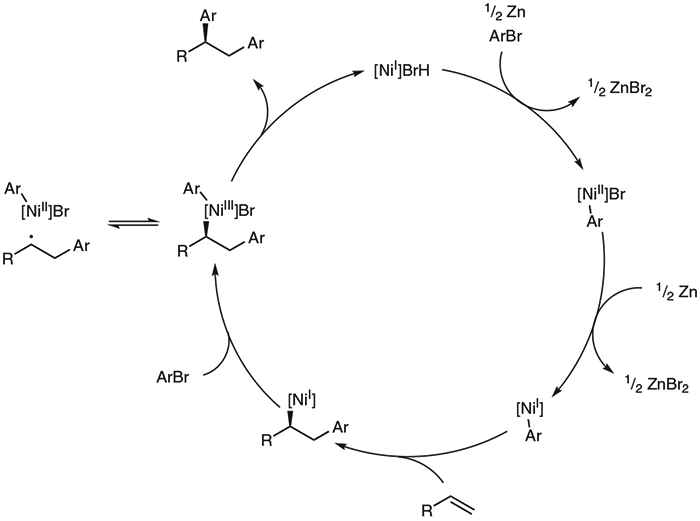
Proposed catalytic cycle
Future Outlook.
We have developed the first intermolecular asymmetric diarylation of vinylarenes using a chiral biOx-nickel catalyst. The enantioselectivity is enhanced by the use of an auxiliary N-oxyl radical ligand, although the precise role of this radical remains obscure. However, we have shown that the radical mechanistic pathways available to nickel can be harnessed to elicit control over reaction outcomes in a different way than palladium commonly operates. We anticipate that the elucidation of the precise role of the N-oxyl additive could provide insight for the development of asymmetric nickel-mediated reactions with mechanisms invoking radical pathways.
Acknowledgment
We would like to thank Dr. Chunhua Hu for X-ray crystallographic analysis and Prof. Jim Canary for sharing chiral HPLC columns.
Funding Information
This work was supported by the National Science Foundation (CHE-1654483) and the National Institutes of Health (NIH) (R01 GM127778). D.A. is supported by the Margaret and Herman Sokol Fellowship. T.D. is a recipient of the Alfred P. Sloan Research Fellowship (FG-2018-10354) and the Camille and Henry Dreyfus Foundation (Camille-Dreyfus Teacher-Scholar Award TC-19-019).
Biography

David Anthony (left) obtained his Bachelor’s degree in chemistry from Cornell University in 2014. As an undergraduate student, he performed research in organic synthesis under the supervision of Dr. Chad A. Lewis. In 2015, he began his graduate studies at New York University under the supervision of Prof. Tianning Diao. His research focuses on the development of novel chemical methods catalyzed by transition metals.
Tianning Diao (right) received her Ph.D. in chemistry from the University of Wisconsin–Madison in 2012 under the supervision of Prof. Shannon Stahl. She conducted her postdoctoral research with Prof. Paul Chirik at Princeton University. In 2014, she joined the faculty of NYU, where she is currently an Assistant Professor of Chemistry.
References
- (1).(a) Giri R; KC SJ Org. Chem 2018, 83, 3013. [DOI] [PubMed] [Google Scholar]; (b) Derosa J; Apolinar O; Kang T; Tran VT; Engle KM Chem. Sci 2020, 11, 4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Chapdelaine MJ; Hulce M Org. React 2004, 38, 225. [Google Scholar]; (b) Ihara M; Fukumoto K Angew. Chem. Int. Ed. Engl 1993, 32, 1010. [Google Scholar]; (c) Wang X; An J; Zhang Z; Tan F; Chen J; Xiao W Org. Lett 2011, 13, 808. [DOI] [PubMed] [Google Scholar]
- (3).Urkalan KB; Sigman MS Angew. Chem. Int. Ed 2009, 48, 3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Liao L; Jana R; Urkalan KB; Sigman MS J. Am. Chem. Soc 2011, 133, 5784. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McCammant MS; Liao L; Sigman MS J. Am. Chem. Soc 2013, 135, 4167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stokes BJ; Liao L; de Andrade AM; Wang Q; Sigman MS Org. Lett 2014, 16, 4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Liu Z; Zeng T; Yang KS; Engle KM J. Am. Chem. Soc 2016, 138, 15122. [DOI] [PubMed] [Google Scholar]
- (6).(a) Wu X; Lin H; Li M; Li L; Han Z; Gong LJ Am. Chem. Soc 2015, 137, 13476. [DOI] [PubMed] [Google Scholar]; (b) Stokes BJ; Liao L; de Andrade AM; Wang Q; Sigman MS Org. Lett 2014, 16, 4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yasui Y; Kamisaki H; Takemoto Y Org. Lett 2008, 10, 3303. [DOI] [PubMed] [Google Scholar]
- (8).(a) Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS Science 2016, 352, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Derosa J; Tran VT; Boulous MN; Chen JS; Engle KM J. Am. Chem. Soc 2017, 139, 10657. [DOI] [PubMed] [Google Scholar]; (c) Shrestha B; Basnet P; Dhungana RK; Kc S; Thapa S; Sears JM; Giri RJ Am. Chem. Soc 2017, 139, 10653. [DOI] [PubMed] [Google Scholar]; (d) Derosa J; van der Puyl VA; Tran VT; Liu M; Engle KM Chem. Sci 2018, 9, 5278. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Huang D; Olivieri D; Sun Y; Zhang P; Newhouse TR J. Am. Chem. Soc 2019, 141, 16249. [DOI] [PubMed] [Google Scholar]; (f) Gao P; Chen LA; Brown MK J. Am. Chem. Soc 2018, 140, 10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Cong H; Fu GC J. Am. Chem. Soc 2014, 136, 3788. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang K; Ding Z; Zhou Z; Kong WJ Am. Chem. Soc 2018, 140, 12364. [DOI] [PubMed] [Google Scholar]; (c) Jin Y; Wang C Angew. Chem. Int. Ed 2019, 58, 6722. [DOI] [PubMed] [Google Scholar]; (d) Tian ZX; Qiao JB; Xu GL; Pang X; Qi L; Ma WY; Zhao ZZ; Duan J; Du YF; Su P; Liu XY; Shu XZ J. Am. Chem. Soc 2019, 141, 7637. [DOI] [PubMed] [Google Scholar]
- (10).(a) Weix DJ Acc. Chem. Res 2015, 48, 1767. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X; Dai Y; Gong H Top. Curr. Chem 2016, 374, 43. [DOI] [PubMed] [Google Scholar]
- (11).Garcia-Dominguez A; Li Z; Nevado CJ Am. Chem. Soc 2017, 139, 6835. [DOI] [PubMed] [Google Scholar]
- (12).Hazari N; Melvin PR; Beromi MM Nat. Rev. Chem 2017, 1, 0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Ozawa F; Kubo A; Hayashi TJ Am. Chem. Soc 1991, 113, 1417. [Google Scholar]; (b) Ashimori A; Overman LE J. Org. Chem 1992, 57, 4571. [Google Scholar]; (c) Ashimori A; Bachand B; Overman LE; Poon DJ J. Am. Chem. Soc 1998, 120, 6477. [Google Scholar]
- (14).Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC J. Am. Chem. Soc 2015, 137, 4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kuang Y; Anthony D; Katigbak J; Marrucci F; Humagain S; Diao T Chem 2017, 3, 268. [Google Scholar]
- (16).Diccianni JB; Diao T Trends Chem. 2019, 1, 830. [Google Scholar]
- (17).Falivene L; Cao Z; Petta A; Serra L; Poater A; Oliva R; Scarano V; Cavallo L Nat. Chem 2019, 11, 872. [DOI] [PubMed] [Google Scholar]
- (18).Anthony D; Lin Q; Baudet J; Diao T Angew. Chem. Int. Ed 2019, 58, 3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lin Q; Diao TJ Am. Chem. Soc 2019, 141, 17937. [DOI] [PMC free article] [PubMed] [Google Scholar]