

SUMMARY
Single-cell transcriptomics has been widely applied to classify neurons in the mammalian brain, while systems neuroscience has historically analyzed the encoding properties of cortical neurons without considering cell types. Here we examine how specific transcriptomic types of mouse prefrontal cortex (PFC) projection neurons relate to axonal projections and encoding properties across multiple cognitive tasks. We found that most types projected to multiple targets, and most targets received projections from multiple types, except PFC→PAG (periaqueductal gray). By comparing Ca2+ activity of the molecularly homogeneous PFC→PAG type against two heterogeneous classes in several two-alternative choice tasks in freely moving mice, we found that all task-related signals assayed were qualitatively present in all examined classes. However, PAG-projecting neurons most potently encoded choice in cued tasks, whereas contralateral PFC-projecting neurons most potently encoded reward context in an uncued task. Thus, task signals are organized redundantly, but with clear quantitative biases across cells of specific molecular-anatomical characteristics.
Graphical Abstract
In Brief
Bridging molecular and systems neuroscience perspectives, Lui et al. examine how specific transcriptomic types of mouse prefrontal cortex projection neurons relate to axonal projections and encoding properties across multiple cognitive tasks
INTRODUCTION
Achieving a “ground truth” understanding of neuronal types is important for dissecting the function of complex neuronal circuits (Jorgenson et al., 2015; Luo et al., 2018). Molecular neuroscience has seen a recent explosion in neuronal cell type classification using single-cell RNA sequencing (scRNAseq) technologies (Zeng and Sanes, 2017). Because transcriptomic data reflects cellular function, is high dimensional, and can be quantitatively compared across brain regions (Tasic et al., 2018) and species (Tosches et al., 2018; Hodge et al., 2019; Kebschull et al., 2020), it is often considered as a foundation to all other properties. However, it is challenging to reconcile cell type definitions from transcriptomic data with those determined by other properties such as developmental history, connectivity patterns, electrophysiological properties, and the encoding of signals related to behavior. Most transcriptomic studies have not investigated the encoding of behaviorally relevant signals in discovered neuronal cell types in vivo. Furthermore, what constitutes a neuronal type in many regions of the mammalian brain is a topic of intense debate (Yuste et al., 2020). Explicitly testing whether a given transcriptomic classification possesses any characteristic anatomical and physiological properties can help determine whether the classification is functionally relevant.
This issue is beginning to be addressed in a variety of neural systems. Applying scRNAseq to systems with a pre-existing ground truth of cell types and function (e.g., mouse retina: Shekhar et al., 2016; fly olfactory system: Li et al., 2017) has resulted in relatively faithful mapping between molecular and functional types. Interrogation of twosubcortically projecting transcriptomic types in mouse anterolateral motor cortex (ALM) during a motor planning task revealed differences in the encoding of preparatory activity (Economo et al., 2018). Profiling of neuronal diversity in the mouse hypothalamus revealed high levels of transcriptomic diversity but little one-to-one matching between transcriptomic types, behavior-specific activation, and connectivity (Moffitt et al., 2018; Kim et al., 2019). Analysis of the mouse striatum revealed that continuous gene expression variation is overlaid on discrete cell types over space, with both continuous and discrete variation contributing to circuit function (Gokce et al., 2016; Stanley et al., 2020). Thus, the level of correspondence between molecular and functional properties can differ substantially across neuron types and brain regions.
Neurons of the mammalian PFC serve at a critical transition between sensation and action, bias diverse sensory signals toward appropriate downstream targets, and underlie cognitive processes such as reward-guided decision-making and behavioral flexibility (Fuster, 2008; Miller and Cohen, 2001; Rushworth et al., 2011; Euston et al., 2012; Zingg et al., 2014). While traditional systems neuroscience techniques have shown how complex task-related signals can be encoded at the single-neuron and population levels (e.g., Asaad et al., 1998; Mante et al., 2013), these studies are typically blind to cell type and projection patterns. Furthermore, cognitive functions operate among a wide range of task demands, and the measured complexity of how PFC encodes task signals depends heavily on the behavioral assays used. It is therefore important to achieve an integrated picture of how specific transcriptomic types relate to their projection patterns, and together underlie a well-defined repertoire of task signals.
To this end, recent studies in mice have suggested that nucleus accumbens-projecting medial PFC (mPFC→NAc) neurons have different roles in the conjunctive encoding of social and spatial targets (Murugan et al., 2017), the restraint of reward seeking (Kim et al., 2017), and the representation of reward predicting cues (Otis et al., 2017). mPFC→PAG neurons were reported to be a key node that dopamine acts on to modulate the encoding of aversive stimuli (Vander Weele et al., 2018), and exhibited activity signatures underlying compulsive alcohol drinking (Siciliano et al., 2019). In a sensory discrimination task, dorsomedial PFC excitatory neurons had diverse task encoding that correlated with different layers containing heterogeneous populations (Pinto and Dan, 2015). As the molecular heterogeneity of these populations is unknown and the behavioral task repertoire is diverse, matching cell type and function is a continuing challenge.
Here, we use the mouse PFC as a case study to address the extent that the encoding of cognitive task-related signals is predicted by transcriptomic and projection properties, starting from the foundation of a transcriptomic analysis. scRNAseq of neurons labeled by Rbp4Cre [most Layer (L) 5 excitatory projection neurons; Gerfen et al., 2013] identified 7 transcriptomic types, but projection mapping revealed that most types projected to multiple targets and most targets received projections from multiple types. We leveraged a unique property that projection-defined mPFC→PAG neurons all derived from a single transcriptomic type, and assessed the diversity of task-related signals present in these neurons by performing Ca2+ imaging in freely moving mice during several two-alternative choice tasks. We contrasted mPFC→PAG neurons to two other classes of PFC neurons: those that project to contralateral PFC (comprising 3 transcriptomic types), and those labeled by Rbp4Cre (comprising all 7 transcriptomic types). The use of scRNAseq data to drive analysis of encoding properties in cognitive tasks bridges an important gap between molecular and systems neuroscience, furthers our understanding of PFC function, and extrapolates principles of how task information is organized in a cell-type framework.
RESULTS
Single-cell transcriptomes of Rbp4Cre-labeled PFC projection neurons
Rodent PFC lacks L4 and contains a thick L5 that targets diverse subcortical and intracortical regions (Gabbott et al., 2005). We thus used the Rbp4Cre line as a foundation for our dataset. To define transcriptomic types and uncover potential differences in gene expression across PFC regions, we broadly profiled dorsomedial (dmPFC), ventromedial (vmPFC), and orbitofrontal (OFC) regions (STAR Methods).
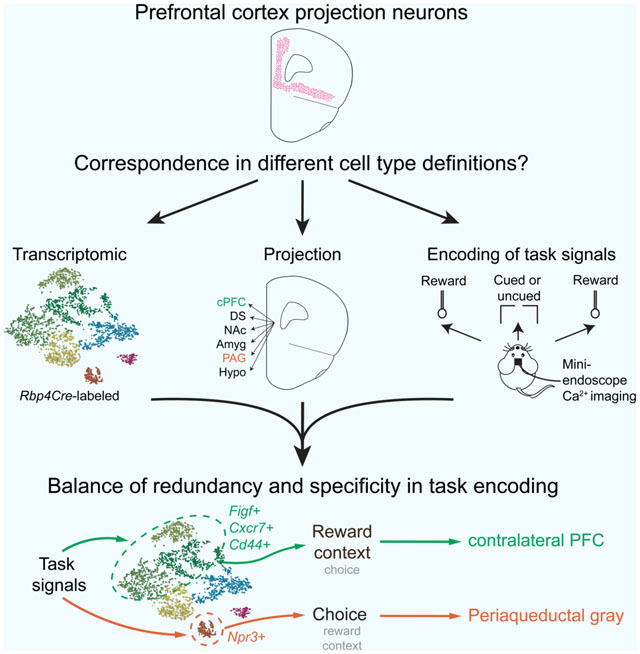
We crossed Rbp4Cre with a reporter mouse expressing tdTomato in Cre+ cells (Ai14; Madisen et al., 2010), dissected and dissociated tissue from postnatal day 34 to 40 (P34–P40) double transgenic progeny, and performed fluorescence-activated cell sorting (FACS) and plate-based scRNAseq on tdTomato+ cells using SMART-Seq2 (Picelli et al., 2014) (Figure 1A). We analyzed 3139 high-quality cells (Figure S1A) pooled from all three regions (dmPFC: 910 cells, n = 3 mice; vmPFC: 1234 cells, n = 4 mice; OFC: 995 cells, n = 4 mice) and performed Seurat unbiased clustering and batch normalization (Butler et al., 2018; Stuart et al., 2019; STAR Methods) at multiple resolutions (Figure 1E). Classification at relatively low clustering resolution gave 7 clusters, each of which was defined based on co-expression of multiple genes. For simplicity, we highlight only one exemplar marker gene from each cluster: Cd44, Figf, Otof, Pld5, Cxcr7, Npr3, and Tshz2, respectively (Figures 1B, 1D, Table S1 Tab 1), but emphasize that these definitions rely on multi-dimensional data.
Figure 1. Transcriptomic map of Rbp4Cre-labeled PFC projection neurons.
(A) Cell isolation from three PFC subregions in Rbp4Cre;Ai14 mice for scRNAseq. Tissue was dissociated, FAC-sorted into plates, and processed with SMART-Seq2. Scale, 1 mm.
(B) Unbiased clustering of 3139 high-quality projection neurons (median ~7000 genes/cell, ~1–2 million reads/cell) based on transcriptomic data, shown in t-distributed stochastic neighbor embedding (tSNE) space, using Seurat with batch correction. The 7 labels are based on marker genes from differential expression analysis across clusters.
(C) Feature plots (top) and violin plots (bottom) showing single-cell gene expression of known markers for excitatory pyramidal neurons (Vglut1+), upper (Cux1) versus deeper (Fezf2) layers, and subcortically projecting neurons (Ctip2). Color scale of feature plots and y axis of violin plots in this and other panels are in the unit of ln[1+ (reads per 10000)]. Dot in each violin plot is the median.
(D) Feature and violin plots similar to (C) for cluster-specific marker genes that best distinguish clusters (see Table S1 Tab 1). Dots in violin plots represent cells.
(E) “Clustree” flowchart (Zappia and Oshlack, 2018) of how cell classifications change across different Seurat clustering resolutions. Arrow intensity indicates the population size moving between levels. The relatively low resolution of 0.3 was chosen because clusters could be distinguished by 1–2 marker genes. Note the relative stability of the Otof, Pld5, Cxcr7, and in particular the Npr3 and Tshz2 clusters.
(F) Determination of Seurat nearest neighbor mapping (Stuart et al., 2019) between Rbp4Cre-labeled types in PFC (defined here) and in ALM or VISp (Tasic et al., 2018). An alluvial diagram (right) shows the mapping of 7 PFC clusters to the 4 ALM and VISp groups (from a full list of 20 types in Figure S1D), with normalization to the same population size for each PFC cluster. IT: intratelencephalic, PT: pyramidal tract (subcortical).
See also Figure S1.
All clusters expressed Slc17a7, which encodes the vesicular glutamate transporter Vglut1, confirming their excitatory neuron identity. These cells could also be divided coarsely into deeply versus superficially located based on expression of marker genes Fezf2 and Cux1 (Greig et al., 2013; Lein et al., 2007), respectively (Figure 1C). Putative Ctip2+ subcortically projecting neurons were further divided into 2 discrete clusters expressing Npr3 or Tshz2. By contrast, gene expression in the remaining 5 clusters had more continuous variation (Figures 1B and 1D). We refer to these 7 clusters as transcriptomic types hereafter. Testing the robustness of this classification using a different method (Tasic et al., 2018) revealed a similar structure (Figures S1B and S1C), with the Figf and Cxcr7 clusters more subdivided.
The majority of the PFC clusters had mixed contributions from all three regions (Figure S1B). This was in contrast to a comparison of ALM versus primary visual (VISp) cortex transcriptomic types, where glutamatergic types were highly region-specific (Tasic et al., 2018). These observations could be explained trivially by continuous spatial differences in gene expression across cortex, which would differentially affect regions that are close together versus far apart. To examine this, we compared Rbp4Cre>tdTomato+ cells among PFC and ALM/VISp (Tasic et al., 2018), and determined the cluster from the Allen Institute annotation that was the nearest neighbor to each PFC cell (Stuart et al., 2019). We found that transcriptomically, PFC neurons were not always nearest neighbors with the more physically proximal ALM cells (Figure 1F, ALM mapping: 62.5%, VISp mapping: 37.5%; Allen clusters pooled based on Figure S1D), indicating that gene expression features defining cortical neuron clusters are not explained by physical distance alone. However, our data do emphasize that the assayed PFC subregions are relatively similar, and the most obvious differences were in subregion contributions to each cluster.
Anatomical locations of transcriptomic types
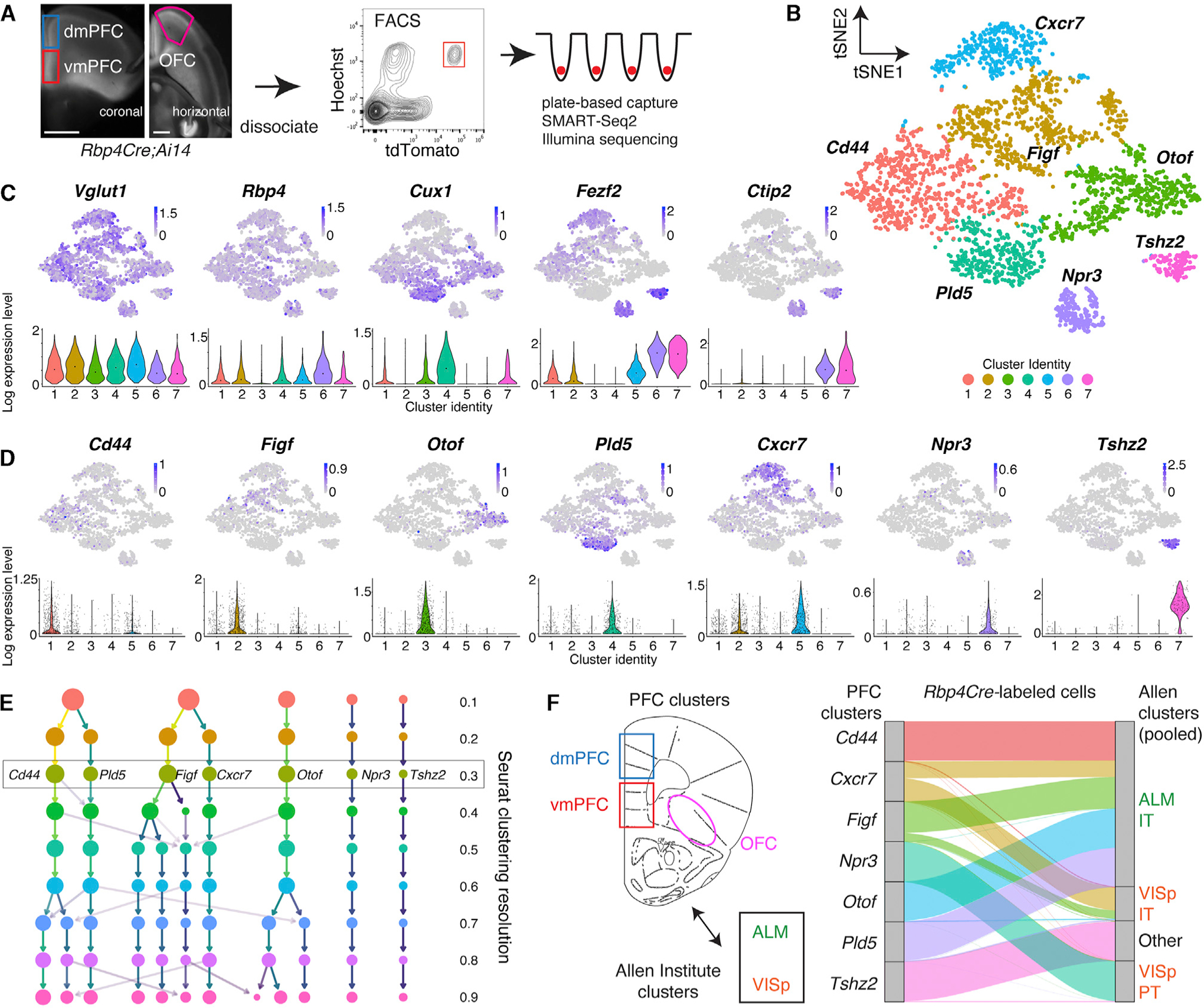
We next examined the spatial location and co-localization of marker expression among the PFC transcriptomic types using hybridization chain reaction-based fluorescence in situ hybridization (HCR-FISH; Choi et al., 2018). Focusing on mPFC, we quantified marker expression within Vglut1-labeled cell soma (Figures 2A, 2B, and S2A). Otof labeling was specific to L2/3 [and negative for Rbp4 in the sequencing data despite being Rbp4Cre>tdTomato+ (Figures 1C and 1D), possibly because Rbp4Cre is transiently expressed in Otof+ cells during development] (Figures 2A, 2B, and S2A). Npr3+ and Tshz2+ cells were located deeper in L5 with similar spatial distributions, but co-localization analysis validated that they were distinct (Figure 2C). Co-labeling of Otof/Figf/Cxcr7 and Otof/Cd44/Cxcr7 demonstrated a variety of single, double, and triple labeled cells distributed throughout L2/3 and 5, consistent with the continuous variation in the sequencing data (Figures 2D and S2B). Cd44/Cxcr7/Tshz2 triple labeling validated the distinctness of Tshz2 with the other markers (Figure S2C). Overall, transcriptomic and spatial organization was similar between dmPFC and vmPFC; the major difference distinguishing OFC was enrichment of Pld5 and Cxcr7(5–2) (Figures S1B and S2D) cluster cells. Because vmPFC is narrower than dmPFC, our data indicate that neuronal heterogeneity in mPFC is best summarized as a complex mixture of laminar expression that is increasingly compressed and intermixed from dorsal to ventral, causing extensive overlap of transcriptomic types in L5.
Figure 2. Anatomical locations of PFC transcriptomic types.
(A) HCR-FISH of cluster-specific marker genes in vmPFC (A–P ~1.95 mm, D–V ~–2.35 mm). Dashed lines are approximate cortical layer boundaries (Allen Atlas; beginning of L2/3, 120 μm; L5a, 230 μm; L5b, 410 μm; L6: 600 μm from midline). Scale, 50 μm.
(B) Laminar distribution of cells expressing cluster-specific markers across vmPFC. Vglut1 was used to segment cell soma to quantify expression of markers. Averaged across n = 4 mice, with 1–2 images per mouse. Layer boundaries are the same as (A) but begin at L2/3.
(C) Double HCR-FISH for Npr3 and Tshz2 in vmPFC. Quantified for both dmPFC and vmPFC and averaged across 4 mice (252 dmPFC, 322 vmPFC cells). Scale, 50 μm.
(D) Triple HCR-FISH for Otof, Cxcr7, and Figf in vmPFC. Quantification similar to (C) (4 mice; 540 dmPFC, 591 vmPFC cells). Scale, 50 μm.
In this and all subsequent figures, stereotactic coordinates are in millimeters (mm) with respect to bregma, and error bars are SEM unless otherwise stated. See also Figures S2A–S2D.
In the remainder of this study we focused only on vmPFC (centered on the infralimbic region) because: 1) it fulfills the traditional definition of PFC in rodents based on dense innervation from mediodorsal thalamus, which is less clearly the case for dmPFC (Rose and Woolsey, 1948; Preuss, 1995; Uylings et al., 2003; Oh et al., 2014); and 2) the full diversity of cell types is more compactly represented in a small space, making tracing and imaging studies more specific.
Complex correspondence between projection targets and transcriptomic types
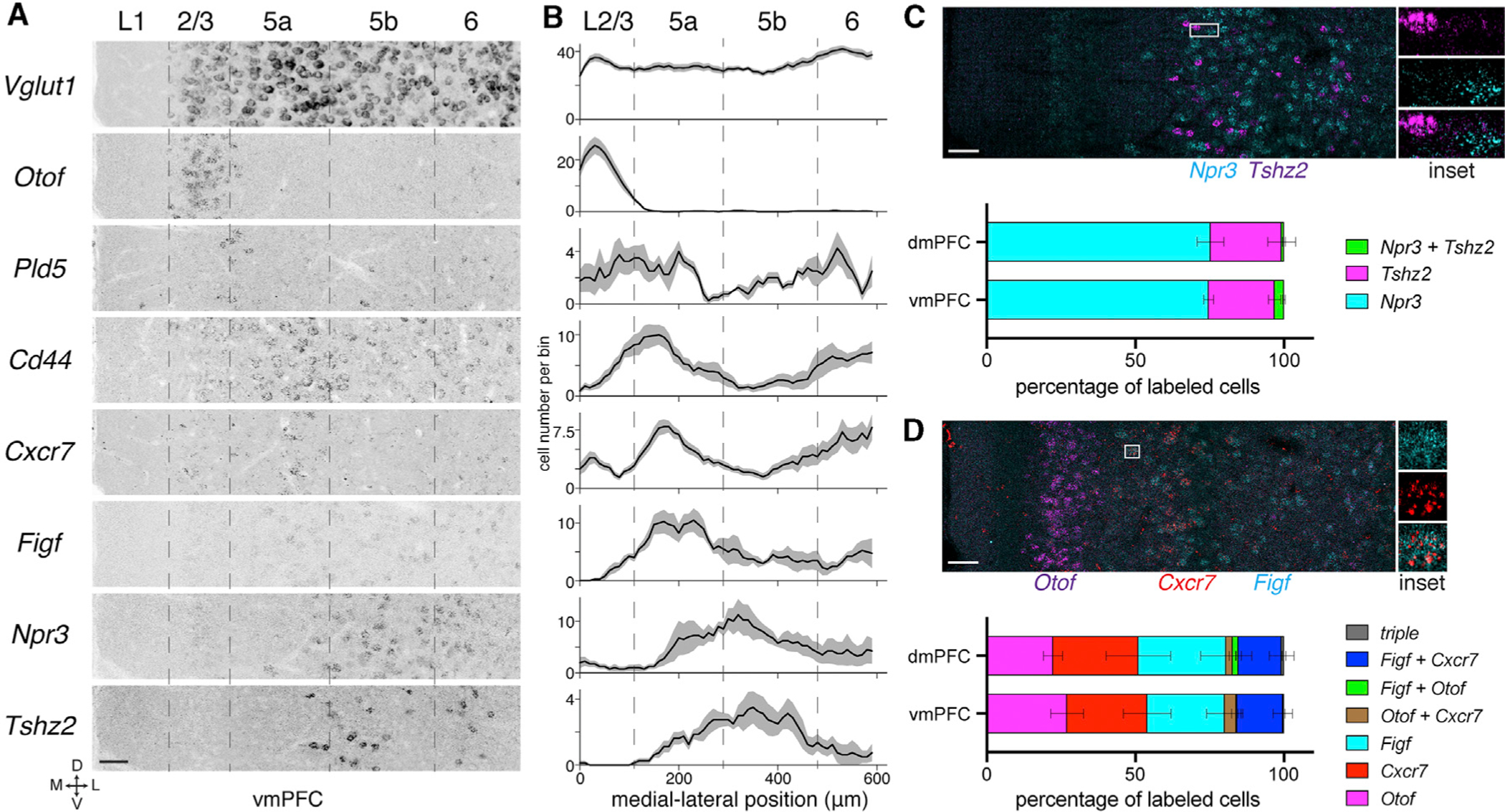
To examine the relationship between transcriptomic types and specific targets of vmPFC projection neurons, we injected retrograde traveling Cre-expressing virus (CAV-Cre or AAVretro-Cre, with the caveat of potential differences in viral tropism, see Limitations of Study section in Discussion), which transduces axon terminals and is transported back to cell bodies, into Ai14 mice at 6 known target sites: ipsilateral dorsal striatum (DS), nucleus accumbens (NAc), hypothalamus (Hypo), periaqueductal gray (PAG), amygdala (Amyg), and contralateral PFC (cPFC) (Figure 3A, see STAR Methods). We dissected vmPFC containing retrogradely labeled tdTomato+ cells (Figure S2E) and performed scRNAseq as before. Appending retrogradely labeled cells to the dataset from Figure 1 roughly recapitulated the previous clustering, with the exception of a new Syt6+ cluster derived mostly from hypothalamus-projecting cells and additional Figf+ cells (Figure S3A). Other than the Syt6+ (L6 marker, Gerfen et al., 2013) cluster, the remaining retrograde cells were classified based on similarity to the 7 reference transcriptomic types of Figure 1 (Stuart et al., 2019).
Figure 3. Relationship between projection patterns and transcriptomic types in vmPFC.
(A) Retrograde labeling from vmPFC targets (red circles indicate injection sites) for scRNAseq. tdTomato+ cells were collected from vmPFC one week after injection. Numbers are distance in mm from bregma (A–P axis). Scale, 500 μm.
(B) Nearest neighbor mapping of retrograde cells collected from vmPFC (n = 440 cPFC, 129 DS, 93 NAc, 290 Amyg, 94 PAG, and 109 Hypo cells) to the 7 transcriptomic types from Figure 1 and Syt6+ L6 cells (Figure S3A), with normalization to the same population size for each target. Mapping to reference datasets with higher clustering resolution (Figure S3B), of only Rbp4+ cells, or of only vmPFC cells gave similar results (data not shown). PL, prelimbic; IL, infralimbic; MO, medial orbital cortex.
(C) Images of retrograde tracing from PAG (red) and HCR-FISH showing that PAG-projecting cells (tdTomato) express Npr3 but not Figf (cyan). HCR-FISH signal was converted to binary puncta and overlaid with tdTomato cell outlines for quantification. Inset is magnification of the boxed region. Scale, 25 μm.
(D) Quantification of retrograde cells (cPFC- or PAG-projecting) that co-localized with markers for different transcriptomic types (Cd44, Figf, Cxcr7, Npr3, n = 3 mice for each).
(E) PAG- (tdTomato) and cPFC-projecting (CTB-488) cells in vmPFC in the same section. Scale, 100 μm.
See also Figures S2E, S2F, and S3.
To analyze how projection-defined cells were distributed among the transcriptomic types, we visualized the mapping between these two categorical variables. Most projection-defined populations consisted of multiple transcriptomic types (Figure 3B, right). Conversely, each transcriptomic type collectively projects to multiple targets (Figure 3B, left, colored by transcriptomic type). Re-mapping retrogradely labeled cells to a higher resolution version of the clusters reached similar conclusions (Figure S3B). Despite the divergence and convergence of projections, the targets of any given transcriptomic type exhibited specific biases. Importantly, our data revealed a special case: >97% of vmPFC→PAG neurons mapped to the Npr3 cluster (15-fold enrichment over random). While we refer to these neurons as “PAG projecting,” it is important to note that PAG is not the only target. Instead, vmPFC→PAG neurons extend collateral branches to multiple subcortical targets, including the hypothalamus as predicted in Figure 3B (Vander Weele et al., 2018; our unpublished data).
We next validated some of these observations by staining sections with PAG- or cPFC-projecting tdTomato+ cells using markers predicted to either co-label or not. cPFC-projecting cells co-labeled with the markers Cd44, Cxcr7, and Figf (Figure S3C) at proportions similar to what was observed in the sequencing data, but not with Npr3 (Figure S3D). Conversely, PAG-projecting cells co-labeled with Npr3 with high frequency, but not with Figf (Figures 3C and 3D) and were distributed throughout mPFC but not OFC (Figure S2F). Finally, double labeling of PAG- and cPFC-projecting cells confirmed that they did not overlap (Figure 3E).
Thus far, our study has parsed out different neuronal populations by their transcriptomic and projection signatures. However, the observation that vmPFC→PAG neurons are highly transcriptomically homogeneous gave a unique opportunity to examine the signal encoding properties of a neuronal population with high molecular homogeneity. As comparisons to the vmPFC→PAG neurons (hereafter the vmPFC→PAG class), we also examined the functional properties of the non-overlapping vmPFC→cPFC class, consisting mainly of 3 transcriptomic types (Figure 3B), as well as all Rbp4Cre-labeled neurons as a third class—thereby applying transcriptomic insights to the study of signal encoding.
Silencing vmPFC interferes with a two-alternative forced choice task
We sought to explore how different vmPFC cell classes contributed to the core function of decision-making by imaging single-cell level neural activity. We adapted a two-alternative forced choice (2AFC) task in freely moving mice (Figure 4A; Uchida and Mainen, 2003; Feierstein et al., 2006), which could reveal differences in how cells represent diverse sensory, motor, and cognitive signals. We trained water-restricted mice to nose poke into a center port, which triggered the immediate release of one of two odor cues. Each odor was associated with a 4 μL water reward delivered from a port either to the left or right side, which the mouse would receive only upon licking the correct port (Figure 4A). The task was self-paced, and mice freely initiated trials and reported decisions. Over a 2–3 week period, mice learned to perform the task (valeric acid or VA → left; 1-hexanol or 1H → right) at high accuracy (> 90%) over hundreds of trials (268 ± 10 trials in a single session, mean ± SEM).
Figure 4. vmPFC is engaged by a two-alternative forced choice task.
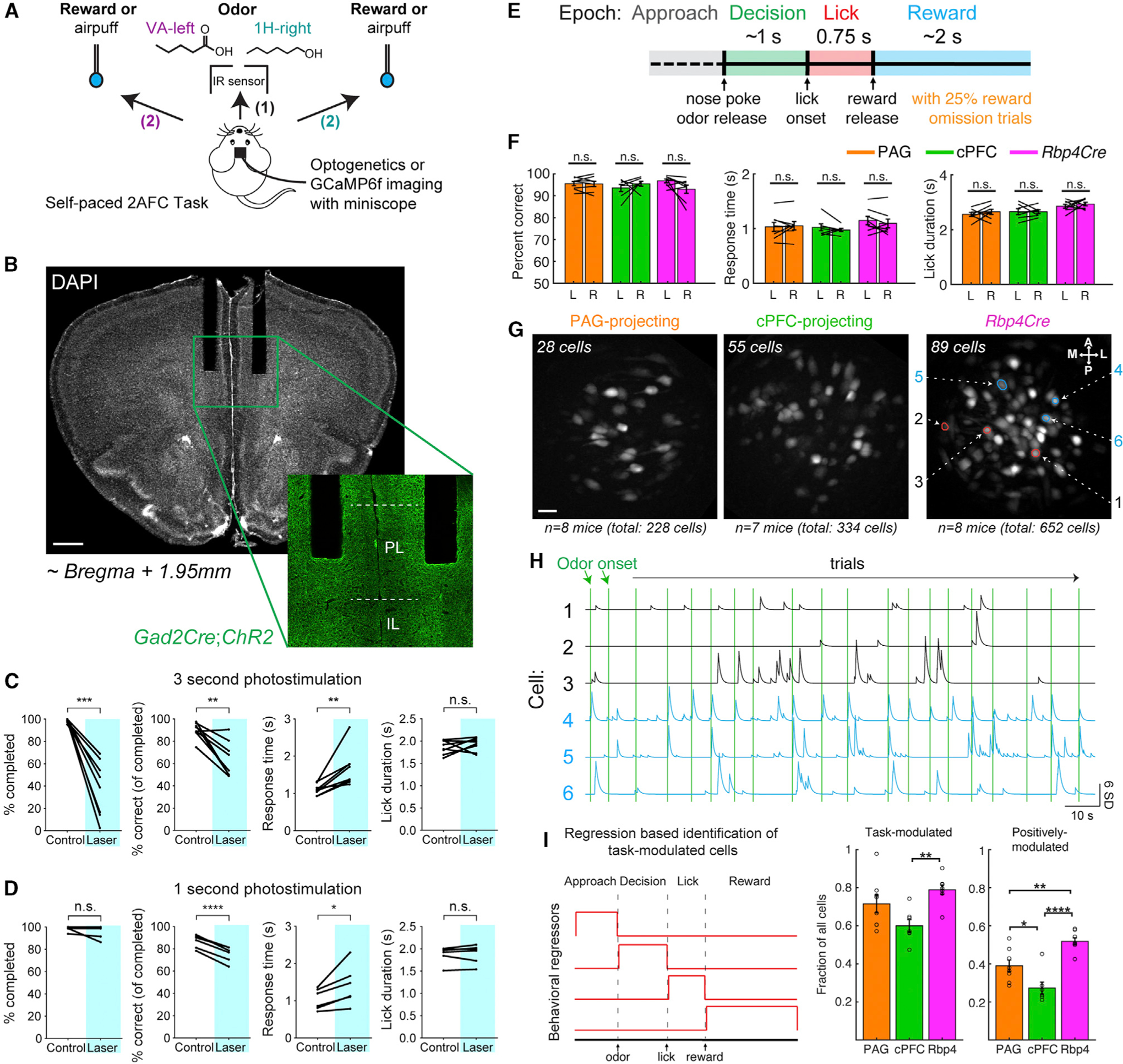
(A) Self-paced 2AFC task for freely moving mice. Mice were trained to nose poke in a center port (1), discriminate between two odor cues (presented for up to 1 s), and move to the correct reward port to obtain a 4 μL water reward (2). Incorrect cue-outcome associations resulted in a brief air puff punishment.
(B) Bilateral optogenetic fibers implanted into PL (A–P: +1.95, M–L: ± 0.35, D–V: −2.3) of mice expressing ChR2(H134R) in all inhibitory neurons (Gad2Cre;Ai32). Scale, 500 μm.
(C, D) Behavioral effects of vmPFC optogenetic inhibition. Photostimulation lasted 3 s (C) or 1 s (D) starting at nose poke, randomly interleaved on 25% of trials. Response time was calculated on all completed trials, and lick duration was calculated only for correct trials. Paired t test was used.
(E) Trial structure and definition of 4 task epochs for imaging.
(F) Performance metrics of mice during imaging for left and right trial types (Rbp4Cre: n = 8, PAG-projecting: n = 8, and cPFC-projecting: n = 7 mice; paired t test). Left: L, Right: R.
(G) Example fields of view for the 3 cell classes, from 2-odor task imaging. Cre-dependent GCaMP6f expression (Ai148) was from CAV-Cre injections at target sites or from crossing to Rbp4Cre. Images are maximum intensity projections from a typical FOV. Rings are example regions-of-interest (ROIs) from CNMF-E. Scale, ~25 μm.
(H) Ca2+ signals (CNMF-E denoised) for 6 highlighted cells (G, right). 3 are time-locked (blue) and 3 are not time-locked (black). Vertical green lines denote odor onset following each voluntary nose poke/trial.
(I) Determination and quantification of cells with task-modulated activity. Four behavioral regressors were used for linear regression, separately for left and right trial types (left). Average fraction of imaged cells that were significantly modulated (middle: positively and negatively; right: only positively) for each cell class (circles represent individual mice, one-way ANOVA, post hoc Tukey’s HSD test).
In this and subsequent figures, mean ± SEM is displayed. n.s.: not significant, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Ca2+ data is represented as the Z score (SD) of the fluorescence intensity signal of single cells.
See also Figure S4.
We first tested whether neural activity in vmPFC of trained mice was important for task performance. Optogenetic activation of cortical inhibitory neurons is an effective strategy to silence specific cortical areas (Pfeffer et al., 2013; Guo et al., 2014b). We implanted bilateral fiber optic cannulae above vmPFC (Figures 4B and S4A) of Gad2Cre;Ai32 double transgenic mice (Taniguchi et al., 2011; Madisen et al., 2012), which expresses channnelrhodopsin (ChR2) in all cortical GABAergic inhibitory neurons. Photostimulation for 3 s beginning at odor onset decreased task performance (Figure 4C). Photostimulation trials had a significantly reduced completion rate: mice often failed to report a choice within the allotted time (4 s) despite receiving an odor cue. Among completed trials, the error rate and response time were increased. The duration of licking for reward on correct trials was not affected, however, arguing against a general motor defect. Photostimulation for 1 s did not affect the proportion of completed trials, but the error rate and response time were similarly increased (Figure 4D), suggesting impaired decision-making. Together, these data suggested that vmPFC is required for proper execution of the 2AFC task (see Limitations of study section and STAR Methods) and prompted us to characterize task-related signals encoded by different vmPFC projection neuron classes.
Imaging task-relevant Ca2+ dynamics in vmPFC classes
We modified the task design for imaging by adding a delay period to de-correlate licking and reward consumption. Mice first approached the center port (Approach epoch, ~1 s), discriminated 2 odors to choose between left and right (Decision epoch, ~1 s), reported their choice by licking (Lick epoch, 0.75 s), and received a water reward 0.75 s after the first lick (Reward epoch, ~2 s) (Figure 4E). To isolate cell activity correlated with reward, we also randomly omitted reward in 25% of the trials (omission trials) and collectively refer to this design as the “2-odor” task. Mice on average did not exhibit significant biases in performance that correlated with the side (Figure 4F).
We utilized a Cre-dependent GCaMP6f mouse (Ai148; Daigle et al., 2018) to label each of the three classes in separate cohorts: PAG-CAV-Cre (1 transcriptomic type), cPFC-CAV-Cre (3 transcriptomic types that exclude the PAG-CAV-Cre type), and Rbp4Cre (7 transcriptomic types including the above two classes). Mice were implanted with a gradient-index lens (GRIN: 500 μm wide) above vmPFC (Figure S4B), through which we performed Ca2+ imaging at cellular resolution using the Inscopix mini-endoscopic system (Ghosh et al., 2011; Stamatakis et al., 2018). Figures 4G and S4C show fields of view (FOV) for all imaged mice. Figure 4H shows example fluorescence traces of 6 cells (FOV in Figure 4G, right panel) whose Ca2+ transients did (blue) or did not correlate (black) with odor onset.
To determine task-relevant activity, we defined a set of behavioral regressors representing the four task epochs for left and right trial types separately (Figure 4I, left) and performed linear regression with the cell activity. Cells were considered task-modulated if at least one regression coefficient was significant when compared to shuffled data that randomized the regressor-activity relationship. Quantifying task-modulated cells as a proportion of all imaged cells per mouse revealed that across all 3 cell classes, 60%–80% of cells were modulated in at least one epoch. However, cPFC-projecting cells were less likely to be modulated compared to the other classes (Figure 4I).
Cell classes are differentially recruited by task epoch
Do different classes have enriched activity in different epochs? We first analyzed the Ca2+ signal of single cells in individual trials and calculated trial-averaged traces aligned to odor onset or first lick. We found cells with activity significantly elevated during each of the four epochs in all three classes (Figures 5A and S5A). Next, we identified positively modulated cells across all mice of each class, and sorted cells by the time of their maximum trial-averaged activity aligned to odor onset and first lick (Figure 5B). In all three cases, peak response times tiled the entire task. This pattern was not an artifact of sorting, as cross validation of averaged data from only even trials but subsequently aligned to odd trials produced nearly identical results (Figure S5B). Thus, there is no absolute relationship between neuron class and the time within the task that it is active; even a molecularly homogeneous class of neurons (PAG projecting) encodes diverse signals.
Figure 5. Differential enrichment of activity across epochs between cell classes.
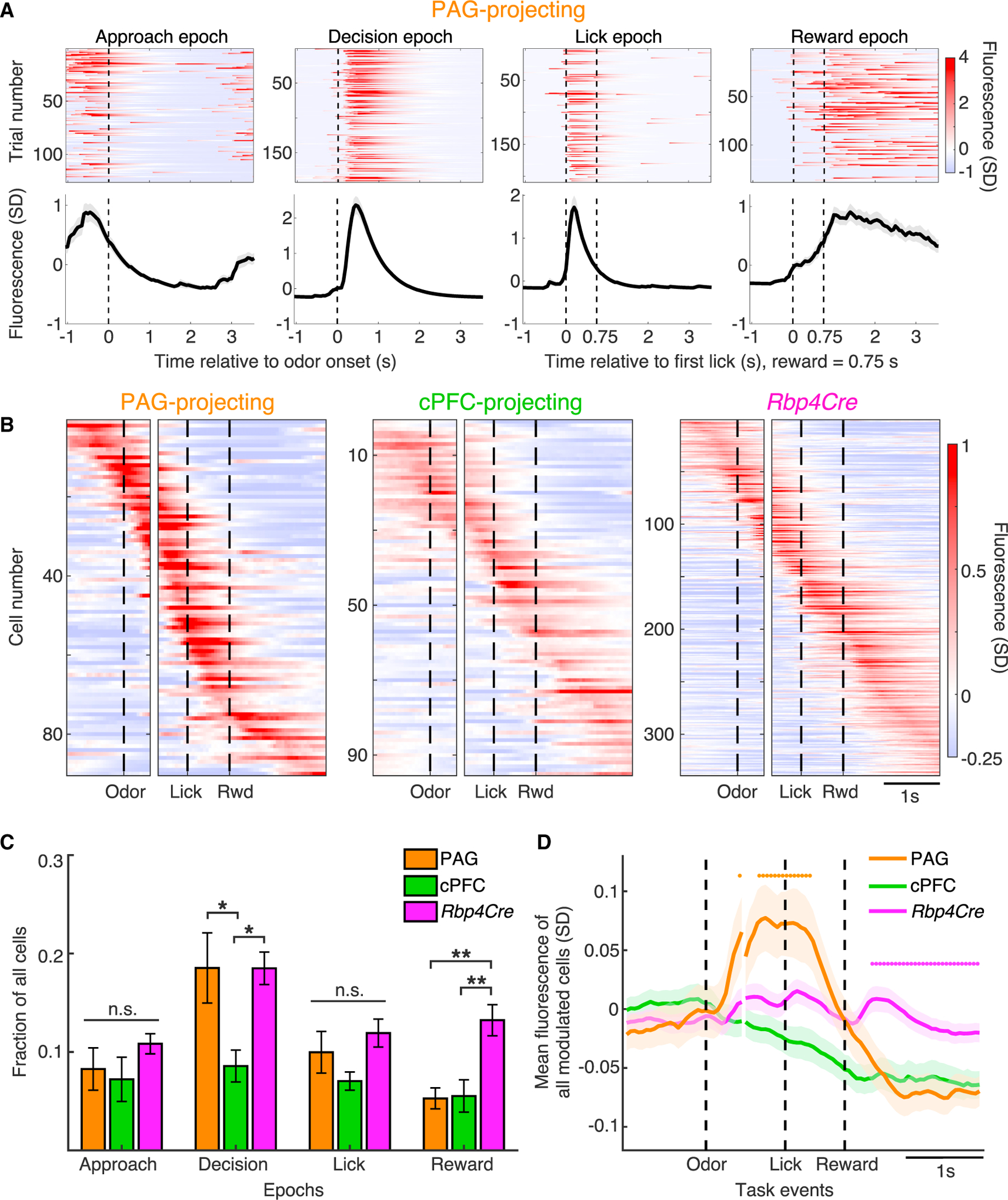
(A) Example single-trial (top) and corresponding trial-averaged activity (bottom) of significantly modulated PAG-projecting cells during the 4 task epochs defined in Figure 4E. Traces include all correct trials. Vertical dashed line in Approach/Decision epochs denotes odor onset. Vertical dashed lines in Lick/Reward epochs denote first lick (left) and reward delivery (right).
(B) Trial-averaged activity of all positively modulated cells sorted by time of maximal activity and grouped by cell class. Panels are aligned to odor onset (left) and first lick (right) (n = 90 PAG-projecting, 95 cPFC-projecting, 339 Rbp4Cre-labeled cells).
(C) Cells positively modulated in each of the four task epochs as a fraction of all imaged cells, on a per-mouse basis (one-way ANOVA, post hoc Tukey’s HSD test).
(D) Average activity trace of task-modulated cells aligned to odor onset and first lick, for each cell class (n = 168 PAG-projecting, 205 cPFC-projecting, 518 Rbp4Cre-labeled cells).
For this and subsequent figures, orange, green, or magenta dots represent PAG-projecting, cPFC-projecting, or Rbp4Cre traces being significantly different than the other two, respectively. Black dots represent where PAG-projecting and cPFC-projecting is significantly different. p < 0.05, one-way ANOVA, post hoc Tukey’s HSD test.
See also Figure S5.
We then classified positively modulated cells based on the epoch wherein statistical significance was first reached. On a per-mouse basis, the PAG-projecting and Rbp4Cre-labeled classes had a greater proportion of Decision epoch-modulated cells compared to the cPFC-projecting class. Conversely, the PAG- and cPFC-projecting classes had a lower proportion of Reward epoch-modulated cells compared to Rbp4Cre (Figure 5C). Thus, although activity of cells in each class tiled the entire trial, the amount of modulation varies in each epoch. To test whether cell classes exhibit quantitative activity differences at the population level, we computed average signals for all task-modulated cells (Figure 5D). The Rbp4Cre trace exhibited two periods of elevated signal: following odor and reward onset. By contrast, the PAG-projecting trace had elevated signal after odor onset, whereas the predominant effect in the cPFC-projecting trace was negative modulation. Furthermore, the magnitude of the PAG-projecting trace was significantly greater than the other two traces during the Decision and Lick epochs, whereas the Rbp4Cre trace was greater during the Reward epoch. Finally, comparing rewarded and reward omission trials (Figures S5C–E) revealed that reward omission was represented in each class by a net loss of activity, with the largest change in Rbp4Cre-labeled cells. These results demonstrate that despite heterogeneity at the single cell level, each class has distinct population level signatures.
vmPFC→PAG neurons contain the most information about choice direction
Choice-specific information has been observed across multiple regions of cortex (Feierstein et al., 2006; Harvey et al., 2012; Guo et al., 2014b; Li et al., 2015; Driscoll et al., 2017; Wagner et al., 2019). Similarly, many vmPFC cells exhibited activity selective not only for the task epoch, but also for left or right choice directions (Figures 6A and S6A). To explore this important task signal, we first analyzed data at the population level by pooling cells from all imaged mice. To visualize choice direction-selective activity over time within a typical trial, we computed trial-averaged activity traces for each cell separately on left and right correct trials, and grouped all cells into a time-varying high-dimensional neural activity trajectory, where each axis represents the activity of a single neuron (Churchland et al., 2006; Shenoy et al., 2013). We used principal component analysis (PCA) and plotted neural activity trajectories using the first 3 PCs, which accounted for ~70% of the variance in the data (Figures 6B and S6B). Prior to odor onset, trajectories of left and right trials were very similar. Upon odor onset, the trajectories rapidly diverged, which persisted through the Reward epoch. Similar results were obtained with each cell class separately visualized in the same PC space (Figure 6C). However, the magnitude of trajectory divergence between left and right trials differed: PAG-projecting cells diverged the most after odor onset, followed by Rbp4Cre-labeled cells, and then cPFC-projecting cells. This suggested that the PAG-projecting class contains the most choice direction-specific information.
Figure 6. Choice direction-specific information differs quantitatively between cell classes.
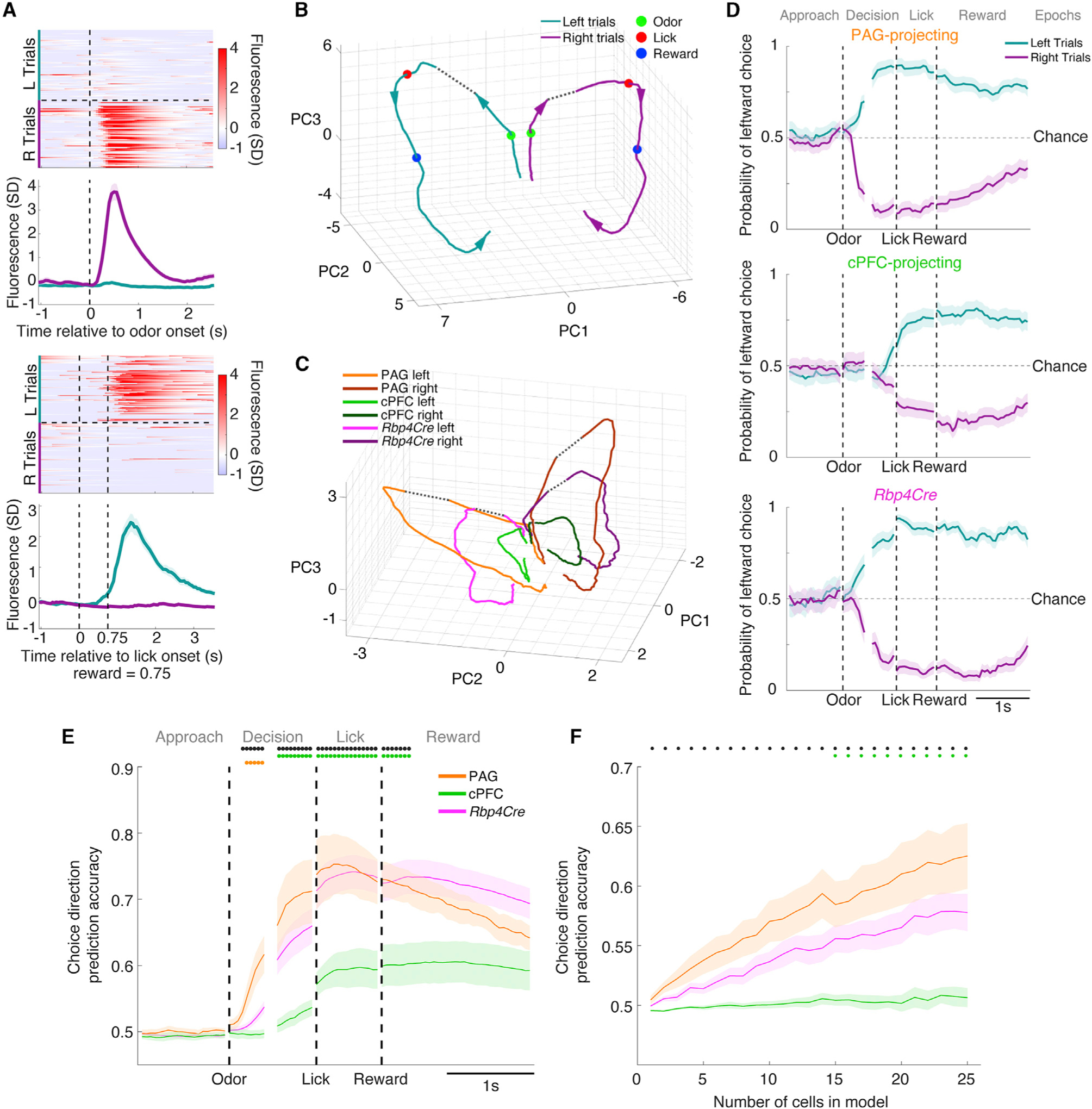
(A) Example single-trial (upper) and corresponding trial-averaged activity (lower) of two choice direction-selective cells.
(B) Population neural activity trajectories of trial-averaged correct left and right trials represented using the first three PCs in activity state space. Arrows denote the direction of time. Green, red, and blue dots represent onset of odor, lick, and reward delivery, respectively. All imaged cells are included (n = 1214). Dotted lines connect data between the two alignment points.
(C) Similar to (B), but neural activity trajectories are subdivided by cell class and randomly subsampled to 200 cells per class.
(D) Choice direction prediction accuracy using a logistic regression model, shown over time across the four epochs, with one example mouse for each cell class. Values toward 1 or 0 indicate accurate left or right choice direction prediction, respectively.
(E) Average choice direction prediction accuracy across mice (n = 5 PAG-projecting, 5 cPFC-projecting, and 8 Rbp4Cre-labeled mice), from data randomly subsampled to 25 cells per mouse (one-way ANOVA, post hoc Tukey’s HSD test).
(F) Average choice direction prediction accuracy during the Decision epoch as a function of the number of cells included in the logistic regression analysis.
See also Figures S6A and S6B.
We next sought to confirm these observations across individual mice where cells were simultaneously imaged. For each mouse, we performed logistic regression to compute a time-varying prediction of choice direction in each epoch (Kiani et al., 2014). Data from PAG-projecting and Rbp4Cre-labeled example mice showed that predictions were at chance levels before odor onset, which increased in accuracy after odor onset and lasted through the Reward epoch. cPFC-projecting cells showed a similar pattern, but prediction accuracy improved later, suggesting less choice direction-specific information in the Decision epoch (Figure 6D). To quantify this and compare cell classes, we randomly subsampled the number of cells used in the regression model to 25 per mouse and calculated the average prediction accuracy for choice direction over time (Figure 6E). This confirmed that the PAG-projecting class contained more information than the cPFC-projecting class, particularly in the Decision epoch, with the Rbp4Cre-labeled class falling in between. Finally, we asked how this information accumulated over increasing numbers of cells. Fewer cells in the PAG-projecting class were needed to reach a given prediction accuracy compared with the cPFC-projecting class, indicating a greater amount of information regardless of population size (Figure 6F). Thus, PAG-projecting cells more potently encode choice direction than cPFC-projecting cells.
Dissecting behavioral variables with two additional tasks
Many of the critical moments of the 2-odor task occur in the Decision epoch, when the mice must interpret the odor identity, make a left versus right choice, and implement their decision by moving to the correct port. However, because the mice are freely moving, we could not dissociate these events. Thus, we devised two additional tasks. First, to determine the extent that different odor cues leading to the same choice are generalized, we designed a 4-odor task: mice were trained to associate one additional odor cue with each side (Left 2: (S)-carvone; Right 2: (R)-carvone; a pair of enantiomers). Second, to determine whether activity observed during the Decision epoch was specific to this task, or if similar activity was evoked whenever a general reward-seeking action toward the left or right ports is made, we designed an uncued task: mice perform similar movements, but do not need to discriminate odors. In this task, nose poke at the center port triggered an immediate reward at the left port for a block of trials, followed by a similar block of trials at the right port.
Mice that contributed to the 2-odor dataset were trained for an additional 2–3 weeks on the 4-odor task (but not the uncued task), and re-imaged while performing the 4-odor task at expert levels. This was immediately followed by the uncued task within the same imaging session (Figures 7A and S6C). Mice transitioned quickly from the 4-odor task to the uncued task and attained high performance (> 80%) within 10–20 trials (Figure S7A), and performed hundreds of trials within one session (165 ± 4 4-odor trials, 50 ± 3 left block trials, 53 ± 3 right block trials, mean ± SEM). The 4-odor task generally recapitulated our previous findings. Sorted, trial-averaged activity in the 4-odor task revealed a similar tiling pattern (Figure 7B) and distribution of task-modulated cells across epochs (Figure S7B) to the 2-odor task (Figures 5B and 5C). Prediction of left versus right choice directions was also similar, with PAG-projecting cells containing more information than cPFC-projecting cells (Figures S7C and S7D).
Figure 7. Two additional cognitive tasks reveal how cell classes differentially encode task signals.
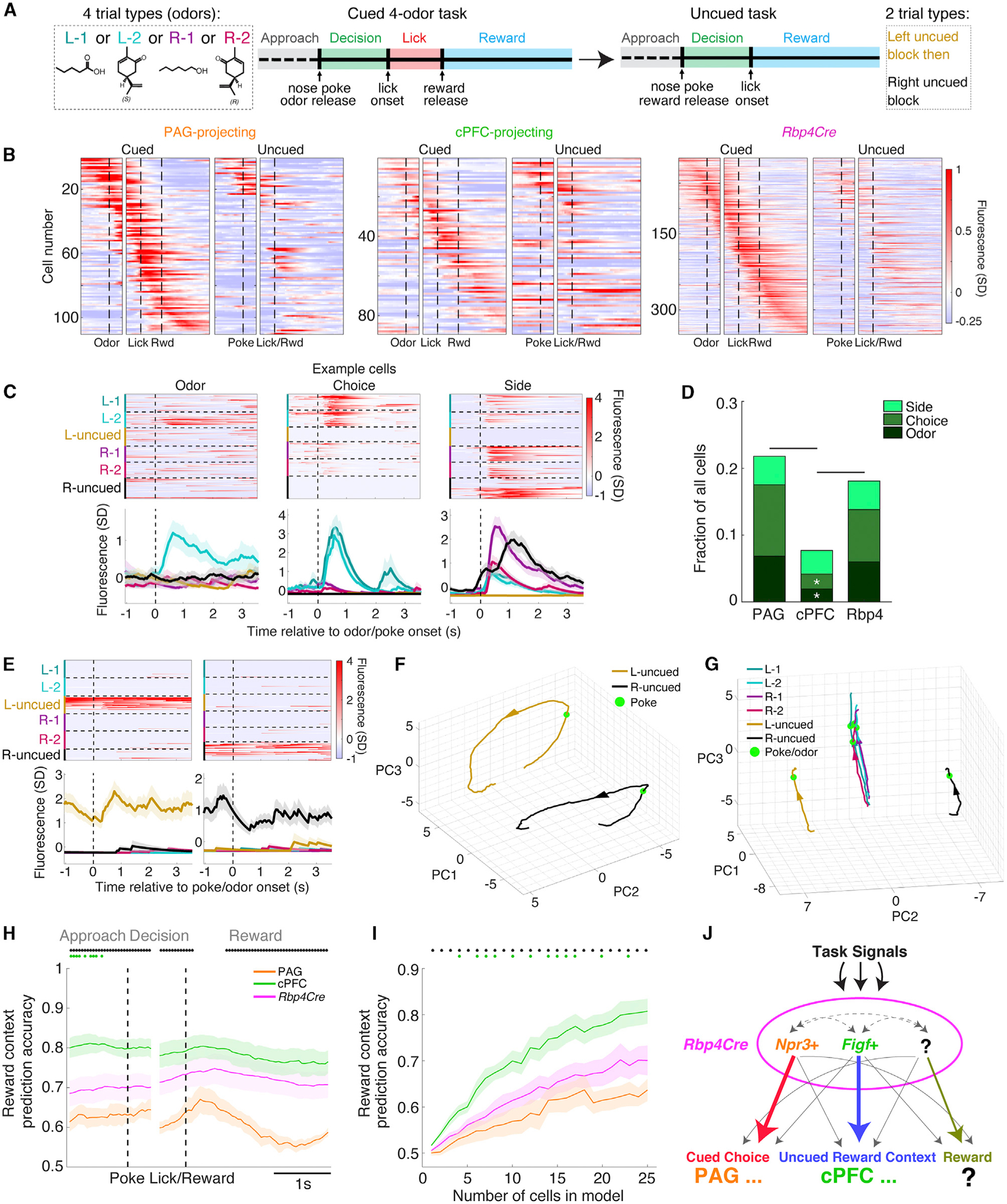
(A) Task design. Mice first discriminated four possible odors to receive a 4μL reward per successful trial. They then immediately switched to an uncued task of repeated left (L-uncued) or right (R-uncued) trials in blocks, resulting in six trial types in the same imaging session.
(B) Trial-averaged activity of all positively modulated cells during the cued task (left) sorted by the time of maximal activity and grouped by cell class (n = 110 PAG-projecting, 89 cPFC-projecting, 348 Rbp4Cre-labeled cells) followed by trial-averaged activity of the same cells during the uncued task (right).
(C) Example Odor- (L-2 only), Choice- (L-1 + L-2), and Side-selective (R-1 + R-2 + R-uncued) cells with single-trial (top) and trial-averaged activity (bottom). Vertical dashed line denotes nose poke/odor onset.
(D) Proportions of cells positively modulated in the Decision epoch, grouped by cell class, and categorized as Odor-, Choice-, or Side-selective using linear regression. Comparison across classes was performed using a permutation test.
(E) Example cells with preferential activity during L-uncued trials (left) or R-uncued trials (right). Vertical dashed line denotes nose poke/odor onset.
(F) Population neural activity trajectories summarizing trial-averaged traces of left versus right uncued trials using the first three PCs in activity state space. All imaged cells were included (n = 1248).
(G) Same as (F) except that all six trial types are plotted, and only the Approach epoch leading up to the nose poke/odor onset is analyzed.
(H) Average reward context prediction accuracy (left versus right block-type) over the course of the uncued task, across mice (n = 4 PAG-projecting, 5 cPFC-projecting, and 8 Rbp4Cre-labeled mice), from data randomly subsampled to 25 cells per mouse (one-way ANOVA, post hoc Tukey’s HSD test).
(I) Average reward context prediction accuracy during the Approach epoch as a function of the number of cells included in the logistic regression analysis.
(J) Schematic summary. Rbp4Cre-labeled cells in vmPFC are divided into cell classes defined by differential gene expression (Npr3+; Figf+/Cxcr7+/Cd44+, simplified as Figf+), which predominantly route different information (cued choice or uncued reward context) to different targets (PAG or cPFC). Note that PAG and cPFC are not the only sites these neurons project to. Our data also suggest that Rbp4Cre-labeled cells contain a subclass that preferentially encodes reward, distinct from PAG- or cPFC-projecting cells.
See also Figures S6C and S7.
However, many cells with task-modulated activity in the 4-odor task exhibited either little task-modulated activity or task-modulated activity with different temporal profiles in the uncued task, supporting the task specificity of the observed neural activity (Figure 7B). Importantly, this difference was not explained by the passage of time, as a comparison of the 4-odor data broken down into first and second halves did not reveal a difference compared with the uncued segment (Figure S7E). Furthermore, inclusion of the uncued data increased the number of PCs required to explain the variance in the dataset, indicating that the uncued task generates activity patterns distinct from the cued task (Figure S7F).
Differential encoding of odor, choice, and side revealed from the 4-odor and uncued tasks
Neural activity enriched in the Decision epoch could be associated with a specific odor (hereafter Odor), both odors for a particular choice direction (hereafter Choice), or movement to one particular side (hereafter Side). The data from the 4-odor and uncued tasks provide us with six contrasting trial types to dissociate these variables. Indeed, we found cells with activity selective for a specific odor, for two odors that predict reward on a specific side, and for movements toward a specific side, corresponding to the Odor, Choice, and Side definitions above (Figure 7C).
To quantify this, for each cell with positive modulation during the Decision epoch, we performed linear regression analysis with six behavioral regressors, each representing one of the six trial types (L-1, L-2, R-1, R-2, L-uncued, R-uncued). We determined which regression coefficient (or group of coefficients) was significant by comparing the true coefficient with those generated from random shuffles of the activity-regressor relationship (STAR Methods). From this, we classified cells into: Odor (modulated by L-1 or L-2 or R-1 or R-2 regressors), Choice (modulated by L-1 and L-2; or R-1 and R-2 regressors), or Side (modulated by L-1 and L-2 and L-uncued; or R-1 and R-2 and R-uncued regressors) categories (Figure 7D). Side-selective cells were in similar proportion across all three classes. Odor- and Choice-selective cells, however, were represented in PAG-projecting and Rbp4Cre-labeled cells at a significantly higher proportion compared to the cPFC-projecting class. These results highlight the complexity of task encoding and validate choice as a more abundantly represented signal in the PAG-compared to cPFC-projecting class.
vmPFC→cPFC neurons preferentially represent reward context in the uncued task
In contrast to the 2- and 4-odor tasks, where the mouse is uncommitted to a choice before odor presentation, choice-specific information is potentially represented even before nose poke in the uncued task. Indeed, we observed cells that exhibited activity only during the uncued trials and specific to one of the two block-types (Figure 7E). In contrast to the transient activity characteristic of the cued trials, some of these cells exhibited heightened activity throughout each trial, including the period prior to nose poke (Figure 7E). Representing the aggregate data during left versus right uncued trials as trajectories in activity state space (Figure 7F), we observed that the trajectories never came close to each other. In addition, comparing trajectories in the Approach epoch leading up to nose poke/odor onset demonstrated clear overlap between the cued trial types, and segregation between the uncued trial types (Figure 7G). Together, these data demonstrate differences in activity state between the cued and uncued tasks, and provide evidence that information about block-type, or the reward context, is present in vmPFC persistently through each trial.
To address how reward context is represented across cell classes in uncued trials, we performed a regression analysis similar to Figures 6D–6F. Left versus right reward context could be predicted from all three classes, but this information was most potent in cPFC-projecting cells (Figures 7H and 7I). The prediction accuracy of reward context by cPFC-projecting cells was consistently higher than that of PAG-projecting cells throughout most of the trial, with Rbp4Cre-labeled cells falling in between (Figure 7H). This was also the case across regression models that included varying numbers of cells (Figure 7I). Thus, despite weak recruitment with no consistent signatures of behavioral encoding in the cued tasks, cPFC-projecting cells are particularly important in signaling the reward context that the mouse is in during the uncued task. Overall, these results emphasize how all three of the cell classes that we studied here have both common information, as well as individual biases in representing specific task signals (Figure 7J).
DISCUSSION
Molecular neuroscience has invested substantial effort to survey transcriptomic heterogeneity of neurons in a variety of brain regions. In contrast, systems neuroscience widely ignores cell-type information when positing and testing mechanisms of information encoding, a practice implicitly justified by the redundant neural activity and recurrent connectivity in higher brain regions (Harris and Mrsic-Flogel, 2013; Shenoy et al., 2013). How do we reconcile these views? Our PFC dataset uncovered transcriptomic types consistent with those from studies that profiled the mouse brain exhaustively (Saunders et al., 2018; Zeisel et al., 2018), other parts of the cortex (Tasic et al., 2018), or PFC more specifically (Bhattacherjee et al., 2019), but we focused on using L5 pyramidal neurons and their projection mapping as a case study to interrogate task encoding properties with a cell-type framework. Using PAG-projecting neurons as an example of molecular homogeneity due to its invariant clustering across multiple resolutions and methods (Figures 1E and S1B), we found that even a homogeneous transcriptomic type encodes diverse information. However, different cell classes preferentially contributed to different aspects of task encoding, suggesting that each transcriptomic type makes quantitatively different contributions to behavior. Given that this mapping is possible even when intentionally focusing on a cortical region known for its complexity, molecular atlas-building efforts will undoubtedly continue to provide a useful framework for analyzing cell function.
Relationship between axon projection patterns and transcriptomic types
Mesoscale axon collateralization patterns of neuron populations (Oh et al., 2014; Zingg et al., 2014; Harris et al., 2019) present reproducible, highly complex innervation patterns to many regions. However, as these populations are subdivided, the extent that projection patterns are subdivided into individual “channels” of output to specific targets or retain an overlapping “broadcast” pattern to multiple targets varies substantially across systems. Projection-defined locus coeruleus norepinephrine neurons have overlapping patterns of output collateralization (Schwarz et al., 2015; Kebschull et al., 2016), whereas ventral tegmental area projection-defined dopamine neurons (Beier et al., 2015) or arcuate AGRP neurons (Betley et al., 2013) have substantially less axon overlap.
A current assumption is that dividing neuron populations based on transcriptomic criteria will be helpful in deciphering the correspondence between transcriptomic versus projection types. Our data indicate that this is not necessarily the case, at least in PFC (Figures 3 and S3). Our finding of a one-to-many and many-to-one mapping between transcriptomic and projection types undoubtedly underlies the complexity of PFC information flow. Evidence from single neuron axon arborization patterns (Lin et al., 2018; Gong et al., 2016; Winnubst et al., 2019; Ren et al., 2019) or MAPseq (Kebschull et al., 2016; Han et al., 2018; Chen et al., 2019) indicate how morphologies of individual neurons can be highly heterogeneous within genetically defined populations. For example, median preoptic nucleus excitatory neurons that signal thirst appear transcriptomically homogeneous, but also target axons to multiple sites with little collateralization (Allen et al., 2017a). One possible explanation could be that transcriptomic heterogeneity in development that is essential for establishing wiring specificity diminishes in adults (Li et al., 2017). Together, these observations suggest a limited extent to which projection patterns can be predicted by transcriptomic data in adult neurons.
Distribution of task information across cell types
Neural responses of individual cells in PFC tend to be highly heterogeneous and represent combinations of experimental and behavioral variables. This feature, referred to as mixed selectivity, has gained prominence as a mechanism for how PFC neurons represent task-related signals in a computationally efficient manner (Fusi et al., 2016; Rigotti et al., 2013). Indeed, all 3 of our examined vmPFC cell classes contained but had relatively small proportions of mixed selective cells based on their representation of choice direction and/or reward context (Figure S7G). Further, task-related signals in goal-directed, cognitive tasks are more generally distributed across the brain than previously thought (Hernández et al., 2010; Allen et al., 2017b and 2019; Steinmetz et al., 2019). Our study extends these perspectives by showing that even within a specific brain region, within a cortical layer, and within a transcriptomic type, a diversity of information is still present in the population and in individual cells, including cells that fulfill criteria of mixed selectivity.
A key feature of our study is our survey of behavioral encoding across multiple tasks. Performance of the uncued task directly after the 4-odor cued task highlights the flexibility of PFC: choice is potently represented by PAG-projecting cells in the cued task, but upon switching to the uncued task, mice adapt within tens of seconds, and reward context is potently represented by cPFC-projecting cells. This is an explicit example of how different cell types come into action in different tasks and underscores the importance of having a diverse behavioral repertoire when testing for cell-type specific encoding. This “division of labor” of cell types is echoed in a study showing that the number of required cortical areas and their dynamics varied across the difficulty of tasks (Pinto et al., 2019).
Neurons within frontal cortices exhibit persistent activity involved in short-term memory (Fuster and Alexander, 1971; Miller et al., 1996). Our finding that reward context is represented preferentially in intracortically projecting neurons suggests that cortico-cortical networks are likely a key player for this activity. While not explicitly tested, our observations may provide a transcriptomic and projection-based cellular context to the literature that has linked PFC to working memory (Spellman et al., 2015; Kamigaki and Dan, 2017; Bolkan et al., 2017; Schmitt et al., 2017) and the representation of decision variables such as value (Bari et al., 2019; Hirokawa et al., 2019), both of which could be in play during our uncued task. In summary, our study has exemplified that integration of transcriptome and axon projection analyses with signal encoding in a dynamic behavioral context bridges molecular and systems neuroscience approaches to studying cognitive functions.
Limitations of study
In this section we acknowledge and discuss several limitations of our study. The extent of spatial spread of retrograde viruses is not possible to quantify precisely, which is a known issue in the field. Because CAV and AAVretro transduce axon terminals as well as axons-in-passage (Schwarz et al., 2015; Tervo et al., 2016), both cell bodies and axons projecting to the injection site from long and short ranges are labeled. Therefore, close to the injection site, it is not possible to determine whether labeling is contributed from local circuitry or from the injection itself. We also acknowledge that both CAV and AAVretro have tropisms in different neuronal classes from different brain regions (Schwarz et al., 2015; Tervo et al., 2016), likely due to the differential expression levels of receptors for these viruses (Li et al., 2018). In general, this problem is likely less severe in our study because it compares the same general class of neurons from the same region (mostly L5 excitatory projection neurons in PFC). However, we cannot rule out that these factors play a role in our data.
Regarding the illumination zone and the extent of the optogenetics effect, we acknowledge that long-range projecting GABAergic cells are expected to exist in PFC, which could exert an inhibitory effect to projection targets outside of PFC (He et al., 2016). However, in our own sequencing data of retrogradely labeled cells, we found that out of 1160 labeled cells from 6 regions, only 5 were GABAergic (~0.4% expressed Gad2). This included 2 cells projecting to contralateral PFC, 2 cells projecting to hypothalamus, and 1 cell projecting to amygdala. This very low percentage in PFC is also consistent with studies from the Allen Institute that quantify other cortical areas (Tasic et al., 2018, also less than 1%). Furthermore, transcriptomic characterization of long-projecting GABA neurons suggest that they are likely very different from canonical inhibitory neurons, using slower neuroendocrine signaling in the place of fast neurotransmission (Paul et al., 2017). Taken together, while we acknowledge that the precise functional contribution of long-range GABAergic neurons in our assay is difficult to estimate, we believe it is unlikely to have exerted a major inhibitory effect brain-wide on the timescale of our assay.
Finally, optogenetic inhibition of projection-defined cells could test for causality in our choice behaviors. We did not pursue this avenue because: (1) Technically, the success of this experiment relies on our ability to label a large fraction of projection-defined cells with retrograde viruses for expressing inhibitory opsins, which may not be easily achievable (based on the relative abundance of PAG-CAV-Cre>tdTomato labeling compared with Npr3 labeling, we estimate that our current strategy labels less than 50%); (2) Conceptually, we already demonstrated a high degree of redundancy in the encoding of task signals across cell types, and under those circumstances, it is not clear what the interpretation of such functional experiments would be. Nevertheless, we acknowledge that these experiments could be a valuable future direction.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Liqun Luo (lluo@stanford.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Sequencing data is deposited at NCBI’s Gene Expression Omnibus, and is publicly available through GEO Series accession number GSE161936. The Ca2+ imaging data and analysis code has not been deposited in a public repository because it is in a non-standard format, but are available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All procedures, husbandry conditions, and housing conditions followed standard animal care and biosafety guidelines approved by Stanford University’s Administrative Panel on Laboratory Animal Care and Administrative Panel on Biosafety in accordance with NIH guidelines. To express tdTomato in Layer 5 projection neurons for sequencing, we crossed Rbp4Cre (Gerfen et al., 2013, mixed background) mice to Ai14 (Rosa-CAG-LSL-tdTomato-WPRE, C57BL/6J background; Madisen et al., 2010) mice. Mice were sacrificed at P34–P40 for single cell isolation and sequencing. A total of 11 mice were used for this purpose.
To express tdTomato in projection-defined neurons for sequencing, we also used Ai14 mice (mixed CD1, C57BL/6J background), injected CAV-Cre (Soudais et al., 2001) at target sites (ipsilateral dorsal striatum, nucleus accumbens, periaqueductal gray, and hypothalamus, as well as contralateral PFC) at P24–P35, and then sacrificed 7 days later at P31–P42, or injected AAVretro-Cre (Tervo et al., 2016) (amygdala) at P34, and then sacrificed at P49, for single cell isolation and sequencing. Each experiment pooled tissue from 2–4 mice, and each site other than dorsal striatum and nucleus accumbens had two separate batches. A total of 29 mice were used for this purpose. Only female mice were used in sequencing experiments.
To express ChR2 in inhibitory neurons for optogenetic silencing experiments, we crossed Gad2Cre (C57BL/6J background; Taniguchi et al., 2011) to Ai32 (Rosa-CAG-LSL-ChR2(H134R)-EYFP-WPRE, C57BL/6J background; Madisen et al., 2012) mice. Following this, we performed cannulae implantation, behavioral training, and optogenetics behavioral experiments. A total of 8 mice (3 males, 5 females) were used for this purpose.
To express the Ca2+ indicator GCaMP6f (Chen et al., 2013) in neocortical Layer 5 pyramidal cells for imaging, we crossed Rbp4Cre (mixed background) to the Cre-dependent GCaMP6f transgenic mouse line Ai148 (TIT2L-GC6f-ICL-tTA2, mixed background; Daigle et al., 2018). A total of 8 mice (6 males, 2 females) were used for this purpose. To express GCaMP6f in projection defined neurons for behavioral training and Ca2+ imaging, we used Ai148 mice, injected CAV-Cre at target sites at P28–P35, and then performed lens implantation surgeries 1 week later. A total of 15 mice (11 males, 4 females) were used for this purpose. The imbalance in male/female ratio was related to surgery survival rates, and was not intentional. For HCR-FISH validation of sequencing data, adult male and female mice aged P35–P60 on a CD1 and C57BL/6J mixed background were used. Prior to their training on the tasks used to generate the datasets in this study, mice were naive to the behavioral task, and gained their task expertise as described in the ‘Behavioral Training’ section below.
All mice used were adults and the reason for different ages was technical. We performed all the sequencing in younger adults so that we did not have to wait for mice longer than absolutely necessary, and also because cell dissociation is relatively easier in younger mice. For the dataset in Figure 1 that we used as a reference, all mice used were very close in age. For the optogenetics and imaging mice, we needed to wait until adulthood to perform surgeries, and they were older by the time their extensive behavioral training, and optogenetics/imaging was completed. We cannot rule out the effect of age in our experiments, but consider it very unlikely to have played a major role.
METHOD DETAILS
Single-Cell Sequencing
The procedure for isolating tdTomato+ cells for single-cell sequencing was identical between those labeled by Rbp4Cre (Figures 1 and S1) or CAV-Cre / AAVretro-Cre (Figures 3 and S3). Mice were briefly anesthetized with isoflurane and decapitated, and the brain was isolated in ice-cold ACSF (2.5 mM KCl, 7 mM MgCl2, 0.5 mM CaCl2, 1.3 mM NaH2PO4, 110 mM choline chloride, 25 mM NaHCO3, 1.3 mM sodium ascorbate, 20 mM glucose, 0.6 mM sodium pyruvate, bubbled in 95% O2 / 5% CO2). Brains were embedded in 3% low-melting point agarose (Fisher BP165–25) in ACSF at 37°C, cooled to 4°C, and then cut on a vibratome (Leica VT1200S) in either the coronal (mPFC dissections) or horizontal (OFC dissections) planes into 350-μm floating sections. To microdissect dmPFC, vmPFC, or OFC, we first identified the two adjacent tissue slices (total 700 μm) that most accurately spanned the following anatomical ranges: A–P: ~bregma 1.6 to 2.3 mm (for dmPFC and vmPFC), or D–V: ~bregma −1.5 to −2.2 mm (for OFC). Next we visualized the fluorescent tdTomato labeling and used the atlas as a guide, to cut out the regions of interest as accurately as possible. For dmPFC and vmPFC, we bisected the medial wall (~2.4 mm height × 1 mm width), into upper and lower portions. For Rbp4Cre-labeled cells, we collected tissue from both sides of the brain. There are no clear anatomical landmarks that delineate PFC subregions, but based on atlas boundaries, we conservatively estimate that the following subregions are contained in each dissection. dmPFC: cingulate and dorsal prelimbic cortex; vmPFC: ventral prelimbic, infralimbic, and medial orbital cortex; OFC: ventral and lateral orbital cortex. For retrogradely labeled cells, we only collected cells from the vmPFC ipsilateral to the injection site, except for contralateral PFC injections. Microdissected tissue was incubated at 37°C in papain enzyme mix (Worthington) + 800 nM kyneurenic acid for 30 min, and triturated gently with a P200 pipette every 15 min thereafter until fully dissociated, usually within 1 h of total incubation time. The cell suspension was spun down at 350 g for 10 min at room temperature, neutralized with ovomucoid inhibitor, spun again, washed in ACSF, stained with Hoechst for 10 min (1:2000, Life Technologies: H3570), washed, filtered (Falcon 532235), and resuspended in 2 mL ACSF.
FACS was performed using the Sony SH800 system with a 130-μm nozzle suitable for the large size of pyramidal neurons. Singlet cells were selected based on low FSC-W, and gated on Hoechst (nuclear stain that penetrates cell membrane) and tdTomato double positivity to identify labeled healthy neurons. Cells fulfilling these criteria were over 100 × brighter than background, and were unambiguously identifiable. Single cells were sorted at a low flow rate (< 100 events/second), and at the highest purity setting (Single Cell) into 96- or 384- well hard shell PCR plates (BioRad HSP9601 or HSP3901) containing 4 or 0.4 μL lysis buffer [0.5 U Recombinant RNase Inhibitor (Takara Bio, 2313B), 0.0625% Triton X-100 (Sigma, 93443–100ML), 3.125 mM dNTP mix (Thermo Fisher, R0193), 3.125 μM Oligo-dT30VN (Integrated DNA Technologies, 5′AAGCAGTGGTATCAACGCAGAGTACT30VN-3′) and 1:600,000 ERCC RNA spike-in mix (Thermo Fisher, 4456740)] in each well, respectively. Following FACS, plates were spun down, sealed and stored at −80°C.
cDNA synthesis and library preparation protocols were adapted from the SMART-Seq2 protocol (Picelli et al., 2014). 96-well versus 384-well processing utilized 4 μL or 0.4 μL starting volumes, respectively, and will hereafter be referred to as 1 unit. Plates were first thawed on ice followed by primer annealing (72°C, for 3 min, then on ice). For reverse transcription, 1.5 units of reaction mix [16.7 U/μL SMARTScribe Reverse Transcriptase (Takara Bio, 639538), 1.67 U/μL Recombinant RNase Inhibitor (Takara Bio, 2313B), 1.67 × First-Strand Buffer (Takara Bio, 639538), 1.67 μM TSO (Exiqon, 5′-AAGCAGTGGTATCAACGCAGAGTGAATrGrGrG-3′), 8.33 mM dithiothreitol (Bioworld, 40420001-1), 1.67 M Betaine (Sigma, B0300-5VL) and 10 mM MgCl2 (Sigma, M1028-10X1ML)], was added to each well either manually (96-well) or with a Formulatrix Mantis liquid handler (384-well). The reaction was then carried out by incubating wells on a thermocycler (Bio-Rad) at 42°C for 90 min, and stopped by heating at 70°C for 5 min. Subsequently, 3.75 units of PCR mix [1.67 × KAPA HiFi HotStart ReadyMix (Kapa Biosystems, KK2602), 0.17 μM IS PCR primer (IDT, 5′-AAGCAGTGGTAT CAACGCAGAGT-3′), and 0.038 U/μL Lambda Exonuclease (NEB, M0262L)] was added to each well. PCR was then performed using the following program: 1) 37°C for 30 min, 2) 95°C for 3 min, 3) 21 cycles of 98°C for 20 s, 67°C for 15 s and 72°C for 4 min, and 4) 72°C for 5 min. For 96-well plate samples, cDNA from every well was purified using 0.7 × AMPure beads (Fisher, A63881), quantified by a Fragment Analyzer (AATI), and diluted to 0.15 ng/μL in Tris-EDTA before tagmentation. For 384-well samples, cDNA from every well was quantified using Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher: P11496), and diluted to 0.4 ng/μL in Tris-EDTA before tagmentation.
For both 96-well and 384- well samples, before tagmentation, we reformatted the samples into a standardized 384-well format, and used the Formulatrix Mantis and Mosquito (TTP Labtech) to automatically perform all liquid handling steps. Tagmentation was performed on double-stranded cDNA using the Nextera XT Library Sample Preparation kit (Illumina, FC-131-1096). Each well was mixed with 0.8 μL Nextera tagmentation DNA buffer and 0.4 μL Tn5 enzyme, then incubated at 55°C for 10 min. The reaction was stopped by adding 0.4 μL Neutralization Buffer and centrifuging at room temperature at 3,220 g for 5 min. Indexing PCR reactions were performed by adding 0.4 μL of 5 μM i5 indexing primer, 0.4 μL of 5 μM i7 indexing primer, and 1.2 μL of Nextera NPM mix. PCR amplification was carried out using the following program: 1) 72°C for 3 min, 2) 95°C for 30 s, 3) 12 cycles of 95°C for 10 s, 55°C for 30 s and 72°C for 1 min, and 4) 72°C for 5 min. After library preparation, wells of each 384-library plate were pooled using a Mosquito liquid handler, and consolidated into one tube. Pooling was followed by two final purifications using 0.8 × AMPure beads (Fisher, A63881).
Library quality was assessed using capillary electrophoresis on a Fragment Analyzer (AATI), and libraries were quantified by qPCR (Kapa Biosystems, KK4923) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Libraries were sequenced on NextSeq 500 or NovaSeq 6000 Sequencing Systems (Illumina) using 2 × 75-bp or 2 × 100-bp paired-end reads, respectively. Sequences were de-multiplexed using bcl2fastq. Reads were aligned to the mouse mm10 genome (with Cre and tdTomato genes added) using STAR version 2.5.4 (Dobin et al., 2013). Gene counts were produced using HTseq version 0.10.0 (Anders et al., 2015), for only exons, with the ‘intersection-strict’ flag.
Histology
Adult mice were perfused transcardially with phosphate buffered saline (PBS) and 4% paraformaldehyde (PFA). Brains were extracted, post-fixed overnight in 4% PFA, cryoprotected in 30% sucrose/PBS for 48 h, embedded in OCT, snap-frozen, and stored at −80°C. For HCR-FISH (Figures 2A, 2C, 2D, S2B, S2C, S2D, 3C, S3C, S3D), 20-μm frozen sections were cut on a cryostat and dried on slides for 4 h. For immunolabeling and other non-HCR-FISH histology (Figures 3E, S2E, S2F, 4B, S4A, S4B), 50-μm floating sections were cut on a cryostat.
For staining HCR-FISH slides, all probe and wash reagents were from Molecular Instruments, and glassware baked at 180°C was used. Samples were treated with 4% PFA for 15 min, PBS for 5 min, Proteinase K buffer (1:100 1M TrisHCl, 1:500 0.5 M EDTA, and 14 μg/mL Proteinase K, in dH2O) for 6 min, 4% PFA for 10 min, and PBS for 10 min. Slides were then placed in a 37°C chamber humidified with a 50:50 formamide:dH2O mixture and 400 μL of probe hybridization buffer was applied to each slide. After 20 min of pre-hybridization, 400 μL of probe mixture (Pld5, Tshz2, Cxcr7, Npr3: 4 nM; Vglut1: 8 nM; Otof: 10 nM; Figf, Cd44: 20 nM) was applied to each slide and incubated for 12–16 h in the humidified chamber at 37°C. Slides were then washed in a series of probe wash buffer and 5 × SSC-T mixtures (1:0, 3:1, 1:1, 1:3, and 0:1) for 15 min each, at 37°C. Slides were washed again in 5 × SSC-T for 5 min before applying 400 μL of amplification buffer to each slide and incubating in the humidified chamber for 30 min. Amplification hairpins were heated quickly and cooled slowly (95°C for 90 s, then 30 min at room temperature shielded from light) and mixed into amplification buffer (Pld5, Tshz2, Cxcr7, Vglut1, Otof: 50 nM; Npr3, Figf, Cd44: 120 nM). 150 μL of amplification mixture was added to each slide, coverslipped, and incubated in a dark humidified chamber at room temperature for 12–16 h. Finally, slides were immersed in 5 × SSC-T for 30 min to remove the coverslips, and washed in 5 × SSC-T, before applying the final coverslip.
For immunolabeling (Figure 4B), 50-μm sections collected into PBS were blocked in 10% normal donkey serum (NDS: Jackson)/PBS/0.3%Triton-X overnight at 4°C, washed 3 × in PBS-T (PBS/0.1%Triton-X), incubated in primary antibody [1:1000 chicken anti-GFP (Aves)/5%NDS/PBS/0.3%Triton-X] for 2 days at 4°C, washed 3 × in PBS-T, incubated in secondary antibody (1:500 anti-chicken Alexa488 (Jackson) /5%NDS/PBS/0.3%Triton-X) overnight at 4°C, washed 3 × in PBS-T, adding 1:10000 DAPI in the final wash, and coverslipped.
Confocal and Slide Scanner Imaging
For images centered on PFC, we focused on bregma A–P: +1.95mm, as dmPFC, vmPFC, and OFC are all well represented at that coronal level. HCR-FISH images were collected on a Zeiss LSM780 confocal microscope at 20 × (Figures 2 and S2) or 40 × (Figures 3 and S3) at 1024 × 1024 or 2048 × 2048 resolution using standard settings, with as many tiles as needed to cover the area for quantification. Images used for quantification in Figures 2 and S2 were single planes. Images used for quantifying co-localization of tdTomato with HCR-FISH in Figures 3 and S3 were maximum intensity projections of 5 images spanning ~20 μm. Other histological images of GFP staining, endogenous tdTomato or endogenous GCaMP6f were collected at 20 × using standard settings. The images in Figure 1A for guiding dissections were collected on a dissecting scope with an epifluorescence camera. The images in Figure S2E for the confirmation of injection location were collected at 5 × on a Leica Ariol slide scanner with the SL200 slide loader.
For quantification of HCR-FISH in laminar analysis, regions of interest capturing dmPFC (300 μm height × 900 μm width, beginning ~1.1 mm below bregma, which corresponds to cingulate cortex) or vmPFC (300 μm height × 750 μm width, beginning ~2.3mm below bregma, which corresponds to infralimbic cortex) were generated from n = 4 mice (2–3 images per mouse). For co-localization, images were generated from n = 3 mice (2–4 images per mouse). For OFC, Pld5 and Vglut1 staining was quantified in the same manner from regions of interest the same size as and from the same section as vmPFC (300 μm height × 750 μm width), and compared in pairwise fashion over n = 4 mice.
Surgical Procedures
We anesthetized mice using isoflurane (1.25%–2.5% in 0.7–1.3 L/min of O2) during surgeries. We immobilized the head in a stereotaxic apparatus (Kopf Instruments), cleaned the skin with Betadine, injected lidocaine (2%, ~0.3 mL) subcutaneously for local anesthesia, cut open the scalp, peeled back connective tissue/muscle and dried the skull. All virus and dye injections were done at a rate of 100 nL/min. After surgery, mice were injected with carprofen (5 mg/kg), 0.9% saline (1–2 mL/100 g body weight), and BuprenorphineSR (0.1 mg/kg) for anti-inflammation, hydration, and pain management, respectively. Mice recovered on a heated pad until ambulatory, were returned to their homecage, and housed in a regular 12 h dark/light cycle with food and water ad libitum, unless otherwise noted.
For retrograde labeling experiments, Ai14 mice were injected with CAV-Cre (200–300 nL, 4.2 × 1012 gc/mL, all sites but amygdala) or AAVretro-Cre (300 nL, 8.7 × 1012 gc/mL, amygdala only) into contralateral PFC (cPFC: M–L/A–P/D–V, −0.6/+2.2/−2.5), ipsilateral dorsal striatum (DS: +1.7/+0.85/−2.8), nucleus accumbens (NAc: +0.6/+1.33/−4.8), amygdala (Amyg: +2.86/−1.3/−4.55), periaqueductal gray (PAG: +0.4/−4.15/−2.8), and hypothalamus (Hypo: +0.55/−1.91/−5.0). For testing injection sites, 100 nL CTB-Alexa488 (Molecular Probes: C-34775) was injected into each site into wild-type mice, which were immediately sacrificed for visualization. The images from Figure S2E (top) are from these experiments.
Regarding the targeting accuracy of the retrograde viruses used for the scRNAseq experiments (Figure 3), we practiced our injections extensively and were confident in our accuracy before the real experiments, and also used injection volumes and concentrations consistent with previous studies (Schwarz et al., 2015; Beier et al., 2015; Ren et al., 2018). However, because the procedure to dissect, dissociate, and FAC-sort the region of interest for sequencing was highly time-sensitive from the standpoints of cell viability and mRNA integrity, we did not prioritize saving the actual tissue from injection sites for presentation in the paper. Thus, we used atlas diagrams to illustrate our intended injection sites (Figure 3A).
The image from Figure 3E results from a dual injection of CAV-Cre into PAG, and CTB-Alexa488 into cPFC, with 1-week survival. For insertion of optogenetic cannulae, Gad2Cre;Ai32 mice [expressing ChR2(H134R)] were implanted with bilateral optical fibers (200-μm core diameter, 0.39NA cannulae, 700-μm spacing that were cut down manually to ~4 mm length, ThorLabs) at the upper border of vmPFC (prelimbic cortex; A–P: +1.95, M–L: ± 0.35, D–V: −2.3), for stimulation centered on infralimbic cortex below. The holes in the skull were covered with Kwik-Sil (WPI) for protection, and the cannulae was then secured with superglue (Loctite Gel Control) and dental Jet Liquid (Lang Dental), sealing all of the exposed skull.
GRIN lens implantation surgeries were performed using Resendez et al., 2016 as a guide. In brief, the skull was thoroughly cleaned and roughened with a scalpel blade, and two small screws (stainless eyeglass screws, 1 mm length) were screwed into the skull over posterior areas of cortex, without penetrating the dura, to lend extra support for the headcap. A ~1 × 1 mm craniotomy was cut over the lens target area, which was cleared of any remaining bone and overlying dura using fine forceps. Bleeding was limited with usage of gelfoam surgical sponge (Pfizer), and no further action was taken until bleeding had completely stopped. To visually identify the implantation location, we inserted an empty glass pipette (typically for viral injections) to 75% of the depth of the lens implantation. This served the purpose of creating a ‘starter’ hole, but no further aspiration of brain tissue was performed. Following starter hole generation, a ProView GRIN lens (500 μm width, 6.1 mm length) was loaded onto the ProView lens holder and attached Inscopix nVoke mini-endoscope. Together, this unit would be inserted into the brain with the camera functioning, to assess whether GCaMP6f-labeled cells were present at the final depth. The lens was then centered on bregma, and lowered slowly into the brain at the target location (in mm) A–P: +1.95, M–L: 0.4, D–V: −2.1, at a rate of ~100 μm/min by the stereotax. Once the intended depth was reached, and GCaMP6f labeled cells were confirmed in the field of view, we then proceeded to finalize lens placement by permanently gluing the lens itself in that location to form the ‘headcap’. First, Kwik-Sil (WPI) was used to cover the remaining exposed brain, and subsequently, liberal amounts of superglue (Loctite Gel Control), which was cured using dental Jet Liquid (Lang Dental), was used to firmly attach the lens to the skull and screws, sealing the skin and lens and skull together. After this, the camera and lens holder were carefully released from the lens, and Kwik-Kast (WPI) was used to protect the exposed surface of the glass. After 2 weeks of recovery, mice were re-examined for the presence of GCaMP6f-labeled cells at the lens tip, and if satisfactory, had a permanent baseplate and removable baseplate cover attached (Loctite Gel Control) to the headcap to serve as an adaptor of fixed focal distance between camera and lens.
To determine the implant locations of optogenetic cannulae and GRIN lenses in mice after experiments (Figures 4B, S4A, and S4B), mice were perfused as described above, but the brain was not isolated from the skull immediately. Rather, the entire head with the cannulae and lens still implanted was processed with 4% PFA and 30% sucrose, to ensure that tracks would remain fixed in place. Hardware was then removed from brains, which were then processed as described above for floating sections. Every section containing a lens or cannulae track was collected, to identify the location of the center point, which was then scored against the standard atlas (Paxinos and Franklin, 2001). Mice for which the center point of the lens or cannulae was outside of prelimbic cortex (not within A–P: 1.7 to 2.1 mm, D–V −1.9 to −2.6 mm) were excluded from analysis.
Behavioral Training
Mice were trained to perform at high levels of accuracy on a two-alternative forced choice task in two groups: 1) cannulae-implanted mice for optogenetics experiments (3 s or 1 s laser session), and 2) lens-implanted mice for Ca2+ imaging (2-odor task, 4-odor task, and uncued task). In general, mice were water restricted to 1 mL per day and monitored daily to ensure general health by visual inspection and maintenance of > 85% of their original weight. Mice were typically able to acquire a minimum amount of water (1 mL) during daily training sessions, and if not, the remainder was supplemented after training. See optogenetics section below for details specific to the optogenetics experiments. Starting one week after the baseplating surgery, all imaging mice were singly housed and chronically wore dummy microscopes with similar weight (1.8 g) and size (8.8 mm × 15 mm × 22 mm) to the Inscopix nVoke miniscope to become accustomed for imaging sessions. The only times the dummy microscopes were removed was to replace with the nVoke miniscope for data collection.
All behavior was performed in custom-built behavioral rigs and controlled by software adapted from the open source Bpod behavioral control system (https://github.com/KepecsLab/Bpod_r0_5). The behavioral box was designed in 3D CAD software with dimensions similar to home cages, and consisted of three main ports: one center odor port and two water delivery ports on the left and right side. The two water ports were 7cm apart, with the odor port in the center. All ports were at 3.5 cm above the bottom of the box (Figure 4A). Odors were delivered using a custom-built olfactometer. Water was delivered through metal ports coupled to a capacitive sensor that recorded licking at 40 Hz. An IR-sensor was placed in front of the center odor port to control trial initiation. Finally, we also included an air puff port immediately adjacent to each water delivery port to provide mild punishments specific to either side. The behavioral training was adapted from Guo et al., 2014a and Feierstein et al., 2006. Behavioral studies were computer-automated without experimenter input. Three weeks post-lens implantation surgery, or one week post-optogenetic cannulae implantation surgery, water-restricted mice were first habituated to the behavioral rig for two days (~45 min per day). During habituation days, mice were given water ad libitum through either water port. Following habituation, mice were trained on the two-alternative forced choice task in stepwise protocols with increasing complexity:
automatic water rewards dispensed whenever nose poke / IR beam break occurred
automatic water rewards coupled to specific odor-side pairings (left with 10% valeric acid in mineral oil and right with 100% 1-hexanol)
water reward dispensed only if mice reported the correct odor-side association by licking, but without any consequence for incorrect choices
incorrect choices punished by air puff, but mice were allowed to recover and report the correct choice after an initial incorrect choice
incorrect choices resulted in a single air puff and termination of the trial
Within ~2–3 weeks of training, mice were able to perform the basic version of the two-alternative forced choice task for optogenetics experiments (Figures 4C and 4D). In this scenario, a typical trial consisted of:
nose poking at the center odor port (trial start)
immediate release of valeric acid or 1-hexanol for up to one second
mice reporting their choice by licking at either the left or right reward port within four seconds from trial start
a correct choice resulting in a 4-μL water reward and an incorrect choice resulting in an air puff punishment. No response within the time allotted (4 s) terminated the trial.
In contrast to mice used for optogenetics experiments, lens-implanted mice used for imaging were additionally trained to expect water rewards to be delivered following a fixed 750-ms delay after the initial lick response. Mice were imaged on the 2-odor task with reward delay, and 25% of rewards were also randomly omitted (omission trials). For these imaging sessions (Figures 4, 5, and 6) mice performed at high accuracy (> 90%) over hundreds of trials (268 ± 10 trials, mean ± SEM) (Figure 4F). Reward omission trials only occurred during imaging sessions, and did not occur during training.
Following successful imaging during the 2-odor task, mice began training on an analogous 4-odor task. The original two odors (10% valeric acid and 100% 1-hexanol) were replaced with 100% (R)-carvone and 100% (S)-carvone. Mice trained on the new odor-side pairings until high performance was reached without reward delay or reward omission (~1–2 additional weeks). Following this, the two original odors were reintroduced, and mice were required to discriminate four odors with valeric acid or (S)-carvone signaling left and 1-hexanol or (R)-carvone signaling right (Figure 7A). Mice typically retained the old odor-side associations. Once high performance was attained, reward delay was reintroduced.
During the 4-odor imaging session, mice performed the 4-odor task at high accuracy (> 88%) for 165 ± 4 trials (mean ± SEM), and were then immediately introduced to a novel task variation consisting of a block of ~50 uncued left trials followed by a block of ~60 uncued right trials to provide a contrasting cognitive context for the mice (Figure 7A). During these uncued blocks, nose pokes did not trigger odor release. Rather, each nose poke triggered the automatic release of a water reward at one port, which was repeated over multiple trials. Within 10–20 trials of the start of each block, mice followed a continuous and stereotyped sequence of nose poking at the center port, approaching the reward port, licking/consuming water, approaching the center port, and so on (Figure S7A). Mice on average performed dozens of trials (50 ± 3 left block trials, 53 ± 3 right block trials, mean ± SEM) during imaging. Most mice were able to successfully complete more than 20 correct trials of each block-type during the first introduction of the uncued task. However, 9 out of 23 mice were unable to complete enough trials on the uncued task the first time. These mice were subsequently re-imaged one or two additional times until at least 20 left and 20 right uncued trials were completed correctly within the imaging session. One mouse was never able to complete enough uncued trials. Only data from the mice and sessions where both cued and uncued trials were completed sufficiently (22/23 mice, 1 session each) were combined for the analysis in Figure 7. A small fraction of mice (5/23) were able to complete more than two blocks, and in this case, trials of the same block-type were pooled together.
We quantified mouse behavior during trials in terms of percent complete, percent correct, response time, and lick duration (Figures 4C, 4D, and 4F). Percent complete is the percentage of trials in which mice reported a response during the allotted 4 s time window after nose poke. Incomplete trials were those where mice nose poked to begin the trial but failed to report a response within 4 s. Percent correct is the percentage of trials, out of completed trials, that mice made the correct choice. Incorrect trials are completed trials with the wrong response, which are non-overlapping with incomplete trials. Response time is the time from odor onset to lick response. Lick duration is the time from first to last lick on correct trials. Unless otherwise specified, we only analyzed ‘correct’ trials from the 2- and 4-odor imaging sessions, where mice made the correct choice and remained licking at the reward port throughout the delay period.
During the uncued task, correct trials were those where mice reported their response by licking at the correct port first and within the allotted 4 s time window. To determine the behavioral performance of mice during the transition from the 4-odor task to the uncued task, we calculated the mean performance of mice as they were performing the left and right blocks. Mice were able to achieve high performance in the first left block within ~10 trials and subsequently in the right block within 20 trials (Figure S7A). The transition between the 4-odor and uncued tasks was immediate (after ~165 4-odor cued trials), and data from both tasks was collected from the same continuous video stream.
Optogenetics
Before each session, expert mice were briefly restrained to attach the optogenetics patch cable, and allowed to recover for > 30 min. During each optogenetics behavioral session, laser stimulation was triggered by the nose poke, and designed to last for 1 or 3 s (separate sessions: Figures 4D and 4C). One second was chosen because the mean response time during normal conditions was approximately one second. Three seconds was chosen as a longer stimulation period that would still allow sufficient time for the mouse to report a choice within the 4 s allotted time. The stimulation laser (473 nm wavelength) was pulsed at 50 Hz with 10 ms pulse durations at an optical power of ~5–6 mW at the output of each fiber tip. Laser stimulation occurred on 25% of randomly interleaved trials during the task and stimulation lengths were kept constant in each session. Behavioral performance was quantified as described above.
Regarding the illumination zone and the extent of the optogenetics effect, we implanted our fiber tips in prelimbic cortex approximately 750 μm above the bottom of infralimbic cortex, the lower boundary of the “intended” illumination zone in vmPFC. Based on the optical settings we selected (200-μm core diameter, 0.39 NA cannulae, 5–6 mW at fiber tip), we estimated light attenuation based on the calculator at optogenetics.org, which indicated that the vast majority of laser light is concentrated within 500 μm of the fiber tip. This is squarely within the region of interest. Furthermore, we used laser power similar to or more conservative than previous mPFC studies in our lab and others (DeNardo et al., 2019; Huang et al., 2018; Selimbeyoglu et al., 2017; Rajasethupathy et al., 2015). Beyond this, however, we do not have empirical data demonstrating the exact extent of the optogenetics effect, but believe it is unlikely for our illuminated tissue volume to have grossly spilled beyond vmPFC.
Collection of Ca2+ Imaging Data
We performed all Ca2+ imaging using the Inscopix nVoke miniscope (Figures 4G, S4C, and S6C), without refocusing the microscopes across the surgeries and two imaging sessions. This ensured that the same field of view was reproduced over time, to the best of our abilities. However, due to the ~2–3 week period between imaging sessions and minor changes to the fields of view during this time, we were not confident in explicitly aligning cells across imaging sessions and so the datasets were analyzed independently. Before imaging, mice were anesthetized with 2% isoflurane for 2 min to attach the miniscope and allowed to recover for 30–60 min. To reduce stress, we did not head-fix mice at any point in this procedure.
Extraction of Ca2+ Imaging Data
Images (360 × 270 pixels) were acquired at 20.01 Hz, LED power of 1 mW/mm2, with a gain of 3.0. Each imaging session typically lasted for 30–40 min, which began with the self-initiated IR beam break in the mouse’s behavior. The resulting imaging data was 2× spatially downsampled and motion corrected using default settings in the Inscopix Data Processing Software. The imaging data was exported, and individual neurons and their respective fluorescence traces were identified using the constrained nonnegative matrix factorization for microendoscopic data (CNMF-E) algorithm (Zhou et al., 2018). For each neuron, the denoised fluorescence trace or the deconvolved Ca2+ events was extracted using the OASIS algorithm [AR(1) model, Friedrich et al., 2017]. Every extracted cell was manually checked for circular spatial footprints and Ca2+ transients characterized by sharp rises and slow decays. Extracted cells were also excluded if they could not be clearly cross-checked between the correlation and maximum intensity images due to low signal. Elongated-shaped ROIs representing dendrites that correlated with the more circular-shaped cell bodies were removed from analysis. All analysis used either the standard deviation of the denoised fluorescence trace of each cell or the corresponding deconvolved Ca2+ events. We used denoised fluorescence traces for the general display of all data over time and logistic regression analysis. We used deconvolved Ca2+ events for the determination of task-modulated cells, as well as for all other linear regression-based analysis in Figures S5D, 7D, and S7G. Under our imaging conditions, GCaMP6f transients are likely the result of multiple spikes (Chen et al., 2013). The results in this study related to Ca2+ imaging were also largely recapitulated using a different cell extraction algorithm (PCA/ICA, Mukamel et al., 2009).
QUANTIFICATION AND STATISTICAL ANALYSIS
Single-Cell Sequencing Data Analysis
To generate the transcriptomic map featured in Figure 1, we used standard procedures for filtering, variable gene selection, dimensionality reduction, and clustering in Seurat v3.0 (Butler et al., 2018; Stuart et al., 2019). The analyzed Rbp4Cre>tdTomato+ cells originated from 11 different samples/batches (dmPFC, n = 3; vmPFC, n = 4; OFC, n = 4). Cells were removed if they expressed fewer than 2000 genes, and genes were removed if they were detected in fewer than 3 cells. Cells expressing the inhibitory neuron marker Gad2 (< 1%) were also removed from consideration. This resulted in a dataset of 3139 cells × 17535 genes. All cells were processed by the same SMART-Seq2 chemistry, but were collected in 96- or 384- format, and sequenced on either the NextSeq 500 or NovaSeq 6000 systems. Thus, we used batch correction within Seurat v3.0 to sequentially define pairwise anchors across the 11 batches (Stuart et al., 2019), and integrate the data together to remove possible batch effects. The assumption that there were physiologically analogous cells across batches should apply because the batches were experimental replicates of adjacent PFC subregions. In brief, each batch/sample was split into its own Seurat object, 3000 variable genes were selected with ‘vst’, anchors were found using FindIntegrationAnchors (k.anchor = 5, k.filter = 50, k.score = 30, dims = 1:15, max.features = 100, anchor.features = 3000) and the data was integrated with IntegrateData (k.weight = 100, dims = 1:15). In the integrated object, counts were log-normalized for each cell using the natural logarithm of (1 + counts per 10000), and scaled using ScaleData while regressing out the effects of the # of genes and the # of reads. Cells were visualized using a 2-dimensional t-distributed Stochastic Neighbor Embedding (tSNE, van der Maaten and Hinton, 2008) of the PC-projected data using the FeaturePlot and VlnPlot functions. PCA was performed on the integrated data (npcs = 15), and 8 PCs were used in FindNeighbors based on a steep dropoff in variance explained by visual inspection of the elbow plot. We used the FindClusters function in Seurat v3.0, and compared the resultant clusters from 9 different levels of resolution (0.1–0.9, 0.1 increments) using the ‘Clustree’ function (Zappia and Oshlack, 2018) to visualize the way in which individual cells either retained or changed their classifications as the resolution parameter was increased (Figure 1E). We eventually settled on a resolution parameter of 0.3 for the majority of the paper, after noting that most clusters classified at this resolution were stable at higher resolution, but could be defined, from a practical standpoint, by individual markers. We note that there are no cases where major and unexpected rearrangements occur in the organization, and that only the Cd44, Figf, and Otof clusters are further split at higher resolution. However, given that in the retrograde mapping studies shown in Figures 3 and S3, we saw one-to-many and many-to-one matching between transcriptomic types and projection types, we did not intentionally further subdivide these clusters.
To generate the transcriptomic map used to visualize intermingling of retrograde (n = 1155) cells and Rbp4Cre>tdTomato+ (n = 3139) cells (Figure S3A), it was not possible to perform a batch correction based on every individual sample because the cell number in some of the retrograde-labeling samples was too low. Therefore, we performed batch correction after segregating the samples by ‘sequencer’, and then regressed out the effect of ‘plate’ (96- versus 384-well) subsequently in the ScaleData function. All other parameters were the same, and this transcriptomic map was primarily for the purpose of visualization.
Differential Expression and Marker Definition
We used the FindAllMarkers function in Seurat v3.0 and applied this to the ‘RNA’ assay in the Seurat object of 3139 high-quality cells classified at Seurat resolution = 0.3 (Figure 1B). This function performs differential expression analysis (Wilcoxon rank sum) between a specific cluster, compared with all other cells not in the cluster, and then iterates through all seven clusters. Because of the immense number of marker genes with statistically significant p values, we filtered genes not only on the basis of differential expression levels and average log fold change (avg_logfc > 0.1), but also on the consistency in which a gene was expressed across different cells in the same cluster or population. ‘PCT’ refers to the percentage of cells within a population expressing a specific gene. We first required that the difference in this membership between the assayed cluster and all other cells (PCTdelta = PCTcluster−PCTother) for each considered gene to be > 0.35. We next required that PCTcluster> 0.5, meaning that a gene must be expressed in greater than 50% of the cells in a cluster, in order to be considered a marker for that cluster. Conversely, PCTother was required to be < 0.25, meaning that at maximum, 25% of cells outside of the cluster can express the marker gene. Together, these requirements enforce that marker genes exhibit close to binary ‘on-off’ expression in and out of the cluster. This resulted in a list of 133 genes (Cluster 1: 6 genes; Cluster 2: 4 genes; Cluster 3: 17 genes; Cluster 4: 7 genes; Cluster 5: 14 genes; Cluster 6: 39 genes; Cluster 7: 46 genes, see Table S1 Tab 1). With this list, we visually inspected available online in situ hybridization resources, further prioritized genes based on expression level differences, binary expression (PCTcluster close to 1, and PCTother close to 0), and practical considerations of whether commercially available HCR probes gave good signal-to-noise ratio in histological tissue. From this, we settled on the combination of genes: Cd44, Figf, Otof, Pld5, Cxcr7, Npr3, and Tshz2 to delineate the seven clusters, but recognize that this is not the only combination that achieves this purpose.
We also computed differentially expressed genes for the same dataset, but at a higher clustering resolution [Seurat resolution = 0.9 (Table S1 Tab 2)]. Here, we were interested more broadly in differentially expressed genes rather than only ‘on-off’ expression. Thus, we lowered the threshold to PCTdelta > 0.25, and removed the requirement for PCTother. This resulted in a longer list of 867 genes. However, we highlighted genes that fulfilled the criteria of Tab 1 (avg_logfc > 0.1, PCTcluster > 0.5, PCTother < 0.25, and PCTdelta > 0.35) in red in Tab 2.
Scrattch.hicat Analysis and Comparison to Seurat
We used scrattch.hicat analysis (Tasic et al., 2018) as a contrasting method to analyze and validate our Rbp4Cre>tdTomato+ PFC clusters (Figures S1B and S1C). As input, we used the same filtered data matrix from Seurat (3139 cells × 17535 genes), and the 0.3 resolution Seurat cluster labels as a reference classification. In brief, raw counts were normalized to log2(1+ counts per million), and the threshold for differential expression and clustering was set based on the following combination of parameters: padj.th = 0.05, lfc.th = 1, low.th = 1, q1.th = 0.5, q2.th = NULL, q.diff.th = 0.7, min.cells = 15, de.score.th = 100. We adhered to these parameters as they were the recommendation by the Allen Institute for data of this size and complexity. We next removed technical artifacts associated with the # of genes, # of reads, type of plate, and the specific sample, to mirror the quality control done in Seurat. Finally, we performed consensus clustering over 20 iterations, which resulted in a new classification that could be compared with the Seurat (resolution = 0.3) classification (Figure S1B), and also visualize the probabilities of cell co-clustering over the different iterations (Figure S1C).
To compare our PFC data to the published Rbp4Cre-labeled cell data from ALM and VISp (Tasic et al., 2018), we first preprocessed the raw data to make it compatible with our data. In brief, we downloaded (https://portal.brain-map.org/atlases-and-data/rnaseq) and considered only SMART-Seq2 data, reads mapped to exons, and cells derived from Rbp4Cre-labeled mice. We filtered cells and detected genes by the same criteria as above (genes detected in > 3 cells, cells expressing > 2000 genes), and then only considered genes expressed across all three datasets. Finally, cells labeled from Allen cluster annotations as ‘doublets’, ‘high intronic’, or ‘low quality’ were removed and not considered. This resulted in a dataset of 14611 considered genes, and n = 3137 PFC, n = 546 ALM, n = 697 VISp neurons, which was used for the mapping analysis in Figure 1F.
Mapping Query and Reference Datasets
We used Seurat v3.0 to assign cells with classification labels based on their proximity to previously classified cells within a reference dataset. For mapping Rbp4Cre-labeled PFC cells to Allen ALM and VISp labels, we first generated a Seurat object containing only the ALM and VISp cells to serve as a reference (data normalization, variable feature finding, data scaling, and PCA done as described above). We next subsetted the PFC data by cluster (Cd44, Figf, Otof, Pld5, Cxcr7, Npr3, or Tshz2), and queried each of these subsets with the reference data. We used FindTransferAnchors (dims = 1:9, k.filter = 25) and TransferData (dims 1:9) to first find anchors between datasets, and then assign classification labels, respectively. To generate Figure 1F, an alluvial diagram was made using the ‘ggalluvial’ R package, based on the number of cells that were assigned to different cluster labels, and normalized to the same number of cells in each PFC cluster. Reference classifications in Figure 1F were pooled from the lists of individual cluster labels shown in Figure S1D. The data in Figures 3B and S3B was generated in a similar way, but query sets were retrogradely-labeled cells from each individual target. The reference classifications were the integrated Rbp4Cre>tdTomato+ Seurat object from Figure 1, at 0.3 (Figure 3B) or 0.9 (Figure S3B) clustering resolutions, and normalized to the number of retrogradely-labeled cells.
HCR-FISH Puncta Finding and Confocal Imaging Data Analysis
To quantify the expression of each marker gene and its co-localization, we used Slc17a7 (Vglut1) as a counterstain because it is a marker of excitatory neurons. We trained the deep learning network U-Net (Falk et al., 2019) to identify and segment Vglut1+ cells into individual ROIs, using a total of 12 (1077–2409 wide × 120–693 pixels tall) manually annotated images as training data (using the default caffemodel as input, 5000 iterations, validation interval of 20, 348 × 348 tile size). For determining marker gene expression levels, fluorescent puncta were identified using the Fiji plugin TrackMate (Tinevez et al., 2017: Laplacian of Gaussian detector with sigma suited to each marker’s punctum size). These puncta were binarized and the individual masks for each neuron from the U-Net segmentation were applied, allowing the area and position of each punctum for each gene to be determined. Using custom python scripts, puncta size was normalized to the minimum size observed in each image, and the number of puncta in a neuron was normalized to the maximum in each image. This resulted in a comparable score of the amount of gene expression in each neuron across images and genes. A threshold was set to consider any neuron to be positive for the marker of interest if it contained enough puncta to be within 75% of the maximum number of puncta observed. Additionally, for stains of multiple markers, co-localization was calculated in the same manner with a percentage breakdown of all single, double, and triple labeled cells. To illustrate the medial–lateral spatial distribution of markers, we counted positive cells using a sliding window of 50 μm width and 10 μm per slide. For each image, we identified the first bin that contained > 17 (vmPFC) or > 11 (dmPFC) cells for 3 consecutive bins, as we found empirically that this was a robust method for defining the transition between the very sparse cells found in L1, and the much denser cells found in L2/3. Subsequently, all per bin cell counts were aligned based on the ‘beginning of L2/3 as an anatomical landmark close to the midline. The traces of each image were then averaged and plotted in Figures 2B and S2A.
Co-localization of marker genes with tdTomato+ cells (Figures 3C, 3D, S3C, and S3D) was processed in a similar manner, but Slc17a7 and U-Net was not used as the basis for defining cell masks. Instead, we thresholded tdTomato signal to include only the cell body, and then converted these into cell outlines using the ‘Analyze Particles’ function in Fiji. Fluorescent puncta were identified as described above with TrackMate, overlaid with tdTomato cell outlines, and the number of puncta was quantified per cell. Similar to above, a cell was considered staining positive if it contained within 75% of the maximum number of puncta observed within a tdTomato+ cell in the image. If there was low to no co-localization, and hence no tdTomato cell clearly contained puncta in the image, then this maximum was calculated based on tdTomato- cells. The 75% threshold was defined from careful inspection of the data, as it was effective in ruling out puncta contributions from slightly overlapping neighboring cells or background staining, but sensitive enough to pick up lowly expressed genes that did fill the cell outlines. For each combination of marker gene and retrograde labeling, 2–4 windows of 440 × 600 μm size were scored for n = 3 mice, in vmPFC.
Analysis of Trial-Averaged Activity and Task-Relevant Modulation
Mice were freely moving and made decisions with slightly variable response times. To assess the temporal specificity of activity in each cell relative to the task structure, we computed trial-averaged activity using the denoised fluorescence trace of each neuron over all considered trials (all correct trials, or all left trials, etc.), and aligned to either the odor onset or the first lick times (Figures 5A, S5A, and 6A). To determine which cells had significant task-modulated activity within a particular trial type (e.g., left trials), we defined a set of behavioral regressors representing the four task epochs (Approach, Decision, Lick, and Reward) for left and right trial types separately (Figure 4I). For each cell, we linearly regressed its deconvolved Ca2+ events during the entire recording period onto the set of eight (2-odor task) or 16 (4-odor task) behavioral regressors. Cells with significant regression coefficients were then considered ‘modulated’ in that epoch. The set of all cells modulated in any task epoch was considered the population of ‘task-modulated’ cells. To determine whether a cell had a significant regression coefficient for any behavioral regressor, we used permutation tests. We randomized the regressor–activity relationship by shuffling the neural activity matrix with respect to the regressors, and performed linear regression to produce coefficients derived from the shuffled data. If the true coefficient was greater (or lower) than the shuffled coefficient for a given cell for over 99% of iterations, it was considered significant. Positively modulated cells were defined as those which exhibited higher regression coefficients than chance and negatively modulated cells were defined as those which exhibited lower regression coefficients than chance. Cells with significant modulation in two consecutive epochs (for example, Decision and Lick) were categorized by the first significant epoch. Cells with significant modulation for multiple trial types (for example, right Decision and left Reward) counted twice for the calculations in Figures 5C and S7B. Trial-averaged heatmaps of positively modulated cell populations were sorted by the time of maximum denoised fluorescence (Figures 5B, S5B, 7B, S7E). When comparing heatmaps between two different trial types, we sorted the activity based on the left most panel (Figures S5B, S6A, 7B, and S7E). All Ca2+ denoised fluorescence traces are z-scored with standard deviation (SD) as the unit.
Neural Activity Trajectory Analysis
To visualize neural activity trajectories, we used PCA for dimensionality reduction on the trial-averaged data of cells. For each cell, we computed and concatenated the trial-averaged trace of left and right trial types such that each cell’s activity was represented as a 2T × 1 dimensional vector, where T is the total number of time points per trial type. For the aggregate trajectory in Figure 6B, we combined all imaged cells into a single matrix with dimensions 2T × N where N is the total number of cells. We used PCA for dimensionality reduction and projected the data onto the first three principal components to visualize left and right neural activity trajectories. For the cell class-specific neural trajectories shown in Figure 6C, we randomly subsampled each cell class to an equal number of cells (200, a number lower than the class with the least number of imaged cells) and used PCA on the resulting matrix with dimensions 2T × (3 × 200). We projected the activity of each cell class on their respective PC loadings [for example, projection of cell class 1 = activity matrix (:,1:200) × PC loadings (1:200,:)] to visualize each cell class’ left and right neural activity trajectories independently. We randomly subsampled cells hundreds of times and the example trajectory in Figure 6C is representative of trajectories commonly observed. For Figure S6B (left) and Figure S7F, we computed the cumulative variance explained as a function of the number of PCs included, for all the data without subsampling. For Figure S6B (right) we randomly subsampled equal amounts of cells for each class. For the aggregate trajectory in Figure 7F, we performed PCA on the concatenated trial-averaged trace of correct left and right uncued trials. For the aggregate trajectory in Figure 7G, we performed PCA on the concatenated trial-averaged trace of the Approach epoch for all six trial types (L-1, L-2, R-1, R-2, L-uncued, R-uncued).
Regression Analysis
We used logistic regression for the analyses in Figures 6D–6F, 7H, 7I, S7C, and S7D, to determine how accurately left versus right choice direction or reward context could be predicted from the neural data. For each mouse, we constructed a data matrix with dimensions N × M × T where N is the number of cells, M is the number of trials and T is the number of time points in the z-scored fluorescence trace. For each mouse, five segments of the trial were analyzed separately [−1 s to −0.05 s (Approach epoch), and 0 s to 0.45 s (early Decision epoch) using the odor onset alignment, −0.5 s to −0.05 s (late Decision epoch), 0 s to 0.7 s (Lick epoch), and 0.75 s to 2.5 s (Reward epoch) using the lick onset alignment] resulting in five data matrices per mouse. To maximize the number of samples to train our classifier, we first reshaped each data matrix along the last 2 dimensions to obtain an N × (M × T) matrix. This allowed us to treat every sample across each epoch equally, meaning we would train only one classifier across all T time points in each epoch. We also constructed a categorical response vector for each epoch with dimensions (M × T) × 1, with left trial time points labeled as 1 and right trial time points labeled as 0.
For each data matrix, the responses of all neurons in 80% of the trials were fit with a logistic model (fitclinear in MATLAB, lasso regularization) and the remaining 20% of the trials were tested using the trained model (predict in MATLAB, 5-fold cross validation) (Figure 6D). The same process was used for each window and the predicted leftward choice accuracy was recorded. This yielded a prediction accuracy trace with 50-ms resolution for each mouse and epoch. Both correct and error trials were included in the analysis of the Approach and Decision epochs while only correct trials were included in the analysis of the Lick and Reward epochs (Figures 6D–6F).
For the example mice in Figure 6D, we randomly subsampled the number of trials to be the same between mice (50 left trials and 50 right trials) but did not subsample the number of cells. To compare the amount of choice direction information between cell classes, we repeated the same analysis as above and randomly subsampled the number of trials and cells to be the same between all mice (50 left trials, 50 right trials, and 25 cells). We then calculated the choice direction prediction accuracy. We repeated this for 30 iterations, for each mouse and for each epoch, and then plotted the mean ± SEM for each cell class (Figure 6E). To quantify the choice direction prediction accuracy as a function of the number of cells included, we repeated the same steps as above for the Decision epoch, and plotted the mean ± SEM as a function of the number of cells included for each cell class (Figure 6F).
Similar methods were used in Figures S7C and S7D for training classifiers to predict left or right choice direction, and Figures 7H and 7I for training classifiers to predict reward context in the uncued task. This required creating a new (M × T) × 1 vector of labels, with left time points labeled as 1 and right time points labeled as 0. In contrast to Figures 6D–6F, S7C, and S7D, only correct trials were included for Figures 7H and 7I because error trials in the uncued task had high behavioral variability. To compare the amount of reward context information between cell classes, we used the last 20 correct left and right trials and randomly subsampled the number of cells to be the same between all mice (25 cells). Here, two segments of the trial were analyzed separately [–1 s to 0.4 s (Approach and early Decision epochs) using the odor onset alignment, and −0.4 s to 2.5 s (late Decision and Reward epochs) using the lick onset alignment] resulting in two data matrices per mouse. To quantify reward context prediction accuracy as a function of the number of cells included, we repeated the same steps as above for the Approach epoch, and plotted the mean ± SEM as a function of the number of cells included for each cell class (Figure 7I).
Analysis on the Effect of Reward Omission
To determine whether a cell was significantly modulated by reward omission, we used linear regression. We defined a set of four behavioral regressors that matched with the trial types: left reward, left reward omission, right reward, and right reward omission that spanned the period of reward/omission onset + 2 s. For each cell, we linearly regressed its deconvolved Ca2+ events during the entire recording period onto the set of four behavioral regressors. Cells with significant regression coefficients were then considered ‘modulated’ in that time window. To determine whether a cell had a significant regression coefficient for any of the four behavioral regressors, we used permutation tests. We randomized the regressor–activity relationship by shuffling the neural activity matrix with respect to the regressors, and performed linear regression to produce coefficients derived from the shuffled data. If the true coefficient was greater than the shuffled coefficient for a given cell in over 99% of iterations, it was considered significant. This allowed us to determine whether a cell was significantly modulated by reward or omission (Figure S5D). To determine whether a cell had selective activity for either reward or omission, we used the same permutation test but calculated whether the difference between the reward regressor coefficient and the omission regressor coefficient was greater than the difference found in the shuffled data. If the true difference was greater than the shuffled difference in over 99% of iterations, then the difference was considered significant. ‘Reduced activity’ cells had a significant reward regressor coefficient, which was also significantly greater than the omission regressor coefficient. ‘Elevated activity’ cells had a significant omission regressor coefficient, which was also significantly greater than the reward regressor coefficient. ‘No change’ cells had a significant reward regressor coefficient, which was not significantly different than the omission regressor coefficient (Figure S5D).
To determine whether the proportion of cells modulated by reward omission (total of ‘Reduced activity’ and ‘Elevated activity’ cells, defined above) differed between cell classes, we first calculated the true difference in the proportion of cells modulated by reward omission between classes. We then pooled all cells and randomly sampled from this pool and calculated the differences in proportions of cells modulated by reward omission between the randomized populations. If the true difference in proportions was greater (or less) than cells drawn randomly from this pool in over 99% of iterations, this was considered significant. To determine the population-level impact of reward omission, we plotted the mean response of all cells during reward and omission trials (Figure S5E).
Classification of Odor-, Choice-, or Side-Selective Cells in the Decision Epoch
To determine whether a cell that was positively modulated during the Decision epoch should be categorized as an Odor-, Choice-, or Side-selective cell, we used linear regression (Figure 7D). We defined a set of six behavioral regressors: left odor 1 (L-1), left odor 2 (L-2), right odor 1 (R-1), right odor 2 (R-2), left uncued (L-uncued), and right uncued (R-uncued) that spanned the decision period (odor onset to first lick). For each cell, we linearly regressed its deconvolved Ca2+ events during the entire recording period onto the set of six behavioral regressors. To determine whether a cell had a significant regression coefficient for any of the six behavioral regressors, we used permutation tests. We randomized the regressor–activity relationship by shuffling the neural activity matrix with respect to the regressors and performed linear regression to produce coefficients derived from the shuffled data. A cell was classified as a Side cell (L-1 and L-2 and L-uncued or R-1 and R-2 and R-uncued) if the lowest of the three left or three right coefficients was greater than the lowest of the three corresponding shuffled coefficients in over 99% of iterations. A cell was classified as a Choice cell (L-1 and L-2 or R-1 and R-2) if the lowest of the two left or two right coefficients was greater than the lowest of the two corresponding shuffled coefficients in over 99% of iterations, and that cell was not already classified as a Side cell. Lastly, a cell was classified as an Odor cell (L-1 or L-2 or R-1 or R-2) if any of the coefficients was greater than the corresponding shuffled coefficients in over 99% of iterations, and that cell was not already classified as a Side or Choice cell.
To determine whether the proportion of Odor, Choice, or Side cells differed between the three cell classes, we first calculated the true difference in the proportion of cells between the PAG-projecting, cPFC-projecting or Rbp4Cre classes. We then pooled all cells and randomly sampled from this pool, and calculated the differences in proportions of cells between the randomized populations. If the true difference in proportions was greater (or less) than cells drawn randomly from this pool in over 99% of iterations, this was considered significant.
Classification of Mixed Selective Cells
To determine whether a cell represented information about choice direction, reward context, or both (mixed selective), we used linear regression (Figure S7G, similar to Mante et al., 2013). We defined a choice regressor that labels all time points within left trials as 1 and all time points within right trials as −1 during the 4-odor task. We similarly defined a reward context regressor during the uncued task. For each cell, we then linearly regressed its deconvolved Ca2+ events onto either the choice or reward context regressor. To determine whether a cell had a significant regression coefficient for either the choice or reward context regressor, we used permutation tests. We randomized left and right trial types and performed linear regression to produce coefficients derived from the shuffled data. A cell was considered to represent information about choice direction if the true coefficient was greater (or less) than the shuffled coefficients in over 99% of iterations. This was performed similarly for reward context. Cells that had significant regressors for both choice direction and reward context were considered mixed selective cells.
To determine whether the proportion of mixed selective cells differed between cell classes, we first calculated the true difference in the proportion of mixed selective cells between classes. We then pooled all cells and randomly sampled from this pool and calculated the differences in proportions of mixed selective cells between the randomized populations. If the true difference in proportions was greater (or less) than cells drawn randomly from this pool in over 99% of iterations, this was considered significant.
Statistical Tests
We used MATLAB for all statistical tests unless otherwise stated. To compare differences between two groups, we used paired or unpaired t tests, as appropriate. To compare differences between three groups, we used the one-way ANOVA or the Kruskal-Wallis test (also non-parametric) with Tukey’s honest significant difference criterion for multiple comparisons correction. In the event that standard statistical tests were not applied, we used custom permutation tests as described within each section above. All significance thresholds were set at p < 0.05 unless otherwise stated.
Supplementary Material
Figure S1. Sequencing quality control, validation of PFC neuron clustering, and other scRNAseq analysis, related to Figure 1
(A) Violin plots showing the distribution of the number of genes detected (left) and the number of reads (right) across single cells derived from each of the three PFC regions. The dots within each violin denote the median of the distribution (~7000 genes, ~1–2 million reads per cell). Effects in the gene expression data driven specifically by differences in the number of detected genes and the number of reads were regressed out from further analysis by the Seurat ScaleData function as part of the standard pipeline.
(B) Comparison of Rbp4Cre>tdTomato+ PFC neuron (n = 3139 cells, as in Figure 1) classification between reference clusters (Figure 1, Seurat resolution = 0.3) and clusters generated from the Allen Institute scrattch.hicat analysis (Tasic et al., 2018). As a quality control measure, principal components correlating with log2 detected genes, # of reads, plate format, and sample replicate were removed, see STAR Methods for additional parameters. Allen clusters are named based on similarity and subdivision from the Seurat clusters. Color scale represents the Jaccard similarity index (intersection divided by the union) of the two groups, and the size of each dot is the square root (sqrt) of the cell number. The percentage contribution of each region to each cluster (normalized to the size of the cell population) is displayed on top, with the same color code as (A). Only one cluster was highly region-specific [Cxcr7(5–2) with > 85% contribution from OFC].
(C) Matrix depicting the robustness of Rbp4Cre>tdTomato+ PFC neuron classification across multiple iterations of consensus clustering in scrattch.hicat. Rows represent cells, columns represent clusters, and color scale represents the probability of being assigned to the cluster based on 20 iterations of consensus clustering.
(D) List of clusters from Tasic et al., 2018 that contained cells derived from Rbp4Cre-labeled mice and met quality control measurements (STAR Methods). Clusters were then grouped into the four larger categories and used for display purposes in Figure 1F.
Figure S2. Additional HCR-FISH validations and example retrograde labeling, related to Figures 2 and 3
(A) Laminar distribution of cells expressing cluster-specific marker genes across dmPFC, analogous to Figure 2B. Dashed lines are approximate cortical layer boundaries according to the Allen Atlas (L5a, 130 μm; L5b, 345 μm; L6, 565 μm, aligned to the beginning of L2/3).
(B) Triple HCR-FISH between Otof, Cd44, and Cxcr7 in vmPFC. Quantification of overlap is shown for both dmPFC and vmPFC, and averaged across n = 4 mice (543 dmPFC, 603 vmPFC cells). Error bars are SEM. Scale bar, 50 μm.
(C) Triple HCR-FISH between Cd44, Cxcr7, and Tshz2 in vmPFC. Quantification of overlap is shown for both dmPFC and vmPFC, and averaged across n = 4 mice (380 dmPFC, 372 vmPFC cells). Error bars are SEM. Scale bar, 50 μm.
(D) Comparison of Pld5 expression in mPFC versus OFC. Sequencing data shows the percentage of cells that belonged to the Pld5 cluster, comparing dmPFC and vmPFC with OFC (dmPFC: 910 cells, n = 3 mice; vmPFC: 1234 cells, n = 4 mice; OFC: 995 cells, n = 4 mice; *p < 0.05, **p < 0.01, unpaired t test). Histology data shows the percentage of Vglut1+ cells that were Pld5+, comparing vmPFC and OFC. Cells were quantified within a 300 × 750 μm area, and a ratio paired t test was used, **p < 0.01. Scale bar, left: 250 μm, insets: 50 μm.
(E) Injection sites of CAV-Cre into the contralateral PFC (−0.6/+2.2/−2.5) and PAG (+0.4/−4.15/−2.8) of Ai14 mice (above). Resulting retrograde labeling of tdTomato+ cells 1 week post injection at bregma A–P +1.95 mm (below). Dotted lines indicate the edge of the tissue slice. Scale bar, 500 μm (top), 100 μm (bottom). Coordinates are expressed in (M–L / A–P / D–V) axes with respect to bregma, in mm.
(F) Overall distribution of PFC cells labeled by CAV-Cre injection into PAG: (+0.4/−4.15/−2.8) in an Ai14 mouse, highlighting presence and absence across PFC subregions. Cg, cingulate; PL, prelimbic; IL, infralimbic; MO, medial orbital; VO, ventral orbital; LO, lateral orbital; AI(D/V): agranular insular (dorsal/ventral); DP (dorsal peduncular) cortices. Scale bar, 500 μm.
Figure S3. Rbp4Cre>tdTomato+ and projection-defined neurons intermingle in tSNE space, and additional histological validation of marker expression in projection-defined neurons, related to Figure 3
(A) tSNE cluster map (top) combining all Rbp4Cre>tdTomato+ neurons from Figure 1 (n = 3139) and retrogradely-labeled vmPFC neurons from all 6 injection sites (n = 1155), labeled by color. Areas where retrograde cells do not intermingle well with Rbp4Cre>tdTomato+ cells are highlighted by arrows. A Syt6 cluster (known Layer 6 marker) is gained, and additional cells are in the Figf cluster. Feature plots of the seven marker genes (bottom) demonstrate that this map still roughly segregates by the marker genes identified in Figure 1, albeit with some interaction between Figf and Cxcr7 clusters. Areas with relatively low retrograde cell intermingling are predominantly in Figf and Syt6 clusters. Color scale represents expression level in the unit of ln[1+ (reads per 10000)].
(B) Nearest neighbor mapping of retrograde cells to transcriptomic cluster identities, at higher resolution, as an alternative to Figure 3B, represented by an alluvial diagram. Using 0.9 resolution in Seurat resulted in 12 clusters + Syt6 as possible transcriptomic types to map retrograde cells to. Under these conditions, differences in the relative contributions of Cd44 and Figf subclusters to cPFC, DS, NAc, or Amyg projections began to emerge, but no one-to-one relationships were observed. While these differences are potentially important, clustering at high resolution did not alter the general conclusions regarding the relation between transcriptomic type and projection target. Furthermore, mapping retrogradely-labeled cells to reference datasets containing only Rbp4+ or only vmPFC cells gave similar results (data not shown). For differentially expressed marker genes of the 12 clusters based on Seurat resolution = 0.9, see Table S1 Tab 2.
(C) Confocal images double-labeled with retrograde tracing from cPFC (red for the tdTomato Cre reporter) and HCR-FISH against Cd44, Cxcr7, or Figf (cyan). Scale bar, 20 μm. HCR-FISH signal was converted to binary puncta and overlaid with tdTomato cell outlines for quantification. Red insets highlight staining-positive cells (within 75% of maximum puncta observed), and cyan insets highlight staining-negative cells (< 25% of maximum puncta observed), at higher magnification.
(D) Confocal images double-labeled with retrograde tracing from cPFC (red for the tdTomato Cre reporter) and HCR-FISH against Npr3 (cyan), showing enrichment of cPFC-projecting neurons in medial (superficial) and Npr3-expressing cells in lateral (deeper) parts of vmPFC Layer 5. Red insets highlight Npr3+ cells and cyan insets highlight Npr3− cells, at higher magnification. Red arrowheads point to Npr3+ cells. Scale bar, 20 μm.
Figure S4. Optogenetic cannulae placement, GRIN lens targeting, and Ca2+ imaging fields of view for the 2-odor task, related to Figure 4
(A) Positions of bilateral optogenetic fibers registered to a standard atlas (Paxinos and Franklin, 2001). For all mice, fibers were located within prelimbic cortex (PL; A–P: +1.95, M–L: ± 0.35, D–V: −2.3). Each different colored dot denotes the location of the tip center for individual mice, and anterior–posterior (A–P) position is summarized from two adjacent atlas outlines. Scale bar, 500 μm.
(B) Coronal sections of GRIN lens tracts (A–P: ~+1.95) and placement (500 μm width, 6.1 mm length, implanted at A–P: +1.95, M–L: +0.4, D–V: −2.4) in the right hemisphere registered to a standard atlas, highlighting GCaMP6f-labeled cells sitting below lens tip. Dots denote the GRIN lens tip center for each mouse, and are colored by cell class (Orange: PAG-projecting, Green: cPFC-projecting, Magenta: Rbp4Cre). Focal plane is ~200–300 μm below the lens tip. Scale bar, 100 μm (left, images), 500 μm (right, atlas). Cg, cingulate cortex; PL, prelimbic cortex; IL, infralimbic cortex.
(C) Correlation image and contour plot of neurons detected by CNMF-E in the fields of view of all imaged mice used in the 2-odor task, labeled with the number of cells analyzed and mouse name (including those in Figure 4G: mouse P1, C1, R2, where maximum intensity projections are displayed and rotated to be in line with the A–P axis). Colored rings are the regions of interest of individual extracted cells. Scale bar, ~25 μm.
Figure S5. Task-relevant Ca2+ activity dynamics vary by cell class in vmPFC, and encoding of reward omission, related to Figure 5
(A) Example single-trial activity (top) and corresponding trial-averaged activity (bottom, mean ± SEM) of four task-modulated cPFC-projecting (left) and Rbp4Cre-labeled (right) cells during each of the four task epochs (Approach, Decision, Lick, and Reward). Traces include all correct rewarded trials. Reward was delivered 0.75 s after the first lick. Vertical dashed line in Approach/Decision epochs denotes odor onset. Vertical dashed lines in Lick/Reward epochs denote first lick and reward release (from left to right).
(B) Trial-averaged activity of odd trials (left) for all positively modulated cells sorted by the time of maximum activity, adjacent to trial-averaged activity of even trials with the same cell sorting order. Heatmap rasters are grouped by cell class, and aligned to odor onset (left) and first lick (right) (n = 90 PAG-projecting cells, n = 95 cPFC-projecting cells, n = 339 Rbp4Cre-labeled cells). Similar patterns of activity observed independently between odd and even trials demonstrate that the tiling pattern is not due to the sorting itself.
(C) Example cells with reduced (left and middle panel) or elevated (right panel) activity during reward omission trials. Single-trial activity (top) and corresponding trial-averaged activity (bottom, mean ± SEM) is shown, color-coded by trial type. Vertical dashed lines denote lick onset (left) and reward release (right).
(D) Fraction of cells that: 1) had elevated activity during reward omission trials, 2) had activity during the Reward epoch that was reduced during reward omission trials, or 3) had activity during the Reward epoch that was unchanged during reward omission trials, across three cell classes. Example cells that had significantly elevated or reduced activity (Figure S5C) during omission trials were observed in all classes, indicating that information about reward omission is broadly present. Comparison of the proportion of cells modulated by reward omission (brackets: total of elevated activity and reduced activity cells) across classes was performed using a permutation test (*p < 0.05). Reduced activity was more common than elevated activity during omission, and Rbp4Cre-labeled cells were most likely to be modulated.
(E) Average population activity of each cell class during rewarded (colored) versus omission trials (gray) (mean ± SEM) in the Lick and Reward epochs. The average signal within each class exhibits a net negative modulation during omission trials in the Reward epoch. Colored dots represent time points in the Reward epoch where average activity in rewarded trials is significantly higher than in omission trials (p < 0.05, paired t test).
Figure S6. Analysis of side-selectivity and dimensionality in the 2-odor task, and imaging fields of view in the 4-odor cued and uncued tasks, related to Figures 6 and 7
(A) Trial-averaged activity of left-selective cells (top) or right-selective cells (bottom). Left-selective cells are sorted by the time of peak activity on left trials and right-selective cells are sorted by that on right trials (left panels) and then compared to activity of the same cell on the opposing trial type (right panels). The loss of activity observed when comparing right panels with their corresponding left panels demonstrate the extent of side-selectivity in all three cell classes.
(B) Fraction of variance explained as a function of the number of principal components included, for all cells regardless of cell class (left, related to Figure 6B) or subdivided by cell class (right, related to Figure 6C). These results indicate that the data derived from each cell class has similar dimensionality, suggesting a similar diversity of activity patterns in all classes.
(C) Correlation image and contour plot of neurons detected by CNMF-E in the fields of view of all imaged mice used in the 4-odor cued and uncued tasks, labeled with the number of cells analyzed and mouse name. Colored rings are the regions of interest of individual extracted cells. Scale bar, ~25 μm.
Figure S7. Neural data is similar between the 2- and 4-odor tasks but differs in the uncued task, and analysis of mixed selectivity, related to Figure 7
(A) Fraction of uncued trials that were correct as mice progressed through the task (mean ± SEM).
(B) Cells positively modulated in each of the four task epochs in the 4-odor task (Approach, Decision, Lick, Reward) as a fraction of all cells, on a per-mouse basis (mean ± SEM; *p < 0.05; one-way ANOVA, post hoc Tukey’s HSD test; n.s., not significant).
(C) Average choice direction prediction accuracy across mice in the 4-odor task (mean ± SEM, PAG-projecting n = 5, cPFC-projecting n = 5, and Rbp4Cre-labeled n = 8 mice). Because of the 4 trial types, data from left (L-1 + L-2) trials and right (R-1 + R-2) trials were pooled for this analysis, and randomly subsampled to 25 cells per mouse (mean ± SEM). Black dots represent time points where the PAG-projecting trace is significantly different than the cPFC-projecting trace. Orange or green dots represent time points where the PAG- or cPFC-projecting trace is significantly different than the other two traces, respectively (p < 0.05, one-way ANOVA, post hoc Tukey’s HSD test). Models were computed independently for each epoch with 5-fold cross validation (randomly selected 80% of trials for training and the remaining 20% for testing).
(D) Average choice direction prediction accuracy during the Decision epoch, as a function of the number of cells included in the logistic regression analysis (mean ± SEM) in the 4-odor task. Black dots represent time points where the prediction accuracy for PAG-projecting cells is significantly higher than for cPFC-projecting cells. Green dots represent time points where the prediction accuracy for cPFC-projecting cells is significantly lower than the other two (p < 0.05, one-way ANOVA, post hoc Tukey’s HSD test).
(E) Trial-averaged activity of all positively modulated cells subdivided into the first half of cued trials, the second half of cued trials, and uncued trials. Heatmap rasters were sorted by the time of maximum activity and grouped by cell class, aligned to odor onset (left) and first lick (right) (n = 110 PAG-projecting cells, n = 89 cPFC-projecting cells, n = 348 Rbp4Cre-labeled cells). The observed similarity in activity between the 1st and 2nd half of the cued trials, and the dissimilarity in activity between the 2nd half of the cued trials and the uncued trials, demonstrates that the difference between uncued and cued trials is not purely due to the passage of time.
(F) Variance in Ca2+ data explained as a function of the number of principal components included, across task type (cued, uncued, and combined). The increase in dimensionality when combining the two datasets indicates that the neural activity patterns found in the two tasks are not the same. For the same imaged cells in the same field of view, novel activity patterns emerge when transitioning from the cued task to the uncued task.
(G) Scatterplot of regression coefficients for choice direction in the 4-odor task versus reward context in the uncued task for every imaged cell. Cells with a significant regression coefficient(s) are colored as follows: choice direction (dark red), reward context (dark blue), or both (mixed selective, turquoise). Bar plot represents the proportion of mixed selective cells grouped by cell class. Comparison of the proportion of mixed selective cells across classes was performed using a permutation test (*p < 0.05).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-GFP chicken polyclonal antibody | Aves Labs | Cat# GFP-1020; RRID: AB_1000240 |
| Slc17a7(Vglut1) probe | Molecular Instruments | NM_182993.2 |
| Cd44 probe | Molecular Instruments | NM_009851.2 |
| Vegfd (Figf) probe | Molecular Instruments | NM_010216.2 |
| Otof probe | Molecular Instruments | NM_031875.2 |
| Pld5 probe | Molecular Instruments | NM_176916.4 |
| Ackr3 (Cxcr7) probe | Molecular Instruments | NM_007722.4 |
| Npr3 probe | Molecular Instruments | NM_008728.2 |
| Tshz2 probe | Molecular Instruments | NM_080455.2 |
| Bacterial and Virus Strains | ||
| CAV-Cre | Soudais et al., 2001 | N/A |
| AAVretro-CMV-Cre-2A-eGFP | Stanford Vector Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Isoflurane | Henry Schein | CAS# 26675-46-7; CHEBI: 6015 |
| Avertin (2,2,2-Tribromoethanol) | Sigma | SKU# T48402 |
| Tissue-Plus O.C.T. Compound | Thermo | Cat# 23-730-571 |
| Cholera Toxin B-Alexa488 | Molecular Probes | C-34775 |
| Hoechst | Life Technologies | H3570 |
| DAPI | Thermo | D1306 |
| Mineral oil | Fisher Scientific | O122–4 |
| Valeric Acid | Sigma-Aldrich | 240370 |
| 1-hexanol | Sigma-Aldrich | H13303 |
| R-carvone | Sigma-Aldrich | 124931 |
| S-carvone | Sigma-Aldrich | 435759 |
| Papain Dissociation System | Worthington | LK003150 |
| SMARTScribe Reverse Transcriptase | Takara | 639538 |
| KAPA HiFi HotStart ReadyMix | Roche | KK2602 |
| AMPure Beads | Fisher | A63881 |
| Critical Commercial Assays | ||
| Quant-iT™ PicoGreen™ dsDNA Assay Kit | Thermo | P11496 |
| Nextera XT Library Sample Preparation kit | Illumina | FC-131–1096 |
| Deposited Data | ||
| RNA-sequencing data | This paper | GSE161936: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE161936 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Rbp4-KL100-Cre | GENSAT | KL100 |
| Mouse: Ai14 (Rosa-CAG-LSL-tdTomato-WPRE) | JAX | 7914 |
| Mouse: Gad2-IRES-Cre | JAX | 10802 |
| Mouse: Ai32 (Rosa-CAG-LSL-ChR2(H134R)-EYFP-WPRE) | JAX | 24109 |
| Mouse Ai148 (TIT2L-GC6f-ICL-tTA2)-D | JAX | 30328 |
| Mouse: C57/Bl6 | JAX | 664 |
| Mouse: CD1 | Charles River | N/A |
| Software and Algorithms | ||
| MATLAB | Mathworks | https://www.mathworks.com |
| Imaris | Oxford Instruments | https://imaris.oxinst.com/ |
| Inscopix Data Collection Software | Inscopix | https://www.inscopix.com |
| Inscopix Data Processing Software | Inscopix | https://www.inscopix.com |
| Python | Python | https://www.python.org/ |
| ImageJ (Fiji) | NIH | https://fiji.sc/ |
| Seurat | New York University | https://github.com/satijalab/seurat |
| scrattch.hicat | Allen Brain Institute | https://github.com/Allenlnstitute/scrattch.hicat |
| Bpod | CSHL/Sanworks | https://github.com/KepecsLab/Bpod_r0_5 |
| Arduino | Arduino | https://www.arduino.cc |
| SOLIDWORKS | Dassault Systèmes | https://www.solidworks.com |
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| CNMF-E | Zhou et al., 2018 | https://github.com/zhoupc/CNMF_E |
| U-Net | UNI Freiburg | https://lmb.informatik.uni-freiburg.de/people/ronneber/u-net/ |
| Ca2+ imaging data and analysis code | This paper | Upon request |
| Other | ||
| White acrylic | Plastic-Craft Products | AC1382 |
| Infrared sensor | Adafruit | 2167 |
| Valves | NResearch | 161T011, 161T031 |
| Flowmeter | Cole-Parmer | EW-03227–18 |
| Arduino Due | Arduino | A000062 |
| Arduino TinkerKit | Arduino | K000001 |
| GRIN lens | Inscopix | 1050–002211 |
| Baseplate | Inscopix | 1050–004201 |
| Baseplate cover | Inscopix | 1050–002193 |
| Dummy microscope | Inscopix | 1050–002196 |
| Kwik-Sil | WPI | N/A |
| Kwik-Kast | WPI | N/A |
| Gel Control Superglue | Loctite | 45198 |
| Jet Liquid | Lang Dental | N/A |
| Gel foam | Pfizer | N/A |
| Dual-core implantable Fiber Optic cannulae | ThorLabs | CFM32L10 |
Highlights.
No 1-to-1 mapping between transcriptomic and projection types for PFC neurons
Even a molecularly homogeneous PFC neuron population encodes diverse task signals
PFC→PAG and PFC→cPFC cells prefer choice and reward context encoding, respectively
Task-related signal encoding across cell types balances redundancy and specificity
ACKNOWLEDGMENTS
We thank E. Kremer for CAV-Cre virus; A. Mizrahi, D. Pederick, and L. DeNardo for critical comments on the manuscript; L. Driscoll and Luo Lab members for helpful discussion and reagents; L. Schwarz for pilot in situ hybridization experiments; L. Murrow for scRNAseq analysis advice; R. Jones and S. Kolluru for technical help with scRNAseq and data management; C. Tsang, L. Cardy, and A. Thomas for Inscopix technical help; and M. Andermann for supporting N.D.N. to work on this project after leaving Stanford. J.H.L. is supported by an NIMH K01-MH114022 grant. S.R.Q. is a Biohub investigator. L.L. is an HHMI investigator. This work was supported by an NIH grant (R01-NS050835) and an NSF NeuroNex grant.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.11.046.
DECLARATION OF INTERESTS
The authors have no competing interests.
REFERENCES
- Allen WE, DeNardo LA, Chen MZ, Liu CD, Loh KM, Fenno LE, Ramakrishnan C, Deisseroth K, and Luo L (2017a). Thirst-associated preoptic neurons encode an aversive motivational drive. Science 357, 1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen WE, Kauvar IV, Chen MZ, Richman EB, Yang SJ, Chan K, Gradinaru V, Deverman BE, Luo L, and Deisseroth K (2017b). Global Representations of Goal-Directed Behavior in Distinct Cell Types of Mouse Neocortex. Neuron 94, 891–907.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen WE, Chen MZ, Pichamoorthy N, Tien RH, Pachitariu M, Luo L, and Deisseroth K (2019). Thirst regulates motivated behavior through modulation of brainwide neural population dynamics. Science 364, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaad WF, Rainer G, and Miller EK (1998). Neural activity in the primate prefrontal cortex during associative learning. Neuron 21, 1399–1407. [DOI] [PubMed] [Google Scholar]
- Bari BA, Grossman CD, Lubin EE, Rajagopalan AE, Cressy JI, and Cohen JY (2019). Stable Representations of Decision Variables for Flexible Behavior. Neuron 103, 922–933.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, Gao XJ, Kremer EJ, Malenka RC, and Luo L (2015). Circuit Architecture of VTA Dopamine Neurons Revealed by Systematic Input-Output Mapping. Cell 162, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betley JN, Cao ZF, Ritola KD, and Sternson SM (2013). Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell 155, 1337–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacherjee A, Djekidel MN, Chen R, Chen W, Tuesta LM, and Zhang Y (2019). Cell type-specific transcriptional programs in mouse prefrontal cortex during adolescence and addiction. Nat. Commun 10, 4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolkan SS, Stujenske JM, Parnaudeau S, Spellman TJ, Rauffenbart C, Abbas AI, Harris AZ, Gordon JA, and Kellendonk C (2017). Thalamic projections sustain prefrontal activity during working memory maintenance. Nat. Neurosci 20, 987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Sun YC, Zhan H, Kebschull JM, Fischer S, Matho K, Huang ZJ, Gillis J, and Zador AM (2019). High-Throughput Mapping of Long-Range Neuronal Projection Using In Situ Sequencing. Cell 179, 772–786.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HMT, Schwarzkopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J, Cunha A, and Pierce NA (2018). Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchland MM, Yu BM, Ryu SI, Santhanam G, and Shenoy KV (2006). Neural variability in premotor cortex provides a signature of motor preparation. J. Neurosci 26, 3697–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Madisen L, Hage TA, Valley MT, Knoblich U, Larsen RS, Takeno MM, Huang L, Gu H, Larsen R, et al. (2018). A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell 174, 465–480.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo LA, Liu CD, Allen WE, Adams EL, Friedmann D, Fu L, Guenthner CJ, Tessier-Lavigne M, and Luo L (2019). Temporal evolution of cortical ensembles promoting remote memory retrieval. Nat. Neurosci 22, 460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll LN, Pettit NL, Minderer M, Chettih SN, and Harvey CD (2017). Dynamic Reorganization of Neuronal Activity Patterns in Parietal Cortex. Cell 170, 986–999.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economo MN, Viswanathan S, Tasic B, Bas E, Winnubst J, Menon V, Graybuck LT, Nguyen TN, Smith KA, Yao Z, et al. (2018). Distinct descending motor cortex pathways and their roles in movement. Nature 563, 79–84. [DOI] [PubMed] [Google Scholar]
- Euston DR, Gruber AJ, and McNaughton BL (2012). The role of medial prefrontal cortex in memory and decision making. Neuron 76, 1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk T, Mai D, Bensch R,Çiçek Ö, Abdulkadir A, Marrakchi Y, Böhm A, Deubner J, Jäckel Z, Seiwald K, et al. (2019). U-Net: deep learning for cell counting, detection, and morphometry. Nat. Methods 16, 67–70. [DOI] [PubMed] [Google Scholar]
- Feierstein CE, Quirk MC, Uchida N, Sosulski DL, and Mainen ZF (2006). Representation of spatial goals in rat orbitofrontal cortex. Neuron 51, 495–507. [DOI] [PubMed] [Google Scholar]
- Friedrich J, Zhou P, and Paninski L (2017). Fast online deconvolution of calcium imaging data. PLoS Comput. Biol 13, e1005423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusi S, Miller EK, and Rigotti M (2016). Why neurons mix: high dimensionality for higher cognition. Curr. Opin. Neurobiol 37, 66–74. [DOI] [PubMed] [Google Scholar]
- Fuster JM (2008). The prefrontal cortex: anatomy, physiology, and neuropsychology of the frontal lobe, Fourth Edition (Philadelphia: Academic Press; ). [Google Scholar]
- Fuster JM, and Alexander GE (1971). Neuron activity related to short-term memory. Science 173, 652–654. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, and Busby SJ (2005). Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J. Comp. Neurol 492, 145–177. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, and Heintz N (2013). GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80, 1368–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh KK, Burns LD, Cocker ED, Nimmerjahn A, Ziv Y, Gamal AE, and Schnitzer MJ (2011). Miniaturized integration of a fluorescence microscope. Nat. Methods 8, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokce O, Stanley GM, Treutlein B, Neff NF, Camp JG, Malenka RC, Rothwell PE, Fuccillo MV, Südhof TC, and Quake SR (2016). Cellular Taxonomy of the Mouse Striatum as Revealed by Single-Cell RNA-Seq. Cell Rep 16, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong H, Xu D, Yuan J, Li X, Guo C, Peng J, Li Y, Schwarz LA, Li A, Hu B, et al. (2016). High-throughput dual-colour precision imaging for brainwide connectome with cytoarchitectonic landmarks at the cellular level. Nat. Commun 7, 12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, and Macklis JD (2013). Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci 14, 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZV, Hires SA, Li N, O’Connor DH, Komiyama T, Ophir E, Huber D, Bonardi C, Morandell K, Gutnisky D, et al. (2014a). Procedures for behavioral experiments in head-fixed mice. PLoS ONE 9, e88678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZV, Li N, Huber D, Ophir E, Gutnisky D, Ting JT, Feng G, and Svoboda K (2014b). Flow of cortical activity underlying a tactile decision in mice. Neuron 81, 179–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Kebschull JM, Campbell RAA, Cowan D, Imhof F, Zador AM, and Mrsic-Flogel TD (2018). The logic of single-cell projections from visual cortex. Nature 556, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, and Mrsic-Flogel TD (2013). Cortical connectivity and sensory coding. Nature 503, 51–58. [DOI] [PubMed] [Google Scholar]
- Harris JA, Mihalas S, Hirokawa KE, Whitesell JD, Choi H, Bernard A, Bohn P, Caldejon S, Casal L, Cho A, et al. (2019). Hierarchical organization of cortical and thalamic connectivity. Nature 575, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Coen P, and Tank DW (2012). Choice-specific sequences in parietal cortex during a virtual-navigation decision task. Nature 484, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Tucciarone J, Lee S, Nigro MJ, Kim Y, Levine JM, Kelly SM, Krugikov I, Wu P, Chen Y, et al. (2016). Strategies and Tools for Combinatorial Targeting of GABAergic Neurons in Mouse Cerebral Cortex. Neuron 91, 1228–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández A, Nácher V, Luna R, Zainos A, Lemus L, Alvarez M, Vázquez Y, Camarillo L, and Romo R (2010). Decoding a perceptual decision process across cortex. Neuron 66, 300–314. [DOI] [PubMed] [Google Scholar]
- Hirokawa J, Vaughan A, Masset P, Ott T, and Kepecs A (2019). Frontal cortex neuron types categorically encode single decision variables. Nature 576, 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WH, Wang DC, Allen WE, Klope M, Hu H, Shamloo M, and Luo L (2018). Early adolescent Rai1 reactivation reverses transcriptional and social interaction deficits in a mouse model of Smith-Magenis syndrome. Proc. Natl. Acad. Sci. USA 115, 10744–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgenson LA, Newsome WT, Anderson DJ, Bargmann CI, Brown EN, Deisseroth K, Donoghue JP, Hudson KL, Ling GS, MacLeish PR, et al. (2015). The BRAIN Initiative: developing technology to catalyse neuroscience discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci 370 10.1098/rstb.2014.0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamigaki T, and Dan Y (2017). Delay activity of specific prefrontal interneuron subtypes modulates memory-guided behavior. Nat. Neurosci 20, 854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull JM, Garcia da Silva P, Reid AP, Peikon ID, Albeanu DF, and Zador AM (2016). High-Throughput Mapping of Single-Neuron Projections by Sequencing of Barcoded RNA. Neuron 91, 975–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull JM, Ringach N, Richman EB, Friedmann D, Kolluru SS, Jones RC, Allen WE, Wang Y, Zhou H, Cho SW, et al. (2020). Cerebellar nuclei evolved by repeatedly duplicating a conserved cell type set. Science, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiani R, Cueva CJ, Reppas JB, and Newsome WT (2014). Dynamics of neural population responses in prefrontal cortex indicate changes of mind on single trials. Curr. Biol 24, 1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CK, Ye L, Jennings JH, Pichamoorthy N, Tang DD, Yoo AW, Ramakrishnan C, and Deisseroth K (2017). Molecular and Circuit-Dynamical Identification of Top-Down Neural Mechanisms for Restraint of Reward Seeking. Cell 170, 1013–1027.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Yao Z, Graybuck LT, Kim TK, Nguyen TN, Smith KA, Fong O, Yi L, Koulena N, Pierson N, et al. (2019). Multimodal Analysis of Cell Types in a Hypothalamic Node Controlling Social Behavior. Cell 179, 713–728.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Li N, Chen TW, Guo ZV, Gerfen CR, and Svoboda K (2015). A motor cortex circuit for motor planning and movement. Nature 519, 51–56. [DOI] [PubMed] [Google Scholar]
- Li H, Horns F, Wu B, Xie Q, Li J, Li T, Luginbuhl DJ, Quake SR, and Luo L (2017). Classifying Drosophila Olfactory Projection Neuron Subtypes by Single-Cell RNA Sequencing. Cell 171, 1206–1220.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, Vaughan A, Sturgill JF, and Kepecs A (2018). A Viral Receptor Complementation Strategy to Overcome CAV-2 Tropism for Efficient Retrograde Targeting of Neurons. Neuron 98, 905–917.e5. [DOI] [PubMed] [Google Scholar]
- Lin R, Wang R, Yuan J, Feng Q, Zhou Y, Zeng S, Ren M, Jiang S, Ni H, Zhou C, et al. (2018). Cell-type-specific and projection-specific brainwide reconstruction of single neurons. Nat. Methods 15, 1033–1036. [DOI] [PubMed] [Google Scholar]
- Luo L, Callaway EM, and Svoboda K (2018). Genetic Dissection of Neural Circuits: A Decade of Progress. Neuron 98, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YW, Garcia AJ 3rd, Gu X, Zanella S, et al. (2012). A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci 15, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mante V, Sussillo D, Shenoy KV, and Newsome WT (2013). Context-dependent computation by recurrent dynamics in prefrontal cortex. Nature 503, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK, and Cohen JD (2001). An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci 24, 167–202. [DOI] [PubMed] [Google Scholar]
- Miller EK, Erickson CA, and Desimone R (1996). Neural mechanisms of visual working memory in prefrontal cortex of the macaque. J. Neurosci 16, 5154–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, Perez JD, Rubinstein ND, Hao J, Regev A, Dulac C, and Zhuang X (2018). Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel EA, Nimmerjahn A, and Schnitzer MJ (2009). Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugan M, Jang HJ, Park M, Miller EM, Cox J, Taliaferro JP, Parker NF, Bhave V, Hur H, Liang Y, et al. (2017). Combined Social and Spatial Coding in a Descending Projection from the Prefrontal Cortex. Cell 171, 1663–1677.e1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Harris JA, Ng L, Winslow B, Cain N, Mihalas S, Wang Q, Lau C, Kuan L, Henry AM, et al. (2014). A mesoscale connectome of the mouse brain. Nature 508, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Namboodiri VM, Matan AM, Voets ES, Mohorn EP, Kosyk O, McHenry JA, Robinson JE, Resendez SL, Rossi MA, and Stuber GD (2017). Prefrontal cortex output circuits guide reward seeking through divergent cue encoding. Nature 543, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul A, Crow M, Raudales R, He M, Gillis J, and Huang ZJ (2017). Transcriptional Architecture of Synaptic Communication Delineates GABAergic Neuron Identity. Cell 171, 522–539.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, and Franklin KBJ (2001). The Mouse Brain in Stereotaxic Coordinates, Second Edition (Academic Press; ). [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ, and Scanziani M (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat. Neurosci 16, 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Pinto L, and Dan Y (2015). Cell-Type-Specific Activity in Prefrontal Cortex during Goal-Directed Behavior. Neuron 87, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L, Rajan K, DePasquale B, Thiberge SY, Tank DW, and Brody CD (2019). Task-Dependent Changes in the Large-Scale Dynamics and Necessity of Cortical Regions. Neuron 104, 810–824.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss TM (1995). Do rats have prefrontal cortex? The rose-woolsey-akert program reconsidered. J. Cogn. Neurosci 7, 1–24. [DOI] [PubMed] [Google Scholar]
- Rajasethupathy P, Sankaran S, Marshel JH, Kim CK, Ferenczi E, Lee SY, Berndt A, Ramakrishnan C, Jaffe A, Lo M, et al. (2015). Projections from neocortex mediate top-down control of memory retrieval. Nature 526, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Friedmann D, Xiong J, Liu CD, Ferguson BR, Weerakkody T, DeLoach KE, Ran C, Pun A, Sun Y, et al. (2018). Anatomically Defined and Functionally Distinct Dorsal Raphe Serotonin Sub-systems. Cell 175, 472–487.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Isakova A, Friedmann D, Zeng J, Grutzner SM, Pun A, Zhao GQ, Kolluru SS, Wang R, Lin R, et al. (2019). Single-cell transcriptomes and whole-brain projections of serotonin neurons in the mouse dorsal and median raphe nuclei. eLife 8, e49424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resendez SL, Jennings JH, Ung RL, Namboodiri VM, Zhou ZC, Otis JM, Nomura H, McHenry JA, Kosyk O, and Stuber GD (2016). Visualization of cortical, subcortical and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat. Protoc 11, 566–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigotti M, Barak O, Warden MR, Wang XJ, Daw ND, Miller EK, and Fusi S (2013). The importance of mixed selectivity in complex cognitive tasks. Nature 497, 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JE, and Woolsey CN (1948). The orbitofrontal cortex and its connections with the mediodorsal nucleus in rabbit, sheep and cat. Res Publ Assoc Res Nerv Ment Dis 27, 210–232. [PubMed] [Google Scholar]
- Rushworth MF, Noonan MP, Boorman ED, Walton ME, and Behrens TE (2011). Frontal cortex and reward-guided learning and decision-making. Neuron 70, 1054–1069. [DOI] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt LI, Wimmer RD, Nakajima M, Happ M, Mofakham S, and Halassa MM (2017). Thalamic amplification of cortical connectivity sustains attentional control. Nature 545, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Miyamichi K, Gao XJ, Beier KT, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ, and Luo L (2015). Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature 524, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, Ramakrishnan C, Fenno LE, Davidson TJ, Wright M, and Deisseroth K (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice . Sci. Transl. Med 9, eaah6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. (2016). Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 166, 1308–1323.e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy KV, Sahani M, and Churchland MM (2013). Cortical control of arm movements: a dynamical systems perspective. Annu. Rev. Neurosci 36, 337–359. [DOI] [PubMed] [Google Scholar]
- Siciliano CA, Noamany H, Chang CJ, Brown AR, Chen X, Leible D, Lee JJ, Wang J, Vernon AN, Vander Weele CM, et al. (2019). A cortical-brainstem circuit predicts and governs compulsive alcohol drinking. Science 366, 1008–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudais C, Laplace-Builhe C, Kissa K, and Kremer EJ (2001). Preferential transduction of neurons by canine adenovirus vectors and their efficient retrograde transport in vivo. FASEB J 15, 2283–2285. [DOI] [PubMed] [Google Scholar]
- Spellman T, Rigotti M, Ahmari SE, Fusi S, Gogos JA, and Gordon JA (2015). Hippocampal-prefrontal input supports spatial encoding in working memory. Nature 522, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis AM, Schachter MJ, Gulati S, Zitelli KT, Malanowski S, Tajik A, Fritz C, Trulson M, and Otte SL (2018). Simultaneous Optogenetics and Cellular Resolution Calcium Imaging During Active Behavior Using a Miniaturized Microscope. Front. Neurosci 12, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley G, Gokce O, Malenka RC, Südhof TC, and Quake SR (2020). Continuous and Discrete Neuron Types of the Adult Murine Striatum. Neuron 105, 688–699.e8. [DOI] [PubMed] [Google Scholar]
- Steinmetz NA, Zatka-Haas P, Carandini M, and Harris KD (2019). Distributed coding of choice, action and engagement across the mouse brain. Nature 576, 266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, et al. (2011). A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervo DG, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron 92, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Tosches MA, Yamawaki TM, Naumann RK, Jacobi AA, Tushev G, and Laurent G (2018). Evolution of pallium, hippocampus, and cortical cell types revealed by single-cell transcriptomics in reptiles. Science 360, 881–888. [DOI] [PubMed] [Google Scholar]
- Uchida N, and Mainen ZF (2003). Speed and accuracy of olfactory discrimination in the rat. Nat. Neurosci 6, 1224–1229. [DOI] [PubMed] [Google Scholar]
- Uylings HB, Groenewegen HJ, and Kolb B (2003). Do rats have a prefrontal cortex? Behav. Brain Res 146, 3–17. [DOI] [PubMed] [Google Scholar]
- van der Maaten LJP, and Hinton GE (2008). Visualizing high dimensional data using t-SNE. J. Mach. Learn. Res 9, 2579–2605. [Google Scholar]
- Vander Weele CM, Siciliano CA, Matthews GA, Namburi P, Izadmehr EM, Espinel IC, Nieh EH, Schut EHS, Padilla-Coreano N, Burgos-Robles A, et al. (2018). Dopamine enhances signal-to-noise ratio in cortical-brainstem encoding of aversive stimuli. Nature 563, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner MJ, Kim TH, Kadmon J, Nguyen ND, Ganguli S, Schnitzer MJ, and Luo L (2019). Shared Cortex-Cerebellum Dynamics in the Execution and Learning of a Motor Task. Cell 177, 669–682.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnubst J, Bas E, Ferreira TA, Wu Z, Economo MN, Edson P, Arthur BJ, Bruns C, Rokicki K, Schauder D, et al. (2019). Reconstruction of 1,000 Projection Neurons Reveals New Cell Types and Organization of Long-Range Connectivity in the Mouse Brain. Cell 179, 268–281.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Hawrylycz M, Aalling N, Aguilar-Valles A, Arendt D, Arnedillo RA, Ascoli GA, Bielza C, Bokharaie V, Bergmann TB, et al. (2020). A community-based transcriptomics classification and nomenclature of neocortical cell types. Nat. Neurosci 23, 1456–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappia L, and Oshlack A (2018). Clustering trees: a visualization for evaluating clusterings at multiple resolutions. GigaScience 7, gigascience/giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, La Manno G, et al. (2018). Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, and Sanes JR (2017). Neuronal cell-type classification: challenges, opportunities and the path forward. Nat. Rev. Neurosci 18, 530–546. [DOI] [PubMed] [Google Scholar]
- Zhou P, Resendez SL, Rodriguez-Romaguera J, Jimenez JC, Neufeld SQ, Giovannucci A, Friedrich J, Pnevmatikakis EA, Stuber GD, Hen R, et al. (2018). Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. eLife 7, e28728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg B, Hintiryan H, Gou L, Song MY, Bay M, Bienkowski MS, Foster NN, Yamashita S, Bowman I, Toga AW, and Dong HW (2014). Neural networks of the mouse neocortex. Cell 156, 1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sequencing quality control, validation of PFC neuron clustering, and other scRNAseq analysis, related to Figure 1
(A) Violin plots showing the distribution of the number of genes detected (left) and the number of reads (right) across single cells derived from each of the three PFC regions. The dots within each violin denote the median of the distribution (~7000 genes, ~1–2 million reads per cell). Effects in the gene expression data driven specifically by differences in the number of detected genes and the number of reads were regressed out from further analysis by the Seurat ScaleData function as part of the standard pipeline.
(B) Comparison of Rbp4Cre>tdTomato+ PFC neuron (n = 3139 cells, as in Figure 1) classification between reference clusters (Figure 1, Seurat resolution = 0.3) and clusters generated from the Allen Institute scrattch.hicat analysis (Tasic et al., 2018). As a quality control measure, principal components correlating with log2 detected genes, # of reads, plate format, and sample replicate were removed, see STAR Methods for additional parameters. Allen clusters are named based on similarity and subdivision from the Seurat clusters. Color scale represents the Jaccard similarity index (intersection divided by the union) of the two groups, and the size of each dot is the square root (sqrt) of the cell number. The percentage contribution of each region to each cluster (normalized to the size of the cell population) is displayed on top, with the same color code as (A). Only one cluster was highly region-specific [Cxcr7(5–2) with > 85% contribution from OFC].
(C) Matrix depicting the robustness of Rbp4Cre>tdTomato+ PFC neuron classification across multiple iterations of consensus clustering in scrattch.hicat. Rows represent cells, columns represent clusters, and color scale represents the probability of being assigned to the cluster based on 20 iterations of consensus clustering.
(D) List of clusters from Tasic et al., 2018 that contained cells derived from Rbp4Cre-labeled mice and met quality control measurements (STAR Methods). Clusters were then grouped into the four larger categories and used for display purposes in Figure 1F.
Figure S2. Additional HCR-FISH validations and example retrograde labeling, related to Figures 2 and 3
(A) Laminar distribution of cells expressing cluster-specific marker genes across dmPFC, analogous to Figure 2B. Dashed lines are approximate cortical layer boundaries according to the Allen Atlas (L5a, 130 μm; L5b, 345 μm; L6, 565 μm, aligned to the beginning of L2/3).
(B) Triple HCR-FISH between Otof, Cd44, and Cxcr7 in vmPFC. Quantification of overlap is shown for both dmPFC and vmPFC, and averaged across n = 4 mice (543 dmPFC, 603 vmPFC cells). Error bars are SEM. Scale bar, 50 μm.
(C) Triple HCR-FISH between Cd44, Cxcr7, and Tshz2 in vmPFC. Quantification of overlap is shown for both dmPFC and vmPFC, and averaged across n = 4 mice (380 dmPFC, 372 vmPFC cells). Error bars are SEM. Scale bar, 50 μm.
(D) Comparison of Pld5 expression in mPFC versus OFC. Sequencing data shows the percentage of cells that belonged to the Pld5 cluster, comparing dmPFC and vmPFC with OFC (dmPFC: 910 cells, n = 3 mice; vmPFC: 1234 cells, n = 4 mice; OFC: 995 cells, n = 4 mice; *p < 0.05, **p < 0.01, unpaired t test). Histology data shows the percentage of Vglut1+ cells that were Pld5+, comparing vmPFC and OFC. Cells were quantified within a 300 × 750 μm area, and a ratio paired t test was used, **p < 0.01. Scale bar, left: 250 μm, insets: 50 μm.
(E) Injection sites of CAV-Cre into the contralateral PFC (−0.6/+2.2/−2.5) and PAG (+0.4/−4.15/−2.8) of Ai14 mice (above). Resulting retrograde labeling of tdTomato+ cells 1 week post injection at bregma A–P +1.95 mm (below). Dotted lines indicate the edge of the tissue slice. Scale bar, 500 μm (top), 100 μm (bottom). Coordinates are expressed in (M–L / A–P / D–V) axes with respect to bregma, in mm.
(F) Overall distribution of PFC cells labeled by CAV-Cre injection into PAG: (+0.4/−4.15/−2.8) in an Ai14 mouse, highlighting presence and absence across PFC subregions. Cg, cingulate; PL, prelimbic; IL, infralimbic; MO, medial orbital; VO, ventral orbital; LO, lateral orbital; AI(D/V): agranular insular (dorsal/ventral); DP (dorsal peduncular) cortices. Scale bar, 500 μm.
Figure S3. Rbp4Cre>tdTomato+ and projection-defined neurons intermingle in tSNE space, and additional histological validation of marker expression in projection-defined neurons, related to Figure 3
(A) tSNE cluster map (top) combining all Rbp4Cre>tdTomato+ neurons from Figure 1 (n = 3139) and retrogradely-labeled vmPFC neurons from all 6 injection sites (n = 1155), labeled by color. Areas where retrograde cells do not intermingle well with Rbp4Cre>tdTomato+ cells are highlighted by arrows. A Syt6 cluster (known Layer 6 marker) is gained, and additional cells are in the Figf cluster. Feature plots of the seven marker genes (bottom) demonstrate that this map still roughly segregates by the marker genes identified in Figure 1, albeit with some interaction between Figf and Cxcr7 clusters. Areas with relatively low retrograde cell intermingling are predominantly in Figf and Syt6 clusters. Color scale represents expression level in the unit of ln[1+ (reads per 10000)].
(B) Nearest neighbor mapping of retrograde cells to transcriptomic cluster identities, at higher resolution, as an alternative to Figure 3B, represented by an alluvial diagram. Using 0.9 resolution in Seurat resulted in 12 clusters + Syt6 as possible transcriptomic types to map retrograde cells to. Under these conditions, differences in the relative contributions of Cd44 and Figf subclusters to cPFC, DS, NAc, or Amyg projections began to emerge, but no one-to-one relationships were observed. While these differences are potentially important, clustering at high resolution did not alter the general conclusions regarding the relation between transcriptomic type and projection target. Furthermore, mapping retrogradely-labeled cells to reference datasets containing only Rbp4+ or only vmPFC cells gave similar results (data not shown). For differentially expressed marker genes of the 12 clusters based on Seurat resolution = 0.9, see Table S1 Tab 2.
(C) Confocal images double-labeled with retrograde tracing from cPFC (red for the tdTomato Cre reporter) and HCR-FISH against Cd44, Cxcr7, or Figf (cyan). Scale bar, 20 μm. HCR-FISH signal was converted to binary puncta and overlaid with tdTomato cell outlines for quantification. Red insets highlight staining-positive cells (within 75% of maximum puncta observed), and cyan insets highlight staining-negative cells (< 25% of maximum puncta observed), at higher magnification.
(D) Confocal images double-labeled with retrograde tracing from cPFC (red for the tdTomato Cre reporter) and HCR-FISH against Npr3 (cyan), showing enrichment of cPFC-projecting neurons in medial (superficial) and Npr3-expressing cells in lateral (deeper) parts of vmPFC Layer 5. Red insets highlight Npr3+ cells and cyan insets highlight Npr3− cells, at higher magnification. Red arrowheads point to Npr3+ cells. Scale bar, 20 μm.
Figure S4. Optogenetic cannulae placement, GRIN lens targeting, and Ca2+ imaging fields of view for the 2-odor task, related to Figure 4
(A) Positions of bilateral optogenetic fibers registered to a standard atlas (Paxinos and Franklin, 2001). For all mice, fibers were located within prelimbic cortex (PL; A–P: +1.95, M–L: ± 0.35, D–V: −2.3). Each different colored dot denotes the location of the tip center for individual mice, and anterior–posterior (A–P) position is summarized from two adjacent atlas outlines. Scale bar, 500 μm.
(B) Coronal sections of GRIN lens tracts (A–P: ~+1.95) and placement (500 μm width, 6.1 mm length, implanted at A–P: +1.95, M–L: +0.4, D–V: −2.4) in the right hemisphere registered to a standard atlas, highlighting GCaMP6f-labeled cells sitting below lens tip. Dots denote the GRIN lens tip center for each mouse, and are colored by cell class (Orange: PAG-projecting, Green: cPFC-projecting, Magenta: Rbp4Cre). Focal plane is ~200–300 μm below the lens tip. Scale bar, 100 μm (left, images), 500 μm (right, atlas). Cg, cingulate cortex; PL, prelimbic cortex; IL, infralimbic cortex.
(C) Correlation image and contour plot of neurons detected by CNMF-E in the fields of view of all imaged mice used in the 2-odor task, labeled with the number of cells analyzed and mouse name (including those in Figure 4G: mouse P1, C1, R2, where maximum intensity projections are displayed and rotated to be in line with the A–P axis). Colored rings are the regions of interest of individual extracted cells. Scale bar, ~25 μm.
Figure S5. Task-relevant Ca2+ activity dynamics vary by cell class in vmPFC, and encoding of reward omission, related to Figure 5
(A) Example single-trial activity (top) and corresponding trial-averaged activity (bottom, mean ± SEM) of four task-modulated cPFC-projecting (left) and Rbp4Cre-labeled (right) cells during each of the four task epochs (Approach, Decision, Lick, and Reward). Traces include all correct rewarded trials. Reward was delivered 0.75 s after the first lick. Vertical dashed line in Approach/Decision epochs denotes odor onset. Vertical dashed lines in Lick/Reward epochs denote first lick and reward release (from left to right).
(B) Trial-averaged activity of odd trials (left) for all positively modulated cells sorted by the time of maximum activity, adjacent to trial-averaged activity of even trials with the same cell sorting order. Heatmap rasters are grouped by cell class, and aligned to odor onset (left) and first lick (right) (n = 90 PAG-projecting cells, n = 95 cPFC-projecting cells, n = 339 Rbp4Cre-labeled cells). Similar patterns of activity observed independently between odd and even trials demonstrate that the tiling pattern is not due to the sorting itself.
(C) Example cells with reduced (left and middle panel) or elevated (right panel) activity during reward omission trials. Single-trial activity (top) and corresponding trial-averaged activity (bottom, mean ± SEM) is shown, color-coded by trial type. Vertical dashed lines denote lick onset (left) and reward release (right).
(D) Fraction of cells that: 1) had elevated activity during reward omission trials, 2) had activity during the Reward epoch that was reduced during reward omission trials, or 3) had activity during the Reward epoch that was unchanged during reward omission trials, across three cell classes. Example cells that had significantly elevated or reduced activity (Figure S5C) during omission trials were observed in all classes, indicating that information about reward omission is broadly present. Comparison of the proportion of cells modulated by reward omission (brackets: total of elevated activity and reduced activity cells) across classes was performed using a permutation test (*p < 0.05). Reduced activity was more common than elevated activity during omission, and Rbp4Cre-labeled cells were most likely to be modulated.
(E) Average population activity of each cell class during rewarded (colored) versus omission trials (gray) (mean ± SEM) in the Lick and Reward epochs. The average signal within each class exhibits a net negative modulation during omission trials in the Reward epoch. Colored dots represent time points in the Reward epoch where average activity in rewarded trials is significantly higher than in omission trials (p < 0.05, paired t test).
Figure S6. Analysis of side-selectivity and dimensionality in the 2-odor task, and imaging fields of view in the 4-odor cued and uncued tasks, related to Figures 6 and 7
(A) Trial-averaged activity of left-selective cells (top) or right-selective cells (bottom). Left-selective cells are sorted by the time of peak activity on left trials and right-selective cells are sorted by that on right trials (left panels) and then compared to activity of the same cell on the opposing trial type (right panels). The loss of activity observed when comparing right panels with their corresponding left panels demonstrate the extent of side-selectivity in all three cell classes.
(B) Fraction of variance explained as a function of the number of principal components included, for all cells regardless of cell class (left, related to Figure 6B) or subdivided by cell class (right, related to Figure 6C). These results indicate that the data derived from each cell class has similar dimensionality, suggesting a similar diversity of activity patterns in all classes.
(C) Correlation image and contour plot of neurons detected by CNMF-E in the fields of view of all imaged mice used in the 4-odor cued and uncued tasks, labeled with the number of cells analyzed and mouse name. Colored rings are the regions of interest of individual extracted cells. Scale bar, ~25 μm.
Figure S7. Neural data is similar between the 2- and 4-odor tasks but differs in the uncued task, and analysis of mixed selectivity, related to Figure 7
(A) Fraction of uncued trials that were correct as mice progressed through the task (mean ± SEM).
(B) Cells positively modulated in each of the four task epochs in the 4-odor task (Approach, Decision, Lick, Reward) as a fraction of all cells, on a per-mouse basis (mean ± SEM; *p < 0.05; one-way ANOVA, post hoc Tukey’s HSD test; n.s., not significant).
(C) Average choice direction prediction accuracy across mice in the 4-odor task (mean ± SEM, PAG-projecting n = 5, cPFC-projecting n = 5, and Rbp4Cre-labeled n = 8 mice). Because of the 4 trial types, data from left (L-1 + L-2) trials and right (R-1 + R-2) trials were pooled for this analysis, and randomly subsampled to 25 cells per mouse (mean ± SEM). Black dots represent time points where the PAG-projecting trace is significantly different than the cPFC-projecting trace. Orange or green dots represent time points where the PAG- or cPFC-projecting trace is significantly different than the other two traces, respectively (p < 0.05, one-way ANOVA, post hoc Tukey’s HSD test). Models were computed independently for each epoch with 5-fold cross validation (randomly selected 80% of trials for training and the remaining 20% for testing).
(D) Average choice direction prediction accuracy during the Decision epoch, as a function of the number of cells included in the logistic regression analysis (mean ± SEM) in the 4-odor task. Black dots represent time points where the prediction accuracy for PAG-projecting cells is significantly higher than for cPFC-projecting cells. Green dots represent time points where the prediction accuracy for cPFC-projecting cells is significantly lower than the other two (p < 0.05, one-way ANOVA, post hoc Tukey’s HSD test).
(E) Trial-averaged activity of all positively modulated cells subdivided into the first half of cued trials, the second half of cued trials, and uncued trials. Heatmap rasters were sorted by the time of maximum activity and grouped by cell class, aligned to odor onset (left) and first lick (right) (n = 110 PAG-projecting cells, n = 89 cPFC-projecting cells, n = 348 Rbp4Cre-labeled cells). The observed similarity in activity between the 1st and 2nd half of the cued trials, and the dissimilarity in activity between the 2nd half of the cued trials and the uncued trials, demonstrates that the difference between uncued and cued trials is not purely due to the passage of time.
(F) Variance in Ca2+ data explained as a function of the number of principal components included, across task type (cued, uncued, and combined). The increase in dimensionality when combining the two datasets indicates that the neural activity patterns found in the two tasks are not the same. For the same imaged cells in the same field of view, novel activity patterns emerge when transitioning from the cued task to the uncued task.
(G) Scatterplot of regression coefficients for choice direction in the 4-odor task versus reward context in the uncued task for every imaged cell. Cells with a significant regression coefficient(s) are colored as follows: choice direction (dark red), reward context (dark blue), or both (mixed selective, turquoise). Bar plot represents the proportion of mixed selective cells grouped by cell class. Comparison of the proportion of mixed selective cells across classes was performed using a permutation test (*p < 0.05).
Data Availability Statement
Sequencing data is deposited at NCBI’s Gene Expression Omnibus, and is publicly available through GEO Series accession number GSE161936. The Ca2+ imaging data and analysis code has not been deposited in a public repository because it is in a non-standard format, but are available from the lead contact upon request.