

Nicotinic signaling regulates top-down circuit maturation and restores attention deficits in mouse model of fragile X syndrome.
Abstract
Cognitive function depends on frontal cortex development; however, the mechanisms driving this process are poorly understood. Here, we identify that dynamic regulation of the nicotinic cholinergic system is a key driver of attentional circuit maturation associated with top-down frontal neurons projecting to visual cortex. The top-down neurons receive robust cholinergic inputs, but their nicotinic tone decreases following adolescence by increasing expression of a nicotinic brake, Lynx1. Lynx1 shifts a balance between local and long-range inputs onto top-down frontal neurons following adolescence and promotes the establishment of attentional behavior in adulthood. This key maturational process is disrupted in a mouse model of fragile X syndrome but was rescued by a suppression of nicotinic tone through the introduction of Lynx1 in top-down projections. Nicotinic signaling may serve as a target to rebalance local/long-range balance and treat cognitive deficits in neurodevelopmental disorders.
INTRODUCTION
The protracted nature of the frontal cortex maturation is important for establishing adult cognitive function, and this process is likely disrupted in neurodevelopmental and psychiatric disorders (1). Currently, little is known about the molecular mechanisms driving this developmental process because of the complexity of frontal cortex circuits and cognitive behavior. Identification of the drivers of circuit maturation in the context of cognitive behavior will likely point to therapeutic targets to reduce cognitive deficits shared across a range of disorders (2).
Of particular significance among frontal cortical circuits participating in cognition are top-down projections from frontal to sensory cortical areas. These neurons are recruited into brain-wide attentional networks conserved across species (3). In mice, frontal-sensory projection neurons from the anterior cingulate area (ACA) to the visual cortex (ACAVIS) regulate the ability to detect visual features, a hallmark feature of visual attention (4). Attentional processing likely depends on developmental integration of local microcircuits and long-range inputs onto top-down frontal-sensory projection neurons up into adulthood. A recent study demonstrated that local excitatory drive onto ACAVIS neurons decreases between adolescence (~p30 to p35) and adulthood (>p60) in mice (5). Attentional behavior in humans also continues to develop into adulthood (6–8). Moreover, deficits in frontal modulation of VIS activity are pervasively reported in neurodevelopmental disorders and often emerge between childhood and adulthood (9, 10). However, the specific mechanisms driving the maturation of top-down circuit integration are poorly understood. As murine ACAVIS neurons are known to causally regulate attention-related processing (4) and undergo a shift in local and long-range input balance following adolescence (5), mechanistic exploration of ACAVIS top-down circuit maturation would facilitate an integrated understanding of molecular-, circuit-, and behavior-level mechanisms. Here, we aim to identify the molecular mechanism driving maturation of frontal circuitry associated with top-down ACAVIS projection neurons in control of attentional behavior.
Neuromodulatory systems that are crucial for adult brain functions, however, are functionally in place early postnatally (11, 12). Multiple neuromodulatory regions project directly to the ACA. Notably, a recent rabies virus–mediated tracing has revealed that basal forebrain (BF) region, where cholinergic neurons are located, is a major input source for adult ACAVIS neurons (13). Here, we use circuit-selective methods in ACAVIS neurons to examine the contribution of cholinergic system to top-down circuit maturation and attentional behavior. We also aim to assess whether these mechanisms are disrupted by a genetic risk of psychiatric disorders with neurodevelopmental origins and to determine whether targeting these mechanisms is sufficient to improve attentional function.
RESULTS
Postadolescent suppression of nicotinic tone in ACAVIS neurons parallels with nicotinic brake Lynx1 increase
ACAVIS neurons undergo a shift in local and long-range input balance between adolescence and adulthood (5). To gain insight into a possible developmental contribution of the cholinergic neuromodulatory system on this late maturation period of ACAVIS projection neurons, we first set out to assess in detail the direct inputs from BFs onto ACAVIS neurons. Rabies virus–mediated input mapping demonstrated this neuromodulatory site as a monosynaptic input source for ACAVIS neurons in the adult mice but did not characterize their cell type or identify their presence during adolescence (13). We thus performed detailed characterization of BF neurons projecting to ACAVIS neurons through rabies virus–mediated input mapping in both adolescent and adult wild-type (WT) mice (Fig. 1A). We used a pseudotyped and genetically modified rabies virus [RbVdG(EnvA)-eGfg] to limit uptake to the ACAVIS neurons expressing the cognate receptor, avian tumor virus receptor A (TVA), for cell entry, and rabies glycoprotein for cell exit (Fig. 1A). We found that both adolescent and adult mice show robust numbers of enhanced green fluorescent protein–positive (eGFP+) input cells in the BF (Fig. 1B). We further demonstrated that most of the inputs from BF were choline acetyltransferase–positive cholinergic neurons (Fig. 1, C and D). These results suggest that BF cholinergic inputs are a major neuromodulatory source for ACAVIS projection neurons present in adolescent development and persist into adulthood.
Fig. 1. BF cholinergic neurons provide robust inputs onto top-down ACAVIS projection neurons.
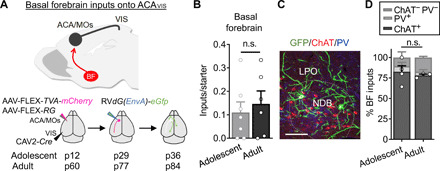
(A) Experimental overview of rabies virus–mediated input mapping of BF inputs onto ACAVIS neurons in adolescent and adult mice. (B) Cre-dependent AAVs encoding TVA-mCherry and Rb-G were injected into ACA and retrograde CAV2-Cre into VIS. After 2.5 weeks, pseudotyped rabies virus was injected into ACA to allow for uptake of rabies virus in ACAVIS neurons to express GFP in input cells over 1 week. AAV injections were initiated at p12 to capture inputs during adolescence (p36) and at p60 to capture inputs during adulthood (p84). (B) Number of BF inputs, when normalized to the number of mCherry + eGFP + starter cells in ACA (t test, t11 = 0.5241, P = 0.611; adolescent: seven mice; adult: six mice). n.s., not significant. (C) Representative image of immunohistochemical characterization of BF GFP + inputs by choline acetyltransferase (ChAT; red) and parvalbumin (PV; blue). Scale bar, 50 μm. LPO, lateral preoptic nucleus; NDB, nucleus of the diagonal band. (D) Inputs from BF are primarily ChAT+ during adolescence (82.5%) and adulthood (78.17%), with a smaller PV+ population (adolescent, 6.775%; adult, 4.467%), and ChAT-PV− (adolescent, 10.73%; adult, 17.37%). The relative proportion of cell types does not differ between adolescent and adult groups (t test, t5 = 0.4904, P = 0.6446; adolescent: four mice; adult: three mice).
To further investigate the developmental contribution of cholinergic signaling to ACAVIS projection neurons, we next examined whether cholinergic tone of ACAVIS neurons changes between adolescence and adulthood. Given that cortical cholinergic tone is limited at nicotinic acetylcholine (ACh) receptors (nAChRs) in adulthood (14, 15), we focused on developmental changes in nicotinic response. We measured ACh-evoked response in the presence of the muscarinic AChR blocker atropine in slice preparations (Fig. 2A). We found that nACh-evoked current detected in adolescent ACAVIS projection neurons became significantly suppressed into adulthood (Fig. 2, B and C). Previous studies show that nACh tone of cortical neurons is suppressed after adolescence because of an increased expression of the glycosylphosphatidylinositol-anchored prototoxin, Lynx1, which brakes nicotinic signaling through nAChR interaction (Fig. 2D) (14, 16). Thus, we examined the expression of Lynx1 in ACAVIS neurons following adolescence by in situ hybridization (Fig. 2E). In this experiment, ACAVIS neurons were labeled by retro adeno-associated virus (AAV) encoding Gfp injected in VIS. We observed an increase in the percentage of Lynx1-expressing ACAVIS neurons between adolescence and adulthood (Fig. 2F). Together, these data demonstrate a dynamic suppression of nicotinic tone in ACAVIS neurons following adolescence, while a nicotinic brake, Lynx1, shows an increase in expression during this developmental period.
Fig. 2. Postadolescent decrease of nicotinic cholinergic tone in ACAVIS neurons parallels with an increase in ACAVIS neurons expressing Lynx1, a nicotinic brake.
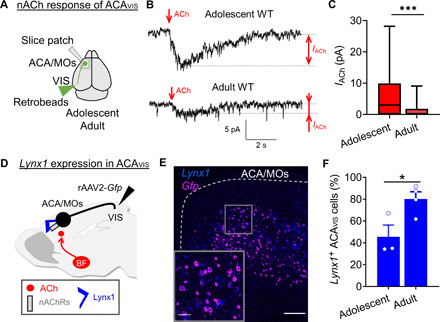
(A to C) nACh tone measurement by whole-cell patch-clamp recordings of ACAVIS neurons. (A) ACAVIS neurons are labeled with retrobeads for whole-cell patch-clamp slice recordings upon ACh puffing in the presence of atropine in adolescent (~p34: p32 to p36) and adult (~p77: p69 to p91) WT mice. (B) Representative recording traces of response to ACh puffing. (C) Adolescent ACAVIS neurons show larger ACh-induced nicotinic currents compared to adult cells [linear mixed model (rank-based), t59 = 4.05, ***P = 0.0002; adolescent: 23 cells from 10 mice; adult: 28 cells from 10 mice). (D to F) Lynx1 expression analysis. (D) ACAVIS neurons were virally labeled with Gfp by retrograde rAAV2-Gfp injected in VIS to examine expression of a nicotinic brake, Lynx1, in adolescent (p31) and adult (p60) groups. (E) Representative image showing Lynx1 mRNA (blue) in Gfp + ACAVIS neurons (pink) (scale bar, 200 μm) by double in situ hybridization of Gfp and Lynx1 in adult mouse. (F) The percentage of Gfp + ACAVIS neurons expressing Lynx1 increases between adolescence and adulthood (unpaired two-tailed t test, t5 = 2.896, *P = 0.0340; adolescent: three mice; adult: four mice).
Lynx1 suppresses postadolescent nicotinic tone to shift local/long-range input balance onto ACAVIS neurons
Having observed the increase in Lynx1-expressing ACAVIS neurons and decrease in nACh tone following adolescence, we hypothesized that Lynx1 is a physiological brake of nicotinic tone in ACAVIS neurons and contributes to the maturation of frontal cortex inputs to ACAVIS projections. We performed whole-cell patch-clamp recordings from ACAVIS neurons fluorescently labeled by retrobeads injected into VIS in adult Lynx1 KO mice. Consistent with the known role of Lynx1 in suppressing nACh tone (16), ACAVIS neurons in adult Lynx1 KO mice exhibited an increased nicotinic tone compared to adult WT mice, which is comparable to the level of adolescent WT mice (Fig. 3, A to C). We next sought to determine whether ACAVIS neurons also maintain adolescent-like input drive in the frontal cortex of adult Lynx1 KO mice by measuring miniature excitatory postsynaptic current (mEPSC) and miniature inhibitory postsynaptic current (mIPSC) in ACAVIS neurons. Compared to adult WT level, ACAVIS neurons in adult Lynx1 KO mice displayed significantly greater mEPSC frequency that resembled adolescent WT levels (5), while the mEPSC amplitude between the groups did not differ (Fig. 3, D to F). All groups display similar mIPSC frequency and amplitude of ACAVIS neurons (Fig. 3, G to I). These data suggest that Lynx1 deletion results in an adolescent-like hyperexcitatory input drive onto ACAVIS neurons maintained even in adulthood.
Fig. 3. Lynx1 deletion leads to hypernicotinic tone and excess excitatory inputs onto ACAVIS neurons in adulthood.
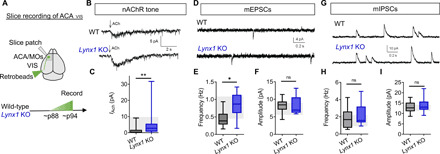
(A) Experimental design: Whole-cell patch-clamp recordings from ACAVIS neurons. (B) Representative traces of nACh response of ACAVIS neurons in adult Lynx1 KO and WT mice. (C) Adult Lynx1 KO mice show increased nicotinic tone in ACAVIS neurons compared to adult WT mice [linear mixed model (rank-based), t28 = 3.33, **P = 0.0025; adult WT: 28 cells per 10 mice; adult Lynx1 KO: 36 cells per 12 mice]. Shaded region indicates the range of adolescent WT level [one to three quartiles from (5)]. (D) Representative traces of mEPSCs. (E and F) Adult Lynx1 KO mice show increased mEPSC frequency compared to adult WT mice [(E) linear mixed model (rank-based), t6.51 = 2.54, *P = 0.0410] but no difference in mEPSC amplitude [(F) linear mixed model (rank-based), t6.22 = −0.58, P = 0.5845]. Adult WT: 15 cells per six mice; adult Lynx1 KO: 10 cells per four mice. Shaded region, range of adolescent WT mice. (G) Representative traces of mIPSCs. (H and I) No changes in mIPSC frequency (H) or mIPSC amplitude (I) were observed between groups [linear mixed model (rank-based), t11.6 = 1.14, P = 0.2789 for frequency; t8.37 = 0.61, P = 0.5551 for amplitude; adult WT: 17 cells per eight mice; adult Lynx1 KO: 16 cells per six mice]. Shaded region, range of adolescent WT mice.
Given that we observed developmental changes in presynaptic excitatory synaptic drive onto ACAVIS neurons and that Lynx1 has been previously reported to regulate dendritic spine dynamics (17, 18), we next assessed the developmental contribution of Lynx1 expression on dendritic spine maturation of ACAVIS neurons in Lynx1 KO and WT mice across development. Dendritic spines of ACAVIS neurons at both adolescent and adult ages were visualized by an eGfp-encoding AAV with retrograde transduction capacity introduced into VIS (Fig. 4A). We observed a significant age (adolescence and adult), genotype (Lynx1 KO and WT), and spine-type (thin, stubby, and mushroom) interaction in dendritic spine density. In WT mice, between adolescence and adulthood, mushroom spine density increases, but thin spine density shows trending yet nonsignificant reduction, reflecting a late maturation of spines. However, in Lynx1 KO mice, mushroom spine density does not increase, while thin and stubby spine density increases following adolescence. While a genotypic effect was not apparent at adolescent age, adult Lynx1 KO mice displayed a higher density of thin and stubby spines compared to adult WT mice (Fig. 4, B and C), consistent with a higher excitatory input drive observed in adult Lynx1 KO mice (Fig. 3E). The distinct developmental trajectory of spine maturation in Lynx1 KO mice indicates a complex combination of late immaturity and deviation from normal developmental trajectory of spine maturations.
Fig. 4. Lynx1 deletion alters developmental trajectory of dendritic spine maturation of ACAVIS neurons.
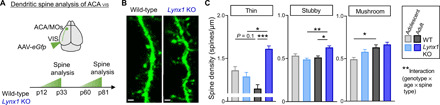
(A) Experimental approach to examine dendritic spines selectively in ACAVIS projection neurons. A cross-sectional approach was applied to both Lynx1 KO and WT mice for adolescent and adult time points. (B) Representative images of dendritic spines of ACAVIS neurons. Scale bar, 1 μm. (C) Dendritic spine density of ACAVIS neurons in WT mice and Lynx1 KO mice during adolescence and adulthood. Doubly repeated measures linear mixed model: spine type × age × genotype interaction; F2,20.6 = 6.42, **P = 0.0068. Thin: adult Lynx1 KO versus adult WT; F1,7.68 = 51.01, ***P = 0.0001; adult Lynx1 KO versus adolescent Lynx1 KO mice; F1,6.22 = 12.74, *P = 0.0111; adolescent WT versus adult WT; F1,7.21 = 3.53, P = 0.1010. Stubby: adult Lynx1 KO versus adult WT; F1,9.85 = 7.45, *P = 0.0215; adult Lynx1 KO versus adolescent Lynx1 KO; F1,9.12 = 14.02, **P = 0.0045. Mushroom: adolescent WT versus adult WT; F1,7.38 = 9.90, *P = 0.0151. Adolescent WT: 68 dendrites per six cells per three mice; adult WT: 56 dendrites per six cells per three mice; adolescent Lynx1 KO: 57 dendrites per six cells per three mice; adult Lynx1 KO: 64 dendrites per six cells per three mice.
To next determine the circuit-level sources of increased inputs onto ACAVIS projection neurons in adult Lynx1 KO mice, we mapped brain-wide monosynaptic rabies inputs onto ACAVIS neurons in adult WT and Lynx1 KO mice (Fig. 5A). To allow for a direct quantitative comparison, we normalized the number of eGFP+ input cells by the number of double eGFP+ mCherry+ starter cells. ACAVIS neurons in Lynx1 KO adults displayed higher connectivity with local inputs from the ACA and adjacent secondary motor cortex (MOs) (Fig. 5, B and C) but no differences in distal long-range input connectivity (Fig. 5D) with similar distribution of input sources (fig. S1). When we calculated the relative percentage of total inputs coming from local areas compared to all other long-range distal brain regions for each animal, ACAVIS neurons display a disrupted local/long-range input balance, with adolescent-like local hyperconnectivity in the adult Lynx1 KO mice (Fig. 5E). These data reveal local hyperconnectivity consistent with an adolescent phenotype (5), suggesting a failure of ACAVIS neurons to reduce local inputs in the absence of Lynx1. Together, these data suggest that postadolescent increase of Lynx1 expression is essential for shifting the balance of local and long-range input onto ACAVIS projection neurons following adolescence by reducing excessive local excitatory inputs.
Fig. 5. Lynx1 deletion leads to excess local inputs onto ACAVIS neurons resulting in imbalanced local and long-range inputs in adulthood.
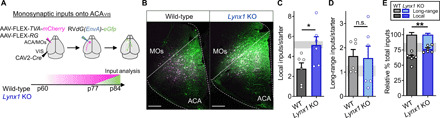
(A) Experimental design to examine monosynaptic inputs onto ACAVIS neurons in adult WT and Lynx1 KO. (B) Representative images of local inputs (green) from ACA and secondary motor cortex (MOs) onto ACAVIS projection neurons in white. Scale bars, 100 μm. (C) The number of inputs onto ACAVIS is higher from local ACA and MOs brain regions in adult Lynx1 KO mice compared to adult WT (t test, t10 = 2.356, *P = 0.0402, six mice each) and comparable to adolescent WT level shown as a shaded region (5), (D) but not from long-range distal brain regions (t test, t10 = 0.1515, P = 0.8826, six mice each), leading to (E) disrupted local/long-range balance with excess local connectivity (t test, t10 = 3.264, **P = 0.0085, six mice each) near an adolescent level shown as a shaded region.
Lynx1 expression in ACAVIS neurons from adolescence is essential for adult attentional behavior
Given that Lynx1 deletion led to excessive local inputs onto ACAVIS projection neurons, we then set out to determine whether this change in circuit connectivity is associated with behavioral consequences. Because ACAVIS projection neurons can enhance the ability to detect visual features in VIS, a hallmark feature of visual attention (4), we hypothesized that Lynx1 expression in ACAVIS is necessary to establish adult top-down visual attentional behavior. We used a freely moving, visual detection task, the five-choice serial reaction time task (5CSRTT) (19) (fig. S2). 5CSRTT is known to require ACA activity (20) and demands more attention compared to a simpler CSRTT with less choice (21). Back-translated from the human continuous performance attention task in humans, the 5CSRTT has been extensively used to interrogate visual attention in rodents. This task assays sustained visual attention by requiring mice to maintain divided attention across five areas in anticipation of the presentation of a brief light stimulus that must be nose-poked to release a reward. Prior studies have shown that attention deficits can be measured quantitatively by decreases in accuracy (19).
We first examined whether germline deletion of Lynx1 affects attentional behavior in adulthood (>p60). The adult Lynx1 KO mice and control adult WT mice first underwent 5CSRTT training sessions to reach the criteria for testing (fig. S3A). During the testing sessions, we found that the genotype (Lynx1 KO/WT) and stimulus duration (2, 1.5, 1, and 0.8 s) interaction for accuracy was suggestive (P = 0.08) but marginally not significant (fig. S3B). We therefore performed post hoc statistical tests as a secondary analysis for a hypothesis testing to further understand how genotype effects differed within subgroups. Post hoc analysis showed a significant reduction of accuracy in Lynx1 KO mice only during 0.8-s stimulus duration trials, suggesting a preferential role of Lynx1 in the most attention-demanding condition. We did not observe any significant impact of Lynx1 deletion in other behavioral readouts (fig. S3, C to H) including an independent assay to test for motivation, the progressive-ratio assay (fig. S3I), suggesting that reduced accuracy is not related to a loss of motivation. Performance deficits cannot also be attributed to vision deficits as adult Lynx1 KO mice are reported to have normal visual acuity (14). Overall, these data support a hypothesis that Lynx1 expression is necessary for proper visual attention behavior.
The mild phenotype in Lynx1 KO mice on task performance may reflect compensational changes due to germline deletion of Lynx1 throughout life. We thus next sought to determine whether attentional behavior deficits can be induced by a selective knockdown (KD) of Lynx1 expression in ACAVIS neurons from adolescence, when Lynx1 expressing-ACAVIS neurons normally increase. We used an intersectional viral approach to achieve Lynx1 KD within ACAVIS projection neurons by bilaterally introducing cre-dependent Lynx1 KD vector (AAV-DIO-Gfp-Lynx1 KD) into ACA and retrograde Cre-encoding virus in VIS (Fig. 6, A and B, and fig. S4A). Suppression of Lynx1 expression was confirmed in eGFP+ cells (Fig. 6, C and D). After going through the 5CSRTT training session (fig. S4B), the KD group showed disrupted visual attentional behavior in the 5CSRTT. This was marked by a decreased accuracy (Fig. 6E) without affecting other behavioral readouts (fig. S4, C to H) including an independent assay to test for motivation, the progressive-ratio assay (fig. S4I). These data reveal that expression of Lynx1 from adolescence in ACAVIS is necessary to establish adult visual attention abilities. The observed attention deficits can either be due to the effect of hypernicotinic tone during behavioral testing in adulthood and/or due to the late developmental role of Lynx1. To examine the former possibility, we acutely infused nAChR agonist (nicotine) through a bilateral cannula targeting ACA in adult WT mice during 5CSRTT. While we used the drug doses known to induce behavioral effect on other behaviors through ACA infusion (22), acute ACA nicotine infusion did not disrupt behavioral readouts of 5CSRTT (fig. S5). Collectively, these results suggest that the behavioral impact of Lynx1 KD is less likely due to the ongoing nicotinic overstimulation during the task but may rather reflect the role of Lynx1 in regulating the maturation of ACAVIS circuitry following adolescence.
Fig. 6. Lynx1 KD in ACAVIS projection neurons from adolescence produces a visual attention deficit in adulthood.
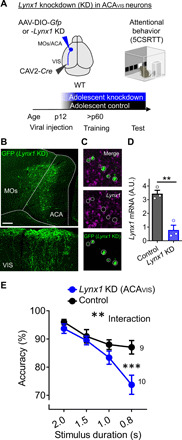
(A) Experimental design: To selectively knock down Lynx1 expression in ACAVIS projection neurons, WT mice were injected with a Cre-dependent AAV for Lynx1 knockdown (KD) (or control cre-dependent Gfp AAV) into ACA and retrograde CAV2-Cre into VIS at p12, and attentional behavior training (5CSRTT) was initiated in adulthood. (B) Representative images of vector expression in ACA and VIS. Scale bar, 200 μm. (C) Validation of Lynx1 KD by in situ hybridization. Representative images of GFP+ virus expressing cells with no Lynx1 mRNA. (D) Compared to control vector, expression of KD construct led to decreased Lynx1 mRNA expression quantified by quantitative polymerase chain reaction (qPCR) (t test, t4 = 6.200, **P = 0.0034; control: three mice; Lynx1 KD: three mice). A.U., arbitrary units. (E) Lynx1 KD from adolescence decreased accuracy of 5CSRTT [two-way repeated measures analysis of variance (ANOVA), group × stimulus duration: F3,51 = 5.723, **P = 0.0019; post hoc Šídák’s multiple comparisons test (0.8 s): ***P = 0.0002; control: 9 mice; Lynx1 KD: 10 mice].
A mouse model of fragile X syndrome shows local and long-range input imbalance and attentional deficits with hypernicotinic tone
Having established that Lynx1-dependent suppression of nACh neuromodulation is essential for top-down circuit maturation and establishing adult attentional behavior, we next sought to determine whether the disruption of this process might be implicated in neurodevelopmental diseases. Informatics-based molecular matching is an effective approach to generate mechanistic hypotheses (23). We thus mapped the adult Lynx1 KO cortical transcriptome signature (23) to publicly available genome-wide association study (GWAS) gene sets curated for neuropsychiatric diseases (24–32). (Fig. 7A). We found greatest enrichment of Lynx1-related signature with the gene set implicated in the autism spectrum disorder (ASD) GWAS dataset (Fig. 7B and table S2). We next sought to determine whether a tighter overlap of the Lynx1 transcriptome signature exists with five more specific publicly available gene sets associated with monogenetic risk of ASD. Notably, there was statistically significant overlap with fragile X mental retardation protein (FMRP)–associated gene sets (P < 0.0001) and gene sets associated with Phelan-McDermid syndrome (Shank3 mutation) (P = 0.0181), with no statistical overlap with gene sets associated with 22q11 deletion, tuberous sclerosis, or Rett syndrome (Fig. 7C and table S2).
Fig. 7. Adult Fmr1 KO mice displays attention deficits with disrupted local/long-range input balance and hypernicotinic tone of ACAVIS neurons.
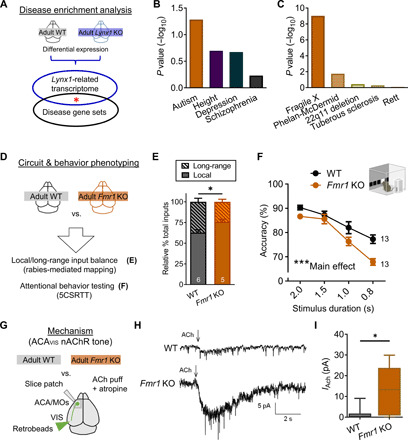
(A) Lynx1-related transcriptional signature (differential expression of adult Lynx1 KO and WT cortex) and GWAS gene sets were analyzed for enrichment. (B) Lynx1 KO signature mapped to GWAS gene sets (ASD: P = 0.0582; height: P = 0.2007; depression: P = 0.2127; schizophrenia: P = 1.0). (C) Lynx1 KO signature mapped to five autism-related gene sets [fragile X syndrome–related FMRP target genes: P = 9.7 × 10−10; Phelan-McDermid syndrome (Shank3KO): P = 0.0181; 22q11 deletion: P = 0.3664; tuberous sclerosis (Tsc2KO): P = 0.5036; Rett syndrome (Mecp2 null mutation): P = 0.8263]. (D) Adult Fmr1 KO mouse was screened for local/long-range input imbalance onto ACAVIS neurons and attentional deficits. (E) Rabies virus–mediated input mapping showed increased local input connectivity ratio for ACAVIS neurons in adult Fmr1 KO mice (t test, t9 = 2.715, *P = 0.0238; WT: five mice; Fmr1 KO: five mice). (F) Adult Fmr1 KO mice showed reduced accuracy with the 5CSRTT [two-way repeated measures ANOVA, main effect (genotype): F1,24 = 14.49, ***P = 0.0009; WT: 13 mice; Fmr1 KO: 13 mice]. (G) Adult Fmr1 KO mice were evaluated for their nicotinic tone of retrobeads labeled ACAVIS by whole-cell patch-clamp recordings. (H) Example trace recordings from ACAVIS neurons upon ACh puffing in the presence of atropine. (I) Adult Fmr1 KO mice show increased nicotinic responses in ACAVIS neurons [linear mixed model (rank-based), t6.94 = 3.40, *P = 0.0116; adult WT: 28 cells per 10 mice; Fmr1 KO: 19 cells per 7 mice).
Given the strongest overlap between Lynx1- and FMRP-associated signatures, we sought to determine whether the loss of function of Fmr1 leads to failed top-down circuit network maturation and attentional behavior deficits similar to the adult Lynx1 KO mice. The Fmr1 KO mouse is considered as a valid model to examine the pathophysiology of fragile X syndrome as mutations observed in humans result in FMRP loss of function. To this end, we examined local and long-range input balance through rabies input mapping and attentional behavior on the 5CSRTT in the adult Fmr1 KO mice (Fig. 7D). After confirming that Fmr1 KO mice displayed similar numbers of starter cells to WT mice (P = 0.2674, unpaired two-tailed t test), monosynaptic input mapping revealed local/long-range imbalance with excess local connectivity, consistent with that of adolescent WT or adult Lynx1 KO connectivity states (Fig. 7E). This balance deficit was driven by combinatorial nonsignificant changes in an absolute level of local inputs and long-range inputs [unpaired two-tailed t test, P = 0.2941 (local), P = 0.3425 (long-range)]. At the behavior level, we found that the adult Fmr1 KO mice display a decrease in accuracy on the attention-demanding 5CSRTT (Fig. 7F and fig. S6). While we also observed an increase in omission and latency to reward in Fmr1 KO mice, the progressive ratio task showed no difference in break point, suggesting that increase in omission likely reflects attention deficits with lesser contribution of motivation deficits (fig. S6). The observed attention deficit is consistent with previous studies that collectively support attention deficits particularly as task difficulty increases in rodent Fmr1 mutant models (33–36) and patients with fragile X syndrome (37, 38).
Given that the circuit and behavioral phenotypes in adult Fmr1 KO mice are similar to those of adult Lynx1 KO mice, we speculated that both mouse lines may show downstream molecular convergence leading to similar circuit and behavior deficits. As previous studies did not detect significant decreases in Lynx1 mRNA and protein expression in Fmr1 KO cortical samples (39, 40), we hypothesized that the Fmr1 KO mice may have an increased nicotinic tone independent of Lynx1 expression change in ACAVIS projection neurons. We therefore performed whole-cell patch-clamp recording in ACAVIS neurons labeled by retrobeads following ACh puffing alongside muscarinic blockade in adult Fmr1 KO mice (Fig. 7G) and found an increased ACh-evoked nicotinic response in adult Fmr1 KO compared to adult WT mice (Fig. 7, H and I). To gain insight into the source of hypernicotinic tone, we additionally performed pharmacological experiments. We found that an increased nicotinic response was more effectively blocked by non-α7 nAChR antagonist dihydro-β-erythroidine (DHβE) over α7 antagonist methyllycaconitine (MLA) (fig. S7). This result suggests contribution of heteromeric nAChR signaling to the hypernicotinic tone in the ACAVIS neurons of Fmr1 KO mice. Together, these data suggest that Fmr1 KO mice have an immature pattern of local/long-range imbalance with attentional behavior consequences by accompanying a juvenile-like excessive nicotinic tone even in adulthood.
Suppression of nicotinic tone in ACAVIS neurons rescues local/long-range imbalance and attentional deficits in Fmr1 KO mice
Having established that Fmr1 KO mice have an increased nicotinic tone and display top-down circuit and behavior deficits similar to Lynx1 KO mice, we hypothesized that Lynx1-mediated suppression of nicotinic tone in ACAVIS projection neurons could ameliorate circuit and behavioral deficits in Fmr1 KO mice. An intersectional viral approach was used to achieve Lynx1 overexpression within ACAVIS projection neurons by bilaterally introducing cre-dependent Lynx1 overexpression vector into ACA and retrograde Cre-encoding virus in VIS (Fig. 8A). After validating viral overexpression of Lynx1 in vivo (Fig. 8, B and C), whole-cell patch-clamp recording was performed from ACAVIS projection neurons. We found that this vector expression in ACAVIS neurons from adolescence resulted in a diminished nicotinic tone in Fmr1 KO mice (Fig. 8D). Adult Fmr1 KO mice were then assessed for local and long-range input balance by rabies virus–mediated monosynaptic labeling in adulthood following suppression of nicotinic tone in ACAVIS neurons from adolescence (fig. S8A). We found that adult Fmr1 KO mice selectively overexpressing Lynx1 in ACAVIS neurons from adolescence resulted in normalization of local and long-range input balance through the reduction of local inputs (Fig. 8E and fig. S8). Moreover, Fmr1 KO mice with top-down Lynx1 overexpression showed an increase of accuracy in 5CSRTT compared to Fmr1 KO mice injected with a Gfp control vector (Fig. 8F and fig. S9) with no changes in other performance metrics (fig. S9). Collectively, these results demonstrated that suppression of nicotinic tone in ACAVIS projection neurons by Lynx1 overexpression from adolescence rescues local and long-range input imbalance and attentional deficits in Fmr1 KO mice.
Fig. 8. Lynx1-mediated suppression of nicotinic tone from adolescence in Fmr1 KO mice restores local/long-range input imbalance of ACAVIS neurons and attentional behavior.
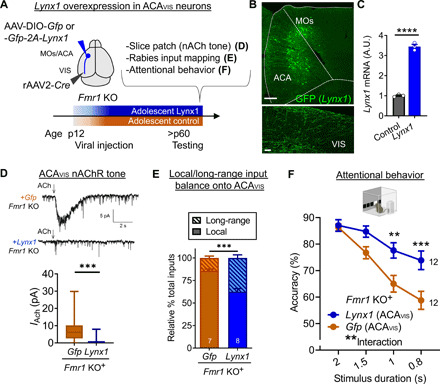
(A) Experimental design: A Cre-dependent Lynx1 overexpression or Gfp control AAV construct was injected in ACA, and retrograde rAAV2-Cre was injected in VIS at p12 in Fmr1 KO mice. (D) nAChR tone, (E) local and long-range input balance, or (F) attentional behavior (5CSRTT) was assessed in adulthood. (B) Representative images showing GFP + ACAVIS neurons in ACA (scale bar, 200 μm) and their terminals in VIS (scale bar, 5 μm). (C) Validation of AAV construct for cre-dependent Lynx1 overexpression. qPCR shows cortical overexpression of Lynx1 mRNA in vivo compared to noninjected native control (t test, t4 = 22.29,****P < 0.0001, three mice each). (D) Top: Representative traces of whole-cell patch recording of ACAVIS neurons overexpressing Lynx1 or a control vector. Bottom: Nicotinic response in adult Fmr1 KO mice [linear mixed model (rank-based), t33 = 4.81,***P < 0.0001; +Gfp: 23 cells per seven mice; +Lynx1: 20 cells per five mice]. (E) Rabies virus–mediated monosynaptic input mapping showed decreased percentage of local connectivity following Lynx1 overexpression from adolescence in adult Fmr1 KO mice (t test, t13 = 5.453,***P = 0.0001; +Gfp: seven mice; +Lynx1: eight mice). (F) Adult Fmr1 KO mice with Lynx1 overexpression in ACAVIS neurons showed an increase in accuracy [two-way repeated measures ANOVA, group × stimulus duration: F3,66 = 5.992, **P = 0.0011; post hoc Šídák’s multiple comparisons test (0.8 s): ***P = 0.0006; 1 s: **P = 0.0055; +Lynx1: 12 mice; +Gfp: 12 mice].
DISCUSSION
In this study, we set out to determine the mechanisms of frontal cognitive circuit maturation and found that dynamic regulation of nicotinic cholinergic tone is a major developmental driver of top-down attentional circuit and behavior. Further, we demonstrated that this process is disrupted in Fmr1 KO mice but can be ameliorated by the top-down circuit-selective suppression of nACh tone (Fig. 9). Previous studies showed that nicotinic signaling is tightly balanced through a multilayered set of mechanisms across early development. nAChR signaling plays a trophic role in developing cortex through regulations of fetal choline level (41), cholinergic innervation from BF (42), and the expression level of nAChRs (43) toward the first three postnatal weeks in rodents (44, 45). However, it was not clear to what extent the nACh system contributes to later stages of cortical maturation. An endogenous inhibitor of nAChRs, Lynx1, provides additional regulation of nicotinic signaling that is necessary to establish cognitive circuit. Our findings emphasize the key role of Lynx1 in late cortical maturation, extending the previously reported role of Lynx1 in limiting developmental cortical plasticity in sensory cortices (14, 17, 18, 46) to establishing cognitive behavior in frontal association cortex.
Fig. 9. Summary scheme.
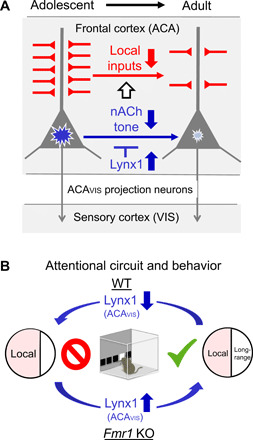
(A) Increased postadolescent Lynx1 expression dampens nicotinic tone (blue blast mark), resulting in necessary reduction in excitatory local inputs (red) onto frontal ACAVIS projection neurons. This process contributes to the shift of local and long-range input balance into adulthood. (B) Removal of Lynx1 from ACAVIS neurons disrupts the balance of local and long-range inputs onto ACAVIS neurons with excessive local inputs and attention deficits. Conversely, hyperlocal inputs onto ACAVIS and attention deficits in Fmr1 KO mice can be prevented by overexpression of Lynx1 in ACAVIS projection neurons from adolescence.
At the circuit and behavior levels, our study suggests that Lynx1 acts as a key molecular mediator of late frontal circuit maturation and cognitive behavior. Specifically, we found that a number of ACAVIS projection neurons expressing the nicotinic brake Lynx1 increases following adolescence. This suppression of nicotinic tone is further associated with a late developmental shift in local/long-range input balance and establishment of adult attentional behavior. Previous studies demonstrated that mice lacking nAChRs show attention deficits (47). Thus, it was, at a glance, unexpected to observe attention deficits by Lynx1 KD as deletion of Lynx1 is associated with elevated nAChR signaling (16) and arousal level (48). However, our series of experiments suggest that dampening of nACh tone by Lynx1 is necessary to limit local excitatory inputs following adolescence, which, in turn, may alter ACAVIS neuron associated network dynamics. Imbalanced local/long-range circuitry in the absence of Lynx1 may prevent finely tuned local computations in the frontal cortex necessary to effectively control distal sensory cortex during cognitive processing in a synchronized manner. As local inputs tend to preferentially target proximal dendrite (49), irrelevant local circuits may also interfere with a transmission from some of long-range inputs targeting distal dendrites to the soma. It should be also noted that attentional behavior deficits caused by Lynx1 KD in ACAVIS neurons can be due to resulting changes in visual input gain in the visual cortex in addition to input balance changes in ACA. One limitation of our study is that our experiments with Lynx1 KD or overexpression from adolescence did not determine whether Lynx1 plays a role during adolescence or/and in adulthood. More temporally selective manipulations of Lynx1 expression are necessary to answer these questions. Similarly, future studies are necessary to examine whether manipulations of Lynx1 expression selectively in ACAVIS projections modulate the local/long-range input balance onto ACAVIS to establish a tighter link between local/long-range input balance and attentional behavior. Another limitation of this study is that we only assessed attentional behavior in adulthood due to the lengthy training and testing period (~2 months) required for touch screen–based 5CSRTT. Future studies should establish a short training protocol to permit assessment of attentional behavior during adolescence to examine to what extent the maturation of ACAVIS circuitry is associated with attentional behavior changes.
Our study provides previously unrecognized insights into the pathophysiology of neurodevelopmental disorders by identifying that Fmr1 KO mice display failed maturation of the ACAVIS projection network leading to attention deficits due to hypernicotinic tone in ACAVIS projection neurons. Our study pieces together the previous findings in rodent models of fragile X syndrome ranging from synaptic-level immaturity (50), connectopathy (51), to late emerging cognitive deficits (33–36, 52) through shared functional deficits in nicotinic signaling. Given that Fmr1 has various functions ranging from regulating the equilibrium between translation and transcription (53, 54), trafficking mRNAs to dendrites without affecting the mRNA/protein level (55), to directing protein-protein interactions (56, 57), future study is warranted to tease apart the cause of hypernicotinic tone in Fmr1 KO mice. While cholinergic dysfunctions have been reported in Fmr1 loss of function, it has been attributed to deficits in muscarinic AChR signaling (58–60). A previous pilot open-label trial for donepezil, an acetylcholinesterase inhibitor, reported that cognitive behavioral function of individuals with fragile X syndrome improved significantly (61), but more recent randomized placebo-controlled trials collectively support an insufficient effect of donepezil in improving cognitive behavior in these individuals (62, 63). Our findings rather point to the dampening of nAChR signaling from adolescent period as an alternative target and time point for interventions. Long-range functional connectivity is commonly impaired in neurodevelopmental disorders and has been hypothesized to arise in the context of excessive short-range connections particularly in the context of ASD (64, 65). Our study stresses a need for future studies to examine the balance between local and long-range connectivity across development in the context of neurodevelopmental disorders. Further, future studies are warranted to examine to what extent a regulation of nicotinic tone, perhaps through modulation of Lynx1-mediated actions, can serve as a possible therapeutic target for impairments of local and long-range input balance in general.
MATERIALS AND METHODS
Animals
Male C57BL/6 mice (Charles River Laboratories), Lynx1 KO mice (gifted from N. Heintz at Rockefeller University) (16), and Fmr1 KO mice (stock no. 003025, the Jackson Laboratory) were used. Mice were housed in a temperature and humidity-controlled vivarium under 12-hour light/dark cycle with ad libitum access to food and water. For behavior testing, mice were allowed access to water for 2 hours each day and maintained approximately 85 to 90% of respective baseline free-feeding weight (5). All animal protocols were approved by the Institutional Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai.
Stereotactic surgery
Following procedures previously described in (5), mice were anesthetized with 2% isoflurane and head-fixed on a heating pad in a mouse stereotactic apparatus (Narishige International USA Inc.). Mice were isolated after surgery until fully awake and immediately returned to their home cage. VIS injection sites relative to lambda are the following: AP (+0.0 mm), ML (+3.0 mm), and DV (−0.4 mm); AP (+0.1 mm), ML (+2.85 mm), and DV (−0.4 mm); AP (+0.1 mm), ML (+3.15 mm), and DV (−0.4 mm). ACA injections sites relative to bregma are the following: AP (+0.7 mm), ML (+0.2 mm), and DV (−0.7 mm); AP (+0.2 mm), ML (+0.2 mm), and DV (−0.7 mm); AP (−0.3 mm), ML (+0.2 mm), and DV (−0.7 mm). For patch-clamp recordings, 500 nl of fluorescent retrobeads (Lumafluor Inc.) were injected into the VIS at least 24 hours before patch recordings to allow visualization of top-down projection neurons. A total of 350 nl of AAV5-hsyn-eGfp-WPRE (University of Pennsylvania Viral Core) was infused into VIS for dendritic spine labeling at p12 for the adolescent group and p60 to p75 for the adult group and allowed to express for 21 days. For rabies-mediated monosynaptic input mapping, 500 nl of a 1:1 volume mixture of AAV8-CA-FLEX-RG and AAV8-EF1a-FLEX-TVAmCherry (University of North Carolina Viral Core) was injected unilaterally in the ACA, and 500 nl of CAV2-Cre (Institut de Genetique Moleculaire de Montpellier) (for Lynx1 KO/WT analysis) or rAAV2-Cre (Boston Children’s Hospital Vector Core) (for Fmr1 KO rescue analysis) was injected in VIS. For an introduction of rabies virus, 2.5 weeks later, 500 nl of pseudotyped EnvA RVdG-Gfp (for Lynx1 KO and Fmr1 KO study) or pseudotyped EnvA RVdG-Bfp (for Fmr1 KO injected with AAV-Gfp/Gfp-Lynx1 in ACAVIS) (Salk Institute Viral Core) was injected into the ACA for local axonal uptake of rabies or VIS for distal axonal uptake of rabies. Mice were euthanized 7 days later. For behavioral studies, 500 nl of AAV8-DIO-Lynx1 KD or AAV8-DIO-Lynx1 (Custom Prep, Boston Children’s Hospital) and 500 nl CAV2-Cre (for Lynx1 KD) or rAAV2-Cre (Boston Children’s Hospital Vector Core) (for Lynx1 overexpression) were injected in the ACA and VIS, respectively, at p12 or p60.
Tissue preparation and histology
Following procedures previously described in (5), animals underwent transcardial perfusion with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. For spine analysis, brains were postfixed in 4% PFA overnight and coronally sectioned at 35 μm for rabies input mapping and viral validation and 100 μm for dendritic spine analysis on a vibratome (Leica VT1000 S). For spine analysis, signal was enhanced by immunohistochemistry. Sections were placed into a blocking solution of 1% bovine serum albumin with 0.8% Triton X-100 in PBS on a rotating shaker for 1 hour at room temperature. The sections were then shaken for 48 hours at room temperature in primary antibody rabbit anti-eGFP (Invitrogen; 1:500), washed three times in blocking solution, and then shaken for 3 hours at room temperature in secondary antibody rabbit anti–Alexa Fluor 488 (Invitrogen; 1:400). The samples were then washed three additional times, twice in blocking solution and once in PBS with 0.01% sodium azide. All slices were then mounted on Superfrost Plus slides using Fluoromount-G mounting medium (SouthernBiotech) and coverslipped. Brains from rabies input mapping and AAV-injected brains for behavioral and other experiments were dissected, postfixed in 4% PFA for 3 hours, and cryoprotected in 30% sucrose in PBS for 24 to 48 hours before embedding in optimum cutting temperature (OCT; Tissue-Tek) and sectioning into 35-μm coronal slices using a cryostat (CM3050, Leica). For rabies input mapping, free-floating sections were washed in PBS before undergoing immunohistochemistry by 1-hour incubation in blocking solution (1% bovine serum albumin in 0.1% Triton X-100 in PBS) followed by overnight incubation with rabbit anti-eGFP antibody (1:1000 in blocking solution; Life Technologies) at room temperature. Sections were washed three times with blocking solution followed by incubation with Alexa Fluor 488–conjugated goat anti-rabbit antibody (Life Technologies) for 2.5 hours, washed three times in blocking solution, and mounted onto slides with 4′,6-diamidino-2-phenylindole (DAPI) Fluoromount-G (SouthernBiotech). Viral injections were validated using an LSM780 confocal microscope (Zeiss) in reference to the Paxinos and Franklin mouse atlas.
Monosynaptic input mapping
Following procedures previously described in (5), samples were imaged using an LSM780 confocal microscope (Zeiss). All slices around the ACA injection regions were stained for starter cell analysis. One of every eight serial sections was stained for input analysis. ImageJ [National Institutes of Health (NIH)] were used to process and analyze all images. Starter cells were defined as both mCherry+ (>50% of soma pixels above 2 SD of the mean image mCherry fluorescence intensity) and eGFP+ (>50% of soma pixels above 3 SD of the mean image GFP fluorescence intensity). Input cells were defined as eGFP+ (>50% of soma pixels above 3 SD of the mean image eGFP fluorescence intensity). To determine the brain region location for all input and starter cells, all brain slices were registered to corresponding Allen Brain Institute coronal maps using anatomical landmarks visualized by DAPI counterstaining and tissue autofluorescence. First, we identified a coronal reference from the Allen Brain Atlas, which is a closest representation of each image. By visually comparing each image with a corresponding atlas, we then manually registered each brain region to each image by drawing boarders to each image using ImageJ. In a small minority of cases, assignment of input neurons to specific brain nuclei may be approximate if eGFP+ cell bodies were located on borders between regions or when anatomical markers were lacking between directly adjacent regions. However, quantitative analyses of input tracing results were performed on anatomical classifications that were at least one hierarchical level broader (as specified by the Allen Brain Atlas) than the discrete brain regions to which input cells were assigned. Notably in many of these cases, directly adjacent nuclei fall together into the same hierarchical group. Both eGFP+ input neurons and eGFP+ mCherry+ starter cells were manually counted using the “Cell Counter” plug-in in ImageJ. For the input analysis, we normalized the total number of input neurons in each brain to the total number of starter cells because the efficiency of rabies uptake by helper cells differed slightly across animals. The investigator performing these analyses remained agnostic of the group identities of animals during image analysis. For input mapping of Fmr1 KO mice injected with AAV-Gfp-Lynx1 or AAV-Gfp, quantification of input neurons was done with blue fluorescent protein+ cells.
Slice electrophysiology
Following procedures previously described in (66), animals were decapitated under isofluorane anesthesia. Brains were quickly removed and transferred into ice-cold artificial cerebrospinal fluid (ACSF) of the following composition: 210.3 mM sucrose, 11 mM glucose, 2.5 mM KCl, 1 mM NaH2PO4, 26.2 mM NaHCO3, 0.5 mM CaCl2, and 4 mM MgCl2. Acute coronal slices of ACA (300 μm) contained both hemispheres. Slices were allowed to recover for 40 min at room temperature in the same solution but with reduced sucrose (105.2 mM) and addition of NaCl (109.5 mM). Following recovery, slices were maintained at room temperature in standard ACSF composed of the following: 119 mM NaCl, 2.5 mM KCl, 1 mM NaH2PO4, 26.2 mM NaHCO3, 11 mM glucose, 2 mM CaCl2, and 2 mM MgCl2. Patch-clamp recordings were performed from fluorescently labeled ACA neurons using borosilicate glass electrodes (3 to 5 megohms). Whole-cell voltage-clamp recordings were obtained with the internal solution containing 120 mM Cs-methanesulfonate, 10 mM Hepes, 0.5 mM EGTA, 8 mM NaCl, 4 mM Mg–adenosine triphosphate (ATP), 1 mM QX-314, 10 mM Na-phosphocreatine, and 0.4 mM Na–guanosine triphosphate (GTP). Current clamp recordings and ACh-evoked currents were obtained with the internal solution containing 127.5 mM K-methanesulfonate, 10 mM Hepes, 5 mM KCl, 5 mM Na-phosphocreatine, 2 mM MgCl2, 2 mM Mg-ATP, 0.6 mM EGTA, and 0.3 mM Na-GTP. Neurons were included in the analysis if input resistance, series resistance, and membrane potential did not change more than 15% during the course of recordings. mIPSC and mEPSC were recorded in the presence of tetrodotoxin (TTX; 1 μM; Abcam). Miniature IPSC and EPSC were separated by holding the neuron at the reversal potential for EPSC or IPSC. To activate nicotinic receptors, ACh (1 mM) was dissolved in ACSF and loaded into a glass pipette connected to a Picospritzer II (Parker Instrumentation, Chicago, IL). Brief localized pulses of ACh (30 ms) were applied with a 60-s interpulse interval close to the soma of recorded neurons (within 50 to 60 μm) using a pressure of 10 psi. To block muscarinic ACh receptors, 1 μM atropine was bath-applied during the entire time of recordings. ACh-evoked currents were averaged across three applications and quantified as the distance between the local peak current and the baseline; the net ACh-evoked charge flow through the membrane was calculated by integrating the area above the baseline-subtracted postsynaptic current after ACh application. For pharmacological experiments, TTX, MLA, and DHβE were diluted in ACSF to 1, 0.1, and 100 μM, respectively, and bath-applied.
Double fluorescence in situ hybridization
Following procedures previously described in (67), cloning for developing RNA probes for in situ hybridization was performed in a pBluescript II SK(−) vector using a polymerase chain reaction (PCR)–based isothermal DNA assembly method. Briefly, primers were designed to linearize the vector and create nonoverlapping overhang sequences. A second set of primers was used to amplify the gene of interest from complementary DNA (cDNA) derived from mouse cortex and create overhangs that were antisense to the vector overhang sequences. Assembly was achieved using Gibson Assembly master mix (New England Biolabs), and the resulting vector was used to transform NEB 5-alpha (New England Biolabs) competent cells. Positive (white) colonies were picked and cultured for miniprep. All miniprep DNA was subjected to restriction digest using Xho I enzyme (New England Biolabs) and examined for insert by gel electrophoresis. Properly inserted colonies were then amplified by 50-ml culture and subsequent midiprep using the HiSpeed Plasmid Midi Kit (QIAGEN). Plasmid DNA was then linearized using Xho I restriction digest and purified via phenol-chloroform and ethanol precipitation. Probes were synthesized using T3 RNA polymerase (Roche), the probe for Lynx1 was labeled with fluorescein (fluorescein isothiocyanate; Roche), and the probe for Gfp was labeled with digoxigenin (DIG; Roche) for double in situ hybridization. Primers used for cloning include the following: Lynx1 (forward), CCGCTCGAGATCCTGTTACCCTGCGTGTG; Lynx1 (reverse), CGGGATCCGCTTCCTCACATC CCACAG; Gfp (forward), GGTATCGATAAGCTTGATATCGTGAGCAAGGGCGAGGA; Gfp (reverse), CCCCGGGCTGCAGGAATCAGCTCGTCCATGCCGA. In situ hybridization of Lynx1 and Gfp was performed as described previously (67). Briefly, mice were anesthetized with isofluorane and cervically dislocated. The brain was removed under ribonuclease-free conditions and quickly frozen in a standard mold with OCT in a chamber of 2-methylbutane on dry ice. After freezing, brains were transferred to −80°C until sectioning. Sectioning was performed on a cryostat at a thickness of 7 μm, and sections were immediately placed onto Tissue Path Superfrost Gold Plus slides (Thermo Fisher Scientific). Sections were allowed to dry and then stored in slide boxes at −80°C. For immunohistochemistry, anesthetized mice were transcardially perfused with cold 4% PFA dissolved in 0.1 M phosphate buffer. The brains were postfixed in 4% PFA at 4°C and cryoprotected in 30% sucrose solution. The frozen brains were sectioned into 40-μm coronal sections using a cryostat. Frozen sections on slides were thawed and fixed in 4% PFA. Following fixation, slides were washed in PBS and incubated for 10 min in acetylation buffer (0.2% HCl, 1.5% triethanolamine, and 0.28% acetic anhydride). Slides were again washed in PBS and then acclimated to hybridization buffer (50% formamide, 5× SSC, 2.5% yeast transfer RNA, 5% salmon sperm DNA, and 10% Denhardt’s solution) in a humidity chamber. Following acclimation, the buffer was replaced with new hybridization buffer containing RNA probes and incubated overnight at 72°C in the humidity chamber. The next day, slides were washed 3 × 30 min in 0.2× SSC at 72°C and blocked in milk solution [tris-buffered saline (TBS) with 0.05% Tween 20 and 1% milk]. Slides were then incubated with milk containing anti-fluorescein-horse-radishperoxidase antibodies (1:2000) for 2 hours at room temperature. Following incubation, slides were washed, subjected to TSA Plus dinitrophenyl (DNP) signal amplification (PerkinElmer), and again washed. Next, slides were again blocked with milk solution and incubated in milk solution containing anti–DIG-alkaline phosphatase and anti–DNP–keyhole limpet hemocyanin–488 (both 1:1000; Roche) for 2 hours at room temperature. Following incubation, slides were washed and acclimated to TBS (pH 8.0) and then incubated in fast red solution (Roche) for 1 hour at room temperature. Last, slides were washed in water and coverslipped using CC/Mount solution (Sigma-Aldrich).
Imaging and quantification
For in situ hybridization colocalization analysis of Lynx1 and Gfp, imaging was performed using LSM780 confocal microscopes (Zeiss) and was processed and analyzed using ImageJ software (NIH). A threshold of light intensity value 9509 was set to define Lynx1 signal and 710 to define Gfp signal. Gfp+ cells were counted as Lynx1+ if they contained one or more puncta of Lynx1 signal. Percentage of colocalization of Lynx1 and Gfp was calculated by Lynx1+ Gfp+ cells/total number of Gfp+ cells. This analysis was done for eight images per animal, from three to four individual mice in each group. For the viral spread validation of mice, mice that had completed behavior testing underwent transcardial perfusion, and slices were collected at specific bregma areas (from 2.06 to −1.78) using the Allen Brain Atlas to analyze the anterior-posterior spread of virally infected cells. Images were acquired using the EVOS FL Imaging System (Thermo Fisher Scientific). Slices were imaged using a 4× lens, and, to ensure consistency across imaging sessions, power was set at 100%. To generate viral spread diagrams, all groups were assigned two levels representing increasing levels of signal intensity. The first level represented the minimum number of mice showing signal in a given area, namely, n = 1. The second level represented more than 50th percentile of mice with overlapping signal in a given area. Using the GNU Image Manipulation Program (GIMP), areas with fluorescent signal were delineated on templates taken from the Paxinos and Franklin mouse atlas. For the validation of AAV spread focused around ACA, we used the Paxinos and Franklin atlas to identify the boundaries of the ACA on any given slice, while the Allen Brain Atlas was used for our whole-brain rabies virus–based input mapping, which allowed us to define anterior-posterior spread better as it has a higher granularity of coronal slices.
Dendritic spine imaging and characterization
Images were acquired using an upright LSM780 Confocal microscope (Carl Zeiss). Following procedures previously described in (5), a nonbiased, whole-neuron approach was taken to characterize dendritic spines. Specifically, two neurons per animal were imaged on both the apical and basal dendrites. To qualify for spine analysis, whole neurons were selected that met the following requirements: (i) The neuron had to be filled and display spines at least up to 200 μm away from the soma on both apical and basal dendrites and (ii) display no overlapping of dendritic processes with other neuron processes. For the chemogenetic study, mCherry+ and mCherry− neurons were defined as having >50% of soma pixels above 3 SD and below 2 SD of the mean image mCherry fluorescence intensity, respectively. Images were taken of sections from two basal dendrites and two apical dendrites from each neuron. Dendritic segments were imaged using a 100× lens (numerical aperture of 1.4; Carl Zeiss) and a zoom of 3.0. Images were taken with a resolution of 1024 × 300, pixel dwell time was 1.58 μm/s, and the line average was set to 8. Pixel size was 0.03 μm in the x-y plane and 0.01 μm in the z plane. To assure consistency of imaging across difference confocal sessions, power was consistently set to 3.0%, and gain was adjusted within the range of 600 to 800 units to achieve consistent light intensity values within the same set of neurons. Images were deconvolved using a three-dimensional resolution enhancement with AutoDeblur software (Media Cybernetics) and then run through the dynamic range filter in Neuron Studio. For quantification, Neuron Studio was used to classify spines as thin, mushroom, or stubby on the basis of the head-to-neck ratio. Spines whose head-to-neck ratio was less than 1 were classified as thin spines, spines whose head-to-neck ratio was approximately 1 were classified as stubby spines, and spines whose head-to-neck ratio was greater than 1 were classified as mushroom spines. To accurately detect dendritic spines from background noise, puncta were only counted as spines if they moved into and out of the z plane with the dendritic branch. Spine density was calculated as the total spine count/dendritic length. All imaging and analysis were all performed by an author who remained agnostic to the group conditions until the analysis was completed.
Behavior: 5CSRTT
Behavior was conducted following procedures previously described in (5) using black plastic trapezoid Bussey-Saksida touch screen chambers [walls: 20 cm high by 18 cm wide (at screen-magazine) by 24 cm wide (at screen) by 6 cm wide (at magazine) (Lafayette Instrument)]. Stimuli were displayed on a touch-sensitive screen (30.7 cm, screen resolution of 600 × 800) divided into five response windows by a black plastic mask (4.0 × 4.0 cm, positioned centrally with windows spaced 1.0 cm apart and 1.5 cm above the floor) fitted in front of the touch screen. Schedules were designed, and data were collected and analyzed using ABET II Touch software (Lafayette Instrument). The inputs and outputs of the multiple chambers were controlled by WhiskerServer software (Lafayette Instrument). Before training on 5CSRTT, mice were initially trained to touch the screen. Mice were acclimated to the chambers for 3 days in 30-min sessions. During this habituation phase, the food magazine was illuminated, and diluted sweetened condensed milk (Eagle Brand) was dispensed every 40 s. Mice were then trained to touch the response windows. If the mouse touched the stimulus, then the milk reward was delivered in conjunction with a tone and magazine light. Touches to nonstimuli had no consequence. After reaching criterion on this phase (20 touches in 30 min for two consecutive days), mice moved onto 5CSRTT training phase. Mice were tested 5 days a week, 100 trials a day (or up to 30 min). Each trial began with the illumination of the magazine light. When the mouse made a nose poke in the food magazine, the stimulus was delivered after an intertrial interval (ITI) period of 5 s. If a mouse touched the screen during this ITI period, then the response was recorded as premature, and the mouse was punished with a 5-s time-out (house light on). After the time-out period, the magazine light illumination and house light switch off signaled onset of the next trial. After the ITI period, a stimulus appeared randomly in one of the five response windows for a set stimulus duration (this varied from 32 to 2 s). A limited-hold period followed by the stimulus duration was 5 s, during which the stimulus was absent but the mouse was still able to respond to the location. Responses during stimulus presence and limited-hold period could be recorded either as correct (touching the stimulus window) or incorrect (touching any other windows). A correct response was rewarded with a tone, as well as milk delivery, indicated by the illumination of the magazine light. A failure to respond to any window over the stimulus and limited-hold period was counted as an omission. Incorrect responses and omissions were punished with a 5-s time-out. In addition, animals could make perseverative responses that are screen touches after a correct or incorrect response. Animals started at stimulus duration of 32 s. With a goal to baseline mice at a stimulus duration of 2 s, the stimulus duration was sequentially reduced from 32, 16, 8, 4, to 2 s. Animals had to reach a criterion (<50 trials, <80% accuracy, and >20% omissions) over two consecutive days to pass from one stage to the next. After reaching baseline criterion with the 2-s stimulus duration (five consecutive days), mice were challenged with an increased attentional demand by reducing the stimulus duration to 2, 1.5, 1, and 0.8 s (reduced stimulus test). They then underwent 4 days of testing. Attention and response control were assessed by measuring the following performance: percent accuracy [100 × correct responses/(correct responses + incorrect responses)], percent omission [100 × omissions/(omissions + correct responses + incorrect responses)], percent premature response [100 × premature responses/(omissions + correct responses + incorrect responses)], percent perseverative response [100 × perseverative responses /(correct responses + incorrect responses)], latency to correct response, and latency to reward collection after correct choices.
Behavior: Progressive-ratio task
Following procedures previously described in (5), behavior was conducted after the completion of the 5CSRTT in touch screen chambers. In initial operant training, mice were trained to nose-touch the visual stimulus (white square, 100% luminance) at the center screen one time in each trial to receive a diluted sweetened condensed milk (Eagle Brand) reward as dispensed in the 5CSRTT. Touches to nonstimuli had no consequence. After reaching criterion of at least 30 completed trials in 30 min, mice moved onto fixed- ratio training. In fixed-ratio training, mice were trained to nose-touch the visual stimulus two, three, and then five times to get a reward. Mice were required to reach criterion for 1 day for fixed ratios of 2:1 and 3:1 and for 3 days for a fixed ratio of 5:1 before proceeding to progressive-ratio training and testing. Task parameters were identical for progressive-ratio training, except completion of each trial; the number of touches required to receive a reward was incremented on a linear +4 basis (i.e., 1, 5, 9, etc.). If no stimulus response or magazine entry in the presence of a delivered reward was detected for 5 min, then the session ended and the animal was removed from the chamber. Otherwise, the session ended after 30 min. Mice had to achieve a stabile “breakpoint” defined as the number of target location responses in the last successfully completed trial for 2 days before proceeding to progressive-ratio testing. In progressive-ratio testing, mice performed the progressive-ratio task using the same parameters as progressive-ratio training but were allowed 60 min to complete the session. Mice were baselined on Monday with progressive-ratio training to ensure consistent performance and tested for four consecutive days. Breakpoint across the 4 days was averaged to give each mouse on breakpoint score.
AAV vector construction
Lynx1 was amplified from a cDNA library derived from mouse cortex and subcloned into a pcDNA3.1(−) vector for an overexpression vector. For Lynx1 KD vector, following procedures previously described in (68), we used short hairpin RNA (shRNA) vector with limited reported off-target effects, in which shRNA is embedded into a larger endogenous microRNA loop that allows robust expression from RNA polymerase 2 promoters of an AAV vector to avoid the recently reported potential off-target effect by the use of acute short hairpin–based KD. Two oligonucleotides including shRNA sequence for Lynx1 were subcloned into pcDNA6.2-GW/EmGfp-miR using BLOCK-iT Pol II miR RNAi Expression Vector Kit with EmGFP (Life Technologies). Subclonings were performed using an isothermal DNA assembly method (Gibson Assembly; New England Biolabs) and transformed into Escherichia coli. Colonies with correct insert were identified through DNA sequencing (Genewiz), cultured, and then isolated using the HiSpeed Midiprep Kit (QIAGEN). Expression was first examined by transfection of N2A cells in vitro to identify one of four sets of shRNAs for the robust Lynx1 KD (CCTGTGAAGCAGTTGTCCATTGTGTCAGTCAGTGGCCAAAACACAATGGAGACA ACTGCTTCAC,TGCTGTGAAGCAGTTGTCTCCATTGTGTTTTGGCCACTGACTGACACAATGGACAACTGCTTCA; Life Technologies). To create the pAAV vector, an inverted bicistronic 2A sequence was inserted into pAAV-Ef1α-DIO-eGfp-WPRE-pA (Addgene no. 37084) upstream of eGfp by PCR linearization and overhang production on pAAV vector. The pcDNA3.1(−)-Lynx1 vector was used as a template for the Lynx1 insert, which was subsequently inserted into the pAAV-DIO-eGfp-2A vector as described above to create a pAAV-DIO-eGfp-2A-Lynx1-WPRE-pA vector (Lynx1 overexpression). pcDNA6.2-mGfp-miR-Lynx1 vector was subcloned into pAAV-DIO-eGfp-2A-Lypd6-WPRE-pA vector to generate pAAV-DIO-mGfp-miR-Lynx1 (Lynx1 KD) vector. After sequence verification, a large culture and Maxiprep isolation produced a purified vector that was sent to the University of North Carolina viral core for viral packaging using an AAV8 serotype. To assay overexpression or KD of Lynx1 through quantitative PCR (qPCR), RNA was isolated using the RNeasy Lipid Tissue Mini Kit (QIAGEN), and cDNA was produced. The cDNA was subjected to qPCR analysis using a TaqMan assay (Life Technologies) at the Icahn School of Medicine at Mount Sinai Quantitative PCR CORE facility to quantify Lynx1 mRNA and β-actin mRNA.
Cannula infusions
Mice were trained on the 5CSRTT before guide cannula implantation surgeries. During surgery, burr holes targeting ACA on both hemispheres (A/P, +0.2 mm and M/L, ±0.2 mm relative to lambda) were made. Twenty-two–gauge guide cannulas (C313GS, Plastics One) were then implanted at the cortical surface and secured using Metabond (Parkell). Dummy cannula were placed into the guide cannula to prevent clogging. Nicotine hydrogen tartrate salt (Sigma-Aldrich, St. Louis, MO) was fully dissolved in physiological saline. Immediately before the 5CSRTT, 500 nl of nicotine solution (0.09 or 0.35 μg per side) or saline was infused bilaterally in ACA at a rate of 500 nl/min through a 28-gauge internal cannula (C313IS, Plastics One) that projected 0.7 mm below the dura. Following infusion, internal cannula was left in place for 1 min to allow for diffusion of drug from infusion site and reduce backflow.
Enrichment analyses
Transcriptomic data of adult Lynx1 KO (>P60) and adult WT C57BL/6 (>P60) mice (n = 3, each group) primary visual cortex (V1) were downloaded (GSE89757), background-corrected, quantile-normalized, and log2-transformed with Limma, and then differential expression was calculated using RankProd. Differentially expressed genes at a false discovery rate of <0.25 were mapped to human using the Mouse Genome Informatics homology reference to yield a 332 genes Lynx1 KO signature. GWAS and autism subtype gene sets were curated from the literature (table S2). The likelihood of genes shared between the Lynx1 KO signature and a given GWAS/autism gene set was assessed by hypergeometric tests (HTSanalyzeR R package), using the intersection of all human orthologous genes expressed on the Lynx1 KO microarray and all human genes (9656 genes) as a background. Enrichment analyses were conducted in R (version 3.2.2).
Statistical methods
Statistical analyses were performed using Prism 6.0 (GraphPad) and SAS v9.4 (SAS). The linear mixed models were used for experiments using patch-clamp recordings and dendritic spine analysis to evaluate the genotype or age differences that involved repeated measures within animals. Specifically, doubly repeated measure-mixed models with respect to neuron and dendrite were used for the dendritic spine models, which included main effects genotype (WT versus KO), age group (adult versus adolescent), spine type (thin, stubby, or mushroom), and the relevant two- and three-way interaction terms. Because the three-way interaction term was highly significant, stratified results by spine type were presented subsequently. For electrophysiological recordings, ranks were used instead of the actual data because of having excess close to zero values. Animals were treated as random effects. In all these models, the variances were allowed to be heterogeneous across different subgroups, as the descriptive data suggested. Degrees of freedom were adjusted on the basis of the Satterthwaite method. For 5SRTT behavior data, the animal specific accuracy rate and omission rate were first summarized for each of the four stimulus durations (2, 1.5, 1, and 0.8 s) and were analyzed using a linear mixed model using a two-way repeated measures analysis with stimulus duration (2, 1.5, 1, and 0.8 s) as within-subject factors, followed by Šídák’s multiple comparisons test. In the analysis of other 5CSRTT data and progressive-ratio testing, an unpaired two-tailed t test was used. For input mapping and ratio comparisons, input numbers were first normalized to the number of starter cells within each animal before analyses. Input mapping analyses were completed by a two-way brain region by genotype models with repeated brain measures. Whenever t test was performed, unpaired two-tailed t test was used. All data are expressed as means ± SEM, and circles represent n values.
Acknowledgments
We thank N. Heintz (Rockefeller University) for providing Lynx1 KO mice, as well as D. Kiraly, M. Baxter, S. Espeso-Gil, S. Akbarian, and members of the Morishita laboratory for helpful feedback. Funding: This work was funded by NIH F30MH111143, Seaver Foundation to E.N.F., NIH F31MH121010 to K.J.N., and NIH R21NS105119, R21MH106919, R01EY024918, and R01MH119523 to H.M. Author contributions: E.N.F. and H.M. designed experiments. E.N.F. and H.M. wrote the manuscript with contributions from all coauthors. Y.G. performed slice electrophysiology. E.N.F. and G.T. performed spine analysis. E.N.F., K.J.N., Y.G., and S.I. performed rabies virus input mapping. S.I., J.S., M.P.D., and E.N.F. performed in situ hybridization. M.P.D. made and validated virus vectors. E.N.F., K.J.N., M.P.D., and Y.G. conducted viral injections. E.N.F., K.J.N., M.P.D., J.S., K.C., S.E.M., C.C., H.K., and P.M. performed behavioral experiments. M.R.S. and E.N.F. conducted informatics analysis. E.N.F., S.I., J.B., L.W., and M.J. performed viral and cannula placement validation. E.N.F., K.J.N., Y.G., H.-M.L., and H.M. performed statistical analysis. H.M. supervised the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All other data and analysis are available from the corresponding author upon reasonable request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabe1527/DC1
REFERENCES AND NOTES
- 1.Paus T., Keshavan M., Giedd J. N., Why do many psychiatric disorders emerge during adolescence? Nat. Rev. Neurosci. 9, 947–957 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee F. S., Heimer H., Giedd J. N., Lein E. S., Šestan N., Weinberger D. R., Casey B. J., Mental health. Adolescent mental health—Opportunity and obligation. Science 346, 547–549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knudsen E. I., Fundamental components of attention. Annu. Rev. Neurosci. 30, 57–78 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Zhang S., Xu M., Kamigaki T., Hoang Do J. P., Chang W.-C., Jenvay S., Miyamichi K., Luo L., Dan Y., Long-range and local circuits for top-down modulation of visual cortex processing. Science 345, 660–665 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nabel E. M., Garkun Y., Koike H., Sadahiro M., Liang A., Norman K. J., Taccheri G., Demars M. P., Im S., Caro K., Lopez S., Bateh J., Hof P. R., Clem R. L., Morishita H., Adolescent frontal top-down neurons receive heightened local drive to establish adult attentional behavior in mice. Nat. Commun. 11, 3983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mollon J., Knowles E. E. M., Mathias S. R., Gur R., Peralta J. M., Weiner D. J., Robinson E. B., Gur R. E., Blangero J., Almasy L., Glahn D. C., Genetic influence on cognitive development between childhood and adulthood. Mol. Psychiatry 26, 656–665 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dye M. W. G., Bavelier D., Differential development of visual attention skills in school-age children. Vision Res. 50, 452–459 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konrad K., Neufang S., Thiel C. M., Specht K., Hanisch C., Fan J., Herpertz-Dahlmann B., Fink G. R., Development of attentional networks: An fMRI study with children and adults. Neuroimage 28, 429–439 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Keehn B. L., Palmer E., Lincoln A. J., Müller R.-A., Functional brain organization for visual search in ASD. Austism Res. 5, 314–330 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Mazaheri A., Coffey-Corina S., Mangun G. R., Bekker E. M., Berry A. S., Corbett B. A., Functional disconnection of frontal cortex and visual cortex in attention-deficit/hyperactivity disorder. Biol. Psychiatry 67, 617–623 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Hoops D., Flores C., Making dopamine connections in adolescence. Trends Neurosci. 40, 709–719 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suri D., Teixeira C. M., Cagliostro M. K. C., Mahadevia D., Ansorge M. S., Monoamine-sensitive developmental periods impacting adult emotional and cognitive behaviors. Neuropsychopharmacology 40, 88–112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang S., Xu M., Chang W.-C., Ma C., Hoang Do J. P., Jeong D., Lei T., Fan J. L., Dan Y., Organization of long-range inputs and outputs of frontal cortex for top-down control. Nat. Neurosci. 19, 1733–1742 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morishita H., Miwa J. M., Heintz N., Hensch T. K., Lynx1, a cholinergic brake, limits plasticity in adult visual cortex. Science 330, 1238–1240 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kassam S. M., Herman P. M., Goodfellow N. M., Alves N. C., Lambe E. K., Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. J. Neurosci. 28, 8756–8764 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miwa J. M., Stevens T. R., King S. L., Caldarone B. J., Ibanez-Tallon I., Xiao C., Fitzsimonds R. M., Pavlides C., Lester H. A., Picciotto M. R., Heintz N., The prototoxin lynx1 acts on nicotinic acetylcholine receptors to balance neuronal activity and survival in vivo. Neuron 51, 587–600 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Bukhari N., Burman P. N., Hussein A., Demars M. P., Sadahiro M., Brady D. M., Tsirka S. E., Russo S. J., Morishita H., Unmasking proteolytic activity for adult visual cortex plasticity by the removal of Lynx1. J. Neurosci. 35, 12693–12702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sajo M., Ellis-Davies G., Morishita H., Lynx1 limits dendritic spine turnover in the adult visual cortex. J. Neurosci. 36, 9472–9478 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbins T., The 5-choice serial reaction time task: Behavioural pharmacology and functional neurochemistry. Psychopharmacology 163, 362–380 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Koike H., Demars M. P., Short J. A., Nabel E. M., Akbarian S., Baxter M. G., Morishita H., Chemogenetic inactivation of dorsal anterior cingulate cortex neurons disrupts attentional behavior in mouse. Neuropsychopharmacology 41, 1014–1023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White M. G., Panicker M., Mu C., Carter A. M., Roberts B. M., Dharmasri P. A., Mathur B. N., Anterior cingulate cortex input to the claustrum is required for top-down action control. Cell Rep. 22, 84–95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raybuck J. D., Gould T. J., The role of nicotinic acetylcholine receptors in the medial prefrontal cortex and hippocampus in trace fear conditioning. Neurobiol. Learn. Mem. 94, 353–363 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith M. R., Burman P., Sadahiro M., Kidd B. A., Dudley J. T., Morishita H., Integrative analysis of disease signatures shows inflammation disrupts juvenile experience-dependent cortical plasticity. eNeuro 3, ENEURO.0240-16.2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schizophrenia Working Group of the Psychiatric Genomics Consortium , Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wray N. R., Ripke S., Mattheisen M., Trzaskowski M., Byrne E. M., Abdellaoui A., Adams M. J., Agerbo E., Air T. M., Andlauer T. M. F., Bacanu S.-A., Bækvad-Hansen M., Beekman A. F. T., Bigdeli T. B., Binder E. B., Blackwood D. R. H., Bryois J., Buttenschøn H. N., Bybjerg-Grauholm J., Cai N., Castelao E., Christensen J. H., Clarke T.-K., Coleman J. I. R., Colodro-Conde L., Couvy-Duchesne B., Craddock N., Crawford G. E., Crowley C. A., Dashti H. S., Davies G., Deary I. J., Degenhardt F., Derks E. M., Direk N., Dolan C. V., Dunn E. C., Eley T. C., Eriksson N., Escott-Price V., Kiadeh F. H. F., Finucane H. K., Forstner A. J., Frank J., Gaspar H. A., Gill M., Giusti-Rodríguez P., Goes F. S., Gordon S. D., Grove J., Hall L. S., Hannon E., Hansen C. S., Hansen T. F., Herms S., Hickie I. B., Hoffmann P., Homuth G., Horn C., Hottenga J.-J., Hougaard D. M., Hu M., Hyde C. L., Ising M., Jansen R., Jin F., Jorgenson E., Knowles J. A., Kohane I. S., Kraft J., Kretzschmar W. W., Krogh J., Kutalik Z., Lane J. M., Li Y., Li Y., Lind P. A., Liu X., Lu L., MacIntyre D. J., MacKinnon D. F., Maier R. M., Maier W., Marchini J., Mbarek H., McGrath P., McGuffin P., Medland S. E., Mehta D., Middeldorp C. M., Mihailov E., Milaneschi Y., Milani L., Mill J., Mondimore F. M., Montgomery G. W., Mostafavi S., Mullins N., Nauck M., Ng B., Nivard M. G., Nyholt D. R., O’Reilly P. F., Oskarsson H., Owen M. J., Painter J. N., Pedersen C. B., Pedersen M. G., Peterson R. E., Pettersson E., Peyrot W. J., Pistis G., Posthuma D., Purcell S. M., Quiroz J. A., Qvist P., Rice J. P., Riley B. P., Rivera M., Mirza S. S., Saxena R., Schoevers R., Schulte E. C., Shen L., Shi J., Shyn S. I., Sigurdsson E., Sinnamon G. B. C., Smit J. H., Smith D. J., Stefansson H., Steinberg S., Stockmeier C. A., Streit F., Strohmaier J., Tansey K. E., Teismann H., Teumer A., Thompson W., Thomson P. A., Thorgeirsson T. E., Tian C., Traylor M., Treutlein J., Trubetskoy V., Uitterlinden A. G., Umbricht D., Van der Auwera S., van Hemert A. M., Viktorin A., Visscher P. M., Wang Y., Webb B. T., Weinsheimer S. M., Wellmann J., Willemsen G., Witt S. H., Wu Y., Xi H. S., Yang J., Zhang F.; eQTLGen; 23andMe, Arolt V., Baune B. T., Berger K., Boomsma D. I., Cichon S., Dannlowski U., de Geus E. C. J., De Paulo J. R., Domenici E., Domschke K., Esko T., Grabe H. J., Hamilton S. P., Hayward C., Heath A. C., Hinds D. A., Kendler K. S., Kloiber S., Lewis G., Li Q. S., Lucae S., Madden P. F. A., Magnusson P. K., Martin N. G., McIntosh A. M., Metspalu A., Mors O., Mortensen P. B., Müller-Myhsok B., Nordentoft M., Nöthen M. M., O’Donovan M. C., Paciga S. A., Pedersen N. L., Penninx B. W. J. H., Perlis R. H., Porteous D. J., Potash J. B., Preisig M., Rietschel M., Schaefer C., Schulze T. G., Smoller J. W., Stefansson K., Tiemeier H., Uher R., Völzke H., Weissman M. M., Werge T., Winslow A. R., Lewis C. M., Levinson D. F., Breen G., Børglum A. D., Sullivan P. F.; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium , Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium , Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 8, 21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marouli E., Graff M., Medina-Gomez C., Lo K. S., Wood A. R., Kjaer T. R., Fine R. S., Lu Y., Schurmann C., Highland H. M., Rüeger S., Thorleifsson G., Justice A. E., Lamparter D., Stirrups K. E., Turcot V., Young K. L., Winkler T. W., Esko T., Karaderi T., Locke A. E., Masca N. G. D., Ng M. C. Y., Mudgal P., Rivas M. A., Vedantam S., Mahajan A., Guo X., Abecasis G., Aben K. K., Adair L. S., Alam D. S., Albrecht E., Allin K. H., Allison M., Amouyel P., Appel E. V., Arveiler D., Asselbergs F. W., Auer P. L., Balkau B., Banas B., Bang L. E., Benn M., Bergmann S., Bielak L. F., Blüher M., Boeing H., Boerwinkle E., Böger C. A., Bonnycastle L. L., Bork-Jensen J., Bots M. L., Bottinger E. P., Bowden D. W., Brandslund I., Breen G., Brilliant M. H., Broer L., Burt A. A., Butterworth A. S., Carey D. J., Caulfield M. J., Chambers J. C., Chasman D. I., Chen Y.-D. I., Chowdhury R., Christensen C., Chu A. Y., Cocca M., Collins F. S., Cook J. P., Corley J., Galbany J. C., Cox A. J., Cuellar-Partida G., Danesh J., Davies G., de Bakker P. I. W., de Borst G. J., de Denus S., de Groot M. C. H., de Mutsert R., Deary I. J., Dedoussis G., Demerath E. W., den Hollander A. I., Dennis J. G., Angelantonio E. D., Drenos F., Du M., Dunning A. M., Easton D. F., Ebeling T., Edwards T. L., Ellinor P. T., Elliott P., Evangelou E., Farmaki A.-E., Faul J. D., Feitosa M. F., Feng S., Ferrannini E., Ferrario M. M., Ferrieres J., Florez J. C., Ford I., Fornage M., Franks P. W., Frikke-Schmidt R., Galesloot T. E., Gan W., Gandin I., Gasparini P., Giedraitis V., Giri A., Girotto G., Gordon S. D., Gordon-Larsen P., Gorski M., Grarup N., Grove M. L., Gudnason V., Gustafsson S., Hansen T., Harris K. M., Harris T. B., Hattersley A. T., Hayward C., He L., Heid I. M., Heikkilä K., Helgeland Ø., Hernesniemi J., Hewitt A. W., Hocking L. J., Hollensted M., Holmen O. L., Hovingh G. K., Howson J. M. M., Hoyng C. B., Huang P. L., Hveem K., Ikram M. A., Ingelsson E., Jackson A. U., Jansson J.-H., Jarvik G. P., Jensen G. B., Jhun M. A., Jia Y., Jiang X., Johansson S., Jørgensen M. E., Jørgensen T., Jousilahti P., Jukema J. W., Kahali B., Kahn R. S., Kähönen M., Kamstrup P. R., Kanoni S., Kaprio J., Karaleftheri M., Kardia S. L. R., Karpe F., Kee F., Keeman R., Kiemeney L. A., Kitajima H., Kluivers K. B., Kocher T., Komulainen P., Kontto J., Kooner J. S., Kooperberg C., Kovacs P., Kriebel J., Kuivaniemi H., Küry S., Kuusisto J., Bianca M. L., Laakso M., Lakka T. A., Lange E. M., Lange L. A., Langefeld C. D., Langenberg C., Larson E. B., Lee I.-T., Lehtimäki T., Lewis C. E., Li H., Li J., Li-Gao R., Lin H., Lin L.-A., Lin X., Lind L., Lindström J., Linneberg A., Liu Y., Liu Y., Lophatananon A., Luan J., Lubitz S. A., Lyytikäinen L.-P., Mackey D. A., Madden P. A. F., Manning A. K., Männistö S., Marenne G., Marten J., Martin N. G., Mazul A. L., Meidtner K., Metspalu A., Mitchell P., Mohlke K. L., Mook-Kanamori D. O., Morgan A., Morris A. D., Morris A. P., Müller-Nurasyid M., Munroe P. B., Nalls M. A., Nauck M., Nelson C. P., Neville M., Nielsen S. F., Nikus K., Njølstad P. R., Nordestgaard B. G., Ntalla I., O’Connel J. R., Oksa H., Olde Loohuis L. M., Ophoff R. A., Owen K. R., Packard C. J., Padmanabhan S., Palmer C. N. A., Pasterkamp G., Patel A. P., Pattie A., Pedersen O., Peissig P. L., Peloso G. M., Pennell C. E., Perola M., Perry J. A., Perry J. R. B., Person T. N., Pirie A., Polasek O., Posthuma D., Raitakari O. T., Rasheed A., Rauramaa R., Reilly D. F., Reiner A. P., Renström F., Ridker P. M., Rioux J. D., Robertson N., Robino A., Rolandsson O., Rudan I., Ruth K. S., Saleheen D., Salomaa V., Samani N. J., Sandow K., Sapkota Y., Sattar N., Schmidt M. K., Schreiner P. J., Schulze M. B., Scott R. A., Segura-Lepe M. P., Shah S., Sim X., Sivapalaratnam S., Small K. S., Smith A. V., Smith J. A., Southam L., Spector T. D., Speliotes E. K., Starr J. M., Steinthorsdottir V., Stringham H. M., Stumvoll M., Surendran P., ‘t Hart L. M., Tansey K. E., Tardif J.-C., Taylor K. D., Teumer A., Thompson D. J., Thorsteinsdottir U., Thuesen B. H., Tönjes A., Tromp G., Trompet S., Tsafantakis E., Tuomilehto J., Tybjaerg-Hansen A., Tyrer J. P., Uher R., Uitterlinden A. G., Ulivi S., van der Laan S. W., Van Der Leij A. R., van Duijn C. M., van Schoor N. M., van Setten J., Varbo A., Varga T. V., Varma R., Velez Edwards D. R., Vermeulen S. H., Vestergaard H., Vitart V., Vogt T. F., Vozzi D., Walker M., Wang F., Wang C. A., Wang S., Wang Y., Wareham N. J., Warren H. R., Wessel J., Willems S. M., Wilson J. G., Witte D. R., Woods M. O., Wu Y., Yaghootkar H., Yao J., Yao P., Yerges-Armstrong L. M., Young R., Zeggini E., Zhan X., Zhang W., Zhao J. H., Zhao W., Zhao W., Zheng H., Zhou W.; EPIC-InterAct Consortium; CHD Exome+ Consortium; ExomeBP Consortium; T2D-Genes Consortium; GoT2D Genes Consortium; Global Lipids Genetics Consortium; ReproGen Consortium; MAGIC Investigators, Rotter J. I., Boehnke M., Kathiresan S., McCarthy M. I., Willer C. J., Stefansson K., Borecki I. B., Liu D. J., North K. E., Heard-Costa N. L., Pers T. H., Lindgren C. M., Oxvig C., Kutalik Z., Rivadeneira F., Loos R. J. F., Frayling T. M., Hirschhorn J. N., Deloukas P., Lettre G., Rare and low-frequency coding variants alter human adult height. Nature 542, 186–190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darnell J. C., Van Driesche S. J., Zhang C., Hung K. Y. S., Mele A., Fraser C. E., Stone E. F., Chen C., Fak J. J., Chi S. W., Licatalosi D. D., Richter J. D., Darnell R. B., FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kong S. W., Sahin M., Collins C. D., Wertz M. H., Campbell M. G., Leech J. D., Krueger D., Bear M. F., Kunkel L. M., Kohane I. S., Divergent dysregulation of gene expression in murine models of fragile X syndrome and tuberous sclerosis. Mol. Autism 5, 1–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ben-Shachar S., Chahrour M., Thaller C., Shaw C. A., Zoghbi H. Y., Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 18, 2431–2442 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Beveren N. J. M., Krab L. C., Swagemakers S., Buitendijk G., Boot E., van der Spek P., Elgersma Y., van Amelsvoort T. A. M. J., Functional gene-expression analysis shows involvement of schizophrenia-relevant pathways in patients with 22q11 deletion syndrome. PLOS ONE 7, e33473 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lanz T. A., Guilmette E., Gosink M. M., Fischer J. E., Fitzgerald L. W., Stephenson D. T., Pletcher M. T., Transcriptomic analysis of genetically defined autism candidate genes reveals common mechanisms of action. Mol. Autism 4, 45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kazdoba T. M., Leach P. T., Silverman J. L., Crawley J. N., Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res. 3, 118–133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Golden C. E. M., Breen M. S., Koro L., Sonar S., Niblo K., Browne A., Burlant N., Di Marino D., De Rubeis S., Baxter M. G., Buxbaum J. D., Harony-Nicolas H., Deletion of the KH1 domain of Fmr1 leads to transcriptional alterations and attentional deficits in rats. Cereb. Cortex 29, 2228–2244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krueger D. D., Osterweil E. K., Chen S. P., Tye L. D., Bear M. F., Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 2587–2592 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moon J., Beaudin A. E., Verosky S., Driscoll L. L., Weiskopf M., Levitsky D. A., Crnic L. S., Strupp B. J., Attentional dysfunction, impulsivity, and resistance to change in a mouse model of fragile X syndrome. Behav. Neurosci. 120, 1367–1379 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Bailey D. B. Jr., Raspa M., Olmsted M., Holiday D. B., Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am. J. Med. Genet. A 146A, 2060–2069 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Cornish K. M., Munir F., Cross G., Differential impact of the FMR-1 full mutation on memory and attention functioning: A neuropsychological perspective. J. Cogn. Neurosci. 13, 144–150 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Xu B., Zhang Y., Zhan S., Wang X., Zhang H., Meng X., Ge W., Proteomic profiling of brain and testis reveals the diverse changes in ribosomal proteins in fmr1 knockout mice. Neuroscience 371, 469–483 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Das Sharma S., Metz J. B., Li H., Hobson B. D., Hornstein N., Sulzer D., Tang G., Sims P. A., Widespread alterations in translation elongation in the brain of juvenile Fmr1 knockout mice. Cell Rep. 26, 3313–3322.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross R. G., Hunter S. K., McCarthy L., Beuler J., Hutchison A. K., Wagner B. D., Leonard S., Stevens K. E., Freedman R., Perinatal choline effects on neonatal pathophysiology related to later schizophrenia risk. Am. J. Psychiatry 170, 290–298 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gould E., Woolf N. J., Butcher L. L., Postnatal development of cholinergic neurons in the rat: I. Forebrain. Brain Res. Bull. 27, 767–789 (1991). [DOI] [PubMed] [Google Scholar]
- 43.Zhang X., Liu C., Miao H., Gong Z.-h., Nordberg A., Postnatal changes of nicotinic acetylcholine receptor α2, α3, α4, α7 and β2 subunits genes expression in rat brain. Int. J. Dev. Neurosci. 16, 507–518 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Proulx E., Piva M., Tian M. K., Bailey C. D. C., Lambe E. K., Nicotinic acetylcholine receptors in attention circuitry: The role of layer VI neurons of prefrontal cortex. Cell. Mol. Life Sci. 71, 1225–1244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heath C. J., Picciotto M. R., Nicotine-induced plasticity during development: Modulation of the cholinergic system and long-term consequences for circuits involved in attention and sensory processing. Neuropharmacology 56 ( suppl. 1), 254–262 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takesian A. E., Bogart L. J., Lichtman J. W., Hensch T. K., Inhibitory circuit gating of auditory critical-period plasticity. Nat. Neurosci. 21, 218–227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guillem K., Bloem B., Poorthuis R. B., Loos M., Smit A. B., Maskos U., Spijker S., Mansvelder H. D., Nicotinic acetylcholine receptor β2 subunits in the medial prefrontal cortex control attention. Science 333, 888–891 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Artoni P., Piffer A., Vinci V., LeBlanc J., Nelson C. A., Hensch T. K., Fagiolini M., Deep learning of spontaneous arousal fluctuations detects early cholinergic defects across neurodevelopmental mouse models and patients. Proc. Natl. Acad. Sci. U.S.A. 117, 23298–23303 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petreanu L., Mao T., Sternson S. M., Svoboda K., The subcellular organization of neocortical excitatory connections. Nature 457, 1142–1145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cruz-Martin A., Crespo M., Portera-Cailliau C., Delayed stabilization of dendritic spines in fragile X mice. J. Neurosci. 30, 7793–7803 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel A. B., Loerwald K. W., Huber K. M., Gibson J. R., Postsynaptic FMRP promotes the pruning of cell-to-cell connections among pyramidal neurons in the L5A neocortical network. J. Neurosci. 34, 3413–3418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asiminas A., Jackson A. D., Louros S. R., Till S. M., Spano T., Dando O., Bear M. F., Chattarji S., Hardingham G. E., Osterweil E. K., Wyllie D. J. A., Wood E. R., Kind P. C., Sustained correction of associative learning deficits after brief, early treatment in a rat model of fragile X syndrome. Sci. Transl. Med. 11, eaao0498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sossin W. S., "Fragile" equilibrium between translation and transcription. Proc. Natl. Acad. Sci. U.S.A. 115, 12086–12088 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bagni C., Zukin R. S., A synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron 101, 1070–1088 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dictenberg J. B., Swanger S. A., Antar L. N., Singer R. H., Bassell G. J., A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926–939 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng P.-Y., Rotman Z., Blundon J. A., Cho Y., Cui J., Cavalli V., Zakharenko S. S., Klyachko V. A., FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown M. R., Kronengold J., Gazula V. R., Chen Y., Strumbos J. G., Sigworth F. J., Navaratnam D., Kaczmarek L. K., Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci. 13, 819–821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D’Antuono M., Merlo D., Avoli M., Involvement of cholinergic and gabaergic systems in the fragile X knockout mice. Neuroscience 119, 9–13 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Chang S., Bray S. M., Li Z., Zarnescu D. C., He C., Jin P., Warren S. T., Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat. Chem. Biol. 4, 256–263 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Thomson S. R., Seo S. S., Barnes S. A., Louros S. R., Muscas M., Dando O., Kirby C., Wyllie D. J. A., Hardingham G. E., Kind P. C., Osterweil E. K., Cell-type-specific translation profiling reveals a novel strategy for treating fragile X syndrome. Neuron 95, 550–563.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kesler S. R., Lightbody A. A., Reiss A. L., Cholinergic dysfunction in fragile X syndrome and potential intervention: A preliminary 1H MRS study. Am. J. Med. Genet. A 149A, 403–407 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bruno J. L., Hosseini S. H., Lightbody A. A., Manchanda M. K., Reiss A. L., Brain circuitry, behavior, and cognition: A randomized placebo-controlled trial of donepezil in fragile X syndrome. J. Psychopharmacol. 33, 975–985 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sahu J. K., Gulati S., Sapra S., Arya R., Chauhan S., Chowdhury M. R., Gupta N., Kabra M., Gupta Y. K., Dwivedi S. N., Kalra V., Effectiveness and safety of donepezil in boys with fragile x syndrome: A double-blind, randomized, controlled pilot study. J. Child Neurol. 28, 570–575 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Barttfeld P., Wicker B., Cukier S., Navarta S., Lew S., Sigman M., A big-world network in ASD: Dynamical connectivity analysis reflects a deficit in long-range connections and an excess of short-range connections. Neuropsychologia 49, 254–263 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Dajani D. R., Uddin L. Q., Local brain connectivity across development in autism spectrum disorder: A cross-sectional investigation. Autism Res. 9, 43–54 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bicks L. K., Yamamuro K., Flanigan M. E., Kim J. M., Kato D., Lucas E. K., Koike H., Peng M. S., Brady D. M., Chandrasekaran S., Norman K. J., Smith M. R., Clem R. L., Russo S. J., Akbarian S., Morishita H., Prefrontal parvalbumin interneurons require juvenile social experience to establish adult social behavior. Nat. Commun. 11, 1003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Demars M. P., Morishita H., Cortical parvalbumin and somatostatin GABA neurons express distinct endogenous modulators of nicotinic acetylcholine receptors. Mol. Brain 7, 75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sadahiro M., Demars M. P., Burman P., Yevoo P., Zimmer A., Morishita H., Activation of somatostatin interneurons by nicotinic modulator Lypd6 enhances plasticity and functional recovery in the adult mouse visual cortex. J. Neurosci. 40, 5214–5227 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabe1527/DC1