

Mapping single-cell RNA-seq data onto IHC images enables the annotation of cell populations at high phenotypic resolution.
Abstract
Highly multiplexed immunohistochemistry (mIHC) enables the staining and quantification of dozens of antigens in a tissue section with single-cell resolution. However, annotating cell populations that differ little in the profiled antigens or for which the antibody panel does not include specific markers is challenging. To overcome this obstacle, we have developed an approach for enriching mIHC images with single-cell RNA sequencing data, building upon recent experimental procedures for augmenting single-cell transcriptomes with concurrent antigen measurements. Spatially-resolved Transcriptomics via Epitope Anchoring (STvEA) performs transcriptome-guided annotation of highly multiplexed cytometry datasets. It increases the level of detail in histological analyses by enabling the systematic annotation of nuanced cell populations, spatial patterns of transcription, and interactions between cell types. We demonstrate the utility of STvEA by uncovering the architecture of poorly characterized cell types in the murine spleen using published cytometry and mIHC data of this organ.
INTRODUCTION
Recently developed technologies for digital imaging and multiplexed immunohistochemistry (mIHC) (1–8) are enabling the field of histology to enter into a quantitative era, allowing for more complex descriptions of tissue architecture. Imaging mass cytometry (5), multiplexed ion beam imaging (6), CO-Detection by indEXing (CODEX) (2), and highly multiplexed quantitative platforms for traditional immunohistochemistry (7, 8) can be used to simultaneously profile the expression level of dozens of proteins in a tissue section with single-cell resolution. Despite this progress, the amount of cell types and states that can be simultaneously identified by mIHC is limited. Computational methods for automated identification of cell populations cluster cells according to expression similarities of the profiled antigens. These clusters are then manually annotated using previous knowledge of cell markers. However, this process is generally partial, subjective, and biased (9). The groups of cells that result from clustering algorithms often differ little in their antigenic profile, and the interpretation of those differences is unclear. Moreover, the design of comprehensive antibody panels that include specific markers for every cell type and state present in the tissue is usually unfeasible. Consequently, the amount of annotated cell populations in mIHC analyses is often substantially smaller than the number of clusters produced by automated methods.
To overcome these limitations and improve the annotation of mIHC data, we propose an approach for enriching mIHC slides with single-cell RNA sequencing (RNA-seq) data. Currently available single-cell RNA-seq technologies can profile the expression level of thousands of genes in each cell, allowing for fine classification of cells based on their gene expression profile. Some of the most recent approaches, like cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) (10), RNA expression and protein sequencing (11), and Abseq (12), allow for augmenting single-cell transcriptomes with concurrent protein measurements by staining single-cell suspensions with oligo-tagged antibodies. These approaches can therefore be used to determine the quantitative relation between gene and antigen expression levels. Here, we build upon CITE-seq and computational methods for the integration of single-cell omics data (13, 14) to identify and annotate cell populations in mIHC images (or, more broadly, in highly multiplexed cytometry datasets) based on single-cell gene expression data of the same tissue. Our method, transcriptomics via epitope anchoring (STvEA), consists of three major steps (Fig. 1). First, it computationally consolidates the protein expression spaces of the mIHC dataset and a matching CITE-seq dataset using a shared antibody panel. This consolidated protein expression space is used to transfer features (e.g., mRNA cell type assignments, gene expression profiles, etc.) from the CITE-seq dataset into the mIHC images. STvEA then finds an optimal clustering of the CITE-seq mRNA expression data such that the resulting cell populations can be accurately mapped into the mIHC images based on their antigenic profile. In this way, STvEA enables the identification and annotation of cell populations in the mIHC data and the study of spatial patterns of transcription.
Fig. 1. Overview of STvEA.
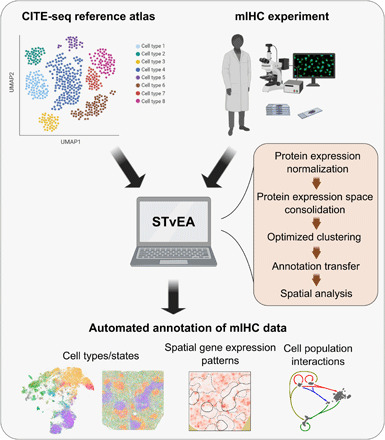
STvEA takes as input an mIHC dataset and a reference CITE-seq atlas and performs automated annotations of the mIHC data based on the reference atlas. It first normalizes and consolidates the protein expression spaces of the mIHC and CITE-seq datasets. Then, it identifies clusters the CITE-seq mRNA data such that the resulting cell populations can be accurately mapped onto the mIHC images. The resulting information is used to annotate cell types and states in the mIHC dataset and predict spatial patterns of gene expression and interactions between cell populations.
We use the murine spleen as a test system to benchmark the stability and performance of STvEA, since well-established antibody panels and high-quality mIHC datasets are readily available for this organ. For that purpose, we have generated a high-quality CITE-seq atlas of the murine spleen and used it with STvEA to annotate published mIHC and mass cytometry datasets of this organ. Our results reveal that STvEA substantially increases the level of phenotypic annotation of these datasets and enables new analyses of highly multiplexed cytometry data. In addition, by systematizing the annotation of cell populations, it improves the reproducibility of the results. We have made this approach available to the entire community as open-source software (Online Methods).
RESULTS
A high-quality CITE-seq atlas of the murine spleen
We aimed to improve and automate the annotation of a published high-resolution mIHC dataset of the murine spleen recently generated with the CODEX technology (2). CODEX uses an in situ polymerization indexing procedure to measure the spatial distribution of a panel of protein markers with submicrometer resolution. To be able to more accurately annotate this dataset, we generated a high-quality CITE-seq dataset of the murine spleen using the same 30-antibody panel (table S1) and mice matching those of the CODEX dataset. In total, we profiled the transcriptome and antigen levels of 7097 cells [with ≥1200 mRNA unique molecular identifiers (UMIs)] using CITE-seq. The median Spearman correlation among the observed expression of mRNAs and the proteins they code for was 0.32, consistent with previous CITE-seq studies (10). We used single-cell variational inference (scVI) (15) to obtain a latent space representation of the mRNA data and clustered the cells in this space using an in-house consensus algorithm (Online Methods). Our analysis found 17 clusters and no noticeable batch effects (Fig. 2A and fig. S1). We performed differential expression analysis to annotate the clusters based on the expression of known marker genes (Fig. 2B and table S2). In addition, we used a spectral graph method (16, 17) to characterize the transcriptomic heterogeneity that originates from the continuous maturation processes occurring in the spleen (Fig. 2C and table S3). This approach allowed us to identify genes with significant gradients of expression within one or several clusters, and we used these results to further annotate the atlas. Overall, we identified 30 cell populations, comprising most of the known splenic cell types (Fig. 2A) (18, 19). These results represent a substantial increase in resolution with respect to previous single-cell RNA-seq atlases of the murine spleen (20–22).
Fig. 2. A high-resolution CITE-seq atlas of the murine spleen.
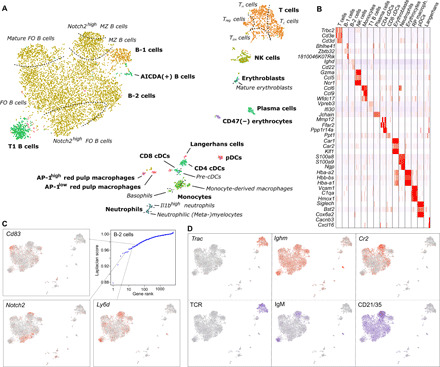
(A) UMAP representation of the mRNA expression data of 7097 cells from the murine spleen profiled with CITE-seq. Cell populations were identified by clustering (represented in different colors) and annotated by differential expression analysis (bold text) and a spectral graph method [italic text; see also (C)]. Dashed lines represent soft transitions in the transcriptome of cells. (B) Heatmap depicting the expression of some of the top differentially expressed genes in each cluster. (C) Analysis of the cellular heterogeneity within the clusters of B-2 cells using a spectral graph approach. Genes were ranked according to their Laplacian score, and statistical significance was assessed for each gene by randomization. In the figure, the expression levels of some of the significant genes are depicted in the UMAP representation. The complete results are provided for all clusters in table S3. (D) mRNA expression levels of Cr2, Ighm, and Trac (top) and the expression levels of the proteins they code for (bottom).
Mapping of the splenic CITE-seq atlas onto histology sections profiled with CODEX
We noticed that most of the cell populations identified in the transcriptomic analysis of the CITE-seq atlas were also localized in the protein expression space (fig. S2). This observation indicates that small differences in cellular epitope levels are often representative of distinct cell populations, even if those differences do not lead to discrete clusters in the protein expression space. Consequently, we reasoned that mapping the CODEX protein expression space into the CITE-seq protein expression space would allow us to survey the CODEX images for the cell populations identified in the transcriptomic analysis. To lessen the technical differences and facilitate the integration of the two spaces, we devised a common approach to background removal and normalization for CODEX and CITE-seq protein expression measurements (fig. S3). In each dataset, we modeled the distribution of protein levels using a two-component mixture model (Online Methods). Our approach led to improved and more consistent protein expression levels across the two datasets (fig. S3). We then used a mutual nearest neighbors anchoring strategy (13, 14) to consolidate the signal component of the two datasets into a common protein expression space (Fig. 3A; Online Methods). By looking at the CODEX neighbors of each CITE-seq cell in the consolidated protein expression space, we were able to identify groups of cells in the mIHC images with similar antigenic profiles to those in the CITE-seq dataset, substantially extending the phenotypic annotation of cell types in the CODEX data. Overall, STvEA led to the annotation of 73% (n = 57,819) of the cells present in the CODEX mIHC images. It correctly recapitulated the known spatial distribution of splenic cell populations, including the partitioning between red pulp, B cell zones, and T cell zones; the location of plasmacytoid dendritic cells (pDCs) in T cell zones; the location of monocytes in the red pulp; and the positioning of CD4 conventional dendritic cells (cDCs) along the bridging channels that connect T cell zones and the red pulp (Fig. 3B) (23). Cell populations that were not annotated by STvEA mostly consisted of stromal cells (CD31+ or ERTR7+ cells) with no representation in the CITE-seq atlas due to the nonenzymatic procedure we used for tissue dissociation, cells without specific marker expression, as noted in the analysis of Goltsev et al. (2), and some T cell subpopulations (fig. S4). CODEX cell population assignments were consistent across CITE-seq replicates (Fig. 3C; median Pearson’s correlation r = 0.998, P < 10−10), and the inferred relative spatial distributions were reproducible across multiple spleens profiled with CODEX (fig. S5). We also tested other approaches for consolidating the normalized protein expression spaces of CODEX and CITE-seq, including Harmony (24) and LIGER (25), and obtained similar results to those obtained by using a mutual nearest neighbors anchoring strategy (fig. S6).
Fig. 3. Mapping of the splenic CITE-seq atlas into histology sections profiled with CODEX.
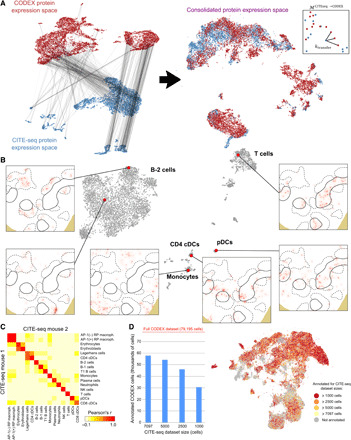
(A) Schematics of the procedure for mapping the CODEX and CITE-seq protein expression spaces. Anchors are identified using mutual nearest neighbors and weighted according to their consistency with the mRNA expression space (left). These anchors are used to consolidate the CODEX and CITE-seq protein expression spaces into a common space (middle). The transfer matrix ℳCITEseq→CODEX is built by looking at the nearest CODEX cells to each CITE-seq cell in this space (right). (B) Mapping of cells from the CITE-seq atlas into a splenic section profiled with CODEX. The figure shows the locations of cells in the section with antigenic profiles similar to those of six cells from the CITE-seq atlas. T and B cell zones are indicated with solid and dashed lines, respectively. (C) Consistency between the annotations of two spleens profiled by CITE-seq and mapped onto the same CODEX dataset. The heatmap shows the correlation between the CODEX cell assignments for each cell population. (D) Number of annotated cells in the CODEX dataset as a function of the number of cells in the CITE-seq atlas. The annotated cells are indicated in a UMAP representation of the CODEX protein expression.
Quantification of mapping uncertainties and stability
To quantitatively assess the magnitude of mapping errors, we looked at the protein expression profile of cells in the CODEX dataset related to the same CITE-seq cells. The average Pearson’s correlation coefficient between the protein expression profiles of CODEX cells related to the same CITE-seq cell was 0.74. This value varied substantially across the CITE-seq atlas (fig. S7), with cells annotated as B-1 B cells, T1 B cells, and dendritic cells having the lowest correlation coefficients (average Pearson’s r = 0.59, 0.55, and 0.56, respectively), possibly due to a lack of specific markers for these populations in the antibody panel. However, these coefficients were substantially larger than the correlation between the protein expression profiles of randomly chosen CODEX cells (fig. S7; mean Pearson’s correlation coefficient r = 0.25).
Assessing the sensitivity and specificity of automated STvEA annotations requires a gold standard to compare with. Although the true cell types are unknown in the CODEX dataset, the expression of highly specific markers by some cell populations, including B220 by B-2 cells, T cell receptor β (TCRβ) by T cells, NKp46 by natural killer (NK) cells, Ly6G by neutrophils and other granulocytes, and ERTR7 by stromal cells, provides a good approximation. Comparing the annotations of STvEA with the expression of these highly specific markers allowed us to estimate the sensitivity and specificity of STvEA annotations across several populations (fig. S8). Our choice of parameters for STvEA favored specificity (for most cell populations, the false-positive rate was <5%) against sensitivity, which, in most cases, was between 60 and 70% (fig. S8). Different parameter choices, however, enabled a higher sensitivity/specificity rate in situations where a higher sensitivity was desirable. For example, increasing the number of neighbors (ktransfer) in the ℳCITEseq → CODEX transfer matrix (Fig. 3A; Online Methods) to 1500 led to the annotation of 97% of the CODEX cells with a specificity >80% for most cell populations (fig. S8). The sensitivity and specificity of STvEA annotations was stable against changes in other parameters (fig. S9).
We next evaluated the stability of STvEA with respect to the number of cells in the CITE-seq atlas. We randomly sampled cells from the CITE-seq dataset to generate smaller datasets and applied STvEA independently to each of these atlases. The percentage of CODEX cells annotated by STvEA decreased from 73% when using the entire CITE-seq atlas (7097 cells) to 38% when using only 1000 cells (Fig. 3D). Expectedly, regions of the CODEX protein expression space with low mapping scores (Online Methods) or less representation in the CITE-seq data were more sensitive to the size of the atlas (fig. S10). However, annotations were highly consistent across atlases of different sizes (median Pearson’s correlation coefficient between the predicted gene expression profile of a CODEX cell when using the original CITE-seq dataset or a down-sampled version with 1000 cells; r = 0.93). These results thus indicate that the size of the CITE-seq atlas mainly affects the percentage of annotated cells in the mIHC images but not the quality of the annotations.
Last, we assessed the stability of the annotations against changes in the size of the antibody panel. We performed logistic lasso regression using the CITE-seq cell populations as response variable to order the antibodies from least to most informative (fig. S11A; Online Methods). The resulting ordering was independent of the amount of correlation between the gene and protein expression levels of each marker in the panel (test of association, P = 0.79). We then successively reduced the size of the antibody panel and applied STvEA. The annotations in the initial analysis were relatively stable against reducing the size of the antibody panel (fig. S11B). In particular, 73% of the annotated cells in the original analysis were still annotated when using a panel with only nine antibodies, and the median Pearson’s correlation coefficient between cell assignments in these two analyses was 0.99, indicating a large degree of consistency for the annotations. Moreover, all the cell types identified in the mRNA analysis were still well represented in the annotations of the CODEX dataset when using nine antibodies (fig. 11C). On the basis of these results, we conclude that STvEA can provide a high level of phenotypic annotation with relatively small antibody panels, as long as antibodies are suitably chosen and cell populations are represented in the reference CITE-seq atlas.
CITE-seq and CODEX are scalable to large antibody panels, and there are published studies (26, 27) using these technologies, respectively, with 198 and 56 antibodies. However, there may be situations where it is unfeasible to design an mIHC experiment with an antibody panel that fully matches that of an existing CITE-seq reference atlas. In those situations, our results indicate that selecting a subset of antibodies for the mIHC panel that maximizes the amount of nonredundant information about the mRNA cell populations in the atlas, as described above, is a suitable approach.
Optimized annotation of cell populations
The analysis described above shows that mapping accuracy is not uniform across the CITE-seq atlas (fig. S7). We therefore reasoned that defining cell populations based exclusively on clustering the mRNA data, without taking into account mapping accuracy, could lead to suboptimal annotation of the mIHC images. In particular, some of the mRNA clusters might not be accurately mapped based on their protein expression profile, whereas other clusters might be split into smaller pieces that could still be accurately distinguished based on their protein expression profile. To overcome this limitation and define cell populations that can be optimally mapped into the mIHC data, we devised a clustering approach of the single-cell mRNA data that takes into account mapping accuracy (Online Methods). Specifically, we used the algorithm HDBSCAN (28) to establish a simplified hierarchical tree of cell populations based on the single-cell mRNA data. A nonuniform cut of this tree was chosen on the basis of the Louvain modularity of the resulting populations in the CODEX protein expression space upon STvEA mapping (Online Methods). This approach partitioned the CITE-seq atlas into 17 phenotypically distinct cell populations that could be accurately mapped onto the mIHC images (Fig. 4, A to C, and fig. S12A). The mapped guided clustering increased the accuracy of STvEA annotations for most cell populations, as determined by the odds ratio between STvEA annotations and the expression of highly specific markers in the CODEX data (fig. S12B). These automated annotations were also consistent with those resulting from manual annotation of the CODEX data (fig. S13; information redundancy R/Rmax = 42%) (2) and included several populations that were not identified in the manual analysis and for which specific markers were not included in the panel, such as pDCs, different stages of erythrocyte maturation, and several B cell subpopulations.
Fig. 4. Transcriptome-guided annotation of cell populations in histology sections profiled with mIHC.

(A) UMAP representation of the protein expression space of a splenic tissue section profiled with CODEX. The representation is labeled by the cell populations identified by STvEA. In total, 17 phenotypically distinct populations were determined in the mRNA CITE-seq data based on their gene expression profile and their mapping into the CODEX dataset. (B) UMAP representation of the CITE-seq gene expression space labeled by the 17 cell populations annotated by STvEA. (C) Image of the tissue section labeled by the 17 cell populations annotated by STvEA.
To estimate the accuracy of STvEA annotations in cases where the antibody panel does not include any specific marker, we removed NK cell or neutrophil markers (NKp46 and Ly6G and CD11b, respectively) from the antibody panel and repeated the automated annotation with STvEA. The specificity to identify NK cells in the absence of specific markers was 97% [that is, only 3% of the NKp46(−) cells were annotated by STvEA as NK cells], whereas the sensitivity was 40%. These values indicate a strong association between STvEA NK cell labels and NKp46(+) CODEX cells (odds ratio = 21.5, Fisher’s exact test P < 10−16). Including NKp46 in the panel boosted the sensitivity to 70% while keeping the specificity at 97%. Similarly, the specificity and sensitivity to identify neutrophils in the absence of specific markers were, respectively, 99 and 44% [odds ratio between STvEA neutrophil labels and Ly6G(+) CD11b(+) cells = 73.8, Fisher’s exact test P < 10−16]. Higher sensitivity (at the cost of reducing specificity) could be obtained by increasing the number of neighbors (ktransfer) in the ℳCITEseq → CODEX transfer matrix (Fig. 3A; Online Methods). Thus, these results suggest that incorporating additional antibodies in the panel can further enhance the annotation of cell populations present in the mRNA CITE-seq data of this analysis. For example, in our specific analysis, we expect that expanding the antibody panel with specific T cell differentiation markers such as CD62L, CD69, and CD103 would increase the phenotypic resolution of STvEA annotations for T cells.
Last, we explored the utility of STvEA as a tool for annotating the clusters produced by some of the existing algorithms for cytometry data analysis, including X-shift (29), SPADE (30), and PhenoGraph (fig. S14) (31). The cell clusters produced by these methods were generally composed of one or two predominant cell types according to the annotations of STvEA. Together, these results show that STvEA is a useful and robust tool for the annotation of mIHC data.
Prediction of spatially resolved gene expression patterns
The mapping of single-cell transcriptomic data onto mIHC images provided by STvEA allowed us to investigate the predicted spatial patterning of any gene in the mRNA dataset (Fig. 5A). To validate some of the spatially resolved gene expression profiles predicted by STvEA, we performed multiplexed RNA fluorescent in situ hybridization (FISH) (32) of several marker genes identified in the differential expression analysis (Fig. 5A and figs. S15 and S16). Specifically, we carried out hybridizations for Bhlhe41, a transcriptional repressor highly expressed by B-1 cells (33) as they mature and migrate from B cell zones into the red pulp (34); and Il1b, expressed by several subpopulations of cDCs, monocytes, macrophages, and neutrophils in the red pulp and T cell zones, but not expressed in B cell zones. In both cases, FISH correctly recapitulated the expression patterns predicted by STvEA (Fig. 5A and figs. S15 and S16), confirming the utility of our computational approach to label mIHC images by gene expression levels. Using this approach, we were also able to resolve in the mIHC images some of the cell differentiation processes that take place in the spleen. For example, the predicted expression patterns of Car1, a marker of immature erythroblasts (35, 36), and Gypa, a marker of intermediate and late stages of erythroblast maturation (36, 37), represented the maturation of erythroblasts in erythroblastic islands of the red pulp (38) and allowed us to annotate erythroblasts in the CODEX dataset according to their stage of maturation (fig. S17).
Fig. 5. Identification of spatially resolved gene expression patterns and interactions between cell populations.
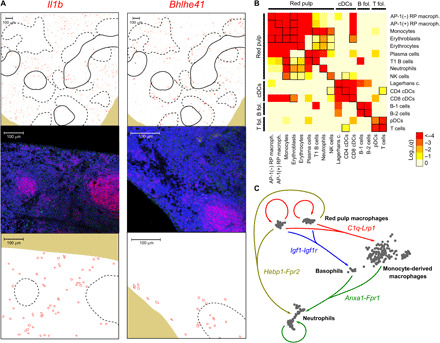
(A) mRNA expression levels predicted by STvEA (top) and measured by RNA FISH (middle and bottom) in murine splenic sections for the genes Il1b (left) and Bhlhe41 (right). Red, Cd79a; green, Il1b/Bhlhe41; blue, DAPI (4′,6-diamidino-2-phenylindole). T and B cell zones in the tissue sections are indicated with solid and dashed lines, respectively. We used B cell zones, highlighted by the expression of Cd79a, as a reference for comparisons between RNA FISH and CODEX tissue sections. The relative location of cells expressing Il1b and Bhlhe41 with respect to B cell zones is indicated at the bottom. (B) Identification of interactions between splenic cell populations. Heatmap showing the significance of the spatial colocalization of splenic cell populations, inferred by STvEA. Significant relations (q ≤ 0.05) that cannot be explained by mapping errors (95% confidence level) are indicated with black squares. (C) Some of the significant potential paracrine interactions among red pulp macrophages, basophils, neutrophils, and monocyte-derived macrophages in the red pulp. Interactions were inferred on the basis of the differential expression of the genes encoding for the ligand and receptor and on their spatial colocalization.
Identification of cell population interactions
Characterizing interactions between cell populations within the context of tissues is a key step toward understanding cell function. Inferring the cell type of individual cells in mIHC images enabled us to survey candidate interactions between cell populations, further expanding the scope of conventional mIHC image analyses. We devised a graph-based approach for assessing the spatial colocalization of cell populations identified in the transcriptomic analysis while accounting for mapping uncertainties (Online Methods). Significant colocalization patterns recapitulated the well-established immune cellular architecture of the spleen, partitioned into red pulp, B cell zones, and T cell zones (Fig. 5B). T cells, pDCs, and CD4 cDCs were recurrently in close proximity within T cell zones. Similarly, red pulp macrophages, erythrocytes, neutrophils, and monocytes were recurrently in close proximity within the red pulp. In addition, several cell populations showed colocalization patterns that spanned multiple splenic compartments (Fig. 5B). Specifically, CD4 cDCs appeared recurrently in close proximity with T cells in T cell zones and with NK cells in the red pulp (Fig. 5B). These inferred relations were reproducible across multiple spleens profiled with CODEX (fig. S18A; Pearson’s correlation coefficient between significance levels, r ≥ 0.98). In addition, an analysis of the same spatial colocalization patterns using the software Giotto (39) led to consistent results (fig. S18B).
To identify molecular cues that potentially mediate the cross-talk between splenic cell populations, we compared differentially expressed genes to a database of receptor-ligand interactions (40) and assessed the relative spatial location of ligand- and receptor-expressing cell populations. Overall, we detected 587 significant interactions based on this approach (Benjamini-Hochberg adjusted q ≤ 0.05; table S4), including the expression of several cues in red pulp macrophages related to the modulation of C1q-dependent phagocytosis, the F2L-mediated priming of neutrophils, and the IGF1-mediated activation of basophils (Fig. 5C and Supplementary Note). These interactions appeared substantially reduced or absent in monocyte-derived macrophages, suggesting the specialization of resident red pulp macrophages in the positive regulation of humoral innate immune responses in the murine spleen.
We validated some of the predicted colocalized spatial patterns of gene expression using multiplexed RNA FISH (fig. S19). In particular, we considered the expression of the ligand-receptor genes Plxnb2 and Sema4c, which, according to our analysis, were significantly colocalized in the tissue sections (Benjamini-Hochberg adjusted q = 10−4; table S4) and expressed by multiple cell populations in the red pulp and T cell zones (fig. S19). As in previous cases, FISH correctly recapitulated the patterns of gene expression inferred by STvEA and confirmed the partial colocalization of Plxnb2- and Sema4c-expressing cells in the spleen (adjacency score P = 0.04; fig. S19).
Annotation of highly multiplexed cytometry data using STvEA
Beyond the realm of mIHC, other technologies also allow for highly multiplexed protein expression profiling of individual cells. Multiparameter flow cytometry and cytometry by time-of-flight (CyTOF) provide high-dimensional proteomic characterizations of single-cell suspensions and are frequently used in immunology and studies of cancer. We reasoned that the same procedure for annotating cell populations on mIHC images could be adapted to these modalities of data. To assess the utility of STvEA in this context, we first applied it to 114,568 wild-type mouse splenocytes profiled with CyTOF in a published study (2). The panel in this study had 22 antibodies in common with our CITE-seq atlas. STvEA correctly consolidated the protein expression spaces of the CyTOF and CITE-seq datasets and annotated 12 cell populations (Fig. 6A and fig. S20), which represented 91% of the cells in the CyTOF dataset. As in our analysis of CODEX data, these annotations included subtle cell populations, such as pDCs and different stages of erythrocyte maturation, which are difficult to identify without specifically tailored antibody panels. For comparison, we also performed more conventional analyses based on the algorithms X-shift, SPADE, and PhenoGraph, followed by manual annotation of the resulting clusters (Fig. 6B and fig. S14). Although the annotations produced by STvEA were consistent with the results of these analyses, STvEA provided an increase in the resolution and number of annotated cell populations with respect to manual annotations. The protein expression levels of cells in the CyTOF and CODEX datasets that were mapped to the same cell in the CITE-seq reference atlas showed a large degree of consistency, with average Pearson’s correlation coefficients ranging from 0.73, 0.61, and 0.57, respectively, for T cells, erythrocytes, and B-2 cells to 0.13 for pDCs (fig. S21).
Fig. 6. Transcriptome-guided annotation of mass cytometry data.
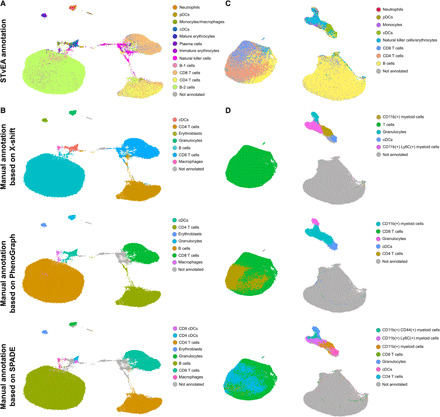
(A) UMAP representation of 114,568 mouse splenocytes profiled with CyTOF by Goltsev et al. (2). The representation is labeled with the cell populations identified by STvEA based on a panel of 22 antibodies. (B) The same representation is labeled according to the manually annotated clusters produced by X-shift, PhenoGraph, and SPADE. Automated, transcriptome-guided annotations are consistent with manual analysis but provide an improvement in resolution and reproducibility. (C) UMAP representation of 146,110 splenocytes from a glioma xenograft model profiled with CyTOF by Dusoswa et al. (41). The representation is labeled with the cell populations identified by STvEA based on a panel of 11 antibodies. (D) The same representation is labeled according to the manually annotated clusters produced by X-shift, PhenoGraph, and SPADE. Annotation of this dataset is particularly challenging due to the small size and high redundancy of the antibody panel. In particular, the panel did not include any marker for B cells, which made it difficult to manually annotate this cell population. Although the annotations provided by STvEA are also limited, they represent an improvement with respect to manual annotation procedures.
We next applied STvEA to 146,110 splenocytes from a glioma mouse model profiled with CyTOF (41). We considered only 11 antibodies shared with our splenic CITE-seq atlas. There was a substantial overlap between the cell populations stained by these antibodies, making the annotation of this dataset particularly challenging. Despite these limitations, STvEA identified and annotated eight phenotypically distinct cell populations (Fig. 6C and fig. S20), accounting for 82% of the cells in the CyTOF dataset. Although these annotations were broader than in other datasets we have analyzed, they still represented a substantial improvement with respect to procedures based on the manual annotation of clusters (Fig. 6D and fig. S14).
DISCUSSION
Methods for simultaneous profiling of protein and gene expression with single-cell resolution are evolving rapidly. Here, we have presented a computational approach for identifying and annotating cell populations in mIHC images by leveraging CITE-seq data of the same or closely related tissues. STvEA enables the optimal transfer of annotations from a CITE-seq dataset onto mIHC images or, more generally, highly multiplexed cytometry data. We have demonstrated the utility of this approach with published mIHC and mass cytometry datasets of the murine spleen, and we have studied interactions between cell populations in this organ based on the inferred spatial patterns of gene expression. The CITE-seq data resource that we have generated for this organ and its integrative spatial analysis can be interrogated through the web interface that accompanies this paper (https://camara-lab.shinyapps.io/stvea). We expect this resource will be of great utility as a reference dataset for this organ.
Our work builds upon some of the recent developments in the integration of single-cell omics data (13, 14) and is similar in spirit to previous studies mapping single-cell RNA-seq data to FISH images (42–46). However, because mapping transcriptomic information onto cytometry data is carried out through the protein expression space, there are multiple conceptual and technical differences with those studies. Specifically, to consolidate protein expression measurements performed with multiple technologies (next-generation sequencing of oligo-tagged antibodies, imaging of fluorescently labeled antibodies, and mass spectrometry of metal-tagged antibodies), we developed tailored normalization schemes for CODEX and CITE-seq. In addition, to account for the inaccuracy introduced by using relatively small antibody panels to map high-dimensional single-cell gene expression spaces, we proposed a new clustering approach to identify cell populations in the CITE-seq data that can be optimally mapped based on their protein expression profile. Last, we introduced graph-based statistical approaches for studying spatial relationships between the inferred patterns of gene expression in the mIHC images.
There are some limitations inherent to STvEA. By design, this approach can only annotate cell populations that are present in the mRNA CITE-seq reference data and that have a blueprint in the expression of profiled protein markers. Moreover, since the CITE-seq and mIHC datasets are generated from different specimens, environmental factors and other sources of biological variability might introduce small artifacts. In this regard, STvEA is complementary to emerging technologies for simultaneous highly multiplexed spatial profiling of proteins and transcripts, such as digital spatial profiling (DSP) (47) and deterministic barcoding in tissue for spatial omics sequencing (DBiT-seq) (48). These technologies perform concurrent spatially resolved proteomic and transcriptomic measurements in a tissue section and are therefore not subjected to mapping uncertainties. However, STvEA also provides a handful of unique advantages, such as its scalability to hundreds of thousands of cells (DSP can be only applied a few dozens of individual cells), its ability to work with submicrometer spatial resolutions (the spatial resolution of DBiT-seq is 20 μm), and its applicability to existing datasets, offering the possibility of performing new analyses of existing datasets. We therefore expect these tools to have complementary domains of applicability and be of great utility to researchers studying the cellular and molecular architecture of tissues, especially in light of the recent explosion of available single-cell RNA-seq and CITE-seq data of tissues.
METHODS
Mouse handling
All animal work was approved by and carried out in compliance with the animal welfare regulations defined by the University of Pennsylvania International Animal Care and Use Committee. Fifteen-week-old female BALB/cJ (stock no. 000651) mice were acquired from The Jackson Laboratory (Bar Harbor, ME). Mice were allowed to age at the University of Pennsylvania Small Animal Facility until they reached approximately 9 months, at which point they were euthanized using CO2 followed by cervical dislocation.
Tissue dissection and preparation of splenic single-cell suspensions
Spleens were removed from mice and mechanically dissociated with a syringe plunger over a 40-μm strainer while being washed with 5 ml of phosphate-buffered saline (PBS) and 10% fetal calf serum. Suspensions were centrifuged briefly to pellet cells. Red blood cells were lysed with an RBC lysis buffer (155 mM NH4Cl, 12 mM NaHCO3, and 0.1 mM EDTA) for 5 min and centrifuged again. Two million cells from the resulting pellet were resuspended in staining buffer (2% bovine serum albumin and 0.01% Tween in PBS) and subsequently incubated with the antibody panel as described below (see “Single-cell CITE-seq library preparation and sequencing”).
CITE-seq antibody conjugation and panel preparation
Antibodies were conjugated to 5′ amino-modified, high-performance liquid chromatography–purified CITE-seq oligonucleiotides purchased from Integrated DNA Technologies. Antibodies were concentrated to 1 mg/ml in PBS (pH 7.4) using 50-kDa cutoff spin columns (UFC505024, Millipore). Oligonucleiotides were resuspended to 1 mg/ml in 1× PBS (pH 7.4) and were subsequently cleaned as suggested in the CITE-seq protocol. Briefly, oligos were heated at 85°C and centrifuged at 17,000g to pellet any debris. For each antibody, 100 μg of antibody and 100 μg of oligo were conjugated using the Thunder-Link PLUS Oligo Conjugation System (SKU: 425-0300, Expedeon). All conjugates were cleaned as described in the CITE-seq protocol and resuspended to their final concentration in the Antibody Resuspension Buffer provided with the kit, with the exception of CD16/32, which was resuspended in 1× PBS. Successful conjugation was validated by running 1 μg of each conjugate on a 2% agarose gel that was subsequently stained with Sybr Gold (S11494, Thermo Fisher Scientific).
To prepare the panel, 1.5 μl of each antibody-oligo conjugate (except CD16/32) were combined in PBS and centrifuged in a 50-kDa cutoff column. After washing, the cleaned panel was recovered by flipping the column upside down and centrifuging. The cleaned panel was resuspended in staining buffer.
Single-cell CITE-seq library preparation and sequencing
CD16/32 antibody-oligo conjugate (1.5 μg) was incubated with the single-cell suspension for 10 min in place of the mouse seroblocker suggested in the CITE-seq protocol. The remaining 29 antibodies were then added to the cell suspension and incubated on ice. After incubation, cells were washed thoroughly, counted on a hemocytometer, and loaded into the 10x Chromium platform (10x Genomics) for single-cell library preparation. Cells were loaded at 1200 cells/μl. Only samples with >80% cell viability were used, profiling a total of two mouse spleens. cDNA libraries were prepared following the standard CITE-seq and 10x Genomics protocols. The resulting antibody-derived tag (ADT) and mRNA libraries were combined at a 1:9 ratio and sequenced with an Illumina HiSeq 2500 at the Center of Applied Genomics, Children’s Hospital of Philadelphia.
Multiplexed RNA FISH of splenic tissue sections
Whole spleens were removed from euthanized mice and immediately submerged in 4% paraformaldehyde (PFA) for 5.5 hours. They were then cryoprotected in a 30% sucrose/70% fixative solution at 4°C until the tissue sank (approximately overnight, ~16 hours). The tissue was embedded in optimal cutting temperature compound (OCT compound, Sakura Finetek Inc., supply no. 4583) on dry ice and frozen at −80°C. Tissue was cut using a cryostat at −20°C into 10-μm-thick sections and frozen again at −80°C. Tissue was used for microscopy within 6 months of fixation and cryoprotection.
RNA FISH experiments were carried out with the RNAscope Multiplex Fluorescence Reagent Kit v2 (Advanced Cell Diagnostics, Hayward, CA, USA; catalog no. 323100). The RNAscope assay for fixed frozen samples was followed per the manufacturer’s protocol with the following two modifications: the postfix incubation was carried out with 4% PFA at room temperature for 90 min, and manual target retrieval with a 5-min sample incubation was performed instead of the steamer method. Probes for mouse Bhlhe41, Il1b, and Plxnb2 (Advanced Cell Diagnostics, catalog nos. 467431, 316891, and 459181) were hybridized with Opal 520 (Akoya Biosciences, catalog no. FP1487001KT), and probes for mouse Cd79a and Sema4c (Advanced Cell Diagnostics, 460181-C2 and 518631-C3) were hybridized with Opal 570 (Akoya Biosciences, catalog no. FP1488001KT). Both dyes were diluted 1:1500 with tyramide signal amplification buffer provided by the RNAscope kit. Channel 2 was diluted in channel 1 1:50 as suggested in the RNAscope protocol. All incubations were carried out using a Stratagene PersonalHyb hybridization oven. Sequential sections were processed alongside the positive and negative controls provided by the RNAscope kit. Immunofluorescence images were acquired using a Leica TCS SP8 Multiphoton confocal microscope.
Single-cell CITE-seq processing
We used Cell Ranger to demultiplex, map to the mouse reference genome (mm10), and count UMIs in the mRNA libraries and CITE-seq-Count to count UMIs in the ADT libraries. We filtered out cells with more than 10% UMIs from mitochondrially encoded genes or less than 1200 mRNA UMIs in total. We used scVI to infer a lower dimensional latent space for visualization and clustering of the mRNA expression data. scVI uses a neural network to fit a zero-inflated negative binomial model to represent the technical variation in scRNA-seq data and create a latent space. We inferred an 18-dimensional latent space representation for the expression data of all genes expressed in at least 15 cells (training size = 0.75, number of epochs = 400, learning rate =1 × 10−3). The dimensionality of the latent space was empirically chosen on the basis of the stability of the resulting representations and was consistent with the elbow of the scree plot. To visualize the mRNA expression, we further reduced the latent space to two dimensions using Uniform Manifold Approximation and Projection (UMAP) with Pearson’s correlation distance.
Clustering and differential expression analysis of single-cell mRNA data
We clustered the cells in the latent space using HDBSCAN and an in-house consensus algorithm. Before clustering, we used UMAP to establish a metric in the 18-dimensional latent space, as suggested by the UMAP Python documentation. Then, we scanned across the min_cluster_size and min_sample parameters of HDBSCAN (min_cluster_size ∈{5,9,13,17}, min_sample ∈{10,13,16,19,22,25,28,31,34,37}) and used cluster-based similarity partitioning to build a consensus matrix
where S is the set of cluster assignments for all parameter configurations that gave rise to clusters with a silhouette score > 0.114, and csi is the cluster ID of cell i in s. For this threshold, about 20% of the initializations of the UMAP metric and parameter scan do not produce any clusters with a satisfactory silhouette score. In this case, the UMAP and parameter scan were reinitialized with a new random seed. We used the above consensus matrix as a dissimilarity matrix among cells to produce a consensus clustering using average linkage agglomerative clustering (inconsistent value ≤0.1). We ran edgeR’s general linear model on the mRNA count data to identify differentially expressed genes between each cluster and all the other cells (fold change threshold > 2).
Laplacian score analysis of single-cell mRNA data
We used the Laplacian score to more accurately annotate the mRNA data by identifying genes that have expression patterns within a cluster that cannot be explained by random variation. For each cluster, we built a graph where nodes represent cells and edges connect pairs of cells that are within ɛ distance, as defined by Pearson’s correlation in the latent space. We took ɛ to be given by the median pairwise distance among cells. For large clusters, we randomly sampled 1000 cells. The Laplacian score ℓ of a gene with expression vector f is defined as
where
and A is the adjacency matrix of the graph. We computed the Laplacian score of the log(1 + TPM ∙ 10−2) expression values for all genes expressed in at least 2% and at most 90% of the cells. To assess the significance of the Laplacian score as compared to random variation, we performed a permutation test by randomizing the cell labels 1000 times.
Normalization of ADT libraries
We fit the distribution of ADT counts for each antibody with a two-component negative binomial mixture model
where khi represents the observed number of ADT UMIs for antigen h in cell i, the mixing parameter bh represents the probability of a measurement of antigen h actually coming from the background, and the signal component is defined as the component of the mixture with the highest median. Upon fitting the model using least-squares estimation, we filtered out the background component of the data by considering the matrix
where the weights wi are introduced to account for differences in the total number of ADT UMIs across cells
These weights can be justified by noting that Prob(r ≤ khi ∣sh ∈ signal) depends linearly on the total number of ADTs except at the tails of the distribution.
We performed batch correction on the values using the mutual nearest neighbors approach of Haghverdi et al. (13) and rescaled the resulting values to be in the [0,1] interval.
We did not consider CD169 in our analysis as it was showing expression in cell populations other than macrophages, possibly reflecting a lack of affinity of the conjugated antibody. In addition, the unimodal distribution of ERTR7 expression values was consistent with the fact that we did not capture stromal cells in our CITE-seq dataset (possibly because of the use of a nonenzymatic dissociation procedure). We therefore assigned all cells to the background component and set .
Processing of CODEX data
We considered the segmented and spillover-compensated CODEX data of the three wild-type mice profiled by Goltsev et al. (2). We filtered out artifacts using a similar gating strategy to that of the CODEX protocol. We removed cells smaller than 1000 or larger than 25,000 voxels. We then identified maximum and minimum cutoffs for blank channels by plotting the expression of one blank channel versus another, as described in the CODEX protocol. We removed cells with intensities above the upper cutoffs in any of the blank channels or below the lower cutoffs in all of the blank channels. Our cutoffs fell around the 99.5 and 0.2 percentiles, respectively. However, we checked that small variations of the specific values did not greatly affect the number of cells removed.
Normalization of CODEX data
We normalized the processed CODEX data by the total levels in each cell
where Mhi is the level of antigen h in cell i before normalization. After this process, antigen levels are well approximated by a two-component Gaussian mixture model
where the Gaussian with the highest median corresponds to the signal component, and the mixing parameter ah represents the probability of a measurement of antigen h actually coming from the background. Upon fitting the model to the data using the expectation-maximization algorithm for maximum likelihood estimation, we filtered out the background component of the data by considering the probabilities
in subsequent analysis.
Mapping of CODEX data into CITE-seq
We mapped the inferred CODEX probabilities p into the CITE-seq space qCITEseq using a modified version of the general strategy proposed by Stuart et al. (14). Specifically, we identified a set of anchors using a mutual nearest neighbors approach with kanchor = 20. We found the nearest neighbors using Euclidean distance in a common 29-dimensional space obtained by canonical correlation analysis (CCA). We then filtered out anchors that do not preserve the structure of the original protein space. For that purpose, we kept only those for which the CODEX cell in the anchor was within the kfilter = 100 nearest CODEX cells to the CITE-seq cell in the anchor, or vice versa, as measured by Pearson’s correlation distance between p and qCITEseq.
Cells in the CODEX dataset were aligned into the CITE-seq protein space using the following transformation
where 𝒜i is the set of kweight = 100 anchors (j1, j2) with smallest Pearson’s correlation distance between vectors and (with components phj2 and phi, respectively), and w(j1, j2), i are weights specifying the effect size of anchor (j1, j2) on the CODEX cell i based on both mRNA and protein data
In this equation, dij2 denotes Pearson’s correlation distance between the vectors and , and c is a parameter specifying the width of the Gaussian kernel. The number of shared neighbors between the two anchor cells, sj1j2, is defined as
where is the set of nearest CITE-seq cells to cell j1 in the mRNA latent space, is the set of nearest CITE-seq cells to cell j2 in the CCA space, is the set of nearest CODEX cells to cell j1 in the CCA space, and is the set of nearest CODEX cells to cell j2 in the CCA space. As before, distances in the mRNA and CCA spaces were measured using Pearson’s correlation and Euclidean distance, respectively. In all cases, the number of nearest neighbors was chosen to be kscore = 80. The values sj1j2 were scaled such that the 0.9 quantile is at 1 and the 0.01 quantile is at 0, and values above or below these quantiles were set to 1 or 0, respectively.
Since randomly sampled sections of the CODEX dataset can be mapped independently and concatenated later, we divided the CODEX dataset into eight random sections of the same size (9900 cells) to provide a speed improvement for the nearest neighbor calculations.
To be able to transfer quantities between the CITE-seq and CODEX datasets, we then built a ℳCITEseq→CODEX transfer matrix
where denotes Pearson’s correlation distance between the vectors and (with components and , respectively), and c is a parameter that specifies the width of the Gaussian kernel. The set contains the nearest CODEX cells to the CITE-seq cell i as measured by , where , and nCODEX is the number of cells in the CODEX dataset. These matrices can be used to transfer quantities across the two datasets. For instance, the inferred mRNA expression level of gene m in the CODEX cell j is given by
where ECITEseq denotes the mRNA expression matrix in the CITE-seq dataset. Similarly, the mRNA cell populations can be mapped to the CODEX data using
where the sum runs over all cells in the CITE-seq dataset and is the indicator function of cluster c. Note that because of the mapping uncertainties, the resulting feature vector is no longer a binary vector. To assess mapping uncertainties (Fig. 3C), we computed the Pearson’s correlation coefficient of the vectors that result from restricting the above sum to cells in each of the two mice profiled with CITE-seq.
Parameter selection
For different values of kanchor, kfilter, and kscore, we evaluated the performance of the algorithm to accurately map a set of “gold standard” cell populations. The populations we considered were B cells, T cells, NK cells, dendritic cells, neutrophils, plasma cells, and red pulp macrophages, as they were general enough to be clearly identifiable in both datasets by clustering and the expression of specific markers. We used the Louvain community detection algorithm in a k = 49 nearest neighbor graph for clustering the CODEX protein data. To quantify the performance of the mapping, we defined the quality scores of a set of anchors 𝒜 as
where ci is the cell type of cell i and 𝒞 is the set of gold standard populations. We sequentially chose the values of kanchor, kfilter, and kscore that maximized these quality scores.
We assessed the behavior of the STvEA mapping with varying values of kanchor, kfilter, kscore, kweight, and ktransfer by measuring how well annotations of certain cell populations aligned with cells highly expressing the corresponding marker. For each value of k, we computed the mapped cluster matrix of the CITE-seq cell types in CODEX. We compared the mapped B-2 cell population to CODEX cells with normalized B220 expression > 0.7, the T cell population to normalized TCR expression > 0.8, the NK cell population to normalized NKp46 expression > 0.9, and the neutrophil population to normalized Ly6G expression > 0.8. We then computed the sensitivity and specificity of each of these true thresholded populations being positively identified by the STvEA mapping.
Quantification of mapping uncertainties and stability
To study the consistency between CODEX cells that are mapped by the same CITE-seq cell, we randomly selected ktransfer pairs of cells among the CODEX cells that map to the same CITE-seq cell and calculated Person’s correlation between antigen levels in each pair of cells. The mean value of the correlation coefficients was taken to represent the mapping uncertainty of each CITE-seq cell, which was shown on the UMAP in fig. S7. To compare these correlation coefficients with the correlation between two random pairs of CODEX cells, we considered 7097 random sets of ktransfer CODEX cells and computed the correlation coefficients using the same method. To assess localized uncertainty in the mapping algorithm, we defined a mapping score for each CODEX cell as and plotted these scores on the UMAP representation of the CODEX protein space (fig. S10). To study how the size of the CITE-seq dataset affects the performance of STvEA, we randomly sampled 5000, 2500, and 1000 cells from the original CITE-seq dataset and ran STvEA using the same default parameters.
To assess the effect of antibody panel selection on STvEA mapping, we used the glmnet R package (49) to perform multinomial logistic lasso regression on the CITE-seq mRNA clusters with respect to the protein expression levels. We identified values of the regularization parameter λ for which a subset of n = 25,21, …,13,9 antibodies had nonzero coefficient and ran STvEA truncating the antibody panel to each of these subsets. The stability of STvEA was evaluated for each antibody panel by computing the Pearson’s correlation coefficient between the vectors of cell population assignments, , for each CODEX cell when using the full and a reduced antibody panel (fig. S11).
Optimized annotation of cell populations
To annotate mIHC images with STvEA while accounting for uncertainty in the mapping algorithm, we sought to identify a clustering of the CITE-seq cells that allows for the highest resolution in number of clusters while maintaining a good modularity of the clusters upon mapping into the CODEX protein expression space. Clustering the CITE-seq mRNA data without taking into account their mapping onto the CODEX images can lead to clusters that cannot be accurately mapped based on the profiled proteins, as well as clusters that could be further divided into pieces that can be still accurately mapped. It is therefore natural to consider a hierarchical clustering approach where cluster splits are only kept if they are mapped into independent clusters in the CODEX protein expression space. We started from the simplified hierarchical tree of an HDSBCAN clustering that passed the silhouette threshold in the original CITE-seq consensus clustering (min_cluster_size = 17, min_samples = 34). For computational simplicity, each CODEX cell was assigned to its closest CITE-seq neighbor in the shared protein space q. Each branch of the hierarchical tree could then be easily mapped onto CODEX. Since HDBSCAN allows some cells to remain unclustered, each bifurcation in the tree may have some “singlets” that are contained in the parent cluster but not the child clusters. To fill out the tree, we imputed these singlets into the closest child cluster based on Pearson’s correlation distance in the protein space. We then implemented an agglomerative clustering approach based on this tree by computing the modularity in the CODEX protein expression space. We considered the k = 50 nearest neighbor graph generated by Pearson’s correlation distance between the protein expression profiles of the CODEX cells. For each bifurcation in the tree, we computed the subgraph spanned by the cells in the two clusters involved in the bifurcation and then computed the Newman-Girvan modularity (50) (also known as Louvain modularity) of the clusters in this subgraph. Starting at the leaves of the tree, two child clusters were merged into their parent if the modularity of the bifurcation was less than a quality threshold tq or if the modularities between the cells in each child cluster versus all other cells in the CODEX dataset were less than a sparsity threshold ts (tq = 0.1, ts = 0.003). In cases where a single-cluster branch in a bifurcation did not pass the sparsity threshold, the cells from this cluster where merged into sibling clusters. Similarly, cells of a single-cluster branch were merged into sibling clusters if this represented an increase in modularity larger than an imputation threshold ti (ti = 5%). After this process, we smoothened the cell assignments in CODEX based on each cell’s neighbors. For each CODEX cell x, we defined a neighborhood of other cells Nx within a protein expression correlation threshold (Pearson > 0.9). The cell assignment of a cell x is then defined as
where Cx is the indicator function of the cluster of x, and ϵ is a constant to control the contribution of neighboring cells (ϵ = 0.01).
Spatial relationship among cell populations
To assess the spatial relationship between two feature vectors f and g defined over the cells in the CODEX dataset, we built a k nearest neighbor graph using Euclidean distance in the CODEX spatial dimensions. We then introduced the adjacency score, defined as
where A is the adjacency matrix of the nearest neighbor graph. This score takes high values when the features take high values in adjacent cells. The scale of the interactions is set by the magnitude of the nearest neighbor parameter k. Features that we have used in this paper include cell population assignments (to assess whether two cell populations colocalize spatially) and mapped gene expression (to assess whether genes encoding for ligands and receptors are expressed in adjacent cells). In our analyses, we used k = 2 and 29, corresponding to median distances of 5 and 13 μm, respectively. The significance of this score was assessed using a null distribution built by permuting the cell IDs. For mutually exclusive binary features (such as cluster assignments), the null distribution can be computed analytically in terms of the hypergeometric distribution Hypergeom(u; N, K, n),
where v and m are the number of nodes and edges in the nearest neighbor graph, respectively, and I is the identity matrix. For nonbinary features, we did not find a closed form for the null distribution, so we approximated it using a normal distribution whose parameters were estimated from 1000 random permutations. We controlled the false discovery rate for multiple hypothesis testing using the Benjamini-Hochberg q value procedure.
To account for the effect of mapping uncertainties on the adjacency score of cell populations, we also computed the overlap score, fT ∙ g, and assessed its significance by randomly permuting the entries of one of the feature vectors. In addition, we evaluated the Pearson’s correlation of the adjacency score q values across the three mice profiled with CODEX (Fig. 4B).
We compared these results to those of Giotto’s cellProximityEnrichment function, which assesses the proximity of cell types by comparing the observed number of shared edges in the network to the expected number. We computed the Benjamini-Hochberg q value using Giotto on a k nearest neighbor network with the same value of k as our adjacency analysis and all other default parameter values.
Identification of paracrine interactions
We used CellPhoneDB (40) to identify significant ligand-receptor pairs within the CITE-seq mRNA expression data. CellPhoneDB identifies genes coding for ligand and receptor pairs that are differentially expressed in one or more cell populations using a curated database of ligands and receptors. Since CellPhoneDB only considers human gene pairs, we generated a mouse ortholog database of ligands and receptors using Ensembl (version 96) (51). For simplicity, this analysis was restricted to only those genes that have a unique ortholog. The 587 significant interactions (CellPhoneDB P ≤ 0.05) identified by this analysis were then filtered using the adjacency score approach described above (see the “Spatial relationship among cell populations” section) with k = 29 (median distance, 13 μm) to identify pairs of genes significantly expressed in adjacent cells. We restricted the adjacency score test to only the pairs of cell populations identified by CellPhoneDB by setting the expression of each gene to 0 outside of the cell population of interest. The expression of any complexes output by CellPhoneDB was calculated as the sum of the expression of their component genes.
Multiplexed single-molecule RNA FISH was performed as described above (see the “Multiplexed RNA FISH of splenic tissue sections” section) using formalin-fixed paraffin-embedded tissue sections, and images were analyzed using ImageJ v2.1.0 (52). We segmented the cells and identified the follicles using tissue autofluorescence. To that end, we used a Gaussian filter (σ = 2) to despeckle the images, and low-intensity regions corresponding to cell nuclei were identified using an adaptive local threshold based on the median intensity in a 50-pixel radial neighborhood (parameter_1 = 0). We filled in holes (iterations = 1, count = 2) and segmented nuclei using the watershed algorithm. Nuclei were defined as regions with an area between 100 and 2500 pixels and a circularity between 0.2 and 1.0. Segmented nuclei with a median pixel intensity above 50 in channel 1 and 30 in channel 2, corresponding to red blood cells, were removed. The remaining segmented nuclei were added to the ImageJ ROI manager and used to tally the number of probes in each nucleus. To identify RNA probes, we used a Gaussian filter (σ = 2) to despeckle the images and a local maxima function (prominence = 30) to distinguish the probes from the background. Probes in channels 1 and 3 separated by less than 15 pixels were filtered out. The adjacency score was run as described above (see the “Spatial relationship among cell populations” section).
Analysis of CyTOF data
We applied STvEA to the CyTOF datasets of Goltsev et al. (2) and Dusoswa et al. (41). The dataset of Goltsev et al. (2) consists of 124,277 cells in total and has 22 antibodies in common with our CITE-seq panel. To preprocess these data, we first removed outlier cells that express fewer than 500 total counts in the 22 antibodies or are within the top 2% of cells for total counts. We applied an arcsinh transform (cofactor 5) to the remaining 114,568 cells and scaled the resulting values to the interval [0,1]. We use STvEA as described above to map our CITE-seq atlas into this dataset. We then applied the optimized clustering approach described above (tq = 0.2, ts = 0.001, ti = 3.5%) to identify 16 clusters in the CyTOF dataset associated with 13 phenotypically different cell populations in the CITE-seq atlas.
For the dataset of Dusoswa et al. (41), we considered all 146,119 cells passing the “Live singlets” gate from the MGL02_Spleen sample. We removed cells with zero expression in the 11 antibodies considered in our analysis, resulting in 146,110 cells in total. We scaled the arcsinh transformed (cofactor 5) values to the interval [0,1] and applied STvEA as described above to map our CITE-seq atlas onto this dataset. We used the optimized clustering approach described above (tq = 0.25, ts = 0.001, ti = 1%) to identify 23 clusters in the CyTOF dataset associated with eight phenotypically different cell populations in the CITE-seq atlas.
Annotation of X-shift, SPADE, and PhenoGraph clusters using STvEA
We used the implementations of these algorithms in the VorteX Java software (https://github.com/nolanlab/vortex), the PhenoGraph Python package (https://github.com/jacoblevine/PhenoGraph), and the SPADE R package (https://github.com/nolanlab/spade). For all datasets, we ran the algorithms using default parameters. We only considered antibodies that were also present in our CITE-seq panel so that any difference between the automated annotations provided by STvEA and the manual annotations were caused by the method and not by differences in the input data. For each dataset, we manually annotated the clusters produced by XShift (VorteX), PhenoGraph, and SPADE using the same biological terms and associated expression markers as in the original publication of the dataset. We merged identically annotated clusters and visualized the annotations using a UMAP representation of the cytometry data (Fig. 6 and fig. S13). To create the pie chart graphs in fig. S14, we started with the output graph produced by SPADE, or we created one from the X-shift and PhenoGraph clusters by computing a minimum spanning tree between cluster centroids using the Pearson’s correlation distance between the protein expression profiles. For each node in the graph, we identified the proportion of cells from each mapped CITE-seq cluster (see the “Optimized annotation of cell populations” section above) in that node and visualized those proportions as a pie chart.
Online database
The complete results of our analysis can be interactively queried through a web application hosted at the URL https://camara-lab.shinyapps.io/stvea.
STvEA software
All algorithms have been implemented and documented in an R package. The package can be downloaded from the URL https://github.com/CamaraLab/STvEA.
Acknowledgments
We would like to thank D. Aldea, R. Aubin, J. Crawford, D. Epstein, Y. Ho, J. Humenik, Y. Kamberov, S. Liebhaber, F. Mafra, M. Peel, R. Pellegrino, Q. Qiu, S. Rakowiecki, J. Smiler, R. Staupe, J. Wherry, H. Wu, and the Zhou lab for scientific discussions and assistance with various aspects of the experiments. Funding: The work of P.G.C. and S.W. is partially funded by Stand-Up-To-Cancer Convergence 2.0. The work of R.G.A. is supported by a NIH NHGRI T32 grant (T32HG00046). Author contributions: E.C.T. performed all the experiments. K.W.G. implemented all the algorithms and performed the analyses. Z.M. and R.G.A. assisted with the analyses. S.W. designed and implemented the web application. P.G.C. conceived the study and supervised the work. All authors contributed to writing the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. CITE-seq raw reads and count matrices have been deposited in the NIH GEO database (accession number: GSE160766). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabc5464/DC1
REFERENCES AND NOTES
- 1.Stack E. C., Wang C., Roman K. A., Hoyt C. C., Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 70, 46–58 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Goltsev Y., Samusik N., Kennedy-Darling J., Bhate S., Hale M., Vazquez G., Black S., Nolan G. P., Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jungmann R., Avendano M. S., Woehrstein J. B., Dai M., Shih W. M., Yin P., Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 11, 313–318 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cornett D. S., Reyzer M. L., Chaurand P., Caprioli R. M., MALDI imaging mass spectrometry: Molecular snapshots of biochemical systems. Nat. Methods 4, 828–833 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Giesen C., Wang H. A. O., Schapiro D., Zivanovic N., Jacobs A., Hattendorf B., Schüffler P. J., Grolimund D., Buhmann J. M., Brandt S., Varga Z., Wild P. J., Günther D., Bodenmiller B., Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Angelo M., Bendall S. C., Finck R., Hale M. B., Hitzman C., Borowsky A. D., Levenson R. M., Lowe J. B., Liu S. D., Zhao S., Natkunam Y., Nolan G. P., Multiplexed ion beam imaging of human breast tumors. Nat. Med. 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banik G., Betts C. B., Liudahl S. M., Sivagnanam S., Kawashima R., Cotechini T., Larson W., Goecks J., Pai S. I., Clayburgh D. R., Tsujikawa T., Coussens L. M., High-dimensional multiplexed immunohistochemical characterization of immune contexture in human cancers. Methods Enzymol. 635, 1–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsujikawa T., Kumar S., Borkar R. N., Azimi V., Thibault G., Chang Y. H., Balter A., Kawashima R., Choe G., Sauer D., El Rassi E., Clayburgh D. R., Kulesz-Martin M. F., Lutz E. R., Zheng L., Jaffee E. M., Leyshock P., Margolin A. A., Mori M., Gray J. W., Flint P. W., Coussens L. M., Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. 19, 203–217 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saeys Y., Van Gassen S., Lambrecht B. N., Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol. 16, 449–462 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P. K., Swerdlow H., Satija R., Smibert P., Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson V. M., Zhang K. X., Kumar N., Wong J., Li L., Wilson D. C., Moore R., McClanahan T. K., Sadekova S., Klappenbach J. A., Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 35, 936–939 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Shahi P., Kim S. C., Haliburton J. R., Gartner Z. J., Abate A. R., Abseq: Ultrahigh-throughput single cell protein profiling with droplet microfluidic barcoding. Sci. Rep. 7, 44447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haghverdi L., Lun A. T. L., Morgan M. D., Marioni J. C., Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M. III, Hao Y., Stoeckius M., Smibert P., Satija R., Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez R., Regier J., Cole M. B., Jordan M. I., Yosef N., Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Govek K. W., Yamajala V. S., Camara P. G., Clustering-independent analysis of genomic data using spectral simplicial theory. PLOS Comput. Biol. 15, e1007509 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He X., Cai D., Niyogi P., Laplacian score for feature selection. Adv. Neural Inf. Process. Syst. 18, 507–514 (2005). [Google Scholar]
- 18.Lewis S. M., Williams A., Eisenbarth S. C., Structure and function of the immune system in the spleen. Sci. Immunol. 4, eaau6085 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bronte V., Pittet M. J., The spleen in local and systemic regulation of immunity. Immunity 39, 806–818 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaitin D. A., Kenigsberg E., Keren-Shaul H., Elefant N., Paul F., Zaretsky I., Mildner A., Cohen N., Jung S., Tanay A., Amit I., Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing group; Supplemental text writing group; Principal investigators , Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S., Saadatpour A., Zhou Z., Chen H., Ye F., Huang D., Xu Y., Huang W., Jiang M., Jiang X., Mao J., Chen Y., Lu C., Xie J., Fang Q., Wang Y., Yue R., Li T., Huang H., Orkin S. H., Yuan G.-C., Chen M., Guo G., Mapping the mouse cell atlas by microwell-seq. Cell 172, 1091–1107.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Eisenbarth S. C., Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 19, 89–103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.-r., Raychaudhuri S., Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welch J. D., Kozareva V., Ferreira A., Vanderburg C., Martin C., Macosko E. Z., Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177, 1873–1887.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian J., Olbrecht S., Boeckx B., Vos H., Laoui D., Etlioglu E., Wauters E., Pomella V., Verbandt S., Busschaert P., Bassez A., Franken A., Bempt M. V., Xiong J., Weynand B., van Herck Y., Antoranz A., Bosisio F. M., Thienpont B., Floris G., Vergote I., Smeets A., Tejpar S., Lambrechts D., A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 30, 745–762 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schurch C. M., Bhate S. S., Barlow G. L., Phillips D. J., Noti L., Zlobec I., Chu P., Black S., Demeter J., McIlwain D. R., Samusik N., Goltsev Y., Nolan G. P., Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell 182, 1341–1359.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McInnes L., Healy J., Astels S., hdbscan: Hierarchical density based clustering. J. Open Source Softw. 2, 205 (2017). [Google Scholar]
- 29.Samusik N., Good Z., Spitzer M. H., Davis K. L., Nolan G. P., Automated mapping of phenotype space with single-cell data. Nat. Methods 13, 493–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu P., Simonds E. F., Bendall S. C., Gibbs K. D. Jr., Bruggner R. V., Linderman M. D., Sachs K., Nolan G. P., Plevritis S. K., Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 29, 886–891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine J. H., Simonds E. F., Bendall S. C., Davis K. L., Amir E.-a., Tadmor M. D., Litvin O., Fienberg H. G., Jager A., Zunder E. R., Finck R., Gedman A. L., Radtke I., Downing J. R., Pe’er D., Nolan G. P., Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang F., Flanagan J., Su N., Wang L.-C., Bui S., Nielson A., Wu X., Vo H.-T., Ma X.-J., Luo Y., RNAscope: A novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 14, 22–29 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kreslavsky T., Vilagos B., Tagoh H., Poliakova D. K., Schwickert T. A., Wohner M., Jaritz M., Weiss S., Taneja R., Rossner M. J., Busslinger M., Essential role for the transcription factor Bhlhe41 in regulating the development, self-renewal and BCR repertoire of B-1a cells. Nat. Immunol. 18, 442–455 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen L., Shinton S. A., Hardy R. R., Hayakawa K., Association of B-1 B cells with follicular dendritic cells in spleen. J. Immunol. 174, 6918–6926 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Villeval J. L., Testa U., Vinci G., Tonthat H., Bettaieb A., Titeux M., Cramer P., Edelman L., Rochant H., Breton-Gorius J., Carbonic anhydrase I is an early specific marker of normal human erythroid differentiation. Blood 66, 1162–1170 (1985). [PubMed] [Google Scholar]
- 36.Welch J. J., Watts J. A., Vakoc C. R., Yao Y., Wang H., Hardison R. C., Blobel G. A., Chodosh L. A., Weiss M. J., Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 104, 3136–3147 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Southcott M. J. G., Tanner M. J. A., Anstee D. J., The expression of human blood group antigens during erythropoiesis in a cell culture system. Blood 93, 4425–4435 (1999). [PubMed] [Google Scholar]
- 38.Dzierzak E., Philipsen S., Erythropoiesis: Development and differentiation. Cold Spring Harb. Perspect. Med. 3, a011601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dries R., Zhu Q., Dong R., Eng C.-H. L., Li H., Liu K., Fu Y., Zhao T., Sarkar A., Bao F., George R. E., Pierson N., Cai L., Yuan G.-C., Giotto, a toolbox for integrative analysis and visualization of spatial expression data. bioRxiv, 701680 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vento-Tormo R., Efremova M., Botting R. A., Turco M. Y., Vento-Tormo M., Meyer K. B., Park J.-E., Stephenson E., Polański K., Goncalves A., Gardner L., Holmqvist S., Henriksson J., Zou A., Sharkey A. M., Millar B., Innes B., Wood L., Wilbrey-Clark A., Payne R. P., Ivarsson M. A., Lisgo S., Filby A., Rowitch D. H., Bulmer J. N., Wright G. J., Stubbington M. J. T., Haniffa M., Moffett A., Teichmann S. A., Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dusoswa S. A., Verhoeff J., Abels E., Méndez-Huergo S. P., Croci D. O., Kuijper L. H., de Miguel E., Wouters V. M. C. J., Best M. G., Rodriguez E., Cornelissen L. A. M., van Vliet S. J., Wesseling P., Breakefield X. O., Noske D. P., Wurdinger T., Broekman M. L. D., Rabinovich G. A., van Kooyk Y., Garcia-Vallejo J. J., Glioblastomas exploit truncated O-linked glycans for local and distant immune modulation via the macrophage galactose-type lectin. Proc. Natl. Acad. Sci. U.S.A. 117, 3693–3703 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satija R., Farrell J. A., Gennert D., Schier A. F., Regev A., Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Achim K., Pettit J.-B., Saraiva L. R., Gavriouchkina D., Larsson T., Arendt D., Marioni J. C., High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol. 33, 503–509 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Karaiskos N., Wahle P., Alles J., Boltengagen A., Ayoub S., Kipar C., Kocks C., Rajewsky N., Zinzen R. P., The Drosophila embryo at single-cell transcriptome resolution. Science 358, 194–199 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Halpern K. B., Shenhav R., Matcovitch-Natan O., Tóth B., Lemze D., Golan M., Massasa E. E., Baydatch S., Landen S., Moor A. E., Brandis A., Giladi A., Stokar-Avihail A., David E., Amit I., Itzkovitz S., Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu Q., Shah S., Dries R., Cai L., Yuan G.-C., Identification of spatially associated subpopulations by combining scRNAseq and sequential fluorescence in situ hybridization data. Nat. Biotechnol. 36, 1183–1190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merritt C. R., Ong G. T., Church S., Barker K., Geiss G., Hoang M., Jung J., Liang Y., McKay-Fleisch J., Nguyen K., Sorg K., Sprague I., Warren C., Warren S., Zhou Z., Zollinger D. R., Dunaway D. L., Mills G. B., Beechem J. M., High multiplex, digital spatial profiling of proteins and RNA in fixed tissue using genomic detection methods. bioRxiv, 559021 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Liu Y., Yang M., Deng Y., Su G., Enninful A., Guo C. C., Tebaldi T., Zhang D., Kim D., Bai Z., Norris E., Pan A., Li J., Xiao Y., Halene S., Fan R., High-spatial-resolution multi-omics atlas sequencing of mouse embryos via deterministic barcoding in tissue. Cell 183, 1665–1681.e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Friedman J., Hastie T., Tibshirani R., Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- 50.Newman M. E. J., Girvan M., Finding and evaluating community structure in networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 69, 026113 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Zerbino D. R., Achuthan P., Akanni W., Amode M. R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C. G., Gil L., Gordon L., Haggerty L., Haskell E., Hourlier T., Izuogu O. G., Janacek S. H., Juettemann T., To J. K., Laird M. R., Lavidas I., Liu Z., Loveland J. E., Maurel T., McLaren W., Moore B., Mudge J., Murphy D. N., Newman V., Nuhn M., Ogeh D., Ong C. K., Parker A., Patricio M., Riat H. S., Schuilenburg H., Sheppard D., Sparrow H., Taylor K., Thormann A., Vullo A., Walts B., Zadissa A., Frankish A., Hunt S. E., Kostadima M., Langridge N., Martin F. J., Muffato M., Perry E., Ruffier M., Staines D. M., Trevanion S. J., Aken B. L., Cunningham F., Yates A., Flicek P., Ensembl 2018. Nucleic Acids Res. 46, D754–D761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rueden C. T., Schindelin J., Hiner M. C., DeZonia B. E., Walter A. E., Arena E. T., Eliceiri K. W., ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogden C. A., deCathelineau A., Hoffmann P. R., Bratton D., Ghebrehiwet B., Fadok V. A., Henson P. M., C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 194, 781–796 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao J.-L., Guillabert A., Hu J., Le Y., Urizar E., Seligman E., Fang K. J., Yuan X., Imbault V., Communi D., Wang J. M., Parmentier M., Murphy P. M., Migeotte I., F2L, a peptide derived from heme-binding protein, chemoattracts mouse neutrophils by specifically activating Fpr2, the low-affinity N-formylpeptide receptor. J. Immunol. 178, 1450–1456 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Sugimoto M. A., Vago J. P., Teixeira M. M., Sousa L. P., Annexin A1 and the resolution of inflammation: Modulation of neutrophil recruitment, apoptosis, and clearance. J. Immunol. Res. 2016, 8239258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hartnell A., Heinemann A., Conroy D. M., Wait R., Sturm G. J., Caversaccio M., Jose P. J., Williams T. J., Identification of selective basophil chemoattractants in human nasal polyps as insulin-like growth factor-1 and insulin-like growth factor-2. J. Immunol. 173, 6448–6457 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Hirai K., Miyamasu M., Yamaguchi M., Nakajima K., Ohtoshi T., Koshino T., Takaishi T., Morita Y., Ito K., Modulation of human basophil histamine release by insulin-like growth factors. J. Immunol. 150, 1503–1508 (1993). [PubMed] [Google Scholar]
- 58.Koketsu R., Suzukawa M., Kawakami A., Komiya A., Ra C., Yamamoto K., Yamaguchi M., Activation of basophils by stem cell factor: Comparison with insulin-like growth factor-I. J. Investig. Allergol. Clin. Immunol. 18, 293–299 (2008). [PubMed] [Google Scholar]
- 59.Ochensberger B., Daepp G. C., Rihs S., Dahinden C. A., Human blood basophils produce interleukin-13 in response to IgE-receptor-dependent and -independent activation. Blood 88, 3028–3037 (1996). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabc5464/DC1