

Summary
Accurate DNA replication is constantly threatened by DNA lesions arising from endogenous and exogenous sources. Specialized DNA replication stress response pathways ensure replication fork progression in the presence of DNA lesions with minimal delay in fork elongation. These pathways broadly include translesion DNA synthesis, template switching, and replication fork repriming. Here, we discuss recent advances toward our understanding of the mechanisms that regulate the fine-tuned balance between these different replication stress response pathways. We also discuss the molecular pathways required to fill single-stranded DNA gaps that accumulate throughout the genome after repriming, and the biological consequences of using repriming instead of other DNA damage tolerance pathways on genome integrity and cell fitness.
Graphical Abstract
eTOC Blurb
Replication stress response pathways allow DNA replication to tolerate obstacles with minimal delay in fork elongation. In this review, Quinet et al. discuss the molecular contexts in which cells choose repriming over template switching and translesion synthesis to tolerate these obstacles, and the consequences of this choice on genome integrity.
High-fidelity DNA replication is constantly challenged by a diverse range of obstacles. These include DNA lesions created by endogenous and exogenous agents and intrinsic replication obstacles such as secondary structures in the DNA template, tightly-bound protein-DNA complexes, and conflicts with the transcription machinery. The transient slowing or stalling of replication forks in response to these challenges is termed “replication stress” (Berti et al., 2020). The accurate processing of stalled or damaged replication forks is central to preserve genome stability and ensure cell survival. Cells have evolved different molecular pathways aimed at preserving the stability of stalled replication forks and promoting their accurate restart. How cells choose between these pathways remains unclear.
DNA damage tolerance (DDT) mechanisms allow replication forks to overcome obstacles with a minimal effect in fork elongation. DDT broadly includes Translesion DNA Synthesis (TLS) and template switching (TS) pathways (Figure 1). A host of specialized polymerases drive TLS by direct bypass of DNA lesions. TLS polymerases (POL) include POL η, REV1, POL κ, POL ι, POL ζ, POL ν, and POL θ, all of which have the ability to replicate through a damaged template, albeit with lower fidelity (Sale, 2013; Vaisman and Woodgate, 2017). TS is generally more accurate than TLS and involves the use of a homologous template, usually the newly synthesized daughter strand on the sister chromatid, to bypass DNA lesions (Adar et al., 2009; Izhar et al., 2013). One version of TS is replication fork reversal, in which replication forks reverse their course by annealing the two daughter strands, leading to the formation of four-way junction structures (Neelsen and Lopes, 2015). Remodeling of replication forks into reversed forks promotes bypass of damage by canonical TS mechanisms, or replication-coupled repair by repositioning the lesion in the double-stranded duplex ahead of the fork (Berti et al., 2020). Moreover, recent studies suggest that fork reversal also occurs at unchallenged forks as a global response to replication stress to hold replication forks in a “standby” mode until replication stress is resolved (Mutreja et al., 2018).
Figure 1. DNA replication stress response mechanisms.
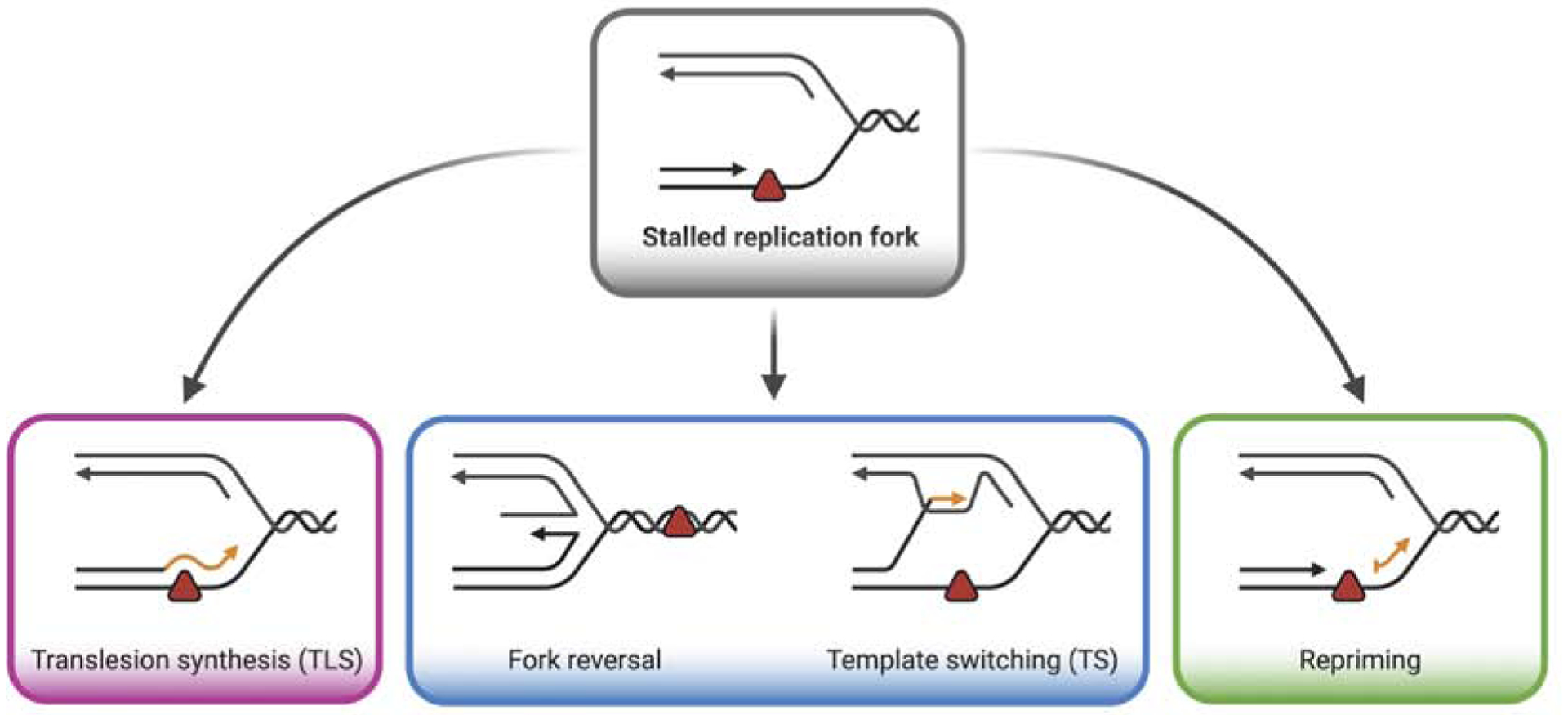
Replication obstacles (represented by the red triangle) transiently stall fork progression. Replication obstacles can be “tolerated” by three distinct pathways to allow resumption of replication fork progression: translesion synthesis (left), template switching or fork reversal (middle), and repriming (right).
In addition to TLS and TS, a third mechanism, termed repriming, can be activated to overcome replication obstacles and ensure DNA replication progression (Figure 1). Repriming involves re-initiation of DNA synthesis beyond a DNA lesion, leaving unreplicated single-stranded DNA (ssDNA) gaps to be filled post-replicatively through either TLS or TS mechanisms. After a brief historical perspective on the repriming mechanisms, we discuss how cells choose between the repriming, TLS, and TS pathways. This has been a long-standing question in the field, and recent papers have provided important clues into how different factors favor one pathway over the other, including the nature of the DNA lesion, extent of DNA damage, PCNA post-translational modifications, as well as changes in the genetic background. Finally, we discuss how the ssDNA gaps that form upon repriming are repaired post-replicatively and the impact of employing repriming versus canonical DDT pathways on cell fitness and genome integrity.
SSDNA GAP FORMATION AND REPRIMING
Early studies suggested that exposure to UV radiation causes minimal delay in DNA replication fork progression but leads to the accumulation of ssDNA discontinuities on the daughter strands in bacteria (Howard-Flanders et al., 1968), mouse (Lehmann and Kirk-Bell, 1972), and human cells (Meneghini, 1976). These daughter-strand ssDNA gaps accumulate on both the lagging and leading strands upon UV irradiation, as observed by electron microcopy in Saccharomyces cerevisae (Lopes et al., 2006). The same ssDNA gaps have been observed in mammalian cells upon exposure to a wide-range of DNA-damaging agents (Diamant et al., 2012; Elvers et al., 2011; Jansen et al., 2009; Quinet et al., 2016). However, the underlying mechanisms leading to the formation of these ssDNA gaps remained mechanistically ill-defined.
Generation of ssDNA gaps in the lagging strand after treatment with DNA-damaging agents can be explained by the discontinuous nature of lagging strand synthesis. In this scenario, synthesis of a new Okazaki fragment ensures replication fork restart and continued replication fork progression, despite blockage of the previous fragment by a DNA lesion. Pioneering studies in bacteria showed that the DnaG primase ensures replication fork restart downstream of a UV lesion in both the lagging and the leading strands (Heller and Marians, 2006) (Figure 2). These findings suggested that the replisome is able to reinitiate DNA synthesis downstream of leading strand lesions and that the repriming activity of DnaG leads to the formation of a ssDNA gap between the lesion and the point where synthesis restarts. These early findings raised several new questions for the field: Is this repriming mechanism conserved in eukaryotes? Is there a human homolog of bacterial DnaG?
Figure 2. Mechanisms of repriming and ssDNA gap formation in different organisms.
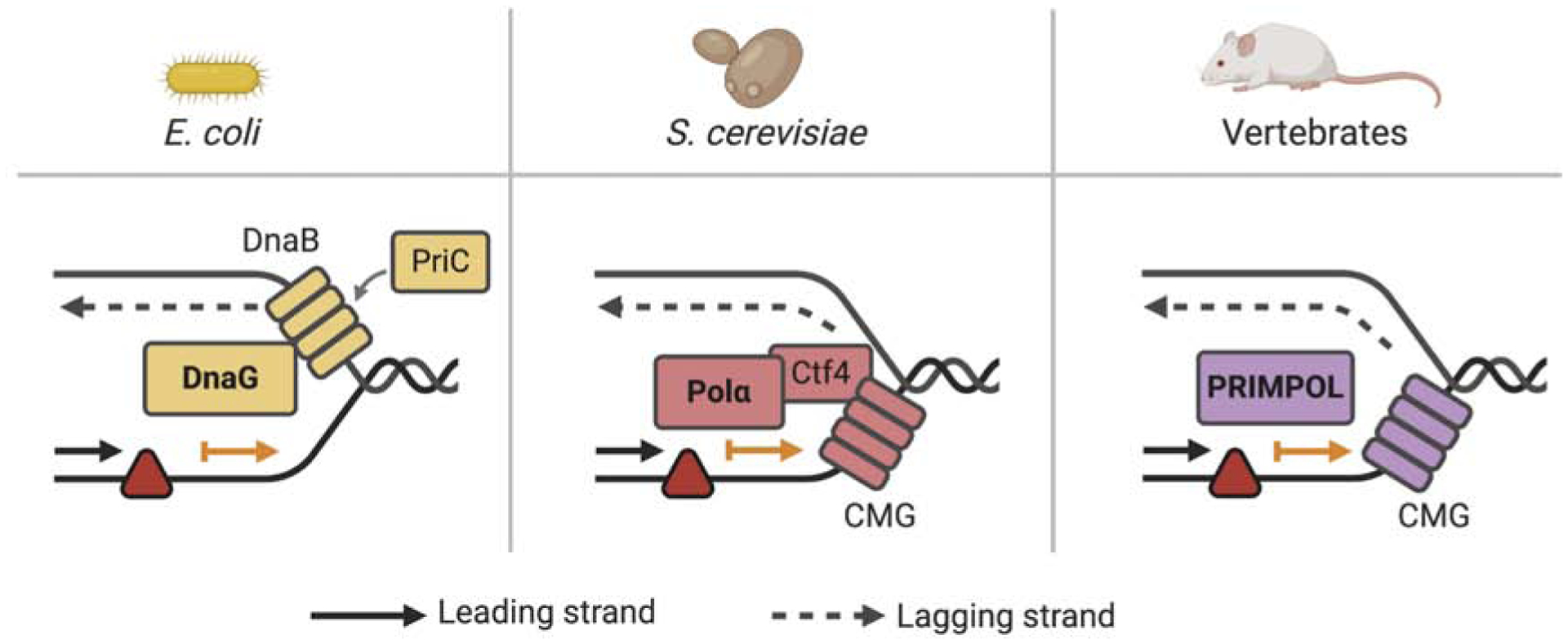
In E. coli, the DnaG primase, as part of the PriC system, interacts with DnaB and promotes repriming in both the leading and lagging strand (left). In budding yeast, repriming is promoted by the Polα/Primase complex and Ctf4, a replisome factor that bridges the MCM component of the CMG helicase and the Polα/Primase complex (middle). In vertebrates, repriming is ensured by PRIMPOL. How recoupling of leading strand synthesis and the CMG helicase occurs after PRIMPOL-mediated repriming in vivo is unknown.
Following these early observations, several reports indicated that Saccharomyces cerevisae uses replication fork repriming to deal with different DNA-blocking lesions (Daigaku et al., 2010; Fumasoni et al., 2015; Karras and Jentsch, 2010; Wong et al., 2020). Repriming in budding yeast is ensured by Polymerase α (Polα)/Primase complex and Ctf4, a replisome factor that bridges the MCM (replicative minichromosome maintenance) helicase and the Polα/Primase complex (Fumasoni et al., 2015) (Figure 2).
The human Primase and DNA-directed Polymerase (PRIMPOL) is a recently discovered enzyme that possesses both primase and polymerase activities. PRIMPOL is a member of the archae-eukaryotic primase (AEP) superfamily (Iyer et al., 2005) and is emerging as a key player in replication stress response in mammalian cells. PRIMPOL synthesizes DNA with limited processivity, rarely incorporating more than four nucleotides on an undamaged template (Keen et al., 2014b). However, all the studies on PRIMPOL polymerase activity have been performed using synthetic DNA substrates and purified recombinant protein, raising the question of whether PRIMPOL polymerase activity has a physiologically relevant function in vivo (Bianchi et al., 2013; Garcia-Gomez et al., 2013; Mouron et al., 2013). While the role of PRIMPOL polymerase activity is still unclear, its primase activity is essential for many of the biologically relevant functions of PRIMPOL in the nucleus (Calvo et al., 2019; González-Acosta et al., 2020; Keen et al., 2014a; Kobayashi et al., 2016; Piberger et al., 2020; Quinet et al., 2020; Schiavone et al., 2016; Svikovic et al., 2019). The functional characterization of its unique primase activity provided the first clues for how repriming and ssDNA gap formation are regulated in mammalian cells (Bianchi et al., 2013; Garcia-Gomez et al., 2013; Mouron et al., 2013; Wan et al., 2013). Following these discoveries, the mechanisms that dictate the choice between the repriming, TLS, and TS pathways became the subject of intensive investigation.
CHOICE BETWEEN REPRIMING, TLS, and TS
Nature of DNA damage and extent of fork stalling.
Some agents challenge DNA replication without inducing DNA damage, such as hydroxyurea (HU) or aphidicolin, whereas others perturb fork progression by introducing a lesion in one or both DNA strands. The nature of the replication challenge is a key determinant of pathway choice (Figure 3). For example, PRIMPOL repriming is favored over TLS when the lesion present on the replication fork is too bulky to be bypassed by canonical TLS polymerases. Exposure to UV-C generates two types of pyrimidine dimers: cyclobutane pyrimidine dimer (CPD) and pyrimidine (6–4) pyrimidone (6–4PP) (Pfeifer et al., 2005). 6–4PPs cause a more pronounced distortion of the DNA double helix compared to CPDs. While CPDs are efficiently bypassed by the TLS polymerase POL η at the replication fork, formation of 6–4PPs leads to ssDNA gap accumulation behind replication forks in DNA repair-deficient mouse embryonic and human fibroblasts, suggesting that tolerance to UV-induced 6–4PPs involves replication fork repriming (Jansen et al., 2009; Quinet et al., 2018).
Figure 3. Factors influencing the choice between the TLS, fork reversal and repriming.
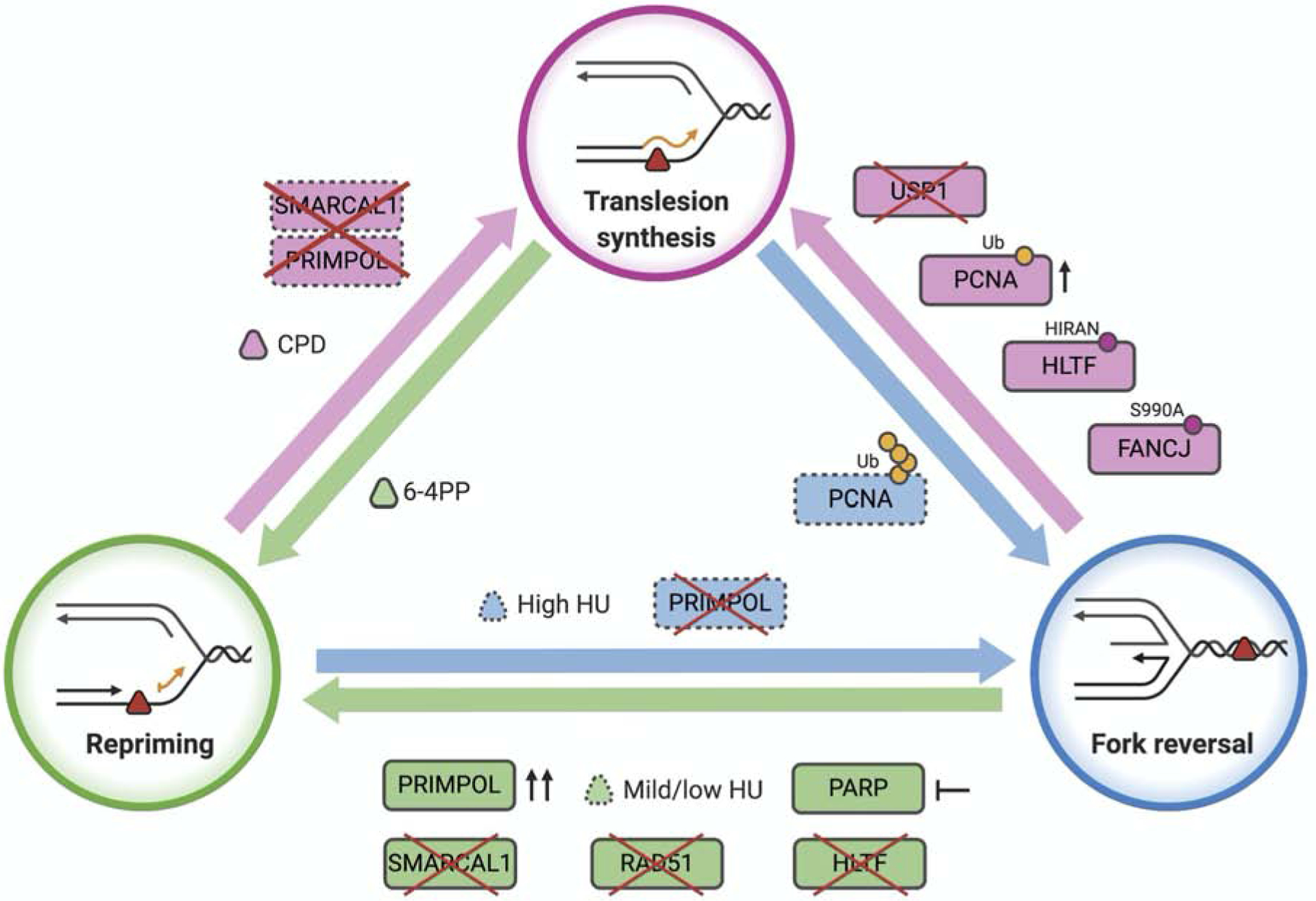
Dashed lines indicate factors that need more investigation. See text for details.
In agreement with the proposed role of repriming in the bypass of bulky 6–4PP, PRIMPOL binding to chromatin increases after treatment with UV-C, and PRIMPOL depletion, or loss of its primase activity, impairs replication fork restart upon UV-C irradiation (Mouron et al., 2013). Moreover, Primpol−/− DT-40 cells complemented with a primase-dead version of the protein are hypersensitive to UV radiation, as well as to treatment with methyl methanesulfonate (MMS) and cisplatin, broadening the spectrum of DNA lesions that can be ”skipped” by PRIMPOL (Kobayashi et al., 2016). Along the same lines, PRIMPOL-mediated repriming is required to rescue replication forks that have been stalled by cisplatin treatment in human cells (Quinet et al., 2020). Although cisplatin mainly generates intra-strand adducts (Poklar et al., 1996), approximately 5% of lesions are inter-strand crosslinks (ICLs) (Deans and West, 2011), which are generally considered an absolute block for replication fork progression. However, ICLs can be “traversed” in a reaction mediated by the FANCM/MHF DNA translocase (Huang et al., 2013). This mechanism relies on the primase activity of PRIMPOL, which leaves the ICL in the ssDNA gap behind the fork to be repaired post-replicatively (González-Acosta et al., 2020). Interestingly, PRIMPOL also plays a role in DNA replication stress response to HU treatment in human cells (Bai et al., 2020; Kobayashi et al., 2016; Mouron et al., 2013; Quinet et al., 2020). Moreover, HU treatment leads to ssDNA gap accumulation in budding yeast, suggesting that repriming is a general mechanism to deal with replication stress even when replication forks do not face DNA lesions (Gallo et al., 2019). These studies suggest that the reduction in the available nucleotide pool caused by HU treatment does not affect the primase activity of PRIMPOL and its ability to rescue stalled replication forks, at least at the HU concentrations used in these experiments. Importantly, PRIMPOL knock-out avian cells are not sensitive to treatment with camptothecin (CPT), γ-rays, or x-rays, suggesting that PRIMPOL is not involved in the repair of DNA breaks (Bianchi et al., 2013; Kobayashi et al., 2016).
Previous findings showed that reversed replication forks accumulate in human cells treated with a variety of genotoxic agents including UV-C, MMS, mitomycin C (MMC), cisplatin, CPT, hydrogen peroxide, and HU (Zellweger et al., 2015). This work raises the question of the frequency of fork reversal events relative to repriming or other replication stress response pathways. Unfortunately, a straightforward answer to this question is complicated by the limitations in the approach used to study this process. Electron microscopy, used to detect reversed forks, only takes a snapshot of this reaction by “freezing” the replication intermediates with the cross-linking step (Vindigni and Lopes, 2017), likely leading to an underestimation of the actual number of replication intermediates that have undergone fork reversal upon drug treatment.
In addition to the type of replication challenge, the extent of fork stalling caused by different concentrations of the same genotoxic agent might affect the equilibrium between different replication stress response pathways. For example, prolonged treatment of BRCA1- or BRCA2-deficient cells with HU concentrations ranging between 2 and 5 mM for 2 to 5 hours leads to reversed fork degradation (Kolinjivadi et al., 2017; Lemacon et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017). This is consistent with the emerging role of BRCA proteins in reversed fork protection. However, treatment of BRCA-deficient cells with lower HU concentrations no longer promotes fork degradation, but leads to the accumulation of ssDNA gaps (Lim et al., 2018; Panzarino et al., 2019). In particular, treatment of BRCA1-null cells with 1 mM HU for 3 hours induces fork degradation only upon depletion of the Ubiquitin Specific Peptidase 1 (USP1), suggesting that USP1 protects forks from degradation at this lower HU concentration (Lim et al., 2018). Moreover, USP1 is not needed for fork protection when BRCA1-deficient cells are treated with an even lower HU concentration (0.5 mM for 2 hours), which instead promotes the accumulation of ssDNA gaps. Along the same lines, Panzarino et al. showed that treatment of BRCA1/2-deficient cells with the same HU concentration (0.5 mM for 2 hours) leads to unrestricted replication fork progression and accumulation of ssDNA gaps on the replicating DNA, without causing fork degradation (Panzarino et al., 2019). These studies show that pathway choice is dictated not only by the type of replication challenge, but also by the concentration of replication inhibitor. In particular, they suggest that repriming is favored over reversal when BRCA-deficient cells are treated with mild HU concentrations. However, future experiments are necessary to determine whether the ssDNA gaps that accumulate in BRCA-deficient cells treated with mild HU concentrations are indeed a consequence of PRIMPOL repriming.
Why would BRCA-deficient cells favor reversal over repriming at higher HU concentrations? First, increasing the HU concentration might cause a drop in the dNTP pool concentration below a threshold needed for efficient PRIMPOL repriming. Second, higher HU doses would increase the extent of fork uncoupling, possibly leading to more RPA bound to ssDNA. On the basis of previous findings that high concentrations of RPA inhibit PRIMPOL recruitment to DNA in vitro (Guilliam et al., 2017; Martinez-Jimenez et al., 2017), we speculate that higher levels of RPA bound to ssDNA might limit replication fork repriming and favor alternative replication stress response pathways, such as fork reversal. Third, increased RPA binding would lead to hyper-activation of the ATR pathway, and ATR activity was recently shown to promote global fork reversal in BRCA-proficient cells in response to ICLs (Mutreja et al., 2018). However, the role of ATR in fork reversal is still controversial because ATR signaling is not necessarily detected in response to all genotoxic agents that induce fork reversal (Zellweger et al., 2015). Moreover, previous findings suggested that ATR activity counteracts, rather than promotes, fork reversal by restraining SMARCAL1 function in reversed fork formation in HU-treated cells (Couch et al., 2013). A possible reason for these contradictory results could be related to the different types of replication challenges used in these studies. A more systematic analysis using different types and concentrations of replication inhibitors would be important to properly address how the extent of replication stress or DNA damage load dictates the choice between replication fork reversal and repriming, as well as the role of ATR in this process. Moreover, whether the function of ATR signaling in fork reversal changes in BRCA-proficient versus -deficient cells remains unknown. As several of these inhibitors are used for the treatment of BRCA-proficient and -deficient tumors, these studies are also crucial to understand the impact of clinically relevant doses of replication inhibitors on pathway choice.
Finally, it is worth mentioning that prolonged fork stalling or replication fork de-protection caused by the loss of BRCA proteins can lead to fork breakage and formation of one-ended double-stranded breaks (DSBs), which require specialized break-induced replication (BIR) pathways to be repaired (Scully et al., 2019). Specific HR factors such as RAD51 are required for reversed fork formation and protection (Berti et al., 2020), but their requirement for BIR is still debated and may reflect mechanistic differences between yeast and human cells (Kramara et al., 2018). Of note, RAD51 function in fork reversal is different from its potential function in BIR because it does not require its strand exchange activity. Moreover, the formation of reversed forks does not require stable RAD51 filaments, which are instead required to protect the already formed reversed forks from nucleolytic processing. As for fork reversal and repriming, the concentrations and timing of drug treatment that promote fork breakage and activate a BIR pathway likely vary as a function of the specific genetic background and type of replication challenge.
Changes in the genetic background.
Recent studies in budding yeast showed that repriming restrains extensive replication fork uncoupling and reversal and that aberrant reversed replication forks accumulate in repriming-deficient Polα/Primase/Ctf4 yeast mutants (Fumasoni et al., 2015). Differently from budding yeast, replication fork reversal is a frequent and physiologically important mechanism used by mammalian cells to cope with replication challenges (Zellweger et al., 2015). This notion is supported by the discovery that different members of the SWI/SNF translocase family, including SMARCAL1, ZRANB3, and HTLF, are capable of converting a three-way junction DNA replication fork into a four-way junction reversed fork in mammalian cells (Bai et al., 2020; Betous et al., 2012; Kile et al., 2015; Vujanovic et al., 2017). Suppression of fork reversal by depletion of SMARCAL1 leads to accumulation of ssDNA gaps in human cells treated with cisplatin, as detected by DNA fiber assay using the ssDNA-specific S1 nuclease (Quinet et al., 2020). In this context, PRIMPOL depletion renders replication tracts insensitive to S1 nuclease cleavage, indicating that the ssDNA gaps formed upon SMARCAL1 loss are PRIMPOL-dependent (Quinet et al., 2020). Along the same line, loss of HLTF promotes daughter-strand ssDNA gap accumulation in human cells treated with low doses of HU (50 or 500 μM) relative to HLTF-proficient cells (Bai et al., 2020; Peng et al., 2018). In addition, depletion of PRIMPOL in HLTF KO cells prevents ssDNA gap accumulation following HU treatment indicating that PRIMPOL repriming is responsible for the observed phenotype (Bai et al., 2020). These data suggest that PRIMPOL-mediated repriming is activated in both SMARCAL1- and HLTF-deficient cells treated with agents that challenge DNA replication by drastically different means (Figure 3). Although both fork remodelers are implicated in fork reversal and their loss promotes repriming, there is also a notable difference because loss of HLTF, but not SMARCAL1, leads to unrestrained fork progression, in addition to promoting ssDNA gap accumulation (Bai et al., 2020; Peng et al., 2018; Quinet et al., 2020). This difference might be related to the distinct genotoxic agents used to challenge DNA replication or to distinct, and yet to be defined, roles of HLTF and SMARCAL1 in replication fork remodeling.
Interestingly, the BRCA1-associated helicase FANCJ (BACH1/BRIP1) is required for unrestrained replication fork progression in HLTF-deficient cells treated with mild doses of HU (Peng et al., 2018). FANCJ is a hereditary breast/ovarian cancer and Fanconi anemia gene functioning in homologous recombination (HR) and replication fork protection (Levitus et al., 2005; Litman et al., 2005). In addition, FANCJ travels with the elongating forks to counteract replication perturbations (Alabert et al., 2014; Sirbu et al., 2011). Combined, the recent findings on the roles of FANCJ (Peng et al., 2018) and PRIMPOL (Bai et al., 2020) in HLTF-deficient cells point to a potential link between FANCJ activity and PRIMPOL repriming in promoting unrestrained fork progression and ssDNA gap formation in this genetic background.
In addition to suppressing fork reversal, loss of another fork remodeler, ZRANB3, leads to unrestrained fork progression upon treatment with different genotoxic agents including CPT, MMC, and UV-C, suggesting that the observed unrestrained fork progression phenotype is independent of the particular kind of replication challenge, at least in the case of ZRANB3-depleted cells (Vujanovic et al., 2017). However, these studies did not determine whether the unrestrained fork progression phenotype of ZRANB3-depleted cells is associated with PRIMPOL-dependent ssDNA gap accumulation, as observed in HTLF-depleted cells.
The notion that suppression of replication fork reversal favors PRIMPOL-dependent repriming is strengthened by the observation that depletion of the central recombinase RAD51, which is required for replication fork reversal (Zellweger et al., 2015), promotes PRIMPOL repriming following UV radiation (Vallerga et al., 2015) or cisplatin treatment (Quinet et al., 2020). Similarly, loss of RAD51 generates daughter-strand ssDNA gaps in Xenopus laevis extracts treated with MMS (Hashimoto et al., 2010). Moreover, preventing reversed fork accumulation by inhibiting PARP activity also leads to PRIMPOL-dependent ssDNA gaps, further supporting the model that preventing replication fork reversal favors repriming and consequent ssDNA gap formation (Quinet et al., 2020). Furthermore, increasing PRIMPOL expression is sufficient to promote replication fork repriming and ssDNA gap formation (Quinet et al., 2020) (Figure 3).
Interestingly, replication forks challenged with cisplatin are still able to progress upon combined loss of PRIMPOL and SMARCAL1, RAD51, or PARP1, at least in the one-hour time window monitored by DNA fiber analysis (Quinet et al., 2020). On the basis of these results, it is tempting to speculate that cells might use an alternative TLS pathway when repriming and reversal are both suppressed, thereby ensuring replication fork progression and damage bypass. However, this does not seem to be the case in HTLF-depleted cells where the suppression of PRIMPOL leads to significantly shorter tracts (Bai et al., 2020), reinforcing the notion that there are important differences between HLTF and SMARCAL1. Interestingly, the HIRAN domain of HLTF is required to restrain fork progression (Kile et al., 2015). Recently, Bai et al. found that cells expressing an HLTF-HIRAN mutant behave differently from HLTF-deficient cells because the unrestrained replication phenotype of the HLTF-HIRAN mutant is not linked to PRIMPOL-dependent ssDNA gap accumulation, but rather relies on TLS activity of the REV1 polymerase (Bai et al., 2020). Similarly, cells expressing FANCJS990A, a FANCJ mutant that promotes POL η-dependent TLS (Xie et al., 2010), display unrestrained replication fork progression upon HU treatment but no accumulation of ssDNA gaps behind the forks (Nayak et al., 2020). Moreover, replication fork reversal was not observed in cells expressing FANCJS990A, suggesting that increased TLS activity restricts fork reversal (Nayak et al., 2020). Altogether these findings point to a finely tuned regulation between repriming, fork reversal, and TLS pathways, which can be affected by changes in the genetic background.
PCNA post-translational modifications.
PCNA monoubiquitination and polyubiquitination are crucial players in the choice between different DDT pathways (Figure 3) (Kanao and Masutani, 2017; Ulrich, 2009). PCNA monoubiquitination facilitates recruitment of specific TLS polymerases through a polymerase switching mechanism (Kannouche et al., 2004), whereas PCNA polyubiquitination has been associated with TS mechanisms, including fork reversal (Hoege et al., 2002; Vujanovic et al., 2017; Xiao et al., 2000). In particular, the ZRANB3 translocase interacts with polyubiquitinated PCNA to promote fork reversal (Ciccia et al., 2012; Vujanovic et al., 2017). More recently, the E3 ubiquitin ligase HLTF has also been shown to be required for replication fork reversal in vivo (Bai et al., 2020; Kile et al., 2015). However, the contribution of HLTF-dependent PCNA polyubiqutination in fork reversal remains unclear, because HLTF fork reversal activity is strictly dependent on its motor ATPase activity and HIRAN domain (Bai et al., 2020; Kile et al., 2015). In this regard, Kile et al. suggested that HLTF-dependent PCNA polyubiquitination might be required to promote efficient recruitment of ZRANB3, which in turn associates with polyubiquitinated PCNA to promote fork reversal (Kile et al., 2015). This model would argue that HLTF acts upstream of ZRANB3 in a common fork reversal pathway mediated by PCNA polyubiquitination.
As already discussed, loss of either ZRANB3 or HLTF leads to unrestrained replication fork progression, and the longer replication tracts of HLTF-deficient cells are characterized by the presence of PRIMPOL-dependent ssDNA gaps behind the advancing replication forks. However, these studies did not establish whether there is a direct link between reduced PCNA polyubiquitination and increased PRIMPOL-dependent repriming because at least two E3 ubiquitin ligases, HLTF and SHPRH, contribute to PCNA polyubiquitination, and polyubiquitination is still observed upon the loss of both proteins (Krijger et al., 2011). Moreover, the connection between PCNA ubiquitination and fork reversal is further complicated by recent studies suggesting that fork reversal still occurs in the PCNA-K164R ubiquitination-defective cells (Thakar et al., 2020). These studies argue that PCNA ubiquitination is required for Okazaki fragment maturation and that reversed forks that accumulate in PCNA-K164R cells contain abnormally long Okazaki fragments.
Loss of USP1, which is required to remove ubiquitin from monoubiquitinated PCNA (Huang et al., 2006) leads to increased levels of monoubiquitinated PCNA and accumulation of ssDNA gaps in BRCA1-deficient cells treated with low HU doses (Lim et al., 2018). How increased levels of monoubiquitinated PCNA lead to ssDNA gap accumulation upon the combined loss of BRCA1 and USP1 remains unclear. Interestingly, depletion of the TLS polymerases POL κ and REV1 suppresses the ssDNA gaps accumulation phenotype observed upon USP1 loss, suggesting that the formation of these gaps is somehow dependent on these TLS enzymes (Lim et al., 2018). An important question for future studies is whether these ssDNA gaps are PRIMPOL-dependent and how POL κ and REV1 are involved in this process. Human FANCD2 and RAD51 also support PCNA monoubiquitination and TLS (Chen et al., 2017). Another key objective for future research will be to investigate whether the proposed roles of these Fanconi anemia and HR proteins in regulating PCNA monoubiquitination affect other replication stress response pathways and whether other signaling events, in addition to the changes in the PCNA ubiquitination status, affect the equilibrium between repriming, TLS, and TS.
ssDNA GAP-FILLING
The ssDNA gaps left behind advancing replication forks as a consequence of repriming need to be properly filled by gap-filling or post-replicative repair mechanisms. As for DDT at stalled replication forks, TLS or TS are the two universal strategies to tolerate DNA lesions opposite to ssDNA gaps and thereby fill in these gaps. The concept of post-replicative repair was proposed by Lehmann and Kirk-Bell studying the effect of UV irradiation in mouse cells (Lehmann and Kirk-Bell, 1972) and by the Prakash group studying post-replicative repair in budding yeast (Prakash, 1981). TS appears to be the main pathway of gap-filling in E. coli (Berdichevsky et al., 2002; Laureti et al., 2015). Early work suggested that TLS predominantly mediates gap filling in budding yeast (Daigaku et al., 2010; Gallo et al., 2019; Karras and Jentsch, 2010). However, this model was challenged by later studies showing that the ssDNA gaps generated upon repriming by the Polα-primase complex are filled by TS or by an alternative HR salvage pathway in S. cerevisiae (Fumasoni et al., 2015; Gonzalez-Huici et al., 2014; Karras et al., 2013). This apparent discrepancy might be due to the difference in the concentration and type of DNA-damaging agent used to promote gap formation (Wong et al., 2020).
Several studies suggest that TLS is the main pathway of ssDNA gap-filling in mammalian cells. The contribution of selected TLS polymerases to this process is still debated. For example, the TLS polymerases REV1 and POL ζ are essential for gap-filling in mammalian cells exposed to UV radiation (Diamant et al., 2012; Jansen et al., 2009). However, the notion that REV1 is required for gap filling in higher eukaryotes is not supported in other studies (Edmunds et al., 2008; Quinet et al., 2016). Along the same lines, Elvers et al. suggested that POL η plays a role in ssDNA gap repair (Elvers et al., 2011), whereas other studies suggested that POL η is primarily involved in TLS at the replication fork (Despras et al., 2010; Quinet et al., 2014; Vallerga et al., 2015). The interpretation of these results may be complicated by the redundancy that exists among TLS polymerases, combined with the fact that most TLS polymerases can act both at the stalled replication forks and at ssDNA gaps.
Defining the contribution of TS in gap-filling in mammalian cells is more challenging due to lack of direct methodologies to investigate homology-mediated mechanisms that do not involve strand transfer. Adar et al. used a gapped plasmid repair assay to directly assess gap-filling in mouse and human cells (Adar et al., 2009). They found that, in addition to TLS, an alternative HR pathway efficiently fills in gaps opposite of a synthetic abasic site or bulky adducts formed upon treatment with benzo[a]pyrene-diol-epoxide (BPDE). Interestingly, RAD51 and NBS1 proteins are required for HR-dependent gap-filling (Adar et al., 2009). These findings agree with a recent report describing a RAD51-dependent HR pathway to repair PRIMPOL-dependent ssDNA gaps opposite to BPDE-induced adducts in human cells (Piberger et al., 2020).
The relative gap filling contribution of TLS versus TS and HR in the human genome remains largely unknown. As for DDT at stalled replication forks, the nature of the DNA damage likely plays an important role in the choice between different gap-filling pathways. The results discussed above suggest that TLS is required to fill ssDNA gaps induced by UV radiation, whereas RAD51-dependent HR mediates post-replicative repair of bulky BPDE-induced adducts (Piberger et al., 2020). Moreover, there is evidence of cross-talk between factors involved in TLS and TS/HR. For example, the TLS polymerases REV1 and POL η were shown to play a role in HR-mediated DSB repair (McIlwraith et al., 2005; Sharma et al., 2012). Conversely, depletion of RAD51 decreases efficiency of both HR and TLS in repairing ssDNA gaps in the plasmid assay (Adar et al., 2009). Along the same line, BRCA1 was proposed to modulate TLS, although whether BRCA1 promotes or inhibits TLS remains unclear (Pathania et al., 2011; Tian et al., 2013).
Two elegant studies showed that PCNA ubiquitination is required for post-replicative ssDNA gap repair in budding yeast (Daigaku et al., 2010; Karras and Jentsch, 2010). However, the role of PCNA ubiquitination in gap-filling in higher eukaryotes remains controversial (Edmunds et al., 2008; Temviriyanukul et al., 2012). Avian DT40 cells expressing the ubiquitination-deficient K164R-PCNA mutant are impaired in gap-filling in response to UV-C irradiation (Edmunds et al., 2008). Conversely, post-replicative repair of ssDNA gaps induced by UV-C is unaffected in mouse embryonic fibroblasts harboring the same K164R-PCNA mutation (Temviriyanukul et al., 2012). Interestingly, avian cells lack any ortholog of HLTF, and Rad5, the HLTF homolog in yeast, promotes ssDNA gap repair by recruiting TLS polymerases to ssDNA gaps (Gallo et al., 2019). These studies open the tantalizing scenario that HLTF might be required for gap-filling in mammalian cells and could explain the discrepancy between the results obtained in chicken and mouse cells.
The cell cycle phase could also influence the choice between different gap-filling pathways. Recent studies proposed that gap-filling by TS is facilitated by PCNA polyubiquitination during the S phase in budding yeast (Branzei and Szakal, 2016; Gonzalez-Huici et al., 2014; Karras et al., 2013). Conversely, TLS at ssDNA gaps is promoted by PCNA monoubiquitination, while counteracted by PCNA polyubiquitination, and may occur preferentially in G2. Branzei and Szakal also proposed that TS error-free pathways act first during the S-phase, whereas the more error-prone mechanisms, such as TLS and salvage HR, preferentially act during G2/M, if the gaps cannot be properly filled during the S-phase (Branzei and Szakal, 2016). Supporting this model, different studies suggested that TLS-dependent gap-filling takes place during the late S and G2 phases in the human genome (Diamant et al., 2012; Elvers et al., 2011; Quinet et al., 2016; Temviriyanukul et al., 2012). Moreover, Adar et al. suggested that HR, facilitated by PCNA SUMOylation and independent of PCNA ubiquitination, preferentially occurs in the G2 phase in mammalian cells (Adar et al., 2009; Branzei and Szakal, 2016). Of note, recombination in G2 would not necessarily be restricted to the sister chromatid, potentially leading to strand transfer and genomic rearrangements. An important topic for future research would be to determine the exact relationship between the TLS, TS, and HR mechanisms of gap filling as a function of the different cell cycle phase.
BIOLOGICAL CONSEQUENCES OF ALTERING THE BALANCE BETWEEN REPRIMING, TLS, AND TS
What are the consequences of employing repriming versus TLS or TS on genome integrity and cell survival? PRIMPOL-knockout mice or human cell lines are viable, suggesting that PRIMPOL is not essential for cell survival (Bailey et al., 2019; Bianchi et al., 2013; Mouron et al., 2013; Quinet et al., 2020). However, human and avian DT40 cells lacking PRIMPOL display increased cellular sensitivity to different DNA-damaging agents, including UV-C, cisplatin, HU, 4NQO, BPDE, MMS, chain-terminating nucleoside analogs, MMC, and trimethyl psoralen activated with UV-A (TMP-UVA) (Bai et al., 2020; Bianchi et al., 2013; Keen et al., 2014a; Kobayashi et al., 2016; Olivieri et al., 2020). Similarly to human cells, PRIMPOL-KO mice are also hypersensitive to MMC and display a significant impairment of bone marrow cell proliferation when treated with this drug (González-Acosta et al., 2020). These studies suggest that PRIMPOL mediates the cellular response to an array of DNA-damaging agents that perturb replication fork progression. Suppprting this notion, loss of PRIMPOL causes defects in replication fork progression and restart, increased sister chromatid exchanges, increased mutagenesis, and micronuclei formation following UV-C radiation and treatment with ICL-inducing agents (Bailey et al., 2019; González-Acosta et al., 2020; Mouron et al., 2013). However, TLS pathways can partially mitigate the replication defects associated with PRIMPOL loss. For example, loss of PRIMPOL significantly affects proliferation and viability in avian DT40 cells only when PRIMPOL is depleted in combination with the TLS polymerases POL η and POL ζ, suggesting that these two TLS polymerases partially compensate for PRIMPOL loss (Kobayashi et al., 2016). Along the same lines, loss of PRIMPOL does not seem to sensitize human cells to UV-C, unless POL η is co-depleted (Bailey et al., 2019), supporting the notion that POL η and PRIMPOL might have complementary roles in response to UV damage. Interestingly, POL κ and POL ζ appear to slow replication forks during the S-phase (Jones et al., 2012; Mehta et al., 2020), suggesting that they might engage on replication forks to prevent faster pathways such as PRIMPOL-mediated repriming from acting in the first place.
Similar to repriming, fork reversal is a physiologically important mechanism to deal with different kinds of replication challenges (Zellweger et al., 2015). However, there are instances when replication fork reversal is deleterious for genome integrity. For example, reversed forks are extensively degraded by nucleases if they are not adequately protected by BRCA proteins (Kolinjivadi et al., 2017; Lemacon et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017). Treatment with multiple cisplatin doses suppresses replication fork reversal and promotes repriming by PRIMPOL in BRCA1-deficient cells (Quinet et al., 2020). These studies suggest that in the absence of BRCA proteins, cells adapt to treatment with multiple cisplatin doses by suppressing fork reversal and upregulating PRIMPOL-mediated repriming as an alternative strategy to cope with cisplatin-induced lesions and prevent pathological reversed fork degradation. Of note, the ATR pathway is a key regulator of the PRIMPOL-dependent adaptive response to cisplatin treatment (Quinet et al., 2020). Moreover, PRIMPOL overexpression decreases BRCA1-deficient cell sensitivity to co-treatments with cisplatin and ATR inhibitors (Quinet et al., 2020), suggesting that the PRIMPOL pathway regulates chemotherapy response to combinatorial treatments with ATR inhibitors, which are currently in clinical trials (NCI-2016–00355).
As already discussed, PRIMPOL-mediated repriming can be more generally activated also in BRCA-proficient cells under conditions of impaired fork reversal, such as loss of the SMARCAL1 or HLTF translocases (Bai et al., 2020; Quinet et al., 2020). Although loss of either SMARCAL1 or HTLF promotes repriming, there are some notable difference among the cellular phenotypes of SMARCAL1 and HLTF-depleted cells. Loss of HLTF increases resistance to HU and MMC (Bai et al., 2020), whereas loss of SMARCAL1 increases cellular sensitivity to HU, CPT, and the DNA polymerase inhibitor aphidicolin (Bansbach et al., 2009). Moreover, loss of HLTF but not SMARCAL1 leads to unrestrained fork progression in a PRIMPOL-dependent manner, as already discussed. The different effects observed upon HLTF and SMARCAL1 loss once again point to important differences between these fork reversal factors, which deserve further investigation.
As fork reversal becomes pathological when reversed forks cannot be adequately protected, PRIMPOL repriming likely leads to increased genomic instability when the ssDNA gaps that form as a consequence of PRIMPOL activity cannot be properly repaired. Indeed, loss of factors involved in gap-filling, such as TLS POL ζ or REV1, significantly increases genomic instability and cell sensitivity to UV-C irradiation (Quinet et al., 2016; Temviriyanukul et al., 2012). When left unrepaired, ssDNA gaps can collapse into DSBs (Elvers et al., 2011; Quinet et al., 2016; Saxena et al., 2019). At least a fraction of these DSBs can be repaired by a HR-mediated mechanism dependent on RAD51, ATR, and the RAD51-paralog XRCC3 (Elvers et al., 2011; Saxena et al., 2019). In the absence of XRCC3, DSBs are not repaired, thereby compromising cell viability (Saxena et al., 2019). In addition, ssDNA gaps activate the ATR/CHK1 pathway in human cells (Jansen et al., 2009; Quinet et al., 2014), and ATR inhibition or depletion increases cell sensitivity to genotoxic agents under conditions of ssDNA gap accumulation (Quinet et al., 2014; Saxena et al., 2019). Moreover, under-replicated DNA that proceeds into mitosis can be converted to DNA lesions, presumably DSBs, and shielded by 53BP1 nuclear bodies in the following G1 phase (Harrigan et al., 2011; Lukas et al., 2011). We speculate that a similar mechanism applies to persistent unrepaired ssDNA gaps and that protection by 53BP1 nuclear bodies would provide a tolerance mechanism until the lesions are repaired.
The idea that ssDNA gaps become toxic intermediates if they are not repaired in a timely fashion is supported by recent studies showing that ssDNA gap accumulation correlates with chemotherapy response in BRCA-deficient tumors. In particular, Cong et al. found that PARP inhibition promotes accumulation of ssDNA gaps in BRCA-deficient cells and that ssDNA gaps are no longer present when the same cells acquire PARP inhibitor resistance (Cong et al., 2019). Along the same lines, Panzarino et al. found that loss of the chromatin remodeling enzyme CHD4 as well as its interaction partners PARP1, EZH2, FEN1, and ZFHX3 suppresses the accumulation of ssDNA gaps in BRCA2-deficient cells while conferring chemoresistance (Panzarino et al., 2019). Moreover, they found an interesting correlation between gap suppression and chemoresistance using BRCA1-null breast cancer patient-derived xenografts with differential sensitivities to cisplatin (Panzarino et al., 2019). The underlying mechanisms of defective gap-filling and the ensuing chemosensitivity in these genetic backgrounds are unknown. In addition, how exactly loss of CHD4 or its interacting partners leads to gap suppression and enhanced cell resistance in BRCA-null cells remains unclear.
The studies on the mechanisms that regulate the choice between repriming, TLS, and TS are central to understand how replicating cells mediate lesion tolerance while maintaining genome stability and also raise several outstanding questions to be addressed in the future: Are there other factors or signaling pathways that influence the balance between repriming, TLS, and TS mechanisms? What is the relative contribution of TLS versus TS and HR to gap filling in the human genome? How does activation of PRIMPOL and the ensuing accumulation of ssDNA gaps correlate with genome instability and cell viability in response to chemotherapeutics? Can we target factors involved in gap-filling to improve DNA-damaging chemotherapy response? For example, overexpression of the REV1 and POL ζ polymerases has been associated with chemotherapy resistance in a variety of cancers such as glioma, cervical cancer, and ovarian carcinoma (Rocha et al., 2018), paving the way to the proposal of inhibition of ssDNA gap filling by TLS as a novel strategy for targeted cancer therapy (Yamanaka et al., 2017). A key question for future studies is to identify other factors involved in gap-filling that could potentially be targeted in cancer therapy. Answering these pressing questions is mandatory to fully define the molecular contexts in which cells choose repriming over TS and TLS activity, as well as to develop efficient strategies to target these pathways in cancer therapy.
ACKNOWLEDGMENTS
We thank K. Cimprich and Y. Ayala for their careful reading of the manuscript and insightful comments. This work was supported by the NCI under grant numbers R01CA237263 (to A.V.) and F30CA254215 (to E.C.). Figures were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adar S, Izhar L, Hendel A, Geacintov N, and Livneh Z (2009). Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res 37, 5737–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, and Groth A (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol 16, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Kermi C, Stoy H, Schiltz CJ, Bacal J, Zaino AM, Hadden MK, Eichman BF, Lopes M, and Cimprich KA (2020). HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol Cell 78, 1237–1251 e1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey LJ, Bianchi J, and Doherty AJ (2019). PrimPol is required for the maintenance of efficient nuclear and mitochondrial DNA replication in human cells. Nucleic Acids Res 47, 4026–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansbach CE, Betous R, Lovejoy CA, Glick GG, and Cortez D (2009). The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev 23, 2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky A, Izhar L, and Livneh Z (2002). Error-free recombinational repair predominates over mutagenic translesion replication in E. coli. Mol Cell 10, 917–924. [DOI] [PubMed] [Google Scholar]
- Berti M, Cortez D, and Lopes M (2020). The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol 21, 633–651. [DOI] [PubMed] [Google Scholar]
- Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, and Cortez D (2012). SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev 26, 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V, Taylor E, Stevanovic I, Green AJ, Stracker TH, Lindsay HD, et al. (2013). PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell 52, 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, and Szakal B (2016). DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair (Amst) 44, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo PA, Sastre-Moreno G, Perpina C, Guerra S, Martinez-Jimenez MI, and Blanco L (2019). The invariant glutamate of human PrimPol DxE motif is critical for its Mn(2+)-dependent distinctive activities. DNA Repair (Amst) 77, 65–75. [DOI] [PubMed] [Google Scholar]
- Chen X, Bosques L, Sung P, and Kupfer GM (2017). A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene 36, 5220. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, et al. (2012). Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell 47, 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong K, Kousholt AN, Peng M, Panzarino NJ, Lee WTC, Nayak S, Krais J, Calvo J, Bere M, Rothenberg E, et al. (2019). PARPi synthetic lethality derives from replication-associated single-stranded DNA gaps. bioRxiv, 781989. [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Davies AA, and Ulrich HD (2010). Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 465, 951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans AJ, and West SC (2011). DNA interstrand crosslink repair and cancer. Nat Rev Cancer 11, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despras E, Daboussi F, Hyrien O, Marheineke K, and Kannouche PL (2010). ATR/Chk1 pathway is essential for resumption of DNA synthesis and cell survival in UV-irradiated XP variant cells. Hum Mol Genet 19, 1690–1701. [DOI] [PubMed] [Google Scholar]
- Diamant N, Hendel A, Vered I, Carell T, Reissner T, de Wind N, Geacinov N, and Livneh Z (2012). DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res 40, 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds CE, Simpson LJ, and Sale JE (2008). PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 30, 519–529. [DOI] [PubMed] [Google Scholar]
- Elvers I, Johansson F, Groth P, Erixon K, and Helleday T (2011). UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res 39, 7049–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumasoni M, Zwicky K, Vanoli F, Lopes M, and Branzei D (2015). Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polalpha/Primase/Ctf4 Complex. Mol Cell 57, 812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo D, Kim T, Szakal B, Saayman X, Narula A, Park Y, Branzei D, Zhang Z, and Brown GW (2019). Rad5 Recruits Error-Prone DNA Polymerases for Mutagenic Repair of ssDNA Gaps on Undamaged Templates. Mol Cell 73, 900–914 e909. [DOI] [PubMed] [Google Scholar]
- Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ, et al. (2013). PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Acosta D, Blanco-Romero E, Mutreja K, Llanos S, Míguez S, García F, Muñoz J, Blanco L, Lopes M, and Méndez J (2020). PrimPol primase mediates replication traverse of DNA interstrand crosslinks. bioRxiv, 2020.2005.2019.104729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Huici V, Szakal B, Urulangodi M, Psakhye I, Castellucci F, Menolfi D, Rajakumara E, Fumasoni M, Bermejo R, Jentsch S, et al. (2014). DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J 33, 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliam TA, Brissett NC, Ehlinger A, Keen BA, Kolesar P, Taylor EM, Bailey LJ, Lindsay HD, Chazin WJ, and Doherty AJ (2017). Molecular basis for PrimPol recruitment to replication forks by RPA. Nat Commun 8, 15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, and Jackson SP (2011). Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 193, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, and Costanzo V (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, and Marians KJ (2006). Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439, 557–562. [DOI] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, and Jentsch S (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141. [DOI] [PubMed] [Google Scholar]
- Howard-Flanders P, Rupp WD, Wilkins BM, and Cole RS (1968). DNA replication and recombination after UV irradiation. Cold Spring Harb Symp Quant Biol 33, 195–207. [DOI] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, and Seidman MM (2013). The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 52, 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, and D’Andrea AD (2006). Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol 8, 339–347. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Koonin EV, Leipe DD, and Aravind L (2005). Origin and evolution of the archaeo-eukaryotic primase superfamily and related palm-domain proteins: structural insights and new members. Nucleic Acids Res 33, 3875–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhar L, Ziv O, Cohen IS, Geacintov NE, and Livneh Z (2013). Genomic assay reveals tolerance of DNA damage by both translesion DNA synthesis and homology-dependent repair in mammalian cells. Proc Natl Acad Sci U S A 110, E1462–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Gali H, Hendel A, Johansson F, Erixon K, Livneh Z, Mullenders LH, Haracska L, et al. (2009). Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol Cell Biol 29, 3113–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MJ, Colnaghi L, and Huang TT (2012). Dysregulation of DNA polymerase kappa recruitment to replication forks results in genomic instability. EMBO J 31, 908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanao R, and Masutani C (2017). Regulation of DNA damage tolerance in mammalian cells by post-translational modifications of PCNA. Mutat Res 803–805, 82–88. [DOI] [PubMed] [Google Scholar]
- Kannouche PL, Wing J, and Lehmann AR (2004). Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14, 491–500. [DOI] [PubMed] [Google Scholar]
- Karras GI, Fumasoni M, Sienski G, Vanoli F, Branzei D, and Jentsch S (2013). Noncanonical role of the 9-1-1 clamp in the error-free DNA damage tolerance pathway. Mol Cell 49, 536–546. [DOI] [PubMed] [Google Scholar]
- Karras GI, and Jentsch S (2010). The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141, 255–267. [DOI] [PubMed] [Google Scholar]
- Keen BA, Bailey LJ, Jozwiakowski SK, and Doherty AJ (2014a). Human PrimPol mutation associated with high myopia has a DNA replication defect. Nucleic Acids Res 42, 12102–12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen BA, Jozwiakowski SK, Bailey LJ, Bianchi J, and Doherty AJ (2014b). Molecular dissection of the domain architecture and catalytic activities of human PrimPol. Nucleic Acids Res 42, 5830–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, Eichman BF, and Cimprich KA (2015). HLTF’s Ancient HIRAN Domain Binds 3’ DNA Ends to Drive Replication Fork Reversal. Mol Cell 58, 1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Guilliam TA, Tsuda M, Yamamoto J, Bailey LJ, Iwai S, Takeda S, Doherty AJ, and Hirota K (2016). Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle 15, 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881 e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramara J, Osia B, and Malkova A (2018). Break-Induced Replication: The Where, The Why, and The How. Trends Genet 34, 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PH, Lee KY, Wit N, van den Berk PC, Wu X, Roest HP, Maas A, Ding H, Hoeijmakers JH, Myung K, et al. (2011). HLTF and SHPRH are not essential for PCNA polyubiquitination, survival and somatic hypermutation: existence of an alternative E3 ligase. DNA Repair (Amst) 10, 438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laureti L, Demol J, Fuchs RP, and Pages V (2015). Bacterial Proliferation: Keep Dividing and Don’t Mind the Gap. PLoS Genet 11, e1005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann AR, and Kirk-Bell S (1972). Post-Replication Repair of DNA in Ultraviolet-Irradiated Mammalian Cells. European Journal of Biochemistry 31, 438–445. [DOI] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, et al. (2005). The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet 37, 934–935. [DOI] [PubMed] [Google Scholar]
- Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA, Ponnienselvan K, Liu JC, Yang C, Kozono D, et al. (2018). USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol Cell 72, 925–941 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, and Cantor SB (2005). BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8, 255–265. [DOI] [PubMed] [Google Scholar]
- Lopes M, Foiani M, and Sogo JM (2006). Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21, 15–27. [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. (2011). 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13, 243–253. [DOI] [PubMed] [Google Scholar]
- Martinez-Jimenez MI, Lahera A, and Blanco L (2017). Human PrimPol activity is enhanced by RPA. Sci Rep 7, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwraith MJ, Vaisman A, Liu Y, Fanning E, Woodgate R, and West SC (2005). Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell 20, 783–792. [DOI] [PubMed] [Google Scholar]
- Mehta KPM, Lovejoy CA, Zhao R, Heintzman DR, and Cortez D (2020). HMCES Maintains Replication Fork Progression and Prevents Double-Strand Breaks in Response to APOBEC Deamination and Abasic Site Formation. Cell Rep 31, 107705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneghini R (1976). Gaps in DNA synthesized by ultraviolet light-irradiated WI38 human cells. Biochim Biophys Acta 425, 419–427. [DOI] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, et al. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat Commun 8, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, Garcia-Gomez S, Chocron S, Blanco L, and Mendez J (2013). Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20, 1383–1389. [DOI] [PubMed] [Google Scholar]
- Mutreja K, Krietsch J, Hess J, Ursich S, Berti M, Roessler FK, Zellweger R, Patra M, Gasser G, and Lopes M (2018). ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep 24, 2629–2642 e2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Calvo JA, Cong K, Peng M, Berthiaume E, Jackson J, Zaino AM, Vindigni A, Hadden MK, and Cantor SB (2020). Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Science Advances 6, eaaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, and Lopes M (2015). Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16, 207–220. [DOI] [PubMed] [Google Scholar]
- Olivieri M, Cho T, Alvarez-Quilon A, Li K, Schellenberg MJ, Zimmermann M, Hustedt N, Rossi SE, Adam S, Melo H, et al. (2020). A Genetic Map of the Response to DNA Damage in Human Cells. Cell 182, 481–496 e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzarino NJ, Krais J, Peng M, Mosqueda M, Nayak S, Bond S, Calvo J, Cong K, Doshi M, Bere M, et al. (2019). Replication gaps underlie BRCA-deficiency and therapy response. bioRxiv, 781955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Nguyen J, Hill SJ, Scully R, Adelmant GO, Marto JA, Feunteun J, and Livingston DM (2011). BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol Cell 44, 235–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Cong K, Panzarino NJ, Nayak S, Calvo J, Deng B, Zhu LJ, Morocz M, Hegedus L, Haracska L, et al. (2018). Opposing Roles of FANCJ and HLTF Protect Forks and Restrain Replication during Stress. Cell Rep 24, 3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP, You YH, and Besaratinia A (2005). Mutations induced by ultraviolet light. Mutat Res 571, 19–31. [DOI] [PubMed] [Google Scholar]
- Piberger AL, Bowry A, Kelly RDW, Walker AK, González-Acosta D, Bailey LJ, Doherty AJ, Méndez J, Morris JR, Bryant HE, et al. (2020). PrimPol-dependent single-stranded gap formation mediates homologous recombination at bulky DNA adducts. Nature Communications 11, 5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poklar N, Pilch DS, Lippard SJ, Redding EA, Dunham SU, and Breslauer KJ (1996). Influence of cisplatin intrastrand crosslinking on the conformation, thermal stability, and energetics of a 20-mer DNA duplex. Proc Natl Acad Sci U S A 93, 7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash L (1981). Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Mol Gen Genet 184, 471–478. [DOI] [PubMed] [Google Scholar]
- Quinet A, Lerner LK, Martins DJ, and Menck CFM (2018). Filling gaps in translesion DNA synthesis in human cells. Mutat Res Genet Toxicol Environ Mutagen 836, 127–142. [DOI] [PubMed] [Google Scholar]
- Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, and Menck CF (2016). Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res 44, 5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Svikovic S, Lemacon D, Carvajal-Maldonado D, Gonzalez-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. (2020). PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol Cell 77, 461–474 e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Vessoni AT, Rocha CR, Gottifredi V, Biard D, Sarasin A, Menck CF, and Stary A (2014). Gap-filling and bypass at the replication fork are both active mechanisms for tolerance of low-dose ultraviolet-induced DNA damage in the human genome. DNA Repair (Amst) 14, 27–38. [DOI] [PubMed] [Google Scholar]
- Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, and Menck CFM (2018). DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics (Sao Paulo) 73, e478s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE (2013). Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb Perspect Biol 5, a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Dixit S, Somyajit K, and Nagaraju G (2019). ATR Signaling Uncouples the Role of RAD51 Paralogs in Homologous Recombination and Replication Stress Response. Cell Rep 29, 551–559 e554. [DOI] [PubMed] [Google Scholar]
- Schiavone D, Jozwiakowski SK, Romanello M, Guilbaud G, Guilliam TA, Bailey LJ, Sale JE, and Doherty AJ (2016). PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Mol Cell 61, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Panday A, Elango R, and Willis NA (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 20, 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Hicks JK, Chute CL, Brennan JR, Ahn JY, Glover TW, and Canman CE (2012). REV1 and polymerase zeta facilitate homologous recombination repair. Nucleic Acids Res 40, 682–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, and Cortez D (2011). Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25, 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svikovic S, Crisp A, Tan-Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, and Sale JE (2019). R-loop formation during S phase is restricted by PrimPol-mediated repriming. EMBO J 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 68, 414–430 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temviriyanukul P, van Hees-Stuivenberg S, Delbos F, Jacobs H, de Wind N, and Jansen JG (2012). Temporally distinct translesion synthesis pathways for ultraviolet light-induced photoproducts in the mammalian genome. DNA Repair (Amst) 11, 550–558. [DOI] [PubMed] [Google Scholar]
- Thakar T, Leung W, Nicolae CM, Clements KE, Shen B, Bielinsky AK, and Moldovan GL (2020). Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat Commun 11, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F, Sharma S, Zou J, Lin SY, Wang B, Rezvani K, Wang H, Parvin JD, Ludwig T, Canman CE, et al. (2013). BRCA1 promotes the ubiquitination of PCNA and recruitment of translesion polymerases in response to replication blockade. Proc Natl Acad Sci U S A 110, 13558–13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich HD (2009). Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair (Amst) 8, 461–469. [DOI] [PubMed] [Google Scholar]
- Vaisman A, and Woodgate R (2017). Translesion DNA polymerases in eukaryotes: what makes them tick? Crit Rev Biochem Mol Biol 52, 274–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallerga MB, Mansilla SF, Federico MB, Bertolin AP, and Gottifredi V (2015). Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc Natl Acad Sci U S A 112, E6624–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindigni A, and Lopes M (2017). Combining electron microscopy with single molecule DNA fiber approaches to study DNA replication dynamics. Biophys Chem 225, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, et al. (2017). Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell 67, 882–890 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Lou J, Xia Y, Su B, Liu T, Cui J, Sun Y, Lou H, and Huang J (2013). hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep 14, 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RP, Garcia-Rodriguez N, Zilio N, Hanulova M, and Ulrich HD (2020). Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol Cell 77, 3–16 e14. [DOI] [PubMed] [Google Scholar]
- Xiao W, Chow BL, Broomfield S, and Hanna M (2000). The Saccharomyces cerevisiae RAD6 group is composed of an error-prone and two error-free postreplication repair pathways. Genetics 155, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie K, Doles J, Hemann MT, and Walker GC (2010). Error-prone translesion synthesis mediates acquired chemoresistance. Proc Natl Acad Sci U S A 107, 20792–20797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chatterjee N, Hemann MT, and Walker GC (2017). Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet 13, e1006842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]