

Abstract
Objective:
To assess whether published scales for measuring upper motor neuron burden (UMNB) show longitudinal change in patients with primary lateral sclerosis (PLS).
Design:
Retrospective calculation of three UMNB scales on a prospectively collected dataset from 53 patients with PLS enrolled in a longitudinal natural history study with at least 2 evaluation visits. UMNB scales were calculated according to their published descriptions. Non-linear trends over time of UMNB scale scores and slopes were calculated for each patient and correlations between UMNB scores and clinical measures were assessed.
Results:
All three UMNB scales exhibited increasing scores over the first 7.8 years of symptoms, with a flattening in slope after approximately 8 years. A scale used in imaging studies (1) and the UPENN UMNS scale (2) provided a better fit to the dataset than the MGH UMNB scale (3). All three UMNB scales exhibited moderate correlations with some clinical measures of movement, such as finger-tapping rate and timed gait. Correlations were strongest for the UPENN UMNS, which was also moderately correlated with the revised ALS functional rating scale.
Conclusion:
In a cohort of PLS patients enrolled in a natural history study, the three UMNB scales exhibited modest linear increases over the first 8 years of symptoms, followed by a plateau. Future clinical trials in PLS should consider stratification of patients by disease duration. UMNB scales may be useful secondary outcomes, but more sensitive primary outcome measures are needed for clinical trials for PLS.
Keywords: Primary lateral sclerosis, outcome measures, natural history, upper motor neuron, spasticity
INTRODUCTION
Primary lateral sclerosis (PLS) is a clinical syndrome characterized by a slowly progressive upper motor syndrome beginning in adult life (4). PLS is classified as a disorder on the spectrum of motor neuron-frontotemporal disorders which is caused by the relatively selective degeneration of corticospinal (upper) motor neurons (UMNs) (5). Because there is no definitive diagnostic test for PLS and symptoms overlap with other disorders, the clinical diagnosis of PLS requires a search for other possible causes and sufficient delay to ensure that patients do not have an upper motor dominant presentation of amyotrophic lateral sclerosis (ALS). This period of diagnostic delay, typically lasting 4 years (6, 7), has limited enrollment of PLS patients in clinical trials at an early stage of their disease. Updated diagnostic criteria were recently proposed to facilitate research and clinical trials in PLS (8). The new criteria designate two levels of diagnostic certainty, with clinically probable PLS defined as the absence of significant lower motor neuron degeneration 2–4 years after symptom onset and clinically definite PLS defined as the absence of significant lower motor neuron degeneration 4 or more years after symptom onset.
Besides the delay in diagnosis, an additional impediment for clinical trials in PLS has been the paucity of valid and sensitive outcome measures of disease severity. A multisite study found that the ALS functional rating scale (ALSFRS-R (9)) was much less sensitive to change over a 3-year time period in established PLS patients than in ALS patients (10). This finding led to the development and validation of a PLS functional rating scale (PLSFRS), which was shown to be reliable and to detect changes with disease progression over 12 months (11). The PLSFRS assesses patients’ ability to carry out daily activities of life, such as communication, self-care, and mobility. Other scales of disease severity have been proposed that are based on clinical examination findings of UMN dysfunction. These upper motor neuron burden (UMNB) scales have been used in cross-sectional imaging studies of ALS and PLS to correlate findings with disease severity (1–3, 12). However, the ability of these UMNB scales to detect longitudinal change has not been assessed.
The primary goal of this study was to evaluate whether three UMNB scales detect change as disease progresses in patients with PLS. The three UMNB scales evaluated were 1) a scale termed here the UK UMN scale used in several imaging studies (1); 2) a UMNB scale used at the Massachusetts General Hospital, the MGH UMNB scale (3); and 3) the Penn Upper Motor Neuron Score (UPENN UMNS) developed in the ALS clinic at the University of Pennsylvania (2). The dataset consisted of clinical measurements that were made prospectively on a cohort of PLS patients followed longitudinally in a natural history study. These data were used to retrospectively calculate UMNB scales. We examined whether rates of change of UMNB scales differ over the duration of disease and whether UMNB scales correlate with functional scales, such as the ALSFRS-R that had been collected in this PLS cohort, or with quantitative measures of motor performance.
METHODS
Patients with PLS were evaluated between July 2000 and April 2019 at the National Institutes of Health (NIH) Clinical Center in Bethesda, Maryland USA. All patients gave written informed consent for the natural history protocol that was approved by the Institutional Review Board (NCT00015444). The protocol consisted of an initial screening visit and longitudinal collection of clinical data, including quantitative measures of movement, for those patients meeting the clinical criteria for PLS (6, 7, 13). Because symptoms begin insidiously in PLS, at the initial visit patients were closely queried to recollect the year and season when the earliest symptom was noted. The midpoint of the 3-month seasonal window when symptoms were first noted was used as the onset of disease in most cases.
Of 136 patients referred for evaluation of PLS during that period, 53 patients who met the following criteria were included in this analysis:
Fulfilled clinical criteria for PLS (6) at their initial visit or upon follow-up visits
An EMG performed 5 years or more after symptom onset showed no active denervation
Came to NIH for at least two evaluations
Had sufficient documentation of measures of reflexes, tone, and pseudobulbar affect to calculate UMNB scales
Patients with symptoms limited to the lower extremities were excluded.
Patients with a family history of PLS, spastic paraparesis, or motor neuron disorder in first degree relatives were excluded.
The dataset consisted of 231 evaluation visits. Of these, the UPENN UMNS could not be calculated for 14 visits because the Ashworth scale had not been obtained. One hundred and twenty-two evaluations were carried out in patients with less than 8 years of symptoms, including 28 patients with initial evaluations prior to 5 years of symptoms. The median number of evaluation visits per patient was 4 (range 2–9).
Calculation of three UMNB scales
UK UMN scale (1)
The UK UMN scale ranges from 0–15. One point is given for each of the following abnormal tendon reflexes: Triceps, Biceps, Brachioradialis (in lieu of supinator), Patellar and Achilles. One point is given for each Hoffman’s sign and each Babinski sign. One point is given for brisk jaw jerk.
MGH UMNB scale (3)
The MGH UMNB scale has a range from 0–45 points. A 4-point scale is used to grade each of the following deep tendon reflexes: Biceps, Brachioradialis, Triceps, Patellar, Ankle. The grades are as follows: 0 if Absent, 1 if Diminished, 2 if Normal, 3 if pathologically brisk/retained in MRC grade <2 muscle, 4 if Clonus. One point is given for each Hoffman’s sign, Babinski sign, and presence of a jaw jerk.
UPENN UMNS score (2)
The UPENN UMNS scale has a range from 0–32 points. One point is given for each of the following abnormal tendon reflexes: Triceps, Biceps, Patellar and Achilles. One additional point is added in each limb if clonus is present. One point is given for each Hoffman’s sign, Babinski sign, presence of a jaw jerk, presence of a facial reflex, and presence of a palmomental sign. One point is given for pseudobulbar affect, using the CNS-Lability scale ≥13 as a cutoff. The Ashworth scale was used to grade the tone in each limb. One point was given for an Ashworth scale grade of 2–3, and 2 points for an Ashworth scale grade of 4–5.
Conversion of NIH clinical data to scores on the three scales
Table 1 shows how each of the three UMNB scales is calculated. To apply each of the three scales retrospectively the data from the NIH PLS cohort were converted as follow. At NIH, deep tendon reflexes had been graded using the NIH myotactic reflex scale (14) with a range from 0–4, in which a grade of 4 indicates a pathologically brisk reflex. In charts, grade 4 reflexes were accompanied by a notation of whether clonus, reflex spread, or crossed adductors were present. To convert to the UK UMN scale, an NIH grade of 4 was given 1 point. For the MGH UMNB scale, reflexes graded as 4 on the NIH score sheet were converted to 3 points, and one point given when clonus was present. For conversion to the UPENN UMNS scale, deep tendon reflexes of Grade 4 were scored as 1 point. The Modified Ashworth scale (MAS) was scored from 0–4 as described by Bohannon and Smith (15). Scores of 1–2 were given 1 point, and MAS scores of 3–4 were given 2 points. Presence of pseudobulbar affect had been noted in charts (16) and was scored 1 point when present.
Table 1.
Calculating the three Upper Motor Neuron Burden Scales
| UMNB Scale | Total Score | Sub-domains | Sub-domain components | Item scoring |
|---|---|---|---|---|
| Penn Upper Motor Neuron Score© (Woo JH et al, 2014) | 0–32 | Reflexes (bilateral) | a. Hyperactive (Biceps, Triceps, Patellar, Ankle) b. Pathological (Jaw jerk, Facial, Palmomental sign, Hoffman sign, Finger flexors, Crossed adductor, Clonus in arm or leg, Babinski sign) |
0 if absent, diminished or normal 1 if pathologically brisk (≥ 3)/retained in weak muscle 0 if absent 1 if present |
| Spasticity | Modified Ashworth scale (MAS) | 0 if no increase in tone 1 if MAS 2 or 3 2 if MAS 4 or 5 |
||
| Pseudobulbar affect | CNS-Lability Scale (CNS-LS) | 0 if CNS-LS ≤ 13 1 if CNS-LS ≥13 |
||
|
Upper Motor Neuron Burden Score
(Zurcher N, et al, 2015) |
0–45 | Reflexes (bilateral) | a.Hyperactive (Biceps, Brachioradialis, Triceps, Patellar, Ankle) b. Pathological (Jaw jerk, Hoffman, Babinski) |
0 if Absent 1 if Diminished 2 if Normal 3 if pathologically brisk/retained in MRC grade ≤2 muscle 4 if Clonus 0 if Absent 1 if Present |
| UK UMN score (Turner et al 2004) | 0–15 | Reflexes (bilateral) | a. Hyperactive (Biceps, Brachioradialis, Triceps, Patellar, Ankle b. Pathological (Jaw jerk, Hoffman, Babinski) |
0 if normal 1 if pathological 0 if Absent 1 if Present |
Clinical and quantitative measures obtained in the NIH cohort
Measures of finger-tapping speed and 20-foot timed gait were obtained at 230 of the 235 evaluation visits. Timed reading of a standard passage (17) and foot-tapping speed were added later to the standard battery of movement measures and had been obtained at 134 evaluation visits.
The ALSFRS-R was obtained as a standard measure at visits beginning in 2006 and was administered at 174 evaluation visits. The ALSFRS-R had been administered to 54 of the 56 PLS patients on at least one evaluation visit. The MiniMental State Exam (18) was administered at 149 evaluation visits and the Montreal Cognitive Assessment (mocatest.org) at 84 visits. At least one MMSE or MOCA was obtained in 48 of the 56 PLS patients.
Statistics
Demographic and clinical measures are reported as means and standard deviations for normally distributed measures. Median values with ranges are reported in Tables for clinical measures that were not normally distributed (timed reading, left finger-tapping speed and right foot-tapping speed).
For each of the three UMNB scales, because of a non-linear change of scores over time (disease duration), the approximate low-rank smoother was applied to capture the trends over time using SAS procedure GLIMMIX (www.SAS.com). Akaike information criterion was used to determine the optimal number and location of the break points (interior knots) by changing the bucket size of the k-d tree (options: type=RSMOOTH, knotmethod=KDTREE, tech=NEWRAP). Since only one optimal breakpoint was selected for all three UMN variables, a piecewise random coefficient model was applied to estimate the slopes for before and after the breakpoint using procedure GLIMMIX. The intercept, slope before breakpoint, and difference between the two slopes were treated as random effects with the covariance structure specified as unstructured (option type=CHOL). The normality assumption was assessed using Shapiro-Wilk test based on the standardized residuals. The standardized residuals <−3 or >3 were treated as outliers. Age and gender did not have significant (p>0.15) effect on any of the three UMNB scales and were not included in the model as covariates. Analysis of covariance (ANCOVA) was performed to examine the correlation (within subjects) between UMNB scales and clinical variables (19), to see, for example, whether within the individual an increase in a UMNB scale score was associated with a decrease in a clinical variable such as the ALSFRS-R score. The significance threshold of p<0.007 (0.05/7 clinical variables) was used with Bonferroni correction for multiple comparisons.
RESULTS
Demographics
The cohort consisted of 28 men and 25 women. The mean age of symptom onset was 49.49 ± 7.88 years (range 33–64 years). Forty-six patients reported that their symptoms began in one or both legs and eight reported a change in speech as the first symptom. Two patients reported that symptoms began in the hand or arm but were noted to have UMN signs in the legs at their first evaluation visit. Forty-seven patients were right-handed and 6 were left-handed. The disease duration at the first evaluation visit ranged from 21.6 months to 23.5 years. The median period for in-person follow-up evaluations was 5.3 years (range 0.8 – 15.8 years), although patients continued to be surveyed annually for changes in diagnosis by mail through 2017. The mean MMSE score was 29.4 ± 1.1. (administered at 149 visits) and the mean MOCA score was 26.3 ± 2 (administered at 84 visits). There was no significant decline in MMSE or MOCA in 37 patients tested on multiple visits. For the 29 patients in whom the ALSFRS-R was administered at their first evaluation visit, the mean score was 40.38 ± 3.8 (range 34–46). The median ALSFRS-R score across all visits was 35 (range 23–45).
Longitudinal changes in UMNB scales
Each of the three UMNB scales showed a breakpoint in the slope of change over time at approximately 7.8 years. For the UK UMN and MGH UMNB scales, the calculated breakpoint occurred at a disease duration of 7.78 years; for the UPENN UMNS scale, the breakpoint occurred at 7.95 years. For simplicity in subsequent analyses, 7.8 years was used as the breakpoint for determining slopes of the two time periods.
All three UMNB scales showed increasing scores in the first 7.8 years of disease duration (Figure 1). Increasing scores indicate that clinical signs of spasticity worsened over this period. After 7.8 years all three UMN scales had slopes that were not significantly different from zero. This flattening of slope indicates that the clinical examination findings of spasticity reach a plateau beyond 8 years of disease. For the UPENN UMNS scale the magnitude of the early and late slopes was significantly different (p=0.025), whereas the magnitude difference between early and late slopes did not reach significance for UK UMN score and the MGH UMNB (p=0.068 and p=0.062 respectively).
Figure 1.
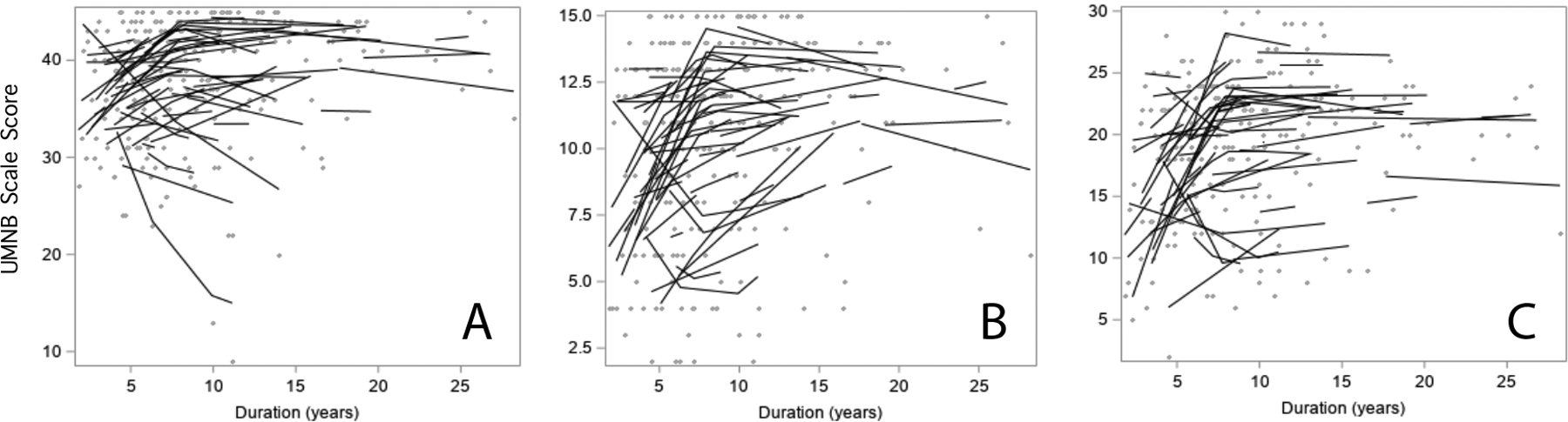
Longitudinal scores for upper motor neuron burden scales for 53 patients with PLS. Solid lines indicate subject-specific intercepts and slopes and gray dots show individual scores A) UK UMN score B) MGH-UMNB scale and C) UPENN UMNS scale. All three scales increase more rapidly in the first 7.8 years, followed by flattening of scores afterwards. Note that many individuals in Panels A and B attain scores near the maximum for the UMNB scale.
Of the three, scales, the UK UMN scale provided the best fit to the data with the smallest residual variance (5.59). The UPENN UMNS gave the second best fit to the data (residual variance 8.41). The average change in the UPENN UMNS score was 1.00 points/year for the first 7.8 years. During this period the UK UMN scores changed by 0.49 points per year, and the MGH UMNB scores changed by 0.63 points per year.
Longitudinal changes in clinical measures
To determine whether the ALS functional rating scale and quantitative measures of movement change over the course of disease in a similar way to the UMNB scales, the piecewise linear mixed effects regression model was applied to these clinical measures. The ALSFRS-R, finger-tapping speed and gait speed worsened over time with steeper slopes before than after 7.8 years (p< 0.007,Table 2). The ALSFRS-R declined by 1.14 points per year in the first 7.8 years, with a non-significant decline of 0.23 points per year thereafter (Figure 2A). Finger-tapping declined by 0.17 taps/s in the right hand and by 0.16 taps/s in the left hand during the first 7.8 years, with non-significant declines thereafter (Figure 2B). Gait speed for 25-foot timed gait declined faster in the first 7.8 years than in later years. Foot-tapping speed was more variable, without clear differences in slope of decline over time. Slopes for change in clinical measures are shown in Table 2.
Table 2.
Change in UMNB scales and clinical measures over time (slopes)
| Change/year First 7.8 years |
Change/year After 7.8 years |
|
|---|---|---|
| UMNB Scales | ||
| UK UMN scale | 0.49 | 0.06 |
| MGH UMNB scale | 0.63 | −0.09 |
| UPENN UMNS | 1.00 | 0.06 |
| Clinical Measures | ||
| ALSFRS-R | −1.14* | −0.23 |
| Right hand finger tapping (taps/s) | −0.17* | −0.02 |
| Left hand finger tapping (taps/s) | −0.16* | 0.02 |
| Timed Gait (ft/s) | −0.16* | −0.03 |
| Timed reading (s) | 5.08 | −0.40 |
| Right foot tapping (taps/s) | −0.07 | 0.00 |
| Left foot tapping (taps/s) | −0.06 | 0.00 |
p < 0.007 (Bonferroni correction)
Figure 2.
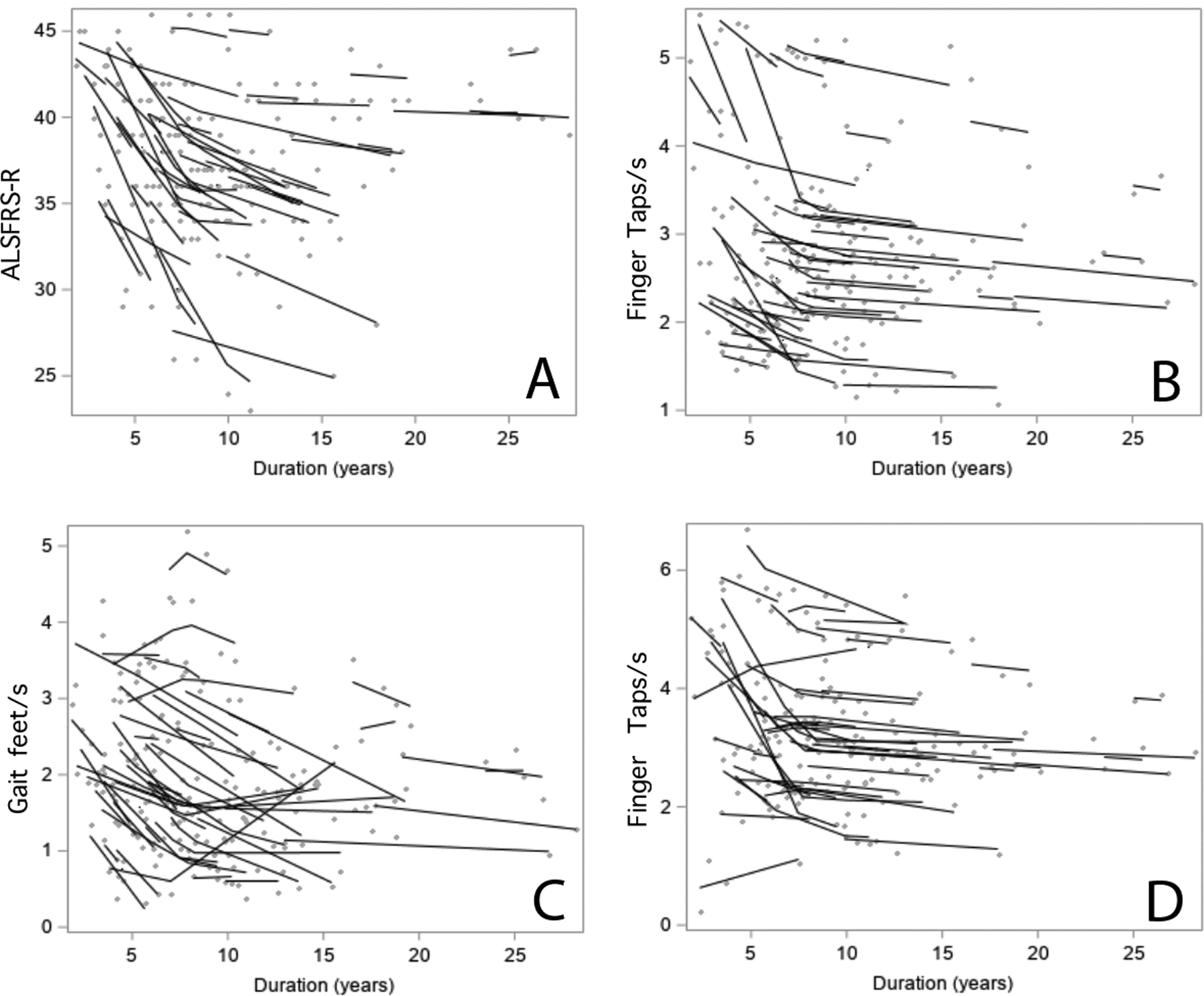
Longitudinal scores on clinical measures for 53 patients with PLS A) ALS Functional rating scale and B) Left hand finger tapping speed C) Gait speed for 25 foot timed walk D) Right hand finger tapping speed. Solid lines indicate slopes for each patient calculated using subject-specific regression models, and gray dots show individual scores. All measures show a change in slope after 7.8 years of disease.
Correlation (within subject) between changes in UMNB scales and clinical measures
Increasing scores on the UMNB scales were moderately correlated with declines in several clinical measures, which differed between scales (Table 3). All three scales were correlated with finger tapping speed. The UPENN UMNS was correlated with all clinical measures except timed speech, and was most strongly correlated with clinical measures, including the ALSFRS-R score.
Table 3.
Within-subject correlation between changes in UMNB scores and clinical measures
| UPENN UMNS | UK UMN scale | MGH UMNB | |
|---|---|---|---|
| ALSFRS-R | −0.42 | n.s. | n.s. |
| Right Finger Taps/s | −0.58 | −0.51 | −0.44 |
| Left Finger Taps/s | −0.64 | −0.43 | −0.40 |
| Timed Gait, ft/s | −0.57 | n.s. | −0.53 |
| Right Foot Taps/s | −0.50 | −0.58 | n.s. |
| Left Foot Taps/s | −0.46 | −0.48 | n.s. |
| Timed reading (s) | n.s. | n.s. | n.s. |
Correlation coefficient with p < 0.007, (Bonferroni correction) n.s. not significant
DISCUSSION
Patients with primary lateral sclerosis often live for decades after symptoms of spasticity begin. In a cohort of PLS patients followed longitudinally, we found that three upper motor neuron burden scales all changed more rapidly during the first approximately 8 years of symptoms than in later years. The clinical examination measures of spasticity on which the UMNB scales are based reach an apparent plateau after about 8 years. In some patients, the plateau was near the maximum score of the UMNB scale, and thus it is possible that continued worsening was masked by the scales’ ceiling. However, the slower decline after 8 years was also seen for a functional rating scale, the ALSFRS-R, as well as for quantitative measures of movement, supporting the explanation that disease progression slows or arrests over time (20). However, it is notable that the ALSFRS-R did continue to show some decline after 8 years, losing 1 point approximately every 4 years, whereas other measures, such as finger tapping speed, remained relatively stable after 8 years. The very slow decline in the ALSFRS-R is consistent with previous reports in patients with PLS (10) and supports using the PLSFRS, a more sensitive scale, in future studies (11).
Among the three UMNB scales evaluated, the UK UMN scale provided the best fit (least variable residuals) to this PLS dataset. A drawback of the UK UMN scale is a potential ceiling effect, with many PLS patients reaching the maximum score of 15. The UPENN UMNS has a bigger range and was the second best fit to the data. Unlike the other scales, the UPENN UMNS incorporates measures of muscle tone and pseudobulbar affect in addition to reflex changes. It requires quantifying muscle tone using the Ashworth scale, which may not be familiar to many neurologists. The MGH UMNB fit these PLS data least well, possibly due to the scoring range for tendon reflexes. Unlike the UPENN UMNS and UK UMN scale which graded reflexes in a binary fashion (hyperactive, not-hyperactive), the MGH UMNB scale includes grades 0–2 for absent, hypoactive, and normal tendon reflexes. The inclusion of these additional grades may be suitable for patients with disorders with mixtures of upper and lower motor signs but increased variability in this dataset. Of the three UMN scales, the magnitude of change in proportion to the range of the scale, was greatest for the UK UMN scale in the first 7.8 years. The UPENN UMNS was most strongly correlated to clinical measures, consistent with a previous report (21).
A potential drawback of all three scales is the inclusion of more than one reflex from each limb, as reflexes in the same limb tend to be highly correlated in PLS. It is likely that medication effects contributed to the variability seen in scores for all three scales in this study. Participants in this longitudinal study received varied medications to treat spasticity as prescribed by their primary providers. This is a limitation for interpreting the variability in performance of scales over time, as medications could change over time, and for generalizing performance of scales in a controlled medication trial.
These findings have implications for the use of an UMNB scale as an outcome measure for interventional studies in PLS. Stratification of patients by disease duration would be needed to account for the change in slope after eight years. A further consideration is that each of the three UMNB scales had a relatively small annual change, even within the first 8 years of disease. However, those changes were correlated with changes in a functional rating scale (ALSFRS-R) and measures of movement. While more sensitive measures are needed for clinical trials in PLS, UMNB scales could potentially contribute to a combined index of disease severity or be suitable as a secondary outcome measure.
ACKNOWLEDGEMENTS
Laura Braun, Carol Hoffman, Zoe Joy, and Jamie Cherup assisted in collating data from charts. We are grateful to the many patients and families who participated in the PLS natural history study over the last two decades.
Footnotes
DECLARATION OF INTEREST
The authors have no conflicts of interest to disclose. This study was supported by the intramural program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke (Z01 NS002976).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author, MK Floeter, at https://data.ninds.nih.gov.
REFERENCES
- 1.Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banati RB. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15(3):601–609. [DOI] [PubMed] [Google Scholar]
- 2.Quinn C, Edmundson C, Dahodwala N, Elman L. Reliable and efficient scale to assess upper motor neuron disease burden in amyotrophic lateral sclerosis. Muscle Nerve. 2020;61(4):508–511. [DOI] [PubMed] [Google Scholar]
- 3.Zurcher NR, Loggia ML, Lawson R, Chonde DB, Izquierdo-Garcia D, Yasek JE, Akeju O, Catana C, Rosen BR, Cudkowicz ME, et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: assessed with [(11)C]-PBR28. NeuroImage Clinical. 2015;7:409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Statland JM, Barohn RJ, Dimachkie MM, Floeter MK, Mitsumoto H. Primary Lateral Sclerosis. Neurol Clin. 2015;33(4):749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Chalabi A, Hardiman O, Kiernan MC, Chio A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 2016;15(11):1182–1194. [DOI] [PubMed] [Google Scholar]
- 6.Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H, Rowland LP. The natural history of primary lateral sclerosis. Neurology. 2006;66(5):647–653. [DOI] [PubMed] [Google Scholar]
- 7.Singer MA, Statland JM, Wolfe GI, Barohn RJ. Primary lateral sclerosis. Muscle Nerve. 2007;35(3):291–302. [DOI] [PubMed] [Google Scholar]
- 8.Turner MR, Barohn RJ, Corcia P, Fink JK, Harms MB, Kiernan MC, Ravits J, Silani V, Simmons Z, Statland J, et al. Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiatry. 2020. Neurol Neurosurg Psychiatry 2020;91(4):373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, Nakanishi A. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169(1–2):13–21. [DOI] [PubMed] [Google Scholar]
- 10.Mitsumoto H, Nagy PL, Gennings C, Murphy J, Andrews H, Goetz R, Floeter MK, Hupf J, Singleton J, Barohn RJ, et al. Phenotypic and molecular analyses of primary lateral sclerosis. Neurology Genetics. 2015;1(1):e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitsumoto H, Chiuzan C, Gilmore M, Zhang Y, Simmons Z, Paganoni S, Kisanuki YY, Zinman L, Jawdat O, Sorenson E, et al. Primary lateral sclerosis (PLS) functional rating scale: PLS-specific clinimetric scale. Muscle Nerve. 2020;61(2):163–172. [DOI] [PubMed] [Google Scholar]
- 12.Woo JH, Wang S, Melhem ER, Gee JC, Cucchiara A, McCluskey L, Elman L. Linear associations between clinically assessed upper motor neuron disease and diffusion tensor imaging metrics in amyotrophic lateral sclerosis. PloS one. 2014;9(8):e105753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain. 1992;115(Pt 2):495–520. [DOI] [PubMed] [Google Scholar]
- 14.Litvan I, Mangone CA, Werden W, Bueri JA, Estol CJ, Garcea DO, Rey RC, Sica RE, Hallett M, Bartko JJ. Reliability of the NINDS Myotatic Reflex Scale. Neurology. 1996;47(4):969–972. [DOI] [PubMed] [Google Scholar]
- 15.Bohannon RW, Smith MB. Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther. 1987;67(2):206–207. [DOI] [PubMed] [Google Scholar]
- 16.Floeter MK, Katipally R, Kim MP, Schanz O, Stephen M, Danielian L, Wu T, Huey ED, Meoded A. Impaired corticopontocerebellar tracts underlie pseudobulbar affect in motor neuron disorders. Neurology. 2014;83(7):620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Riper C Speech correction 4th ed. Englewoood Cliffs (NJ):Prentice Hall; 1963. [Google Scholar]
- 18.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. [DOI] [PubMed] [Google Scholar]
- 19.Bland JM, Altman DG. Calculating correlation coefficients with repeated observations: Part 1--Correlation within subjects. BMJ. 1995;310(6977):446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floeter MK, Mills R. Progression in primary lateral sclerosis: a prospective analysis. Amyotroph Lateral Scler. 2009;10(5–6):339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finegan E, Chipika RH, Li Hi Shing S, Doherty MA, Hengeveld JC, Vajda A, Donaghy C, McLaughlin RL, Pender N, Hardiman O, et al. The clinical and radiological profile of primary lateral sclerosis: a population-based study. J Neurol. 2019;266(11):2718–2733. [DOI] [PubMed] [Google Scholar]