

Abstract
The main inherited cardiac arrhythmias are long QT syndrome, short QT syndrome, catecholaminergic polymorphic ventricular tachycardia and Brugada syndrome. These rare diseases are often the underlying cause of sudden cardiac death in young individuals and result from mutations in several genes encoding ion channels or proteins involved in their regulation. The genetic defects lead to alterations in the ionic currents that determine the morphology and duration of the cardiac action potential, and individuals with these disorders often present with syncope or a life-threatening arrhythmic episode. The diagnosis is based on clinical presentation and history, the characteristics of the electrocardiographic recording at rest and during exercise and genetic analyses. Management relies on pharmacological therapy, mostly β-adrenergic receptor blockers (specifically, propranolol and nadolol) and sodium and transient outward current blockers (such as quinidine), or surgical interventions, including left cardiac sympathetic denervation and implantation of a cardioverter–defibrillator. All these arrhythmias are potentially life-threatening and have substantial negative effects on the quality of life of patients. Future research should focus on the identification of genes associated with the diseases and other risk factors, improved risk stratification and, in particular for Brugada syndrome, effective therapies.
The field of inherited arrhythmic disorders is bursting with novel information and data, ranging from genetic findings to advances in diagnosis and risk stratification to progress in personalized — even gene-specific — management. Currently, the greatest interest and challenge concerns the so-called ion channelopathies — inherited conditions related to primary electrical disorders in the setting of a structurally normal heart. These diseases are caused by mutations in genes encoding ion channels. Of note, other types of inherited arrhythmias exist, such as hereditary atrial fibrillation and arrhythmogenic right ventricular cardiomyopathy. This Primer focuses on the four major channelopathies: long QT syndrome (LQTS), short QT syndrome (SQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT) and Brugada syndrome (BrS). These four clinical entities share several features: they have an overall low prevalence, their diagnosis is not always simple and they can be fatal. The fact that all these diseases have the potential to trigger life-threatening arrhythmias increases the responsibilities and the concerns of the clinicians who see patients with these conditions only occasionally owing to their rarity, and are, therefore, often ill at ease in taking clinical decisions that may be difficult or impossible to reverse.
Following a brief overview of epidemiology, genetics and underlying electrophysiological mechanisms, this Primer focuses on the clinical aspects of diagnosis, risk stratification and therapy, including — whenever appropriate — gene-specific management. The pregnancy-associated risks are considered, as well as the effects of these diseases on the quality of life of the patients. Finally, a large reference section provides the interested readers with the sources for the statements presented in this document.
Epidemiology
Studies investigating sudden cardiac death (SCD) in young individuals (of <35 years of age), conducted in the USA, Denmark and the Netherlands1–3, have estimated an incidence in this age group of 1.3–3.2 per 100,000 person-years. A 2016 3-year, prospective, population-based study of SCD among persons of 1 to 35 years of age in Australia and New Zealand uncovered a higher prevalence of SCD in males than females and found that SCD occurred predominantly during sleep (38%) or at rest (27%)4. At autopsy, a structurally normal heart was found in 40% of cases, suggesting a possible primary electrical disorder caused by ion channel dysfunction, such as LQTS, SQTS and CPVT, as the cause of death4. Coronary artery disease and inherited cardiomyopathies were established as probable cause of death in 24% and 16% of cases, respectively4. The incidence of SCD in this age group (<35 years) is much lower than that observed in the general population. Studies in the USA and the Netherlands have reported a yearly incidence of 0.6 to >1.4 per 1,000 individuals in the general population5,6. At the general population level, the average age at SCD is 65 years, around 75% of individuals are male7, and coronary artery disease and its consequences, including acute myocardial ischaemia and heart failure, are the cause of SCD in about 80% of cases8.
The cardiac channelopathies that can be associated with SCD are rare, especially SQTS. Systematic population-based studies are lacking, and the real prevalence of these disorders is largely unknown, with the notable exception of LQTS. As a consequence, the numbers reported in articles and reviews, including guidelines (for example, the figure of 1 per 10,000 individuals for CPVT9), are at best educated guesses. Regarding LQTS, a prospective electrocardiogram (ECG) study was performed in 44,596 infants of 15–25 days of age (of whom 43,080 were white individuals)10. Whenever the first corrected QT (QTc) interval measured exceeded 450 ms, a second ECG was repeated within 1–2 weeks. If QT prolongation was confirmed and QTc exceeded 470 ms (at the beginning of the study) or 460 ms (towards the end of the study, when the cut-off threshold for genetic tests was lowered), genetic analysis was performed. Disease-causing mutations were identified in 29% of the infants with a QTc between 451 and 460 ms and in 49% of those with a QTc >470 ms. Overall, 17 of 43,080 white infants were diagnosed with LQTS, demon-strating a prevalence of at least 1 in 2,534 apparently healthy live births (95% CI, 1 in 1,583 to 1 in 4,350). As genetic analyses were not performed for infants with a QTc between 450 and 470 ms, it was suggested that the actual prevalence of LQTS with a positive ECG phenotype is at least 1 in 2,000 individuals10.
Founder mutations causing primary electrical disorders have been described in certain geographical regions. For example, LQTS founder mutations have been reported in Finland11, South Africa12, Canada13, the Netherlands14 and Sweden15. In such regions, the prevalence of the disorder is expected to be higher than in other areas. Knowledge concerning ethnic differences in the prevalence of these arrhythmias is limited, as most of the patient cohorts studied originated from the Western world and, to a lesser extent, from Asian countries. Ethnic differences have been observed for BrS, as ECG studies showed that the prevalence is higher among Asians (0.0–0.94%) and Japanese–Americans (0.15%) than in Europeans (0.0–0.02%) and North Americans (0.005–0.1%)16. However, complete figures regarding precise estimates of BrS are lacking, as in individuals with suspected BrS but without a spontaneous type I ECG, the diagnosis cannot be confirmed without additional investigations (such as, for example, drug challenge during ECG recording)17 (see Diagnosis, screening and prevention).
Mechanisms/pathophysiology
The great majority of genes identified to date as being associated with the primary electrical disorders encode the crucial cardiac ion channel pore-forming α-subunits or proteins that interact with and regulate ion channels (also known as channel-interacting proteins or ChIPs) (FIG. 1; TABLE 1).
Fig. 1 |. Genes and proteins involved in the pathogenesis of inherited cardiac arrhythmias.
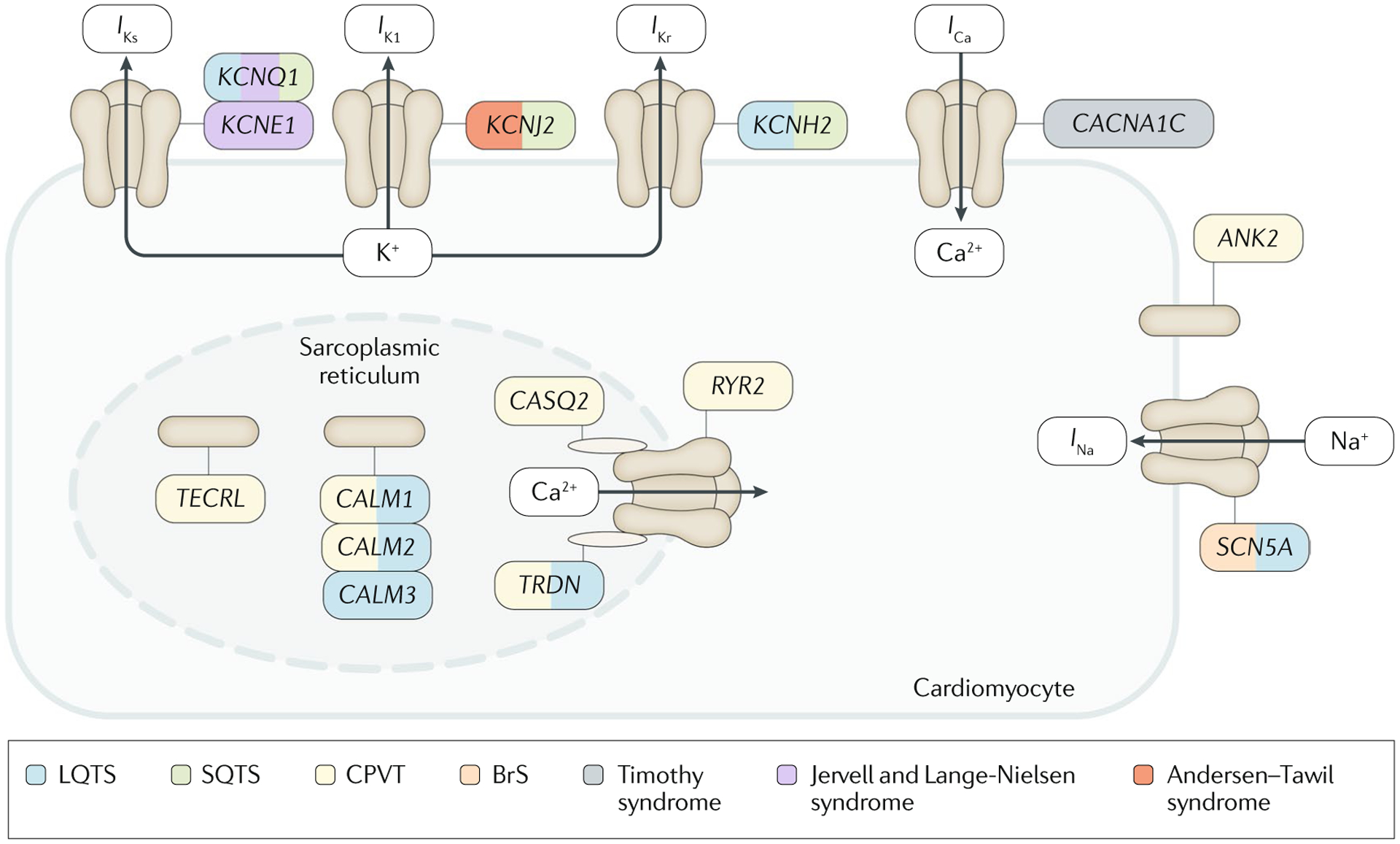
The figure shows the transmembrane ionic channels that are responsible for the potassium (IKs, IK1 and IKr), calcium (ICa) and sodium (INa) currents that contribute to the cardiac action potential. Proteins of the sarcoplasmic reticulum that are involved in calcium handling (encoded by CASQ2, RYR2, CALM1, CALM2, CALM3 and TRDN) are also shown. Other proteins that can be mutant in inherited cardiac arrhythmias are encoded by ANK2 and TECRL. The genes that encode all these proteins or their subunits are coloured according to the disease with which they have been associated. BrS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; LQTS, long QT syndrome; SQTS, short QT syndrome.
Table 1 |.
Genes frequently associated with inherited cardiac arrhythmias
| Gene | Protein | Function | Associated disorder | Inheritance |
|---|---|---|---|---|
| KCNQ1 | Potassium voltage-gated channel subfamily KQT member 1 (also known as KV7.1) | Subunit of the voltage-gated potassium channel responsible for the IKs current | LQTS (LQT1) | AD |
| Jervell and Lange-Nielsen syndrome | AR | |||
| SQTS (SQT2) | AD | |||
| KCNH2 | Potassium voltage-gated channel subfamily H member 2 (also known as KV11.1) | Pore-forming subunit of the voltage-gated potassium channel responsible for the IKr current | LQTS (LQT2) and SQTS (SQT1) | AD |
| KCNE1 | Potassium voltage-gated channel subfamily E member 1 | Subunit of the potassium channel responsible for the IKs current | Jervell and Lange-Nielsen syndrome | AR |
| KCNJ2 | Inward rectifier potassium channel 2 (Kir2.1) | Potassium channel responsible for the IK1 current | Andersen–Tawil syndrome and SQTS (SQT3) | AD |
| SCN5A | Sodium channel protein type 5 subunit-α (also known as NaV1.5) | Subunit of the voltage-gated sodium channel responsible for the INa current | LQTS (LQT3) | AD |
| BrS | Complex inheritance | |||
| CALM1 | Calmodulin 1 | Calcium-binding protein | LQTS and CPVT | AD |
| CALM2 | Calmodulin 2 | Calcium-binding protein | LQTS and CPVT | AD |
| CALM3 | Calmodulin 3 | Calcium-binding protein | LQTS | AD |
| ANK2 | Ankyrin B | Protein involved in the localization and membrane stabilization of ion transporters and ion channels | CPVT | AD |
| TRDN | Triadin | Sarcoplasmic reticulum component of the calcium release unit | LQTS and CPVT | AR |
| CACNA1C | Voltage-dependent L-type calcium channel subunit α1C (also known as CaV1.2) | Pore-forming subunit of the calcium channel responsible for L-type calcium currents | Timothy syndrome | AD |
| RYR2 | Ryanodine receptor 2 | Sarcoplasmic reticulum calcium channel | CPVT | AD |
| CASQ2 | Calsequestrin 2 | Component of the sarcoplasmic reticulum calcium release unit | CPVT | AR |
| TECRL | Trans-2,3-enoyl-CoA reductase-like | Endoplasmic reticulum protein | CPVT | AR |
AD, autosomal dominant; AR, autosomal recessive; BrS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; LQT1, type 1 long QT syndrome; LQTS, long QT syndrome; SQT1, type 1 short QT syndrome; SQTS, short QT syndrome.
LQTS genetics
LQTS is most commonly inherited as an autosomal dominant disorder (initially known as Romano–Ward syndrome). Inherited genetic variations in one of the three major LQTS susceptibility genes underlie the disorder in ~90% of patients in whom a causative mutation was identified. Type 1 LQTS (LQT1), caused by genetic variants in KCNQ1 (REF.18) encoding the subunit KV7.1 of the voltage-gated potassium channel that is responsible for the outward potassium current IKs, accounts for ~30–35% of cases19. LQT2, caused by genetic variants in KCNH2 encoding the pore-forming α-subunit KV11.1 of the voltage-gated potassium channel responsible for the inward rectifying potassium current IKr20, accounts for ~25–40% of cases. LQT3 is caused by genetic variants in SCN5A21 encoding the subunit-α NaV1.5 of the voltage-gated sodium channel responsible for the inward sodium current INa and represents ~5–10% of cases of LQTS. Approximately 15–20% of patients with a definite clinical diagnosis of LQTS remain genotype-negative after extensive genetic testing22. Single-nucleotide variants, small insertions or small deletions are the most commonly encountered disease-causing variations in these genes19. However, large gene rearrangements have also been described23. About 5% to 10% of patients with LQTS host multiple mutations in these genes, and symptoms typically present at a younger age and with a more severe phenotype in these patients than in patients with a single mutation24–26.
Genotype–phenotype correlations.
Genotype–phenotype studies in the KCNQ1 and KCNH2 genetic subtypes have identified relationships between the severity of the disorder and the type of mutation, the location of the mutation and the extent of functional derangement of the ion channel. Studies conducted in patients with KCNQ1 defects have identified that variants at sequences encoding transmembrane regions of the KV7.1 channel are associated with a higher risk of cardiac events than variants in sequences encoding the carboxy terminus, and variants that result in a severe reduction of channel function due to a dominant-negative effect are also associated with a higher risk of cardiac events than variants that result in haploinsufficiency25. In patients with KCNH2 defects, missense variants in the sequence encoding the pore region of the KV11.1 channel have been associated with more severe clinical manifestations and higher cardiac event rates than those associated with mutations27 localized outside the pore-coding sequence.
Other LQTS-associated genes.
Several other genes encoding either ion channel subunits (KCNE1, KCNE2, KCNJ5 and SCN4B) or proteins that regulate ion channel function (CALM1, CALM2, CALM3, AKAP9, CAV3, ANK2, SNTA1 and TRDN) have also been implicated in LQTS causality28. However, it should be noted that most of these genes have been implicated using a hypothesis-driven, candidate gene approach, and, therefore, the strength of evidence supporting their causality varies widely28. In fact, some of these genes received a disputed-evidence (KCNE2, KCNJ5, SCN4B, SNTA1, AKAP9 and ANK2) or limited-evidence (CAV3, KCNE1 and KCNJ2) gene designation on application of the gene-disease association framework of the Clinical Genome (ClinGen) Resource28–30. By contrast, CALM1, CALM2, CALM3 and TRDN were found to have strong (TRDN) or definitive (CALM1, CALM2 and CALM3) evidence.
Patients with LQTS harbouring variants in CALM1, CALM2 or CALM3 (encoding calmodulin 1, calmodulin 2 and calmodulin 3, respectively) present symptoms early in life with profound QTc interval prolongation, which may be accompanied by 2:1 atrioventricular block and a high predisposition for cardiac arrest and sudden death31,32. Calmodulin is a calcium-sensing, signal-transducing protein that regulates many calcium-dependent processes and modulates the function of several cardiac ion channels. CALM1, CALM2 and CALM3 are located on distinct chromosomes, have a nucleotide sequence homology of 85%, and yet code for a completely identical, 149 amino acid calmodulin protein. Despite this high redundancy, one mutant allele out of six is sufficient to cause LQTS. Relevant information is provided by the International Calmodulinopathy Registry33 on 74 patients. In a series of 29 patients whose family members were genetically screened, the culprit CALM variant occurred de novo in 93% of cases, and in the remaining cases germline mosaicism was present in one of the parents33. Patients with homozygous or compound heterozygous pathogenetic variants in TRDN may manifest either a predominant LQTS or CPVT phenotype34,35.
Extracardiac manifestations.
Three clinical variants of LQTS manifest with extracardiac features besides QT interval prolongation. The autosomal recessive Jervell and Lange-Nielsen syndrome, characterized by sensori-neural deafness and high arrhythmic risk, is caused by homozygous or compound heterozygous mutations in KCNQ1 (REF.36) or KCNE1 (REF.37); KCNE1 mutations are associated with a less severe phenotype than KCNQ1 mutations38. Timothy syndrome presents with multiorgan dysfunction including webbing of fingers and toes, congenital heart defects, immune deficiency, hypo-glycaemia, cognitive abnormalities and autism39. The same recurrent sporadic de novo missense mutation in CACNA1C (encoding the pore-forming subunit-α CaV1.2 of the voltage-gated cardiac calcium channel that is responsible for long-lasting (L-type) calcium currents), which results in the G406R amino acid substitution, accounts for many of the cases reported to date40, although other missense mutations in this gene have also been reported. However, mutations in CACNA1C have also been described for autosomal dominant LQTS in the absence of extracardiac features41. The Andersen–Tawil syndrome, which presents with facial dysmorphism and hypokalaemic periodic paralysis, is caused by dominantly inherited loss-of-function mutations in KCNJ2, which encodes the inward rectifier potassium channel Kir2.1 (which is responsible for the IK1 current)42. Although Andersen–Tawil syndrome was initially presented as part of the LQTS spectrum, it has been argued that the QT interval prolongation in this disorder is erroneously diagnosed by the inclusion of the prominent U wave abnormality in the QTc calculations28; accordingly, this arrhythmic syndrome should not be regarded as part of LQTS28.
SQTS genetics
SQTS is inherited as an autosomal dominant disorder. Mutations in three genes encoding potassium channels, namely, KCNH2 (REF.43), KCNQ1 (REF.44) and KCNJ2 (REF.45), have been implicated in the disorder and are associated with its subtypes SQT1, SQT2 and SQT3, respectively. In contrast to loss-of-function variants associated with LQTS, SQTS-causing variants in KCNH2, KCNQ1 and KCNJ2 lead to a gain-of-function defect of the affected potassium channel. Mutations in the L-type calcium channel subunit genes CACNA1C, CACNB2 and CACNA2D1 (REF.46) have been described in patients presenting with a shorter than normal QTc or an overlapping phenotype that combines an abbreviated QTc and a BrS ECG phenotype47, yet evidence for their causality is limited. These mutations are expected to cause a loss of channel function, thereby also abbreviating the action potential (AP). The same missense mutation in SLC4A3, encoding the anion exchange protein 3 (also known as solute carrier family 4 member 3), has been described in a large family with SQTS and a smaller, unrelated family48.
CPVT genetics
CPVT is commonly inherited in an autosomal dominant manner. In 65% of CPVT probands, the disorder is caused by a mutation in RYR2, which encodes the ryanodine receptor 2 (RYR2), a calcium release channel located on the sarcoplasmic reticulum (SR) of cardiomyocytes49–51, and these individuals have CPVT type 1. RYR2 mutations are typically missense and cluster predominantly at sequences encoding specific regions of RYR2, although some mutations still occur outside these clusters52,53. Mutations in CASQ2, which encodes calsequestrin 2, a protein that binds to free calcium inside the SR, cause CPVT type 2, a much rarer but more severe autosomal recessive form of CPVT54. Other genes involved in calcium homeostasis, namely CALM1 (REF.55) and TRDN56, have also been implicated as a cause of CPVT. Mutations in CALM2 have been described in patients with overlapping features of LQTS and CPVT57. Mutations in ANK2 and KCNJ2 that also cause, respectively, Ankyrin B syndrome (initially called LQT4) and Andersen–Tawil syndrome have been described in a few patients with normal QTc and exercise-induced arrhythmias58,59. Furthermore, recessive mutations in TECRL, encoding the trans-2,3-enoyl-CoA reductase-like protein expressed in the endoplasmic reticulum, have been associated with a clinical phenotype that has overlapping features of LQTS and CPVT60. The CPVT genetic test panel should include at least examination of the entire 105 translated exons of RYR2, CASQ2, KCNJ2, CALM1, CALM2, CALM3, TRDN, TECRL and PKP2 (encoding plakophilin 2)61. Notably, PKP2-mediated arrhythmogenic right ventricular cardiomyopathy can have a pre-cardiomyopathic electrical phase of the disease that mimics CPVT62.
BrS genetics
The only gene thus far unequivocally implicated in BrS is SCN5A63,64. As opposed to LQTS-associated variants, BrS-associated SCN5A variants are loss of function. At least 20 other genes have been reported for BrS; however, these genes have almost exclusively been discovered through candidate gene studies in single individuals or small families. Furthermore, the recent reappraisal of all reported BrS susceptibility genes (either by testing for an increased burden of rare genetic variants in patients with BrS compared with controls65 or by the application of the ClinGen evidence-based gene curation framework64) supports only the involvement of rare variations in SCN5A, which are found in ~20% of probands65. The largely sporadic presentation of the disorder and the low disease penetrance in families with rare variants in SCN5A, as well as the observation of phenotype-positive genotype-negative individuals in such families, has suggested that BrS is a disorder with an inheritance more complex than the Mendelian inheritance that was previously considered66. A genome-wide association study that identified common small-effect susceptibility variants in the vicinity of SCN5A and HEY2 provided evidence in support of this complex inheritance67.
Genetic modifiers
As for most Mendelian disorders, the management of patients with Mendelian primary electrical disorders is complicated by the variability in clinical severity among patients, even those carrying the same mutation. Such variability within families is evidenced by low disease penetrance (the proportion of individuals carrying a variant who show phenotypic effects), variable expressivity (different severity observed among individuals carrying the familial genetic defect) and pleiotropy (the phenomenon of a single gene affecting several phenotypic traits). Variability is observed both in the extent of the ECG abnormality and in the occurrence of arrhythmic events. Cascade screening and genotyping68,69 has disclosed that variability in clinical manifestations can be profound, even between siblings with the same mutations, ranging from a normal ECG and no symptoms to a full-blown phenotype with life-threatening arrhythmias. An example is the large variability in QTc duration among individuals harbouring the A341V founder mutation in KCNQ1 causing LQTS12 (FIG. 2). A study showed a large spectrum of QTc values in a population of individuals carrying this mutation, including some values within the physiological range; if QTc prolongation were determined only by the A341V mutation, the QTc values should be uniformly prolonged with modest variability12. This observation points to the presence of additional factors affecting ventricular repolarization.
Fig. 2 |. Variability in baseline QTc.
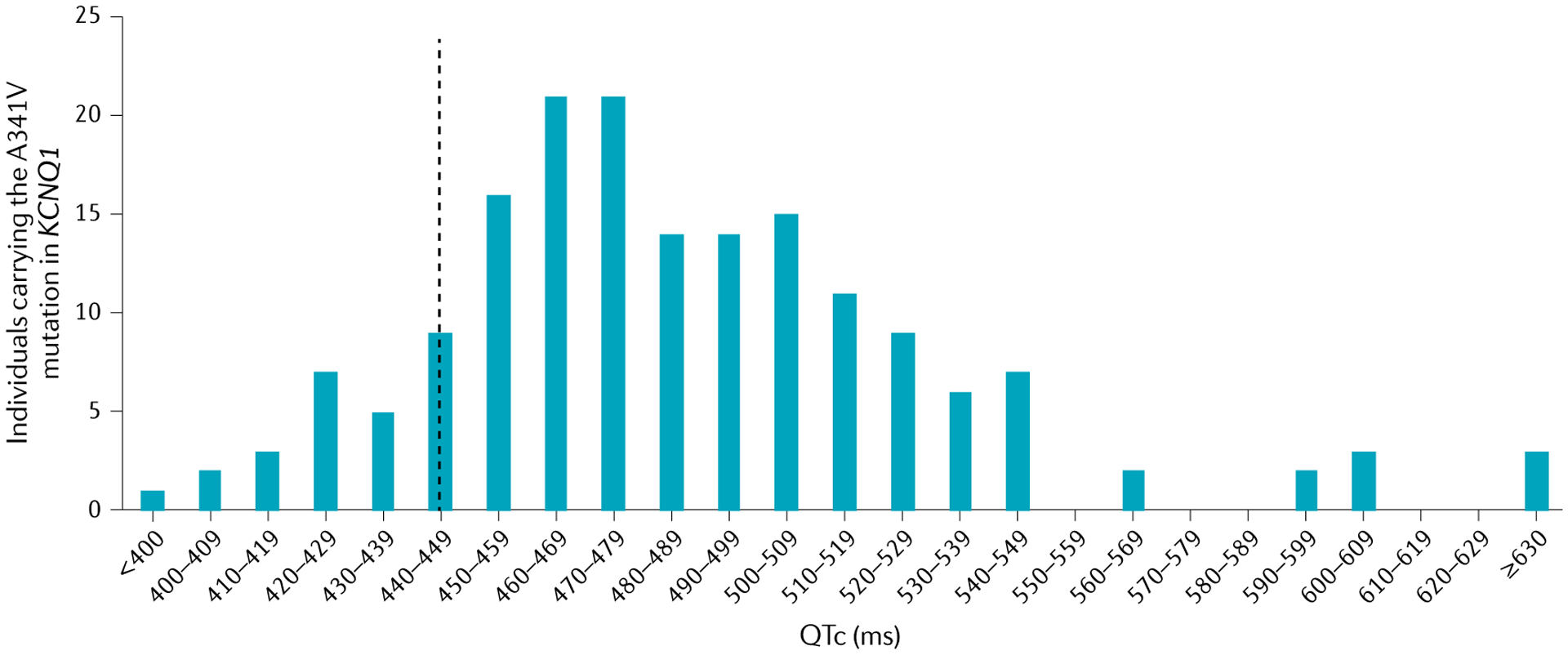
Distribution of the duration of heart rate corrected QT interval (QTc) in individuals who carry the same KCNQ1 mutation and who belong to a LQT1 South African founder population. The vertical dashed line represents the upper limit of normal values for men (440 ms). If the QTc prolongation were determined only by the A341V mutation, the QTc values should be uniformly prolonged with modest variability. The large spectrum of QTc values points to the presence of additional variants affecting ventricular repolarization. Data from REF.12.
Factors that modify the clinical expression of Mendelian primary electrical disorders can be non-genetic, such as age, sex70 and, according to some studies, features of the cardiac tissues, such as, for example, fibrosis. In addition to non-genetic factors, the inheritance of other genetic variants, commonly referred to as genetic modifiers, contributes to the variability in the phenotype71. Genetic modifiers may exacerbate or temper the effect of the disease-causing mutation, and the net effect of the mutation and genetic and non-genetic modifiers determines the severity of the ECG defect and/or occurrence of arrhythmias.
Research into genetic modifiers in these primary electrical disorders is still in its infancy, yet some associations have started to emerge71, primarily in LQTS71. Moreover, the mechanism of action of modifier genes is beginning to be unravelled72. The identification of these associations has been in part facilitated by the availability of large founder populations, wherein the presence of the same disease-causing mutation favours the identification of modulatory variants, as it enables the exclusion of interindividual variation stemming from different primary genetic defects73–76. As more modulatory variants are identified, combined genotyping for disease-causing mutations as well as for modifiers may enable more refined risk stratification.
Pathophysiology of the individual arrhythmias
Characteristics of the cardiac AP.
The cardiac cell, similar to all excitable cells, maintains a voltage difference between the exterior and interior of the cell — the membrane potential — of typically −60 to −90 mV, depending on the cell type. Thus, the interior of the cell has a negative voltage relative to the exterior. When the voltage-gated sodium (in atrial and ventricular cardiomyocytes) or calcium channels (in nodal cardiac cells) open, the membrane depolarizes (the membrane potential becomes more positive), giving rise to an AP. The cardiac AP has a key role in coordinating the contraction of the heart. The cardiac cells of the sinoatrial node provide the pacemaker potential, which determines the heart rate. The APs of these cells propagate through the atria to the atrioventricular node, which is normally the only conduction pathway between the atria and the ventricles. APs from the atrioventricular node travel through the bundle of His and then to the Purkinje fibres system to activate the ventricles in an orderly fashion. Abnormalities in the cardiac AP (due to congenital mutations, exposure to toxins or drugs or injury) can lead to the development of cardiac arrhythmias.
There are four phases to the cardiac AP in the contracting atrial or ventricular cardiac cells (FIG. 3). The sharp depolarization (phase 0) is due to inward movement of sodium ions, whereas repolarizations (phases 1 and 3) are principally due to outward movement of potassium ions. During the phase 2 plateau period, the cells are maintained in a depolarized state achieved through a balance between the inward movement of calcium ions and the outward movement of potassium ions.
Fig. 3 |. Ventricular action potential and ionic currents.
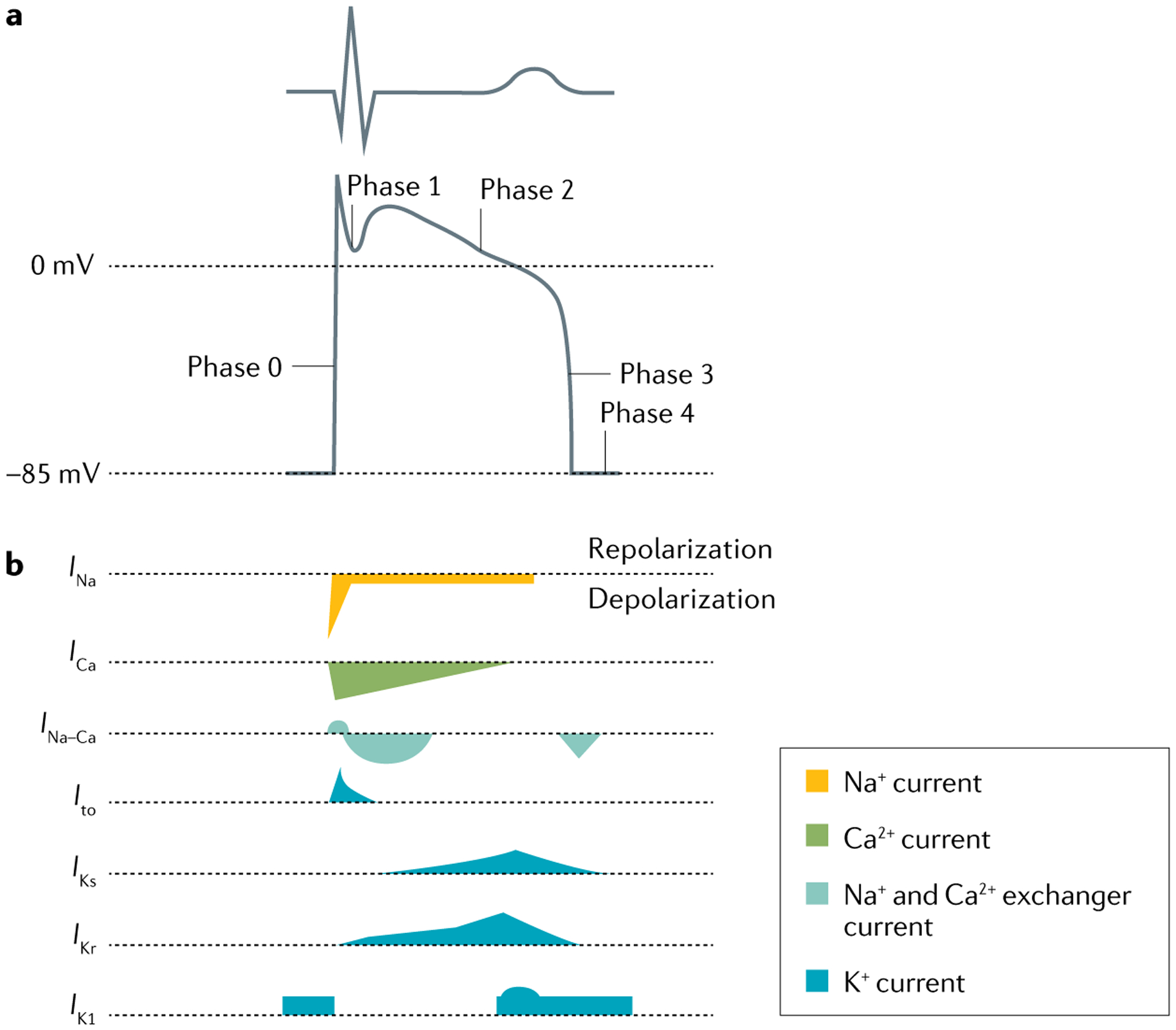
a | Schematic showing a normal electrocardiogram trace and the corresponding phases of the ventricular action potential (0, 1, 2, 3 and 4) that determine the shape of the trace. b | The major transmembrane ionic currents that generate the ventricular action potential. Inward currents that contribute to depolarization are oriented downwards, and outward currents that contribute to repolarization are oriented upwards; the shapes of the currents indicate their relative intensity. Ito, transient outward potassium current.
SDR as a common link in arrhythmogenesis.
The four channelopathies thus far discussed differ with respect to the characteristics of the QT interval. In LQTS and in SQTS, the QT interval prolongs or shortens, respectively, as a result of the disease, whereas in BrS and early repolarization the QT interval remains largely unchanged or abbreviates. However, these four syndromes have in common an amplification of the spatial dispersion of repolarization (SDR)77, which results in the development of polymorphic ventricular tachycardia (VT) and fibrillation when dispersion of repolarization and refractoriness reach the threshold for reentry. When polymorphic VT occurs in the setting of long QT, it is referred to as Torsades de Pointes according to its specific morphology. The threshold for reentry decreases as the AP duration and refractoriness are reduced and the conduction path length required for establishing a reentrant depolarization wave is progressively reduced77. Of note, the conduction path length required for establishing reentry is also importantly influenced by conduction velocity, such that reentry is facilitated when conduction is slowed. It has to be said that the hypothesis supporting a major role of SDR has largely originated from experiments in canine wedge preparations78, with interpretations that are not uniform; indeed, several electrophysiologists have different views as to the arrhythmic substrate in some of these conditions79.
LQTS.
In LQTS80–82, a reduction of net repolarizing current secondary to loss of function of outward potassium channel currents or gain of function of inward currents underlies the prolongation of the myocardial AP and QT interval that attend both congenital and acquired LQTS83–85. Acquired LQTS is most of the time secondary to drugs that have an IKr-blocking effect86. Accentuation of SDR and refractoriness within the ventricular myocardium, secondary to exaggerated trans-septal or transmural dispersion of repolarization, has been identified as the principal arrhythmogenic substrate in LQTS87,88. This exaggerated intrinsic heterogeneity together with early afterdepolarization-induced triggered activity, both caused by a reduction in net repolarizing current, underlie the substrate and trigger the development of Torsades de Pointes observed in LQTS87. The mechanism underlying Torsades de Pointes has been a matter of debate for many years. In addition to reentrant activity, early afterdepolarization-mediated focal activity has been proposed. Although short episodes of Torsades de Pointes may be due to focal activity, longer and non-terminating episodes are always maintained by reentrant activity89.
SDR is further exaggerated by sympathetic influences, especially in LQT1 and LQT2, accounting for the great sensitivity of these patients to adrenergic stimuli. It is important to keep in mind that neurally mediated sympathetic activation acts through release of noradrenaline at the neural terminals in the ventricles, which has consequences different from those produced by blood-borne catecholamines, and the effects of noradrenaline as a neurotransmitter are different from those produced by circulating catecholamines. Locally released noradrenaline increases the heterogeneity of repolarization and facilitates ventricular fibrillation90; this effect is very different from that of adrenaline and, experimentally, of isoprenaline (a β-adrenergic receptor agonist).
SQTS.
SQTS is an extremely rare channelopathy, characterized by pathologically accelerated repolarization resulting in very short QT intervals and the appearance of tall peaked T waves on the ECG. The augmented Tpeak–Tend interval associated with this ECG feature suggests that SDR underlies the arrhythmogenic substrate in the ventricles. Experimental studies suggest that the abbreviation of AP duration in SQTS is heterogeneous, owing to preferential abbreviation in the epicardium and resulting in an increase in SDR. Dispersion of repolarization and refractoriness serve as substrate for reentry, as they promote unidirectional conduction block. Marked abbreviation of wavelength is an additional factor promoting the maintenance of reentry. Abbreviation of AP duration and effective refractory period and amplification of SDR also predispose to the development of atrial fibrillation by creating the substrate for reentry91.
Recent studies involving computational modelling of SQTS92, induced pluripotent stem cell cardiomyocytes (iPS-CMs) isolated from patients with SQTS that recapitulate the SQTS cellular phenotype93–96 and data from a transgenic rabbit model of SQT1 have provided useful insights into arrhythmogenesis and arrhythmia substrates in SQTS consistent with previous findings. Nevertheless, there is no clear understanding of the mechanism underlying the premature ventricular contractions (PVCs) that precipitate polymorphic VT in SQTS.
CPVT.
Mutations in RYR2, calsequestrin 2 (a calcium-binding protein that acts as a calcium buffer within the SR), calmodulin and triadin, which underlie CPVT, all lead to abnormal regulation of cellular calcium homeostasis97,98. This disruption leads to excessive calcium accumulation (overload) in the SR and spontaneous release of calcium from the SR in the cytoplasm, causing augmentation of transient inward current and the development of delayed afterdepolarizations. Several lines of evidence point to delayed afterdepolarization-induced triggered activity as the mechanism underlying mono-morphic or bidirectional VT in patients with CPVT. Sympathetic influences greatly amplify the calcium dysregulation, leading to precipitation of episodes of CPVT during exercise and accounting for the respon-siveness of patients to β-adrenergic receptor blockade and sympathetic denervation99,100.
Several studies have reported that abnormal calcium handling can result from missense mutations or post-translational modification, including oxidation and S-nitrosylation, and several plausible mechanisms have been proposed — for example, defective RYR2 interaction with peptidyl-prolyl cis–trans isomerase (FKBP) proteins owing to phosphorylation of RYR2 at serine 2808 by cAMP-dependent protein kinase A, phosphorylation of RYR2 at serine 2814 by calcium/calmodulin-dependent protein kinase type II, store overload-induced Ca2+ release, defective intrasubunit domain interaction in RYR2 (domain unzipping) or disruptions in calsequestrin 2 (REF.53). Finally, defective calmodulin binding to RYR2 has also been shown to be crucial to induce CPVT101–103.
The ectopic activity originating from the epicardium is associated with an increased Tpeak–Tend interval and augmented SDR due to reversal of the normal activation sequence across the ventricular wall. The increased SDR in turn creates the substrate for reentry, permitting the induction of polymorphic VT104. Propranolol (a β-adrenergic receptor blocker) and verapamil (an L-type calcium channel blocker) suppressed all arrhythmic activity in experimental wedge preparations104. Thus, an accentuation of SDR may play a key part in the transition of bidirectional VT to polymorphic VT and ventricular fibrillation in the setting of CPVT. However, a different conclusion comes from a study in transgenic mice, which provided evidence that the His–Purkinje system can be an important source of delayed afterdepolarization-induced triggered activity giving rise to focal arrhythmias in CPVT98,105. Currently, we are left with these two hypotheses.
BrS.
Prominent J waves in the ECG are associated with BrS and early repolarization. The electrocardiographic J wave is the result of a transmural voltage gradient created by the presence of a transient outward potassium current (Ito) mediated by the potassium voltage-gated channel subfamily D member 3 (also known as KV4.3) that creates a notch in the shape of the AP — that is, in the first part of phase 2, in the ventricular epicardium but not the endocardium106–108 (FIG. 4). The transmural gradient and associated J wave is much greater in the right ventricle than in the left ventricle, particularly in the region of the right ventricular outflow tract (RVOT), because of a more prominent Ito-mediated AP notch in the right ventricular epicardium109. This distinction explains why BrS is a right ventricular disease. The RVOT origin of BrS could also be attributed to the reduced expression of gap junctions and sodium channels110. By contrast, in the left ventricle, Ito is most prominent in the inferior wall, accounting for why early repolarization occurs in the left ventricle, with the most prominent J point elevation appearing in the inferior ECG leads111. The cellular mechanism underlying early repolarization is similar to that of BrS111. The principal difference is the region most involved in generating the arrhythmogenic substrate: the epicardial cells in the left ventricular inferior wall are most susceptible to early repolarization owing to a high density of KV4.3 channels111. Using ECG imaging techniques, a study demonstrated markedly abbreviated activation–recovery intervals and marked dispersion of repolarization in the inferior and lateral regions of the left ventricle in patients with early repolarization112. However, recent epicardial mapping data demonstrate abnormal conduction (fractionated epicardial signals — that is, multiple deflections in the QRS complex) in 75% of the patients with early repolarization79. The authors concluded that these data demonstrate that two distinct substrates — delayed depolarization and abnormal early repolarization — underlie inferolateral J wave syndromes (A.A.M.W.). This interpretation is challenged by data showing that, in experimental models of the J wave syndromes, fractionated ECGs giving rise to J waves are due to delayed repolarization, as discussed below (by C.A.).
Fig. 4 |. Spatial dispersion of repolarization.
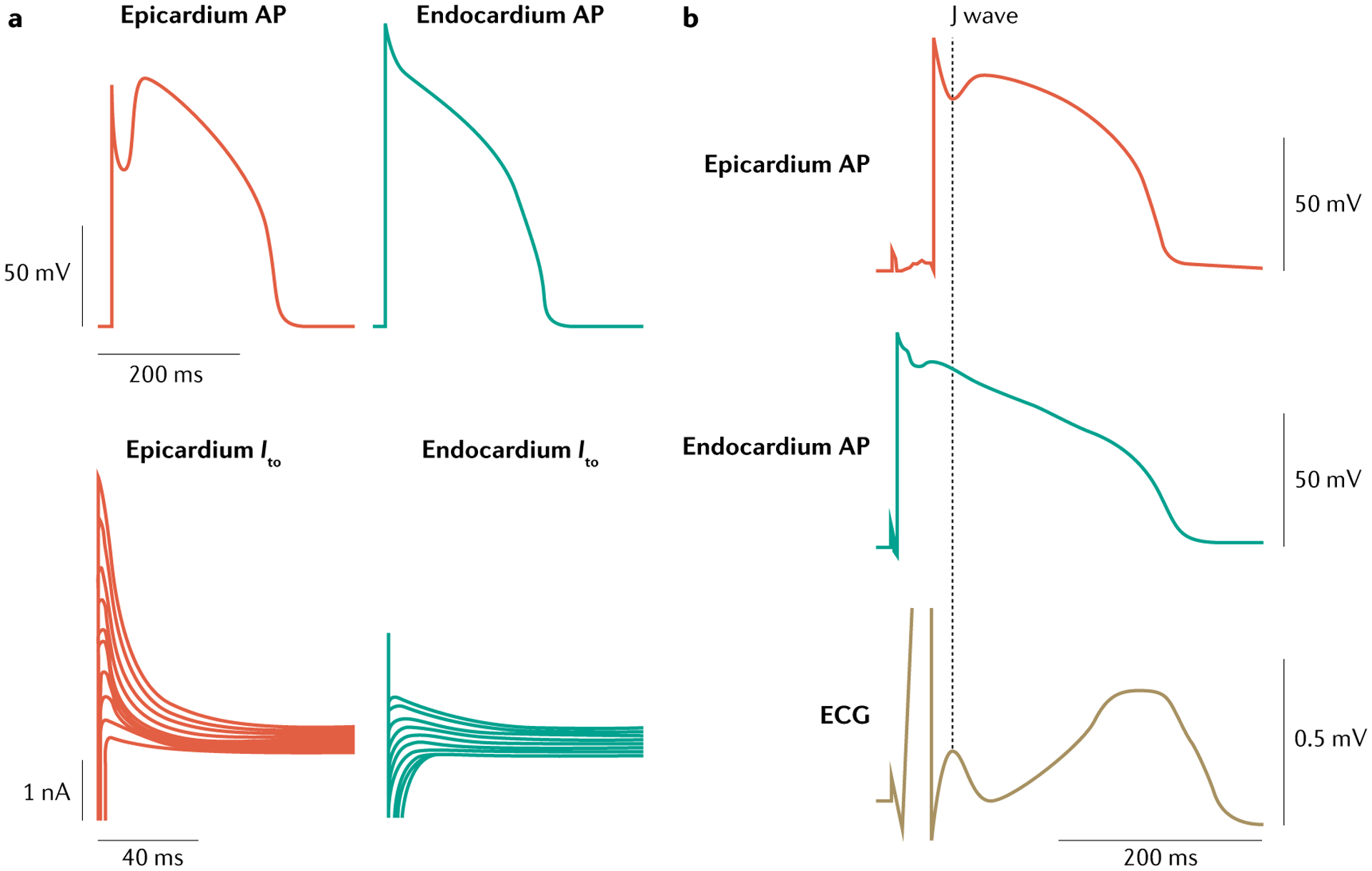
a | A prominent transient outward current (Ito) is responsible for phase 1 of the action potential (AP), giving rise to a prominent AP notch in the epicardium but not in the endocardium, where this current is relatively small. b | The presence of a prominent notch in the epicardium but not the endocardium gives rise to a transmembrane voltage gradient. Heterogeneous transmural distribution of the Ito-mediated AP notch is responsible for the inscription of the J wave on the electrocardiogram (ECG). Part a adapted from REF.253, Liu, D. W., Gintant, G. A. & Antzelevitch, C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ. Res. 72(3), 671–687 (https://www.ahajournals.org/journal/res). Part b adapted from REF.108, Yan G. X. & Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation 93(2), 372–379 (https://www.ahajournals.org/journal/circ).
The ionic and cellular mechanisms underlying BrS have long been a matter of debate113,114. Two principal hypotheses have been proposed, the repolarization hypothesis and the depolarization hypothesis. These two hypotheses are next presented by their two proponents, respectively C.A. and A.A.M.W. These arguments and observations notwithstanding, it stands to reason that the repolarization and depolarization hypotheses are not mutually exclusive and may indeed be synergistic. The repolarization hypothesis, also alluded to in the previous paragraph, asserts that an outward shift in the balance of currents at the end of phase 1 of the right ventricular epicardial AP, due to genetic mutations in genes encoding ion channels, generates the BrS ECG phenotype and the underlying repolarization abnormalities (that is, a heterogeneous loss of the epicardial AP dome) responsible for the development of both the epicardium to endocardium gradient and transmural SDR. These repolarization abnormalities could lead to the development of reentry during phase 2 of the AP, thereby generating closely coupled premature beats, which can trigger polymorphic VT or ventricular fibrillation. By contrast, the depolarization hypothesis maintains that fibrosis and reduced expression of gap junction α1 protein (also known as connexin 43) and sodium channels in the RVOT110 lead to discontinuities in conduction that are responsible for the development of the ECG and arrhythmic manifestations of BrS.
C.A.
In BrS, the amplitude of J waves, typically appearing as type I ST segment elevation, diminishes at fast heart rates, owing to reduced availability of Ito, consistent with the repolarization hypothesis113,115. The ability of agents such as quinidine (a sodium channel blocker and a blocker of Ito) to reduce ST segment elevation and exert an ameliorative effect in BrS is likewise attributable to a reduction in Ito. These observations lend support to the repolarization hypothesis, as does the demonstration of a lack of conduction delay in the RVOT in a whole heart experimental model of BrS116 and the demonstration of an ameliorative effect following radiofrequency ablation of RVOT, a procedure that destroys the epicardial cells with the most prominent Ito-mediated AP notch117–119.
A.A.M.W.
In every debate on the pathophysiological substrate of the ST segment elevation in the right precordial ECG leads, the ability of quinidine to reduce ST segment elevation is prominently mentioned (see also the previous paragraph). This argument seems valid, given that the sodium blocking activity of quinidine is expected to be detrimental (that is, increasing the level of ST segment elevation), but it is also important to consider that the AP morphology is a crucial determinant for the safety of conduction. This factor is particularly relevant in areas where conduction is potentially hampered — for example, in areas with substantial fibrosis (such as the RVOT in patients with BrS)119 and in Purkinje–ventricular junctions. Hence, every intervention that alters the AP morphology (for example, decreases in Ito or any other early potassium current or an increase in ICa, among others) can improve the safety of conduction120,121 and, therefore, can be compatible with the depolarization hypothesis. Quinidine blocks Ito and, therefore, can increase the safety of conduction in regions in which conduction is compromised.
Diagnosis, screening and prevention
Clinical manifestations
LQTS.
Patients with LQTS may remain without symptoms throughout their life or may become symptomatic. Most symptomatic patients have their first arrhythmic event during the first 20 years of life, individuals with LQT1 or Jervell and Lange-Nielsen syndrome earlier than those with LQT2 or LQT3; at variance with males, females remain at risk throughout life122. The main clinical manifestations are syncopal episodes, cardiac arrest and SCD123,124. These events are due to Torsades de Pointes VT, which often degenerates in ventricular fibrillation. The occurrence or absence of arrhythmic events in a patient with a disease-causing mutation does not predict what may happen in their offspring125, with the exceptions of specific genetic variants that are associated with very high126 or very low127 clinical severity.
Most symptomatic patients show a distinct prolongation of the QT interval, which is often accompanied by a bizarre morphology of the T wave128,129. The arrhythmic risk increases significantly when QTc is >500 ms (REF.70) and especially when episodes of T wave alternans are observed130. Specific triggers for arrhythmic events in patients with LQTS have been identified; namely, swimming, running, unexpected noises (such as the telephone ringing, an alarm clock and thunder) or being frightened131. The identification of the LQTS-associated genes has revealed that, at variance with all other channelopathies, there is a strong genotype–phenotype correlation in the three LQTS subtypes, a correlation that is especially important for the recognition of the conditions that may trigger the arrhythmias122. Patients with LQT1 are at increased risk whenever sympathetic activity increases, as during emotional or physical stresses, especially swimming122,132. Patients with LQT2 are at increased risk when exposed to sudden noises, especially if they are at rest or asleep and are woken up abruptly; of note, sleep is not a homogeneously quiet state, as during rapid eye movement sleep there are bursts of both vagal and sympathetic activity that can be quite arrhythmogenic. Individuals with LQT2 are also very sensitive to low plasma levels of potassium, and female patients are at high risk during the postpartum period133,134, probably because of sleep disruption. Finally, patients with LQT3 are at risk primarily when they are at rest or asleep. Independently of the genotype, infants who have a cardiac event in the first year of life are at an especially high risk of mortality and often are not protected by the traditional therapies135. LQTS, as well as all other channelopathies, can contribute to sudden death in infancy. Almost 10% of infants who die suddenly in the first year of life carry LQTS-causing mutations136,137, and a prolonged QT interval in newborn babies increases the risk for sudden death138. It is evident that, without genetic testing, an infant who died suddenly in the first months of life would be labelled as a case of sudden infant death syndrome139,140. These data have conceptual implications. On one hand, they strengthen the rationale for wide-spread ECG screening in the first month of life10,138, with the objective to identify infants with LQTS who are at risk of dying in the first year of life or later141; on the other hand, they call for great restraint before assuming that recurrent sudden deaths in infancy always imply infanticide.
SQTS.
To date, just over 200 patients have been reported to have SQTS, and most clinical data come from two relatively large studies142,143. In both studies, a male predominance was reported, but the risk of cardiac arrest was similar in both sexes. Although the majority of patients with SQTS are symptomatic, SQTS may also be diagnosed in asymptomatic patients during family screening or as an incidental finding during routine ECG or during sport pre-participation screening. One study142 reported the long-term outcome of 53 patients with SQTS from the European Short QT registry (75% male, median age 23 years, follow-up period 64 ± 27 months); 62% of patients were symptomatic at presentation. The most common presenting symptom was cardiac arrest (33%). Most cardiac arrest events (>90%) occurred between 14 and 40 years of age in male patients. Thirteen patients (24%) had palpitations, and in six of these patients atrial fibrillation or atrial flutter were documented; eight patients (15%) had syncope. Atrial fibrillation was noted already in newborn babies and young children.
The second study143 included 73 patients with SQTS (84% male; mean age 26 ± 15 years, follow-up 60 ± 41 months); 53% of patients (n = 39) were symptomatic at presentation, and SCD or non-fatal sudden cardiac arrest (40%) were the most common presenting symptoms143. The second most common symptom was syncope (19%). The rate of cardiac arrest was 4% in the first year of life. The annual event rate was 1.3% between 20 and 40 years of age, and 40% of patients experienced a cardiac arrest by 40 years of age (cumulative probability of an annual event rate 0.9%). There was no reported trigger of cardiac arrest, and in 83% of patients cardiac arrest occurred during rest.
CPVT.
Phenotypically, CPVT most closely mimics LQT1, with adrenergic-triggered syncopes, seizures, sudden cardiac arrest or SCD. In fact, many patients with CPVT had been previously misdiagnosed with either atypical LQTS or exercise-triggered epilepsy49,99. Although CPVT can manifest at any time, sentinel events are most likely to occur during the first two decades of life and are uncommon after 40 years of age. Thus, CPVT should be suspected in any patient with a structurally normal heart and a normal resting ECG with a normal QT interval who experiences a sudden faint, seizure or cardiac arrest during exercise, exertion or emotional stress (positive or negative). In fact, in these settings, CPVT would be more plausible as the root cause than LQT1 with a normal QT interval.
Although it has been reported, CPVT is rarely the cause of sudden infant death syndrome136,137,144. By contrast, CPVT is one of the most common genetically identifiable explanations for sudden unexplained death in individuals of 1–35 years of age in whom autopsy results did not show structural heart defects, especially if the death occurred during exertional activity, particularly swimming145,146. In an otherwise unexplained drowning or near drowning, CPVT should be explored as a possibility. In addition, CPVT should be investigated in patients diagnosed previously with an exertion-triggered generalized seizure disorder, especially if the epilepsy diagnosis had been accompanied by a normal electro-encephalogram. CPVT also contributes to idiopathic ventricular fibrillation as shown in a genetic study in 76 survivors that documented that disease-causing mutations were present in 7% of cases147.
BrS.
The most frequent clinical manifestations of BrS include a type 1 ECG (see Diagnosis) and malignant ventricular arrhythmias148,149, more specifically rapid polymorphic VT or ventricular fibrillation, typically initiated by extrasystoles from the RVOT region. Rarely, isolated ectopic beats (most commonly with a left bundle branch block or inferior axis deviation morphology on the ECG) or non-sustained ventricular arrhythmias are observed. However, some patients with BrS may report syncopal events, which are probably caused by self-terminating arrhythmias and lead to an increased risk of malignant arrhythmias (see below). Arrhythmic events occur typically during the night150. Supraventricular arrhythmias, most often atrial fibrillation, have been recognized as part of the BrS phenotype since they were first described as a separate clinical entity148. The prevalence of atrial fibrillation may be as high as almost 40% in certain cohorts151. The first symptoms typically occur in the third to fourth decade of life (both in men and women); however, they can also occur in children and in individuals of >50 years of age152. Fever is a specific trigger for symptoms, particularly in the paediatric subgroup153,154. Males are more frequently affected than females, as shown by every study.
Three morphologies of the BrS ECG pattern have been described. Type 1 is characterized by a high J point with a coved-type ST segment in the right precordial ECG leads, often followed by a terminal negative T wave. Type 2 ECG has a saddle-back-shaped ST segment, with an elevation of ≥2 mm, and type 3 has a saddle-back type or a coved ST segment with an elevation of <2 mm. Another characteristic ECG feature is the presence of minor conduction delay at all cardiac levels, manifested by slight PR prolongation, QRS widening (fractionation) and an abnormal electrical axis, which can be either leftward (manifesting with a wide “S” in lead I) or rightward. Conduction delay occurs in particular in the presence of a loss-of-function sodium channel mutation155, which underlies the disease in approximately 20% of cases156.
Risk stratification in asymptomatic patients with BrS is ill defined. Most studies are retrospective in nature, and no single parameter, with the exception of a spontaneous (that is, not induced by administration of sodium channel blockers or fever) type 1 ECG, has consistently been shown to predict risk of malignant arrhythmias157. An asymptomatic patient with a spontaneous type 1 ECG has a yearly risk of ~1% to present with malignant arrhythmias, and such risk may or may not be considered sufficient to warrant the implant of an implantable cardioverter–defibrillator (ICD), whereas the risk in symptomatic patients is ~3–7% per year, depending on the presenting symptom. Probably the most reliable risk factor is a (progressive) fractionated ECG in the right precordial leads157. Programmed electrophysiological stimulation (that is, stimulation of the heart by intracardiac catheters following a predesigned stimulation protocol) may help to estimate the risk, provided a low-intensity protocol is used158.
Diagnosis
LQTS.
The diagnosis of typical cases (such as a youngster who experienced a syncope during emotional or physical stress and has a prolonged QT interval) is quite straightforward. The upper limits of normal QTc (with Bazett’s correction) are 440 ms for males and 460 ms for females. The normal mean QTc is 400 ± 20 ms. Accidental diagnoses are often triggered by medical visits and ECG ahead of participation in sports, especially in countries such as Italy where these visits are mandatory by law. In asymptomatic individuals, especially when the QT interval is only modestly prolonged, the diagnosis is more complex and requires experience and additional tests. To help clinicians, a diagnostic score has been developed and upgraded over the years80,124,159,160 (BOX 1). The accurate measurement of the QTc duration at rest is most informative both for diagnosis and prognosis. More and more frequently the so-called tangent method is used, on the basis that it facilitates the identification of the point where the descending limb of the T wave crosses the baseline. However, this method is actually misleading whenever the descending limb of the T wave has a slow component, which happens often when there is an impairment of IKs and IKr. The tangent method can, therefore, lead to an underestimation of the QT interval and should not be used when there is a suspicion of LQTS. The morphology of the T wave is also indicative, especially in the precordial leads, and often suggests the probable underlying genotype (FIG. 5). The exercise stress test almost never induces arrhythmias, at striking variance with CPVT, but can provide three important pieces of information about QT interval adaptation (or lack of it) to R–R interval shortening, T wave changes in the first minutes of recovery161,162 — both useful parameters to support the diagnosis of LQTS — and the rapidity of the reduction in heart rate during the first minute following cessation of exercise163, which is important for risk stratification. The 24-h, 12-lead Holter ECG recording provides almost invaluable information, especially about cardiac activity during the night, because it is typical of LQTS to present even gross, albeit sudden and brief, changes in T wave morphology. Finally, in several patients, there are specific mechanical alterations, which can be identified by echocardiography164–169.
Box 1 |. 1993–2011 long QT syndrome diagnostic criteria.
ECG findings
Electrocardiogram (ECG) findings can be included in the score in the absence of medications or disorders known to affect ECG features. Corrected QT (QTc) is calculated by Bazett’s formula, in which QTc = QT/√RR.
QTc 450–459 ms (in male individuals) (1 point)
QTc 460–479 ms (2 points)
QTc ≥480 ms (3 points)
QTc on fourth minute of recovery from exercise stress test ≥480 ms (1 point)
Torsades de Pointes (2 points)
T wave alternans (1 point)
Notched T wave in three leads (1 point)
Resting heart rate below the second percentile for the patient’s age (0.5 point)
Clinical history
Syncopea with stress (2 points)
Syncopea without stress (1 point)
Congenital deafness (0.5 point)
Family history
Family members with definite long QT syndrome (LQTS) diagnosisb (1 point)
Unexplained sudden cardiac death before 30 years of age among first-degree relativesb (0.5 point)
A score of ≤1 point indicates a low probability of LQTS, a score of 1.5–3 points indicates an intermediate probability of LQTS, and a score of ≥3.5 points indicates a high probability of LQTS.
aMutually exclusive. bMutually exclusive. Adapted from REF.160, Schwartz P. J. & Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation 124(20), 2181–2184 (https://www.ahajournals.org/journal/circ).
Fig. 5 |. Examples of ECG traces.
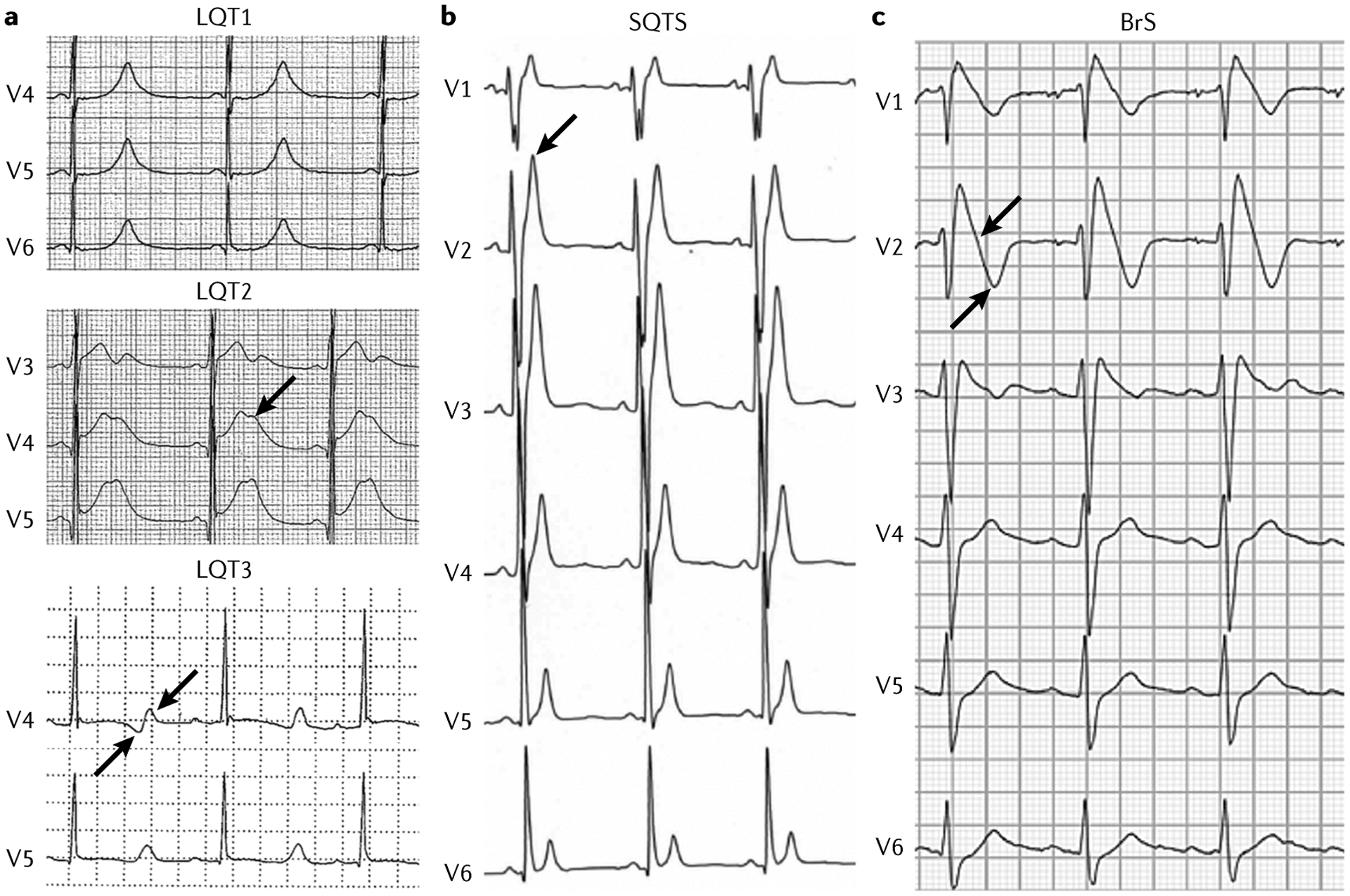
a | Example of electrocardiogram (ECG) traces of patients with long QT syndrome (LQTS). In LQTS type 1 (LQT1), the QT interval is extremely prolonged, with a tall peaked T wave. In LQT2, a very prolonged QT interval is present, with a clear and typical notch on the T wave (arrow). In LQT3, a very prolonged QT interval is followed by a late-onset diphasic (arrows) T wave. b | In a patient with short QT syndrome (SQTS), the QT interval is extremely short, with a tall peaked T wave (arrow). c | A patient with Brugada syndrome (BrS) has an ECG trace with a typical type 1 pattern. There is a coved-type ST segment (arrow) in the right precordial leads followed by a terminal negative T wave (arrow).
Since the late 1990s, genetic testing has not only aided the diagnosis of LQTS but has also enabled the identification of the specific genotype and family members of patients who also carry the causative mutation (and are therefore at risk) but have a borderline QT interval duration and would probably escape diagnosis and not receive protective measures. Of course, the results of genetic tests must be interpreted correctly, and physicians who care for these patients should know the difference between a disease-causing mutation and a variant of uncertain significance170. Once the disease-causing mutation has been identified in the proband, the entire family should undergo cascade genetic screening, which, within 2–3 weeks, can identify the relatives who are genotype-positive and those who are genotype-negative68,69.
SQTS.
The hallmark of SQTS is a very short QT interval. SQTS was initially recognized in patients with a QTc <300 ms. Subsequently, patients with slightly or moderately shortened QT intervals (≤360 ms) and similar clinical presentations were also described47,142,171,172. Several population and cohort studies showed that the prevalence of individuals with QTc <360 ms is very low. In a large Finnish cohort, the prevalence of patients with a QTc <340 ms was 0.4%, and that of patients with a QTc <320 ms was 0.1%173. In a paediatric population, the prevalence of QTc ≤340 ms was 0.05%174. These studies revealed that short QT interval alone was not associated with increased risk of arrhythmic events. Thus, the cut-off value of QT interval for defining SQTS remains a matter of debate, as there is an overlap between healthy individuals and patients with SQTS. In the 2015 guidelines of the European Society of Cardiology175, a cut-off value of QTc ≤340 ms was recommended for the diagnosis of SQTS (class I, level of evidence C). SQTS should be also considered in patients with QTc ≤360 ms in the presence of one or more of the following factors: a confirmed pathogenetic mutation; a family history of SQTS; a family history of sudden death before 40 years of age; and non-fatal VT or ventricular fibrillation episodes in the absence of structural heart disease (class IIa, level of evidence C).
Owing to the impaired QT adaptation to heart rate changes in patients with SQTS, experts recommend measuring the QT interval on ECG with a heart rate between 50 and 70 beats per min. Other prevalent ECG patterns that could be helpful in establishing the diagnosis of SQTS include PQ segment deviation (observed in 81% of patients with SQTS), a pattern of tall and symmetrical T waves with a very short or missing ST segment, early repolarization (in 65% of patients with SQTS), frequent presence of apparent U waves (especially in precordial leads) and impaired adaptation of the QT interval during exercise176–180. Furthermore, overlapping syndromes have been described in patients who had non-fatal sudden cardiac arrest and presented with a BrS-like ECG pattern and a shorter than normal QT interval. As in LQTS, genetic testing is recommended for patients with suspected SQTS. However, similarly to BrS, a causative mutation is identified only in ~20% of cases and, therefore, genetic test results must be interpreted very carefully.
CPVT.
In a child, adolescent or young adult with the clinical manifestation described earlier, CPVT should always be considered. In CPVT, the heart is virtually always structurally normal, and, in fact, identification of a structural abnormality should rule out a CPVT diagnosis. Similarly, the 12-lead ECG at rest is essentially normal in CPVT99,181. The diagnosis of CPVT is established in a clinically symptomatic patient who has a structurally normal heart by cardiac imaging and a normal ECG at rest but an abnormal exercise stress test (FIG. 6). The appearance during exercise of bidirectional VT is regarded as pathognomonic for CPVT. Although bidirectional VT provoked in this setting is extremely specific for CPVT, it is seen only rarely.
Fig. 6 |. Catecholaminergic polymorphic ventricular tachycardia exercise test.
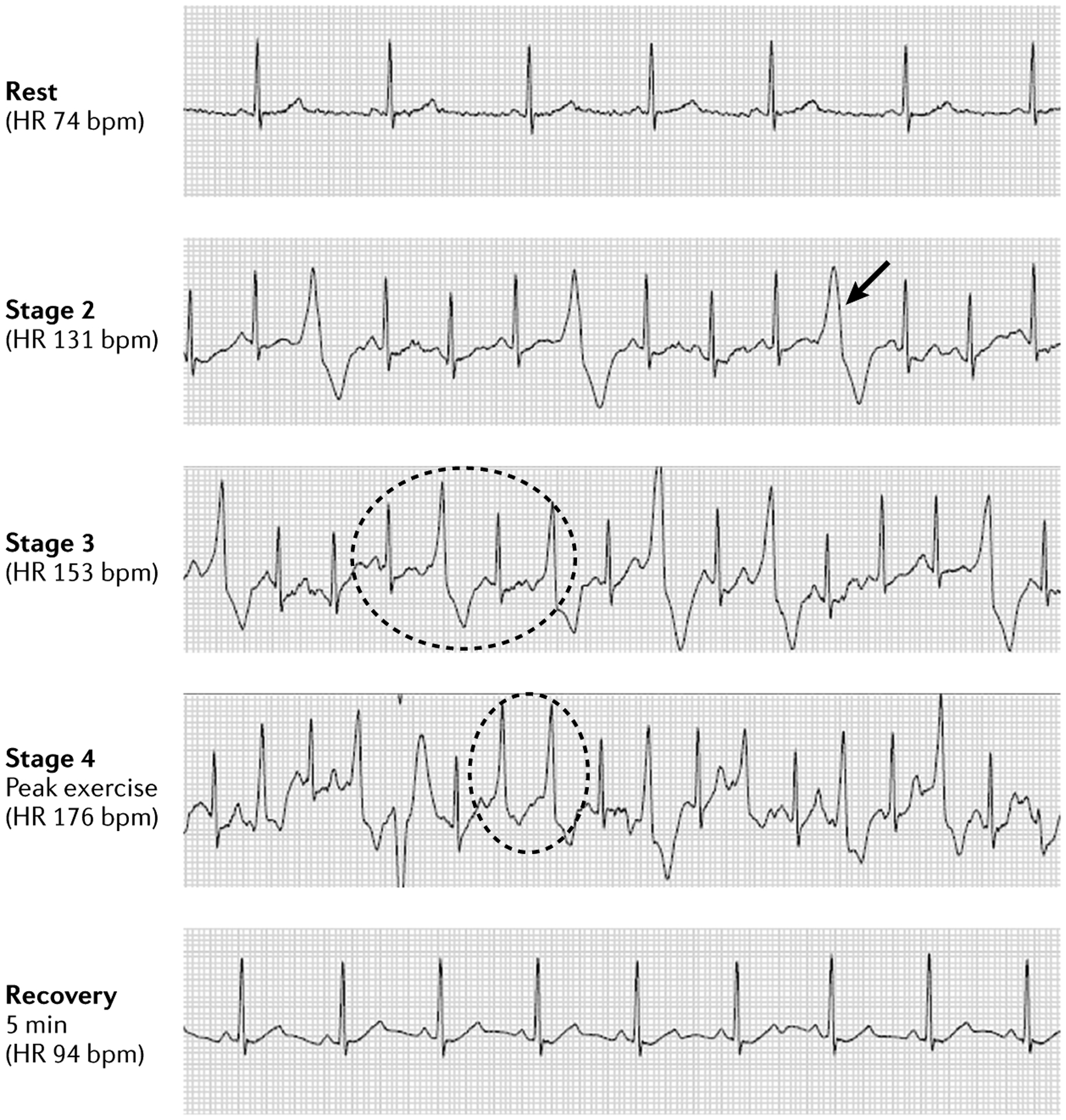
Exercise stress test in a patient with catecholaminergic polymorphic ventricular tachycardia as confirmed by genetic analysis. Sinus rhythm was normal at rest, with ectopy increasing with exercise. The patient demonstrated single premature ventricular contractions (PVCs) (arrow) with increasing frequency as the heart rate (HR) progressively increased, transitioning to PVCs in bigeminy (dotted circle in stage 3) with couplets (dotted circle in stage 4) and triplets at peak exercise. Ectopy disappears after 5 min of recovery, and normal sinus rhythm is restored. bpm, beats per minute.
The characteristic stress test profile of most patients with CPVT involves normal sinus rhythm at rest, with the onset of single ventricular extrasystoles (PVCs) starting when the sinus heart rate reaches ~110–130 beats per minute. As the heart rate increases, the single PVCs (which often originate from the RVOT) increase in frequency and then progress to PVCs in bigeminy. Next, the bigeminal pattern gives way to PVC couplets, including bidirectional couplets, and then occasionally non-sustained VT, polymorphic VT and, rarely, bidirectional VT at higher heart rates. Sometimes, the arrhythmic pattern that manifests during exercise often ceases at the highest heart rates, and normal sinus rhythm then persists throughout the recovery phase, or, if arrhythmia persisted at the highest heart rates, the arrhythmia ceases almost immediately in the recovery phase. Accordingly, in the patient with a history consistent with CPVT and normal echocardiography and ECG, the presence of exercise-induced arrhythmia in the pattern described that culminates in bidirectional PVC couplets means that the diagnosis of CPVT is very likely to be correct, even though bidirectional VT did not occur.
In a patient with suspected CPVT on the basis of clinical manifestations and cardiological testing, CPVT genetic testing is indicated22. The vast majority of CPVT cases stem from pathogenetic missense variants in RYR2, and a rare missense variant can be identified in 60–70% of patients with a robust clinical diagnosis of CPVT52. Of note, a rare missense variant in RYR2 can also be found in 3% of healthy, asymptomatic individuals52. In other words, when the clinical evidence for a CPVT diagnosis is compelling, identification of a rare missense variant in RYR2 has at least a 20:1 chance of being the pathogenetic mutation of CPVT type 1. In accordance with the 2013 HRS/EHRA/APHRS guidelines, the identification of a CPVT type 1 causative mutation is deemed equivalent to a clinical diagnosis of CPVT that should compel cascade, variant-specific genetic testing of all appropriate relatives and the possible initiation of prophylactic β-adrenergic receptor blocker therapy even in asymptomatic family members with a normal stress test who are positive for the variant9.
BrS.
BrS is diagnosed in the presence of a characteristic ECG pattern including a spontaneous type 1 pattern with a coved ST segment elevation in one or both of the right precordial leads V1 and V2, positioned in the second, third or fourth intercostal space17. A type 1 ECG is mandatory for the diagnosis; only when a type 2 or type 3 ECG converts into a type 1 pattern (that is, with a drug test) can the diagnosis of BrS be made. As described earlier, type 1 morphology reflects a coved-type ST elevation of ≥2 mm (0.2 mV) in the right precordial leads followed by a negative terminal ST segment182 (FIG. 5). In the absence of a spontaneous abnormal ECG with the V1 and V2 leads in the standard positions, the diagnostic sensitivity of the ECG can be increased by placing the right precordial leads higher, administering a sodium channel blocker (the drug challenge test, with ajmaline being the most effective drug, although the sensitivity and specificity of these tests is unknown) or increasing the vagal tone (that is, by recording the ECG after the patient has had a meal or exercised)183. These procedures (preferably a drug challenge test or, alternatively, a ‘heavy meal test’ or exercise test) should be performed in any patient with a reasonable suspicion of BrS with a non-diagnostic baseline ECG. Additional clinical criteria are required to reach the diagnosis in cases in which a type 1 ECG is only observed after drug challenge test or during fever17, and a 12-lead 24-h Holter ECG recording performed at regular intervals can be useful. Of note, several differential diagnoses are possible even when the ECG shows the typical type 1 form184.
Management
Pharmacological therapy
LQTS.
The pharmacological therapy of LQTS is rather straightforward. With very few exceptions (see below), β-adrenergic receptor blockers should be given to every diagnosed patient, because the risk of a fatal first event is high. The only two β-adrenergic receptor blockers that are effective beyond doubt are propranolol and nadolol124. Metoprolol should not be used185, as it is not effective; concerns about efficacy exist also for atenolol186, and available data about the other β-adrenergic receptor blockers are insufficient. There is no justification to risk the patients’ lives with drugs of uncertain efficacy. The few patients in whom not starting β-adrenergic receptor blocker therapy may be considered are asymptomatic males with LQT1 off therapy by 25 years of age122 and genotype-positive but phenotype-negative (that is, with a normal QTc) individuals. Incorrect considerations have led for several years to the misconception that β-adrenergic receptor blockers would not be useful, and potentially dangerous, for patients with LQT3; it is now evident that these drugs are very effective also for these patients135,187.
The understanding of the mechanism of action of SCN5A mutations has led to the proposal of using the sodium channel blocker mexiletine to shorten the QT interval in patients with LQT3 (REF.188). This first example of gene-specific therapy was successful and was confirmed by recent studies189. Whenever mexiletine shortens the QTc by >40 ms in patients with a baseline QTc >500 ms, this drug should be added to therapy as recommended in 2005 (REF.190) and also by the HRS/EHRA/APHRS guidelines9. Very recently, groups in the Mayo Clinic and Milan have observed that mexiletine effectively shortens QTc also in a small series of patients with LQT2, and this assessment has now become routine in these two large referral centres191. A practical point to be kept in mind is the simplicity and rapidity by which it is possible to determine whether or not mexiletine is effective in a patient, even without knowing the functional characteristics of the mutation that the patient carries. Indeed, the therapeutic plasma concentration is reached within 90–120 minutes from administration of half of the daily oral dose, and, by monitoring the ECG for 2 hours, it is evident whether or not QTc shortens by at least 40 ms; if the QTc does shorten, the patient is considered a responder, and mexiletine can be added to the therapeutic regimen. Despite high hopes, there are insufficient data about the efficacy of ranolazine and other new sodium channel blockers to recommend their use.
Finally, data on patient-specific iPS-CMs raise the possibility that the lumacaftor–ivacaftor combination (a drug that corrects trafficking defects and is used clinically for cystic fibrosis) might be useful in patients with LQT2 with mutations causing trafficking defects192. Indeed, the initial clinical data in the same patients whose iPS-CMs responded to lumacaftor seem to support the experimental observation193.
SQTS.
The main predictor for recurrent arrhythmic events in patients with SQTS is a non-fatal cardiac arrest event. Asymptomatic patients with QTc 300–360 ms should be monitored and followed up without any prophylactic medication194. Patients with markedly shortened QTc (≤300 ms) may be at increased risk of SCD, especially during sleep or rest. The only pharmacological therapy that leads to lengthening of the QTc and reduction of arrhythmic events is quinidine. Quinidine should be considered on a case-by-case basis in patients with increased risk of SCD and strong family history of SCD as a primary prevention (class IIb, level of evidence C)175. In patients with SQTS and recurrent ICD shocks, quinidine has been shown to prevent further ICD discharges142. Finally, emergency isoprenaline infusion can be effective in patients with an electrical storm or refractory ventricular fibrillation to restore and maintain sinus rhythm164.
CPVT.
β-Adrenergic receptor blocker therapy is the standard, first-line therapy in all patients with symptomatic CPVT who manifested self-limiting syncope or seizures as their sentinel event9,194. Nadolol is the preferred β-adrenergic receptor blocker in CPVT195,196. Preliminary evidence suggests that carvedilol may be a suitable alternative, but only if nadolol is either not available or not tolerated197. Combination drug therapy with a β-adrenergic receptor blocker and flecainide is increasingly utilized. In addition to its sodium channel blocker activity, flecainide may help to reduce diastolic calcium SR overload by stabilizing the ‘leaky’ RYR2s that stem from RYR2 defects198,199. A small, randomized, placebo-controlled trial confirmed the ability of flecainide to decrease the burden of exercise-triggered ventricular ectopy and arrhythmias200.
In general, among the largest CPVT centres throughout the world, the proxy to therapeutic success is the normalization of the patient’s stress test, with general tolerance for the occasional presence of PVCs in bigeminy that persist on therapy. If bidirectional PVC couplets or worse arrhythmias persist during follow-up stress testing, medication doses are increased, combination drug therapy is initiated201 or additional non-pharmacological therapies are considered.
BrS.
The options for pharmacological therapy are limited in BrS. Antiarrhythmic drugs can be life-saving in the acute phase of an arrhythmic storm. Intravenous isoprenaline is the most effective treatment, with an immediate effect on the arrhythmia burden and the ECG pattern, which may normalize. The drug that has received the greatest interest so far and on which multiple retrospective studies have yielded encouraging reports is quinidine202. Cilostazol and milrinone (inotropic agents) are other drugs with reported beneficial effects in some patients with BrS with arrhythmias17. Importantly, there is a long list of drugs that have to be avoided in patients with BrS203 (Brugadadrugs.org), including virtually all cardiac sodium channel blockers except quinidine.
Non-pharmacological therapy
LQTS.
Left cardiac sympathetic denervation (LCSD) and ICD are the two established non-pharmacological therapies that are the pillars for the management of LQTS, and genetic progress has enabled gene-specific management. Family members of patients with LQTS should receive cardiopulmonary resuscitation training, and, in particular, they should be taught the importance of precordial thump, as this manoeuvre, if correctly performed within 1 minute from the onset of loss of consciousness, almost invariably restores sinus rhythm. For children with LQTS, the acquisition of an automatic external defibrillator may be considered204.
Since the early 1970s, patients in whom β-adrenergic receptor blockers are not sufficient to prevent arrhythmic events have benefited from LCSD123,205–207. The rationale for this surgical intervention is well under-stood208 and largely hinges on the interruption of the release of noradrenaline from the left cardiac sympathetic nerves, which are quantitatively dominant at the ventricular level, and on the increase in the ventricular fibrillation threshold, which means that it is more difficult for a VT to degenerate into ventricular fibrillation209. As LCSD is a preganglionic denervation, there is no reinnervation or postdenervation supersensitivity210. The currently preferred surgical approach is by thoracoscopy124,211–213, which is simpler and less invasive than the traditional retropleural approach214. As LCSD has few to no contraindications, it is performed increasingly often. The main indication remains for patients not protected by β-adrenergic receptor blockers; however, LCSD is also indicated in primary prevention when a patient, albeit asymptomatic on β-adrenergic receptor blockers, keeps showing signs of high risk, such as a QTc >500 ms or T wave alternans207. LCSD is also effective in preventing multiple shocks in patients with an ICD207 and in low-risk patients who are intolerant to β-adrenergic receptor blockers. In the few cases of LCSD failure, it is recommended to proceed with right cardiac sympathetic denervation to obtain a complete bilateral sympathectomy. This sequential approach has been used with success since the late 1980s206,207. There is no justification for performing a bilateral cardiac sympathectomy in patients with LQTS or CPVT without having first assessed whether unilateral LCSD has failed; otherwise, the patients would undergo a longer and unnecessary surgery, doubling the risk of complications, and would be deprived without reason of an important component of the adrenergic control of their cardiac function. Importantly, the most common reason for post-LCSD breakthrough events is a suboptimal surgical procedure whereby only the left stellate ganglion is removed100,207. Indeed, to be effective, LCSD requires section of the lower half of the left stellate ganglion together with the first four thoracic ganglia208.
As a general rule, an ICD should be implanted whenever a patient has survived a cardiac arrest. However, as per the 2013 guidelines9, after a careful assessment, young patients with LQT1 who experienced a cardiac arrest while not on β-adrenergic receptor blocker therapy could be treated only with β-adrenergic receptor blockers and/or LCSD215. In all other patients, the pros and cons of ICDs should be very carefully considered, and the alternative option of LCSD should always be presented to the patients and their families216,217. The largest study on ICDs in LQTS has shown that a staggering 31% of patients have adverse events (for example, infection, lead dislodgement and tricuspid valve insufficiency) within 5 years from the implant, and the younger the patient, the higher the number of ICD substitutions that will be required over time218. A correct strategy is often to use both approaches — LCSD to prevent arrhythmic episodes and the ICD as a safety net.
Finally, the understanding of gene-specific triggers for arrhythmic events has led to gene-specific management for LQTS190. Patients with LQT1 should limit exposure to physical and, if possible, emotional stress; they should be allowed to swim but under the supervision of an adult who can swim. Their sport participation is questionable, depends on the local laws, and different views coexist219–222. Patients with LQT2 are at risk especially when exposed to sudden noises when they are at rest or asleep; alarm clocks and telephones should be avoided in their bedrooms. These individuals are also very sensitive to decreases in their plasma potassium levels. Potassium supplements should be given after repeated episodes of diarrhoea and potassium-sparing agents to patients with chronically low potassium levels. Women with LQT2 are at increased risk in the postpartum period and should avoid sleep deprivation, if at all possible. Their partners should take care of feeding the infant during the night-time, when breast-feeding can be avoided. The indications for patients with LQT3 are more uncertain, as arrhythmic events in these patients tend to occur at night. As night-time deaths in both LQT2 and LQT3 are often not silent223, it is recommended to have an intercom in the room if the patient is a child, and that adults sleep with another adult, as these measures would enable recognition of gasping noises and prompt intervention.
SQTS.
Patients with SQTS with a history of cardiac arrest or with documented spontaneous sustained VT are at increased risk of recurrent arrhythmic events (with an estimated risk of recurrent cardiac arrest of 10% per year) and, therefore, should receive an ICD for secondary prevention. Quinidine is recommended to reduce the number of ICD shocks. In patients who refuse ICD implantation, treatment with quinidine may be considered9,176.
CPVT.
The most rational and recommended non-pharmacological therapeutic option for CPVT is videoscopic LCSD surgery100,211,224. The LCSD surgical procedure is the same previously reported for LQTS. LCSD is indicated as a treatment intensification option for patients with recurrent sustained VT during stress testing or recurrent syncope despite receiving adequate or maximally tolerated β-adrenergic receptor blocker therapy194 or combination therapy.
The other option is the placement of an ICD. Because CPVT has a higher lethality per cardiac event than LQTS, the management of both symptomatic and asymptomatic patients with CPVT tends to be more aggressive, with increased ICD use. However, CPVT is the only entity in which ICD itself may contribute to not only morbidity but also mortality225–227. Indeed, ICD-associated comorbidities are far more severe than those of either pharmacological therapy or LCSD, and inappropriate shocks are an inherent painful and stressful experience that may elicit a lethal arrhythmic storm. In such situations, an initial inciting shock occurs inappropriately owing to sinus tachycardia or atrial fibrillation and activates the CPVT substrate, thereby precipitating a severe and ultimately fatal electrical storm225–227. The most recent analysis of the effect of ICD implants in patients with CPVT has concluded that ICD use should be limited as much as possible and LCSD should be favoured228. Presently, the largest and more experienced CPVT centres throughout the world increasingly aim to reserve ICD only for patients with CPVT with a clinical manifestation of sudden cardiac arrest that required external defibrillation resuscitation. However, even in these centres, if the sentinel event of sudden cardiac arrest occurred while the individual was undiagnosed and, therefore, untreated, it may still be possible to successfully implement a non-ICD treatment programme with triple therapy (nadolol, flecainide and LCSD) that may confer the same survival benefit as an ICD but without its comorbidities.
BrS.
Non-pharmacological therapy options consist of ICD implant and, more recently, epicardial ablation of the arrhythmogenic substrate (that is, areas that generate fractionated ECG) in the RVOT area. Patients who have survived a cardiac arrest or who have demonstrated ventricular arrhythmias have a class I indication for an ICD implant9. Epicardial ablation of the RVOT was first performed in nine severely symptomatic patients; after ablation of an area with abnormal low amplitude QRS voltages and late to very late (>200 ms) fractionated activity, in eight patients the ECG normalized, and recurrence of arrhythmic events was successfully prevented229. This epicardial approach was investigated in a larger cohort, which also included asymptomatic patients; the ECG manifestations disappeared after the procedure and could no longer be elicited by ajmaline or flecainide exposure, and in the 39 symptomatic patients no arrhythmia recurred in the short (10 months) follow-up period230. The exact role for epicardial ablation has to be established; of note, given the potential complications of this procedure (for example, tamponade or damage to the coronary arteries) and the relatively low event rate in asymptomatic patients with BrS with a type 1 ECG, a prophylactic epicardial ablation in asymptomatic patients raises major ethical questions and should be discouraged at present231.
Pregnancy-associated risk
This section focuses primarily on LQTS, as this channelopathy is the only one with adequate studies during pregnancy. The only study in CPVT to date indicates no augmented maternal risk during or after pregnancy232. Similarly, arrhythmic event rates in women with BrS do not seem to be elevated during pregancy233; thus, the management of BrS in pregnancy is limited to aggressive fever management and avoidance of medications that may provoke arrhythmias.
Maternal risk of adverse events.
Surveillance during pregnancy in women with channelopathies focuses on primary arrhythmia prevention with adequate β-adrenergic receptor blockade, monitoring for breakthrough arrhythmias and identification of high-risk women (that is, those with a history of syncope or cardiac arrest). There are abundant data supporting the safety of commonly used β-adrenergic receptor blockers in pregnancy134,234,235. The β-adrenergic receptor blockers with proven efficacy in LQTS are nadolol and propranolol. Atenolol should not be used in pregnancy, as it is associated with significant intrauterine growth restriction (pregnancy class D)236. The genetic subtype is also an important modulator of pregnancy-related arrhythmia risk. Postpartum cardiac event rates are <1% in LQT1 but reach 16% in patients with LQT2 (REF.133), and this observation mandates specific cautionary measures as described; there are no data for LQT3 (REF.133). Recent studies have also shown that the risks of miscarriage and stillbirth are increased in LQTS pregnancies, and the probability of fetal death is greater if the mother, rather than the father, carries the pathogenetic mutation (or mutations) (24.4% and 3.4%, respectively)237.
Labour and delivery management.
The management plan for labour and delivery should be individualized. Low-risk women (with no previous arrhythmic events) who are adequately treated with β-adrenergic receptor blockers can safely proceed with spontaneous vaginal delivery unless there are maternal or fetal indications for assisted or Caesarean delivery. Although active pushing increases the maximal heart rate, the heart rate response is expected to be blunted in a woman with adequate β-adrenergic receptor blockade. In pregnant women with LQTS, intrapartum rhythm monitoring is not necessary, as event rates are extremely low. However, cardiac tele metry is advisable during labour in women with CPVT, as arrhythmia can occur and progress from isolated PVCs to more complex and unstable ventricular arrhythmias. Caesarean delivery with maternal telemetry should be considered in pregnant women with poorly controlled arrhythmias or a history of cardiac arrest238.
Any pharmacological therapy in women with LQTS or BrS should be reviewed239. Oxytocin, commonly used during labour and delivery, has some potential for QT prolongation240, but this effect should not preclude the use of oxytocin when indicated241. A reasonable approach to oxytocin use is to optimize serum potassium and magnesium levels and to obtain an ECG at baseline and 1–2 h after oxytocin is initiated. The drug should be discontinued if the QTc exceeds 500 ms or increases beyond 60 ms from baseline86. Decisions regarding analgesia, including neuronal axial blockade, should be made according to maternal preference and estimated obstetric risk.
Management of the newborn baby.
Neonatal genetic screening for the familial pathogenetic variant should be performed. Because the QT interval is often prolonged in healthy newborn babies for the first 7–10 days242,243, 12-lead ECG screening of the neonate who is at risk of LQTS is unreliable for the diagnosis of LQTS until after the second week of life10, unless QTc of the newborn baby exceeds 500 ms. The Bazett method to calculate QTc is valid and useful also in infants244. Until the genetic test results are available, or if a variant of unknown significance is detected, ongoing clinical follow-up is recommended.
Pregnancy and risk of disease transmission.
When one of the parents is affected by a channelopathy and the disease-causing mutation is known, the cardiologist is often asked whether it is possible to prevent transmission of this potentially life-threatening disorder to the offspring. The cardiologist has the responsibility of providing the essential information on in vitro fertilization and pre-implantation genetic screening of the embryo, as this approach allows the possibility of favouring the development of the embryo without the mutation, thereby ensuring the offspring will not have the disease present in the family245–247.
Quality of life
The quality of life of the patients should be a primary concern for their doctors. For channelopathies, there are three main aspects to consider: the negative effects of certain therapies (mainly ICDs), the effect on sports participation and the specific concerns of patients with BrS.
ICDs are undoubtedly life-saving devices but, in these disorders, when physicians opt for an ICD they might be influenced to some extent by their own wish to avoid possible negligence investigations. ICDs often have a very negative effect on the quality of life, especially in young patients. In particular, patients with LQTS or CPVT are very sensitive to catecholamines, and the pain and fear generated by every ICD shock often contribute to initiate a vicious cycle of recurrent VT and/or ventricular fibrillation, which can result in an electrical storm with multiple ICD shocks. Experienced investigators limit the use of ICDs as much as possible in these patients124, and, barring a previous cardiac arrest, whenever feasible they use LCSD instead. Indeed, LCSD has been associated with a documented improvement in quality of life248,249.
For a youngster involved in competitive sports, having to stop practising following the diagnosis of a channelopathy is a major negative event with substantial psychological effects. Physician recommendation on whether it is appropriate to continue the sporting activity should take into account the local legislature (as in some regions the law forbids competitive sports for individuals with these diseases), the type of sports and the specific characteristics of the individual patient250. Owing to the pressure to perform by the other players, team sports are more dangerous than individual sports in which players may decide how much to push themselves. Phenotypic and genotypic features should also be considered (for example, patients with LQT2 or LQT3 are at somewhat lower risk than individuals with LQT1). The European Society of Cardiology is updating its previous consensus document251, which provided different perspectives compared with the more recent and more liberal consensus document by the American Heart Association and the American College of Cardiology250.
Asymptomatic patients with BrS and a spontaneous type 1 ECG have a unique problem, as they are usually told that their major risk is to die suddenly at night, and their options are limited to implanting an ICD, which would potentially be used in ~1% of them yearly, whereas the others would remain without cardiac events. Such a situation causes anxiety in these patients and their families. Thus, the mildly encouraging data with quinidine are welcome, as they enable doctors to encourage and reassure patients. A future therapeutic option could be prophylactic ablation of the RVOT area with abnormal QRS complexes. However, a multicentre randomized trial to demonstrate that the benefits out-weigh the complication rate of the ablation procedure in patients who are still asymptomatic is mandatory before this procedure enters regular clinical practice.
Outlook
LQTS
The current approach to LQTS management is overall very satisfactory, and sudden death among properly treated patients is rare nowadays, with the exception of infants presenting with cardiac events in the first year of life because of their poor response to therapy. However, too many patients who would benefit from LCSD do not receive it, as even in large centres there are few surgeons capable of performing LCSD or else this option is not offered to the patient. This same problem occurs in CPVT.
Two areas of ongoing active research have the potential of providing a very useful clinical fallout and, therefore, should be expanded. One is the field investigating genetic modifiers71, as increased knowledge of these factors could help to improve risk stratification, and understanding their mechanisms of action72 might lead to novel therapies. The second area involves the use of patient-specific iPS-CMs, as recent data193,252 raise the possibility that they might represent a useful tool to identify drugs that are effective against mutation-specific arrhythmogenic mechanisms.
SQTS
Patients with SQTS are very rare, as <200 cases have been reported; accordingly, data on risk stratification are lacking. The European Short QT registry should be expanded to better understand the natural course of this disease. Furthermore, we have only limited information on overlap syndromes such as the combination of a shorter than normal QT and BrS. Data on long-term treatment with quinidine are also lacking, and adherence to and tolerability of quinidine therapy remain a problem. Perhaps in the future the use of iPS-CM research will help to identify effective patient-specific drugs that can restore the functions of ion channels.
CPVT
Numerous advances are needed in the diagnosis, prognosis and therapy of CPVT. In one-third of individuals with a clinical diagnosis of CPVT, a pathogenetic defect cannot be identified, suggesting that some CPVT-associated genes have not yet been identified. In addition, long delays persist in the diagnosis of CPVT. In particular, young patients presenting with a sentinel event of sudden cardiac arrest are often recommended ICD implantation as a secondary prevention measure after completing a cardiological evaluation to rule out both structural and arrhythmic causes of SCD. However, such evaluation often fails to include an exercise stress test, thereby failing to test for possible CPVT. This must change.
The identification of patients who are at negligible risk remains difficult, and, therefore, universal β-adrenergic receptor blocker therapy remains the general recommendation. Nevertheless, treating all patients with CPVT with lifelong prophylactic β-adrenergic receptor blocker therapy on the basis solely of a positive CPVT genetic test is wholly unsatisfying, and much progress is needed in risk stratification. In addition, there is substantial heterogeneity throughout the world in the treatment of patients with previously symptomatic CPVT, ranging from β-adrenergic receptor blocker therapy only, to combination drug therapy with β-adrenergic receptor blockers and flecainide, combination therapy and LCSD, and ICD therapy only. We must increase awareness that, among these options for symptomatic patients, β-adrenergic receptor blockade monotherapy is probably insufficient and ICD monotherapy should never be recommended228. In the future, perhaps a patient who presents with sudden cardiac arrest and is subsequently diagnosed with CPVT may be effectively treated with combination drug therapy and LCSD rather than with ICD. Finally, regarding quality of life, continued observational studies are needed to determine whether, akin to some cases of LQTS, sports participation in a well-treated patient with CPVT might be a reasonable consideration.
BrS
BrS is probably one of the disease entities with the most controversial issues and open questions. The most pressing questions concern the pathogenesis (including the role of genetics), risk stratification and the best treatment options. Thus, future research should be directed towards these areas. Large patient registries, with detailed clinical data, will be needed to ultimately identify the clinical parameters that contribute to risk and to quantify the risk of individual patients, in particular asymptomatic patients. To date, conflicting data exist on most proposed risk factors157, and a pooled analysis of all BrS registries may be the way forwards. When combined with genetic data, such analysis could also identify genetic factors that contribute to risk stratification. The presence of more than four (out of six) risk alleles, identified through genome-wide association studies, is more likely to result in a type 1 ECG pattern67. This dataset is currently being expanded, and, in a much larger set of patients, additional risk alleles might be identified and more statistical power might be present to successfully associate the number of risk alleles with the risk of cardiac events.
Finally, the role of targeted epicardial ablation should be studied through multicentre randomized trials before individual centres start performing epicardial ablation of RVOT in asymptomatic patients. Invasive ablation procedures should be performed by very experienced clinicians in high-capacity centres and should also be used to gain more insight into the electrophysiological mechanisms of the ECG characteristics. Specific pacing protocols may be one way to provide this much needed insight.
RElatED links.
Brugadadrugs.org: www.brugadadrugs.org
European Reference Network for rare and Low Prevalence Complex Diseases of the Heart (ERN GUARD-HEART): http://guardheart.ern-net.eu
Acknowledgements
The authors thank P. De Tomasi (Istituto Auxologico Italiano, IRCCS, Center for Cardiac Arrhythmias of Genetic Origin, Milan, Italy) for an extraordinary editorial support. C.A. acknowledges support from NHLBI (HL47678, HL138103 and HL152201), the W.W. Smith Charitable Trust and the Martha and Wistar Morris Fund; C.R.B., P.J.S. and A.A.M.W. acknowledge the support of ERN GUARD-Heart; C.R.B. and A.A.M.W. acknowledge the support of the Netherlands Heart Foundation (CVON Predict2 project) and Leducq Foundation for Cardiovascular Research grant 17CVD02 “The sodium channel as a therapeutic target for prevention of lethal cardiac arrhythmias”; P.J.S. acknowledges the support of Leducq Foundation for Cardiovascular Research grant 18CVD05 “Towards Precision Medicine with Human iPSCs for Cardiac Channelopathies” and of ESCAPE-NET project (European Union’s Framework Horizon 2020 programme under grant agreement no. 733381). M.B. acknowledges support from the German Center for Cardiovascular Research (DZHK) and Hector Foundation. M.J.A. acknowledges support from the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program.
Glossary
- U wave
A small deflection immediately following the T wave and usually in the same direction.
- Polymorphic ventricular tachycardia
Rapid ventricular rhythm with a continuously varying QRS complex morphology.
- Refractoriness
Also known as refractory period. The period of depolarization and repolarization of the cell membrane after excitation during which the cell cannot respond to a second electrical stimulus.
- Reentry
A self-sustaining rhythm abnormality that occurs when the propagating electric impulse fails to conclude and the action potential propagates in a closed loop manner.
- Torsades de Pointes
A specific form of polymorphic ventricular tachycardia characterized by a gradual change in the amplitude and twisting of the QRS complexes around the isoelectric line.
- Conduction velocity
The speed at which an action potential is distributed throughout the myocardium; conduction velocity is still the primary metric for quantifying the spread of electrical activity in cardiac muscle.
- Transmural dispersion of repolarization
The difference between the longest and the shortest repolarization times measured at two points across the cardiac wall.
- Early afterdepolarization
Spontaneous diastolic depolarization that occurs during phase 2 and/or phase 3 of the cardiac action potential, thereby delaying repolarization.
- Triggered activity
The generation of spontaneous action potentials outside the sinus node as a result of afterdepolarizations.
- Focal activity
Abnormal formation of a depolarizing impulse outside the sinus node.
- Wavelength
The distance travelled by the excitation wave during its refractory period.
- Delayed afterdepolarizations
Spontaneous diastolic depolarizations that occur after repolarization is complete.
- Bidirectional VT
A ventricular tachycardia (VT) in which the QRS axis (as seen in the frontal electrocardiogram leads) shifts 180° with each alternate beat.
- J waves
Dome-like deflections of the electrocardiogram trace between the QRS complex and the ST segment.
- Right ventricular outflow tract
(RVOT). an extension of the infundibulum (a conical pouch of the right ventricle) in the ventricular cavity through which the blood flows towards the pulmonary artery.
- T wave alternans
Beat-to-beat variation in the polarity or amplitude of T waves; this phenomenon indicates an unevenness in the refractoriness of the myocardium and points to elevated cardiac electrical instability.
- Electrical axis
The net direction of the depolarization activity, resulting from the sum of all the electrical vectors on ECG.
- Bigeminy
Heart rhythm characterized by two beats close together with a pause following each pair of beats; the first beat in the pair is the sinus beat, whereas the second one is a premature contraction. bigeminy rhythm is often associated with the sensation of the heart skipping a beat.
- PVC couplets
Two premature ventricular contractions (PVCs) in consecutive heart beats.
Footnotes
Competing interests
M.J.A. is a consultant for Audentes Therapeutics, Boston Scientific, Gilead Sciences, Invitae, Medtronic, MyoKardia and St. Jude Medical, and holds equity/royalties of AliveCor, Blue Ox Health and StemoniX. C.A. is a consultant for Novartis Institutes for BioMedical Research, Inc. and Trevena, Inc. All other authors declare no competing interests.
Peer review information
Nature Reviews Disease Primers thanks J. Kanters, R. Kass, H. Morita, S. Nattel, S. Ohno, Y. Rudy, K. Shivkumar, M. Yano and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Meyer L et al. Incidence, causes, and survival trends from cardiovascular-related sudden cardiac arrest in children and young adults 0 to 35 years of age: a 30-year review. Circulation. 126, 1363–1372 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Winkel BG et al. Nationwide study of sudden cardiac death in persons aged 1–35 years. Eur. Heart J 32, 983–990 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Bardai A et al. Incidence, causes, and outcomes of out-of-hospital cardiac arrest in children. A comprehensive, prospective, population-based study in the Netherlands. J. Am. Coll. Cardiol 57, 1822–1828 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Bagnall RD et al. A prospective study of sudden cardiac death among children and young adults. N. Engl. J. Med 374, 2441–2452 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Kong MH et al. Systematic review of the incidence of sudden cardiac death in the United States. J. Am. Coll. Cardiol 57, 794–801 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Vreede-Swagemakers JJM et al. Out-of-hospital cardiac arrest in the 1990s: a population-based study in the Maastricht area on incidence, characteristics and survival. J. Am. Coll. Cardiol 30, 1500–1505 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Bardai A et al. Epilepsy is a risk factor for sudden cardiac arrest in the general population. PLoS One 7, e42749 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huikuri HV, Castellanos A & Myerburg RJ Sudden death due to cardiac arrhythmias. N. Engl. J. Med 345, 1473–1482 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Priori SG et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC inJune 2013. Heart Rhythm. 10, 1932–1963 (2013). [DOI] [PubMed] [Google Scholar]; An important consensus document on genetic arrhythmias.
- 10.Schwartz PJ et al. Prevalence of the congenital long-QT syndrome. Circulation 120, 1761–1767 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the study that provided the data-based prevalence of LQTS.
- 11.Piippo K et al. A founder mutation of the potassium channel KCNQ1 in long QT syndrome: implications for estimation of disease prevalence and molecular diagnostics. J. Am. Coll. Cardiol 37, 562–568 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Brink PA et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation. 112, 2602–2610 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Arbour L et al. A KCNQ1 V205M missense mutation causes a high rate of long QT syndrome in a first nations community of northern British Columbia: a community-based approach to understanding the impact. Genet. Med 10, 545–550 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Postema PG et al. Founder mutations in the Netherlands: SCN5a 1795insD, the first described arrhythmia overlap syndrome and one of the largest and best characterised families worldwide. Neth. Heart J 17, 422–428 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winbo A et al. Origin of the Swedish long QT syndrome Y111C/KCNQ1 founder mutation. Heart Rhythm. 8, 541–547 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Refaat MM, Hotait M & Scheinman M Brugada syndrome. Card. Electrophysiol. Clin 8, 239–245 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Antzelevitch C et al. J-wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Heart Rhythm. 13, e295–e324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; A ‘must read’ report on J wave syndromes.
- 18.Wang Q et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet 12, 17–23 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Tester DJ, Will ML, Haglund CM & Ackerman MJ Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2, 507–517 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Curran ME et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 80, 795–803 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Wang Q et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum. Mol. Genet 4, 1603–1607 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Ackerman MJ et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm. 8, 1308–1339 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Koopmann TT et al. Long QT syndrome caused by a large duplication in the KCNH2 (HERG) gene undetectable by current polymerase chain reaction-based exon-scanning methodologies. Heart Rhythm. 3, 52–55 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Schwartz PJ, Priori SG & Napolitano C How really rare are rare diseases? The intriguing case of independent compound mutations in the long QT syndrome. J. Cardiovasc. Electrophysiol 14, 1120–1121 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Moss AJ et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 115, 2481–2489 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westenskow P, Splawski I, Timothy KW, Keating MT & Sanguinetti MC Compound mutations: a common cause of severe long-QT syndrome. Circulation. 109, 1834–1841 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Shimizu W et al. Genotype–phenotype aspects of type 2 long QT syndrome. J. Am. Coll. Cardiol 54, 2052–2062 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giudicessi JR, Wilde AAM & Ackerman MJ The genetic architecture of long QT syndrome: a critical reappraisal. Trends Cardiovasc. Med 28, 453–464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strande NT et al. Evaluating the clinical validity of gene–disease associations: an evidence-based framework developed by the clinical genome resource. Am. J. Hum. Genet 100, 895–906 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adler A et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 141, 418–428 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crotti L et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127, 1009–1017 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boczek NJ et al. Spectrum and prevalence of CALM1-, CALM2-, and CALM3-encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome-associated calmodulin missense variant, E141G. Circ. Cardiovasc. Genet 9, 136–146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crotti L et al. Calmodulin mutations and life-threatening cardiac arrhythmias: insights from the International Calmodulinopathy Registry. Eur. Heart J 40, 2964–2975 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; The analysis of the genetic and clinical knowledge on the effects of calmodulin mutations.
- 34.Altmann HM et al. Homozygous/compound heterozygous triadin mutations associated with autosomal-recessive long-QT syndrome and pediatric sudden cardiac arrest: elucidation of the triadin knockout syndrome. Circulation 131, 2051–2060 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Clemens DJ et al. International Triadin Knockout Syndrome Registry. Circ. Genom. Precis. Med 12, e002419 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Neyroud N et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet 15, 186–189 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Schulze-Bahr E et al. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat. Genet 17, 267–268 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Schwartz PJ et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation 113, 783–790 (2006). [DOI] [PubMed] [Google Scholar]; A comprehensive review of the Jervell and Lange-Nielsen syndrome.
- 39.Splawski I et al. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Dufendach KA et al. Clinical outcomes and modes of death in Timothy syndrome: a multicenter international study of a rare disorder. JACC Clin. Electrophysiol 4, 459–466 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Boczek NJ et al. Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circ. Cardiovasc. Genet 6, 279–289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Plaster NM et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105, 511–519 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Brugada R et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 109, 30–35 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Bellocq C et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109, 2394–2397 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Priori SG et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res 96, 800–807 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Templin C et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J 32, 1077–1088 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antzelevitch C et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 115, 442–449 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thorsen K et al. Loss-of-activity-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat. Commun 8, 1696 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Priori SG et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103, 196–200 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Lieve KV, van der Werf C & Wilde AA Catecholaminergic polymorphic ventricular tachycardia. Circ. J 80, 1285–1291 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Laitinen PJ et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103, 485–490 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Medeiros-Domingo A et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J. Am. Coll. Cardiol 54, 2065–2074 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Priori SG & Chen SRW Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res 108, 871–883 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lahat H, Pras E & Eldar M A missense mutation in CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Ann. Med 36, 87–91 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Nyegaard M et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet 91, 703–712 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roux-Buisson N et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet 21, 2759–2767 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Makita N et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet 7, 466–474 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tester DJ et al. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm. 3, 800–805 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Mohler PJ et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl Acad. Sci. USA 101, 9137–9142 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Devalla HD et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol. Med 8, 1390–1408 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tester DJ et al. Plakophilin-2 truncation variants in patients clinically diagnosed with catecholaminergic polymorphic ventricular tachycardia and decedents with exercise-associated autopsy negative sudden unexplained death in the young. JACC Clin. Electrophysiol 5, 120–127 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cerrone M et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun 8, 106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Q et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296 (1998). [DOI] [PubMed] [Google Scholar]
- 64.Hosseini SM et al. Reappraisal of reported genes for sudden arrhythmic death. Circulation 138, 1195–1205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le Scouarnec S et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum. Mol. Genet 24, 2757–2763 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Probst V et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ. Cardiovasc. Genet 2, 552–557 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Bezzina CR et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet 45, 1044–1049 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; Novel insights on the complexity of the genetics of BrS.
- 68.Hofman N, Tan HL, Alders M, van Langen IM & Wilde AAM Active cascade screening in primary inherited arrhythmia syndromes. Does it lead to prophylactic treatment? J. Am. Coll. Cardiol 55, 2570–2576 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Schwartz PJ Cascades or waterfalls, the cataracts of genetic screening are being opened on clinical cardiology. J. Am. Coll. Cardiol 55, 2577–2579 (2010). [DOI] [PubMed] [Google Scholar]
- 70.Priori SG et al. Risk stratification in the long-QT syndrome. N. Engl. J. Med 348, 1866–1874 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Schwartz PJ, Crotti L & George AL Modifier genes for sudden cardiac death. Eur. Heart J 39, 3925–3931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive review on modifier genes associated with sudden death.
- 72.Lee Y-K et al. MTMR4 SNVs modulate ion channel degradation and clinical severity in congenital long QT syndrome: insights in the mechanism of action of protective modifier genes. Cardiovasc. Res 10.1093/cvr/cvaa019 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schwartz PJ Sudden cardiac death, founder populations, and mushrooms: what is the link with gold mines and modifier genes? Heart Rhythm. 8, 548–550 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Crotti L et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation 112, 1251–1258 (2005). [DOI] [PubMed] [Google Scholar]
- 75.Crotti L et al. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation 120, 1657–1663 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.de Villiers CP et al. AKAP9 is a genetic modifier of congenital long-QT syndrome type 1. Circ. Cardiovasc. Genet 7, 599–606 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Antzelevitch C & Oliva A Amplification of spatial dispersion of repolarization underlies sudden cardiac death associated with catecholaminergic polymorphic VT, long QT, short QT and Brugada syndromes. J. Intern. Med 259, 48–58 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ueda N, Zipes DP & Wu J Prior ischemia enhances arrhythmogenicity in isolated canine ventricular wedge model of long QT 3. Cardiovasc. Res 63, 69–76 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Haïssaguerre M et al. Depolarization versus repolarization abnormality underlying inferolateral J-wave syndromes: new concepts in sudden cardiac death with apparently normal hearts. Heart Rhythm. 16, 781–790 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schwartz PJ Idiopathic long QT syndrome: progress and questions. Am. Heart J 109, 399–411 (1985). [DOI] [PubMed] [Google Scholar]
- 81.Moss AJ et al. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 84, 1136–1144 (1991). [DOI] [PubMed] [Google Scholar]
- 82.Zipes DP The long QT interval syndrome. A Rosetta stone for sympathetic related ventricular tachyarrhythmias. Circulation 84, 1414–1419 (1991). [DOI] [PubMed] [Google Scholar]
- 83.Dumaine R & Antzelevitch C Molecular mechanisms underlying the long QT syndrome. Curr. Opin. Cardiol 17, 36–42 (2002). [DOI] [PubMed] [Google Scholar]
- 84.Roden DM Drug-induced prolongation of the QT interval. N. Engl. J. Med 350, 1013–1022 (2004). [DOI] [PubMed] [Google Scholar]; A comprehensive review on drug-induced LQTS.
- 85.Itoh H et al. The genetics underlying acquired long QT syndrome: impact for genetic screening. Eur. Heart J 37, 1456–1464 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schwartz PJ & Woosley RL Predicting the unpredictable: drug-induced QT prolongation and Torsades de Pointes. J. Am. Coll. Cardiol 67, 1639–1650 (2016). [DOI] [PubMed] [Google Scholar]
- 87.Antzelevitch C & Shimizu W Cellular mechanisms underlying the long QT syndrome. Curr. Opin. Cardiol 17, 43–51 (2002). [DOI] [PubMed] [Google Scholar]
- 88.Sicouri S, Glass A, Ferreiro M & Antzelevitch C Transseptal dispersion of repolarization and its role in the development of Torsade de Pointes arrhythmias. J. Cardiovasc. Electrophysiol 21, 441–447 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vandersickel N et al. Short-lasting episodes of Torsade de Pointes in the chronic atrioventricular block dog model have a focal mechanism, while longer-lasting episodes are maintained by re-entry. JACC Clin. Electrophysiol 3, 1565–1576 (2017). [DOI] [PubMed] [Google Scholar]
- 90.Han J & Moe GK Nonuniform recovery of excitability in ventricular muscle. Circ. Res 14, 44–60 (1964). [DOI] [PubMed] [Google Scholar]
- 91.Nof E, Burashnikov A & Antzelevitch C Cellular basis for atrial fibrillation in an experimental model of short QT1: implications for a pharmacological approach to therapy. Heart Rhythm. 7, 251–257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Whittaker DG, Colman MA, Ni H, Hancox JC & Zhang H Human atrial arrhythmogenesis and sinus bradycardia in KCNQ1-linked short QT syndrome: insights from computational modelling. Front. Physiol 9, 1402 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guo F et al. Patient-specific and gene-corrected induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of short QT syndrome. Circ. Res 124, 66–78 (2019). [DOI] [PubMed] [Google Scholar]
- 94.Shinnawi R et al. Modeling reentry in the short QT syndrome with human-induced pluripotent stem cell-derived cardiac cell sheets. J. Am. Coll. Cardiol 73, 2310–2324 (2019). [DOI] [PubMed] [Google Scholar]
- 95.El-Battrawy I et al. Modeling short QT syndrome using human-induced pluripotent stem cell-derived cardiomyocytes. J. Am. Heart Assoc 7, e007394 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Odening KE et al. Transgenic short-QT syndrome 1 rabbits mimic the human disease phenotype with QT/action potential duration shortening in the atria and ventricles and increased ventricular tachycardia/ventricular fibrillation inducibility. Eur. Heart J 40, 842–853 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Wehrens XHT The molecular basis of catecholaminergic polymorphic ventricular tachycardia: what are the different hypotheses regarding mechanisms? Heart Rhythm. 4, 794–797 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cerrone M, Napolitano C & Priori SG Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca2+ regulation. Heart Rhythm. 6, 1652–1659 (2009). [DOI] [PubMed] [Google Scholar]
- 99.Hayashi M et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 119, 2426–2434 (2009). [DOI] [PubMed] [Google Scholar]; A thorough analysis of the clinical manifestations of CPVT.
- 100.De Ferrari GM et al. Clinical management of catecholaminergic polymorphic ventricular tachycardia: the role of left cardiac sympathetic denervation. Circulation 131, 2185–2193 (2015). [DOI] [PubMed] [Google Scholar]; The definite evidence for the role of left cardiac sympathetic denervation in CPVT.
- 101.Nakamura Y et al. Ryanodine receptor-bound calmodulin is essential to protect against catecholaminergic polymorphic ventricular tachycardia. JCI insight 4, e126112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu X et al. Defective calmodulin binding to the cardiac ryanodine receptor plays a key role in CPVT-associated channel dysfunction. Biochem. Biophys. Res. Commun 394, 660–666 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Klipp RC et al. EL20, a potent antiarrhythmic compound, selectively inhibits calmodulin-deficient ryanodine receptor type 2. Heart Rhythm. 15, 578–586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nam G-B, Burashnikov A & Antzelevitch C Cellular mechanisms underlying the development of catecholaminergic ventricular tachycardia. Circulation. 111, 2727–2733 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cerrone M et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ. Res 101, 1039–1048 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Litovsky SH & Antzelevitch C Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ. Res 62, 116–126 (1988). [DOI] [PubMed] [Google Scholar]
- 107.Litovsky SH & Antzelevitch C Differences in the electrophysiology of ventricular epicardium and endocardium as the basis for the Osborne wave. Circulation 80, II–129 (1989). [Google Scholar]
- 108.Yan GX & Antzelevitch C Cellular basis for the electrocardiographic J wave. Circulation 93, 372–379 (1996). [DOI] [PubMed] [Google Scholar]
- 109.Di Diego JM, Sun ZQ & Antzelevitch C I(to) and action potential notch are smaller in left vs. right canine ventricular epicardium. Am. J. Physiol 271, H548–H561 (1996). [DOI] [PubMed] [Google Scholar]
- 110.Boukens BJ et al. Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ. Res 113, 137–141 (2013). [DOI] [PubMed] [Google Scholar]
- 111.Koncz I et al. Mechanisms underlying the development of the electrocardiographic and arrhythmic manifestations of early repolarization syndrome. J. Mol. Cell Cardiol 68, 20–28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ghosh S et al. Early repolarization associated with sudden death: insights from noninvasive electrocardiographic imaging. Heart Rhythm. 7, 534–537 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wilde AAM et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J. Mol. Cell Cardiol 49, 543–553 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; A thorough analysis of the pathophysiology of BrS.
- 114.Morita H, Zipes DP & Wu J Brugada syndrome: insights of ST elevation, arrhythmogenicity, and risk stratification from experimental observations. Heart Rhythm. 6, S34–S43 (2009). [DOI] [PubMed] [Google Scholar]
- 115.Antzelevitch C & Yan G-X J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm. 12, 1852–1866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Di Diego J et al. In a whole-heart model of the Brugada syndrome, delayed conduction in the RVOT “does not” contribute to inscription of the electrocardiographic J wave/ST segment elevation. Heart Rhythm. 15, S242 (2018). [Google Scholar]
- 117.Brugada J et al. Brugada syndrome phenotype elimination by epicardial substrate ablation. Circ. Arrhythmia Electrophysiol 8, 1373–1381 (2015). [DOI] [PubMed] [Google Scholar]
- 118.Szél T & Antzelevitch C Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of Brugada syndrome. J. Am. Coll. Cardiol 63, 2037–2045 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nademanee K et al. Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome. J. Am. Coll. Cardiol 66, 1976–1986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rohr S & Kucera JP Involvement of the calcium inward current in cardiac impulse propagation: induction of unidirectional conduction block by nifedipine and reversal by Bay K 8644. Biophys. J 72, 754–766 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huelsing DJ, Spitzer KW, Cordeiro JM & Pollard AE Conduction between isolated rabbit Purkinje and ventricular myocytes coupled by a variable resistance. Am. J. Physiol 274, H1163–H1173 (1998). [DOI] [PubMed] [Google Scholar]
- 122.Schwartz PJ et al. Genotype–phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103, 89–95 (2001). [DOI] [PubMed] [Google Scholar]; The evidence for the importance of the genotype in the triggering of arrhythmic events in LQTS.
- 123.Schwartz PJ, Periti M & Malliani A The long Q-T syndrome. Am. Heart J 89, 378–390 (1975). [DOI] [PubMed] [Google Scholar]
- 124.Schwartz PJ & Ackerman MJ The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur. Heart J 34, 3109–3116 (2013). [DOI] [PubMed] [Google Scholar]; An updated view on the clinical management of LQTS.
- 125.Kimbrough J et al. Clinical implications for affected parents and siblings of probands with long-QT syndrome. Circulation 104, 557–562 (2001). [DOI] [PubMed] [Google Scholar]
- 126.Crotti L et al. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation 116, 2366–2375 (2007). [DOI] [PubMed] [Google Scholar]
- 127.Lim ET et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 10, e1004494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jervell A & Lange-Nielsen F Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval, and sudden death. Am. Heart J 54, 59–68 (1957). [DOI] [PubMed] [Google Scholar]
- 129.Schwartz PJ & Priori SG in Cardiac electrophysiology. From cell to bedside 4th edn (eds Zipes DP & Jalife J) 711–719 (Saunders, 2004). [Google Scholar]
- 130.Schwartz PJ & Malliani A Electrical alternation of the T-wave: clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long Q-T syndrome. Am. Heart J 89, 45–50 (1975). [DOI] [PubMed] [Google Scholar]
- 131.Schwartz PJ, Zaza A, Locati E & Moss AJ Stress and sudden death. The case of the long QT syndrome. Circulation 83, II71–II80 (1991). [PubMed] [Google Scholar]
- 132.Ackerman MJ, Tester DJ & Porter CJ Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin. Proc 74, 1088–1094 (1999). [DOI] [PubMed] [Google Scholar]
- 133.Khositseth A, Tester DJ, Will ML, Bell CM & Ackerman MJ Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm. 1, 60–64 (2004). [DOI] [PubMed] [Google Scholar]
- 134.Seth R et al. Long QT syndrome and pregnancy. J. Am. Coll. Cardiol 49, 1092–1098 (2007). [DOI] [PubMed] [Google Scholar]
- 135.Schwartz PJ, Spazzolini C & Crotti L All LQT3 patients need an ICD: true or false? Heart Rhythm. 6, 113–120 (2009). [DOI] [PubMed] [Google Scholar]
- 136.Arnestad M et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation 115, 361–367 (2007). [DOI] [PubMed] [Google Scholar]
- 137.Tester DJ et al. Cardiac genetic predisposition in sudden infant death syndrome. J. Am. Coll. Cardiol 71, 1217–1227 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Schwartz PJ et al. Prolongation of the QT interval and the sudden infant death syndrome. N. Engl. J. Med 338, 1709–1714 (1998). [DOI] [PubMed] [Google Scholar]
- 139.Schwartz PJ et al. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N. Engl. J. Med 343, 262–267 (2000). [DOI] [PubMed] [Google Scholar]
- 140.Schwartz PJ Molecular diagnosis in a child with sudden infant death syndrome. Lancet 358, 1342–1343 (2001). [DOI] [PubMed] [Google Scholar]
- 141.Saul JP et al. Rationale and objectives for ECG screening in infancy. Heart Rhythm 11, 2316–2321 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Giustetto C et al. Long-term follow-up of patients with short QT syndrome. J. Am. Coll. Cardiol 58, 587–595 (2011). [DOI] [PubMed] [Google Scholar]
- 143.Mazzanti A et al. Novel insight into the natural history of short QT syndrome. J. Am. Coll. Cardiol 63, 1300–1308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tester DJ et al. A mechanism for sudden infant death syndrome (SIDS): stress-induced leak via ryanodine receptors. Heart Rhythm. 4, 733–739 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Choi G et al. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation 110, 2119–2124 (2004). [DOI] [PubMed] [Google Scholar]
- 146.Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM & Ackerman MJ Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin. Proc 87, 524–539 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Leinonen JT et al. The genetics underlying idiopathic ventricular fibrillation: a special role for catecholaminergic polymorphic ventricular tachycardia? Int. J. Cardiol 250, 139–145 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Brugada P & Brugada J Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol 20, 1391–1396 (1992). [DOI] [PubMed] [Google Scholar]; The original description of BrS.
- 149.Mizusawa Y & Wilde AAM Brugada syndrome. Circ. Arrhythmia Electrophysiol 5, 606–616 (2012). [DOI] [PubMed] [Google Scholar]
- 150.Matsuo K et al. The circadian pattern of the development of ventricular fibrillation in patients with Brugada syndrome. Eur. Heart J 20, 465–470 (1999). [DOI] [PubMed] [Google Scholar]
- 151.Morita H et al. Atrial fibrillation and atrial vulnerability in patients with Brugada syndrome. J. Am. Coll. Cardiol 40, 1437–1444 (2002). [DOI] [PubMed] [Google Scholar]
- 152.Milman A et al. Age of first arrhythmic event in Brugada syndrome: data from the SABRUS (Survey on Arrhythmic Events in Brugada Syndrome) in 678 patients. Circ. Arrhythm. Electrophysiol 10, e005222 (2017). [DOI] [PubMed] [Google Scholar]
- 153.Michowitz Y et al. Fever-related arrhythmic events in the multicenter survey on arrhythmic events in Brugada syndrome. Heart Rhythm. 15, 1394–1401 (2018). [DOI] [PubMed] [Google Scholar]
- 154.Mizusawa Y et al. Prognostic significance of fever-induced Brugada syndrome. Heart Rhythm. 13, 1515–1520 (2016). [DOI] [PubMed] [Google Scholar]
- 155.Smits JPP et al. Genotype–phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J. Am. Coll. Cardiol 40, 350–356 (2002). [DOI] [PubMed] [Google Scholar]
- 156.Kapplinger JD et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 7, 33–46 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Adler A et al. Risk stratification in Brugada syndrome: clinical characteristics, electrocardiographic parameters, and auxiliary testing. Heart Rhythm. 13, 299–310 (2016). [DOI] [PubMed] [Google Scholar]
- 158.Sroubek J et al. Programmed ventricular stimulation for risk stratification in the Brugada syndrome: a pooled analysis. Circulation 133, 622–630 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Schwartz PJ, Moss AJ, Vincent GM & Crampton RS Diagnostic criteria for the long QT syndrome. An update. Circulation 88, 782–784 (1993). [DOI] [PubMed] [Google Scholar]
- 160.Schwartz PJ & Crotti L QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation 124, 2181–2184 (2011). [DOI] [PubMed] [Google Scholar]
- 161.Schwartz PJ & Crotti L in Cardiac Electrophysiology. From Cell to Bedside 7th edn. (eds Zipes DP & Jalife J) 893–904 (Elsevier, 2009). [Google Scholar]
- 162.Horner JM, Horner MM & Ackerman MJ The diagnostic utility of recovery phase QTc during treadmill exercise stress testing in the evaluation of long QT syndrome. Heart Rhythm. 8, 1698–1704 (2011). [DOI] [PubMed] [Google Scholar]
- 163.Crotti L et al. Vagal reflexes following an exercise stress test: a simple clinical tool for gene-specific risk stratification in the long QT syndrome. J. Am. Coll. Cardiol 60, 2515–2524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nador F et al. Unsuspected echocardiographic abnormality in the long QT syndrome. Diagnostic, prognostic, and pathogenetic implications. Circulation 84, 1530–1542 (1991). [DOI] [PubMed] [Google Scholar]; The first evidence for a mechanical abnormality in LQTS.
- 165.De Ferrari GM et al. Effect of calcium channel block on the wall motion abnormality of the idiopathic long QT syndrome. Circulation 89, 2126–2132 (1994). [DOI] [PubMed] [Google Scholar]
- 166.De Ferrari GM & Schwartz PJ Vox clamantis in deserto. We spoke but nobody was listening: echocardiography can help risk stratification of the long-QT syndrome. Eur. Heart J 36, 148–150 (2015). [DOI] [PubMed] [Google Scholar]
- 167.Ter Bekke RMA et al. Electromechanical window negativity in genotyped long-QT syndrome patients: relation to arrhythmia risk. Eur. Heart J 36, 179–186 (2015). [DOI] [PubMed] [Google Scholar]
- 168.Haugaa KH et al. Left ventricular mechanical dispersion by tissue Doppler imaging: a novel approach for identifying high-risk individuals with long QT syndrome. Eur. Heart J 30, 330–337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.De Ferrari GM & Schwartz PJ Long QT syndrome, a purely electrical disease? Not anymore. Eur. Heart J 30, 253–255 (2009). [DOI] [PubMed] [Google Scholar]
- 170.Ackerman MJ Genetic purgatory and the cardiac channelopathies: exposing the variants of uncertain/unknown significance issue. Heart Rhythm. 12, 2325–2331 (2015). [DOI] [PubMed] [Google Scholar]; A practical and insightful view of the complexities of the genetics of channelopathies.
- 171.Viskin S et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm. 1, 587–591 (2004). [DOI] [PubMed] [Google Scholar]
- 172.Schimpf R, Wolpert C, Gaita F, Giustetto C & Borggrefe M Short QT syndrome. Cardiovasc. Res 67, 357–366 (2005). [DOI] [PubMed] [Google Scholar]; An expert review on SQTS.
- 173.Anttonen O et al. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 116, 714–720 (2007). [DOI] [PubMed] [Google Scholar]
- 174.Guerrier K et al. Short QT interval prevalence and clinical outcomes in a pediatric population. Circ. Arrhythm. Electrophysiol 8, 1460–1464 (2015). [DOI] [PubMed] [Google Scholar]
- 175.Priori SG & Blomström-Lundqvist C 2015 European Society of Cardiology guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death summarized by co-chairs. Eur. Heart J 36, 2757–2759 (2015). [DOI] [PubMed] [Google Scholar]
- 176.Wolpert C et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J. Cardiovasc. Electrophysiol 16, 54–58 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Anttonen O et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm. 6, 267–271 (2009). [DOI] [PubMed] [Google Scholar]
- 178.Schimpf R et al. Electromechanical coupling in patients with the short QT syndrome: further insights into the mechanoelectrical hypothesis of the U wave. Heart Rhythm. 5, 241–245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tülümen E et al. PQ segment depression in patients with short QT syndrome: a novel marker for diagnosing short QT syndrome? Heart Rhythm. 11, 1024–1030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Watanabe H et al. High prevalence of early repolarization in short QT syndrome. HeartRhythm. 7, 647–652 (2010). [DOI] [PubMed] [Google Scholar]
- 181.Postma AV et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet 42, 863–870 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wilde AAM et al. Proposed diagnostic criteria for the Brugada syndrome. Eur. Heart J 23, 1648–1654 (2002). [DOI] [PubMed] [Google Scholar]
- 183.Makimoto H et al. Augmented ST-segment elevation during recovery from exercise predicts cardiac events in patients with Brugada syndrome. J. Am. Coll. Cardiol 56, 1576–1584 (2010). [DOI] [PubMed] [Google Scholar]
- 184.Baranchuk A et al. Brugada phenocopy: new terminology and proposed classification. Ann. Noninvasive Electrocardiol 17, 299–314 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chockalingam P et al. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: higher recurrence of events under metoprolol. J. Am. Coll. Cardiol 60, 2092–2099 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Chatrath R, Bell CM & Ackerman MJ Beta-blocker therapy failures in symptomatic probands with genotyped long-QT syndrome. Pediatr. Cardiol 25, 459–465 (2004). [DOI] [PubMed] [Google Scholar]
- 187.Wilde AAM et al. Clinical aspects of type 3 long-QT syndrome: an international multicenter study. Circulation 134, 872–882 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; The study that proved that β-adrenergic receptor blockers are effective also for patients with LQT3.
- 188.Schwartz PJ et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 92, 3381–3386 (1995). [DOI] [PubMed] [Google Scholar]; The first suggestion for gene-specific therapy.
- 189.Mazzanti A et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J. Am. Coll. Cardiol 67, 1053–1058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Schwartz PJ Management of long QT syndrome. Nat. Clin. Pract. Cardiovasc. Med 2, 346–351 (2005). [DOI] [PubMed] [Google Scholar]
- 191.Bos JM et al. Mexiletine shortens the QT interval in patients with potassium channel-mediated type 2 long QT syndrome. Circ. Arrhythm. Electrophysiol 12, e007280 (2019). [DOI] [PubMed] [Google Scholar]
- 192.Mehta A et al. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur. Heart J 39, 1446–1455 (2018). [DOI] [PubMed] [Google Scholar]
- 193.Schwartz PJ et al. From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome type 2. Eur. Heart J 40, 1832–1836 (2019). [DOI] [PubMed] [Google Scholar]; The first attempt to verify clinically the responses to drugs made in iPS-CMs from patients with LQTS.
- 194.Al-Khatib SM et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary. J. Am. Coll. Cardiol 138, e210–e271 (2018). [DOI] [PubMed] [Google Scholar]
- 195.Ackerman MJ et al. Beta-blocker therapy for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia: are all beta-blockers equivalent? Heart Rhythm. 14, e41–e44 (2017). [DOI] [PubMed] [Google Scholar]
- 196.Leren IS et al. Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with β1-selective β-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 13, 432–440 (2016). [DOI] [PubMed] [Google Scholar]
- 197.Zhou Q et al. Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat. Med 17, 1003–1009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Watanabe H et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med 15, 380–383 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.van der Werf C et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J. Am. Coll. Cardiol 57, 2244–2254 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kannankeril PJ et al. Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia: a randomized clinical trial. JAMA Cardiol. 2, 759–766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Watanabe H et al. Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 10, 542–547 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Belhassen B Management of Brugada syndrome 2016: should all high risk patients receive an ICD? Alternatives to implantable cardiac defibrillator therapy for Brugada syndrome. Circ. Arrhythm. Electrophysiol 9, e004185 (2016). [DOI] [PubMed] [Google Scholar]
- 203.Postema PG et al. Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website (www.brugadadrugs.org). Heart Rhythm. 6, 1335–1341 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pundi KN, Bos JM, Cannon BC & Ackerman MJ Automated external defibrillator rescues among children with diagnosed and treated long QT syndrome. Heart Rhythm. 12, 776–781 (2015). [DOI] [PubMed] [Google Scholar]
- 205.Moss AJ & McDonald J Unilateral cervicothoracic sympathetic ganglionectomy for the treatment of long QT interval syndrome. N. Engl. J. Med 285, 903–904 (1971). [DOI] [PubMed] [Google Scholar]
- 206.Schwartz PJ et al. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome. A worldwide report. Circulation 84, 503–511 (1991). [DOI] [PubMed] [Google Scholar]
- 207.Schwartz PJ et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation 109, 1826–1833 (2004). [DOI] [PubMed] [Google Scholar]
- 208.Schwartz PJ Cardiac sympathetic denervation to prevent life-threatening arrhythmias. Nat. Rev. Cardiol 11, 346–353 (2014). [DOI] [PubMed] [Google Scholar]; The updated review on cardiac sympathetic denervation in the prevention of lethal arrhythmias.
- 209.Schwartz PJ, Snebold NG & Brown AM Effects of unilateral cardiac sympathetic denervation on the ventricular fibrillation threshold. Am. J. Cardiol 37, 1034–1040 (1976). [DOI] [PubMed] [Google Scholar]
- 210.Schwartz PJ & Stone HL Left stellectomy and denervation supersensitivity in conscious dogs. Am. J. Cardiol 49, 1185–1190 (1982). [DOI] [PubMed] [Google Scholar]
- 211.Collura CA, Johnson JN, Moir C & Ackerman MJ Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 6, 752–759 (2009). [DOI] [PubMed] [Google Scholar]
- 212.Bos JM, Bos KM, Johnson JN, Moir C & Ackerman MJ Left cardiac sympathetic denervation in long QT syndrome: analysis of therapeutic nonresponders. Circ. Arrhythm. Electrophysiol 6, 705–711 (2013). [DOI] [PubMed] [Google Scholar]
- 213.Schwartz PJ, De Ferrari GM & Pugliese L Cardiac sympathetic denervation 100 years later: Jonnesco would have never believed it. Int. J. Cardiol 237, 25–28 (2017). [DOI] [PubMed] [Google Scholar]
- 214.Odero A, Bozzani A, De Ferrari GM & Schwartz PJ Left cardiac sympathetic denervation for the prevention of life-threatening arrhythmias: the surgical supraclavicular approach to cervicothoracic sympathectomy. Heart Rhythm. 7, 1161–1165 (2010). [DOI] [PubMed] [Google Scholar]
- 215.Vincent GM et al. High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation 119, 215–221 (2009). [DOI] [PubMed] [Google Scholar]
- 216.Horner JM et al. Implantable cardioverter defibrillator therapy for congenital long QT syndrome: a single-center experience. Heart Rhythm. 7, 1616–1622 (2010). [DOI] [PubMed] [Google Scholar]
- 217.Schwartz PJ Efficacy of left cardiac sympathetic denervation has an unforeseen side effect: medicolegal complications. Heart Rhythm. 7, 1330–1332 (2010). [DOI] [PubMed] [Google Scholar]
- 218.Schwartz PJ et al. Who are the long-QT syndrome patients who receive an implantable cardioverter–defibrillator and what happens to them? Data from the European Long-QT Syndrome Implantable Cardioverter–Defibrillator (LQTS ICD) registry. Circulation 122, 1272–1282 (2010). [DOI] [PubMed] [Google Scholar]
- 219.Johnson JN & Ackerman MJ Competitive sports participation in athletes with congenital long QT syndrome. JAMA 308, 764–765 (2012). [DOI] [PubMed] [Google Scholar]
- 220.Johnson JN & Ackerman MJ Return to play? Athletes with congenital long QT syndrome. Br. J. Sports Med 47, 28–33 (2013). [DOI] [PubMed] [Google Scholar]
- 221.Pelliccia A Long QT syndrome, implantable cardioverter defibrillator (ICD) and competitive sport participation: when science overcomes ethics. Br. J. Sports Med 48, 1135–1136 (2014). [DOI] [PubMed] [Google Scholar]
- 222.Schwartz PJ in Exercise and Sport Cardiology Vol. 3 (eds Thompson P & Fernandez A) 269–278 (World Scientific Publishing Europe, 2018). [Google Scholar]
- 223.Schwartz PJ The congenital long QT syndromes from genotype to phenotype: clinical implications. J. Intern. Med 259, 39–47 (2006). [DOI] [PubMed] [Google Scholar]
- 224.Wilde AAM et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N. Engl. J. Med 358, 2024–2029 (2008). [DOI] [PubMed] [Google Scholar]
- 225.Miyake CY et al. Efficacy of implantable cardioverter defibrillators in young patients with catecholaminergic polymorphic ventricular tachycardia: success depends on substrate. Circ. Arrhythm. Electrophysiol 6, 579–587 (2013). [DOI] [PubMed] [Google Scholar]
- 226.Roses-Noguer F, Jarman JWE, Clague JR & Till J Outcomes of defibrillator therapy in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 11, 58–66 (2014). [DOI] [PubMed] [Google Scholar]
- 227.Roston TM et al. Implantable cardioverter–defibrillator use in catecholaminergic polymorphic ventricular tachycardia: a systematic review. Heart Rhythm. 15, 1791–1799 (2018). [DOI] [PubMed] [Google Scholar]
- 228.van der Werf C et al. Implantable cardioverter–defibrillators in previously undiagnosed patients with catecholaminergic polymorphic ventricular tachycardia resuscitated from sudden cardiac arrest. Eur. Heart J 40, 2953–2961 (2019). [DOI] [PubMed] [Google Scholar]; A large study on the disquieting consequences of the ICDs in patients with CPVT.
- 229.Nademanee K et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 123, 1270–1279 (2011). [DOI] [PubMed] [Google Scholar]
- 230.Pappone C et al. Electrical substrate elimination in 135 consecutive patients with Brugada syndrome. Circ. Arrhythmia Electrophysiol 10, 1–13 (2017). [DOI] [PubMed] [Google Scholar]
- 231.Viskin S Radiofrequency ablation of asymptomatic Brugada syndrome: don’t go burning my heart. Circulation 137, 1883–1884 (2018). [DOI] [PubMed] [Google Scholar]
- 232.Cheung CC et al. Pregnancy in catecholaminergic polymorphic ventricular tachycardia. JACC Clin. Electrophysiol 5, 387–394 (2019). [DOI] [PubMed] [Google Scholar]
- 233.Rodríguez-Mañero M et al. The clinical significance of pregnancy in Brugada syndrome. Rev. Esp. Cardiol 67, 176–180 (2014). [DOI] [PubMed] [Google Scholar]
- 234.Heradien MJ et al. Does pregnancy increase cardiac risk for LQT1 patients with the KCNQ1-A341V mutation? J. Am. Coll. Cardiol 48, 1410–1415 (2006). [DOI] [PubMed] [Google Scholar]
- 235.Ishibashi K et al. Arrhythmia risk and β-blocker therapy in pregnant women with long QT syndrome. Heart 103, 1374–1379 (2017). [DOI] [PubMed] [Google Scholar]
- 236.Duan L, Ng A, Chen W, Spencer HT & Lee M-S Beta-blocker subtypes and risk of low birth weight in newborns. J. Clin. Hypertens 20, 1603–1609 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Cuneo BF et al. Mothers with long QT syndrome are at increased risk for fetal death: findings from a multicenter international study. Am. J. Obstet. Gynecol 222, 263.e1–263.e11 (2020). [DOI] [PubMed] [Google Scholar]; The risk of fetal death in LQTS families.
- 238.Vogl SE et al. Mode of delivery is associated with maternal and fetal endocrine stress response. BJOG 113, 441–445 (2006). [DOI] [PubMed] [Google Scholar]
- 239.Fazio G, Vernuccio F, Grutta G & Re GL Drugs to be avoided in patients with long QT syndrome: focus on the anaesthesiological management. World J. Cardiol 5, 87–93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Bodi I et al. Postpartum hormones oxytocin and prolactin cause pro-arrhythmic prolongation of cardiac repolarization in long QT syndrome type 2. Europace 21, 1126–1138 (2019). [DOI] [PubMed] [Google Scholar]
- 241.Martillotti G, Talajic M, Rey E & Leduc L Long QT syndrome in pregnancy: are vaginal delivery and use of oxytocin permitted? A case report. J. Obstet. Gynaecol. Can 34, 1073–1076 (2012). [DOI] [PubMed] [Google Scholar]
- 242.Schwartz PJ et al. Guidelines for the interpretation of the neonatal electrocardiogram. A task force of the European Society of Cardiology. Eur. Heart J 23, 1329–1344 (2002). [DOI] [PubMed] [Google Scholar]
- 243.Walsh SZ Electrocardiographic intervals during the first week of life. Am. Heart J 66, 36–41 (1963). [DOI] [PubMed] [Google Scholar]
- 244.Stramba-Badiale M et al. For neonatal ECG screening there is no reason to relinquish old Bazett’s correction. Eur. Heart J 39, 2888–2895 (2018). [DOI] [PubMed] [Google Scholar]
- 245.Dahdouh EM et al. Technical update: preimplantation genetic diagnosis and screening. J Obstet. Gynaecol. Can 37, 451–463 (2015). [DOI] [PubMed] [Google Scholar]
- 246.Treff NR & Zimmerman RS Advances in preimplantation genetic testing for monogenic disease and aneuploidy. Annu. Rev. Genomics Hum. Genet 18, 189–200 (2017). [DOI] [PubMed] [Google Scholar]
- 247.Regitz-Zagrosek V et al. 2018 ESC guidelines for the management of cardiovascular diseases during pregnancy. Eur. Heart J 39, 3165–3241 (2018). [DOI] [PubMed] [Google Scholar]
- 248.Antiel RM et al. Quality of life after videoscopic left cardiac sympathetic denervation in patients with potentially life-threatening cardiac channelopathies/cardiomyopathies. Heart Rhythm. 13, 62–69 (2016). [DOI] [PubMed] [Google Scholar]
- 249.Schwartz PJ When the risk is sudden death, does quality of life matter? Heart Rhythm. 13, 70–71 (2016). [DOI] [PubMed] [Google Scholar]
- 250.Ackerman MJ, Zipes DP, Kovacs RJ & Maron BJ Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 10: the cardiac channelopathies: a scientific statement from the American Heart Association and American College of Cardiology. J. Am. Coll. Cardiol 66, 2424–2428 (2015). [DOI] [PubMed] [Google Scholar]
- 251.Heidbüchel H et al. Recommendations for participation in leisure-time physical activity and competitive sports of patients with arrhythmias and potentially arrhythmogenic conditions. Part 2: ventricular arrhythmias, channelopathies, and implantable defibrillators. Europace 10.1093/europace/euaa106 (2020). [DOI] [PubMed] [Google Scholar]
- 252.Wu JC et al. Towards precision medicine with human iPSCs for cardiac channelopathies. Circ. Res 125, 653–658 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Liu DW, Gintant GA & Antzelevitch C Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ. Res 72, 671–687 (1993). [DOI] [PubMed] [Google Scholar]