

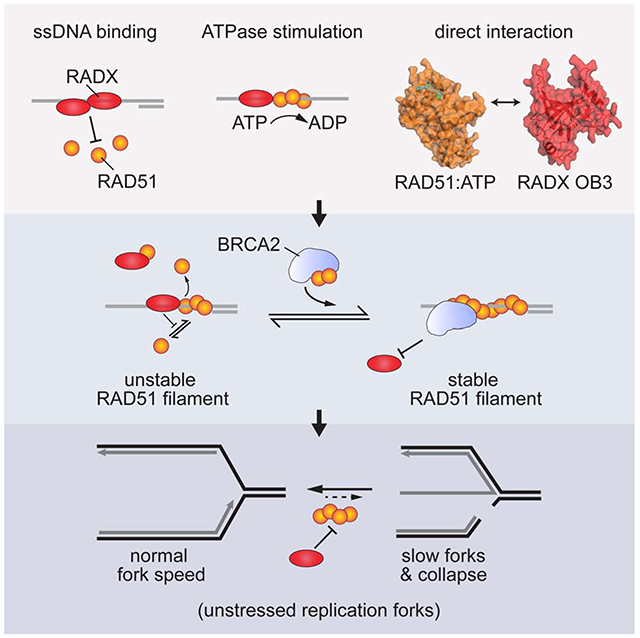
Summary
The RAD51 recombinase forms nucleoprotein filaments to promote double-strand break repair, replication fork reversal, and fork stabilization. The stability of these filaments is highly regulated since both too little and too much RAD51 activity can cause genome instability. RADX is a single-strand DNA (ssDNA) binding protein that regulates DNA replication. Here we define its mechanism of action. We find that RADX inhibits RAD51 strand exchange and D-loop formation activities. RADX directly and selectively interacts with ATP-bound RAD51, stimulates ATP hydrolysis, and destabilizes RAD51 nucleofilaments. The RADX interaction with RAD51, in addition to its ssDNA binding capability, is required to maintain replication fork elongation rates and fork stability. Furthermore, BRCA2 can overcome the RADX-dependent RAD51 inhibition. Thus, RADX functions in opposition to BRCA2 in regulating RAD51 nucleofilament stability to ensure the right level of RAD51 function during DNA replication.
Graphical Abstract
eToc
Adolph et al. identify a direct interaction between RADX and ATP-bound RAD51. The interaction and RADX ssDNA binding combine to destabilize RAD51 nucleofilaments. This activity prevents inappropriate RAD51 action at elongating replication forks to maintain fork speed and stability.
Introduction
The RAD51 recombinase maintains genome stability by promoting double-strand break (DSB) repair and regulating replication fork stability (Bhat and Cortez, 2018). At breaks, it promotes homologous recombination (HR) repair by catalyzing DNA strand exchange and homology searches. At stalled replication forks, RAD51 helps remodel the DNA in a process called fork reversal and protects nascent DNA from nuclease-mediated degradation (Hashimoto et al., 2010; Schlacher et al., 2011; Zellweger et al., 2015). These RAD51 activities depend on its ability to bind DNA to form a helical nucleoprotein filament (nucleofilament).
The generation and stability of the RAD51 nucleofilament are key points of regulation. When single-strand DNA (ssDNA) is exposed, it is tightly bound by the ssDNA binding protein, Replication Protein A (RPA) (Bhat and Cortez, 2018). RPA binding is a pre-requisite for RAD51 filament formation since it helps to remove DNA secondary structures; however, RPA also excludes RAD51, which has a much lower affinity for ssDNA (Chen and Wold, 2014; Kowalczykowski, 2015; Ma et al., 2017). Therefore, RAD51 requires the action of mediator proteins like BRCA2 to replace RPA on ssDNA and generate the nucleofilament (Kowalczykowski, 2015; Liu et al., 2010). BRCA2 also helps direct RAD51 to ssDNA instead of double-strand DNA (dsDNA) (Jensen et al., 2010). RAD51 filament assembly is tightly regulated. For example, anti-recombinases that translocate on DNA such as BLM, RECQ5, and FBH1 can remove RAD51 (Bugreev et al., 2007; Hu et al., 2007; Simandlova et al., 2013). The RAD51 filament must be disassembled for downstream processes of HR; however, inappropriate disruption of RAD51 filaments frees ssDNA, making it vulnerable to degradation. During replication, metastable RAD51 binding to stalled replication forks is thought to promote fork reversal, and a BRCA2-stabilized RAD51 filament formed on the reversed fork prevents nascent-strand degradation (Hashimoto et al., 2010; Schlacher et al., 2011; Zellweger et al., 2015).
RADX was recently identified as a ssDNA binding protein at replication forks that is needed to maintain fork stability (Dungrawala et al., 2017; Schubert et al., 2017). RADX-deficient cells accumulate DSBs during replication and have slow replication elongation rates. Inactivation of fork reversal enzymes or RAD51 rescues these phenotypes suggesting that RADX prevents inappropriate replication fork reversal when cells are not experiencing replication stress (Dungrawala et al., 2017). This RADX activity depends on its ability to bind ssDNA. Based on the genetic relationship between RADX and RAD51, RADX was proposed to be a RAD51 antagonist (Dungrawala et al., 2017). A balance between RADX and RPA also is important to prevent replication problems (Schubert et al., 2017).
Here, we sought to understand how RADX regulates RAD51 and DNA replication. We found that RADX stimulates RAD51-dependent ATP hydrolysis and identified a direct protein-protein interaction between RADX and RAD51 mediated by the oligosaccharide binding domain-3 (OB3) of RADX. The interactions of RADX with RAD51 and ssDNA inhibit RAD51 nucleofilament formation and filament-mediated processes including strand exchange and D-loop formation. The BRC motifs of BRCA2 overcome the RADX-mediated RAD51 inhibition. Finally, a RADX mutant that cannot interact with RAD51 is unable to maintain replication fork stability. Thus, RADX regulates the RAD51 nucleofilament to control DNA replication.
Results
RADX blocks RAD51 from binding ssDNA
Flag-RADX purified from human cells was shown to outcompete RAD51 for ssDNA (Bhat et al., 2018). We confirmed that this is also the case for MBP-HIS-RADX purified from insect cells even when 2000-fold less RADX is added to the binding assay than RAD51. (Figures 1A and 1B).
Figure 1. RADX inhibits RAD51 assembly on DNA.
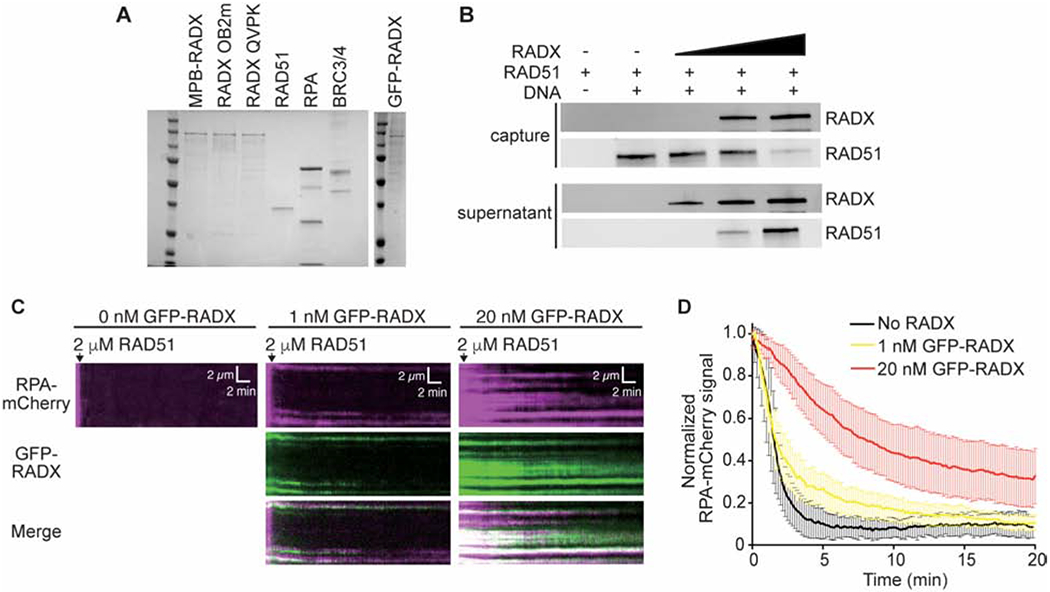
(A) Coomassie stained SDS-PAGE gel showing purified proteins used in this study. Each lane contains ~200 nM of purified protein. Actual amounts used is specified in each experiment. (B) Pull-down experiments showing MBP-RADX (0.5, 1, and 2nM) outcompetes RAD51 (4μM) for binding ssDNA. (C) Kymographs showing the assembly of RAD51 on ssDNA molecules coated with RPA-mCherry and 0 nM (left), 1 nM (middle), and 20 nM GFP-RADX (right). Assembly was initiated by injecting 2 μM RAD51 together with 2 mM ATP and 5 mM Ca2+ into sample chambers containing ssDNA molecules coated with RPA-mCherry and GFP-RADX while monitoring the loss of RPA-mCherry signal. (D) Graphs showing the normalized RPA-mCherry signal intensity integrated over entire ssDNA molecules during the assembly of the RAD51 filaments. Error bars represent 68% confidence intervals (n=30 ssDNA molecules).
RADX ssDNA binding was also shown to regulate RPA in cells (Schubert et al., 2017). To better characterize the relationship of RADX, RPA, and RAD51, we examined their association with ssDNA curtains using total internal reflection fluorescence microscopy (TIRFM) (Qi and Greene, 2016). Specifically, we monitored RPA-mCherry, GFP-RADX, and RAD51, which is not fluorescently labeled. In this assay, RAD51 forms filaments in the presence of RPA-mCherry which can be monitored through the rapid loss of RPA-mCherry signal intensity (Figures 1C and 1D). When increasing concentrations of RADX are present, the RAD51 assembly is decreased in a concentration dependent manner (Figures 1C and 1D). In the absence of RADX, RAD51 filaments are assembled at a rate of 10.3 ± 1.4 ×10−3 s−1. This rate decreases slightly to 6.9 ± 0.7 ×10−3 s−1 in the presence of 1 nM RADX and decreases significantly to 2.6 ± 0.2 ×10−3 s−1 in the presence of 20 nM RADX (Figure 1D). GFP-RADX co-localizes with the RPA-mCherry protein on the DNA suggesting little displacement of RPA by RADX at these concentrations. Thus, ssDNA that is occupied by RADX and RPA acts as a significant barrier to RAD51 filament assembly.
RADX stimulates ATP turnover by RAD51
Since RADX can exclude RAD51 from ssDNA we predicted it would also inhibit the DNA-dependent ATPase activity of RAD51. We measured RAD51-dependent ATP hydrolysis using a spectrophotometric assay that couples hydrolysis of ATP to the oxidation of NADH, resulting in a decrease in absorbance (Supplementary Figure 1). As predicted, when RADX is pre-bound to ssDNA it inhibits the ATP hydrolysis rate of RAD51 in a concentration-dependent manner (Figure 2A). RPA also inhibits RAD51 ATP hydrolysis consistent with previously published results for RPA and RAD51, as well as the functionally related E. coli proteins SSB and RecA (Kowalczykowski, 2015; Kowalczykowski et al., 1987; Sugiyama et al., 1997). However, in marked contrast to the inhibition observed when RADX is added prior to RAD51, when RAD51 is added to the ssDNA prior to RADX, RADX stimulates RAD51-dependent ATP hydrolysis (Figure 2B). The increased rate of ATP hydrolysis is not due to any intrinsic ATPase activity in the RADX protein preparation since it does not hydrolyze ATP on its own either in the absence or presence of ssDNA, and it does not stimulate ATP hydrolysis by the RAD51 K133R mutant that lacks ATPase activity (Supplementary Figures 1A and 1B). The stimulation of RAD51 ATP hydrolysis also is not due to removal of secondary structures in the DNA that would allow more RAD51 to bind since we utilized a poly(dT) ligand that has little secondary structure, and RPA addition does not have the same effect (Figure 2B). In addition, RADX does not stimulate RAD51 ATPase activity when no DNA is present (Supplementary Figure 1B). We conclude that there is a unique activity of RADX on the RAD51-ssDNA nucleofilament that is unlikely to be due solely to competition for ssDNA.
Figure 2. RADX prevents RAD51 nucleofilament assembly by promoting ATP hydrolysis.
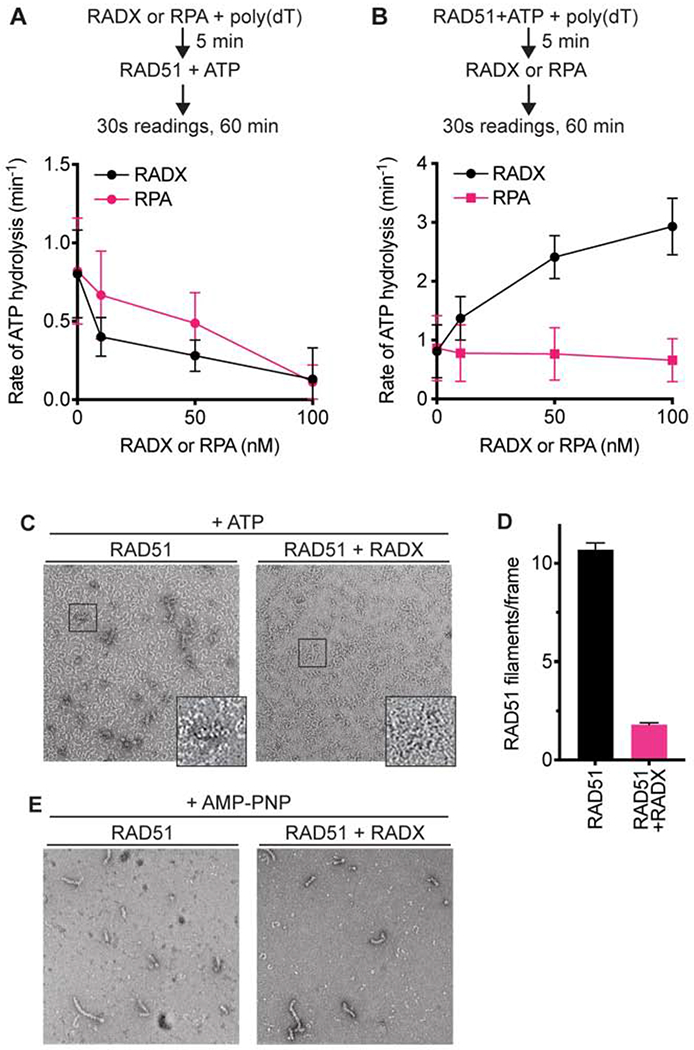
(A) RAD51 ATPase assay with increasing concentrations of RADX or RPA added to the reaction prior to the addition of RAD51. (B) RAD51 ATPase assay with RADX or RPA added to the assay after RAD51. n=3, +/− SD. See also Supplementary Figure 1. (C) Representative negative stain EM images of nucleofilaments formed in the presence and absence of RADX. The images are of samples fixed at a 10-minute timepoint after the addition of RADX. While RAD51 protofilaments are seen in both images, RAD51-DNA nucleofilaments are seen only in the absence of RADX. (D) Quantitation of RAD51 nucleofilaments formed in the presence or absence of RADX. The assessment was done using n=183 and n=195 micrographs collected randomly over the grid in the absence and presence of RADX, respectively (Mean+/−SEM). (E) Representative negative stain EM images of nucleofilaments formed in the presence and absence of RADX, with AMP-PNP in the reaction buffer.
RADX prevents the formation of RAD51-ssDNA nucleofilaments
ATP hydrolysis releases RAD51 from DNA (Kowalczykowski, 2015). We used negative-stain electron microscopy (EM) to test whether the increase in ATP hydrolysis by RAD51 in the presence of RADX affects RAD51 nucleofilament formation. RAD51 was pre-bound to a 72 base ssDNA oligonucleotide in the presence of ATP and magnesium, and samples were fixed to EM grids at timepoints after RADX addition. Under these conditions RAD51 is able to begin the filament formation process before the addition of RADX. The grids show two classes of particles – the first are small, thin, often spiral structures formed by RAD51, referred to as protofilaments. The second are the helical nucleofilament particles formed by RAD51 and DNA that match previously published data (Zhao et al., 2018). Filament formation in the presence or absence of RADX was monitored by counting the number of nucleofilaments present in over 150 images per condition. As expected, in the absence of RADX, RAD51 forms filaments; whereas, a striking reduction in the number of nucleofilaments is seen in the presence of RADX (Figures 2C and 2D). In contrast, when RAD51 is incubated with DNA in the presence of the non-hydrolysable ATP analog AMP-PNP, RADX is unable to disrupt filament formation, allowing RAD51 to generate longer helical filaments (Figure 2E). We did not detect a difference in the appearance, or pitch, of the RAD51 filament when RADX was present. Therefore, RADX inhibits RAD51 filament formation in a manner dependent on RAD51 ATP hydrolysis.
RADX inhibits RAD51-mediated strand exchange and D-loop formation
Since RADX stimulates RAD51 ATPase activity and inhibits filament formation we asked whether it also inhibits other ATP-dependent RAD51 activities. First, we employed an assay that measures the ability of RAD51 to catalyze DNA strand exchange in the presence of RPA to generate joint molecules and eventually a nicked circular dsDNA product (Figure 3A). 10μM RAD51 (a 3:1 stoichiometry to the ssDNA) is added to facilitate strand exchange in this assay. Since RAD51 is at a 24-170-fold molar excess over RADX in different cell types (Bhat et al., 2018), we employed a concentration of RADX 100-fold lower than RAD51. Addition of 100nM RADX completely inhibited product formation when RADX was added prior to the initiation of the reaction (Figure 3B). RADX was also able to inhibit product formation when added at a later time point, though the degree of inhibition was reduced (Figure 3C).
Figure 3. RADX prevents RAD51 mediated strand-exchange and D-loop formation.
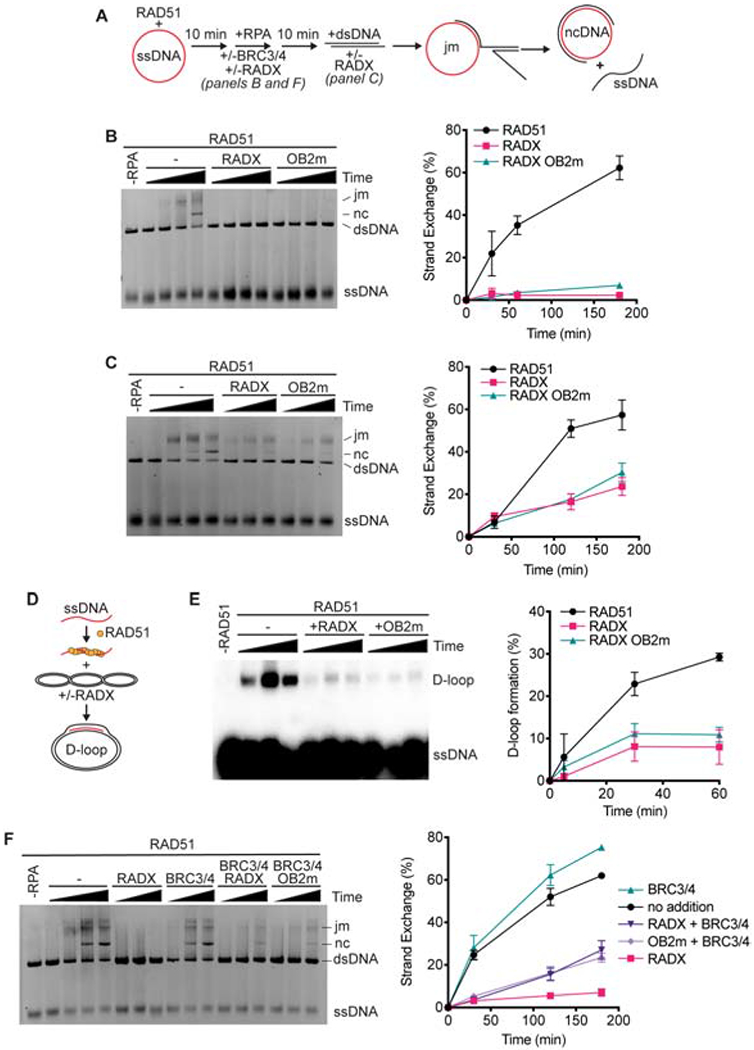
(A) Schematic of RAD51-mediated strand exchange assay (jm, joint molecules; nc, nicked circular dsDNA). RADX was added either 10 minutes prior to the dsDNA in panels B and F or at the same time as dsDNA in panel C. (B) A representative gel and quantitation from n=3 strand exchange assays are shown. Error bars are SD. (C) Strand exchange assay in which RADX or RADX OB2m was added at the same time as the dsDNA. Representative gel and quantitation from n=3 experiments. (D) Schematic of D-loop formation assay. (E) A representative gel of the D-loop assay and quantitation (n=3, +/− SD). (F) Strand exchange assay with BRC3/4 and/or RADX proteins added at the same time as RPA.
The reduction of product formation in the presence of RADX could be due to RADX antagonizing one or more of the multiple steps of the strand exchange reaction, such as strand invasion or branch migration. RADX reduced joint molecule or nicked circle product formation to a similar degree suggesting that the earliest steps of strand invasion may be inhibited (Supplementary Figure 2A). To test this hypothesis directly, we utilized a displacement loop (D-loop) assay, which uses similar reaction conditions but only measures the first strand invasion step and does not depend on branch migration (Figure 3D). Consistent with RADX destabilizing the RAD51 nucleofilament and acting at the early steps of strand invasion, we observe a marked reduction in D-loop product formation in the presence of RADX (Figure 3E).
To test if the inhibition of RAD51-mediated strand exchange and D-loop formation could be due solely to RADX excluding RAD51 from the ssDNA we utilized a RADX ssDNA binding mutant, RADX OB2m, which greatly reduces the ssDNA binding affinity of RADX and does not compete for ssDNA with RAD51 (Figure 1A; Bhat et al., 2018). Surprisingly, RADX OB2m markedly inhibited RAD51 functions in both the strand-exchange and D-loop assays to the same extent as wild-type RADX (Figures 3B–3E), although additional experiments in the presence of the BRC motifs of BRCA2 do reveal a difference between wild-type and OB2m RADX (see below). Thus, RADX regulates RAD51 via a mechanism that is not solely dependent on its ability to outcompete RAD51 for ssDNA.
In cells, factors that promote RAD51 nucleofilament stability, like BRCA2, may counteract the inhibitory action of RADX. The BRC3/4 motifs of BRCA2 stabilize the RAD51 filament in vitro by reducing RAD51 ATP hydrolysis and stimulating RAD51-mediated strand exchange activity (Carreira and Kowalczykowski, 2011; Jensen et al., 2010). Therefore, we investigated whether the BRCA2 BRC3/4 motifs could overcome inhibition by RADX. As previously reported, BRC3/4 stimulates the formation of strand exchange products mediated by RAD51 (Figure 3F and Supplementary Figure 2B). When BRC3/4 and RADX are added to the reaction simultaneously, the presence of BRC3/4 reduces the inhibition observed by RADX as well as RADX OB2m (Figure 3F). This partial neutralization by the BRC3/4 motifs is also observed if BRC3/4 is allowed to interact with the RAD51 filament prior to the addition of RADX, and BRC3/4 almost fully neutralizes the RADX OB2m protein in this condition suggesting that impairing RADX DNA binding does reduce its ability to inhibit RAD51 when BRCA2 is present (Supplementary Figure 2B). Since BRC3/4 acts to stabilize the nucleofilament, this ability of BRC3/4 to rescue RAD51 function in vitro further supports the idea that the mechanism of RADX-mediated RAD51 antagonism is via regulation of nucleofilament stability.
RADX directly interacts with RAD51
Since ssDNA binding does not fully explain how RADX regulates RAD51, we tested whether RADX antagonism of RAD51 is mediated by a direct interaction. Indeed, in an in vitro binding assay using purified proteins, we observed a direct interaction between RADX and RAD51 (Figure 4A). Strikingly, the direct interaction is observed only when ATP or the ATP analog AMP-PNP is bound to RAD51 since we were unable to observe an interaction with nucleotide-free RAD51 or when RAD51 was bound to ADP (Figure 4A). RADX also interacts with the RAD51 K133R protein which binds, but cannot hydrolyze ATP (Supplementary Figure 3A). In contrast, RADX has greatly reduced interaction with RAD51 K133A which has approximately 100-fold lower binding affinity to ATP (Chi et al., 2006; Forget et al., 2007). Moreover, the RAD51-RADX interaction was not altered by addition of ssDNA, and RADX OB2m also binds RAD51-ATP, confirming that the interaction is independent of DNA (Supplementary Figure 3B). These results suggest the interaction of RADX with RAD51 is facilitated by an ATP-induced change in the conformation of RAD51.
Figure 4. RADX interacts directly with RAD51 through residues in the RADX OB3 domain.
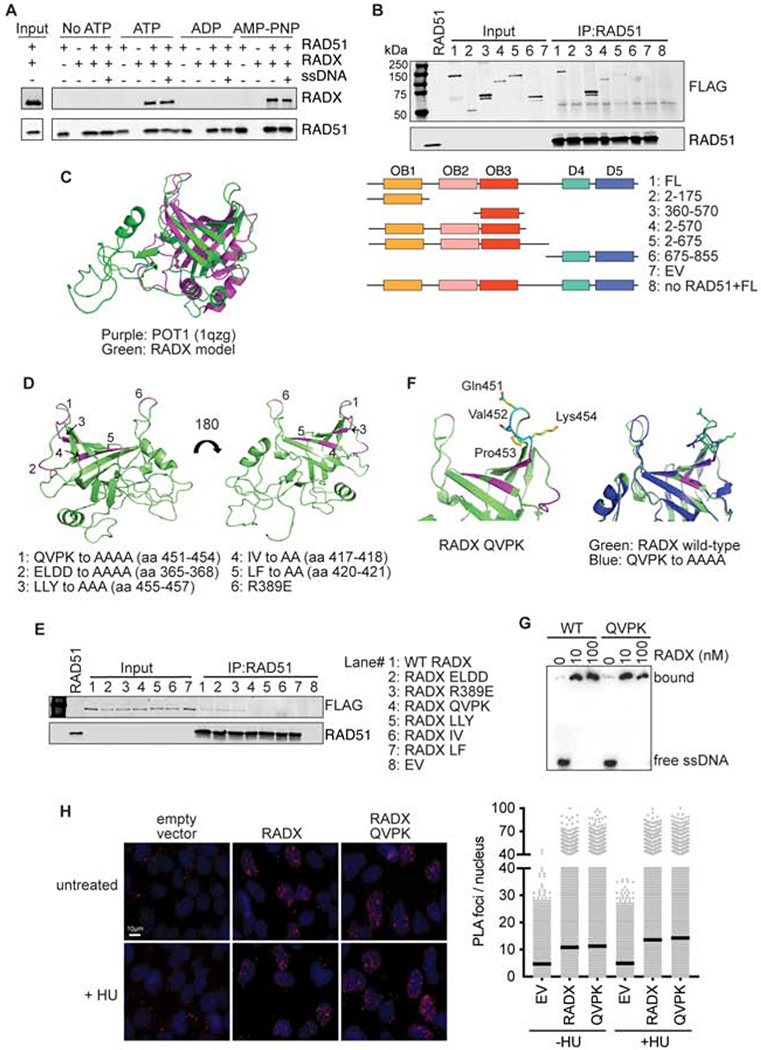
(A) Direct interaction of RADX and RAD51 was assessed using RAD51 antibody conjugated to Protein G beads to pull down purified RAD51 and RADX in the presence of the indicated nucleotide with and without ssDNA. Each binding reaction contains equimolar amounts of RADX and RAD51. After washing, the beads were boiled in SDS loading buffer and eluted protein detected by immunoblotting. (B) Interaction of purified RADX fragments with RAD51 in the presence of ATP. FL, full length (C) Homology model of RADX OB3 domain based on the OB-fold domain in POT1 (PDBID 1qzg). (D) Location of the surfaces of RADX OB3 tested for interactions with RADX. (E) Interaction of purified RADX OB3 mutants with RAD51 in the presence of ATP. (F) Location of the QVPK residues on the predicted loop of OB3 (left panel) and comparison of the models of wild-type and QVPK mutant OB3 domains (right panel). (G) Electrophoretic mobility shift assay of purified RADX and RADX QVPK mutant protein with poly (dT60) ssDNA. (H) Proximity ligation assay between Flag-RADX and EdU. Cells were labeled for 10 minutes with EdU and treated with hydroxyurea (HU) for two hours where indicated. An image and quantitative data from a representative experiment is shown. (EV, empty vector)
The interaction of RADX with RAD51-ATP is reduced when the BRC3/4 motifs of BRCA2 are included in the binding reaction (Supplementary Figure 3C). RAD51 binds BRC3/4 in these conditions, while RADX does not (Supplementary Figure 3D). Thus, the BRC motifs of BRCA2 compete with RADX for RAD51 binding suggesting that they may interact with a similar binding surface on RAD51. These results are consistent with the ability of the BRC3/4 motifs to neutralize the ability of RADX to inhibit RAD51 strand exchange activity.
To determine a specific region of RADX responsible for interacting with RAD51, we used a series of RADX deletion mutants purified as FLAG-RADX proteins from HEK293T cells for RAD51 binding assays. The deletions were designed based on the prediction that RADX contains three N-terminal oligonucleotide/oligosaccharide (OB) binding domains and possibly two structured domains (D4 and D5) in its C-terminus (Figure 4B)(Dungrawala et al., 2017). The pull-down experiments showed that fragments of RADX containing amino acid residues 360-570 are necessary and sufficient to mediate the interaction with RAD51 (Figure 4B and Supplementary Figure 3E).
Residues 360-570 are within the predicted OB3 domain, which aligns best to the OB-fold of POT1 (Figure 4C). Using the structural model of RADX OB3 threaded through the structure of the POT1-OB domain and sequence conservation of residues within this region, we generated a series of mutations across the OB3 domain and examined the ability of these mutants to interact with RAD51 (Figure 4D). Alanine mutations of residues 451-454, 455-457, 417-418, or 420-421 were each found to disrupt the interaction between RAD51 and RADX (Figure 4E and Supplementary Figure 3F). These mutations are predicted to alter residues that help form the cleft of the OB fold. In contrast, a charge reversal R389E mutation, or a 4-residue alanine mutation of residues 365-368 in a region more distal from the cleft retained interaction with RADX (Figure 4E). We selected the 451-454 mutation (QVPK to AAAA, RADX QVPK) for further analysis since these mutations are predicted to be in a loop region that would not be expected to disrupt packing of the hydrophobic core (Figure 4F). Purified RADX QVPK retains ssDNA binding similar to that of wild-type RADX (Figure 4G). In addition, this mutant localizes to replication forks in cells similar to wild-type RADX (Figure 4H). These results imply that this OB3 mutation does not grossly disrupt protein folding or other RADX functions.
RADX interactions with RAD51 and ssDNA are required to destabilize the RAD51 nucleofilament.
To determine if the interaction between RAD51 and RADX is responsible for inhibiting RAD51, we tested whether the RADX QVPK mutant could stimulate ATP hydrolysis by RAD51 using the spectrophotometric assay. RADX QVPK retains a reduced but still significant ability to stimulate RAD51 ATP turnover (Figure 5A). In contrast, the RADX OB2m mutant that has reduced ssDNA binding affinity is unable to stimulate RAD51 ATP hydrolysis. Thus, the reaction conditions of this ATP assay may be mostly reflecting how effectively RADX replaces RAD51 on the ssDNA, leading to the observed increase in ATP hydrolysis by RAD51 as it dissociates.
Figure 5. RADX must interact with RAD51 to inhibit strand exchange and D-loop formation.
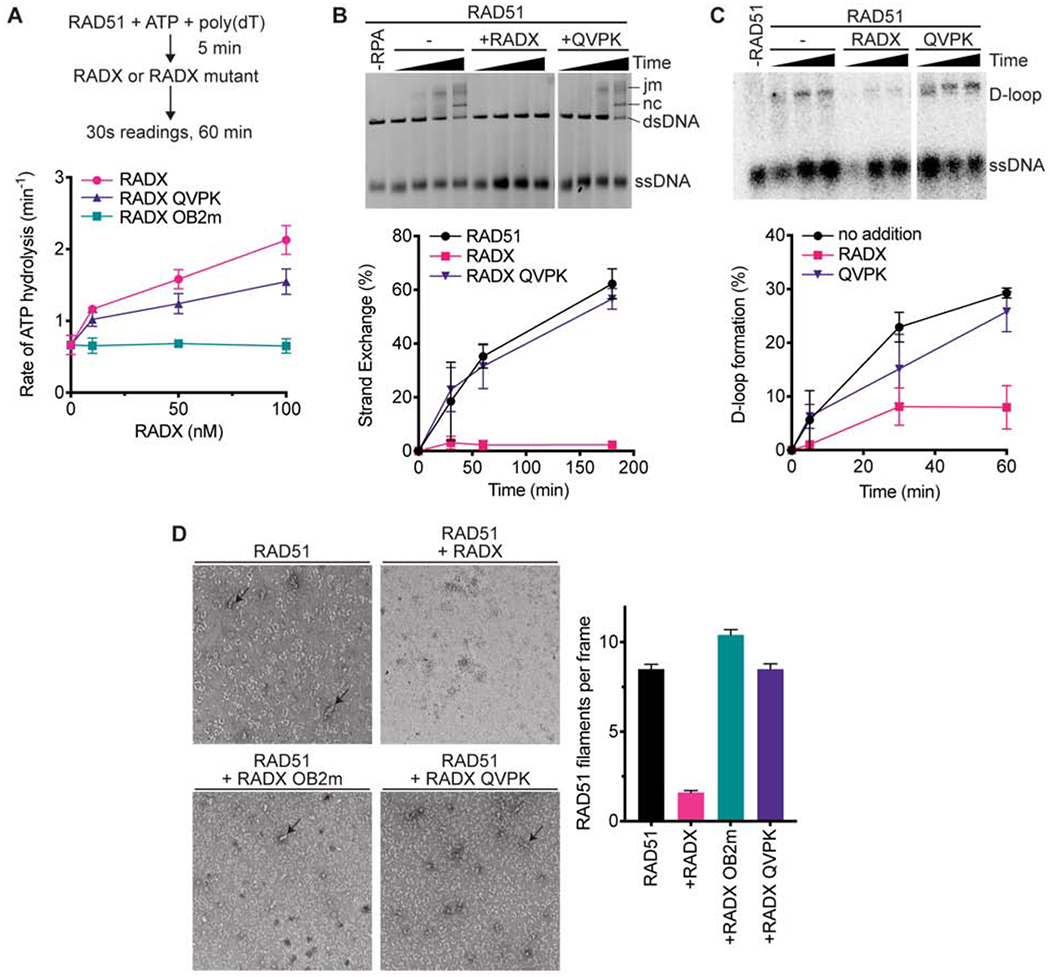
(A) The ssDNA-dependent ATPase activity of RAD51 was measured in the presence of wild-type, QVPK, and OB2m RADX proteins (n=3 +/− SD). (B) Strand exchange assay in which RADX wild-type and QVPK mutant were added at the same time as RPA (n=3 +/−SD). (C) D-loop formation assay with representative gel and quantitation (n=3 +/−SD). (D) Nucleofilament formation by RAD51 without RADX (n=178) in the presence of RADX (n=163), RADX OB2m (n=194), or RADX QVPK (n=208). (Error bars are SEM). The panels are representative micrographs.
We then tested whether RAD51 binding is important for RADX to inhibit other RAD51-functions. In contrast to wild-type RADX and RADX OB2m, the RADX QVPK protein is completely unable to inhibit RAD51 strand exchange activity (Figure 5B). RADX QVPK is also largely unable to inhibit RAD51-mediated D-loop formation (Figure 5C). These results suggest that the interaction with RADX contributes to the disruption of the RAD51 nucleofilament. Indeed, unlike wild-type RADX, both the QVPK and OB2m mutants fail to destabilize RAD51 nucleofilaments (Figure 5D). Together, these data show that RAD51- and ssDNA-binding by RADX antagonizes RAD51 filament formation, although only the RAD51 interaction is essential to block RAD51-mediated strand exchange and D-loop formation.
The RADX interaction with RAD51 is essential to maintain replication fork stability
We next asked whether the RAD51 interaction is important for RADX function in cells. To this end, RADXΔ cells derived using CRISPR-Cas9 targeting of the RADX gene in U2OS cells were complemented with lentivirus encoding GFP-tagged RADX or RADX QVPK. As we reported previously, wild-type RADX expression in this system is attenuated after a few passages presumably because of toxicity associated with its overexpression (Figure 6A) (Dungrawala et al., 2017). In contrast, the RADX QVPK mutant expressed at high levels through at least 14 passages suggesting it does not cause any growth impairment (Figure 6A).
Figure 6. RADX interacts with RAD51 to maintain replication fork stability.
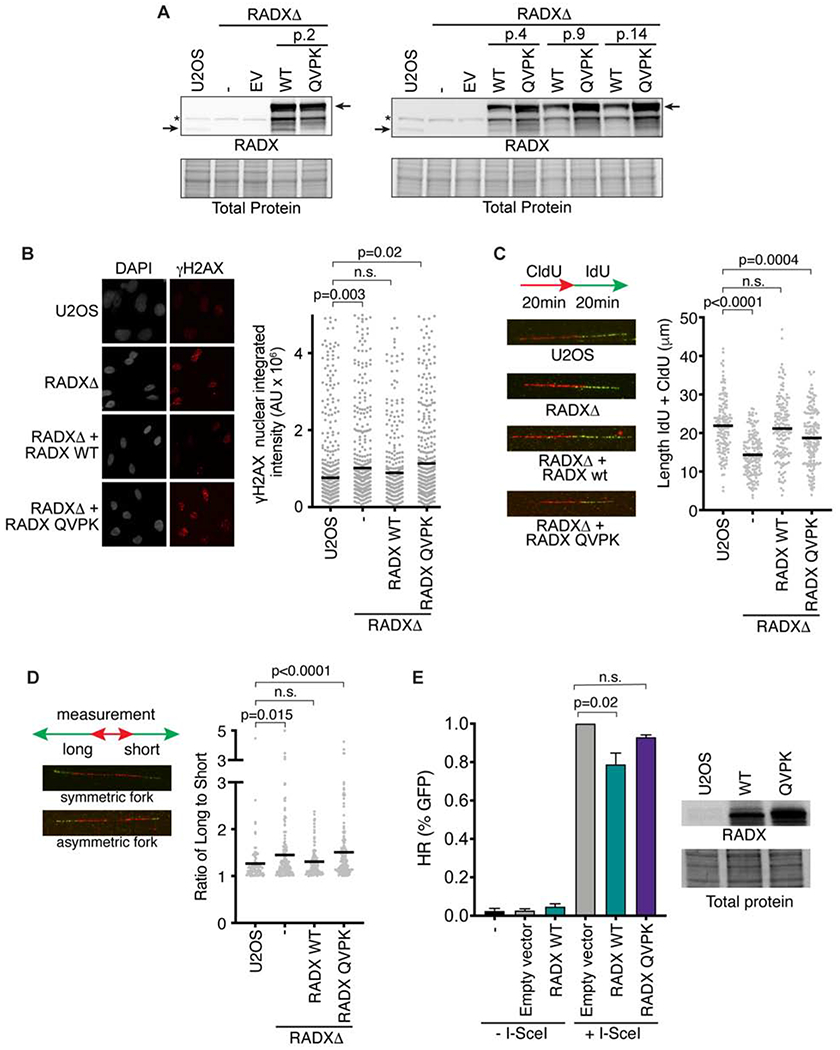
(A) Immunoblots of U2OS, or RADXΔ cells infected with lentivirus expressing the wild-type (WT) RADX or RADX QVPK (EV = empty vector). The passage number after infection and selection is indicated. (B) Cells were imaged for γH2AX. P values were derived from a one-way ANOVA with Dunnett’s multiple comparison test. (C) Cells were labeled with CldU and IdU as indicated and then analyzed by DNA combing. Representative images and experiment are shown with P values derived from a one-way ANOVA with Dunnett’s multiple comparison test. (D) Fork symmetry was assessed as indicated. At least 177 DNA fibers were measured from four biological replicates. P values were derived from a Kruskal-Wallis test. (E) The percentage of DR-GFP-positive U2OS cells after overexpression of empty vector, RADX WT or RADX QVPK and I-Scel expression vector. Immunoblots show the level of overexpression of RADX WT and RADX QVPK (Mean+/−SEM, n=3 in which 25,000 cells were scored per experiment; p-value derived from One-way ANOVA with Dunnett’s multiple comparisons test).
RADXΔ cells have elevated levels of the DNA damage marker γH2AX in S-phase (Figure 6B). Expression of wild-type RADX rescues this phenotype; however, the RADX QVPK mutant expressing cells retain elevated γH2AX. Inactivating RADX also causes decreased rates of replication fork elongation, and an increase in asymmetric sister replication forks (Figures 6C and 6D). Again, expression of wild-type but not the QVPK mutant RADX protein can partially or completely rescue these defects (Figures 6C and 6D). These results were the same irrespective of cell line passage number. Finally, as reported previously (Dungrawala et al, 2017), overexpression of RADX modestly reduces the efficiency of HDR as measured by the DR-GFP assay, but this affect is not observed with overexpression of RADX QVPK (Figure 6E).
Thus, we conclude that RADX prevents DNA replication-associated DNA damage, maintains replication elongation, and prevents fork collapse by directly binding RAD51 and decreasing its ability to form nucleofilaments.
Discussion
RAD51 has multiple DNA repair and replication functions and must be highly regulated to maintain genome integrity. We find that RADX interferes with the stability of the RAD51 nucleofilament through a direct interaction with the ATP-bound form of RAD51. Along with ssDNA binding, the RADX interaction with RAD51 regulates RAD51-mediated functions in vitro and is needed to maintain replication fork stability in cells. The BRC domains of BRCA2 can counteract the inhibition of RADX on RAD51. RADX inactivation can also partially suppress the hypersensitivity of BRCA2-deficient cells to chemotherapeutic agents and PARP inhibitors (Dungrawala et al., 2017). Thus, a balance of BRCA2, RADX, and other RAD51 regulatory factors maintains the appropriate level of RAD51 filament formation and function.
RADX specifically targets ATP-bound RAD51, stimulates its ATPase activity, and inhibits its ATP-dependent functions. Stimulation of ATP turnover may promote RAD51 release from ssDNA increasing access for RADX. The nucleotide binding pocket of one RAD51 molecule is in direct contact with the ATPase domain of the adjacent molecule when it forms a filament (Conway et al., 2004). The physical interaction with RADX may alter the orientation of these surfaces and reconfigure the active site to promote ATP turnover. A similar mechanism was proposed for yeast Rad51 and the anti-recombinase Srs2, where the direct interaction allows for access to the ssDNA by Srs2 to promote clearing of the Rad51 filament (Antony et al., 2009; Colavito et al., 2009; Seong et al., 2009).
While some motor proteins such as BLM, RECQL5 and FBH1 can inhibit RAD51 through an active translocation along ssDNA to disrupt bound RAD51 (Bugreev et al., 2007; Hu et al., 2007; Simandlova et al., 2013), other regulators of RAD51 such as FIGNL1 and PARI do so without utilizing enzymatic activities (Matsuzaki et al., 2019; Moldovan et al., 2012). Interestingly, both FIGNL1 and PARI are unable to destabilize RAD51 filaments when RAD51 is unable to hydrolyze ATP, a mechanism shared by RADX. Still other RAD51 regulators target other nucleotide-bound forms of RAD51. BCCIPβ and XRCC2 promote conformational changes within RAD51 that reduce the RAD51-ADP bound state to promote active RAD51-ATP filaments (Kelso et al., 2017; Shim et al., 2004). Similarly the interaction with BRCA2 occludes the ATP binding site of RAD51, preventing occupancy by ATP or preventing the conformational change required for ATP hydrolysis (Carreira et al., 2009; Pellegrini et al., 2002).
RADX may also bind within a region of the ATPase core domain of RAD51, and this could be a rationale for the nucleotide dependence of the RAD51 interaction. Indeed, the ability of the BRC3/4 motifs of BRCA2 to compete with RADX for RAD51 binding suggests their binding surfaces on RAD51 may overlap. The BRC repeats are required for HR but are dispensable for the activity of BRCA2 in fork protection (Schlacher et al., 2011). Nonetheless, RADX seems to largely function at replication forks instead of double-strand breaks. Since overexpression of RADX can inhibit HDR, its apparent specificity may be because of the relative concentrations of RAD51 regulators at forks and at DSBs, as well as the amount of ssDNA present at these locations. However, further studies of RADX in HR may be warranted since overexpression of RADX can modestly reduce HR. In any case, our results indicate that RADX joins the complex web of RAD51 regulators.
The interaction between RAD51 and RADX is crucial to maintain replication fork stability. RADX-deficient, S-phase cells have elevated levels of the DNA damage marker γH2AX, reduced fork elongation rates, and a higher frequency of asymmetric sister replication forks. These phenotypes can be attributed to excessive RAD51 activity and inappropriate fork reversal, as depleting RAD51 or the fork reversal enzyme ZRANB3 from RADX-deficient cells rescues elongation rates and prevents fork collapse (Dungrawala et al., 2017). Neither the RADX QVPK or OB2m mutant proteins can complement the RADX-deficient phenotypes (Dungrawala et al., 2017; Schubert et al., 2017), and both inhibit RAD51 filament stability. A recent paper reported that RADX inhibits RAD51 by condensing ssDNA to prevent RAD51 binding, and this function is inactivated by the OB2m mutation (Zhang et al., 2020). However, we find that the RADX OB2m can still inhibit some RAD51 functions in vitro including strand exchange and D-loop formation. Its reduced ability to counteract BRCA2 in the strand exchange assay does suggest impairment in function. RADX OB2m retains some DNA binding activity perhaps because ssDNA binding is multivalent and only one site is mutated (Dungrawala et al., 2017). The incomplete inactivation of ssDNA binding may mask the importance of this interaction in some biochemical assays, but cellular assays indicate that RADX binding to both RAD51 and ssDNA regulates RAD51 function and maintains genome stability during replication.
In cells experiencing persistent replication stress, RAD51 must be able to act at stalled forks to promote reversal and protection of nascent strands. Yet, RADX accumulates at stalled forks. Thus, further studies are needed to understand how the inhibitory activity of RADX towards RAD51 is either neutralized or beneficial in conditions of replication stress.
Limitations of Study
Finally, this study has limitations. The cellular assays were performed in a single cancer cell line in the absence of added replication stress. RADX could function differently in other cell types, and the function of RADX binding to RAD51 at stalled forks was not investigated. The biochemical assays also have limitations. The purified, tagged RADX protein may not retain post-translational modifications or protein-protein interactions that would be present in cells. In addition, the RAD51 biochemical assays do not fully recapitulate the physiological conditions of the cell, and are sensitive to factors such as buffer conditions and concentrations of the proteins.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Cortez (david.cortez@vanderbilt.edu)
Materials Availability
Plasmids and cell lines generated in this study are available upon request from the lead contact.
Data and Code Availability
Unprocessed blots, gels and microscopy images are available on Mendeley Data at https://dx.doi.org/10.17632/g3rfkjw225.1
Experimental model and subject details
Cell culture
U2OS, and HEK293T cells were cultured in DMEM with 7.5% fetal bovine serum (FBS). U2OS RADXΔ were described previously (Dungrawala et al., 2017). Complementation of RADXΔ cells with cDNA expression vectors was completed by lentiviral infection and selection for the linked puromycin resistance cassette as described (Dungrawala et al., 2017). Plasmid and siRNA transfections were performed with polyethylenimine and Dharmafect1 (Dharmacon) respectively.
Method Details
Plasmids
The RADX cDNA was obtained from the ThermoScientific Open Biosystems Human ORFeome collection (Catalog number OHS-1770). The mutant RADX OB2m was described previously (Dungrawala et al., 2017). Mutations in the OB3 domain were constructed using by site-directed mutagenesis and were sequence-verified. In addition to RADX R389E, five different multiple alanine mutants were generated substituting for residues 365-368, 417-418, 420-421,451-454, and 455-457.
Protein Purification
His-MBP-RADX, -RADXOB2m and -RADXQVPK and His-GFP-RADX were purified from baculovirus infected Sf9 cells. Briefly, cells were lysed in buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 10 mM NaF, 10 mM sodium phosphate monobasic, 10 mM sodium pyrophosphate, 1% Triton X-100, 10% glycerol, 1 mM DTT, and a cOmplete protease inhibitor cocktail tablet (Roche). After high-speed centrifugation, the cleared lysates were incubated with Talon metal affinity resin for 2 hours at 4 °C. The beads were washed once in buffer containing 1% Triton X-100, 500 mM NaCl in PBS and then washed in buffer containing 50 mM Tris (pH 8.0), 300 mM NaCl and increasing amounts of imidazole (5-20 mM). The bound proteins were eluted in 50 mM Tris (pH 8.0), 300 mM NaCl, 10% glycerol, and 1 mM EDTA and 200 mM imidazole. The fractions containing protein were then subjected to size exclusion chromatography on a Superdex 200 10/300 Increase GL (GE Healthcare) in elution buffer with added protease inhibitors. Human RAD51 was expressed in E. coli and purified using a protocol that exploits its affinity for BRCA2 BRC repeat 4 as previously reported (Bhat et al., 2018). RPA-mCherry was purified as previously described (Ma et al., 2017). GST-BRC3/4 was obtained from Dr. Patrick Sung and purified as described (San Filippo et al., 2006).
ATPase assay
The ATPase activity of RAD51 was measured using a NADH-coupled microplate photometric assay. ATPase activity is measured as the rate of NADH decomposition (molecules of NADH decomposed per minute per molecule of RAD51). RADX (0-100 nM) or RPA (0-100 nM) were incubated with 10 μM (nucleotides) of poly(dT) ssDNA for 5 minutes at 30 °C prior to the addition of RAD51 (3 μM) and ATP (3 mM) to initiate the reaction. The reaction buffer contained 20 mM Tris pH 7.5, 10 mM MgCl2, 0.44 mM KCl, 1 mM DTT and 5% glycerol. The reaction was allowed to proceed for one hour at 30 °C with readings at 340 nm every 30 seconds.
Alternatively, RAD51 or RAD51 K133R (3 μM) was incubated with 10 μM (nucleotides) of poly(dT) ssDNA for 5 minutes at 30 °C prior to the addition of RADX (0-100 nM) or RPA (0-100 nM). ATP (3 mM) was included in the initial reaction with RAD51 and ssDNA. The reaction was allowed to proceed for 1hr at 30 °C with readings at 340 nm every 30 seconds. Additionally, the reaction above was also performed in the absence of ssDNA, and RAD51 or RAD51 K133R were incubated in the presence of RADX (0-100 nM) with readings taken as described above. A control reaction of no NADH, in which all reaction components were added except for NADH was used to subtract the background absorbance at 340 nm. Additional reaction controls for RADX alone (no RAD51 added) and no RAD51 added were included and are shown in Supplementary Figure 1.
RAD51 filament formation and negative stain electron microscopy
RAD51 filament formation in the presence or absence of RADX was assessed as follows. The initial reaction contained RAD51 (4 μM), ATP/AMP-PNP (4 μM), dT72 ssDNA (6 μM) in reaction buffer of 25 mM HEPES, 25 mM KCl, 4 mM MgCl2 (pH7.5). This reaction mix was incubated at 37 °C for 45 minutes. An initial aliquot (time-point 0) was taken to estimate the filament formation, the sample was split and RADX (40 nM) was added to one aliquot. The reaction was then allowed to proceed for 15 min at 37 °C, at which point aliquots were taken from both reactions. The samples were quenched with 5mM EDTA and fixed on grids for negative stain electron microscopy. Samples were applied to glow-discharged continuous carbon coated grids, and negatively stained using 2% uranyl acetate. All grids were screened and imaged on a ThermoFisher FEI Morgagni microscope operating at 100kV with an AMT 1k × 1k CCD camera. The grids used to quantitate the filament formation were further imaged on a ThermoFisher FEI Tecnai F20 operating at 200 kV with a 4k × 4k CCD camera. Randomized and spatially distanced areas on each of these grids (with and without RADX) were imaged. Data processing and analysis was done using the Scipion suite of software (de la Rosa-Trevin et al., 2016).
Single molecule DNA curtain assays
ssDNA curtains were prepared and visualized by total internal reflection fluorescence microscopy (TIRFM) equipped with a 488-nm laser (Coherent Sapphire, 200 mW), a 561-nm laser (Coherent Sapphire, 200 mW), and two Andor iXon EMCCD cameras. Flowcells and ssDNA curtains were prepared as previously described (Crickard et al., 2020). In brief, lipid bilayers were built upon a quartz glass with nanofabricated chromium barriers and pedestals. The ssDNA substrate was generated using rolling circle replication with a biotinylated primer, a circular M13 ssDNA template, and phi29 DNA polymerase. The biotinylated ssDNA was injected into the sample chamber and attached to the bilayer through a biotin–streptavidin linkage. A custom–built shuttering system was used to avoid signal bleed–through during image acquisition. With this system, images from the green (GFP-RADX) and the red (RPA-mCherry) channels are recorded independently, these recordings are offset by 100 milliseconds such that when one camera records the red channel image, the green laser is shuttered off, and vice versa.
RAD51 filament assembly kinetics, with or without RADX, were measured as follows. Biotinylated ssDNA was aligned at the barriers by application of flow in BSA buffer (40 mM Tris-HCl [pH 8.0], 1 mM MgCl2, 1 mM DTT, 0.2 mg/mL BSA) at 37°C with a flow rate of 1 mL/min. Secondary structure was reduced with a 500 μL injection of 7 M urea, immediately followed by 5–10 mL of BSA buffer containing 100 pM RPA-mCherry. Then, 1 or 20 nM GFP-RADX was injected into the flow cell through a 150 μL loop, buffer flow was terminated when protein reached the flow cell, and the reactions were incubated at 37°C for 5-10 minutes. After that, GFP-RADX was flushed out with RAD51 HR buffer (20 mM Tris-HCl [pH 7.5], 1 mM MgCl2, 5 mM CaCl2, 100 mM KCl, 2 mM ATP, 1 mM DTT, 0.2 mg/mL BSA) at 1 ml/min for 2 min. Then, 2 μM RAD51 in a 150 μL loop was injected into the flow cell, buffer flow was terminated when protein reached the flow cell, and the reactions were incubated at 37°C for 20 minutes. The loss of RPA fluorescence signal was monitored to verify recombinase filament assembly. The time-increase decrease in the normalized RPA-mCherry fluorescence intensity (integrated over entire ssDNA molecules) reflects the disassembly of RPA-mCherry, which is used for the calculation of RAD51 assembly. The resulting intensity curves were fitted to a simple exponential decay function: I= A×e−k*t, where I is the normalized fluorescence intensity, k is the observed disassembly rate of RPA, which is equal to RAD51 assembly rate.
Strand Exchange Assay
The strand exchange assay was performed as described and conditions adapted from (Bugreev and Mazin, 2004). Briefly, ϕX174 circular ssDNA (30 μM) was incubated with RAD51 (10 μM) for 10 minutes at 37 °C in buffer containing 25 mM TrisOAc pH 7.5, 250 mM NaCl, 1 mM MgCl2, 2 mM CaCl2, 2 mM ATP and 1 mM DTT. Then, RPA was added to the reaction at a final concentration of 2 μM and further incubated for 10 minutes. The reaction was initiated by the addition of 30 μM ϕX174 dsDNA, linearized by ApaL1, and the reaction proceeded for 180 minutes. The addition of RADX (0-100 nM) or BRC3/4 (300 nM) is indicated in the reaction scheme in Figure 3A. Time points were taken at 0, 30,120 and 180 minutes by removal of 7 ul of reaction mixture into 3 μl of 0.5% SDS and 0.5 μg/mL proteinase K, and incubated for 20 minutes at 37 °C. After addition of loading dye, the deproteinized samples were loaded onto a 1.0% agarose gel in 1X TAE buffer and electrophoresed at 20V for 16 hr. The products were visualized after one-hour ethidium bromide staining and quantified using ImageLab software (BioRad) Total percent strand exchange was calculated using the integrated intensities of the dsDNA and product bands, and the formula (JM/1.5)+NC/((JM/1.5)+NC)+ dsDNA (Liu et al., 2011).
Displacement Loop Assay
32P-labeled oligonucleotide D1 (3 μM) which is complementary to positions 1932–2022 of pBluescript SK DNA was incubated with RAD51 (1 μM) in buffer containing 25 mM TrisOAc pH 7.5, 250 mM NaCl, 1 mM MgCl2, 2 mM CaCl2, 2 mM ATP and 1 mM DTT for 5 min at 37 °C. RADX, RADX OB2m, or RADX QVPK were added to the reactions at the same time as the addition of supercoiled pBluescript SK (35 μM base pairs) to initiate the reaction. Reactions were incubated at 37 °C and at the indicated time point (0, 5, 30 min) an aliquot of the reaction was removed and added to SDS (0.5%) and Proteinase K (5 μg/ml) to deproteinize the reactions, followed by a 20 min incubation at 37 °C. All reaction products were resolved on a 1.0% agarose gel, dried and visualized using a phosphorimager (Typhoon FLA 7000, GE Healthcare) and quantified using ImageLab (BioRad).
RADX RAD51 pulldown assays
HEK293T cells were transfected using FLAG-GFP-RADX, or FLAG-GFP-RADX fragment or FLAG-GFP-RADX OB3 mutant constructs. Cells were lysed in NETN buffer (20 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5% NP40, cOmplete protease inhibitor cocktail) for 30 minutes at 4 °C. Clarified lysates were incubated with Anti-FLAG M2 magnetic beads (Sigma) for 2 hours at 4 °C. Beads were washed 2X in lysis buffer, 2X in LiCl buffer (lysis buffer containing 0.3 M LiCl) and twice in elution buffer (50 mM Tris pH 8.0, 10% glycerol, 200 mM KCl, 1.0 mM EDTA, 1 mM DTT, 0.2 mM PMSF). The bound proteins were eluted in the elution buffer containing 0.25 mg/mL Flag peptide on ice for 90 minutes. For the RAD51 pulldown, RAD51 mouse monoclonal antibody (14B4, Abcam) was incubated with Protein G magnetic beads for 1 hour at room temperature. The resin was washed once in binding buffer (50 mM Tris pH 8.0, 200 mM NaCl, 2 mM CaCl2, 2 mM ATP, 1 mM DTT). 100 nM of purified RAD51, RAD51 K133R, or RAD51 K133A was added to the beads and incubated further for 1 hour at room temperature. The resin was washed twice in binding buffers before the addition of the purified RADX protein for 1 hour. Resin was washed twice in binding buffer before the addition of 2X SDS buffer and resolved by SDS PAGE and immunoblotting. For the RADX pulldowns presented in Supplementary Figure 3, conditions were identical to above, except the pulldown was performed with protein A conjugated to GFP antibody (Santa Cruz, sc-8334) for RADX fragment mutants or RADX antibody (Novus Biologicals NBP2-13887) for the RADX OB3 mutants. For the BRC3/4 competition experiment, conditions were identical to the RAD51 pulldowns above, except that 20-100 nM of purified GST-BRC3/4 was added to the RAD51 pulldown prior to the addition of RADX. For the RADX pulldown of GST-BRC3/4, protein A was conjugated to RADX antibody (Novus Biologicals, NBP2-13887) and 100 nM of purified RADX was added to the beads as above before the addition of 100 nM of GST-BRC3/4.
Generation of homology model
Homology models of RADX OB3 domain and the QVPK mutant were generated by threading using the coordinates of the POT1 OB-fold domain (PDBID 1qzg) using the program MODELLER v9.16 (Eswar et al., 2006). The images in panels C, D and F of Figure 4 were generated in Pymol (Schrodinger LLC).
DNA Binding assays
The RADX electrophoretic mobility shift assay was performed largely as previously described (Bhat et al., 2018). Briefly, 1.0 nM 32P-labeled oligo-dT60 ssDNA was incubated with RADX in 20 μl reactions of binding buffer containing 50 mM Tris pH 7.5, 300 mM NaCl, 10% glycerol and 1 mM DTT. The reactions were incubated at room temperature for 30 minutes and were separated on an 8% 1X TBE 37.5:1 gel at 60V for 120 minutes at 4 °C. Gels were dried and quantified using a Typhoon. The biotin-ssDNA pull-down assays utilized 4 μM RAD51 pre-bound to the DNA in the presence of 50 mM Tris pH 7.5, 300 mM NaCl, 2 mM ATP, 2 mM CaCl2, 10% glycerol and 1 mM DTT. Increasing amounts of RADX was added and reactions were incubated for 30 minutes prior to separation of the DNA-bound and supernatant fractions and analysis by SDS-PAGE and immunoblotting.
Immunofluorescence
Cells plated in 96-well clear-bottom plates were incubated with media containing 10 μM EdU for 20 minutes, pre-extracted for 5 minutes on ice in 20 mM HEPES, pH 7.0, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose, and 0.5%Triton X-100 followed by fixation in 3% paraformaldehyde. Cells were blocked for 1 hour in PBS containing 5% BSA. EdU was labeled by addition of 2 mg/mL sodium ascorbate, 2 mM copper sulfate, and 5 μM Alexa Fluor 647 conjugated azide in PBS for 30 minutes. Primary antibody incubation was performed for 1 hour in 1% BSA in PBS followed by a 45-minute incubation in secondary antibody. Nuclei were stained with a 5-minute incubation with DAPI in PBS. Plates were imaged on a Molecular Devices ImageXpress system and integrated nuclear intensity of γH2AX of EdU-positive cells quantitated using the Molecular Devices software.
Proximity ligation assay
To determine nascent chromatin localization, cells were plated in a 96-well plate and labeled with 10μM EdU for 20 minutes. Cells were permeabilized using 0.5% Triton X-100 solution (20mM HEPES, 50mM NaCl, 3mM MgCl2, 300mM Sucrose and 0.5% Triton X-100) and fixed in 3% paraformaldehyde minutes on ice. Cells were then incubated in 10% goat serum followed by antibodies to FLAG (Sigma F3165) and anti-biotin to recognize EdU after conjugation to biotin azide (Cell Signaling 5597). Proximity ligation was completed according to the manufacturer’s protocol (Sigma) and images were obtained and quantified using a Molecular Devices ImageXpress instrument.
DNA molecular combing
Cell were labeled with 20 μM CldU (Sigma, C6891) followed by 100 μM IdU (Sigma, l7125), for the time indicated. Cells were embedded in agarose plugs and the DNA was applied to coverslips using Genomic Vision’s combing instrument. The DNA was stained with antibodies that recognize IdU and CldU for 1 h, washed in PBS, and probed with secondary antibodies for 45 minutes. Images were obtained using a 40X oil objective (Nikon Eclipse Ti). Analysis of fiber lengths performed using Nikon Elements software with the investigator blinded to sample identity.
GFP assays
DR-GFP U2OS reporter cells were used as described previously (Nakanishi et al., 2005). Briefly, 2.5×105 cells were transfected with 2 μg total DNA of either wild type RADX, RADX QVPK, empty vector or I-SceI expressing plasmid pCBASceI using FuGENE HD (Promega). After 72 hours, cells were analyzed by flow cytometry.
Immunoblotting
Whole-cell lysates were extracted using Igepal lysis buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 1% Igepal CA-630, 1 mM EDTA, pH 8.5) enriched with sodium fluoride (1 mM), sodium vanadate (1 mM), protease inhibitor cocktail (Roche), phenylmethylsulfonyl fluoride (PMSF, 1 mM), 25 units of Pierce Universal Nuclease, and 1 mM MgCl2. Proteins were analyzed by SDS-PAGE and immunoblotting.
Quantification and statistical analysis
All statistical analyses are described in the figure legends and were completed with Prism. Investigators were blinded to sample identities and all experiments were completed at least three times unless otherwise indicated.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-RADX | Novus Biologicals | Cat#NBP2-13887; RRID: AB_2687552 |
| Mouse monoclonal anti-RAD51 (14B4) | Abcam | Cat# ab213, RRID:AB_302856 |
| Mouse monoclonal anti FLAG M2 | Sigma Aldrich | Cat#F3165; RRID:AB_259529 |
| Rabbit polyclonal anti-GFP | Santa Cruz | Cat#sc-8334; RRID:AB_641123 |
| Rabbit polyclonal to GST | Abcam | Cat# ab111947 AB_10861266 |
| Mouse monoclonal anti-phospho-Histone H2A.X (Ser139) | Millipore | Cat#16-193, RRID:AB_310795 |
| Mouse monoclonal anti BrdU | BD | Cat#347580; RRID:AB_10015219 |
| Rat monoclonal anti BrdU | Abcam | Cat#ab6326; RRID:AB_305426 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Life Technologies | Cat# A32723; RRID:AB_2633275 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Life Technologies | Cat# A11007; RRID:AB_10561522 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CldU | Sigma Aldrich | Cat#C6891 |
| IdU | Sigma Aldrich | Cat#l7125 |
| EdU | VICB synthesis core | N/A |
| Alexa Fluor 647 azide | Thermo Fisher | Cat#A10277 |
| Hydroxyurea | Sigma Aldrich | Cat#H8627 |
| MBP-RADX | This study | N/A |
| MBP-RADX OB2m | This study | N/A |
| MBP-RADX QVPK | This study | N/A |
| GFP-RADX | This study | N/A |
| RAD51 | Provided by M.M. | N/A |
| RAD51 K133R | Provided by M.M. | N/A |
| RAD51 K133A | Provided by M.M. | N/A |
| RPA | Provided by W.J.C. | N/A |
| GST-BRC3/4 | Provided by Patrick Sung | N/A |
| RPA-mCherry | Provided by E.J.C. | N/A |
| Critical Commercial Assays | ||
| Duolink In Situ PLA Probe Anti-Rabbit PLUS | Sigma Aldrich | Cat#DU092002 |
| Duolink In Situ PLA Probe Anti-Rabbit MINUS | Sigma Aldrich | Cat#DU092004 |
| Duolink In Situ Detection Reagents, Red | Sigma Aldrich | Cat#DU092008 |
| Coverslips | MicroSurfaces Inc. | N/A |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC | Cat#HTB-96; RRID:CVCL_0042 |
| HEK293T | ATCC | Cat#CRL-11268; RRID:CVCL_1926 |
| U2OS ΔRADX | (Dungrawala et al., 2017) | N/A |
| Oligonucleotides | ||
| dT60: 5’-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3’ | IDT | N/A |
| dT72: 5’-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3′ | IDT | N/A |
| OligoD1: 5’-AAATCAATCTAAAGTATATATGAGTAAACTTGGTCTGACAGTTACCAATGCTTAATCAGTGAGGCACCTATCTCAGCGATCTGTCTATTT-3’ | IDT | N/A |
| Recombinant DNA | ||
| Plasmid pDC1272 = pLEFP-FLAG-RADX | This study | N/A |
| Plasmid pMA32 = pLEGFP FLAG-RADX QVPK | This study | N/A |
| Plasmid pMA33 = pLEGFP-FLAG-RADX IV | This study | N/A |
| Plasmid pMA34 = pLEGFP-FLAG-RADX LLY | This study | N/A |
| Plasmid pMA35 = pLEGFP-FLAG-RADX R389E | This study | N/A |
| Plasmid pMA36 = pLEGFP-FLAG-RADX LF | This study | N/A |
| Plasmid pMA37 = pLEGFP-FLAG-RADX ELDD | This study | N/A |
| Plasmid pKB06 = pEF1a-RADX | This study | N/A |
| Plasmid pKB29= pEF1a-RADX OB2m | This study | N/A |
| Plasmid pMA39= pEF1a-RADX-QVPK | This study | N/A |
| Software and Algorithms | ||
| GraphPad Prism8/9 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_000306 |
| ImageLab | BioRad | http://www.biorad.com/en-us/sku/1709690-image-lab-software; RRID:SCR_014210 |
| MODELLER v9.16 | (Eswar et al., 2006) | https://salilab.org/modeller; RRID:SCR_008395 |
| PyMOL Version 2.0 | The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. | https://www.pymol.org/; RRID:SCR_000305 |
| Scipion | (de la Rosa-Trevin et al., 2016) | http://scipion.i2pc.es; RRID:SCR_016738 |
| Deposited Data | ||
| Unprocessed blots, gels and microscopy images | This paper | https://dx.doi.org/10.17632/g3rfkjw225.1 |
Highlights.
RADX interacts directly with ATP-bound RAD51
RADX antagonizes RAD51 functions by destabilizing RAD51 nucleofilaments
Interaction between RADX and RAD51 is required to maintain fork elongation rates
BRC motifs of BRCA2 neutralize RADX inhibition of RAD51
Acknowledgements
We thank Dr. Patrick Sung and Dr. Youngho Kwon for providing reagents and critical comments. This research was supported by NIH grant R01 GM116616 to D.C., R35 GM118089 to W.J.C., and R01 CA221858 to E.C.G. M.M. is supported by the French League against Cancer (équipe labéllisée). FM is supported by a Ph.D. fellowship from the Paoli-Calmettes Institute. RADX proteins were generated with the assistance of the SBDR EMB core (P01 CA092584). Additional funding was received from the Breast Cancer Research Foundation and the Vanderbilt-Ingram Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Antony E, Tomko EJ, Xiao Q, Krejci L, Lohman TM, and Ellenberger T (2009). Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol Cell 35, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, and Cortez D (2018). RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol 25, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KP, Krishnamoorthy A, Dungrawala H, Garcin EB, Modesti M, and Cortez D (2018). RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep 24, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugreev DV, and Mazin AV (2004). Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc Natl Acad Sci U S A 101, 9988–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugreev DV, Yu X, Egelman EH, and Mazin AV (2007). Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev 21, 3085–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MK, Venkitaraman AR, and Kowalczykowski SC (2009). The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 136, 1032–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira A, and Kowalczykowski SC (2011). Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc Natl Acad Sci U S A 108, 10448–10453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, and Wold MS (2014). Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 36, 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Van Komen S, Sehorn MG, Sigurdsson S, and Sung P (2006). Roles of ATP binding and ATP hydrolysis in human Rad51 recombinase function. DNA Repair (Amst) 5, 381–391. [DOI] [PubMed] [Google Scholar]
- Colavito S, Macris-Kiss M, Seong C, Gleeson O, Greene EC, Klein HL, Krejci L, and Sung P (2009). Functional significance of the Rad51-Srs2 complex in Rad51 presynaptic filament disruption. Nucleic Acids Res 37, 6754–6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway AB, Lynch TW, Zhang Y, Fortin GS, Fung CW, Symington LS, and Rice PA (2004). Crystal structure of a Rad51 filament. Nat Struct Mol Biol 11, 791–796. [DOI] [PubMed] [Google Scholar]
- Crickard JB, Moevus CJ, Kwon Y, Sung P, and Greene EC (2020). Rad54 Drives ATP Hydrolysis-Dependent DNA Sequence Alignment during Homologous Recombination. Cell 181, 1380–1394 e1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa-Trevin JM, Quintana A, Del Cano L, Zaldivar A, Foche I, Gutierrez J, Gomez-Blanco J, Burguet-Castell J, Cuenca-Alba J, Abrishami V, et al. (2016). Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J Struct Biol 195, 93–99. [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, and Cortez D (2017). RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Molecular cell 67, 374–386 e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, and Sali A (2006). Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5, Unit-5 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forget AL, Loftus MS, McGrew DA, Bennett BT, and Knight KL (2007). The human Rad51 K133A mutant is functional for DNA double-strand break repair in human cells. Biochemistry 46, 3566–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, and Costanzo V (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P, et al. (2007). RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 21, 3073–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB, Carreira A, and Kowalczykowski SC (2010). Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso AA, Goodson SD, Watts LE, Ledford LL, Waldvogel SM, Diehl JN, Shah SB, Say AF, White JD, and Sehorn MG (2017). The beta-isoform of BCCIP promotes ADP release from the RAD51 presynaptic filament and enhances homologous DNA pairing. Nucleic acids research 45, 711–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC (2015). An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harbor perspectives in biology 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Doty T, Gibson B, and Heyer WD (2010). Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol 17, 1260–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sneeden J, and Heyer WD (2011). In vitro assays for DNA pairing and recombination-associated DNA synthesis. Methods in molecular biology 745, 363–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CJ, Gibb B, Kwon Y, Sung P, and Greene EC (2017). Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic acids research 45, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K, Kondo S, Ishikawa T, and Shinohara A (2019). Human RAD51 paralogue SWSAP1 fosters RAD51 filament by regulating the anti-recombinase FIGNL1 AAA+ ATPase. Nature communications 10, 1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ, and D’Andrea AD (2012). Inhibition of Homologous Recombination by the PCNA-Interacting Protein PARI. Molecular cell 45, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, and Jasin M (2005). Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci U S A 102, 1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, and Venkitaraman AR (2002). Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 420, 287–293. [DOI] [PubMed] [Google Scholar]
- Qi Z, and Greene EC (2016). Visualizing recombination intermediates with single-stranded DNA curtains. Methods 105, 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J, Chi P, Sehorn MG, Etchin J, Krejci L, and Sung P (2006). Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J Biol Chem 281, 11649–11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert L, Ho T, Hoffmann S, Haahr P, Guerillon C, and Mailand N (2017). RADX interacts with single-stranded DNA to promote replication fork stability. EMBO reports 18, 1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong C, Colavito S, Kwon Y, Sung P, and Krejci L (2009). Regulation of Rad51 recombinase presynaptic filament assembly via interactions with the Rad52 mediator and the Srs2 anti-recombinase. J Biol Chem 284, 24363–24371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim KS, Schmutte C, Tombline G, Heinen CD, and Fishel R (2004). hXRCC2 enhances ADP/ATP processing and strand exchange by hRAD51. J Biol Chem 279, 30385–30394. [DOI] [PubMed] [Google Scholar]
- Simandlova J, Zagelbaum J, Payne MJ, Chu WK, Shevelev I, Hanada K, Chatterjee S, Reid DA, Liu Y, Janscak P, et al. (2013). FBH1 helicase disrupts RAD51 filaments in vitro and modulates homologous recombination in mammalian cells. J Biol Chem 288, 34168–34180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Schaub JM, and Finkelstein IJ (2020). RADX condenses single-stranded DNA to antagonize RAD51 loading. Nucleic acids research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Xu J, Zhao W, Sung P, and Wang HW (2018). Determining the RAD51-DNA Nucleoprotein Filament Structure and Function by Cryo-Electron Microscopy. Methods in enzymology 600, 179–199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed blots, gels and microscopy images are available on Mendeley Data at https://dx.doi.org/10.17632/g3rfkjw225.1