

Abstract
Previous work has suggested androgen receptor (AR) signaling mediates prostate cancer progression in part through the modulation of autophagy. However, clinical trials testing autophagy inhibition using chloroquine derivatives in men with castration-resistant prostate cancer (CRPC) have yet to yield promising results, potentially due to the side effects of this class of compounds. We hypothesized that identification of the upstream activators of autophagy in prostate cancer could highlight alternative, context-dependent targets for blocking this important cellular process during disease progression. Here, we used molecular, genetic and pharmacological approaches to elucidate an AR-mediated autophagy cascade involving Ca2+/calmodulin-dependent protein kinase kinase 2 (CAMKK2; a kinase with a restricted expression profile), 5’-AMP-activated protein kinase (AMPK) and Unc-51 like autophagy activating kinase 1 (ULK1), but independent of canonical mechanistic target of rapamycin (mTOR) activity. Increased CAMKK2-AMPK-ULK1 signaling correlated with disease progression in genetic mouse models and patient tumor samples. Importantly, CAMKK2 disruption impaired tumor growth and prolonged survival in multiple CRPC preclinical mouse models. Similarly, an inhibitor of AMPK-ULK1 blocked autophagy, cell growth and colony formation in prostate cancer cells. Collectively, our findings converge to demonstrate that AR can co-opt the CAMKK2-AMPK-ULK1 signaling cascade to promote prostate cancer by increasing autophagy. Thus, this pathway may represent an alternative autophagic target in CRPC.
Keywords: prostate cancer, autophagy, CAMKK2, AMPK, ULK1, androgen, androgen receptor (AR)
Introduction
Prostate cancer is the second leading cause of cancer mortality among men in the United States (1). While most prostate cancers can be treated effectively with surgery and/or radiation, a significant number of men present with de novo metastatic disease or progress following initial treatment. The standard of care for advanced prostate cancer is androgen deprivation therapy (ADT) due to the central role of the androgen receptor (AR) in almost all prostate cancers (2). Although ADT is initially effective in slowing the cancer, it often fails within 2-3 years, after which the disease progresses to a stage referred to as castration-resistant prostate cancer (CRPC). There is currently no cure for CRPC. Interestingly, despite the failure of ADT in CRPC, the overwhelming majority of prostate cancers are still driven by AR as a result of a variety of AR reactivation mechanisms (ex. increased intratumoral androgen synthesis, AR gene and enhancer amplifications, splice variants, etc) (2). As such, AR and processes downstream of the receptor remain viable therapeutic targets in CRPC.
In an effort to identify downstream effectors of AR signaling in prostate cancer, we demonstrated CAMKK2, encoding the Ca2+/calmodulin-dependent protein kinase kinase 2 (CAMKK2) protein, to be a direct AR target gene in prostate cancer (3). These data were soon validated by several other groups (4–6). CAMKK2 expression correlated with both initial response to ADT and transition to CRPC in multiple clinical cohort tissue microarrays (TMAs) (4). In addition, CAMKK2 tracked with Gleason grade and was elevated in different genetically engineered mouse models (GEMMs) of prostate cancer (5, 7). The specific AR binding site we first identified that regulates CAMKK2 expression (3) was later confirmed by others and shown to be one of the most robust AR binding sites in CRPC patient samples (6). Functionally, CAMKK2 is required for maximum AR-mediated prostate cancer cell growth, migration and invasion in cell culture and tumor growth in xenograft and GEMMs (3–5, 7, 8).
Androgens, in a CAMKK2-dependent manner, increased the phosphorylation of AMP-activated protein kinase (AMPK) on threonine-172 of its α catalytic subunit’s activation loop. Threonine-172 p-AMPK levels correlated with prostate cancer relative to benign prostatic tissue and were further elevated in biochemically recurrent disease (9). Importantly, we previously demonstrated that many of the pro-cancer effects of CAMKK2 in prostate cancer are mediated through the activation of AMPK (3). Accordingly, knockdown of AMPK impaired AR-mediated prostate cancer cell growth (9). These data indicate that AR-CAMKK2 signaling can promote prostate cancer in part through AMPK, a known regulator of macroautophagy (10).
Macroautophagy, herein referred to as autophagy, is a highly conserved process whereby cellular components are captured and delivered to a double membrane vesicle known as an autophagosome, and subsequently degraded by the lysosomal system (11). Autophagy can function as a survival mechanism in response to stress by recycling the lysosomal breakdown products towards essential processes. Autophagy can also serve as a cellular quality control mechanism by removing damaged organelles and toxins. Therefore, autophagy is of importance in physiological processes as well as diseases such as cancer (12). However, the role of autophagy in cancer is complicated and context dependent (13–16). For example, autophagy can protect cells and tissues from damage and impair malignant transformation (17, 18). Conversely, in more advanced cancers, autophagy can enable cells to evade apoptosis in hypoxic and nutrient-deficient environments as well as promote drug resistance (19–22). In prostate cancer, studies from our laboratory and others using cell lines, xenografts and genetic mouse models indicate that autophagy can promote disease progression (23–30). This included various studies demonstrating that chloroquine, a compound known to block autophagic flux, could inhibit prostate cancer progression (23, 26, 29, 31). These preclinical data provided the rationale for a series of clinical trials (NCT04011410, NCT00726596, NCT00786682, NCT03513211, NCT01828476, NCT02421575, NCT01480154) that tested the efficacy of chloroquine derivatives such as hydroxychloroquine in men with prostate cancer (32). Chloroquine and hydroxychloroquine were chosen because they 1) are already FDA-approved for the treatment of malaria and rheumatological disorders and 2) have been demonstrated to impair autophagic flux by increasing lysosomal pH and decreasing autophagosome-lysosome fusion (33, 34). Hence, chloroquine and hydroxychloroquine represented potential clinical grade inhibitors of autophagy that could be rapidly repurposed for the treatment of cancer. To date, however, these trials, as well as similar trials in other tumors types, have yielded mixed results (14). To that end, a major challenge has been achieving high enough concentrations of chloroquine or hydroxychloroquine in patients to consistently block autophagy without major side effects (15). The chloroquine-mediated side effects may in part be due to the mechanism of action of this drug. Chloroquine-like compounds inhibit autophagy by blocking the late lysosomal step. Since lysosomal function is required for processes beyond autophagy, this indicates that chloroquine is not specific for autophagy. Hence, we speculate that targeting other steps in autophagy could provide an improved therapeutic window.
We previously demonstrated that androgens, in an AR-dependent manner could increase autophagy and autophagic flux through multiple mechanisms (23, 24). These included indirect activation of autophagy through increases in reactive oxygen species (ROS) and the expression of several core components of the autophagic machinery. Given AMPK’s known link to autophagy (35–37) and our previous findings that AR could increase AMPK activity in a CAMKK2-dependent manner (3, 8), we sought to determine whether the increased CAMKK2 observed in AR+ prostate cancer could be driving disease progression in part through activating autophagy. We also reasoned that delineation of this signaling cascade, combined with the restricted expression profile of CAMKK2 and tolerance for its systemic inhibition in mice (38, 39), could nominate alternative ways to safely block autophagy in men with prostate cancer.
Results
Chloroquine impairs CRPC xenograft growth
To initially assess the effect of chloroquine in a preclinical model of CRPC, castrated NSG mice were injected with CRPC 22Rv1 cells stably expressing firefly luciferase (22Rv1-fLuc). When tumors became palpable, mice were randomized to PBS/control and chloroquine treatment groups (Fig. 1A). The tumors were monitored by bioluminescence imaging (BLI) and caliper measurements until the maximum allowable size. In the first two weeks, the BLI clearly showed inhibition of tumor growth by chloroquine (Fig. 1B). However, the bioluminescence intensity lost its sensitivity once the tumors grew large (data not shown), which likely resulted from a lack of oxygen or necrosis in the center of the large tumors. Despite this, the tumor volume demonstrated that chloroquine treatment decreased tumor growth rate as expected (Fig. 1C). The decreased tumor growth rate corresponded to a prolonged survival (Fig. 1D). Compared to the vehicle group, fewer cell nuclei were stained, suggesting the breakdown of chromatin and therefore fewer viable cells (Fig. 1E). As expected, chloroquine treatment increased both p62 (Fig. 1E) and LC3B accumulation (Supplementary Fig. S1), indicating impaired autophagic flux. The reduced tumor growth appeared to be a product of reduced proliferation and increased cell death as assessed by BrdU and TUNEL staining, respectively (Fig. 1E). These observations suggest that inhibiting autophagy using chloroquine can reduce proliferation and increase cell death, ultimately decreasing CRPC growth and prolonging survival.
Fig 1. Chloroquine inhibits castration-resistant prostate cancer (CRPC) growth in vivo.
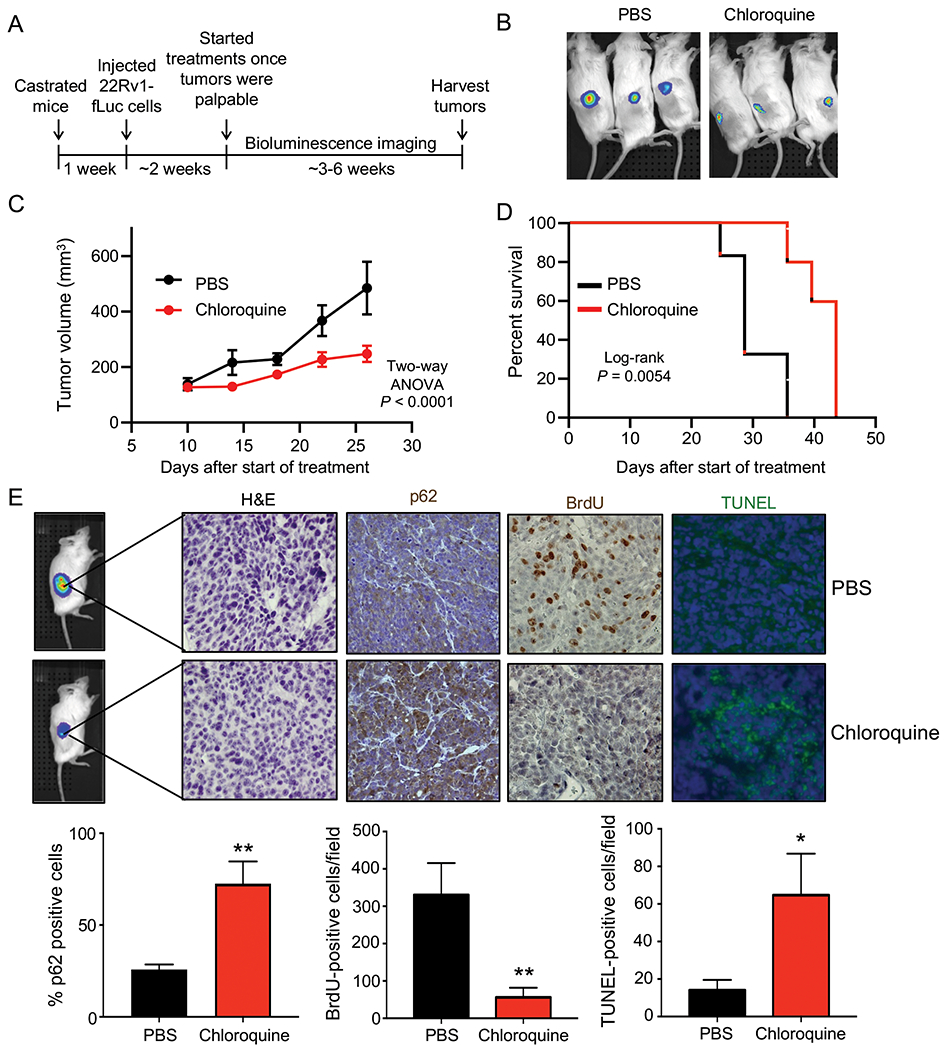
(A) Schematic of xenograft study using CRPC 22Rv1-fLuc cells in castrated NSG mice treated via intraperitoneal injections (IP) once/day, 6 days/week with vehicle (PBS) or 60 mg/kg/day chloroquine (PBS: n=6, chloroquine: n=7). (B) Bioluminescence imaging of six representative mice bearing tumors. PBS = vehicle. (C) Tumor growth curves of 22Rv1-fLuc xenograft mice treated with vehicle (PBS) or chloroquine. P values were calculated using two-way ANOVA. (D) Kaplan-Meier survival curve of 22Rv1-luc xenograft mice following chloroquine treatment. P value was calculated using log-rank test. (E) H&E, p62, BrdU and TUNEL staining in the xenograft tumors (top). Quantification of p62, BrdU and TUNEL staining (bottom). P values were calculated using two-tailed t test. *P < 0.05, **P < 0.01.
CAMKK2 promotes autophagy and autophagic flux in prostate cancer
Although chloroquine derivatives have been tested in cancer clinical trials, the high dosage needed in patients to maintain autophagy inhibition remains a challenge that limits the therapeutic window of this class of compounds. We propose that targeting upstream regulators of autophagy may offer safer, alternative options for inhibiting autophagy. CAMKK2 has previously been shown to be a direct transcriptional target of AR in prostate cancer that promotes the phosphorylation and activation of AMPK (3, 9). Given the critical role of AMPK in autophagy (10, 36, 37, 40), we investigated whether CAMKK2 augmented autophagy in prostate cancer. To do this, we first engineered hormone-sensitive LNCaP cells to inducibly express CAMKK2 in the presence of doxycycline (DOX) (LNCaP-CAMKK2). We then examined via immunoblot the effect of CAMKK2 overexpression on AMPK phosphorylation and the accumulation of phosphatidylethanolamine-conjugated LC3B (LC3BII), a marker of autophagy (Fig. 2A). CAMKK2 overexpression increased p-AMPK and conversion of LC3BI to LC3BII (Fig. 2A). Likewise, CAMKK2 overexpression increased GFP-LC3 puncta formation, indicative of increased autophagosome formation (Fig. 2B). To further confirm the effects on autophagy, transmission electron microscopy (TEM) was used to verify the increased number of autophagic vesicles (autophagosomes and autophagolysosomes) following CAMKK2 expression (Fig. 2C). Given the high expression of CAMKK2 in AR+ CRPC (3, 4, 6, 41), we next knocked out CAMKK2 in C4-2 cells, an LNCaP-derived CRPC model, using CRISPR-Cas9 to assess the effects of CAMKK2 disruption in CRPC. Two CAMKK2 knockout (KO) clones were selected (Supplementary Figs. S2A–B) and compared to control (Cas9 only) cells to examine effects on autophagy (Figs. 2D–F). Both CAMKK2 KO clones exhibited substantially reduced AMPK phosphorylation and LC3B conversion (Fig. 2D) as well as decreased LC3 puncta (Fig. 2E). Compared to control C4-2 Cas9 cells, it was also difficult to find autophagic vesicles in CAMKK2 knockout cells by TEM (Fig. 2F). However, apoptotic bodies were clearly detectable (Fig. 2F). We confirmed the effects of CAMKK2 inhibition on autophagy using an independent model of CRPC, 22Rv1 cells, in which we created a stable derivative that could express shRNA targeting CAMKK2 in the presence of DOX. Similar to CAMKK2 genetic KO in C4-2 cells, the inducible knockdown of CAMKK2 in 22Rv1 cells inhibited autophagy (Figs. 2G–I).
Fig 2. CAMKK2 increases autophagy in prostate cancer cells.
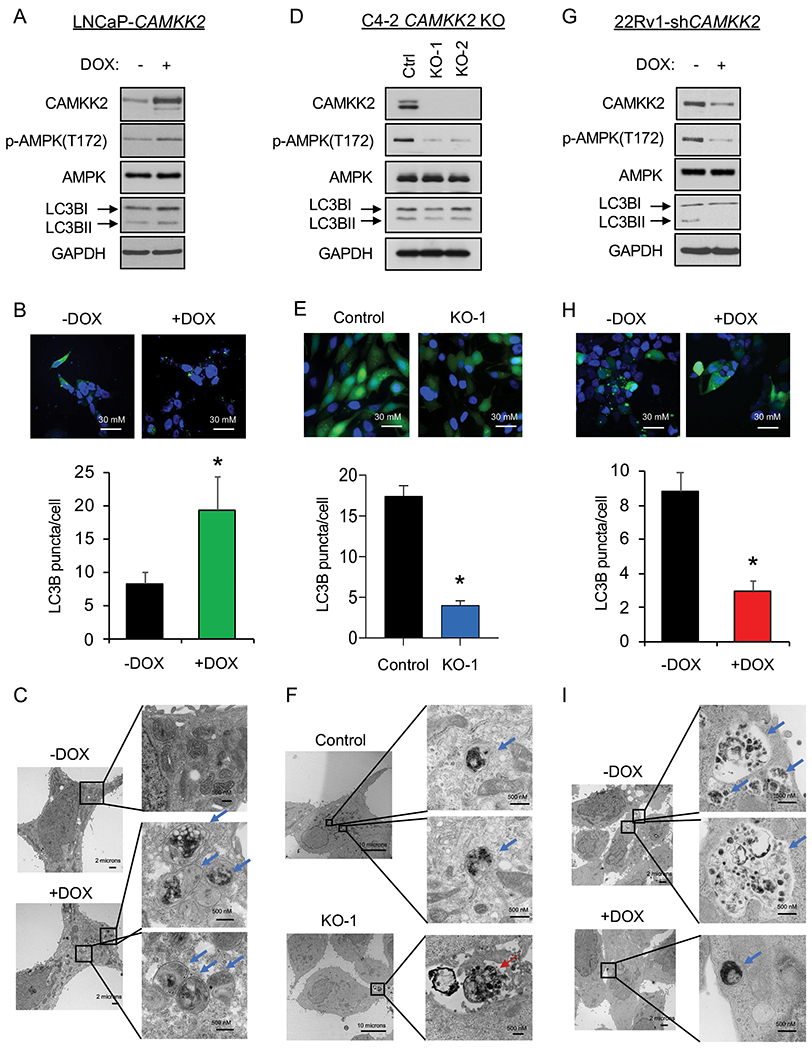
(A) Immunoblot analysis of doxycycline (DOX)-inducible LNCaP stable cells (LNCaP-CAMKK2) that express CAMKK2 upon addition of 50 ng/ml DOX for 48 hours. (B) LNCaP-CAMKK2 cells were transiently transfected with GFP-LC3 (green) and then treated ± 50 ng/ml DOX for 48 hours. Representative images (top). GFP-LC3 puncta (green) were quantified as the average number of GFP-LC3 puncta per cell ± SEM (bottom). The nuclei are stained with DAPI (blue) for reference. P value was calculated using a two-tailed t test. *P < 0.05. (C) LNCaP-CAMKK2 cells were treated ± 50 ng/ml DOX for 48 hours and imaged using transmission electron microscopy (TEM). Two magnifications of ultrastructures are shown. Blue arrows indicate autophagosomes and autolysosomes. (D) Immunoblot analysis of two independent clones of CRISPR-modified C4-2 CAMKK2 knockout (KO) cells compared with their parental C4-2 Cas9 control cells (Ctrl). (E) GFP-LC3 was expressed in C4-2 Cas9 control and CAMKK2 KO cell derivatives. GFP-LC3 puncta (representative images; top) and quantification (bottom) are shown as in B. (F) C4-2 control and C4-2 CAMKK2 KO cells were imaged using TEM as in C. Red arrows indicate apoptotic bodies. (G) Immunoblot analysis of DOX-inducible 22Rv1 stable cells that express shRNA targeting CAMKK2 (22Rv1-shCAMKK2) with 800 ng/ml DOX treatment for 72 hours. (H) 22Rv1-shCAMKK2 cells were transiently transfected with GFP-LC3 and then treated ± 800 ng/ml DOX for 72 hours. GFP-LC3 puncta (representative images; top) and quantification (bottom) are shown as in B. (I) 22Rv1-shCAMKK2 cells were treated ± 800 ng/ml DOX for 72 hours and imaged with TEM as in C.
There are several sequential steps involved in autophagy, including initiation, autophagosome formation, autolysosome fusion and degradation. Hence, CAMKK2-mediated increases in LC3 lipidation and relocalization could result from either increased autophagic entry or decreased autophagic flux (23). We therefore used a tandem mCherry-GFP-LC3B fusion protein to evaluate CAMKK2’s role in autophagic flux. LC3B fusion protein is represented as a yellow signal due to the equal expression of both mCherry and GFP basally (diffuse signal) and during early autophagy/prelysosomal fusion (puncta). However, after lysosomal fusion (late autophagy), the acidic environment of the lysosome quenches the GFP signal but retains mCherry, resulting in the colorimetric shift from yellow to red. Consistent with our previous studies (23, 24), androgens increased overall LC3B puncta and GFP−mCherry+ LC3B puncta (red) (Fig. 3A). We also observed that CAMKK2 overexpression has a similar result as androgen treatment, which significantly elevated total and red puncta (Fig. 3A). This indicates that CAMKK2, an AR target, can promote autophagic flux similar to androgen treatment. To further validate these findings, we used a lysosomal block assay (23, 42). As described above, chloroquine is a lysosomotropic agent that can block the lysosomal turnover of LC3B. Therefore, in the presence of chloroquine, LC3BII would only be increased if there was greater entry into autophagy. We observed androgens or CAMKK2 expression further increased LC3BII levels in the presence of a lysosomal block, while knockdown of CAMKK2 decreased LC3B conversion (Fig. 3B and Supplementary Fig. S3), suggesting that CAMKK2 enhanced autophagic flux by increasing autophagy initiation.
Fig 3. CAMKK2 promotes autophagic flux.
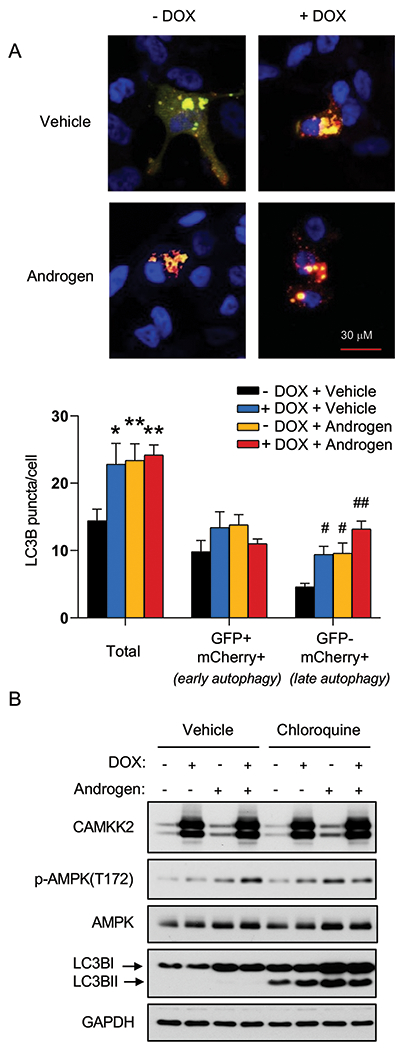
(A) LNCaP-CAMKK2 cells were transfected with an mCherry-GFP-LC3 plasmid and treated ± 10 nM R1881 (androgen) ± 50 ng/ml DOX. Representative fluorescence images of the cellular localization of autophagic puncta (top) and quantification (bottom). P values were calculated using one-way ANOVA with Dunnett’s test. *P < 0.05, **P < 0.01, compared to vehicle group in total. #P < 0.05, ##P < 0.01, compared to vehicle group in GFP-mCherry+. (B) LNCaP-CAMKK2 cells were treated ± 10 nM R1881 (androgen) ± 50 ng/ml DOX ± 20 μM chloroquine (lysosomal block/inhibitor of autophagic flux) for 72 hours. Cell lysates were then subjected to immunoblot analysis.
CAMKK2 is required for CRPC cell growth in vivo
Previous studies using the pharmacological inhibitor STO-609 have suggested the potential role of CAMKK2 in CRPC growth (4). However, STO-609 has multiple kinase targets (43–45). Here, we used a genetic approach to assess the role of CAMKK2 in CRPC tumorigenesis and progression in vivo. Castrated NSG mice were subcutaneously injected with C4-2 Cas9 control and C4-2 CAMKK2 KO cells (Fig. 4A). CAMKK2 ablation had a profound effect on CRPC tumor growth (Fig. 4B). In fact, when the average tumor size of the control group was ~500 mm3, no tumors could even be detected in the KO groups. Accordingly, CAMKK2 KO also dramatically prolonged survival (Fig. 4C). Immunohistochemical (IHC) analysis of tumor tissues determined both a reduction in proliferation and increase in cell death in CAMKK2 KO groups compared to control (Fig. 4D). In these endpoint samples, we were unable to detect consistent increased apoptosis between CAMKK2 KO groups as assessed by cleaved caspase-3 (Supplementary Fig. S4). While this may be due to missing the apoptotic window, at this time we cannot rule out the involvement of other forms of cell death (ex. necrosis). To validate our findings in a second model of CRPC and test what would happen if we decreased CAMKK2 after tumor implantation, we leveraged our DOX-inducible 22Rv1-shCAMKK2 cell model (Fig. 4E and Supplementary Fig. S5A–B). Consistent with the C4-2 CAMKK2 KO xenograft results, knockdown of CAMKK2 in 22Rv1 tumors decreased tumor burden over time and consequently increased overall survival (Figs. 4F–G). Moreover, CAMKK2 knockdown-mediated tumor growth reduction was again correlated to lower proliferation (BrdU) and more cell death (TUNEL) (Fig. 4H). Consistent with a pro-survival role of CAMKK2-mediated autophagy, we also observed inducible CAMKK2 knockdown tumors displayed increased necrosis, but clear regions of perivascular tumor sparing (Fig. 4H, +DOX H&E (high magnification)). Collectively, these data suggest that CAMKK2 is required for maximum CRPC tumorigenesis and progression in vivo, potentially by enabling cells to withstand the harsh, nutrient-deficient tumor microenvironment.
Fig 4. CAMKK2 is required for CRPC tumor growth in vivo.
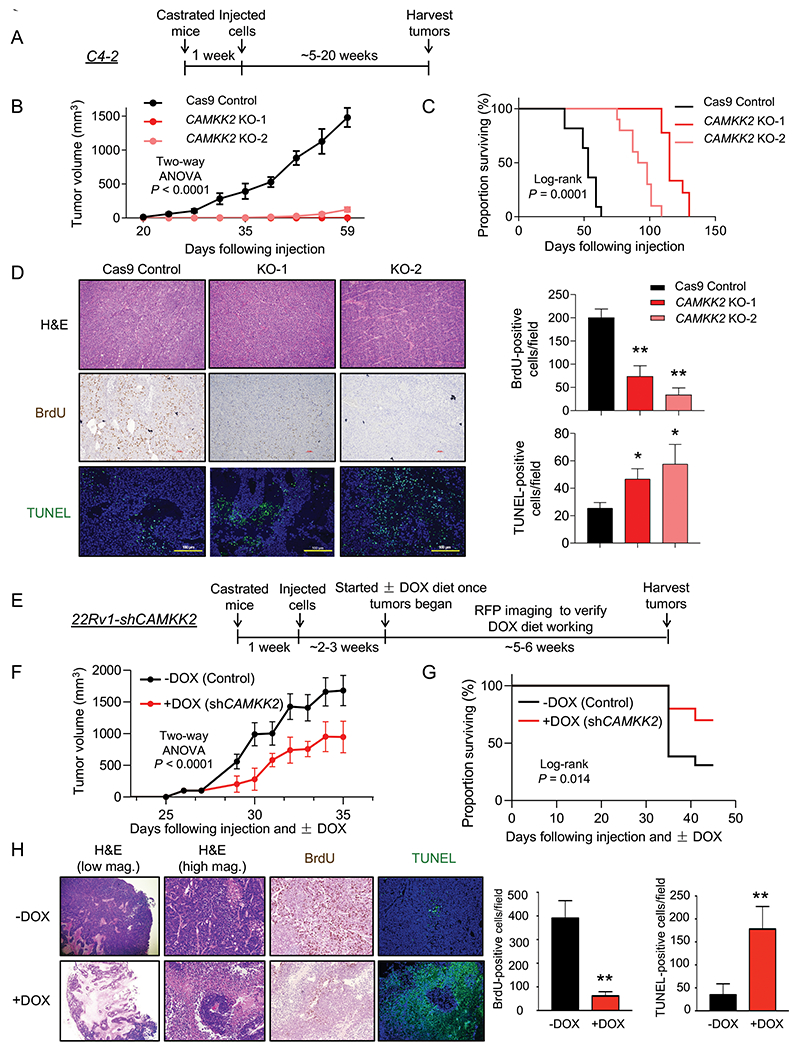
(A) Schematic of xenograft study using CRPC C4-2 Cas9 control and CAMKK2 CRISPR knockout (KO) cell derivatives in castrated NSG mice. (B) Tumor growth curves of C4-2 Cas9 control and C4-2 CAMKK2 KO xenografts in castrated NSG mice (n = 10/group). P values were calculated using two-way ANOVA. (C) Kaplan-Meier survival curve of C4-2 Cas9 control and C4-2 CAMKK2 KO xenograft mice. P values were calculated using the log-rank test. (D) C4-2 xenograft tumor samples were stained with H&E, BrdU and TUNEL. Representative images (left) and quantifications of BrdU and TUNEL staining (right). *P < 0.05, **P < 0.01 by one-way ANOVA with Dunnett’s test. (E) Schematic of xenograft study using DOX-inducible CRPC 22Rv1-shCAMKK2 cells in castrated NSG mice. (F) Tumor growth curves of 22Rv1-shCAMKK2 xenografts in castrated NSG mice fed control or DOX-enriched (625 mg/kg) chow. P value was calculated using two-way ANOVA. (G) Kaplan-Meier survival curve of 22Rv1-shCAMKK2 xenograft mice ± DOX. P value was calculated using the log-rank test. (H) 22Rv1-shCAMKK2 xenograft tumor samples were stained with H&E, BrdU and TUNEL. Representative images (left) and quantifications of BrdU and TUNEL staining (right). Note, evidence of perivascular tumor sparing in DOX-treated tumors (H&E high magnification (mag.)). **P < 0.01 by t test.
AR-CAMKK2-AMPK signaling enhanced autophagy through phosphorylation of ULK1 at serine 555 independent of mTOR-mediated ULK phosphorylation of S757
Since AR-CAMKK2 signaling promotes autophagic flux, we further explored the mechanism by which it initiated autophagy. A key protein involved in autophagy initiation is the serine/threonine protein kinase Unc-51 like autophagy activating kinase 1 (ULK1), which functions as part of a complex to transduce upstream signals to the downstream core autophagy machinery (46). AMPK is a known ULK1 upstream regulator by phosphorylating and activating ULK1 at multiple sites in a context dependent-manner (40, 47, 48). Thus, we speculated that AR-CAMKK2 activated autophagy through ULK1 in prostate cancer. To explore this possibility, we co-treated LNCaP cells with androgens and the CAMKK2 inhibitor STO-609. Androgens increased the levels of CAMKK2, p-AMPK and LC3BII, an effect that could be abrogated by STO-609 (Fig. 5A). Androgens also increased ULK1 phosphorylation at serine 555, an effect that was again reversed by STO-609 (Fig. 5A). This was of interest because serine 555 has been shown to be a critical phosphorylation site necessary for AMPK-mediated autophagy in vitro and in vivo (40, 49–51). To exclude the non-specific effects of STO-609, we next tested p-ULK1(S555) status in cells following genetic or molecular modification of CAMKK2 and AMPK. In LNCaP-CAMKK2 cells, DOX alone increased CAMKK2 expression level, resulting in a similar increase in p-AMPK, p-ULK1 and LC3BII levels compared to androgen treatment alone (Fig. 5B). These increases could be reversed upon knockdown of AMPKα1, the predominant AMPK α catalytic subunit in prostate cancer (3, 9, 52, 53). The requirement for AMPK was confirmed with three independent siRNAs (Fig. 5C). To verify that ULK1 phosphorylation was necessary for CAMKK2-mediated autophagy, we transfected LNCaP-CAMKK2 cells with constructs expressing vector control, WT ULK1 or ULK1 4SA mutant, an AMPK non-phosphorylatable ULK1 (40). Cells treated with DOX (CAMKK2 expression) and expressing WT ULK1 had increased LC3BII levels, indicating an increase of autophagy, while 4SA mutant blocked CAMKK2-mediated autophagy (Fig. 5D). The requirement of CAMKK2 for AMPK-ULK1-mediated autophagy was confirmed in both the C4-2 and 22Rv1 CRPC models (Figs. 5E–F). Notably, phosphorylation of S757, previously shown to be the major mTOR inhibitory target site on ULK1 (54), was unchanged following modulation of CAMKK2-AMPK signaling (Supplementary Fig. S6). Moreover, mTOR signaling remained largely intact in our models suggesting that in prostate cancer, AMPK and mTOR may be able to coexist. In fact, in 22Rv1 cells, knockdown of CAMKK2 even decreased p-S6 levels (Supplementary Fig. S6C), suggesting that under some contexts CAMKK2 may even promote mTOR activity. Whether this is due to the PTEN intact status of these cells (LNCaP and C4-2 are PTEN mutated) is unknown. Together, these findings demonstrated that AR-CAMKK2 triggers AMPK phosphorylation and activation, and in turn phosphorylates ULK1 at serine 555, which ultimately stimulates autophagy independent of mTOR regulation.
Fig 5. AR-CAMKK2-AMPK signaling increases autophagy by phosphorylating ULK1 at serine 555.
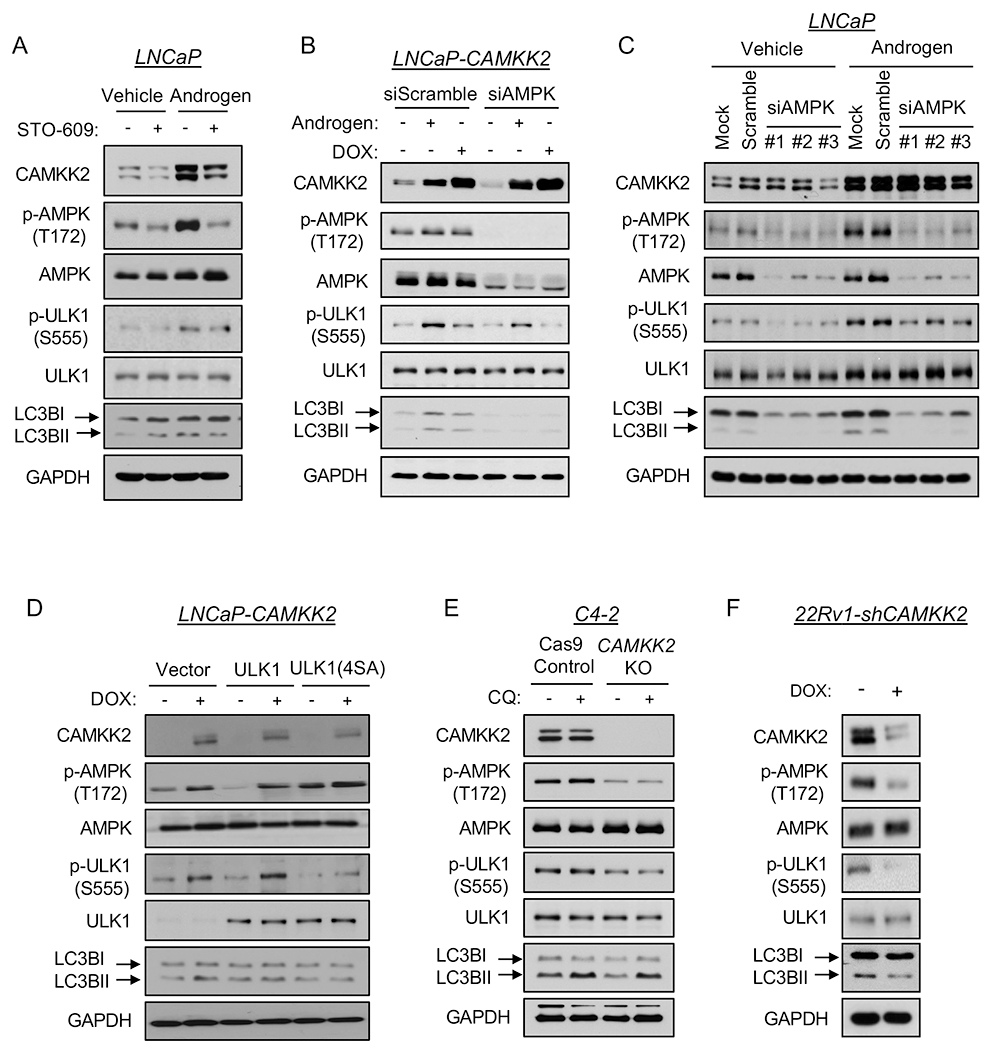
(A) LNCaP cells were treated ± 10 nM R1881 (androgen) ± 30 μM STO-609 for 72 hours. (B) LNCaP-CAMKK2 cells were transfected with siRNAs targeting scramble control or the α1 catalytic subunit of AMPK (siAMPK) and then treated with androgen for 72 hours or DOX (50 ng/ml) for 48 hours. Cell lysates were subjected to immunoblot analysis. (C) Parental LNCaP cells were transfected with siRNAs targeting scramble control or three different regions of the α1 catalytic subunit of AMPK (siAMPK) and then treated with vehicle or androgen for 72 hours. Cell lysates were subjected to immunoblot analysis. (D) LNCaP-CAMKK2 cells were transfected with empty vector, ULK1 or ULK1 (4SA) expression constructs and then treated ± DOX for 48 hours. Cell lysates were subjected to immunoblot analysis. (E) Immunoblot analysis of C4-2 Cas9 control and CAMKK2 KO derivative cells treated with vehicle or chloroquine (20 μM). (F) Immunoblot analysis of 22Rv1-shCAMKK2 cells ± 800 ng/ml DOX treatment for 72 hours.
ULK1 correlates with poor patient prognosis in men with prostate cancer
To examine the clinical association between ULK1 and patient prognosis, we analyzed two well-annotated, publicly available patient databases. The expression level of ULK1 mRNA was inversely correlated with disease-free survival in both TCGA (55) (Fig. 6A) and Taylor et al. 2010 (56) (Fig. 6B) clinical cohorts. Consistent with these clinical correlations, previous histological studies have linked high ULK1 levels to biochemical recurrence and PSA levels (57, 58). Unfortunately, the ULK1 antibody used in these studies is no longer commercially available and validation studies using other commercially available ULK1 or p-ULK1(S555) antibodies revealed these antibodies were not suitable for ICC/IF or IHC (Supplementary Figs. S7–9), precluding the evaluation of ULK1 or p-ULK1 protein levels in patient samples.
Fig 6. High ULK1 tumor expression predicts poor patient prognosis in independent clinical cohorts of men with prostate cancer.
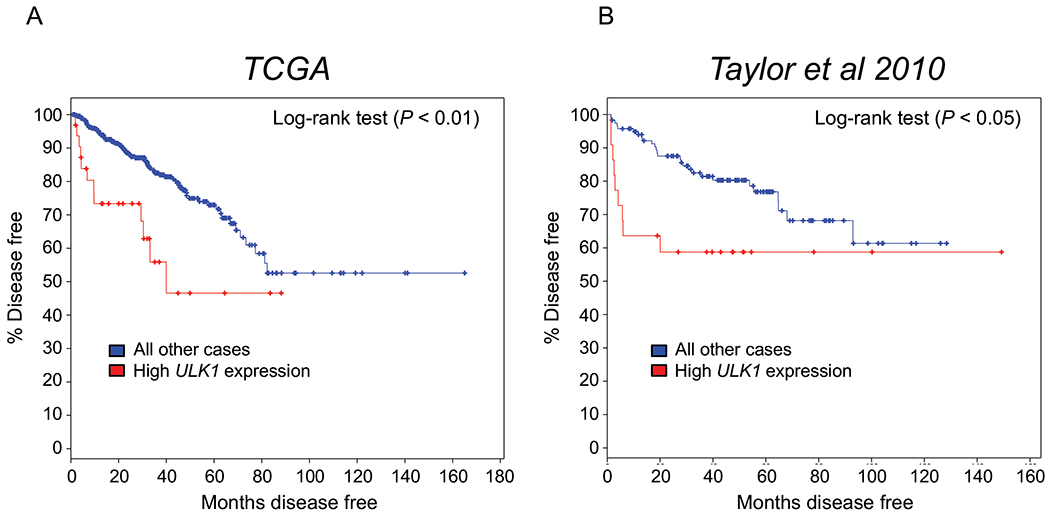
Kaplan-Meier estimates of disease-free survival in The Cancer Genome Atlas (TCGA) and Taylor et al. 2010 clinical cohorts based on ULK1 expression. Data were generated from cBioPortal. The high expression ULK1 group was defined by ULK1 mRNA expression > 2 SD above the mean.
Pharmacological targeting of ULK1 inhibits prostate cancer cell growth
We next wanted to determine if ULK1 was a potential therapeutic target in prostate cancer. To test this, we leveraged SBI-0206965 (6965), a recently described ULK1 inhibitor that has shown anti-cancer effects in lung cancer cells under nutrient deprivation (Fig. 7A) (59). To validate 6965’s antagonistic effects in prostate cancer cells, we first used the known ULK1 substrate VPS34 to determine whether 6965 could block ULK1 activity. 6965 decreased both basal and androgen-induced phosphorylation of VPS34 at serine 249, in alignment with the reduction of LC3BII (Fig. 7B). In 22Rv1 cells, 6965 also resulted in inhibition of p-VPS34 and LC3BII accumulation (Supplementary Fig. S10). Interestingly, we noticed an increase of p-AMPK after 6965 treatment, consistent with the previously described negative feedback loop that exists between ULK1 and AMPK (60). Next, to assess the efficacy of 6965 on prostate cancer cell growth, we treated LNCaP-CAMKK2 cells with androgens, DOX and/or 6965 for 7 days. Although 6965 did not significantly inhibit basal LNCaP cell growth, it blocked androgen- and/or DOX-mediated LNCaP cell growth (Fig. 7C), consistent with our previous findings that siRNA-mediated knockdown of ULK1 blocked androgen-mediated cell growth (24). Interestingly, the LNCaP-derived CRPC derivative C4-2 cells were more sensitive to 6965 treatment, showing a ~70% reduction in growth (Fig. 7D). In 22Rv1 cells, ~50% growth inhibition was observed (Fig. 7E). To evaluate the long-term effects of 6965 on cell proliferation, we performed clonogenic assays. All three cells were very sensitive to prolonged 6965 treatment with almost 100% inhibition in clonogenic potential (Figs. 7F–H). Collectively, these data indicate that ULK1 is a potentially druggable target for the treatment of prostate cancer. Future studies would need to explore the safety of such an approach in vivo.
Fig 7. The ULK1 inhibitor SBI-0206965 represses prostate cancer cell growth.
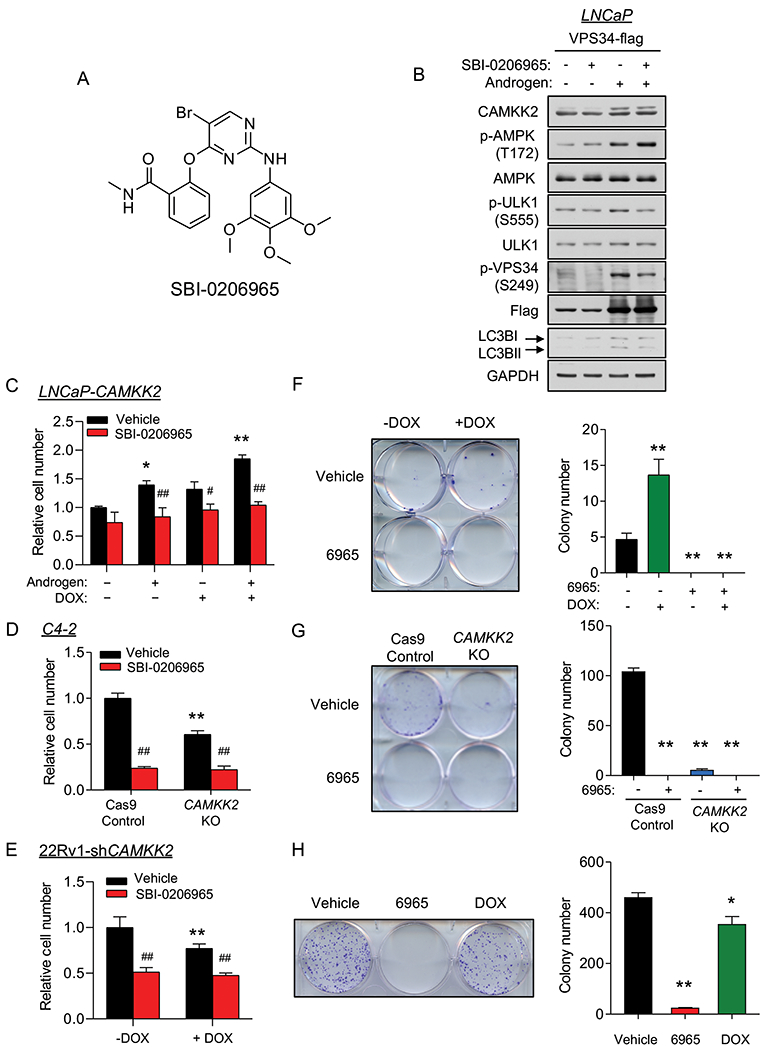
(A) Chemical structure of the ULK1 inhibitor SBI-0206965. (B) LNCaP cells were transfected with VPS34-FLAG following 72 hours 10 nM R1881 (androgen) treatment. Cell lysates were collected 2 hours after vehicle or SBI-0206965 (10 μM) treatment and subjected to immunoblot analysis. (C) Cell growth of LNCaP-CAMKK2 cells following 7 days R1881 (androgen, 10 nM), DOX (50 ng/ml) and/or SBI-0206965 (10 μM) treatment. *P < 0.05, **P < 0.01 compared to no androgen/DOX/SBI-0206965 treatment group. #P < 0.05, ##P < 0.01, compared to corresponding vehicle (SBI-0206965) treatment group. (D) Cell growth of C4-2 Cas9 control and C4-2 CAMKK2 KO derivative cells ± SBI-0206965 (10 μM). **P < 0.01 compared to C4-2 control cells. ##P < 0.01, compared to vehicle treatment group. (E) Cell growth of 22Rv1-shCAMKK2 cells treated for 7 days ± DOX (800 ng/ml) ± SBI-0206965 (10 μM). *P < 0.05, **P < 0.01 compared to no DOX treatment group. ##P < 0.01, compared to corresponding vehicle (SBI-0206965) treatment group. (F) Colony formation assay of LNCaP-CAMKK2 cells following 28-day DOX and/or SBI-0206965 (10 μM) under 100 pM R1881 (androgen) treatment (required for LNCaP colony formation). Representative image (left). Quantification of three independent experiments (right). **P < 0.01, compared to double-vehicle treatment group. (G) Colony formation assay of C4-2 Cas9 control and C4-2 CAMKK2 KO derivative cells ± SBI-0206965 (10 μM) for 21 days. Representative image (left). Quantification of three independent experiments (right). **P < 0.01, compared to C4-2 control vehicle treatment group. (H) Colony formation assay of 22Rv1-shCAMKK2 cells treated for 21 days ± DOX (800 ng/ml) or SBI-0206965 (10 μM). Representative image (left). Quantification of three independent experiments (right). *P < 0.05, **P < 0.01, compared to vehicle treatment group.
Discussion
Although autophagy has context-dependent roles in cancer (16, 61–64), our data support a pro-cancer role for this cellular process in prostate cancer. These findings are consistent with our previous work (23, 24) and the work of others in the field (25, 26, 29, 31, 63, 65–67). As presented in our previous reports, blocking autophagy by molecular or pharmacological approaches resulted in decreased androgen-mediated prostate cancer cell growth (23, 24). Mechanistically, androgens stimulate AR to promote autophagy through multiple mechanisms including the indirect accumulation of intracellular ROS and more directly through the transcription of several core autophagy genes (23, 24). In this study, we revealed a novel mechanism underlying how AR regulates autophagy. Our data demonstrated that an AR-CAMKK2-AMPK signaling cascade can drive autophagy through the phosphorylation of ULK1, an important initiator of autophagy, at serine 555. This phosphorylation activates the ULK1 complex and ultimately initiates autophagy and autophagic flux in an mTOR-ULK1(S757)-independent manner for prostate cancer cell proliferation and survival in vitro and in vivo, promoting CRPC progression (Fig. 8). Our finding not only provides a novel mechanistic insight into AR’s regulation of autophagy, but highlights potential new avenues for therapeutic targeting of autophagy in prostate cancer. A non-AR-mediated regulation of autophagy has been reported as a resistance mechanism to treatment with the anti-tumor compound triptolide in prostate cancer (68). As a result, chloroquine was applied to overcome triptolide resistance, enhancing the anti-tumor effect of triptolide in much the same way chloroquine enhanced the effect of hormone ablation in our own CRPC models (Fig. 1). Despite differences identified in the ULK1 phosphorylation sites, our results agree with the overall concept that CAMKK2-AMPK-induced ULK1 activation and autophagy provides an important survival mechanism for prostate cancer cell growth. Likewise, in a genetic mouse model of Pten- and Tp53-deficient prostate cancer or AR-indifferent prostate cancer cells, ULK1-mediated autophagy (ULK1 phosphorylation was not examined) inhibited apoptosis (27). However, unlike these prior studies, our data suggest that AR signaling can promote CAMKK2-AMPK-mediated ULK1 activation and autophagy independent of any inhibition of mTOR activity. Moreover, our data directly demonstrate key roles for CAMKK2 and ULK1 not only in cell survival, but also in proliferation.
Fig 8. Working model depicting how AR-CAMKK2-AMPK signaling regulates autophagy by ULK1 phosphorylation and activation in prostate cancer.
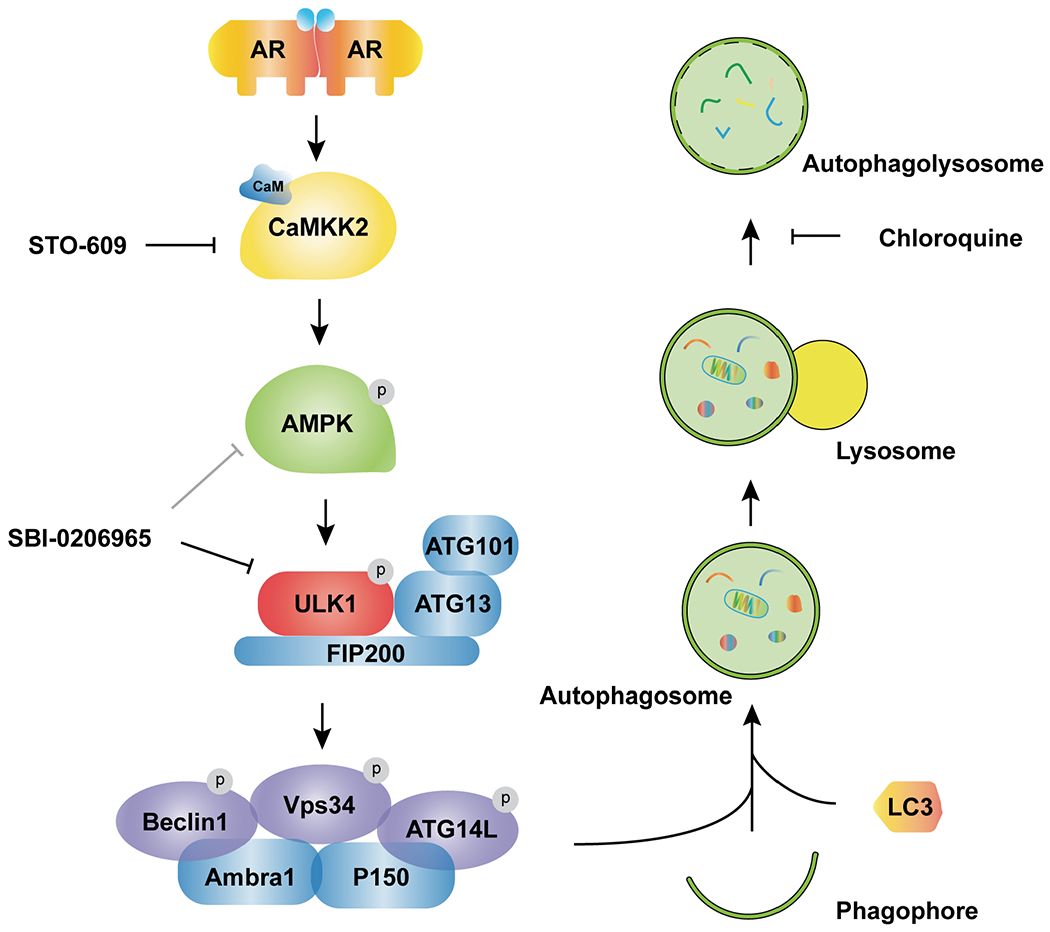
AR increases the expression of CAMKK2 which in turn phosphorylates and activates AMPK at threonine 172. As a result, AMPK phosphorylates ULK1 at serine 555 which activates the ULK1 complex and initiates autophagy in an mTOR-independent manner, supporting prostate cancer growth. This growth and survival mechanism can be blocked at several steps and as such, offers alternative strategies for targeting autophagy in prostate cancer.
Despite agreement that autophagy promotes prostate cancer progression, how this process is regulated by AR is still debated (16, 23, 24, 26, 31, 35, 52, 63, 67, 69). These discrepancies may be attributable to differences in the duration of upstream signals, reliance on indirect or nonselective modulators of autophagy or treatment conditions. As we and others have shown, androgens, in an AR-dependent mechanism, can directly and indirectly increase autophagy through a variety of mechanisms including elevating intracellular ROS levels and transcription of core autophagy genes (23–25, 63). As shown here, there is also a clear, direct AR regulation of AMPK-mediated autophagy through the expression of CAMKK2. The mechanism underlying how antiandrogens can, like androgens, paradoxically also can increase autophagy is less clear. But these different observations may speak to the potential benefit of targeting downstream effector processes like autophagy that can be activated under a variety of conditions to drive disease progression. Our data presented here provides evidence that targeting CAMKK2-AMPK-ULK1 signaling may be an effective, alternative strategy to block protective autophagy in advanced prostate cancer.
Under glucose or amino acid starvation, ULK1 is well characterized to be regulated by AMPK. AMPK binds to the serine/proline-rich domain and can phosphorylate ULK1 at multiple sites (S317, S467, S555, T575, S637 and S777) which subsequently change ULK1 conformation and enhance its kinase activity. In addition, AMPK can indirectly promote ULK1 activity through the inhibition of mTOR and subsequent decrease of mTOR-mediated ULK1 phosphorylation on S757—an inhibitory posttranslational modification (54). These phosphorylation events, in turn, promote the formation of the ULK1 complex (ULK1, ATG13, ATG101, and FIP200) (46, 54). Activated ULK1 can further phosphorylate downstream VPS34 complex members to induce autophagic entry (46). In this study, we first demonstrated the S555 site of ULK1 as a downstream target of AMPK in response to androgen treatment. S555 was increased under androgen treatment but could not be activated when cells were subjected to AMPK siRNA (Fig. 5B&C). When cells were reconstituted with a non-phosphorylatable ULK1 mutant (4SA), they were defective in autophagy following AMPK activation (Fig. 5D). Surprisingly, the AR-CAMKK2-AMPK activation of ULK1-mediated autophagy was independent of any inhibition of mTOR signaling and subsequent ULK1(S757) phosphorylation (Supplementary Fig. S6) (54). Although we cannot exclude contributions from other phosphorylation sites, these findings together suggest the importance of ULK1 S555 phosphorylation by AMPK in AR-mediated autophagy induction and support prior reports that S555 is functionally one of the most important AMPK target sites on ULK1 (40, 50, 51).
Interestingly, we observed a negative feedback loop between AMPK and ULK1 similar to what has been described before in HEK293 cells under starvation (60). While non-phosphorylatable ULK1 mutants impaired autophagy, they significantly increased p-AMPK (T172) (Fig. 5D). Likewise, when cells were treated with the ULK1 inhibitor SBI-0206965, a robust enhancement of p-AMPK was detected (Figs. 7B and Supplementary Fig. S10). It is unclear at this time if this translates to other AMPK-mediated processes being hyperactivated and therefore influencing prostate cancer cell pathobiology.
The efficacy of autophagy inhibition in preclinical models of cancer has paved the way for new clinical trials investigating the efficacy of autophagy inhibition in patients, particularly in combination with traditional anti-cancer treatments. Chloroquine and its derivative hydroxychloroquine, as FDA-approved drugs, have been favored and repurposed in prostate cancer. Previous studies indicated that chloroquine in combination with other therapeutic agents including anti-androgens, chemotherapy and kinase inhibitors can induce greater cytotoxicity than single agent treatment alone (14, 26, 29, 31, 67, 70–72). Likewise, our data indicate anti-cancer effects for chloroquine in combination with androgen deprivation therapy in vivo (Fig. 1). Although a series of clinical trials in prostate cancer have been started to test the efficacy of chloroquine analogs, thus far, limited clinical efficacy has been observed. This is believed to be due in large part to an inability to achieve the drug concentration needed for sustained inhibition of autophagy within tumors prior to the onset of significant side effects (15). Despite the fact that hydroxychloroquine is safer than chloroquine, a micromolar concentrations are required to maintain autophagy inhibition in patients (70). Even so, variable effects on autophagy are still being observed, possibly due to inconsistencies in cell penetration that are in part dependent on the individual’s tumor microenvironment (14). Thus, long-term and high-dosage treatments will inevitably reduce the therapeutic window. Given the potential challenges in the use of lysosomotropic agents, which are not even specific for autophagy, targeting other steps in autophagy, such as ULK1, may provide alternative solutions.
ULK1 expression is highly correlated with patient disease-free time, biochemical recurrence, Gleason score, and metastasis (Fig. 6 & (57, 58)). Currently, three studies have investigated the ULK1 inhibitor SBI-0206965 and showed potent and selective inhibition on ULK1 activity (59, 73, 74). In agreement with other reports, our data showed that SBI-0206965 inhibited ULK1 activity as evidenced by the reduction of p-VPS34 (S249) (Figs. 7B and Supplementary Fig. S10). Moreover, SBI-0206965 exhibited its anti-growth activity in both hormone-sensitive and CRPC cells (Figs. 7C–H). A recent study suggested that SBI-0206965 is a dual inhibitor of AMPK and ULK1 (75). While this would potentially be beneficial for blocking two important nodes of AR-CAMKK2-AMPK-ULK1 signaling, we did not observe a consistent decrease of p-ULK1 after SBI-0206965 treatment, suggesting SBI-0206965 may not function as an AMPK inhibitor in our models. However, we acknowledge that this interpretation may be convoluted due to the above-described feedback mechanism between AMPK and ULK1 (60). In addition, the efficacy and pharmacokinetic profile of SBI-0206965 in vivo are still largely unknown.
Given that systemic blocking or genetic ablation of CAMKK2 appears well-tolerated in mouse models and CAMKK2 has a more restricted expression profile but is elevated in prostate cancer, we propose targeting CAMKK2 may be a viable alternative. Unfortunately, the use of STO-609 as used in this study is likely not a clinically viable option due to its off-target effects on other kinases and pharmacokinetic limitations (43–45). There are, however, ongoing efforts to develop next-generation CAMKK2 inhibitors (44, 76–78).
In summary, our results provide a novel mechanism that links AR signaling and protective autophagy in prostate cancer. Targeting CAMKK2 decreases the AMPK-mediated phosphorylation of ULK1 at serine 555, which in turn stalls the initiation of autophagy and impairs prostate cancer cell growth. These findings not only add a mechanistic layer of complexity to shed light on AR’s regulation of autophagy, but also provides new opportunities for inhibiting autophagy in prostate cancer that we postulate warrant being tested to determine if they can overcome the existing limitations of chloroquine.
Materials and methods
Cell culture, plasmids and reagents
A detailed list of cell lines, plasmids and reagents can be found in the Supplementary Materials and Methods.
Xenografts, histology and immunostaining
All animal experiments were approved by and conducted under the Institutional Animal Care and Use Committee (IACUC) at the University of Texas MD Anderson Cancer Center (MDACC) and the University of Houston according to NIH and institutional guidelines. Tumor volume was calculated by the formula: length x width2/2. Further details are included in the Supplementary Materials and Methods.
Plasmid and small interfering RNA (siRNA) transfections
Transfections were conducted as previously described (8, 23) using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific, Waltham, PA, USA). The siRNAs were purchased from Sigma-Aldrich (St. Louis, MO, USA) and transfected using DharmaFECT 1. Sequences of the shRNA and siRNAs are listed in Supplementary Table 1.
Generation of CRISPR/Cas9 CAMKK2 knockout cells.
Inducible Cas9/CRISPR cells were created using pCW-Cas9 and pLX-sgRNA lentiviral constructs. The gRNAs targeting CAMKK2 were designed by http://crispor.tefor.net/ (79) and synthesized by Sigma-Aldrich (listed in Supplementary Table 1) and Thermo Fisher Scientific. Further details are included in the Supplementary Materials and Methods.
Western blot analysis
Western blot analysis was performed as before (8, 9, 23, 24). A list of antibodies used can be found in the Supplementary Materials and Methods.
Immunofluorescence microscopy
Immunofluorescence microscopy details can be found in Supplementary Materials and Methods.
Transmission electron microscopy (TEM)
TEM details can be found in the Supplementary Materials and Methods.
Proliferation assays
Proliferation assays were carried out as previously described (23).
Clonogenic assays
Clonogenic assay details can be found in the Supplementary Materials and Methods.
Statistical analysis
Statistical analyses were performed using Microsoft Excel 2013 (Redmond, WA, USA) and GraphPad Prism 8 (San Diego, CA, USA). Bioinformatic analyses were generated from cBioPortal (80, 81). One-way or two-way ANOVAs, Student t-tests were used to determine the significance among groups as indicated in the figures or figure legends. Log-rank test was used to determine the significance of Kaplan-Meier curves. Grouped data are presented as mean ± SEM unless otherwise noted. P values are indicated in figures or figure legends.
Supplementary Material
Acknowledgments
We thank Drs. Mondira Kundu, Christopher Counter and Nancy Weigel for plasmids and reagents, Kenneth Dunner, Jr. in the High Resolution Electron Microscopy Core Facility at MDACC for help with TEM, and Dr. Jeff Spencer for help with sgRNA design. This work was supported by grants from the National Institutes of Health (NIH R01CA184208 to D.E.F.), American Cancer Society (RSG-16-084-01-TBE to D.E.F.), an Institutional Research Grant (to D.E.F.) and generous philanthropic contributions to The University of Texas MD Anderson Moon Shots Program (to D.E.F.). This work was also supported by an Antje Wuelfrath Gee and Harry Gee, Jr. Family Legacy Scholarship, Robert Hazelwood Graduate Fellowship for Cancer Research (to C.L.) and MD Anderson Odyssey fellow supported by the CFP foundation (to A.M.B). Electron microscopy was performed by the CCSG-funded MDACC High Resolution Electron Microscopy Facility while histology was performed with the CCSG-funded MDACC Research Histology Core Laboratory, NIH grant P30CA0166772.
Conflict of Interest
D.E.F. has received research funding from GTx, Inc and has a familial relationship with Hummingbird Bioscience, Maia Biotechnology, Alms Therapeutics, Hinova Pharmaceuticals and Barricade Therapeutics. The other authors report no potential conflicts of interest.
REFERENCES
- 1.American Cancer Society. Cancer Facts & Figures 2020. Atlanta: American Cancer Society. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaturvedi AP, Dehm SM. Androgen Receptor Dependence. Adv Exp Med Biol. 2019;1210:333–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frigo DE, Howe MK, Wittmann BM, Brunner AM, Cushman I, Wang Q, et al. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011;71(2):528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30(13):2719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karacosta LG, Foster BA, Azabdaftari G, Feliciano DM, Edelman AM. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J Biol Chem. 2012;287(29):24832–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23(1):35–47. [DOI] [PubMed] [Google Scholar]
- 7.Penfold L, Woods A, Muckett P, Nikitin AY, Kent TR, Zhang S, et al. CAMKK2 Promotes Prostate Cancer Independently of AMPK via Increased Lipogenesis. Cancer Res. 2018;78(24):6747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White MA, Tsouko E, Lin C, Rajapakshe K, Spencer JM, Wilkenfeld SR, et al. GLUT12 promotes prostate cancer cell growth and is regulated by androgens and CaMKK2 signaling. Endocr Relat Cancer. 2018;25(4):453–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene. 2014;33(45):5251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardie DG. AMPK and autophagy get connected. EMBO J. 2011;30(4):634–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Klionsky DJ. Autophagy and disease: unanswered questions. Cell Death Differ. 2020;27(3):858–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amaravadi RK, Kimmelman AC, Debnath J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019;9(9):1167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fabio Gabrriele CM, Sergio Comincini. Prostate cancer cells at a therapeutic gunpoint of the autophagy process. Journal of Cancer Metastasis and Treatment. 2018;4(17). [Google Scholar]
- 17.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4(7):773–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan Q, Wang M, Yu M, Zhang J, Bristow RG, Hill RP, et al. Role of Autophagy as a Survival Mechanism for Hypoxic Cells in Tumors. Neoplasia. 2016;18(6):347–55. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Daskalaki I, Gkikas I, Tavernarakis N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front Cell Dev Biol. 2018;6:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Y, Han JJ, Tennakoon JB, Mehta FF, Merchant FA, Burns AR, et al. Androgens promote prostate cancer cell growth through induction of autophagy. Mol Endocrinol. 2013;27(2):280–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blessing AM, Rajapakshe K, Reddy Bollu L, Shi Y, White MA, Pham AH, et al. Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy. 2017;13(3):506–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, DiPaola RS. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016;30(4):399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen HG, Yang JC, Kung HJ, Shi XB, Tilki D, Lara PN Jr., et al. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene. 2014;33(36):4521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Wang J, Xiong H, Wu F, Lan T, Zhang Y, et al. Co-targeting hexokinase 2-mediated Warburg effect and ULK1-dependent autophagy suppresses tumor growth of PTEN- and TP53-deficiency-driven castration-resistant prostate cancer. EBioMedicine. 2016;7:50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett HL, Stockley J, Fleming JT, Mandal R, O’Prey J, Ryan KM, et al. Does androgen-ablation therapy (AAT) associated autophagy have a pro-survival effect in LNCaP human prostate cancer cells? BJU Int. 2013;111(4):672–82. [DOI] [PubMed] [Google Scholar]
- 29.Kaini RR, Hu CA. Synergistic killing effect of chloroquine and androgen deprivation in LNCaP cells. Biochem Biophys Res Commun. 2012;425(2):150–6. [DOI] [PubMed] [Google Scholar]
- 30.Lamoureux F, Thomas C, Crafter C, Kumano M, Zhang F, Davies BR, et al. Blocked autophagy using lysosomotropic agents sensitizes resistant prostate tumor cells to the novel Akt inhibitor AZD5363. Clin Cancer Res. 2013;19(4):833–44. [DOI] [PubMed] [Google Scholar]
- 31.Kranzbuhler B, Salemi S, Mortezavi A, Sulser T, Eberli D. Combined N-terminal androgen receptor and autophagy inhibition increases the antitumor effect in enzalutamide sensitive and enzalutamide resistant prostate cancer cells. Prostate. 2019;79(2):206–14. [DOI] [PubMed] [Google Scholar]
- 32.Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17(4):654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625(1-3):220–33. [DOI] [PubMed] [Google Scholar]
- 35.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marcelo KL, Means AR, York B. The Ca(2+)/Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol Metab. 2016;27(10):706–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem. 2012;287(38):31658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dadwal UC, Chang ES, Sankar U. Androgen Receptor-CaMKK2 Axis in Prostate Cancer and Bone Microenvironment. Front Endocrinol (Lausanne). 2018;9:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–5. [DOI] [PubMed] [Google Scholar]
- 43.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408(3):297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Byrne SN, Scott JW, Pilotte JR, Santiago ADS, Langendorf CG, Oakhill JS, et al. In Depth Analysis of Kinase Cross Screening Data to Identify CAMKK2 Inhibitory Scaffolds. Molecules. 2020;25(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kukimoto-Niino M, Yoshikawa S, Takagi T, Ohsawa N, Tomabechi Y, Terada T, et al. Crystal structure of the Ca(2)(+)/calmodulin-dependent protein kinase kinase in complex with the inhibitor STO-609. J Biol Chem. 2011;286(25):22570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;61(6):585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J, Guan KL. Regulation of the autophagy initiating kinase ULK1 by nutrients: roles of mTORC1 and AMPK. Cell Cycle. 2011;10(9):1337–8. [DOI] [PubMed] [Google Scholar]
- 48.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5(11):e15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J. 2011;440(2):283–91. [DOI] [PubMed] [Google Scholar]
- 50.Tian W, Li W, Chen Y, Yan Z, Huang X, Zhuang H, et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 2015;589(15):1847–54. [DOI] [PubMed] [Google Scholar]
- 51.Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8(1):548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khan AS, Frigo DE. A spatiotemporal hypothesis for the regulation, role, and targeting of AMPK in prostate cancer. Nat Rev Urol. 2017;14(3):164–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berglund L, Bjorling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7(10):2019–27. [DOI] [PubMed] [Google Scholar]
- 54.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7(6):643–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell. 2018;173(2):291–304 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang HY, Ma YD, Zhang Y, Cui J, Wang ZM. Elevated levels of autophagy-related marker ULK1 and mitochondrion-associated autophagy inhibitor LRPPRC are associated with biochemical progression and overall survival after androgen deprivation therapy in patients with metastatic prostate cancer. J Clin Pathol. 2017;70(5):383–9. [DOI] [PubMed] [Google Scholar]
- 58.Liu B, Miyake H, Nishikawa M, Tei H, Fujisawa M. Expression Profile of Autophagy-related Markers in Localized Prostate Cancer: Correlation With Biochemical Recurrence After Radical Prostatectomy. Urology. 2015;85(6):1424–30. [DOI] [PubMed] [Google Scholar]
- 59.Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59(2):285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7(7):696–706. [DOI] [PubMed] [Google Scholar]
- 61.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15(17):5308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mathew R, White E. Autophagy, stress, and cancer metabolism: what doesn’t kill you makes you stronger. Cold Spring Harb Symp Quant Biol. 2011;76:389–96. [DOI] [PubMed] [Google Scholar]
- 63.Chhipa RR, Wu Y, Ip C. AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cell Signal. 2011;23(9):1466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.White E Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Niture S, Ramalinga M, Kedir H, Patacsil D, Niture SS, Li J, et al. TNFAIP8 promotes prostate cancer cell survival by inducing autophagy. Oncotarget. 2018;9(42):26884–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sha J, Han Q, Chi C, Zhu Y, Pan J, Dong B, et al. Upregulated KDM4B promotes prostate cancer cell proliferation by activating autophagy. J Cell Physiol. 2020;235(3):2129–38. [DOI] [PubMed] [Google Scholar]
- 67.Mortezavi A, Salemi S, Kranzbuhler B, Gross O, Sulser T, Simon HU, et al. Inhibition of autophagy significantly increases the antitumor effect of Abiraterone in prostate cancer. World J Urol. 2019;37(2):351–8. [DOI] [PubMed] [Google Scholar]
- 68.Zhao F, Huang W, Zhang Z, Mao L, Han Y, Yan J, et al. Triptolide induces protective autophagy through activation of the CaMKKbeta-AMPK signaling pathway in prostate cancer cells. Oncotarget. 2016;7(5):5366–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Munkley J, Rajan P, Lafferty NP, Dalgliesh C, Jackson RM, Robson CN, et al. A novel androgen-regulated isoform of the TSC2 tumour suppressor gene increases cell proliferation. Oncotarget. 2014;5(1):131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Solitro AR, MacKeigan JP. Leaving the lysosome behind: novel developments in autophagy inhibition. Future Med Chem. 2016;8(1):73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verbaanderd C, Maes H, Schaaf MB, Sukhatme VP, Pantziarka P, Sukhatme V, et al. Repurposing Drugs in Oncology (ReDO)-chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience. 2017;11:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Y, Luo P, Leng P. Effect of Autophagy Inhibitor Hydroxychloroquine on Chemosensitivity of Castration-resistant Prostate Cancer. Sichuan Da Xue Xue Bao Yi Xue Ban. 2019;50(3):323–7. [PubMed] [Google Scholar]
- 73.Martin KR, Celano SL, Solitro AR, Gunaydin H, Scott M, O’Hagan RC, et al. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience. 2018;8:74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petherick KJ, Conway OJ, Mpamhanga C, Osborne SA, Kamal A, Saxty B, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290(18):11376–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dite TA, Langendorf CG, Hoque A, Galic S, Rebello RJ, Ovens AJ, et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J Biol Chem. 2018;293(23):8874–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Price DJ, Drewry DH, Schaller LT, Thompson BD, Reid PR, Maloney PR, et al. An orally available, brain-penetrant CAMKK2 inhibitor reduces food intake in rodent model. Bioorg Med Chem Lett. 2018;28(10):1958–63. [DOI] [PubMed] [Google Scholar]
- 77.Asquith CRM, Godoi PH, Counago RM, Laitinen T, Scott JW, Langendorf CG, et al. 1,2,6-Thiadiazinones as Novel Narrow Spectrum Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) Inhibitors. Molecules. 2018;23(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Profeta GS, Dos Reis CV, Santiago ADS, Godoi PHC, Fala AM, Wells CI, et al. Binding and structural analyses of potent inhibitors of the human Ca(2+)/calmodulin dependent protein kinase kinase 2 (CAMKK2) identified from a collection of commercially-available kinase inhibitors. Sci Rep. 2019;9(1):16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17(1):148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.