

Summary.
The major effect of allosteric HIV integrase (IN) inhibitors (ALLINIs) is observed during virion maturation, where ALLINI treatment interrupts IN-RNA interactions via drug-induced IN aggregation, leading to the formation of aberrant virions. To understand the structural changes that accompany drug-induced aggregation, we determined the soft matter properties of ALLINI-induced IN aggregates. Using small-angle neutron scattering (SANS), scanning electron microscopy (SEM), and rheology, we have discovered that the higher-order aggregates induced by ALLINIs have the characteristics of weak three-dimensional gels with a fractal-like character. Their formation is inhibited by the host factor LEDGF/p75, as well as ex vivo resistance substitutions. Mutagenesis and biophysical analyses reveal that homomeric carboxy-terminal domain (CTD) interactions are required to achieve the branched polymer nature of the ALLINI-induced aggregates. These studies provide key insight into the mechanisms of ALLINI action and resistance in the context of the crowded virion environment where ALLINIs exert their effect.
Keywords: HIV, ALLINI, retroviral integration, polymer network, oligomerization, rheology, small-angle neutron scattering, small-angle X-ray scattering, host factors, analytical ultracentrifugation
Graphical Abstract
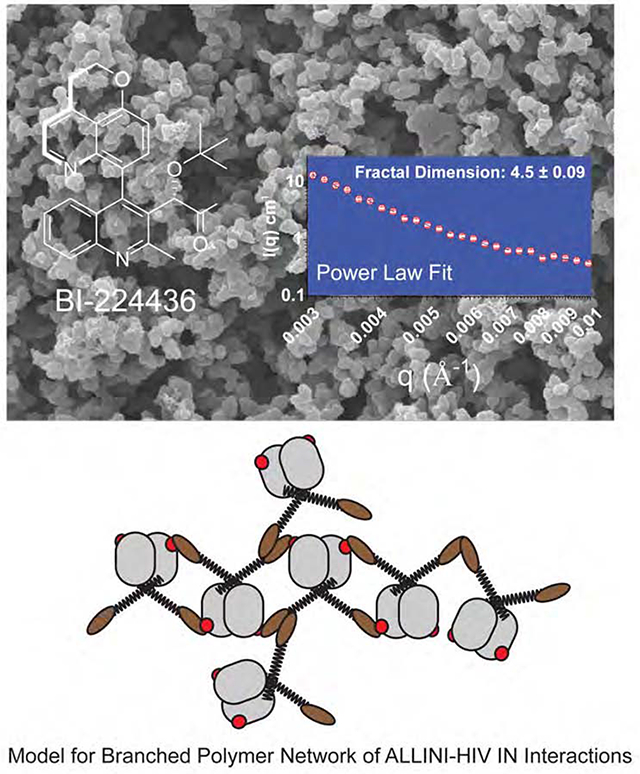
eTOC.
Allosteric HIV integrase (IN) inhibitors (ALLINIs) are potent antivirals that induce the inappropriate aggregation of IN. We report on the soft matter properties of these aggregates, discovering a branched polymer with fractal-like character and that homomeric carboxy-terminal domain interactions are required to form these 3-D networks.
Introduction.
The global burden of the Human Immunodeficiency Virus/Acquired Immune Deficiency Syndrome (HIV/AIDS) pandemic and the sustained emergence of drug-resistance continues to drive the demand for novel antivirals (Arts and Hazuda, 2012; Cihlar and Fordyce, 2016). Integrase (IN) is one of three virally encoded enzymes, all of which are established pharmaceutical targets. Compounds such as the second-generation integrase inhibitors Dolutegravir and Bictegravir are part of the drug class known as strand transfer inhibitors (INSTIs). These drugs target the IN active site within nucleoprotein assemblies called intasomes, blocking the initial covalent attachment of viral cDNA to a host chromosome. Our understanding of the structure and mechanism of retroviral integration has matured with crystallographic and electron cryo-microscopy (cryo-EM) studies of intasomes from a variety of retrovirus, including HIV (Ballandras-Colas et al., 2017; Hare et al., 2011; Maertens et al., 2010; Maskell et al., 2015; Passos et al., 2017; Wilson et al., 2019; Yin et al., 2016), informing potential mechanisms of resistance in the clinical setting. A second novel class of IN inhibitors have been identified that targets a distal allosteric site on the protein and acts late in viral replication (Christ et al., 2012; Christ et al., 2010; Engelman et al., 2013; Feng et al., 2013; Fontana et al., 2015; Gupta et al., 2014; Jurado et al., 2013; Le Rouzic et al., 2013; Molteni et al., 2001; Tsiang et al., 2012); these are known as the allosteric inhibitors of IN (ALLINIs, also referred to as LEDGINs, NCINIs, INLAIs, or multimodal inhibitors). In contrast to the INSTIs, the mechanism of action of the ALLINIs is less well-understood, and these compounds have yet to achieve approval for use in humans. However, continued investigation and development remains strongly motivated by their potency against virus in cell culture.
IN is comprised of three distinct structural domains (Figure 1A): an N-terminal domain (NTD, residues 1-50) that binds Zn2+, a catalytic core domain (CCD, residues 50-212) with conserved RNase H superfamily fold and D,D-35-E active site motif, and a C-terminal domain (CTD, 212-288) with an SH3-like fold. A characteristic feature of recombinant HIV IN in solution is its self-association, with reports of the enzyme sampling a variety of oligomeric states ranging from monomers to tetramers to higher-order species (Deprez et al., 2000; Deprez et al., 2001; Gupta et al., 2014; Gupta et al., 2010; Gupta et al., 2016; Jenkins et al., 1996). Like the full-length enzyme, the isolated domains of IN are oligomeric and have been observed to participate in a variety of homomeric and heteromeric intra- and intermolecular interactions (Cai et al., 1998; Cai et al., 1997; Chen et al., 2000; Dyda et al., 1994; Goldgur et al., 1998; Hare et al., 2009a; Hare et al., 2009b; Lodi et al., 1995; Wang et al., 2001).
Figure 1. HIV-1 Integrase.

A. Domain Map of HIV-1 Integrase. B. Chemical structures of small molecules used in these studies. C. Open polymer configuration of ALLINI-bound HIV integrase as observed in the IN-GSK1264 crystal structure. D. ALLINI binding site and CCD-CTD interface.
The discovery of the functional interaction between the cellular co-factor LEDGF/p75 (product of the PSIP1 gene) and IN (Craigie and Bushman, 2012; Engelman and Cherepanov, 2012) inspired the subsequent development of the ALLINI class of small molecules (Figure 1B). The IN-binding domain of LEDGF/p75 (IBD) binds tightly to the dimer interface of the CCD, facilitating HIV integration at active transcription units within the host genome (Ciuffi et al., 2005; Schroder et al., 2002; Shun et al., 2007). Over the past decade, a number of molecules have been developed that specifically target this protein-protein interaction and display potent antiviral activity (Balakrishnan et al., 2013; Christ et al., 2012; Christ et al., 2010; Gupta et al., 2014; Jurado et al., 2013; Le Rouzic et al., 2013; Sharma et al., 2014; Tsiang et al., 2012). A distinctive and surprising feature of this class of molecules is their mode of action. In contrast to INSTIs, ALLINIs only modestly affect early replication but very potently disrupt late replication events, including virion assembly and maturation (Balakrishnan et al., 2013; Fontana et al., 2015; Gupta et al., 2014; Jurado et al., 2013; Le Rouzic et al., 2013). The most striking effect is the induction of aberrant IN polymerization, which correlates with the inhibition of IN-RNA interactions (Kessl et al., 2016; Madison et al., 2017) and the formation of defective virions with reduced infectivity (Desimmie et al., 2013; Fontana et al., 2015; Gupta et al., 2014; Gupta et al., 2016; Jurado et al., 2013). ALLINI-induced IN aggregation is readily reproduced in vitro with purified components (Gupta et al., 2014; Gupta et al., 2016).
We previously reported the first crystal structure of full-length HIV-1 IN in complex with the quinoline-derived ALLINI GSK1264, explaining prior biochemical observations and providing the first molecular insights into the basis for ALLINI-induced polymerization of IN (Gupta et al., 2016). In this structure, the ALLINI is entirely buried between the CCD of one IN dimer and the CTD of an adjacent IN dimer, in an open polymer configuration within the crystal lattice (Figure 1C). Introduction of ALLINI resistance mutations into the CCD-CTD interface disrupts IN oligomerization and ALLINI-dependent aggregation of IN. However, important questions remained unanswered. While the crystal structure reveals the initiating events of the drug-induced aggregation phenomenon, it is not clear how the crystallographic linear polymer of IN relates to the ALLINI-induced polymers and insoluble aggregates that are formed in solution and in HIV virions.
To develop a more complete understanding ALLINI action, we have probed the properties of ALLINI-induced protein aggregates with a battery of methods to determine their soft matter properties. Using the first-generation ALLINI molecules BI-D and BI-224436 (Fader et al., 2016; Fenwick et al., 2014), we have applied scanning electron microscopy, rheology, and small-angle neutron scattering (SANS) to determine that the ALLINI-induced aggregates have the properties of weak three-dimensional gels with a branched, fractal-like character. The formation of these gels at high concentrations is readily inhibited by ex vivo resistance mutations and host factors that interrupt binding between the CCD and CTD of IN. We have also found using mutagenesis and solution biophysics that CTD-CTD interactions between IN dimers are required to form branched polymers in ALLINI-induced aggregates.
Results.
Kinetics of ALLINI-induced aggregation by BI-224436 and BI-D.
The ALLINIs efficiently promote aggregation of recombinant HIV IN, resulting in the dose-dependent formation of insoluble aggregates with an opaque appearance at high micromolar concentrations (Figure 2A). To quantify the kinetics and dose-response of this drug-induced aggregation, light scattering at 405 nm can be monitored as a function of time (Figure 2B) (Gupta et al., 2014; Gupta et al., 2016). For the characterization of ALLINI-induced effects on purified HIV-IN, we employed preparations of INY15A,F185H. Substitutions at F185 are known to improve solubility (Jenkins et al., 1996) and viruses containing the F185H substitution are replication-competent (Engelman et al., 1997). The Y15A substitution selects for a single conformation of the isolated NTD (Nomura et al., 2006) that is correlated with diminished oligomerization of full-length IN (Gupta et al., 2016; Hare et al., 2009a). Recombinant IN is typically purified in buffer containing 1 M NaCl and the detergent CHAPS to improve solubility. In the absence of drug, no protein aggregation was observed (Figure 2A, left). In the presence of ALLINIs, the rate and extent of aggregation observed was strongly dependent on protein concentration and ionic strength (data not shown). A concentration of 15 μM monomer and ambient temperature was chosen for assays to optimize signal-to-noise and the ability to observe initial kinetics without the confounding effects of heat-induced aggregation expected at higher concentrations. Aggregation of INY15A,F185H by BI-D and BI-224436 is time-dependent (Figure 2B) and maximal aggregation is dependent on drug concentration (Figure 2C) and can be described by a modified Hill equation (Bocanegra et al., 2013; del Alamo et al., 2005; Rincon et al., 2011). Over a thirty-minute time course and in 1M NaCl, a ~50% greater extent of aggregation is observed in the presence of BI-224436 (t1/2=162.9 ± 2.5) versus BI-D (t1/2=579.0 ± 5.2). In both cases, aggregation reached a plateau within 30 minutes. In a dose-response experiment (Figure 2C), an EC50 for BI-224436-induced aggregation of 4.1 ± 0.5 μM was determined with a Hill coefficient of 1.3; maximal aggregation was achieved at concentrations of drug near the total protein concentration of 15 μM.
Figure 2. Properties of ALLINI-Induced Aggregation.

A. Concentration-dependent aggregation of HIV Integrase with BI-224436. In the absence of drug, protein at 221 μM concentration appears transparent. With the increasing addition of BI-224436, the evolution of an opaque aggregate is readily apparent. B. Time-course kinetics of drug-induced aggregation for BI-224436 (open black circles) and BI-D (grey circles). Protein aggregation without drug is shown with closed black circles. C. Dose response profile for BI-224436 after a 1-hour incubation at room temperature. D. Scanning Electron Microscopy (SEM) analyses of BI-224436 and BI-D-induced IN aggregates. E. Dynamic Light Scattering (DLS) analyses of drug-induced aggregates after an overnight incubation in 1 mM drug.
SEM analysis of drug induced aggregates.
To interrogate the physical nature of ALLINI-IN aggregates, we performed transmission and scanning electron microscopy (TEM and SEM). Based on the PDB 5HOT ALLINI-IN crystal structure, where linear open polymers were observed, it was predicted that aggregates in solution would be composed of fibrils. However, no fibrils were observed by TEM analysis (data not shown). At lower magnifications, SEM revealed a homogenous three-dimensional network with fractal-like appearance for both BI-D and BI-224436 (Figure 2D). Dynamic light scattering (DLS) indicates that the BI-D and BI-224436 induced aggregates have a large spatial extent, with hydrodynamic radii approaching ~1 micron (Figure 2E).
Rheological properties of ALLINI-induced aggregates.
We next examined the rheological properties of the drug induced IN aggregations to determine if the three-dimensional network seen by SEM could be verified by an effect on the viscoelasticity of ALLINI-containing suspensions. In the absence of BI-224436 the viscosity of the HIV-IN solution was indistinguishable from that of water. After addition of BI-224436 both the viscosity and the elastic resistance of the suspension increase in a manner consistent with the formation of a branched network stabilized by non-covalent bonds. Figure 3A shows that as the ALLINI-IN sample is increasingly deformed at a constant strain rate, the resulting resistance, quantified by the shear stress, first rises linearly with strain up to approximately 35% and then abruptly drops to a steady level at larger strains, suggesting that the sample responds initially like an elastic solid, but then the network ruptures at large deformation. The shear modulus, a measure of elastic stiffness, calculated from the initial slope of the stress-strain plot is 24 Pa, is characteristic of a weak solid (Chen et al., 2010). When apparent viscosity is calculated from the torque resulting from steady deformation at different shear rates, the viscosity is a power law function of the shear rate and shows no tendency to reach a steady magnitude at either the high or low range of the accessible shear rates (Fig 3B). These characteristics are also consistent with a dynamic network held by labile inter-particle interactions that are disrupted to different extents at different shear rates (Buxbaum et al., 1987).
Figure 3. Viscoelastic Properties of Drug-Induced Aggregates.
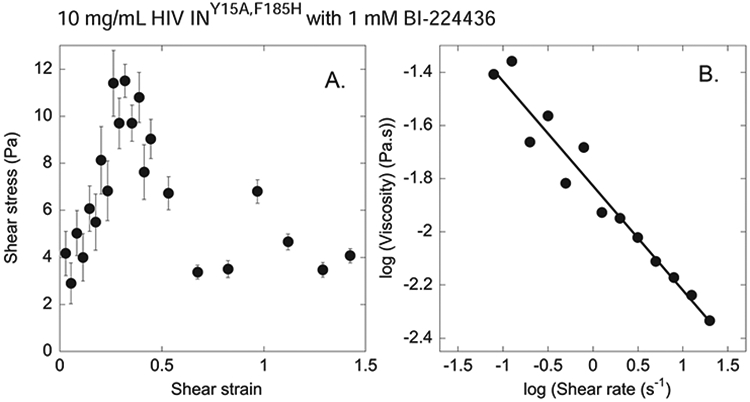
A. Shear stress at increasing levels of shear strain for a sample of 10 mg/mL HIV-1 INF185H•1 mM BI-224436 deformed at a constant shear rate of 0.15 s−1 showing the initial elastic response followed by an abrupt fracture of the sample. B. Viscosity vs shear rate at steady shear deformation for very large strains at which intermolecular bonds are continuously broken and reforming, and elongated structures orient in the direction of flow to an extent that depends on the rate of deformation.
Probing the physical nature of ALLINI-induced aggregates by SANS.
While techniques like the optical turbidity, sedimentation assays, and dynamic light scattering provide reliable methods to detect the formation of aggregates, they provide little insight into the nature of the structure of these aggregates at different length scales. To probe the physical nature of these materials more rigorously, we applied small-angle neutron scattering (SANS). Experimental neutrons are ideal for the study of higher order soft matter and biomaterials because, unlike X-rays, neutrons do not induce aggregation and damage in proteins, and they penetrate deeply into thick materials without attenuation of signal. Small-angle scattering is additionally well-suited to probe the fractal structure of a polymer where the length scales of interest fall below ~1000 Å (Teixeira, 1988). We examined IN concentrations in the range of 20-200 μM for each of three model ALLINIs: BI-D, BI-224436, and CX04328 (Christ et al., 2010) across a range of time courses from fifteen minutes to 16 hours at 20°C, with near identical results obtained in each case (Figure 4 and STable 1). For each ALLINI/IN mixture, the scattering observed was isotropic, indicating a homogenous sample ordered in three dimensions (Figure 4A).
Figure 4. SANS Analysis of ALLINI-induced aggregates of HIV IN.

A. Two-dimensional Neutron Scattering of BI-224436-induced aggregates of HIV-1 INF185H. B. Porod Models for Polymer Networks with different shapes. The slope of the Porod region can be directly related to different surface and mass fractal dimensions. For mass fractals, a lower figure correlates with Gaussian chains where larger figures indicate collapsed polymer coils; values of 3-4 indicated branched systems (gels). For surface fractals, values between 3-4 indicate rough interfaces. C. Representative small-angle neutron scattering (SANS) of BI-224436 induced drug aggregate of HIV INF185H. Shown in grey is the experimental data where 0.003 A−1 < q < 0.84 Å−1. Shown in inset is a Power law fit to data obtained from 100 μM INF185H-BI-224436, in the range where 0.003 < q < 0.01 Å−1. Shown in the main plot is the same data, fit with a Fractal model in the range where 0.003 < q < 0.05 Å−1. A summary of the parameters derived from model fitting of SANS data can be found in STable 1. D. Schematic representation of the mesoscopic structure of the network formed by the ALLINI-mediated branched polymers of HIV IN, where ζ is the correlation length obtained from SANS data at 20°C. The small spheres are a simplified view of the basic units of the network backbone (IN dimers). The scattering centers (grey circles) are formed by regions of clustered protein that give rise to the scattering curves presented. The properties derived from SANS of INY15A,F185H in the absence of drug are provided in STable 2. E. Dose-response curve for the drug-induced aggregation of 80 μM HIV INY15A,F185H. Each data point represents one recorded SANS profile. Using the zero-angle intensity from each experiment, a dose-response curve could be obtained, revealing the concentration for half-maximal aggregation (EC50). The data were fit with a standard Hill equation, yielding an apparent EC50 of 22.1 μM ± 2.33 and a cooperativity factor of 5.6 ± 2.35. Error bars shown on the graph represent the error associated with the determination of I(0).
Consistent with the ~1 μm_dimensions inferred from DLS, a Guinier region in the data was not experimentally accessible, so a precise determination of the radius of gyration (Rg) could not be performed. In all cases, the scattering data can be described as comprised of two components: a low to mid-q signal down to ~0.05 Å−1, and a high-q signal comprising the rest of the scattering profile (qmax=0.84 Å−1). Data in the low to mid-q regime were readily fit to empirical relationships by two different methods. In the first method, we used a basic power law function: the simplest analysis of the decay of intensity in the Porod region (0.003 Å−1 < q < 0.01 Å−1) indicates a fractal structure, where ~3 < Df < ~4 (Bale and Schmidt, 1984), and Df is the fractal dimension (Fig. 4B-C & STable 1). These values are characteristic of globular protein hydrogels, which typically have rough, corrugated surfaces. In contrast, a fibril or linear polymer would be expected to have a fractal dimension of 1 to 2 (Bale and Schmidt, 1984).
In the second fitting method, we used a model that approximates the scattering from fractal-like aggregates of spherical building blocks (Teixeira, 1988). Using this method, the correlation length (ζ) can be derived, which for gels can be described as the size of the scattering centers (Fig 4C-D). The fit to the data yielded ζ-values ranging from 705 to 2364 Å for 0.003 < q < 0.05. In this analysis, the basic “block” unit was assigned as a IN dimer, using Dmax approximated by SANS on unaggregated IN as a fixed parameter. The parameters derived from this fitting for ALLINI-induced aggregates of IN are shown in STable 1 and a physical interpretation of the results is shown in Figure 4D. These data are consistent with a mesoscopic structure of the network formed by the ALLINI-mediated branched polymers of HIV IN.
Zero-angle (I(0)) scattering from SANS data is directly proportional to the size and concentration of the material being scattered. Using I(0) as a readout of total aggregation, we next assessed the dose-dependence of aggregation for BI-224436. Scattering data were measured on samples of 80 μM INY15A/F185H with increasing amounts of BI-224436 or BI-D after 30 minutes of incubation at 20°C. A plot of zero-angle scattering as a function of drug concentration for BI-224436 is shown in Fig 4E. The data are readily fit to a Hill equation, resulting in an EC50 of 22±2.3 μM and an exponent of 5.6±2.4. Since two ALLINIs can bind to an IN dimer, saturation of IN binding would occur at 80 μM BI-224436. However, maximum I(0) signal is achieved at ~40 μM drug, indicating that sub-stoichiometric quantities of drug are enough to completely aggregate IN. This implies that not every binding site needs to be occupied to elicit maximal aggregation.
Effects of ex vivo resistance mutations and host factors on drug-induced aggregation.
We next sought to relate the structure of IN bound to ALLINIs to the observable aggregation properties in vitro and further to effects on viral replication. We thus tested whether amino acid substitutions that make HIV resistant to ALLINIs in tests of viral replication in culture also affected the types of aggregates induced by IN. W131C and N222K are replication-competent mutations that arise during culture in the presence of isoquinoline-derived ALLINIs including GSK1264 (Gupta et al., 2016; Tsiang et al., 2012). We have previously shown that these substitutions greatly diminish oligomerization and drug-response at lower concentrations of protein (Gupta et al., 2016). In the CTD-CCD interface observed in the 5HOT crystal structure, both residues are located at the interface of the CCD α3 helix and the CTD (Fig 5A). As anticipated, we find that aggregation measured by zero-angle scattering is almost entirely abrogated by these substitutions in equimolar mixtures of BI-224436 with >200 μM INW131C or INN222K (Fig 5B-D). Similar results were obtained for BI-D (data not shown). Hence, substitutions that arise in virus in response to ALLINI treatment potently counter ALLINI-induced aggregation in vitro.
Figure 5. Effects of ex vivo resistance mutations and host factors on drug-induced aggregation.

A. W131C and N222K at the CCD-CTD-ALLINI interface. B-D. Small-angle neutron scattering profiles for 212 μM INY15A,F185H (B, red), 205 μM inW131C,F185H (C, blue), and 200 μM INF185H,N222K (D, pink) in the presence and absence of BI-224436 (DMSO controls are shown in grey). Arrows indicate I(0) in each condition. E. The LEDGF(IBD)•CCD•NTD interface, as seen in the PDB 3F9K HIV-2 structure. F-G. SANS profiles for 100 μM INF185H or 100 μM co-expressed INF185H•LEDGF(IBD) in the presence of 100 μM BI-224436 (F) or 100 μM BI-D (G). Arrows indicate I(0) in each condition.
We next tested the ability of BI-224436 and BI-D to aggregate INF185H in the presence and absence of the cofactor LEDGF. The LEDGF integrase binding domain (IBD) binds at the same site on the IN CCD dimer as do the ALLINIs, but unlike ALLINIs the IBD does not recruit an IN CTD from a different IN dimer (Fig. 5E). In prior work, it was shown that at sub-micromolar concentrations, the drug-induced aggregation of IN is readily reversed by the introduction of the IBD (Feng et al., 2016), which is expected to compete with the CTD-CCD-drug interactions promoting polymerization. When micromolar concentrations of IN are pre-incubated with IBD, the aggregation response as observed by turbidity is attenuated. At the higher micromolar concentrations used in SANS experiments, we observe that the introduction of purified LEDGF exogenously or by co-expression does attenuate but does not fully reverse the induced aggregation (Figure 5F-G), in line with the expectation that the ALLINI competes with IBD binding (Gupta et al., 2014).
A role for homomeric CTD interactions in branched polymer formation.
The crystal structure of an ALLINI bound to full length IN revealed a one-dimensional open polymer of HIV IN mediated by CCD-CTD interactions (Figure 1C and D). However, the data presented here imply a more complex three-dimensional network, suggesting that the CTD-CCD interaction observed in the crystal structure only partially explain the branched three-dimensional polymer (Figure 6A). IN constructs containing only the CTD and CCD are able to support drug-induced aggregation with GSK1264 (Gupta et al., 2014; Gupta et al., 2016; Shkriabai et al., 2014), suggesting that additional inter-domain interactions within the aggregates may involve these domains. In prior studies of HIV IN by NMR, it was reported that the CTD can self-associate with an apparent Kd of ~1 μM (Lodi et al., 1995). We thus considered a model where branching occurs via self-association of the CTD (Fig 6A). In this model, both drug-stabilized CCD-CTD interactions and CTD self-interactions would underlie the polymer network. Indeed, a superposition of the 5HOT crystal structure with the NMR model (Lodi et al., 1995) or the unpublished crystal structure of the HIV CTD dimer available in the Protein Data Bank (PDB 5TC2) reveals that the CTD-CTD dimerization interface is distal to the CTD-ALLINI-CCD interface, supporting a model where both modes of interaction could occur simultaneously (Fig. 6B).
Figure 6. Disruption of the CTD-CTD Dimer interface.

A. Models for linear vs. branched polymer networks of ALLINI-induced HIV IN interactions. B. Superposition of the CTD Dimer (PDB 1IHV) with the CCD-CTD-ALLINI interaction observed in the IN-ALINI crystal structure (PDB 5HOT). C. CTD-CTD dimer interface. The quaternary structure of the CTD dimer was confirmed by SEC-SAXS (see SFig1A and STable 3) and was best captured by PDB 5TC2. Shown in spheres are residues that were mutated for effects on oligomerization. D. SEC-MALS analyses of His6-FLAG-SUMO (HFS)-tagged CTD220-270 to assess the effects of interfacial mutations on oligomerization. Relative to wild-type CTD fusion (dimers, red), the mutants L241A (orange), L242A (green), and the more extensive quadruply mutated W243Q/V250T/I257N/V259T (blue) disrupted dimerization of the isolated domain. E. Sedimentation velocity (SV-AUC) analysis of isolated CTD220-270(L242A) (green), indicating a single species with a calculated mass consistent with a monomer.
CTD dimerization substitutions ablate ALLINI-dependent effects.
We purified the IN CTD and verified its ability to form dimers using size-exclusion chromatography in-line with synchrotron small-angle scattering (SEC-SAXS). Data were analyzed using singular value decomposition with evolving factor analysis (SVD-EFA)(Meisburger et al., 2016) to deconvolute the dataset into its minimal components with maximal redundancy. In both methods, both monomers and dimers were readily identified, and the decomposed scattering profiles were consistent with known CTD dimer structures (SFig 1A). The CTD is composed of five β-strands arranged as a sandwich, where the two exposed faces of the sandwich each contain two strands and one shared strand (Fig. 6C). The dimer interface is defined by interactions between β-strands 2, 3, and 4, and is predominantly hydrophobic, burying ~400 Å2 of total accessible surface between both subunits. To further interrogate this dimerization interface, we introduced the L241A or L242A substitutions (Eijkelenboom et al., 1999; Lu et al., 2005; Lutzke and Plasterk, 1998), which are IN class II mutations that have been previously shown to affect replication, enzyme activity, and infectivity in virus (Lu et al., 2005). Both substitutions readily disrupted dimerization, as evidenced by SEC-MALS analysis (Figure 6D). Introduction of a more extensive remodeling of the dimerization interface (W243Q/V250T/I257N/V259T) also led to a monomeric CTD (Fig. 6D). Further analysis of the L242A mutant using sedimentation velocity analytical ultracentrifugation (SV-AUC) confirmed a monomeric species (Fig 6E), but a weak ability to dimerize at high concentrations was still evident from SEC-SAXS analysis (STable 3).
To study the effects of CTD dimerization on IN aggregation, we introduced the L242A substitution into full-length INF185H. In contrast to the INF185H control, which forms oligomers ranging from monomers to tetramers (Gupta et al., 2014; Gupta et al., 2016), the INF185H,L242A construct is hypo-oligomeric, forming primarily monomers and dimers at micromolar concentrations (SFig 1B). When we tested the ability of BI-224436 to induce aggregation of IN containing the L242A substitution, we found the IN mutant was entirely resistant, as evidenced by a lack of detectable turbidity in the presence of a six-fold excess of drug (Fig. 7A). A similar result was obtained when the quadruply-substituted (W243Q/V250T/I257N/V259T) IN was tested for ALLINI-induced aggregation (data not shown). These results indicate that the CTD dimerization interface is required for ALLINI aggregation.
Figure 7. Role of the CTD in branched polymer formation.

A. Turbidity time courses for INF185H,L242A in the presence of BI-224436. No aggregation was observed in absence of drug, nor the presence of up to three-fold excess of ALLINI. Shown in green is INY15A,F185H. B. SV-AUC analysis of 15 μM INF185H,L242A in the presence (purple) and absence (orange) of 100 μM BI-224436. In the absence of drug, species consistent with monomers and dimers are observed (confirmed by SEC-MALS; see SFig1B). In the presence of ALLINI, a ladder of species is observed consistent with the formation of higher-order oligomers. C. Shown is a cartoon schematic depicting the SV-AUC analysis of drug-mediated CCD-CTD interactions using Alexafluor647-labelled HFS-CTD. D. SV-AUC results at 657 nm is shown, with control data in the absence of any drug for both HFS-CTDL242A alone (red) and a mixture with excess CCD (orange). No evidence of complex formation is observed. F. In the presence of BI-224436, a new ~5S species is formed that is consistent with a higher order HFS-CTD2•CCD2•ALLINI2 complex (~77.8 kD suggested by SE-AUC analysis (See SFig 2 and STable 4). The breadth of the species observed in the presence of drug are consistent with an associating system.
Based on the structures shown in Fig 6B and C, the L242A substitution should not affect the ability of ALLINIs to form the one-dimensional polymer shown in Fig. 6B. To test this prediction, we performed SV-AUC on the INF185H,L242A protein in the presence and absence of 100 μM BI-224436 (Fig. 7B). In the absence of drug, the protein forms monomers and dimers, consistent with the results from SEC-MALS analysis. In the presence of BI-224436, a new c(S) distribution is seen that is consistent with a mixture of oligomers formed by self-association of dimers. This result supports the idea that the L242A substitution does not block the ability of ALLINIs to induce CCD-CTD interaction between IN dimers.
To further test the hypothesis that IN L242A is resistant to ALLINI-induced aggregation but can still form ALLINI-induced oligomers, we probed the CCD-CTD interaction using isolated domains. Based on SEC-MALS, sedimentation equilibrium (SE-AUC), SV-AUC, and SEC-SAXS, the IN CCDF185K is predominantly a dimer in solution at 10-100 μM concentrations (SFig1 and STable 3). The complexes predicted to form between IN CCD and monomeric IN CTDL242A in the presence of ALLINIs should therefore have stoichiometries of 1:2 and 2:2 CTD:CCD, since each CCD dimer can bind two ALLINI-bridged CTDs. Using SE-AUC analyses of CCD/CTD mixtures, the weight-average molecular weight (Mw) increases in the presence of BI-224436; the data are well described by an 2A + B ↔ BA2 association model with apparent Kd of 0.62 μM (SFig 2 and STable 4). In the absence of BI-224436, the same association model requires Kd >> 1 mM.
To verify inclusion of the CTD in these complexes, Alexafluor647-labelled CTDL242A was mixed with unlabeled CCDF185K (Fig. 7C) and the mixture was analyzed by SV-AUC in the presence and absence of excess BI-224436 In the absence of ALLINI, the CTD sediments at ~2.5S and does not form larger species in the presence of the CCD dimer (Fig. 7D). In the presence of ALLINI and CCD, a new species is formed that is consistent with a 2:2 CTD:CTD stoichiometry (Fig. 7E), consistent with results from sedimentation equilibrium, confirming the presence of the CTD in the drug-induced species.
Discussion.
In this study, we report the soft matter properties of ALLINI-induced aggregates of HIV-IN and the contributions of homomeric CTD interactions to the formation of three-dimensional branched polymers. The results reported herein and in prior studies together support a unique mode-of-action for ALLINIs. By analogy to classical polymerization models, the ALLINI acts as an “initiator’ for polymerization, bringing base units (IN dimers) together to form multimers. This is then followed by propagation of the polymer, not only in one dimension, but at branch points mediated by homomeric CTD interactions (Figure 6A). This mode-of-action resembles that of Taxol, which stabilizes polymers of microtubules. Indeed, like the polymers of ALLINI-induced HIV IN, microtubules also branch via native protein-protein interfaces (Dent, 2020). We have shown that ablation of the IN CTD•CTD interface still allows for propagation of polymers along one dimension, but without the formation of insoluble three-dimensional polymers. In contrast to recent proposals of the role of tetramers and dimers in ALLINI action (Koneru et al., 2019), this open polymerization model argues for a continuum of oligomers that start with a base dimer. In our study, we employed ambient temperatures and buffer additives like 1M NaCl to lower the rate of ALLINI-induced aggregation. In in vivo conditions and at physiological temperatures (37°C), it would be expected that aggregation would be even more efficient.
In contrast to other published reports, our studies were conducted at high protein concentrations (~20-200 μM), which should more closely model the environment in virions in which the ALLINIs exert their strongest effect. The crowded environment of a nascent virion is estimated to have a total macromolecular concentration exceeding 500 mg/ml (Benjamin et al., 2005; Briggs et al., 2004; del Alamo et al., 2005). Electron cryo-tomography studies of HIV-1 virus-like particles (VLPs) has revealed particle volumes that imply that viral RT and IN enzymes exist at concentrations in the millimolar regime (Benjamin et al., 2005), in contrast to the expected nanomolar concentrations of IN in the cytoplasm of an infected T-cell. For the ALLINIs, it can be argued that this difference in local environment partially underlies the difference in potency observed in early vs late replication, and our results further illustrate how potently ex vivo resistance substitutions abrogate this aggregation even at elevated concentrations. It is also possible that host proteins in virions may influence aggregation--host heat shock proteins (HSPs) HSP60, HSP70 and HSP90AB1 have been identified in assembled virions (Bartz et al., 1994; Linde et al., 2013), and HSP60 has been shown to interact with HIV-IN (Parissi et al., 2001).
Our experimental results highlight the unique functional properties of the CTD, which is poorly conserved across retroviruses. The domain has been identified as a site for post-translational modification and host-factor interactions (Allouch and Cereseto, 2011; Allouch et al., 2011; Cereseto et al., 2005; Kiernan et al., 1999; Lammers et al., 2010; Larue et al., 2012; Ying et al., 2012) and is implicated in a variety of functional processes during viral replication including intasome formation, integration, histone interactions (Mauro et al., 2019) and reverse transcription (Tekeste et al., 2015). Here we linked the dimerization properties of the CTD to the ability of ALLINIs to induce three-dimensional branched polymers. In the absence of ALLINIs, we observe only very weak interaction between the isolated CCD and CTD domains of IN, although the hypo-oligomeric properties of ALLINI resistance mutants mapped to this interface argues that weak interactions do contribute to the properties of IN in solution. The existence of many resistance mutations and polymorphs mapping to this interface argues against a functional role in viral replication for the CCD-CTD interaction exploited by ALLINIs.
Lastly, our findings have several implications for future development of this inhibitor class. Small-angle neutron scattering allowed us to fit empirical models from a panel of different ALLINI-induced aggregates, revealing differences in correlation length. A physical interpretation of these differences may be that the proportion of CCD-CTD vs CTD-CTD interactions occurring in an aggregate alters the geometry of interactions between IN dimer subunits. In the crystal structure of the IN-ALLINI complex, an idealized linear polymer was observed, whereas in aggregates, three-dimensional polymeric networks are formed, and our findings argue that CTD-CTD interactions are key to these differences. It might be surmised that larger ALLINI scaffolds could perturb the idealized polymer observed in crystallo, instead favoring a different proportion of CTD-CTD interactions once aggregation is initiated. This informs future drug design efforts, as this attribute of ALLINI-induced effects may be a tunable property. In addition, using isolated domains, we further dissected the drug-induced interaction, allowing us to determine the affinity of an ALLINI-induced IN-IN interaction. This allows for future structure-activity relationship studies where Kd can be related to ALLINI chemical structure and possibly other tunable properties. These observations inform ongoing optimization of the ALLINI•CCD interface needed, and the additional considerations of avidity and other participating CTD•CTD interactions that drive the formation of IN branched polymers.
STAR Methods.
Resource Availability
Lead Contact.
Further information and requests for either resources or reagents should be directed and will be fulfilled by the lead contacts Gregory Van Duyne (vanduyne@pennmedicine.upenn.edu) and Frederic Bushman (bushman@pennmedicine.upenn.edu).
Materials Availability.
Plasmids generated in these studies are available on request to the lead contacts Gregory Van Duyne (vanduyne@pennmedicine.upenn.edu) and Frederic Bushman (bushman@pennmedicine.upenn.edu.
Data and Code Availability.
All data generated in these studies are available on request from the lead contacts Gregory Van Duyne (vanduyne@pennmedicine.upenn.edu) and Frederic Bushman (bushman@pennmedicine.upenn.edu). Structures used in our analyses including 5HOT, 5TC2, 3F9K, and 1IHV, are publicly available in the Protein Databank (www.rcsb.org).
Experimental Model Details
Bacterial Strains.
For expression of proteins, E.coli bacterial strain BL21(DE3) (Novagen) was cultured at 37°C in 2xYT media.
Method Details
Inhibitor Preparation.
The inhibitors INH2 (Christ et al., 2010), BI-D (Fader et al., 2016; Fenwick et al., 2014) and BI-224436 (Fenwick et al., 2014) were purchased from MedChemExpress (Monmouth Junction, NJ, U.S.A.). To create stock solutions for this study, drugs were resuspended in acetonitrile at 1- or 10-mM concentrations, aliquoted, and lyophilized. Dried aliquots were stored at −80°C until use. Inhibitors were then resuspended to initial volumes using sample buffers for subsequent experimentation.
Protein Expression and Purification.
Full-length and truncated HIV-1 IN(NL4-3), co-expressions with LEDGF(IBD)346-471 constructs, and purified LEDGF(IBD)326-530 were expressed and purified as described previously, with some modifications (Ciuffi et al., 2006; Diamond and Bushman, 2006; Gupta et al., 2010; Gupta et al., 2016). IN-only constructs encoding the F185H solubility mutation were expressed from a pETDuet-derived (Novagen) vector in which the IN construct was inserted in-frame with a C-terminal Mxe intein (New England Biolabs) containing a chitin binding domain and hexahistidine tag. Proteins were purified using nickel-nitrilotriacetic acid (NTA) (Qiagen) and chitin (New England Biolabs) resins. After fusion proteins were liberated by intein cleavage in 50 mM dithiothreitol (DTT) overnight at 4°C, IN preparations were further purified using a Superdex 75 HiLoad 16/60 column at room temperature, eluted isocratically in 20 mM HEPES-NaOH pH 7.5, 1 M NaCl, 7 mM CHAPS, 10 μM ZnOAc2, and 10 mM β-ME or DTT
IN domain constructs described herein (CCD, CTD) were expressed from an expression vector containing an in-frame hexahistidine-FLAG-SUMO (HFS) fusion with Ulp1 protease cleavage site. Proteins were purified using nickel-NTA resin, followed by overnight cleavage at 4°C with Ulp1. These materials were subjected to a second nickel-NTA column to capture liberated fusion partner, yielding purified domain. For experiments using the intact HFS-IN fusion, proteins were immediately purified after nickel-NTA elution using a Superdex 75 HiLoad 16/60 column at room temperature, eluted isocratically in 20 mM HEPES-NaOH pH 7.5, 300 mM NaCl, and 1 mM DTT.
The QuikChange method (Wang and Malcolm, 1999) was used to generate the point mutations described in this study. Purified proteins were concentrated at 4°C in a YM-3 or YM-10 Centricon (Millipore) and aliquots with 10% glycerol were flash-frozen in liquid nitrogen for storage at −80°C
Aggregation Assays.
Assays were performed as previously described (Gupta et al., 2014), with the following modification: turbidity assays were performed using VICTOR3V™ 1420 multilabel counter (PerkinElmer, Waltham, Massachusetts, U.S.A.) by measuring the absorbance of the reaction solution at 405 nm. Final reaction conditions were 20 mM HEPES, pH 7.3, 375-505 mM NaCl, 3.75-5.05 mM CHAPS, 10 mM DTT, 10 μM ZnAc2 with inhibitor concentrations ranging from 0.08 μM to 88 μM at 24-27°C. For the graphical representation of aggregation, the baseline (IN stock buffer + drug buffer) has been subtracted from the results. Time-course data were analyzed as previously described (del Alamo et al., 2005) using an empirical Hill function as implemented in Origin 7.0 (Northampton, Massachusetts, U.S.A.):
where OD is the optical density at incubation time t; ODf is the optical density at infinite time; t50 is the time at which the OD is equal to one-half the ODf; and n is a Hill coefficient/cooperativity factor.
Scanning Electron Microscopy.
HIV INY15A,F185H was isolated by gel filtration using a Superdex 75 26/60 column at 1 ml/min in 20 mM HEPES•NaOH pH 7.5, 1 M NaCl, 10 mM dithiothreitol (DTT), 7mM CHAPS, and 10 μM ZnOAc2. Protein were concentration to 6.6 mg/ml (205 μM monomer and drug added to two-fold excess (410 μM). After brief vortexing, mixtures were nutated 4°C for an hour, then centrifuged at 13,000 rpm at 4°C for 5 minutes. After aspiration of the supernatant, 500 μl of a 2.5% glutaraldehyde solution was then added for 2.5 hours at room temperature, after which the supernatant was aspirated. Pellets were stored at 4°C in 10 mM sodium cacodylate buffer until data collection. Images were obtained on a FEI Quanta 250 Scanning Electron Microscope at the University of Pennsylvania Electron Microscopy Resource Laboratory.
Dynamic Light Scattering.
Measurements of the hydrodynamic radii of aggregated IN preparations were performed using a Wyatt DynaPro Nanostar dynamic light scattering instrument (Wyatt Technology, Santa Barbara, CA) at 20°C. The data were analyzed using Dynamics 7.0 software. A Rayleigh sphere model was used for the analysis of autocorrelation functions, meaning that the hydrodynamic radii calculated were sphere-equivalent radii. By measuring the fluctuations in the laser light intensity scattered by the sample, the instrument can determine the rate at which the particles diffuse through the medium. This value is converted to hydrodynamic radius using the Stokes-Einstein relation:
where D is the is the diffusion coefficient, k is the Boltzmann constant, T is the absolute temperature, η0 is the viscosity and Rh is the sphere-equivalent hydrodynamic radius (Einstein, 1905).
Rheology.
The viscoelastic properties of 10 mg/ml HIV INY15A,F185H with 1 mM BI-224436 were measured with an RFS3 strain-controlled rheometer with the same held between parallel plates at room temperature. The shear elastic modulus was calculated from the slope of shear stress vs shear strain for a sample deformed at a constant strain rate of 0.15 s−1. The viscosity of the sample was measured by steady shear deformation over a range of strain raters from 0.05 s−1 to 50 s−1.
SEC-MALS.
Absolute molecular weights were determined by multiangle light scattering coupled with refractive interferometric detection (Wyatt Technology, Santa Barbara, California, US) and a Superdex 75 10/300 GL column (GE Healthcare) at room temperature as previously described (Gupta et al., 2010).
Sedimentation Equilibrium (SE) and Sedimentation Velocity (SV) Analytical Ultracentrifugation (AUC).
Analytical ultracentrifugation experiments were performed with an XL-A analytical ultracentrifuge (Beckman-Coulter) and a TiAn60 rotor with six-channel (for SE) or two-channel (for SV) charcoal-filled epon centerpieces and quartz windows. Complete sedimentation velocity profiles were recorded every 30 seconds for 200 boundaries at 40,000 rpm and 20°C. Data were fit using the c(S) distribution model of the Lamm equation as implemented in SEDFIT (25). After optimizing meniscus position and fitting limits, sedimentation coefficients (S) and frictional ratios (f/f0) were determined by iterative least squares fitting of the Lamm equation, with all fit RMSDs less than 0.01. SE data were collected at 4oC with detection at 280 nm for 1-3 sample concentrations. SE analyses were carried out using global gits to data acquired at multiple speeds for each concentration with strict mass conservation using the program SEDPHAT (Vistica et al., 2004) Error estimates for equilibrium constants were determined from a 1,000-iteration Monte Carlo simulation. The partial specific volume (), solvent density (ρ), and viscosity (η) were derived from chemical composition by SEDNTERP (Laue et al., 1992). SE figures (Supplemental Figure 2) were prepared using the program GUSSI (Brautigam, 2015).
Small-angle Neutron Scattering.
SANS data were collected at the Bio-SANS beam line CG-3 of the High Flux Isotope Reactor at ORNL using a single instrument configuration with 7-meter sample-to-detector distance and two cameras. Data were collected at 20°C using 1 mm quartz cells and a neutron wavelength of 6 Å ± 0.15. The range of momentum transfer q used was 0.003 < q < 0.84 Å, where q = 4πsin(θ)/λ, where θ is the scattering angle and λ is the neutron wavelength. Additional descriptions of the instrument and setup have been previously published (Berry et al., 2012; Heller et al., 2014). The recorded scattering data were circularly averaged and reduced to one-dimensional scattering profiles using MantidPlot software (Taylor et al., 2012). Calibration of the SANS data to an absolute scale was performed by measuring a porous silica standard with known intensity at zero angle, and blank buffers containing the same percentage D2O as the samples were similarly measured and subtracted from the sample scattering for background correction. For each measurement, equimolar ALLINI was added to 20-200 μM recombinant IN in a final buffer of 20 mM HEPES•NaOH pH 7.5, 300 mM NaCl, 7 mM CHAPS, 5 mM DTT and 90% D2O (Cambridge Isotopes Laboratories, Tewksbury, MA) (0.2 micron filtered) and allowed to incubate for 30 minutes at room temperature before data collection.
One-dimensional scattering profiles were fit to one of two empirical relationships using the program SasView 5.0.1 (http://www.sasview.org/). The first was a basic power law model, where:
And the second, a model that approximates the scattering from fractal-like aggregates of spheres was used:
where ϕ is the volume fraction of the spherical “building block” particles of radius R0, Vblock is the volume of a single building block, ρsolvent is the scattering length density of the solvent, ρblock is the scattering length density of the building blocks, and P(q) and S(q) are the scattering from randomly distributed spherical particles (the building blocks) and the interference from such building blocks organized in a fractal-like clusters (Teixeira, 1988). In the fitting, scattering length densities for solvent and protein were calculated using the program MuLCH(Whitten et al., 2008) and fixed during the fitting.
Small-angle X-ray Scattering.
Size-exclusion chromatography (SEC)-SAXS data were collected at beamline 16-ID (LiX) of the National Synchrotron Light Source II (Upton, NY) (DiFabio et al., 2016). Data were collected at a wavelength of 1.0 Å in a three-camera configuration, yielded accessible scattering angle where 0.006 < q < 3.0 Å−1, where q is the momentum transfer, defined as q = 4π sin(θ)/λ, where λ is the X-ray wavelength and 2θ is the scattering angle; data to q<0.5 Å−1 were used in subsequent analyses. 100 μL of 5-10 mg/mL CTD220-288, CTD220-270, or CTD220-270(L242A), was injected and isocratically eluted at 0.5 ml/min from a Superdex 200 10/300 sizing column (G.E. Healthcare) equilibrated in 20 mM HEPES-NaOH pH 7.5, 300 mM NaCl, and 1 mM DTT at room temperature. Eluent from the column flowed into a 1 mm capillary for subsequent X-ray exposures at 1-s intervals. Plots of intensity from the forward scatter closely correlated to in-line UV and refractive index (RI) measurements.
SAXS Analysis.
SVD-EFA analysis of the SEC-SAXS data sets were performed as previously described (Meisburger et al., 2016), as implemented in the program RAW (Hopkins et al., 2017). Buffer subtracted profiles were analyzed by singular value decomposition (SVD) and the ranges of overlapping peak data determined using evolving factor analysis (EFA)(Maeder, 1987). The determined peak windows were used to identify the basis vectors for each component and the corresponding SAXS profiles were calculated. When fitting manually, the maximum diameter of the particle (Dmax) was incrementally adjusted in GNOM (Semenyuk and Svergun, 1991) to maximize the goodness-of-fit parameter, to minimize the discrepancy between the fit and the experimental data, and to optimize the visual qualities of the distribution profile. The theoretical SAXS profiles for atomic models were created using the CRYSOL program (Svergun et al., 1995). To facilitate comparison of atomic models to small-angle scattering data, atomistic models representing the complete composition of the protein were constructed and the resulting model was gradually relaxed by energy minimization in a box of water the programs VMD (Humphrey et al., 1996) and NAMD (Phillips et al., 2005), using CHARMM42 force fields. The models were rendered using the program PYMOL (DeLano, 2004).
Quantification and Statistical Analysis
Data from aggregation assays (Figure 2) were analyzed with Origin Software Version 7 (OriginLab Corporation, North Hampton, MA). These experiments were performed in triplicate. For kinetic trials, representative data is shown and for dose response experiments, data points were shown as the mean ± standard deviations for three replicates. Error estimates for sedimentation equilibrium constants were determined from a 1,000-iteration Monte Carlo simulation. In Figure 4, error bars shown on the graph represent the error associated with the determination of I(0). Errors determined for the radius of gyration (Rg) by SAXS represent the error associating with least-squares fitting of the primary data in Guinier Plot analysis.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dithiothreitol (DTT) | RPI | Cat#D110000-50.0 |
| Isopropyl-β-D-thiogalactopyranoside (IPTG) | Enzo | Cat#582-001-6100 |
| Ni-NTA-Superflow | Qiagen | Cat#30430 |
| Chitin Binding Resin | New England Biolabs | Cat#S665 |
| Superdex 75 Increase 10/300 | G.E. Healthcare | Cat#17-5171-01 |
| Superose 6 Increase 10/300 | G.E. Healthcare | Cat#29-0915-96 |
| HEPES | Sigma-Aldrich | Cat#H3375 |
| Sodium Chloride | Fisher Scientific | Cat#S271 |
| CHAPS | Sigma-Aldrich | Cat#C3023 |
| Zinc(II) Sulfate | Fisher Scientific | Cat#S25642A |
| Glycerol | Fisher Scientific | Cat#L-13751 |
| Amicon MWCO 10 kD | Millipore Sigma | Cat#UFC901024 |
| Amicon MWCO 3 kD | Millipore Sigma | Cat#UFC900324 |
| Ulp1 SUMO Protease | Lifesensors | Cat#SP-4010 |
| BI-D | MedChemExpress | Cat#HY-18601 |
| BI-224436 | MedChemExpress | Cat#HY-18595 |
| CX04328 | MedChemExpress | Cat#HY-10522 |
| INF185H | (Gupta et al., 2010) | N/A |
| INY15A,F185H | (Gupta et al., 2016) | N/A |
| INW131C,F185H | (Gupta et al., 2016) | N/A |
| INN222K,F185H | (Gupta et al., 2016) | N/A |
| INN222K,F185H | (Gupta et al., 2016) | N/A |
| INY99H,L129F,N222K | (Gupta et al., 2016) | N/A |
| INF185H•LEDGF347-471(IBD) | (Gupta et al., 2010) | N/A |
| IN(CCD)F185K | (Gupta et al., 2014) | N/A |
| IN(CTD)220-280 | This study | N/A |
| IN(CTD)220-270 | This study | N/A |
| IN(CTD)220-270(L242A) | This study | N/A |
| HFS-IN(CTD)220-280(L242A) | This study | N/A |
| Deposited Data | ||
| C-terminal domain of HIV-1 Integrase, crystal structure | this paper | PDB: 5TC2 |
| Two-domain fragment of HIV-2 Integrase in complex with LEDGF(IBD) | (Hare et al., 2009b) | PDB: 3F9K |
| Solution Structure of the DNA Binding Domain Of HIV-1 Integrase, NMR, Minimized Average Structure | (Lodi et al., 1995) | PDB: 1IHV |
| HIV-1 Integrase bound with GSK1264 | (Gupta et al., 2016) | PDB: 5HOT |
| Recombinant DNA | ||
| pETDuet (Novagen)-C-terminal Mxe Intein-CBD-6xHis Fusion Expression Vector | (Gupta et al., 2010) | N/A |
| pCDFDuet (Novagen) in-frame with an N-terminal His6-FLAG-Smt3 affinity tag Expression Vector | (Gupta et al., 2017) | N/A |
| Software and Algorithms | ||
| NAMD | (Phillips et al., 2005) | https://www.ks.uiuc.edu/Research/namd/ |
| PyMOL | Schrodinger, LLC | https://pymol.org |
| BioXTAS RAW | (Hopkins et al., 2017) | https://sourceforge.net/projects/bioxtasraw/ |
| ATSAS | (Franke et al., 2017) | https://www.embl-hamburg.de/biosaxs/software.html |
| Origin Software | OriginLab Corporation | https://www.originlab.com |
| VMD | (Humphrey et al., 1996)) | https://www.ks.uiuc.edu/Research/vmd/ |
| Mantidplot | Mantid Project | https://download.mantidproject.org/ |
| SEDFIT | (Schuck, 2000) | http://www.analyticalultracentrifugation.com/download.htm |
| SEDPHAT | (Vistica et al., 2004) | http://www.analyticalultracentrifugation.com/download.htm |
| SEDNTERP | Biomolecular Interaction Technologies Center (BITC) | http://bitcwiki.sr.unh.edu/index.php/Main_Page |
| ASTRA | Wyatt Technology | https://www.wyatt.com/products/software/astra.html |
| DYNAMICS | Wyatt Technology | https://www.wyatt.com/products/software/dynamics.html |
| ScÅtter | ALS SIBYLS | https://bl1231.als.lbl.gov/scatter/ |
| SASView | NSF DANSE | http://www.sasview.org/download/ |
Highlights.
Allosteric Inhibitors drive the inappropriate aggregation of HIV Integrase.
Drug-induced aggregates behave like weak 3D-gels with a fractal character.
Homomeric C-terminal domain interactions underlie this branched-polymer behavior.
Acknowledgements.
This work was supported by NIH grants R01-AI129661 (FB and GVD) and the Center for AIDs Research at the University of Pennsylvania (2P30-AI045008 (KG)), and a grant from ViiV healthcare (FB and GVD). KG additionally acknowledges support of the Johnson Research Foundation. DLS, SEC-MALS, and AUC measurements were performed at the Johnson Research Structural Biology and Biophysics Core Facility (Philadelphia, PA, U.S.A). The SEC-SAXS data were obtained at 16-ID (LIX) at the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The research at ORNL’s High Flux Isotope Reactor was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests. The authors declare that they have no competing interests with the contents of this article.
References.
- Allouch A, and Cereseto A (2011). Identification of cellular factors binding to acetylated HIV-1 integrase. Amino acids 41, 1137–1145. [DOI] [PubMed] [Google Scholar]
- Allouch A, Di Primio C, Alpi E, Lusic M, Arosio D, Giacca M, and Cereseto A (2011). The TRIM family protein KAP1 inhibits HIV-1 integration. Cell host & microbe 9, 484–495. [DOI] [PubMed] [Google Scholar]
- Arts EJ, and Hazuda DJ (2012). HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med 2, a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan M, Yant SR, Tsai L, O'Sullivan C, Bam RA, Tsai A, Niedziela-Majka A, Stray KM, Sakowicz R, and Cihlar T (2013). Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PloS one 8, e74163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale HD, and Schmidt PW (1984). Small-Angle X-Ray-Scattering Investigation of Submicroscopic Porosity with Fractal Properties. Physical Review Letters 53, 596–599. [Google Scholar]
- Ballandras-Colas A, Maskell DP, Serrao E, Locke J, Swuec P, Jonsson SR, Kotecha A, Cook NJ, Pye VE, Taylor IA, et al. (2017). A supramolecular assembly mediates lentiviral DNA integration. Science 355, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz SR, Pauza CD, Ivanyi J, Jindal S, Welch WJ, and Malkovsky M (1994). An Hsp60 related protein is associated with purified HIV and SIV. J Med Primatol 23, 151–154. [DOI] [PubMed] [Google Scholar]
- Benjamin J, Ganser-Pornillos BK, Tivol WF, Sundquist WI, and Jensen GJ (2005). Three-dimensional structure of HIV-1 virus-like particles by electron cryotomography. J Mol Biol 346, 577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry KD, Bailey KM, Beal J, Diawara Y, Funk L, Steve Hicks J, Jones AB, Littrell KC, Pingali SV, Summers PR, et al. (2012). Characterization of the neutron detector upgrade to the GP-SANS and Bio-SANS instruments at HFIR. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment 693, 179–185. [Google Scholar]
- Bocanegra R, Alfonso C, Rodriguez-Huete A, Fuertes MA, Jimenez M, Rivas G, and Mateu MG (2013). Association equilibrium of the HIV-1 capsid protein in a crowded medium reveals that hexamerization during capsid assembly requires a functional C-domain dimerization interface. Biophys J 104, 884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA (2015). Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol 562, 109–133. [DOI] [PubMed] [Google Scholar]
- Briggs JA, Simon MN, Gross I, Krausslich HG, Fuller SD, Vogt VM, and Johnson MC (2004). The stoichiometry of Gag protein in HIV-1. Nat Struct Mol Biol 11, 672–675. [DOI] [PubMed] [Google Scholar]
- Buxbaum RE, Dennerll T, Weiss S, and Heidemann. (1987). F-actin and microtubule suspensions as indeterminate fluids. Science 235, 1511. [DOI] [PubMed] [Google Scholar]
- Cai M, Huang Y, Caffrey M, Zheng R, Craigie R, Clore GM, and Gronenborn AM (1998). Solution structure of the His12 --> Cys mutant of the N-terminal zinc binding domain of HIV-1 integrase complexed to cadmium. Protein Sci 7, 2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, and Gronenborn AM (1997). Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat Struct Biol 4, 567–577. [DOI] [PubMed] [Google Scholar]
- Cereseto A, Manganaro L, Gutierrez MI, Terreni M, Fittipaldi A, Lusic M, Marcello A, and Giacca M (2005). Acetylation of HIV-1 integrase by p300 regulates viral integration. EMBO J 24, 3070–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DTN, Wen Q, Janmey PA, Crocker JC, and Yodh AG (2010). Rheology of Soft Materials. Annual Review of Condensed Matter Physics 1, 301–322. [Google Scholar]
- Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, and Stroud RM (2000). Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proceedings of the National Academy of Sciences of the United States of America 97, 8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ F, Shaw S, Demeulemeester J, Desimmie BA, Marchand A, Butler S, Smets W, Chaltin P, Westby M, Debyser Z, et al. (2012). Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrobial agents and chemotherapy 56, 4365–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, et al. (2010). Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nature chemical biology 6, 442–448. [DOI] [PubMed] [Google Scholar]
- Cihlar T, and Fordyce M (2016). Current status and prospects of HIV treatment. Curr Opin Virol 18, 50–56. [DOI] [PubMed] [Google Scholar]
- Ciuffi A, Diamond TL, Hwang Y, Marshall HM, and Bushman FD (2006). Modulating target site selection during human immunodeficiency virus DNA integration in vitro with an engineered tethering factor. Human gene therapy 17, 960–967. [DOI] [PubMed] [Google Scholar]
- Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, and Bushman F (2005). A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med 11, 1287–1289. [DOI] [PubMed] [Google Scholar]
- Craigie R, and Bushman FD (2012). HIV DNA Integration. Cold Spring Harb Perspect Med 2, a006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Alamo M, Rivas G, and Mateu MG (2005). Effect of macromolecular crowding agents on human immunodeficiency virus type 1 capsid protein assembly in vitro. Journal of virology 79, 14271–14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2004). Use of PYMOL as a communications tool for molecular science. Abstr Pap Am Chem S 228, U313–U314. [Google Scholar]
- Dent EW (2020). Dynamic microtubules at the synapse. Curr Opin Neurobiol 63, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deprez E, Tauc P, Leh H, Mouscadet JF, Auclair C, and Brochon JC (2000). Oligomeric states of the HIV-1 integrase as measured by time-resolved fluorescence anisotropy. Biochemistry 39, 9275–9284. [DOI] [PubMed] [Google Scholar]
- Deprez E, Tauc P, Leh H, Mouscadet JF, Auclair C, Hawkins ME, and Brochon JC (2001). DNA binding induces dissociation of the multimeric form of HIV-1 integrase: a time-resolved fluorescence anisotropy study. Proceedings of the National Academy of Sciences of the United States of America 98, 10090–10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, et al. (2013). LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 10, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond TL, and Bushman FD (2006). Role of metal ions in catalysis by HIV integrase analyzed using a quantitative PCR disintegration assay. Nucleic Acids Res 34, 6116–6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFabio J, Chodankar S, Pjerov S, Jakoncic J, Lucas M, Krywka C, Graziano V, and Yang L (2016). The Life Science X-ray Scattering Beamline at NSLS-II. Aip Conf Proc 1741. [Google Scholar]
- Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, and Davies DR (1994). Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 266, 1981–1986. [DOI] [PubMed] [Google Scholar]
- Eijkelenboom AP, Sprangers R, Hard K, Puras Lutzke RA, Plasterk RH, Boelens R, and Kaptein R (1999). Refined solution structure of the C-terminal DNA-binding domain of human immunovirus-1 integrase. Proteins 36, 556–564. [DOI] [PubMed] [Google Scholar]
- Einstein A (1905). Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flussigkeiten suspendierten Teilchen. Annalen der Physik 322, 549–560. [Google Scholar]
- Engelman A, and Cherepanov P (2012). The structural biology of HIV-1: mechanistic and therapeutic insights. Nature reviews. Microbiology 10, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Kessl JJ, and Kvaratskhelia M (2013). Allosteric inhibition of HIV-1 integrase activity. Current opinion in chemical biology 17, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Liu Y, Chen H, Farzan M, and Dyda F (1997). Structure-based mutagenesis of the catalytic domain of human immunodeficiency virus type 1 integrase. Journal of virology 71, 3507–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader LD, Bailey M, Beaulieu E, Bilodeau F, Bonneau P, Bousquet Y, Carson RJ, Chabot C, Coulombe R, Duan J, et al. (2016). Aligning Potency and Pharmacokinetic Properties for Pyridine-Based NCINIs. ACS Med Chem Lett 7, 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Dharmarajan V, Serrao E, Hoyte A, Larue RC, Slaughter A, Sharma A, Plumb MR, Kessl JJ, Fuchs JR, et al. (2016). The Competitive Interplay between Allosteric HIV-1 Integrase Inhibitor BI/D and LEDGF/p75 during the Early Stage of HIV-1 Replication Adversely Affects Inhibitor Potency. ACS Chem Biol 11, 1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Sharma A, Slaughter A, Jena N, Koh Y, Shkriabai N, Larue RC, Patel PA, Mitsuya H, Kessl JJ, et al. (2013). The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV-1 integrase inhibitors. The Journal of biological chemistry 288, 15813–15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick C, Amad M, Bailey MD, Bethell R, Bos M, Bonneau P, Cordingley M, Coulombe R, Duan J, Edwards P, et al. (2014). Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrobial agents and chemotherapy 58, 3233–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J, Jurado KA, Cheng N, Ly NL, Fuchs JR, Gorelick RJ, Engelman AN, and Steven AC (2015). Distribution and Redistribution of HIV-1 Nucleocapsid Protein in Immature, Mature, and Integrase-Inhibited Virions: a Role for Integrase in Maturation. Journal of virology 89, 9765–9780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, et al. (2017). ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr 50, 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, and Davies DR (1998). Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proceedings of the National Academy of Sciences of the United States of America 95, 9150–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Brady T, Dyer BM, Malani N, Hwang Y, Male F, Nolte RT, Wang L, Velthuisen E, Jeffrey J, et al. (2014). Allosteric inhibition of human immunodeficiency virus integrase: late block during viral replication and abnormal multimerization involving specific protein domains. The Journal of biological chemistry 289, 20477–20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Diamond T, Hwang Y, Bushman F, and Van Duyne GD (2010). Structural properties of HIV integrase. Lens epithelium-derived growth factor oligomers. The Journal of biological chemistry 285, 20303–20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Sharp R, Yuan JB, Li H, and Van Duyne GD (2017). Coiled-coil interactions mediate serine integrase directionality. Nucleic Acids Res 45, 7339–7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Turkki V, Sherrill-Mix S, Hwang Y, Eilers G, Taylor L, McDanal C, Wang P, Temelkoff D, Nolte RT, et al. (2016). Structural Basis for Inhibitor-Induced Aggregation of HIV Integrase. PLoS Biol 14, e1002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Di Nunzio F, Labeja A, Wang J, Engelman A, and Cherepanov P (2009a). Structural basis for functional tetramerization of lentiviral integrase. PLoS Pathog 5, e1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Shun MC, Gupta SS, Valkov E, Engelman A, and Cherepanov P (2009b). A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog 5, e1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Smith SJ, Metifiot M, Jaxa-Chamiec A, Pommier Y, Hughes SH, and Cherepanov P (2011). Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol Pharmacol 80, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller WT, Urban VS, Lynn GW, Weiss KL, O'Neill HM, Pingali SV, Qian S, Littrell KC, Melnichenko YB, Buchanan MV, et al. (2014). The Bio-SANS instrument at the High Flux Isotope Reactor of Oak Ridge National Laboratory. Journal of Applied Crystallography 47, 1238–1246. [Google Scholar]
- Hopkins JB, Gillilan RE, and Skou S (2017). BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J Appl Crystallogr 50, 1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27-38. [DOI] [PubMed] [Google Scholar]
- Jenkins TM, Engelman A, Ghirlando R, and Craigie R (1996). A soluble active mutant of HIV-1 integrase: involvement of both the core and carboxyl-terminal domains in multimerization. The Journal of biological chemistry 271, 7712–7718. [DOI] [PubMed] [Google Scholar]
- Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, et al. (2013). Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proceedings of the National Academy of Sciences of the United States of America 110, 8690–8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl JJ, Kutluay SB, Townsend D, Rebensburg S, Slaughter A, Larue RC, Shkriabai N, Bakouche N, Fuchs JR, Bieniasz PD, et al. (2016). HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 166, 1257–1268 e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, Calomme C, Burny A, Nakatani Y, Jeang KT, et al. (1999). HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J 18, 6106–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koneru PC, Francis AC, Deng N, Rebensburg SV, Hoyte AC, Lindenberger J, Adu-Ampratwum D, Larue RC, Wempe MF, Engelman AN, et al. (2019). HIV-1 integrase tetramers are the antiviral target of pyridine-based allosteric integrase inhibitors. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers M, Neumann H, Chin JW, and James LC (2010). Acetylation regulates cyclophilin A catalysis, immunosuppression and HIV isomerization. Nature chemical biology 6, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue R, Gupta K, Wuensch C, Shkriabai N, Kessl JJ, Danhart E, Feng L, Taltynov O, Christ F, Van Duyne GD, et al. (2012). Interaction of the HIV-1 intasome with transportin 3 protein (TNPO3 or TRN-SR2). The Journal of biological chemistry 287, 34044–34058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue TM, Shah BD, Ridgeway TM, and Pelletier SL (1992). Analytical ultracentrifugation in biochemistry and polymer science. Harding SE, Rowe AJ, and Horton JC, eds. (Cambridge [England] :, Royal Society of Chemistry; ). [Google Scholar]
- Le Rouzic E, Bonnard D, Chasset S, Bruneau JM, Chevreuil F, Le Strat F, Nguyen J, Beauvoir R, Amadori C, Brias J, et al. (2013). Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 10, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde ME, Colquhoun DR, Ubaida Mohien C, Kole T, Aquino V, Cotter R, Edwards N, Hildreth JE, and Graham DR (2013). The conserved set of host proteins incorporated into HIV-1 virions suggests a common egress pathway in multiple cell types. J Proteome Res 12, 2045–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi PJ, Ernst JA, Kuszewski J, Hickman AB, Engelman A, Craigie R, Clore GM, and Gronenborn AM (1995). Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry 34, 9826–9833. [DOI] [PubMed] [Google Scholar]
- Lu R, Ghory HZ, and Engelman A (2005). Genetic analyses of conserved residues in the carboxyl-terminal domain of human immunodeficiency virus type 1 integrase. Journal of virology 79, 10356–10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzke RA, and Plasterk RH (1998). Structure-based mutational analysis of the C-terminal DNA-binding domain of human immunodeficiency virus type 1 integrase: critical residues for protein oligomerization and DNA binding. Journal of virology 72, 4841–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison MK, Lawson DQ, Elliott J, Ozanturk AN, Koneru PC, Townsend D, Errando M, Kvaratskhelia M, and Kutluay SB (2017). Allosteric HIV-1 Integrase Inhibitors Lead to Premature Degradation of the Viral RNA Genome and Integrase in Target Cells. Journal of virology 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder M (1987). Evolving Factor-Analysis for the Resolution of Overlapping Chromatographic Peaks. Anal Chem 59, 527–530. [Google Scholar]
- Maertens GN, Hare S, and Cherepanov P (2010). The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 468, 326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskell DP, Renault L, Serrao E, Lesbats P, Matadeen R, Hare S, Lindemann D, Engelman AN, Costa A, and Cherepanov P (2015). Structural basis for retroviral integration into nucleosomes. Nature 523, 366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro E, Lesbats P, Lapaillerie D, Chaignepain S, Maillot B, Oladosu O, Robert X, Fiorini F, Kieffer B, Bouaziz S, et al. (2019). Human H4 tail stimulates HIV-1 integration through binding to the carboxy-terminal domain of integrase. Nucleic Acids Res 47, 3607–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisburger SP, Taylor AB, Khan CA, Zhang S, Fitzpatrick PF, and Ando N (2016). Domain Movements upon Activation of Phenylalanine Hydroxylase Characterized by Crystallography and Chromatography-Coupled Small-Angle X-ray Scattering. J Am Chem Soc 138, 6506–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molteni V, Greenwald J, Rhodes D, Hwang Y, Kwiatkowski W, Bushman FD, Siegel JS, and Choe S (2001). Identification of a small molecule binding site at the dimer interface of the HIV integrase catalytic domain. Acta Crystallogr. D. Biol. Crystallogr 57, 536–544. [DOI] [PubMed] [Google Scholar]
- Nomura Y, Masuda T, and Kawai G (2006). Structural analysis of a mutant of the HIV-1 integrase zinc finger domain that forms a single conformation. Journal of biochemistry 139, 753–759. [DOI] [PubMed] [Google Scholar]
- Parissi V, Calmels C, De Soultrait VR, Caumont A, Fournier M, Chaignepain S, and Litvak S (2001). Functional interactions of human immunodeficiency virus type 1 integrase with human and yeast HSP60. Journal of virology 75, 11344–11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos DO, Li M, Yang R, Rebensburg SV, Ghirlando R, Jeon Y, Shkriabai N, Kvaratskhelia M, Craigie R, and Lyumkis D (2017). Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science 355, 89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon V, Bocanegra R, Rodriguez-Huete A, Rivas G, and Mateu MG (2011). Effects of macromolecular crowding on the inhibition of virus assembly and virus-cell receptor recognition. Biophys J 100, 738–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder ARW, Shinn P, Chen HM, Berry C, Ecker JR, and Bushman F (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110, 521–529. [DOI] [PubMed] [Google Scholar]
- Schuck P (2000). Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenyuk AV, and Svergun DI (1991). Gnom - a Program Package for Small-Angle Scattering Data-Processing. Journal of Applied Crystallography 24, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Slaughter A, Jena N, Feng L, Kessl JJ, Fadel HJ, Malani N, Male F, Wu L, Poeschla E, et al. (2014). A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS Pathog 10, e1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkriabai N, Dharmarajan V, Slaughter A, Kessl JJ, Larue RC, Feng L, Fuchs JR, Griffin PR, and Kvaratskhelia M (2014). A critical role of the C-terminal segment for allosteric inhibitor-induced aberrant multimerization of HIV-1 integrase. The Journal of biological chemistry 289, 26430–26440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, and Engelman A (2007). LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes & development 21, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D, Barberato C, and Koch MHJ (1995). CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. Journal of Applied Crystallography 28, 768–773. [Google Scholar]
- Taylor J, Arnold O, Bilheaux J, Buts A, Campbell S, Doucet M, Draper N, Fowler R, Gigg M, Lynch V, et al. (2012). Mantid, A high performance framework for reduction and analysis of neutron scattering data. p. W26.010. [Google Scholar]
- Teixeira J (1988). Small-angle scattering by fractal systems. Journal of Applied Crystallography 21, 781–785. [Google Scholar]
- Tekeste SS, Wilkinson TA, Weiner EM, Xu X, Miller JT, Le Grice SF, Clubb RT, and Chow SA (2015). Interaction between Reverse Transcriptase and Integrase Is Required for Reverse Transcription during HIV-1 Replication. Journal of virology 89, 12058–12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiang M, Jones GS, Niedziela-Majka A, Kan E, Lansdon EB, Huang W, Hung M, Samuel D, Novikov N, Xu Y, et al. (2012). New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. The Journal of biological chemistry 287, 21189–21203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistica J, Dam J, Balbo A, Yikilmaz E, Mariuzza RA, Rouault TA, and Schuck P (2004). Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal Biochem 326, 234–256. [DOI] [PubMed] [Google Scholar]
- Wang JY, Ling H, Yang W, and Craigie R (2001). Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J 20, 7333–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, and Malcolm BA (1999). Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques 26, 680–682. [DOI] [PubMed] [Google Scholar]
- Whitten AE, Cai S, and Trewhella J (2008). MULCh: modules for the analysis of small-angle neutron contrast variation data from biomolecular assemblies. Journal of Applied Crystallography 41, 222–226. [Google Scholar]
- Wilson MD, Renault L, Maskell DP, Ghoneim M, Pye VE, Nans A, Rueda DS, Cherepanov P, and Costa A (2019). Retroviral integration into nucleosomes through DNA looping and sliding along the histone octamer. Nat Commun 10, 4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Shi K, Banerjee S, Pandey KK, Bera S, Grandgenett DP, and Aihara H (2016). Crystal structure of the Rous sarcoma virus intasome. Nature 530, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Zhang Y, Zhou X, Qu X, Wang P, Liu S, Lu D, and Zhu H (2012). Selective histonedeacetylase inhibitor M344 intervenes in HIV-1 latency through increasing histone acetylation and activation of NF-kappaB. PloS one 7, e48832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in these studies are available on request from the lead contacts Gregory Van Duyne (vanduyne@pennmedicine.upenn.edu) and Frederic Bushman (bushman@pennmedicine.upenn.edu). Structures used in our analyses including 5HOT, 5TC2, 3F9K, and 1IHV, are publicly available in the Protein Databank (www.rcsb.org).