

SUMMARY
Squamous cell carcinoma (SCC) is a global public health burden originating in epidermal stem and progenitor cells (ESPCs) of the skin and mucosa. To understand how genetic risk factors contribute to SCC, studies of ESPC biology are imperative. Children with Fanconi anemia (FA) are a paradigm for extreme SCC susceptibility caused by germline loss-of-function mutations in FA DNA repair pathway genes. To discover epidermal vulnerabilities, patient-derived pluripotent stem cells (PSCs) conditional for the FA pathway were differentiated into ESPCs and PSC-derived epidermal organotypic rafts (PSC-EOR). FA PSC-EORs harbored diminished cell-cell junctions and increased proliferation in the basal cell compartment. Furthermore, desmosome and hemidesmosome defects were identified in the skin of FA patients, and these translated into accelerated blistering following mechanically-induced stress. Together, we demonstrate that a critical DNA repair pathway maintains the structure and function of human skin and provide 3D epidermal models wherein SCC prevention can now be explored.
Keywords: Fanconi anemia, pluripotent stem cells, epidermal stem and progenitor cells, epidermal organotypic rafts, epidermis, skin integrity, desmosome, hemidesmosome, blistering, squamous cell carcinoma
Graphical Abstract
eTOC Blurb
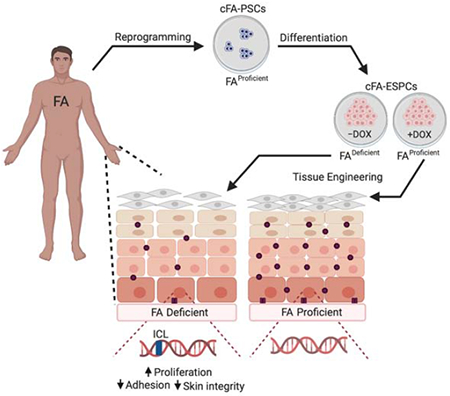
Ruiz-Torres and colleagues implicate a key DNA repair pathway in sustained epidermal structure and function. Directed differentiation of pluripotent stem cells into 3D-epidermis demonstrates that conditional loss of the Fanconi anemia (FA) pathway stimulates basal cell proliferation and impairs cell-cell junctions. Correspondingly, patients with FA are vulnerable to mechanically-induced blistering.
INTRODUCTION
Epidermal stem and progenitor cells (ESPCs) give rise to stratified epithelium during embryonic development (Belokhvostova et al., 2018). Both epidermis and mucosa represent key physical barriers and the first line of organismal defense against environmental insults via tightly connected layers of keratinocytes. Exposure to carcinogens, such as ultraviolet (UV) light, alcohol, and tobacco, or human papillomavirus (HPV) infection are risk factors for SCC, but constitutional genetic and cellular alterations underlying SCC predisposition remain poorly understood.
Fanconi anemia (FA) is an inherited disorder caused by germline loss-of-function mutations in over 20 genes involved in DNA interstrand crosslink (ICL) repair, resulting in susceptibility to DNA damage and heightened sensitivity to DNA crosslinking agents (Bogliolo and Surralles, 2015, Rodriguez and D’Andrea, 2017, Sumpter and Levine, 2017, Kottemann and Smogorzewska, 2013, Auerbach, 2009). Hematopoietic stem and progenitor cell death and resulting bone marrow failure (BMF) are clinical hallmarks, sometimes progressing to leukemia (Li et al., 2007). Affected individuals are also dramatically predisposed to early-onset and aggressive SCCs of the head and neck region, primarily in the oral cavity and esophagus, the anogenital tract (Kutler et al., 2016, Kutler et al., 2003a, Rosenberg et al., 2008, Kutler et al., 2003b), and skin (Kutler et al., 2003a, Fund, 2014). In our cohort of 105 FA patients, 11.4% reported a history of skin cancer at a mean age of 37 ± 11 years (Table 1). In contrast to the hematopoietic compartment (Muller and Williams, 2009), these tumors occur in the absence of epidermal stem cell exhaustion. However, the underlying vulnerabilities of ESPCs in FA remain elusive due to the lack of developmental models. We hypothesized that unexplored FA epidermis-specific pathologies -beyond genome instability- exist based on two lines of clinical data. First, bone marrow transplantation to treat BMF does not decrease and in fact further increases the risk of SCC (Rosenberg et al., 2005), likely as a consequence of tissue injury. Second, analyses of data from The Cancer Genome Atlas (TCGA) (Cerami et al., 2012, Gao et al., 2013, Nalepa and Clapp, 2018) reveal associations between somatic non-synonymous FA gene mutations and head and neck (21%) and skin SCC (59%), but not leukemia (1.5%) (Figure S1 and Table S1).
Table 1: Skin cancer prevalence in adults with Fanconi Anemia.
Characteristics of skin cancer cases in individuals with FA. Of 105 FA patients between the ages of 19 to 51 years old, 12 (11.4%) self-reported a diagnosis of skin-related cancer. Patients were diagnosed with skin cancer at a mean age of 37 ± 11 years old. Abbreviations: Comp Group= Complementation Group; NR= Not reported; BMT= Bone Marrow Transplant; BCC=Basal Cell Carcinoma; SCC= Squamous Cell Carcinoma; M= Male; F= Female. See also Figure S1 and Table S1.
| ID | Gender | Age (years) | Status | Comp Group | BMT | Cancer Type | Site of SCC |
|---|---|---|---|---|---|---|---|
| 1 | M | 19 | Alive | NR | Yes | SCC | Mucosa (tongue) |
| 2 | F | 24 | Alive | NR | No | Melanoma | Skin (breast) |
| 3 | F | 27 | Alive | Other | No | SCC, BCC | Skin (face) |
| 4 | M | 30 | Alive | A | No | SCC | Skin (face), mucosa (throat, esophagus) |
| 5 | M | 31 | Alive | Other | Yes | SCC, BCC | Skin (face) |
| 6 | M | 35 | Alive | A | Yes | 50+SCC, BCC | Skin (face, thumb, back, torso, scalp) |
| 7 | F | 40 | Alive | NR | No | SCC, BCC | Skin, mucosa (lip, gums, vulva) |
| 8 | F | 42 | Deceased | Other | No | SCC | Skin (hand) |
| 9 | F | 42 | Deceased | A | Yes | SCC | Mucosa (lip, salivary gland, thyroid) |
| 10 | F | 51 | Deceased | NR | No | SCC | Skin (leg), mucosa (oral, anal) |
| 11 | F | 51 | Alive | NR | No | SCC | Skin (face, head) |
| 12 | M | 51 | Deceased | A | No | SCC | Skin (ear) |
Herein we subjected previously published pluripotent stem cells (PSCs), cultured from 2 individuals with FA and conditionally complemented (Chlon et al., 2016), to directed differentiation into cellular ESPC and PSC-derived epidermal organotypic raft (PSC-EOR) tissue models. FA pathway loss did not affect the growth and differentiation of ESPCs. However, once engineered into PSC-EORs, electron microscopy (EM) analyses demonstrate an unexpected role for the FA pathway in sustaining tissue adhesion and suppressing basal cell proliferation. Functional consequences were then explored in human cohorts, where similar ultrastructural tissue pathologies were linked to numerical and structural defects in desmosomes and hemidesmosomes. Finally, functional significance for the observed tissue defects was directly tested in a cohort of FA compared to control individuals. Our data demonstrate unequivocally that FA individuals are prone to skin blistering in response to mechanical stress and implicate DNA damage in the observed tissue fragility. We hypothesize that inherited structural defects of the FA epidermis, and perhaps mucosa, cause vulnerabilities to mechanical and/or environmental stress, thus further compounding the existing genome instability to promote SCC. The above models of epidermal development can now be used to uncover the full range of clinical phenotypes in FA and relevant molecular mechanisms, with the goal of identifying effective therapies to prevent and reverse SCC susceptibility.
RESULTS AND DISCUSSION
Engineering PSC-derived epidermal organotypic rafts with a conditional FA pathway
Elucidation of the molecular and cellular consequences of FA pathway loss in the normal epidermis has been challenging since FA mouse models do not recapitulate human phenotypes spontaneously (Bakker et al., 2013). To model the development and differentiation of human FA epidermis in vitro, a personalized medicine approach was chosen. We used PSCs with a conditional FA pathway (cFA-PSCs) cultured from individuals with FANCA mutations, which were conditionally FANCA-corrected by the addition of doxycycline (DOX) (Chlon et al., 2016). This system served as the source of isogenic FA pathway-proficient (+DOX) and deficient (−DOX) ESPCs to discover unreported phenotypes in the FA pathway-deficient epidermis. The cFA-PSC lines had been rigorously evaluated previously, demonstrating that the FA pathway is essential for PSC self-renewal, but not for differentiation into ectoderm, endoderm, and mesoderm (Chlon et al., 2016). Two cFA-PSC lines from 2 independent donors were differentiated into ESPCs in the presence and absence of DOX, using the approach shown in Figure 1A. Addition of RA and BMP4 initiated cFA-PSC differentiation into ectodermal lineages (Itoh et al., 2011, Petrova et al., 2014, Kogut et al., 2014). On day 4, the cells were switched to keratinocyte culture media and DOX was either maintained or withdrawn from the cultures. A ROCK GTPase inhibitor (Y-27632) was added on day 10 to maintain keratinocyte specification into ESPCs and prevent their terminal differentiation. In contrast to previous data where the FA pathway was essential for sustained proliferation of PSCs and PSC-derived hematopoietic progenitors in vitro under standard conditions (Muller and Williams, 2009, Chlon et al., 2016, Yung et al., 2013, Raya et al., 2009, Suzuki et al., 2015, Muller et al., 2012), FA pathway loss in ESPCs did not impair cellular growth and morphology (Figure 1B). As expected, monoubiquitinated FANCD2 foci -which mark an active FA DNA repair machinery- were undetectable in −DOX ESPCs despite γH2AX-marked DNA damage (Figure 1C), and these cells did not express detectable FANCA nor FANCD2 monoubiquitination (Figure 1D). Both +DOX and −DOX ESPCs exhibited basal cell characteristics defined by keratin 14 (K14) and ΔNp63 expression as well as sustained cell cycle progression in monolayer cultures (Figure 1E and Figure S2). The cFA-ESPCs were used to generate PSC-EORs (Hoskins et al., 2009) that formed 3D engineered epidermis with proper basal/suprabasal epidermal differentiation, as shown by K14 and keratin 10 (K10) staining (Figure 1F). Together, these findings support the use of the ESPC and PSC-EOR systems as a viable approach for phenotype discovery in the human FA epidermis.
Figure 1. Engineering PSC-derived epidermal organotypic rafts with a conditional FA pathway.
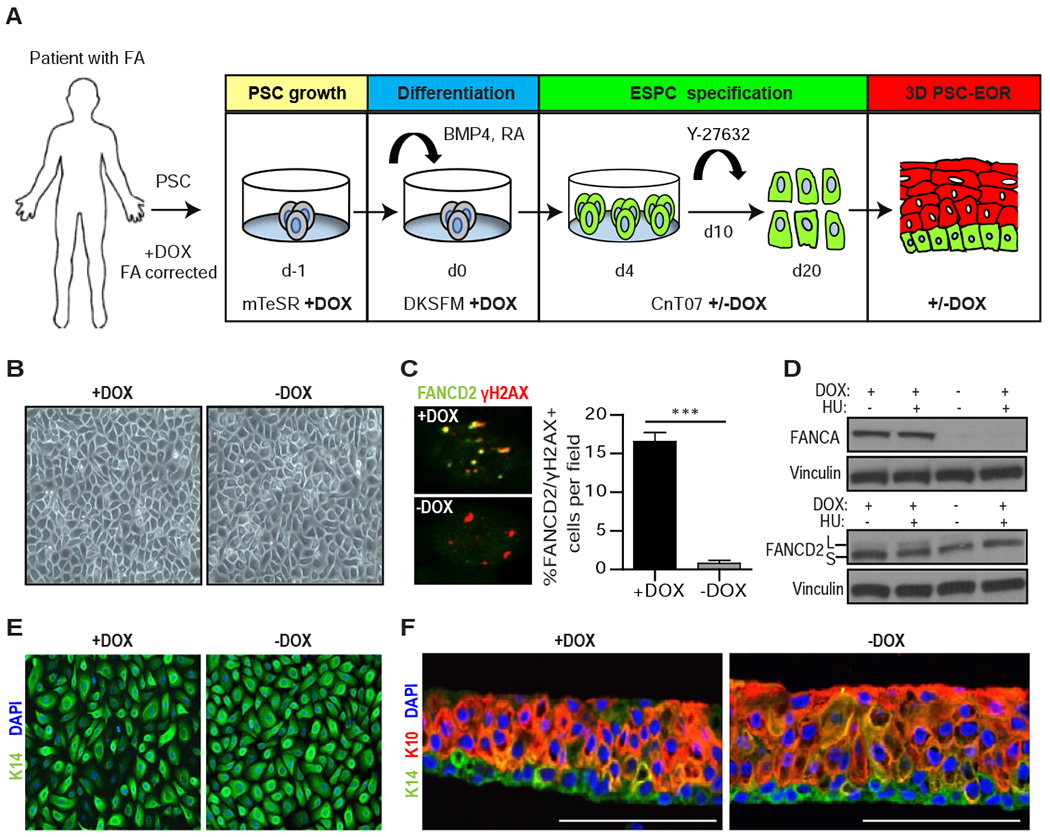
(A) Schematic of the 20-day protocol used for the directed differentiation of patient-derived cFA-PSCs into ESPCs and 3D engineered epidermis, with cells cultured in the presence of DOX starting on day −1. To generate FA pathway-deficient ESPCs, DOX was removed on day 4. mTeSR: embryonic/PSC medium; DKSFM: defined keratinocyte serum-free media; CnT07: progenitor-cell-targeted epidermal-keratinocyte media; Y-27632: ROCK kinase inhibitor. (B) Bright field images of cFA-ESPCs at passage 2 showing similar formation of cohesive sheets of cells; one representative example is shown. (C) Immunofluorescence (IF) analysis and proportion of FANCD2 and γH2AX+ cells in +/−DOX ESPCs treated with 300 nM MMC for 16 hours prior to fixation; ***P<0.0001, two-tailed Student’s t-test; n=3. Error bars represent SEM. (D) Western blot analysis of whole-cell lysates from +/−DOX ESPCs that were untreated or DNA-damaged with 2 mM hydroxyurea (HU) for 24 hours. FANCD2 monoubiquitination demonstrates the inducibility of the FA pathway. L and S: monoubiquitinated and unmodified forms of FANCD2, respectively. Vinculin, loading control; n=2, one representative example is shown. (E) IF analysis for the basal cell marker K14 and 4’6-diamidine-2-phenylindole (DAPI) in ESPCs at passage 2, demonstrating basal cell characteristics; n=3; Magnification, 40x. (F) PSC-EORs derived from +/−DOX ESPCs were processed for IF to image K14+ basal and K10+ suprabasal cells, and DAPI staining. Scale bar, 100 μm. See also Figure S2.
The FA-dependent ESPC transcriptome suggests tissue adhesion defects
To uncover FA-dependent biological processes, we performed RNA-Sequencing (RNA-Seq) analyses of +/−DOX ESPCs in monolayer cultures. Expression levels of 120 genes were differentially regulated in the −DOX compared to the +DOX populations (p<0.05, log-fold change >0.5; Figure 2A and 2B). Classification of the most significantly deregulated ontologies supported relevance for cell adhesion (Figure 2C), in line with our published data from SCC cells where FA pathway loss stimulated epithelial-to-mesenchymal-like transition (EMT), with cytoskeletal reorganization, diminished cell-cell adhesion, and stimulated invasion (Romick-Rosendale et al., 2016). Validation of the transcriptome analyses by RT-qPCR in 2 independent experiments demonstrated increased levels of proliferation, differentiation, and cell fate genes (NOTCH1 and VWCE) and decreased levels of genes involved in cell junction complexes (LGALS4, CLN9, PCDHB6; Figure 2D). These results indicated a possible role for the FA pathway in regulating cellular adhesion, and we tested such a role by engineering ESPCs into 3D PSC-EORs.
Figure 2. FA-dependent ESPC transcriptome suggests cell adhesion defects.

mRNA from ESPCs derived from 2 distinct donors with conditional functionality of the FA pathway were submitted for RNA-Seq analysis. (A) Scatterplot of genes by the log-fold change (x-axis) of their expression in −DOX compared to +DOX conditions, and the corresponding p-values (y-axis) based on paired t-test. 120 genes with absolute log-fold change > 0.5 and p-value < 0.05 are highlighted. (B) Heatmap of relative expression (row-normalized) of the 120 genes in (A) in the four samples. (C) Top biological processes altered in ESPCs upon FA pathway loss. Genes common to both FA pathway-deficient ESPCs were analyzed using ToppGene server to identify significantly altered biological processes. (D) RT-qPCR validation of genes deregulated in −DOX compared to +DOX ESPCs using an independent biological ESPC replicate for each donor. Red, upregulated. Blue, downregulated.
PSC-EORs uncover adhesion and proliferation abnormalities in the FA pathway-deficient epidermis
Skin function depends on the proper formation of cell junctional complexes between keratinocytes. Included in such cell structures are desmosomes and hemidesmosomes, specialized cell-cell and cell-substrate junctional complexes that are key for sustained skin integrity and resistance to mechanical stress (Getsios et al., 2004, Brooke et al., 2012, Walko et al., 2015) (Figure 3A). Hence, we set out to investigate junctional defects in the FA epidermis. For this, the cFA-ESPCs were further differentiated into PSC-EORs and examined for tissue ultrastructure by EM. Interestingly, the −DOX engineered epidermis had fewer and shortened desmosomes with less mature plaques (Figure 3B). FA pathway deficiency was reported to diminish quiescence and increase proliferation in hematopoietic stem cells under physiological stress (Zhang et al., 2016) and in basal epidermal cells of the murine skin and tongue (Park et al., 2013 and Park et al., 2010). On the other hand, repression of desmosome components was reported to stimulate cell proliferation (Wan et al., 2007). To test a potential correlation between desmosome defects and proliferative gains, 5’ethynyl-2’-deoxyuridine (EdU) incorporation experiments were performed (Figure 3C). The −DOX versus +DOX engineered epidermis harbored significantly more EdU+ proliferating basal keratinocytes, despite normal-appearing morphology by hematoxylin and eosin (H&E) staining (Figure 3D). These findings are consistent with in vivo studies showing that FA pathway loss stimulates proliferation subtly, but significantly, in the normal epidermis and dramatically in the HPV16 E7-transgenic murine epidermis (Park et al., 2010) and with our in vitro studies of human HPV-positive EORs (Hoskins et al., 2009).
Figure 3. Cell junctional defects and increased proliferation in the FA epidermis.
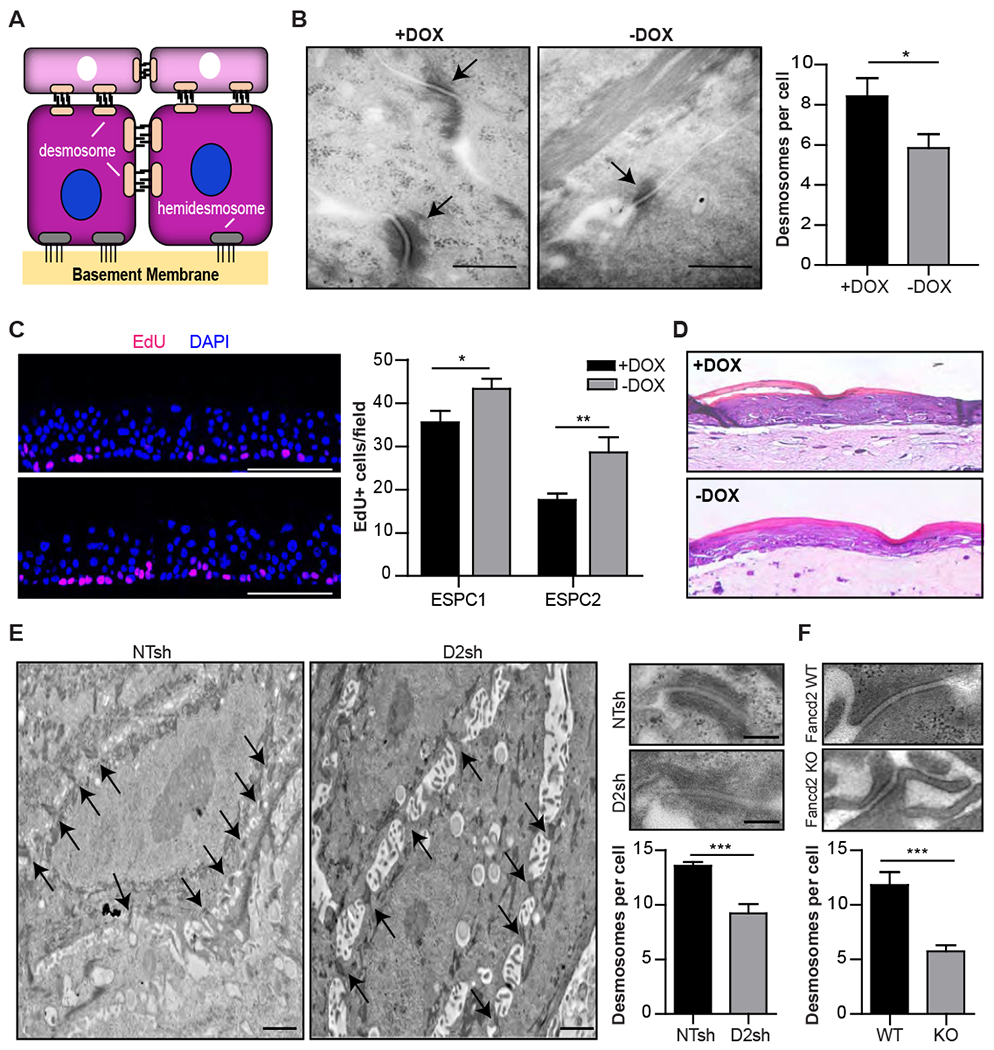
(A) Schematic representation showing cell-cell desmosome and cell-basal lamina hemidesmosome junctions on basal keratinocytes. (B) Sections from 3D +/−DOX PSC-EORs were used for EM. Images and quantification of desmosomes per cell are shown; n=3, *P=0.03. Arrows: desmosomes. Scale bar, 500 nm. Error bars represent SEM. (C) Quantification of EdU+ cells in engineered epidermis generated from +/−DOX ESPC1 and ESPC2 cell populations; n=2 biological replicates for ESPC1, n=2 technical replicates for ESPC2. *P=0.04 (ESPC1) and **P=0.0091 (ESPC2). Scale bar, 100 μm. Error bars represent SEM. (D) H&E staining showing normal overall architecture on FA-ESPCs-derived engineered epidermis cultured in presence or absence of DOX; representative images are shown. Magnification, 40x. (E) EM images and quantification of desmosomes present per cell on 3D epidermis derived from NTsh or FANCD2sh NIKS. Magnified insets show the representative structure of one desmosome; n=3. Scale bars, 2 μm and 250 nm (insets). ***P=0.0002. Error bars represent SEM. (F) Representative structure and quantification of desmosomes present per cell in tongues from Fancd2 WT (WT) and Fancd2 KO (KO) mice; n=2. ***P<0.0001. Error bars represent SEM. See also Figure S3.
Ultrastructural desmosome defects observed in −DOX PSC-EORs were validated by EM in 3D epidermis engineered from near-diploid immortalized keratinocytes derived from skin (NIKS) transduced with short-hairpin RNA (shRNA) targeting FANCD2 (FANCD2sh) or with non-targeting shRNA (NTsh) vectors (Hoskins et al., 2009) (Figure 3E). Multiple adhesion abnormalities, particularly shortened and fewer desmosomes, and pronounced intercellular spaces were again observed in FANCD2sh compared to NTsh engineered tissues. Proliferative gains were also observed in FANCD2sh compared to NTsh EORs derived from normal cutaneous NIKS and normal oral keratinocytes (NOKS) (Figure S3A and S3B). Finally, abnormal and fewer desmosomes were observed in the tongues of Fancd2 knockout (Fancd2 KO) mice compared to their Fancd2 wild-type (Fancd2 WT) counterparts (Figure 3F). Collectively, these data from diverse epidermal models and gene modulations support the concept that germline or somatic FA pathway loss diminishes keratinocyte adhesion and stimulates proliferation in the context of normal stratified tissue.
Defective cell junctions in persons with FA
The goal of the preclinical studies described above was to discover unreported epidermal phenotypes in FA. To validate these in patient populations, skin biopsies from affected versus healthy control individuals were obtained and analyzed. Importantly, these FA samples were representative of mutations in 4 distinct FA genes, FANCA, FANCB, FANCC, and FANCD1/BRCA2, thus ruling out FANCA-specific activities and implicating the contribution of the entire pathway. No morphological defects were noted in the epidermis upon blinded histological analyses (Figure 4A). However, FA specimens harbored accumulation of melanin pigment in the upper dermis indicative of pigment incontinence, which was not observed in control samples (Figure 4A bottom panels and 4B). The increased dermal pigment could not be attributed to race (Table S2) nor gender (67% in females, 33% in males; P= 0.5), and has been linked to basal cell injury by others (Davis and Callender, 2010). Furthermore, EM analyses revealed reduced numbers of both desmosomes and hemidesmosomes, and structural anomalies in FA versus control specimens (Figure 4C and 4D). Alterations in cell adhesion molecules are often associated with epidermal differentiation defects (Lopez-Pajares et al., 2013). However, FA skin could not be distinguished from control skin based on blinded analysis of K14/K10 stained specimens (Figure 4E and Figure S4), thus demonstrating the existence of differentiation-independent skin vulnerabilities in FA.
Figure 4. Defective cell junctions in patients with FA.

(A) Skin biopsies from affected individuals (FA) and gender-matched controls were H&E stained. Arrows on magnified insets: melanin present in the upper dermis. Scale bar, 100 μm and 50 μm (insets). (B) Pigment incontinence defined as the accumulation of melanin pigment in the upper dermis was assessed in biopsies from 9 FA and 9 unaffected individuals. Quantification on the right shows incidence of pigment incontinence only in FA compared to control individuals. + = pigment incontinence present; − = pigment incontinence not present. (C) EM images and quantification of desmosomes per cell on skin-punch biopsies from FA and control populations. Arrows: desmosomes. Magnified insets: structure of one representative desmosome; Data from control (n=4) and individuals with FA (n=9) was quantified on the right. No donor had received a bone marrow transplant. ***P<0.0001. Error bars represent SEM. Scale bar, 2 μm and 200 nm (insets). (D) EM images and quantification of hemidesmosomes present per 1 μm of basement membrane on the same donors; n=4. Arrows: hemidesmosomes. *P=0.03. Error bars represent SEM. Scale bar, 500 nm. (E) Sections from skin punch biopsies of controls and individuals with FA were stained for the differentiation markers K14/K10 and could not be distinguished from each other based on examination by a pathologist blinded to FA status. Magnification, 40x. See also Figure S4, Table S2, and Table S3.
Accelerated blistering is an unexplored epidermal vulnerability trait in FA
Desmosome and hemidesmosome defects are linked to blistering disorders that are also associated with SCC risk (Lopez-Pajares et al., 2013) (e.g., bullous pemphigoid or pemphigus vulgaris) but spontaneous blistering has not been reported in FA. Therefore, we tested individuals with FA versus healthy controls directly for susceptibility to mechanically-induced blistering. The forearm of individuals with FA and age-matched normal controls (Table 2) was subjected to controlled negative pressure and the time for blister formation was recorded (Figure 5A). Exposure to mechanical stress uncovered strikingly accelerated blistering in FA skin, which occurred within half the time needed for the onset of blistering in control skin. Altogether, these data are the first to demonstrate that in humans, an intact FA pathway is critical for sustained skin structure and function (Figure 5B).
Table 2: Accelerated blistering in FA skin.
Time from negative pressure initiation to first appearance of skin blisters in FA patients (n=8) or age-matched controls (n=9). FA patients showed shorter time to blister formation (median: 21 minutes) compared to matched-controls (median: 41 minutes). Two of the FA patients had undergone bone marrow transplantation but showed no difference in blister formation compared to those FA individuals who did not have a bone marrow transplant. Abbreviations: Comp Group= Complementation Group, BMT= Bone Marrow Transplant; M= Male; F= Female.
| Subject | Gender | Age (years) | Control/FA | Comp Group | BMT | Blistering Time (minutes) |
|---|---|---|---|---|---|---|
| 1 | F | 18 | Control | 31 | ||
| 2 | F | 18 | Control | 41 | ||
| 3 | M | 13 | Control | 48 | ||
| 4 | M | 18 | Control | 41 | ||
| 5 | M | 22 | Control | 33 | ||
| 6 | M | 21 | Control | 48 | ||
| 7 | F | 17 | Control | 45 | ||
| 8 | F | 15 | Control | 40 | ||
| 9 | M | 18 | Control | 43 | ||
| 10 | F | 15 | FA | A | No | 22 |
| 11 | F | 21 | FA | A | Yes | 20 |
| 12 | F | 13 | FA | A | Yes | 22 |
| 13 | M | 12 | FA | C | No | 23 |
| 14 | M | 13 | FA | A | No | 19 |
| 15 | F | 23 | FA | A | No | 16 |
| 16 | M | 23 | FA | A | No | 20 |
| 17 | F | 25 | FA | G | No | 24 |
Figure 5. Loss of the FA pathway results in impaired epidermal integrity.
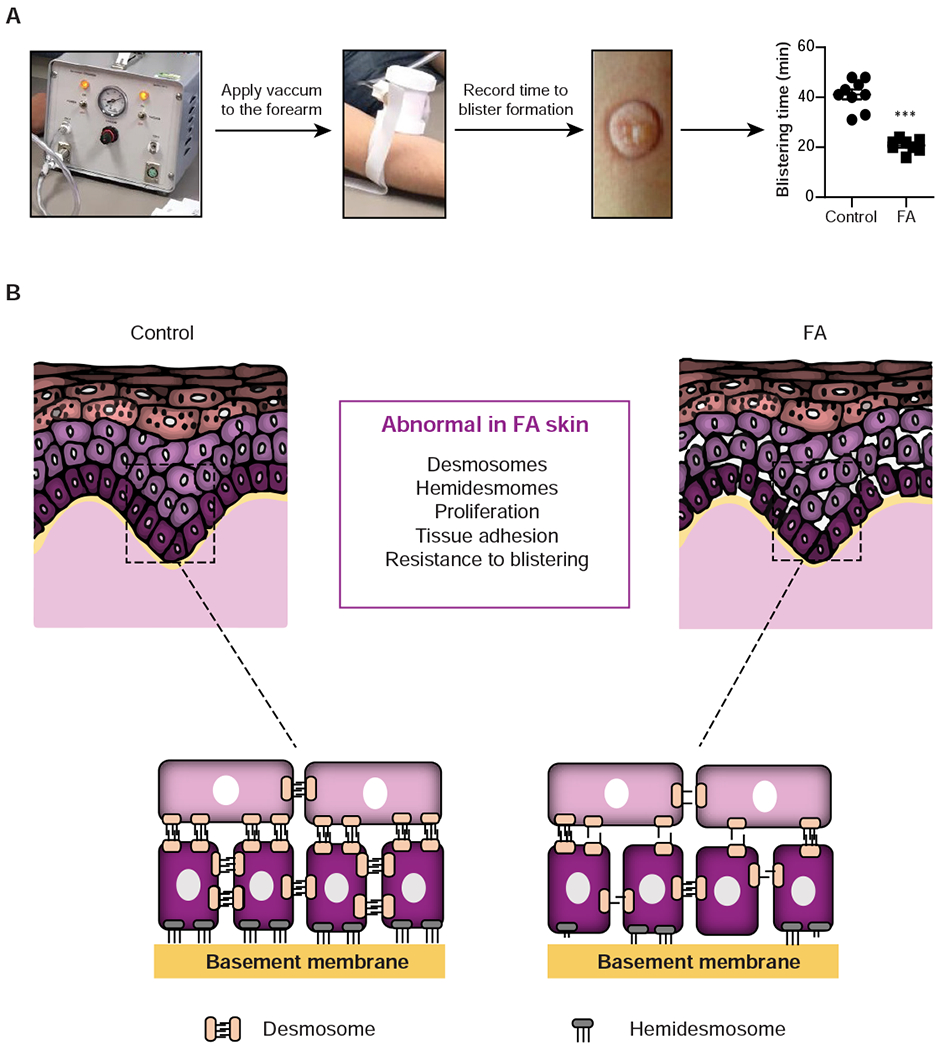
(A) A Negative Pressure Cutaneous Suction System (Electronic Diversities) was applied to consented individuals following IRB approval and time to blister formation was recorded as a measure of skin fragility. Minimal time required for blister formation in the skin of control (n=9) and FA (n=8) populations is graphed. ***P<0.0001. Error bars represent SEM. (B) Working model showing FA versus normal epidermis. Strong desmosome and hemidesmosome connections that normally maintain human epidermal integrity are impaired in the FA skin, in conjunction with weakened cell adhesion and stimulated proliferation phenotypes which may be active contributors to the increased SCC susceptibility observed in FA. See also Figure S5.
Due to the known role of the FA pathway in DNA crosslink repair, we asked whether a mild, nontoxic dose of the DNA crosslinker mitomycin C (MMC) is sufficient to stimulate proliferation and diminish desmosomes as observed in the FA epidermis. NIKS transduced with NTsh or FANCD2sh vectors were cultured in presence of vehicle or 500 pg/mL of MMC. When compared to untreated NIKS, this low dose of MMC did not cause cellular toxicity, nor reduce cellular growth. As expected, γH2AX staining revealed elevated DNA damage in untreated FANCD2sh compared to control cells (Figure S5A). Addition of MMC resulted in increased damage in NTsh cells comparable to that in untreated FANCD2sh cells. This level of damage was further exacerbated upon FANCD2 loss (Figure S5A). The resulting cell populations were then engineered into 3D epidermis with continuous MMC or vehicle exposure that did not impair tissue morphology (Figure S5B). As expected from Figure 3C and Figure S3A, FANCD2 loss stimulated proliferation in basal and some suprabasal cells (Figure S5C). MMC treatment of control NTsh cells was sufficient to stimulate proliferation much like FA pathway loss, including inappropriate proliferation in suprabasal cells. The combination of MMC treatment with FANCD2 loss reduced the overall proliferation down to baseline levels (Figure S5C) in the absence of apoptosis (data not shown), suggesting that cells respond to a further increase in DNA damage by undergoing cell cycle arrest. We next detected the desmosome component desmoplakin (DSP) by confocal microscopy as a proxy for desmosomes (Figure S5D). As expected from Figures 3B, 3E, and 3F, FANCD2 loss diminished DSP foci. Importantly, MMC treatment of control NTsh epidermis was sufficient to reduce DSP foci much like FA pathway loss. On the other hand, MMC treatment in combination with FANCD2 loss restored DSP foci in 3D epidermis, thus potentially placing desmosome function downstream of proliferation in the context of FA-related DNA damage.
In this study, we identified structural and functional defects in the skin of individuals with FA. FA is an inherited disorder characterized by cellular and organismal sensitivity to crosslinkers, congenital abnormalities, progressive BMF, and an extreme incidence of SCC. FA arises from a germline loss-of-function in one of 22 known FA genes associated with the FA pathway, which functions in the repair of DNA ICLs. Other activities reported for this pathway include the suppression of nonhomologous end-joining, stabilization of stalled replication forks, control of cytokinesis, and selective autophagy (Pace et al., 2010, Ceccaldi et al., 2016). ICLs occur endogenously during the S phase of the cell cycle or following exposure to crosslinkers, including chemotherapeutics or aldehydes (Stone et al., 2008). These lesions block transcription and replication, and their continuous removal is therefore required for sustained cellular survival. While all cells in the body have evolved sophisticated mechanisms to repair DNA damage, the consequences of defective repair are distinct for different organs and are likely rooted in developmental processes that remain poorly understood. Correspondingly, in the intensely studied FA hematopoietic compartment, loss of this pathway results in stem and progenitor cell exhaustion and aplastic anemia (Bagby, 2018). However, in stark contrast to the hematopoietic system, a similar decline in ESPC fitness in the skin of humans or mice has not been reported. Moreover, despite close associations between FA and SCC susceptibility, the pathological consequences of FA pathway loss in human epidermis have remained elusive due to the lack of appropriate developmental models.
To retrace the developmental history of FA pathway-deficient (versus proficient) epidermis, we generated ESPCs and PSC-EORs by modifying previously described strategies involving the use of defined culture conditions and the manipulation of RA, Wnt, and BMP signaling pathways (Itoh et al., 2011, Petrova et al., 2014, Kogut et al., 2014). Similar to other protocols, RA was added to induce differentiation into ectoderm, which gives rise to the nervous system and skin (Petrova et al., 2014). BMP4 was added to promote epidermal differentiation at the expense of neural differentiation by blocking the response to fibroblast growth factor signaling, thus mimicking known downstream effects of Wnt signaling in driving ectodermal fate (Itoh et al., 2011, Petrova et al., 2014). In contrast to other protocols for keratinocyte differentiation (Itoh et al., 2011), our system uses feeder-free conditional patient-derived PSCs, where the functionality of the FA pathway is restored by the addition of DOX. This allows an isogenic comparison of FA pathway-proficient and deficient ESPCs and tissues, thus ensuring that the observed phenotypes are due to the loss of the FA pathway and independent of the genetic landscape of the donor. Our protocol does not require continuous addition of RA and BMP4 for specification/selection of keratinocytes, nor sequential changes in oxygen conditions to achieve high yields of cells (Petrova et al., 2014). Finally, our system overcomes the spontaneous cell death that occurs post Day 10 using previously described protocols by adding the ROCK GTPase inhibitor, Y-27632. This additional step rescued cellular growth, maintained the cells in a basal state, and prevented their terminal differentiation (Yugawa et al., 2013). The combination of ROCK GTPase inhibitor with the use of a progenitor-targeted defined media allowed the generation of functional ESPCs within a shorter time frame yet with similar yield. Using cFA-PSC-derived ESPCs, derivative PSC-EORs, and primary human skin specimens from FA and control populations, we demonstrate that FA pathway loss is fully compatible with epidermal differentiation, in agreement with sustained homeostasis of skin and mucosa in individuals with FA.
Human epidermis comprises the outermost layer of the skin and is a stratified squamous epithelium composed predominantly of keratinocytes. These provide adherence and self-renewal capacity in the face of constant physical insults and environmental exposures. ESPCs in the basal cell layer are the only keratinocytes with proliferative capacity and these are anchored to the basement membrane and underlying dermis via hemidesmosomes. Upon exit from basal into suprabasal keratinocyte layers, ESPCs undergo cell cycle arrest and continue their upward migration to differentiate terminally prior to being shed. Intercellular junctional complexes, including desmosomes and hemidesmosomes, are key for tight adhesion and communication within the epidermis. Desmosomes also regulate morphogenesis, proliferation, and differentiation (Nekrasova and Green, 2013, Brooke et al., 2012, Broussard et al., 2015, Ishida-Yamamoto and Igawa, 2014), and harbor tumor suppressive activities (Zhou et al., 2017). Hemidesmosomes connect basal cells to the basal lamina, thus anchoring the epidermis to the underlying dermis. Mutations in structural components of desmosomes and hemidesmosomes cause inherited blistering disorders of the skin and mucosa that are further linked to SCC (Montaudie et al., 2016). Our data from isogenic FA ESPC, PSC-EOR, and FA knockdown and knockout model systems all demonstrate diminished desmosome and hemidesmosome junctional complexes, accompanied by increased proliferation, epidermal fragility, and susceptibility to induced blistering upon FA pathway loss. This surprising finding uncovers subclinical phenotypes in the FA skin that are intrinsically oncogenic. The observed desmosome defects in Fancd2 knockout mouse tongue (Figure 3F) and increased proliferation in human oral keratinocytes (Figure S3) support similarly compromised tissue integrity in the FA mucosa. Furthermore, decreased adhesion in FANCD2sh keratinocytes also supports broader relevance for the general population where somatic FA gene mutations in the epidermis may promote SCC susceptibility and early-onset and aggressive disease. This hypothesis is in agreement with the association of sporadic tumors with FA mutations (Figure S1) and our previously published studies where FA gene depletion in SCC cells diminished cancer cell adhesion and promoted invasion in vitro and in ovo (Romick-Rosendale et al., 2016).
In summary, we set out to identify a functional role for the FA pathway in the developing epidermis and have shown it to be essential for sustained keratinocyte adhesion and appropriate restraint of proliferation. Our data suggest that children with FA are born with subclinical dermatologic vulnerabilities, offering a cell-autonomous explanation for the extreme risk of SCC in children and young adults with biallelic-germline mutations in the FA pathway and the common observation of somatic mutations in FA genes in individuals with SCC in the general population. The cFA-ESPC system represents a unique personalized model wherein the embryonic transition from PSCs to ESPCs and stratified epidermis can be recapitulated in the susceptible FA host. We have identified cell junctional defects as hallmarks of FA skin (Figure 4C and 4D), together with epidermal fragility (Figure 5A). These EMT-like phenotypes, coupled with proliferative gains, represent intrinsic mechanisms for SCC susceptibility. Importantly, we found that exposure to MMC was sufficient to induce proliferation and repress desmosome foci in the normal, FA pathway-proficient epidermis (Figure S5C and S5D). This data raises the provocative hypothesis that exposure to low dose crosslinkers may diminish skin integrity in the general population akin to FA pathway loss. DSP foci restoration in epidermis with combined FANCD2 loss and MMC treatment now requires further study. We posit that the observed suppression of proliferation (Figure S5C) due to the added stress of DNA damage is responsible, supporting a model wherein excessive proliferation is upstream of diminished desmosome function.
Our system will serve as a valuable screening tool for drugs that prevent FA pathway-associated sporadic and inherited SCC at the earliest stages of life, and to identify epidermis-specific vulnerabilities relevant to SCC susceptibility in the general population. We have discovered unexpected pathologies in the structure and function of FA pathway-deficient epidermis. Relevant mechanisms that connect FA pathway loss to aberrant cell junction deregulation and proliferation can now be investigated. Furthermore, possible defects in other intercellular junctions, including gap, tight, and adherens junctions, have yet to be defined. Finally, the current cFA-PSC-EOR system was designed for exclusive focus on the keratinocyte compartment. Other FA pathway-deficient cell types, including stromal and immune cells, may be incorporated in the future to assess possible contributions of the epidermal microenvironment to disease phenotypes and cancer susceptibility in FA.
Limitations of Study
We have identified subclinical pathologies in the FA epidermis and skin, however, relevant molecular mechanisms that connect FA pathway loss to aberrant proliferation and cell junction defects remain to be elucidated. Exposure of the normal epidermis to the DNA crosslinker MMC appears to mimic FA phenotypes, but the effects of combined FA pathway loss and MMC will require further study. While our experiments focused on desmosomes and hemidesmosomes, hemidesmosome abnormalities were observed only in patient biopsies. These could not be further studied in 3D engineered epidermis as the EOR system lacks a basement membrane. Additionally, a limitation of the cFA-EOR system is that it was designed to study keratinocyte biology and function and does not incorporate other cell types such as immune and stromal cells. Finally, we describe cFA-PSC and cFA-ESPC systems derived from patients with mutations in FANCA and it is possible that other phenotypes may emerge in response to the loss of function of other FA genes. Nevertheless, adhesion defects were detectable in patient tissue from several FA complementation groups.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Susanne Wells (susanne.wells@cchmc.org).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The datasets generated during this study have been deposited in NCBI’s Gene Expression Omnibus and are available through GEO Series accession number GSE117362: (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117362)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Skin Tissue
Human skin biopsies were collected with the approval of the Cincinnati Children’s Hospital Medical Center (CCHMC) Institutional Review Board (IRB) (CCHMC IRB# 2008-0899 and CCHMC IRB# 2017-7007 previously CR1_2008-1331), in accordance with the Declaration of Helsinki principles. Their use for research was in accordance with the terms of the informed consent provided. Participants involved both males and females, ages 3 to 37 years-old for the pigment incontinence studies (Table S2) and ages 2 to 35 years-old for EM analyses (Table S3). No data was excluded. Patients undergoing clinically-indicated annual bone marrow aspirations were asked to provide both bone marrow and skin biopsies (taken from the same site as the bone marrow) for research purposes. Donors for FA skin samples included patients with a confirmed diagnosis of FA visiting the CCHMC FA Comprehensive Care Center. Donors for non-FA samples included healthy adult volunteers with no history of BMF or aplastic anemia and no history of malignancy involving the skin. Sample selection was done randomly based on the number of samples available to reduce any potential self-selection bias. All immunostained sections from human skin biopsies were reviewed and scored by a pathologist in a blinded manner without knowledge of FA or control status.
Human Skin Fragility Study
Human skin fragility studies were performed with the approval of the CCHMC IRB (CCHMC IRB# 2016-2846), in accordance with Declaration of Helsinki principles and their research use was in accordance with the terms of the informed consent provided. The study was deemed minimal risk by the IRB. Participants involved both males and females, ages 12 to 25 years old (Table 2). No data was excluded. Patients with a confirmed diagnosis of FA and healthy controls were invited to participate. Subjects were age-matched to controls +/− 5 years to allow for direct comparison between groups. Healthy controls were volunteers without an active skin infection, any skin blistering disorder, or diagnosis of FA. FA patients with an active skin infection or graft-versus-host disease of the skin were excluded from the study. All patients (or legal guardians in subjects <18 years-old) gave written informed consent and were enrolled in institutional protocols approved by CCHMC.
Human SCC Risk Factor Study
Human skin risk factor studies were performed with the approval of the CCHMC IRB (CCHMC IRB# 2010-3354), in accordance with Declaration of Helsinki principles and their research use was in accordance with the terms of the informed consent provided. Self-reported data from FA patients (N=105) between the ages of 19 and 51 years was obtained using a survey. No data was excluded. Written informed consent was obtained by trained research staff from participants after a verbal description of the study. Patients visiting the CCHMC FA Comprehensive Care Center or attending the Fanconi Anemia Research Fund sponsored annual adult and family meetings were invited to participate. Individuals of all ages were eligible to participate if they reported a diagnosis of FA and were willing to complete study-related surveys. Self-report or parent surveys were primarily completed online but paper surveys were offered upon request.
Human cFA-PSC
Human PSC lines transduced with a DOX-inducible vector (Meerbrey et al., 2011) to conditionally correct the FANCA mutation were generated by our laboratory with the support of the CCHMC Pluripotent Stem Cell Facility, as previously published (Chlon et al., 2016), and were IRB approved at CCHMC. The cFA-PSC1 line (previously published as FA-A#1 IND-iPS1) is female, while the cFA-PSC2 line (previously published as FA-A#2 IND-iPS3) is male. Both cFA-PSC lines were checked for and determined to have a normal karyotype and tested with an in vivo teratoma assay. Lines were directly differentiated into cFA-ESPCs, generated in our laboratory and validated by western blot for the expression of FANCA and activation of FANCD2. cFA-PSC and ESPC lines were not authenticated.
Human NIKS
Unmodified human NIKS were obtained from the Lambert Laboratory at University of Wisconsin (Madison, USA) and transduced with NTsh and FAND2sh vectors. The NIKS line is male, isolated from human neonatal foreskin (Park et al., 2010). The cell line was not authenticated.
Human NOKS
Unmodified human NOKS were obtained from the Münger Laboratory at Tufts University (Massachusetts, USA) and transduced with NTsh and FAND2sh vectors. The sex of the cell line has not been reported (Piboonniyom et al., 2002). The cell line was not authenticated.
Animals
All mice were housed in the animal facility at CCHMC in accordance with NIH Guidelines for the Care and Use of Laboratory Animals. Animals were maintained on a 12 hr light-dark cycle with access to water and standard chow ad libitum. Healthy mice from both genders between 4 and 6 months-old were used for the experiments. Fancd2 wild-type (WT) and mutant knockout (KO) mice were used for ultrastructural analyses of oral mucosa. The mice were not involved in any other study/or procedure. All experiments were performed under the approval of the Institutional Animal Care and Use Committee (IACUC) of CCHMC (IACUC2017-0004).
METHOD DETAILS
Experimental Design
To confirm the reproducibility of generating ESPCs and EORs from PSCs, this method has been used by 3 distinct researchers in the Wells laboratory. Sample size was chosen after taking into consideration the means of the values between the experimental group and the control group, the standard error, and the statistical analysis used. No data was excluded from analysis. For each experiment, the number of biologically independent replicates is noted in the figure legends. Gene expression analysis by RNA-Seq was performed once using two distinct cFA-ESPC lines from two different donors, and then validated by RT-qPCR with an additional independent biological cFA-ESPC sample from each line/donor. For randomization, plates of cells and EORs were randomly divided into 2 groups (either +/−DOX for ESPCs or NTsh/D2sh for NIKS and NOKS). For the DNA damage analyses, plates of NIKS NTsh/D2sh cells were further randomly divided into Vehicle and MMC-treated groups. Groups were exposed to the same environment and culture conditions and processed identically. Analyses of the in vitro studies were not blinded.
Skin Tissue Samples
Skin punch biopsies (less than 3 millimeters in diameter) were obtained from FA patients undergoing a bone marrow aspirate and from gender-matched normal controls. Biopsies were fixed in 4% paraformaldehyde (PFA), paraffin embedded, sectioned, and stained by H&E or analyzed by IF. A clinical pathologist who was blinded to FA status evaluated the tissues. Additional biopsies were fixed in 2% glutaraldehyde and processed for EM as described below. Sample selection was done randomly based on the number of samples available to reduce any potential self-selection bias. No data was excluded.
Skin Fragility Study
Patients with a confirmed diagnosis of FA (12 to 25 years of age) and age-matched (+/− 5 years) healthy controls were invited to participate in this clinical study which was deemed minimal risk. Healthy controls were volunteers without an active skin infection, any skin blistering disorder, or diagnosis of FA. FA patients with an active skin infection or graft-versus-host disease of the skin were excluded from the study. A full demographic/clinical characterization of subjects is provided in tabular format in Table 2. Variables collected included demographic (age, gender) and clinical characteristics including FA complementation group, skin health, and date/age at bone marrow transplant (if any). The time to blister formation on the skin was measured using a negative pressure instrument (Electronic Diversities), as previously reported (Tidman and Eady, 1986). Briefly, a negative pressure of 200 mmHg was applied to the subjects’ forearms. The skin was carefully monitored for suction blister formation until the first sign of skin blistering was observed. At this moment, the time to blister was recorded. No data was excluded. No skin biopsies were collected after blister formation, in accordance with the terms of the approved IRB.
SCC Risk Factor Surveys
Analysis of cutaneous SCC prevalence in FA patients was performed using self-reported data (primarily online via Research Electronic Data Capture software (Harris et al., 2009) from FA patients (N=105) between the ages of 19 and 51 years. A full demographic/clinical characterization of subjects with a history of cancer is provided in tabular format in Table 1. Variables collected included demographic (age, race, economic and living status) and clinical characteristics including FA complementation group, current or previous cancer diagnoses, skin health, and date/age at bone marrow transplant (if any). For CCHMC patients, information provided by the patient was verified using the Electronic Medical Record whenever possible. No data was excluded.
TCGA Analysis
The results depicted here are in whole or part based upon data available at the TCGA Research Network (http://www.cbioportal.org/) (Cerami et al., 2012, Gao et al., 2013) using the following published studies: AML- TCGA, NEJM 2013; HNSCC- TCGA, Nature 2015; and Cutaneous SCC- DFCI, Clinical Cancer Research 2015.
Directed Differentiation of PSCs into ESPCs
Generation of FA patient-derived PSCs with conditional expression of FANCA (cFA-PSCs) has been described previously (Chlon et al., 2016). The two PSC lines used here were previously published as FA-A#1 IND-iPS1 and FA-A#2 IND-iPS3. cFA-PSCs were maintained in mTeSR1 medium (Stem Cell Technologies) supplemented with 100 ng/mL DOX and passaged every 4 days. For directed differentiation into epidermal lineages, some elements of published differentiation technologies were incorporated into the following protocol (Itoh et al., 2011, Petrova et al., 2014, Kogut et al., 2014). The cFA-PSCs were transferred into matrigel-coated plates at low-density and cultured in mTeSR1 supplemented with 100 ng/mL DOX (Sigma-Aldrich) for 1 day. To initiate differentiation (day 0), cells were cultured for 4 days in defined keratinocyte serum-free media (DKSFM, Thermo Fisher Scientific) supplemented with 1% penicillin/streptomycin, 1 μM retinoic acid (RA, Sigma-Aldrich), 25 ng/mL bone morphogenic protein 4 (BMP4, Stemgent), and 100 ng/mL DOX. On day 4, cells were washed with phosphate buffered saline (PBS) and the medium was changed to CnT-07 (CELLnTEC) supplemented with 1% penicillin/streptomycin in the presence or absence of 100 ng/mL DOX. On day 10, the CnT-07 medium was also supplemented with 10 μM Y-27632 (Enzo Life Sciences). After 20 days, the cells were transferred to collagen IV-coated plates (Sigma-Aldrich) for expansion in CnT-07 media supplemented with 10 μM Y-27632 in the presence or absence of 100 ng/mL DOX. Media was removed and replaced at 2-day intervals.
Cell Culture
NIKS transduced with shRNA to knockdown FANCD2 (FANCD2sh) or non-targeting shRNA (NTsh) were cultured on irradiated J2-3T3 feeder cells as previously described (Hoskins et al., 2009, Schurr et al., 2012) and maintained in F-media supplemented with 1 μg/mL puromycin for continued vector selection. NOKS were carried as previously described (Piboonniyom et al., 2002). NOKS were transduced with NTsh or FANCD2sh vectors (Hoskins et al., 2009) and cultured in keratinocyte serum-free medium (Thermo Fisher Scientific) supplemented with 1 μg/mL puromycin for sustained selection of vector-containing cells. shRNA-expressing vectors were obtained through the Sigma™ MISSION® shRNA program (Sigma-Aldrich): NTsh (SHC002V) and FANCD2sh (TRCN0000082841). For DNA damage analyses, NTsh and FANCD2sh NIKS were pretreated with vehicle or a nontoxic dose of MMC (500 pg/mL) for at least 2 passages with no change in cellular morphology or growth. Cells were then engineered into 3D epidermis with continuous exposure to vehicle or MMC until harvest.
3D EOR Cultures
Human epidermal organotypic rafts were generated as previously described (Hoskins et al., 2009). Briefly, 1×106 human immortalized keratinocytes (NIKS or NOKS) were plated on a 24 mm collagen matrix harboring embedded J2-3T3 mouse fibroblasts to simulate the dermis. For PSC-EORs generated using cFA-ESPCs, 2×106 cells were plated. EORs were cultured at the liquid-air interface to generate a stratified epithelium with differentiation properties that reflect human epidermis. After 14 days, rafts were fixed in 4% PFA and embedded in paraffin. Sections were stained with H&E and examined for histopathology by routine microscopy.
Mouse models
All animal experiments were approved by the IACUC Committee at CCHMC, and the mice were housed in the CCHMC animal facility. Fancd2+/− (C57Bl/6J background) mice were provided by Dr. Markus Grompe (Oregon Health & Sciences University) (Houghtaling et al., 2003). Fancd2 WT and KO mice were obtained from breeding Fancd2+/− mice. Tongue from male or female Fancd2 WT or KO mice (4 to 6 months-old) were harvested after CO2 euthanasia followed by cervical dislocation.
Transmission Electron Microscopy
EORs and human skin biopsy tissues were fixed in 2% glutaraldehyde, 4% PFA, and 2 mM CaCl2 in 0.05 M sodium cacodylate buffer, pH 7.2 at 4°C until processing. Tissues were transferred to 0.2 M sodium cacodylate buffer, osmicated, dehydrated, infiltrated, and embedded in LX-112 resin (Ladd Research Industries). After polymerization at 60°C for 3 days, ultrathin sections were cut using a Reichert-Jung Ultracut E microtome and stained in 2% aqueous uranyl acetate and Reynolds lead citrate. Images were visualized with a transmission electron microscope (Hitachi H-6750) equipped with a digital camera (AMT BioSprint16). The number of desmosomes was counted for a minimum of 10 FA pathway-proficient and deficient cells. The number of hemidesmosomes was counted per 1 μm of basement membrane of skin biopsy using ImageJ software (Schneider et al., 2012).
Immunofluorescence
Cells were fixed on coverslips with 4% PFA, washed with PBS, permeabilized with 0.2% Triton X-100 in PBS for 3 minutes, blocked with 5% goat serum (Jackson ImmunoResearch) for 30 minutes, incubated with primary antibodies for 1 hour in a humidified chamber at 37 °C, washed with PBS, incubated with the appropriate fluorescent secondary antibodies (Thermo Fisher Scientific) for 30 minutes at 37 °C, washed in PBS, mounted in ProLong Gold antifade/DAPI (Thermo Fisher Scientific), and imaged using a confocal microscope Nikon A1 LUNA. Where indicated, 300 nM Mitomycin C (MMC, Sigma-Aldrich) was added to the medium 16 hours before fixation. For IF in 3D EORs, tissue was fixed in 4% PFA for 24 hours prior to dehydration and embedding into paraffin blocks. Embedded EORs were sectioned onto glass slides, deparaffinized, and subjected to antigen retrieval in 10 mM sodium citrate for 40 seconds, followed by standard IF protocols. For EdU staining, EORs were incubated with 10 μM EdU for 24 hours, while PSC-EORs were incubated for 48 hours, before harvest and fixation. EORs were subsequently embedded, sectioned, and the Click-it EdU Cell Proliferation Assay kit (Thermo Fisher Scientific) was used per the manufacturer’s instructions, followed by IF as described above. For DNA damage analyses, NIKS NTsh and FANCD2sh cells were stained for γH2AX. A minimum of 170 γH2AX+ cells were counted manually per condition. Sections from derivative EORs were stained for DSP 1/2 and the cell membrane marker wheat germ agglutinin (WGA). Automated quantification of DSP foci was performed using ImageJ/Fiji’s Spot Counter plugin, using user-defined box size 10 and noise tolerance 100. Table S4 lists the antibodies used. All antibodies have been validated previously.
Flow Cytometry
For cell-cycle analyses of +/−DOX ESPCs in monolayer, cells were incubated with 10 μM EdU for 45 minutes, then enzymatically collected with trypsin, neutralized with DMEM supplemented with 10% FBS, and pelleted by gentle centrifugation. The cell pellet was fixed in PFA for 15 minutes at room temperature, mixed with PBS, and pelleted by gentle centrifugation. The Click-it EdU Cell Proliferation Assay kit (Thermo Fisher Scientific) and DAPI were used for staining the single cell suspensions. Samples were acquired on a BD LSR II Instrument (BD Biosciences, San Jose, CA) and the results were analyzed using FlowJo software v10.0.7 (Tree Star). Data concerning the percent of cells in different phases of the cell cycle were reported as frequency within the live cells. Live cells were gated on FSC-H, FSC-W gate to exclude doublets.
Western Blot Analysis
Whole cell lysates from cFA-ESPCs were prepared in NETN-100 buffer (200 mM Tris-HCL, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40) containing 50 mM NaF and 200 μM Na2VO3. Whole cell lysates from NIKS and NOKS were prepared in Laemmli buffer containing 10% beta-mercaptoethanol. 30-50 μg of protein was loaded per lane on 7.5% or 10% Bis-Tris gels (BioRad), and protein was transferred to PVDF membranes at 500 mA for 3 hours or 1 hour, respectively. Membranes were blocked in 5% milk and incubated with antibodies according to the manufacturer’s protocol. Table S4 lists all antibodies that were used. All antibodies were previously validated.
RNA isolation and RT-qPCR
cFA-ESPCs were enzymatically collected with trypsin (Thermo Fisher Scientific), neutralized with DMEM (Thermo Fisher Scientific) supplemented with 10% FBS, washed with PBS, and pelleted by gentle centrifugation. RNA was extracted from cells using the RNAEasy Mini Kit and Qiashredder Columns (Qiagen), following the manufacturer’s protocol. cDNA was synthesized from 1 μg of RNA using the QuantiTect Reverse Transcription Kit (Qiagen). 40 ng of cDNA was used per RT-qPCR reaction with SYBR Green PCR Mastermix for ΔNp63 or SYBR Green PowerUp Mastermix for RNA-Seq validation (Thermo Fisher Scientific) and analyzed on the ABI7500 Real-time PCR system. Relative gene expression values were determined by using the ΔΔCT method with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the housekeeping gene. Table S5 lists all primers that were used.
RNA-Sequencing
cFA-ESPCs were washed three times with 1X PBS, scraped in 1X PBS, and collected. Total RNA was extracted from cFA-ESPCs +/−DOX in culture using the High Pure RNA Isolation kit (Roche) according to the manufacturer’s protocol. Isolated RNA was quantified (Qubit RNA assay kit) and quality controlled for degradation. RNA-Seq was conducted by the CCHMC DNA sequencing and Genotyping Core. Briefly, RNAseq libraries were prepared with the Illumina TruSeq RNA preparation kit. The libraries were sequenced on an Illumina Hi-Seq 2500 sequencer to obtain 20 million of 75 bp paired-end stranded reads per sample. Quantification of mRNA expression level was determined based on the ToppHat/Cufflinks pipeline. Reads were aligned to the human hg19/GRCh37 reference genome using ToppHat. The BAM files containing the aligned reads were used to quantify mRNA expression level using Cufflinks with the UCSC known gene reference annotation. RNA expression values were normalized by the fragments per kilobase per megabase calculation (FPKM). Gene ontology enrichment analysis was performed using Toppgene (https://toppgene.cchmc.org/) (Chen et al., 2009), using the ToppFun function with the Biological Process database. Settings used were Correction = FDR, p-Value cutoff = 0.05. Gene Limits = 5 ≤ n ≤ 300.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism. Student’s t-test analysis with two-tailed distribution or one-way ANOVA was used to determine significance, which was set at p≤0.05. All data are represented as mean ± standard error of the mean (SEM). For each experiment, the number of biologically independent samples and p-values are noted in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Krt14 | BioLegend | Cat#905301; RRID:AB_2565048 |
| Mouse anti-phospho-Histone H2A.X Ser139 | Millipore | Cat#05-636; RRID:AB_309864 |
| Mouse anti-cytokeratin 10 | Abcam | Cat#ab9025; RRID:AB_2134556 |
| Rabbit anti-FANCD2 | Novus | Cat#NB100-182; RRID:AB_10002867 |
| Mouse anti-Vinculin | Sigma-Aldrich | Cat#V9131; RRID:AB_477629 |
| Rabbit anti-FANCA | Cascade BioScience | Cat#ABP-6201; RRID: AB_2810978 |
| Mouse anti-Desmoplakin 1/2, clone DP447, supernatant | Progen Biotechnik | Cat#651155; RRID: AB_2861410 |
| Wheat Germ Agglutinin, tetramethylrhodamine conjugate | Thermo Fisher Scientific | Cat# W849; |
| Donkey anti-rabbit IgG HRP-conjugated | GE Healthcare | Cat#NA934; RRID:AB_772206 |
| Sheep anti-mouse IgG HRP-conjugated | GE Healthcare | Cat#NA931; RRID:AB_772210 |
| AlexaFluor Goat anti-rabbit 488 | Thermo Fisher Scientific | Cat#A-11008; RRID: AB_143165 |
| AlexaFluor Goat anti-mouse 568 | Thermo Fisher Scientific | Cat#A-11004; RRID: AB_2534072 |
| Biological Samples | ||
| Human Skin Tissue Samples | CCHMC | Tables S2 and S3 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline | Sigma-Aldrich | Cat#D9891 |
| Puromycin | Sigma-Aldrich | Cat#P8833-10MG |
| hESC-qualified Matrigel | BD Biosciences | Cat#354277 |
| mTeSR1 media | Stem Cell Technologies | Cat#5850 |
| Dispase | Thermo Fisher Scientific | Cat#17105-041 |
| DMEM/F-12, HEPES | Thermo Fisher Scientific | Cat#11330-302 |
| Y-27632 Dihydrochloride | Enzo Life Sciences | Cat#ALX-270-333-M005 |
| Human recombinant BMP4 | StemGent | Cat#03-0007 |
| PCT Epidermal Keratinocyte Medium, Defined | ZenBio (CELLnTEC) | Cat#CnT-07 |
| 1x Defined Keratinocyte-SFM media | Thermo Fisher Scientific | Cat#10744-019 |
| Retinoic acid | Sigma-Aldrich | Cat#R2625 |
| Collagen Type IV | Sigma-Aldrich | Cat#C5533-5MG |
| Keratinocyte SFM media | Thermo Fisher Scientific | Cat#17005042 |
| Fetal bovine serum | GE Healthcare | Cat#SH30396.03 |
| DMEM | Thermo Fisher Scientific | Cat#11965-092 |
| F12 Media | Thermo Fisher Scientific | Cat#11765-054 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat#15140-122 |
| Human recombinant insulin | Thermo Fisher Scientific | Cat#12585-014 |
| Adenine | Sigma-Aldrich | Cat#A2786-5G |
| Cholera toxin | Millipore | Cat#227036 |
| Hydrocortisone | Sigma-Aldrich | Cat#H0888-1G |
| Fungizone (Amphotericin B) | Omega Scientific | Cat#FG-70 |
| EGF | Sigma-Aldrich | Cat#E4127-.1MG |
| Collagen Type I, rat tail | EMD Millipore | Cat#08-115 |
| Ham’s F12 Nutrient Mix, powder | Thermo Fisher Scientific | Cat#21700075 |
| 1,2-dioctanoyl-sn-glycerol | Cayman Chemicals | Cat#62225 |
| 0.25% Trypsin-EDTA | Thermo Fisher Scientific | Cat#25200-056 |
| 0.05% Trypsin-EDTA | Thermo Fisher Scientific | Cat#25300-054 |
| Mitomycin C | Sigma-Aldrich | Cat#M4287-2MG |
| Triton X-100 | Fisher Scientific | Cat#BP151-500 |
| Tween 20 | Fisher Scientific | Cat#BP337-500 |
| Glacial acetic acid | Fisher Scientific | Cat#A38-500 |
| ProLong Gold antifade Mountant with DAPI | Thermo Fisher Scientific | Cat#P36931 |
| Normal goat serum | Jackson ImmunoResearch | Cat#005-000-121 |
| Pierce™ ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat#32106 |
| SuperSignal Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat#34095 |
| Western Lightening Plus ECL | Perkin Elmer | Cat#NEL103001EA |
| Critical Commercial Assays | ||
| Click-It Edu Alexa Fluor 647 Imaging Kit | Thermo Fisher Scientific | Cat#C10340 |
| Click-It Edu Alexa Fluor 488 Flow Cytometry Kit | Thermo Fisher Scientific | Cat#C10425 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23227 |
| RNeasy Mini kit | Qiagen | Cat#74104 |
| QuantiTect reverse transcription kit | Qiagen | Cat#205313 |
| SYBR Green PCR master mix | Thermo Fisher Scientific | Cat#4309155 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | Cat#A25777 |
| High Pure RNA Isolation Kit | Roche | Cat#11828665001 |
| Deposited Data | ||
| Raw and analyzed RNA-Seq data | This paper | GSE117362 |
| Experimental Models: Cell Lines | ||
| Human: cFA-PSCs | CCHMC Pluripotent Stem Cell Facility | Chlon et al. 2016 |
| Human: cFA-ESPCs | This paper | N/A |
| Human: NIKS | Lambert Lab | Park et al., 2010 |
| Human: NOKS | Münger Lab | Piboonniyom et al., 2002 |
| Oligonucleotides | ||
| See Table S5 for RT-qPCR primers | IDT DNA | N/A (synthesized) |
| Recombinant DNA | ||
| pINDUCER20 | Meerbrey et al., 2011 | Addgene plasmid 44012 |
| Mission pLKO.1-puro shRNA Control | Sigma-Aldrich | SHC002V |
| Mission pLKO.1-puro shRNA FANCD2 | Sigma-Aldrich | TRCN0000082841 |
| Software and Algorithms | ||
| AMT BioSprint16 Image Capture Engine V700 | Advanced Microscopy Techniques Corp. | N/A |
| NIS Elements AR, version 4.40 | Nikon | N/A |
| StepOne Software v2.3 | Applied Biosystems | https://www.thermofisher.com/us/en/home/technical-resources/software-downloads/StepOne-and-StepOnePlus-Real-Time-PCR-System.html |
| GraphPad Prism, version 5.01 | GraphPad | https://www.graphpad.com/ |
| FlowJo software v10.0.7 | FlowJo | https://www.flowjo.com/ |
| ImageJ software | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| ToppGene Suite | Chen et al., 2009 | https://toppgene.cchmc.org/ |
| Research Electronic Data Capture software | Harris et al., 2009 | https://projectredcap.org/software/ |
Highlights.
Differentiation of conditional patient-derived PSCs models FA and normal epidermis
FA pathway loss promotes proliferative gains, desmosome and hemidesmosome defects
Epidermal vulnerabilities translate into accelerated blistering in patients with FA
A functional FA pathway sustains the structure and function of human skin
ACKNOWLEDGMENTS
We thank all members of the Wells laboratory for reagents and discussions, and Dr. James Wells, Dr. Christopher Mayhew, and Amy Pitstick from the Pluripotent Stem Cell Core for reagents and technical assistance. We thank Dr. Matthew Kofron for hands-on support in the Confocal Imaging Core Facility and Georgianne Ciraolo for support by the Pathology Core Facility in the use of EM. This work was supported by the Environmental Carcinogenesis and Mutagenesis Training Program T32ES007250-26 (PI: Stambrook), NCI R01 CA223790, NCI R01 CA228113, and a grant from the Fanconi Anemia Research Fund (PI: Wells), NHLBI R01HL108102 (PI: Butsch Kovacic), Center for Clinical and Translational Science and Training Grant 1UL1TR001425-01, and NIAMS F31AR070008 (PI: Ruiz-Torres).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS: The authors declare no competing interests.
REFERENCES
- AUERBACH AD 2009. Fanconi anemia and its diagnosis. Mutat Res, 668, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAGBY G 2018. Recent advances in understanding hematopoiesis in Fanconi Anemia. F1000Res, 7, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAKKER ST, DE WINTER JP & TE RIELE H 2013. Learning from a paradox: recent insights into Fanconi anaemia through studying mouse models. Dis Model Mech, 6, 40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELOKHVOSTOVA D, BERZANSKYTE I, CUJBA AM, JOWETT G, MARSHALL L, PRUELLER J & WATT FM 2018. Homeostasis, regeneration and tumour formation in the mammalian epidermis. Int J Dev Biol, 62, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOGLIOLO M & SURRALLES J 2015. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev, 33, 32–40. [DOI] [PubMed] [Google Scholar]
- BROOKE MA, NITOIU D & KELSELL DP 2012. Cell-cell connectivity: desmosomes and disease. J Pathol, 226, 158–71. [DOI] [PubMed] [Google Scholar]
- BROUSSARD JA, GETSIOS S & GREEN KJ 2015. Desmosome regulation and signaling in disease. Cell Tissue Res, 360, 501–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CECCALDI R, SARANGI P & D’ANDREA AD 2016. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol, 17, 337–49. [DOI] [PubMed] [Google Scholar]
- CERAMI E, GAO J, DOGRUSOZ U, GROSS BE, SUMER SO, AKSOY BA, JACOBSEN A, BYRNE CJ, HEUER ML, LARSSON E, ANTIPIN Y, REVA B, GOLDBERG AP, SANDER C & SCHULTZ N 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov, 2, 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN J, BARDES EE, ARONOW BJ & JEGGA AG 2009. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res, 37, W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHLON TM, RUIZ-TORRES S, MAAG L, MAYHEW CN, WIKENHEISER-BROKAMP KA, DAVIES SM, MEHTA P, MYERS KC, WELLS JM & WELLS SI 2016. Overcoming Pluripotent Stem Cell Dependence on the Repair of Endogenous DNA Damage. Stem Cell Reports, 6, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS EC & CALLENDER VD 2010. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol, 3, 20–31. [PMC free article] [PubMed] [Google Scholar]
- FUND FAR 2014. Fanconi Anemia: Guidelines for Diagnosis and Management, Eugene, OR. [Google Scholar]
- GAO J, AKSOY BA, DOGRUSOZ U, DRESDNER G, GROSS B, SUMER SO, SUN Y, JACOBSEN A, SINHA R, LARSSON E, CERAMI E, SANDER C & SCHULTZ N 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal, 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GETSIOS S, HUEN AC & GREEN KJ 2004. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol, 5, 271–81. [DOI] [PubMed] [Google Scholar]
- HARRIS PA, TAYLOR R, THIELKE R, PAYNE J, GONZALEZ N & CONDE JG 2009. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform, 42, 377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOSKINS EE, MORRIS TA, HIGGINBOTHAM JM, SPARDY N, CHA E, KELLY P, WILLIAMS DA, WIKENHEISER-BROKAMP KA, DUENSING S & WELLS SI 2009. Fanconi anemia deficiency stimulates HPV-associated hyperplastic growth in organotypic epithelial raft culture. Oncogene, 28, 674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOUGHTALING S, TIMMERS C, NOLL M, FINEGOLD MJ, JONES SN, MEYN MS & GROMPE M 2003. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev, 17, 2021–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISHIDA-YAMAMOTO A & IGAWA S 2014. Genetic skin diseases related to desmosomes and corneodesmosomes. J Dermatol Sci, 74, 99–105. [DOI] [PubMed] [Google Scholar]
- ITOH M, KIURU M, CAIRO MS & CHRISTIANO AM 2011. Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci U S A, 108, 8797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOGUT I, ROOP DR & BILOUSOVA G 2014. Differentiation of human induced pluripotent stem cells into a keratinocyte lineage. Methods Mol Biol, 1195, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOTTEMANN MC & SMOGORZEWSKA A 2013. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature, 493, 356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUTLER DI, AUERBACH AD, SATAGOPAN J, GIAMPIETRO PF, BATISH SD, HUVOS AG, GOBERDHAN A, SHAH JP & SINGH B 2003a. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg, 129, 106–12. [DOI] [PubMed] [Google Scholar]
- KUTLER DI, PATEL KR, AUERBACH AD, KENNEDY J, LACH FP, SANBORN E, COHEN MA, KUHEL WI & SMOGORZEWSKA A 2016. Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up. Laryngoscope, 126, 870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUTLER DI, SINGH B, SATAGOPAN J, BATISH SD, BERWICK M, GIAMPIETRO PF, HANENBERG H & AUERBACH AD 2003b. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood, 101, 1249–56. [DOI] [PubMed] [Google Scholar]
- LI J, SEJAS DP, ZHANG X, QIU Y, NATTAMAI KJ, RANI R, RATHBUN KR, GEIGER H, WILLIAMS DA, BAGBY GC & PANG Q 2007. TNF-alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J Clin Invest, 117, 3283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOPEZ-PAJARES V, YAN K, ZARNEGAR BJ, JAMESON KL & KHAVARI PA 2013. Genetic pathways in disorders of epidermal differentiation. Trends Genet, 29, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEERBREY KL, HU G, KESSLER JD, ROARTY K, LI MZ, FANG JE, HERSCHKOWITZ JI, BURROWS AE, CICCIA A, SUN T, SCHMITT EM, BERNARDI RJ, FU X, BLAND CS, COOPER TA, SCHIFF R, ROSEN JM, WESTBROOK TF & ELLEDGE SJ 2011. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A, 108, 3665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONTAUDIE H, CHIAVERINI C, SBIDIAN E, CHARLESWORTH A & LACOUR JP 2016. Inherited epidermolysis bullosa and squamous cell carcinoma: a systematic review of 117 cases. Orphanet J Rare Dis, 11, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLER LU, MILSOM MD, HARRIS CE, VYAS R, BRUMME KM, PARMAR K, MOREAU LA, SCHAMBACH A, PARK IH, LONDON WB, STRAIT K, SCHLAEGER T, DEVINE AL, GRASSMAN E, D’ANDREA A, DALEY GQ & WILLIAMS DA 2012. Overcoming reprogramming resistance of Fanconi anemia cells. Blood, 119, 5449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLER LU & WILLIAMS DA 2009. Finding the needle in the hay stack: hematopoietic stem cells in Fanconi anemia. Mutat Res, 668, 141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NALEPA G & CLAPP DW 2018. Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer, 18, 168–185. [DOI] [PubMed] [Google Scholar]
- NEKRASOVA O & GREEN KJ 2013. Desmosome assembly and dynamics. Trends Cell Biol, 23, 537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PACE P, MOSEDALE G, HODSKINSON MR, ROSADO IV, SIVASUBRAMANIAM M & PATEL KJ 2010. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science, 329, 219–23. [DOI] [PubMed] [Google Scholar]
- PARK E, KIM H, KIM JM, PRIMACK B, VIDAL-CARDENAS S, XU Y, PRICE BD, MILLS AA & D’ANDREA AD 2013. FANCD2 activates transcription of TAp63 and suppresses tumorigenesis. Mol Cell, 50, 908–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARK JW, PITOT HC, STRATI K, SPARDY N, DUENSING S, GROMPE M & LAMBERT PF 2010. Deficiencies in the Fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer. Cancer Res, 70, 9959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETROVA A, CELLI A, JACQUET L, DAFOU D, CRUMRINE D, HUPE M, ARNO M, HOBBS C, CVORO A, KARAGIANNIS P, DEVITO L, SUN R, ADAME LC, VAUGHAN R, MCGRATH JA, MAURO TM & ILIC D 2014. 3D In vitro model of a functional epidermal permeability barrier from human embryonic stem cells and induced pluripotent stem cells. Stem Cell Reports, 2, 675–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIBOONNIYOM SO, TIMMERMANN S, HINDS P & MUNGER K 2002. Aberrations in the MTS1 tumor suppressor locus in oral squamous cell carcinoma lines preferentially affect the INK4A gene and result in increased cdk6 activity. Oral Oncol, 38, 179–86. [DOI] [PubMed] [Google Scholar]
- RAYA A, RODRIGUEZ-PIZA I, GUENECHEA G, VASSENA R, NAVARRO S, BARRERO MJ, CONSIGLIO A, CASTELLA M, RIO P, SLEEP E, GONZALEZ F, TISCORNIA G, GARRETA E, AASEN T, VEIGA A, VERMA IM, SURRALLES J, BUEREN J & IZPISUA BELMONTE JC 2009. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature, 460, 53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODRIGUEZ A & D’ANDREA A 2017. Fanconi anemia pathway. Curr Biol, 27, R986–R988. [DOI] [PubMed] [Google Scholar]
- ROMICK-ROSENDALE LE, HOSKINS EE, PRIVETTE VINNEDGE LM, FOGLESONG GD, BRUSADELLI MG, POTTER SS, KOMUROV K, BRUGMANN SA, LAMBERT PF, KIMPLE RJ, VIRTS EL, HANENBERG H, GILLISON ML & WELLS SI 2016. Defects in the Fanconi Anemia Pathway in Head and Neck Cancer Cells Stimulate Tumor Cell Invasion through DNA-PK and Rac1 Signaling. Clin Cancer Res, 22, 2062–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENBERG PS, ALTER BP & EBELL W 2008. Cancer risks in Fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica, 93, 511–7. [DOI] [PubMed] [Google Scholar]
- ROSENBERG PS, SOCIE G, ALTER BP & GLUCKMAN E 2005. Risk of head and neck squamous cell cancer and death in patients with Fanconi anemia who did and did not receive transplants. Blood, 105, 67–73. [DOI] [PubMed] [Google Scholar]
- SCHNEIDER CA, RASBAND WS & ELICEIRI KW 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods, 9, 671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHURR MJ, FOSTER KN, LOKUTA MA, RASMUSSEN CA, THOMAS-VIRNIG CL, FAUCHER LD, CARUSO DM & ALLEN-HOFFMANN BL 2012. Clinical Evaluation of NIKS-Based Bioengineered Skin Substitute Tissue in Complex Skin Defects: Phase I/IIa Clinical Trial Results. Adv Wound Care (New Rochelle), 1, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STONE MP, CHO YJ, HUANG H, KIM HY, KOZEKOV ID, KOZEKOVA A, WANG H, MINKO IG, LLOYD RS, HARRIS TM & RIZZO CJ 2008. Interstrand DNA cross-links induced by alpha,beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc Chem Res, 41, 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUMPTER R JR. & LEVINE B 2017. Emerging functions of the Fanconi anemia pathway at a glance. J Cell Sci, 130, 2657–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUZUKI NM, NIWA A, YABE M, HIRA A, OKADA C, AMANO N, WATANABE A, WATANABE K, HEIKE T, TAKATA M, NAKAHATA T. & SAITO MK. 2015. Pluripotent cell models of fanconi anemia identify the early pathological defect in human hemoangiogenic progenitors. Stem Cells Transl Med, 4, 333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIDMAN MJ & EADY RA 1986. Structural and functional properties of the dermoepidermal junction in obligate heterozygotes for recessive forms of epidermolysis bullosa. Arch Dermatol, 122, 278–81. [PubMed] [Google Scholar]
- WALKO G, CASTANON MJ & WICHE G 2015. Molecular architecture and function of the hemidesmosome. Cell Tissue Res, 360, 363–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAN H, SOUTH AP & HART IR 2007. Increased keratinocyte proliferation initiated through downregulation of desmoplakin by RNA interference. Exp Cell Res, 313, 2336–44. [DOI] [PubMed] [Google Scholar]
- YUGAWA T, NISHINO K, OHNO S, NAKAHARA T, FUJITA M, GOSHIMA N, UMEZAWA A & KIYONO T 2013. Noncanonical NOTCH signaling limits self-renewal of human epithelial and induced pluripotent stem cells through ROCK activation. Mol Cell Biol, 33, 4434–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YUNG SK, TILGNER K, LEDRAN MH, HABIBOLLAH S, NEGANOVA I, SINGHAPOL C, SARETZKI G, STOJKOVIC M, ARMSTRONG L, PRZYBORSKI S & LAKO M 2013. Brief report: human pluripotent stem cell models of fanconi anemia deficiency reveal an important role for fanconi anemia proteins in cellular reprogramming and survival of hematopoietic progenitors. Stem Cells, 31, 1022–9. [DOI] [PubMed] [Google Scholar]
- ZHANG H, KOZONO DE, O’CONNOR KW, VIDAL-CARDENAS S, ROUSSEAU A, HAMILTON A, MOREAU L, GAUDIANO EF, GREENBERGER J, BAGBY G, SOULIER J, GROMPE M, PARMAR K & D’ANDREA AD 2016. TGF-beta Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell Stem Cell, 18, 668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU G, YANG L, GRAY A, SRIVASTAVA AK, LI C., ZHANG G & CUI T. 2017. The role of desmosomes in carcinogenesis. Onco Targets Ther, 10, 4059–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study have been deposited in NCBI’s Gene Expression Omnibus and are available through GEO Series accession number GSE117362: (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117362)