

SUMMARY
R-2-hydroxyglutarate (R-2HG), a metabolite produced by mutant isocitrate dehydrogenases (IDH), was recently reported to exhibit anti-tumor activity. However, its effect on cancer metabolism remains largely elusive. Here, unlike its reported inhibitory effect on ATP synthase in glioma, we show R-2HG mainly attenuates aerobic glycolysis, a hallmark of cancer metabolism, in (R-2HG-sensitive) leukemia cells. Mechanistically, R-2HG abrogates FTO/m6A/YTHDF2-mediated post-transcriptional upregulation of PFKP and LDHB (two critical glycolytic genes) expression, and thereby suppresses aerobic glycolysis. Knockdown of FTO, PFKP, or LDHB recapitulates R-2HG-induced glycolytic inhibition in (R-2HG-sensitive) leukemia cells but not normal CD34+ hematopoietic stem/progenitor cells, and inhibits leukemogenesis in vivo, while their overexpression reverses R-2HG-induced effects. R-2HG also suppresses glycolysis and downregulates FTO/PFKP/LDHB expression in human primary IDH-wildtype AML cells, demonstrating the clinical relevance. Collectively, our study reveals previously unrecognized effects of R-2HG and RNA modification on aerobic glycolysis in leukemia, highlighting the therapeutic potential of targeting cancer epitranscriptomics and metabolism.
Graphical Abstract
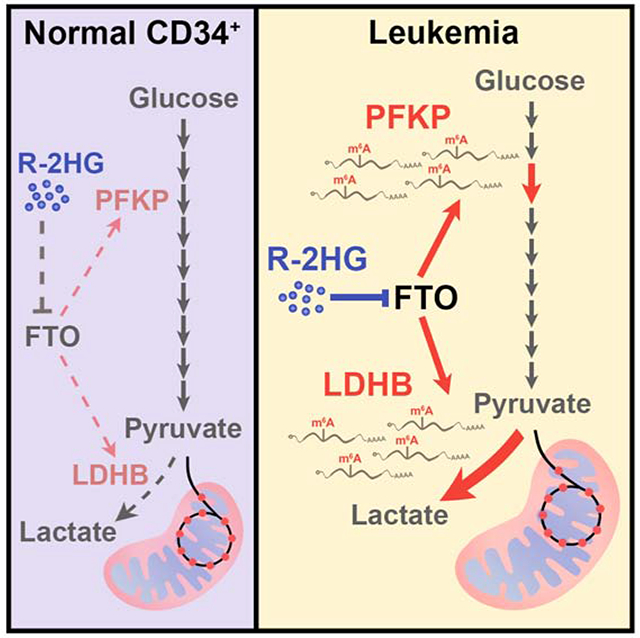
In Brief (eTOC blurb)
Qing et al. demonstrate that R-2HG, a metabolite produced by mutant IDH, significantly suppresses aerobic glycolysis in sensitive (IDH-wildtype) leukemia cells but not in normal hematopoietic stem/progenitor cells. R-2HG exerts glycolytic inhibitory effects by targeting the FTO/m6A/YTHDF2 signaling to downregulate PFKP and LDHB expression, contributing to its overall anti-tumor activity.
INTRODUCTION
An emerging hallmark of cancer is reprogramming of cellular metabolism (Hanahan and Weinberg, 2000, 2011; Hsu and Sabatini, 2008; Pavlova and Thompson, 2016). In normal healthy cells, glucose is broken down to pyruvate via glycolysis, and pyruvate will be further converted into acetyl-CoA to fuel the tricarboxylic acid (TCA) cycle (or Krebs cycle), which can generate NADH and FADH2 for the electron transport chain to effectively generate energy. Conversely, tumor cells rely substantially on the relatively inefficient glycolysis to meet energy demand even in normoxic conditions, a phenomenon termed as ‘aerobic glycolysis’ or ‘Warburg effect’ (Phan et al., 2014; Warburg, 1956). Thus, targeting glycolysis represents an attractive and promising approach for clinical cancer therapy.
Isocitrate dehydrogenases (IDHs), critical enzymes in the TCA cycle, reversibly catalyze the conversion of isocitrate to α-ketoglutarate (α-KG). IDH1/2 is mutated in ~80% of lower-grade glioma and 10-20% of acute myeloid leukemia (AML) patients (Cancer Genome Atlas Research et al., 2015; Medeiros et al., 2017; Papaemmanuil et al., 2016; Yan et al., 2009). IDH mutations occur at the conserved arginine residues that are important for enzymatic activity: IDH1R132, IDH2R140, and IDH2R172. Mutant IDH1/2 lose the activity of the wild-type form and gain the neomorphic function that converts α-KG to the R enantiomer of 2-hydroxyglutarate (R-2HG) (Dang et al., 2009; Ward et al., 2010; Xu et al., 2011).
R-2HG was previously considered an oncometabolite, leading to the clinical application of IDH mutant inhibitors (DiNardo et al., 2018; Rohle et al., 2013; Stein et al., 2019; Wang et al., 2013). However, paradoxically, IDH-mutant glioma and glioblastoma (GBM) patients had substantially longer overall survival compared to IDH-wildtype patients, and several clinical studies showed a similar trend for AML patients (Cancer Genome Atlas Research et al., 2015; Green et al., 2011; Patel et al., 2012). In addition, only 40% or less of IDH-mutant AML patients responded to IDH inhibitor therapy (DiNardo et al., 2018; Stein et al., 2017). In accordance with this, our recent work provides compelling evidence that R-2HG exhibits an intrinsic anti-tumor activity in the vast majority of IDH-wildtype leukemia and GBM cell lines by targeting the fat mass and obesity-associated protein (FTO) (Su et al., 2018). As the first discovered RNA demethylase of N6-methyladenosine (m6A) (Jia et al., 2011), the most prevalent internal modification in eukaryotic mRNAs (Deng et al., 2018), FTO plays an essential oncogenic role in AML (Li et al., 2017). Interestingly, as a metabolite itself, R-2HG has been reported to alter cellular metabolism in GBM cells by inhibiting ATP synthase of the mitochondrial electron transport chain, resulting in its growth suppressive effects and rendering the cells vulnerable to glucose starvation (Fu et al., 2015). Thus, R-2HG mainly targets oxidative phosphorylation (OXPHOS) in GBM cells (Fu et al., 2015). However, little is known about whether and how R-2HG modifies the cellular metabolism in leukemia cells.
In the present study, we observed that R-2HG primarily suppresses glycolysis in leukemia cells and thereby rewires leukemic metabolism. R-2HG induces the glycolytic inhibition by downregulating the expression of two glycolytic enzymes, phosphofructokinase platelet (PFKP) and lactate dehydrogenase B (LDHB), in an FTO/m6A-dependent manner, and such metabolic regulation contributes to the anti-tumor activity of R-2HG.
RESULTS
Cellular metabolism is suppressed by R-2HG in sensitive leukemia cells
Among the 27 human leukemia cell lines we previously tested, NOMO-1 and U937 cells show highest sensitivity whereas NB4 and K562 cells display lowest sensitivity to cell-permeable R-2HG treatment (Su et al., 2018), which was confirmed herein (Figures S1A–S1E). To comprehensively assess the effects of R-2HG on cellular metabolism in leukemia cells, we performed metabolomic analysis in NOMO-1 (R-2HG-sensitive) and NB4 (R-2HG-resistant) cells after PBS or R-2HG treatment, aiming to detect R-2HG-regulated metabolic pathways (Figure 1A; Tables S1 and S2). Principal component analysis (PCA) of all metabolites demonstrated a sharp difference between PBS- and R-2HG-treated NOMO-1 cells, but not between PBS- and R-2HG-treated NB4 cells (Figure 1B). We next identified 51 metabolites that were significantly changed upon R-2HG treatment in NOMO-1 cells, without significant changes in NB4 cells. Most of the 51 metabolites exhibited decreased levels in NOMO-1 cells upon R-2HG treatment (Figure 1C), indicating that R-2HG exhibits an overall inhibitory effect on cellular metabolism in sensitive leukemia cells. In contrast, the 51 metabolites showed minimal changes in resistant cells (i.e., NB4) upon R-2HG treatment (Figure 1D). Notably, the metabolomic analysis was performed at 48 hours post R-2HG treatment, at which time the cell viability of sensitive cells was not significantly impaired yet, suggesting the metabolic changes in the R-2HG-treated cells were unlikely consequences of general cell death (Figure S1F).
Figure 1. R-2HG differentially regulates cellular metabolism in sensitive and resistant leukemia cells.
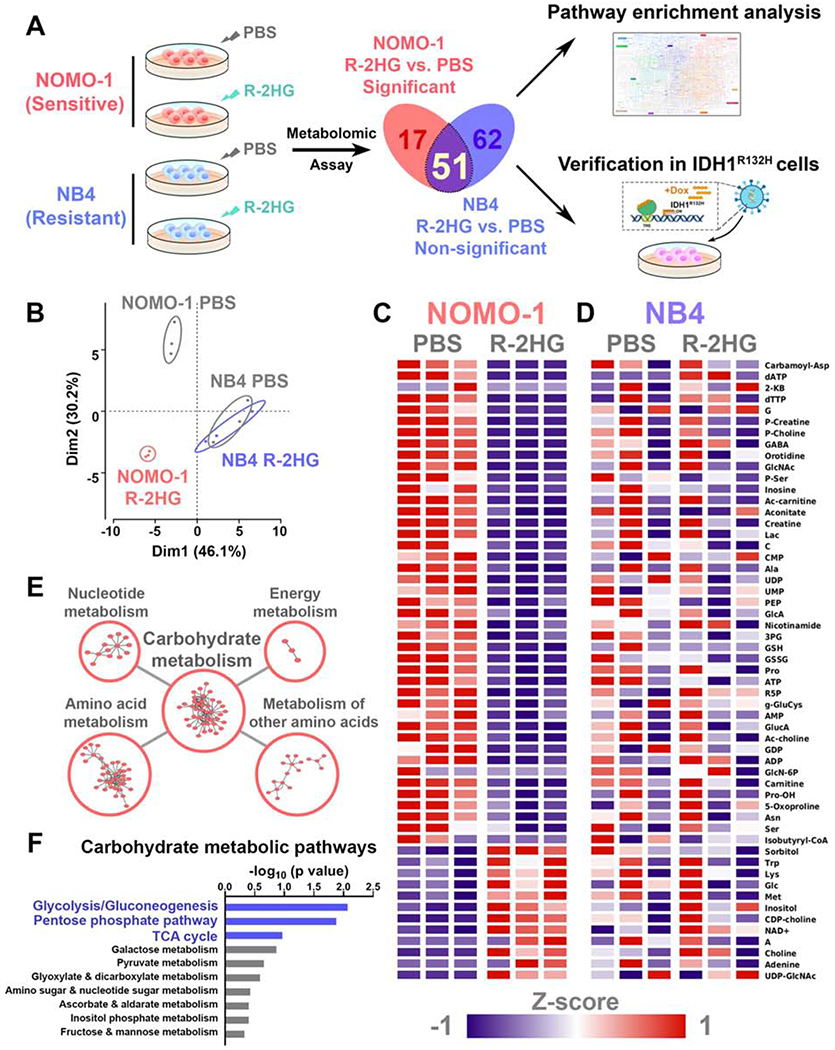
(A) Flow chart for the identification and verification strategy of differentially R-2HG-regulated metabolic pathways between NOMO-1 and NB4 cells. Graphical element is adapted from KEGG pathway map (Kanehisa, 2019).
(B) PCA plots of all the detected metabolites in PBS- or R-2HG-treated NOMO-1 and NB4 cell samples.
(C and D) Heatmaps showing relative intracellular level changes of the 51 overlapping metabolites in PBS- or R-2HG-treated NOMO-1 (C) and NB4 (D) cells. Data are represented as Z-score normalized intracellular levels.
(E) Top 5 enriched KEGG metabolic pathway classes in the 51 overlapping metabolites. The p values for the 5 classes are: Nucleotide metabolism, p = 2.69×10−7; Amino acid metabolism, p = 0.00414; Metabolism of other amino acids, p = 0.00652; Carbohydrate metabolism, p = 0.0126; Energy metabolism, p = 0.0836.
(F) Top 10 enriched KEGG carbohydrate metabolic pathways in the 51 overlapping metabolites. Pathways are arranged by their −log10 (p value).
We then conducted pathway enrichment analysis using the 51 metabolites. First, enrichment analysis for all KEGG metabolic pathway classes identified 5 top enriched classes (Figure 1E). Among them, carbohydrate metabolism is significantly enriched. Given that carbohydrate metabolism plays a key role in cancer cell metabolism and comprises multiple specific metabolic pathways with crucial functions (Hay, 2016; Vander Heiden and DeBerardinis, 2017), we conducted a further enrichment analysis for all KEGG carbohydrate metabolic pathways with the 51 metabolites (Figure 1F). The top 3 enriched carbohydrate metabolic pathways are glycolysis/gluconeogenesis, pentose phosphate pathway (PPP), and TCA cycle, all of which were suppressed in NOMO-1 cells upon R-2HG treatment (Figures 1C and 1F).
To verify the above discoveries, we utilized a more physiologically relevant model established previously where leukemia cell lines were transduced with doxycycline-inducible IDH1R132H to produce endogenous R-2HG (Su et al., 2018). As expected, endogenous R-2HG produced by ectopic IDH1R132H exerts a similar effect to exogenous R-2HG on cell proliferation/growth in leukemia cells (Figures S1C–S1E). The expression of IDH1R132H was confirmed by Western blot (Figure S1G), and the increase in intracellular R-2HG levels in IDH1R132H expressing cells was comparable to or even greater than that in cells treated with exogenous R-2HG (Figure S1H). By comparing metabolites downregulated in exogenous-R-2HG-treated NOMO-1 cells with those downregulated in NOMO-1 cells upon endogenous R-2HG production, we found that the majority are shared by the two models, with 48 overlapping metabolites identified (Figures S1I and S1J; Tables S1 and S3). Pathway enrichment analysis of the 48 metabolites showed that carbohydrate metabolism is highly enriched, while glycolysis/gluconeogenesis is among the top 3 enriched carbohydrate metabolic pathways (Figures S1K and S1L). Collectively, our data suggest that both exogenous and endogenous R-2HG exert a similar and broad inhibitory effect on cellular metabolism in sensitive leukemia cells.
Glycolysis is the major R-2HG-regulated metabolic pathway in sensitive leukemia cells
The top 3 enriched carbohydrate metabolic pathways (i.e., glycolysis, TCA cycle, and PPP) are orchestrated and connected tightly in cell metabolism. To further test the effect of R-2HG on the energy producing glycolysis and TCA cycle, we measured glycolytic rates and mitochondrial respiration (OXPHOS) in the sensitive and resistant leukemia cells. In sensitive cell lines (NOMO-1 and U937) we tested, R-2HG treatment reduced both basal and compensatory glycolytic rates in a dose-dependent manner at an early time point (i.e. 24 hours post R-2HG treatment; Figures 2A and S2A). Intriguingly, although R-2HG has been reported previously to inhibit ATP synthase and thus suppress mitochondrial respiration in GBM cells (Fu et al., 2015), mitochondrial respiration remained unaffected by R-2HG in the two sensitive leukemia cell lines (Figures 2B and S2B). To further corroborate the effect of R-2HG on glycolysis in leukemia cells, we detected glycolytic rates by a second method, radioactive glycolysis assay, as well as determining lactate levels and ATP levels. Consistently, R-2HG treatment significantly decreased glycolytic rates and reduced lactate and ATP levels in both NOMO-1 and U937 cells (Figures 2C–2E and Figures S2C–S2E), indicating a close correlation between glycolytic rates and ATP levels as well as the presence of aerobic glycolysis in sensitive cells. In contrast to sensitive leukemia cells, neither NB4 nor K562 cells displayed significantly reduced glycolytic rates, OXPHOS, or generation of lactate or ATP after R-2HG treatment (Figures 2F–2J and Figures S2F–S2J).
Figure 2. R-2HG suppresses glycolysis in sensitive, but not resistant leukemia cells.
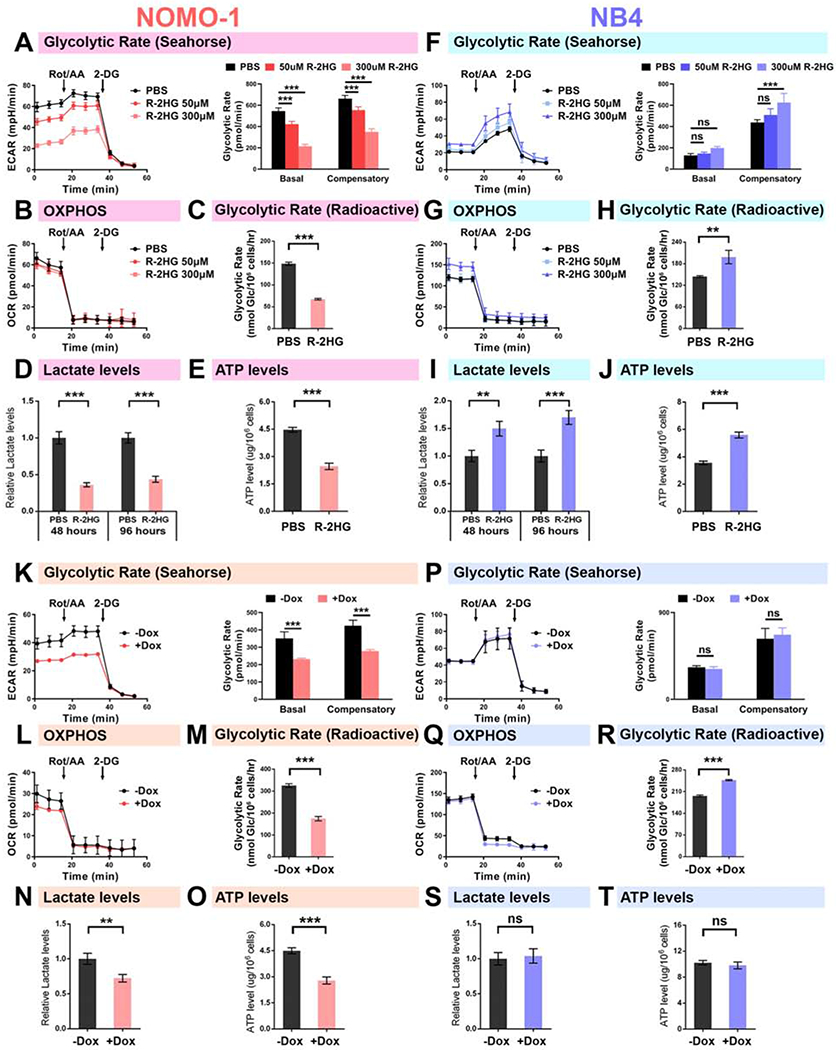
(A) Effects of R-2HG on glycolytic rates (as determined by Seahorse Glycolytic Rate Assay) in NOMO-1 cells upon treatment with R-2HG for 24 h. The extracellular acidification rate (ECAR) over time (left panel) and the calculated glycolytic rates at different stages of measurement (right panel) are shown.
(B) Effects of R-2HG on oxygen consumption rates (OCR) in NOMO-1 cells.
(C-E) Effects of R-2HG on glycolytic rates (C; as detected by radioactive glycolysis assay), lactate levels (D), and ATP levels (E) in NOMO-1 cells treated with 300 μM R-2HG for 48 h.
(F and G) Effects of R-2HG on glycolytic rates (F) and OCR (G) in NB4 cells treated with R-2HG for 24 h.
(H-J) Effects of R-2HG on glycolytic rates (H), lactate levels (I), and ATP levels (J) in NB4 cells treated with 300 μM R-2HG for 48 h.
(K) Effects of endogenous R-2HG on glycolytic rates in NOMO-1 cells, as determined by the Seahorse Glycolytic Rate Assay. Cells were treated with doxycycline (Dox) for 48 h to induce IDH1R132H expression.
(L) Effects of endogenous R-2HG on OCR in NOMO-1 cells.
(M-O) Effects of endogenous R-2HG on glycolytic rates (M; as detected by radioactive glycolysis assay), lactate levels (N), and ATP levels (O) in NOMO-1 cells.
(P and Q) Effects of endogenous R-2HG on glycolytic rates (P; as determined by the Seahorse Glycolytic Rate Assay) and OCR (Q) in NB4 cells.
(R-T) Effects of endogenous R-2HG on glycolytic rates (R; as detected by radioactive glycolysis assay), lactate levels (S), and ATP levels (T) in NB4 cells.
Data are represented as mean ± SD. ns, not significant (p ≥ 0.05); **, p < 0.01; ***, p < 0.001.
See also Figure S2.
We also confirmed all the results observed in exogenous R-2HG treated cells with the IDH1R132H cell model. Similarly, endogenous R-2HG significantly suppressed glycolysis and reduced lactate and ATP levels in sensitive but not resistant cells, whereas showing little effects on mitochondrial respiration in either sensitive or resistant cells (Figures 2K–2T and Figures S2K–S2P). Meanwhile, we replicated the experiment conducted by Fu et al. and found very similar results to theirs (Fu et al., 2015): the GBM cell line U87 cells demonstrated impaired mitochondrial respiration and enhanced glycolysis upon R-2HG treatment (Figure S2Q), indicating that R-2HG imposes different effects on metabolism between leukemia and GBM cells. Together, our data suggest that glycolysis is the primary metabolic pathway suppressed by both exogenous and endogenous R-2HG in sensitive leukemia cells.
Suppression of FTO contributes to R-2HG-induced glycolytic inhibition in sensitive cells
We have previously shown that FTO is a critical target of R-2HG in sensitive leukemia cells (Su et al., 2018). To determine whether FTO mediates the metabolic effect of R-2HG, we knocked down FTO with small hairpin RNAs (shRNAs) in both sensitive and resistant cells (Figures 3A and S3A). As expected, knockdown (KD) of FTO in sensitive leukemia cells (NOMO-1 and U937) inhibited cell proliferation/growth (Figures 3B and S3B) and promoted apoptosis and cell-cycle arrest (at G0/G1 phase) (Figures 3C and S3C), resembling the phenotype observed in R-2HG-treated sensitive cells (Su et al., 2018). Furthermore, forced expression of FTO almost completely rescued/reversed the R-2HG-induced inhibition on cell proliferation/growth in sensitive cells (Figure S3D), confirming the critical role of FTO in mediating the growth-inhibitory effect of R-2HG. Metabolomic analysis of FTO-KD NOMO-1 cells displayed an overall reduction in all metabolite levels compared to the control group (Table S4), and the majority of downregulated metabolites in FTO-KD NOMO-1 cells coincided with those downregulated in R-2HG treated NOMO-1 cells (Figure 3D). Further pathway enrichment analysis revealed that the shared downregulated metabolites significantly enriched for carbohydrate metabolism from all metabolic pathway classes, and glycolysis/gluconeogenesis from carbohydrate metabolic pathways (Figures 3E and 3F).
Figure 3. FTO mediates the glycolytic inhibitory effect of R-2HG in sensitive leukemia cells.
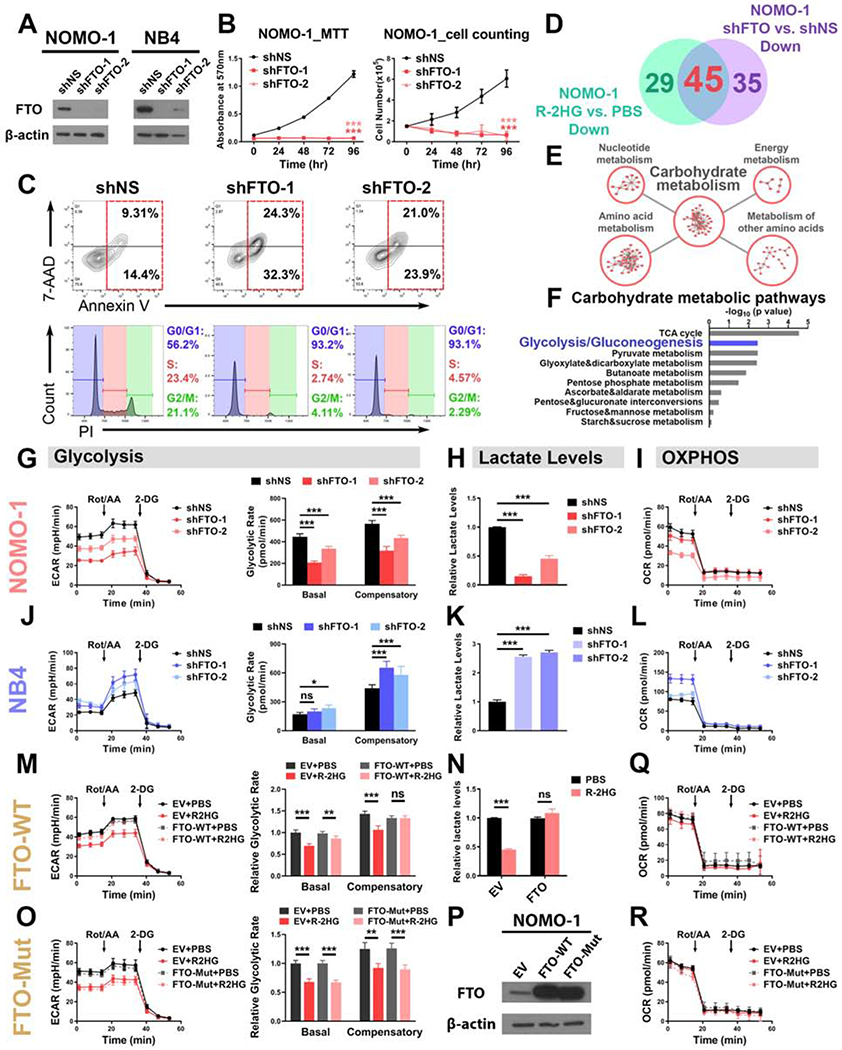
(A) Verification of FTO KD efficiency in NOMO-1 and NB4 cells.
(B) FTO KD in NOMO-1 cells inhibited cell proliferation/growth, as determined by MTT (left panel) and cell counting (right panel) assays.
(C) Effects of FTO KD on apoptosis (upper panel) and cell cycle (lower panel) in NOMO-1 cells.
(D) Venn diagram showing numbers of metabolites downregulated in NOMO-1 cells upon R-2HG treatment and FTO KD.
(E) The top 5 enriched KEGG metabolic pathway classes detected by the pathway enrichment analysis of the shared downregulated metabolites.
(F) List of the top 10 enriched carbohydrate metabolic pathways.
(G-I) Effects of FTO KD on glycolytic rates (G), lactate levels (H), and mitochondrial respiration (I) in NOMO-1 cells.
(J-L) Effects of FTO KD on glycolytic rates (J), lactate levels (K), and mitochondrial respiration (L) in NB4 cells.
(M and N) Overexpression of wild-type FTO rescued the glycolytic rates (M) and lactate levels(N) in 50 μM R-2HG-treated NOMO-1 cells.
(O) Overexpression of catalytic-dead FTO failed to rescue the glycolytic inhibition induced by R-2HG (50 μM) in NOMO-1 cells.
(P) Confirmation of wild-type and catalytic-dead FTO overexpression efficiency. EV: empty vector.
(Q and R) Effects of wild-type (Q) and catalytic-dead (R) FTO on mitochondrial respiration in NOMO-1 cells with and without R-2HG (50 μM) treatment.
Data are represented as mean ± SD. ns, not significant (p ≥ 0.05); *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Similar to R-2HG treatment, FTO KD also significantly reduced glycolytic rates and lactate levels, whereas exhibiting relatively limited effects on mitochondrial respiration in sensitive cells (Figures 3G–3I and Figures S3E–S3G). Similar to R-2HG treatment, FTO KD did not decrease glycolytic rates or lactate levels, or mitochondrial respiration in resistant cells either (Figures 3J–3L, S3H–S3J). Moreover, forced expression of wild-type FTO but not mutant FTO (which loses its m6A demethylase activity) largely rescued the glycolytic inhibitory effect of R-2HG in NOMO-1 cells, indicated by the restored glycolytic rates and lactate levels (Figures 3M–3P), suggesting the requirement of FTO m6A demethylase activity for the regulation of glycolytic flux. Mitochondrial respiration was not significantly altered by R-2HG treatment and/or FTO overexpression (Figures 3Q and 3R). Consistently, our recently identified effective FTO inhibitor, CS1 (Su et al., 2020), was also able to drastically reduce glycolytic rates in NOMO-1 cells without significantly altering mitochondrial respiration at low nanomolar concentration (200 nM) (Figures S3K and S3L), implying the potential of pharmacologically targeting FTO to manipulate cancer cell metabolism for leukemia therapy. Taken together, our data indicate that R-2HG-mediated glycolysis suppression is largely owing to its inhibition on the m6A demethylase activity of FTO in sensitive leukemia cells.
PFKP and LDHB are major glycolytic enzymes that are regulated by the R-2HG/FTO axis
To identify the downstream metabolic target genes of the R-2HG/FTO axis, we subjected glycolysis- and gluconeogenesis-related genes (Table S5) to the screening strategy shown in Figure 4A: 31 out of the total 35 genes were found to contain m6A modifications in their transcripts; expression of 16 out of the 31 genes was suppressed by FTO KD; and 7 out of the 16 genes were also downregulated in expression by R-2HG treatment (Huang et al., 2019; Linder et al., 2015; Su et al., 2018). We then examined the expression of these 7 genes as well as almost all the remaining glycolytic enzymes by RT-qPCR in sensitive and resistant cells, with or without exogenous R-2HG treatment (Figure 4B) or doxycycline-induced endogenous R-2HG production (Figures S4A and S4B). One screened out gene, GAPDHS, is not expressed in leukemia cells and was below detection limit of RT-qPCR. Among all the genes tested, PFKP and LDHB were found to be consistently and most significantly downregulated by exogenous and endogenous R-2HG in sensitive cells (Figures 4B and S4A). In contrast, neither PFKP nor LDHB displayed significant downregulation in resistant cells (Figures 4B and S4B). Next, we demonstrated that FTO KD also significantly downregulated the expression of PFKP and LDHB in sensitive U937 cells, but much less likely in resistant K562 cells (Figure S4C), likely due to the much lower abundance of FTO in resistant cells than in sensitive cells (Su et al., 2018). Our Western blot assays confirmed that both PFKP and LDHB (but not LDHA) were consistently downregulated in both exogenous and endogenous R-2HG treated sensitive cells (Figures 4C and S4D), in contrast to their minimal changes in similarly treated resistant cells (Figures 4D and S4E). While our results showed that the catalytic activity of FTO is important for its regulation of cell metabolism (see Figures 3M–3O), the protein level of FTO is also downregulated by extended R-2HG treatment in sensitive cells (especially after 96-hour treatment; see Figures 4C and S4D). Thus, we sought to determine which event (i.e., FTO catalytic activity suppression or FTO expression downregulation) is the primary one induced by R-2HG in sensitive cells. We found that the m6A level was already increased at an early time point (24 hours) after R-2HG treatment, when the protein level of FTO was not decreased yet (Figure 4E), indicating that R-2HG-induced inhibition of FTO’s m6A demethylase activity occurs before the downregulation of FTO expression. This is consistent with our previous finding that R-2HG inhibits FTO catalytic activity to downregulate the expression of transcription factor CEBPA in an m6A-dependent manner, which in turn inhibits FTO transcription via a feedback mechanism and thereby reduce its protein level subsequently (on and especially after 48 hours post R-2HG treatment) (Su et al., 2018). Moreover, here we observed that the protein levels of the key metabolic targets and glycolytic rates were already decreased at 24 hours post R-2HG treatment (Figures 2A, S2A and 4E). Collectively, our data suggest that R-2HG-induced suppression of FTO catalytic activity is the primary event that mediates R-2HG’s metabolic effects by inhibiting expression of key glycolytic enzymes (i.e., PFKP and LDHB) and thereby glycolysis in sensitive leukemia cells; the subsequent downregulation of FTO expression can further enhance R-2HG’s metabolic effects.
Figure 4. The R-2HG/FTO axis modulates PFKP and LDHB expression to regulate glycolysis in sensitive leukemia cells.
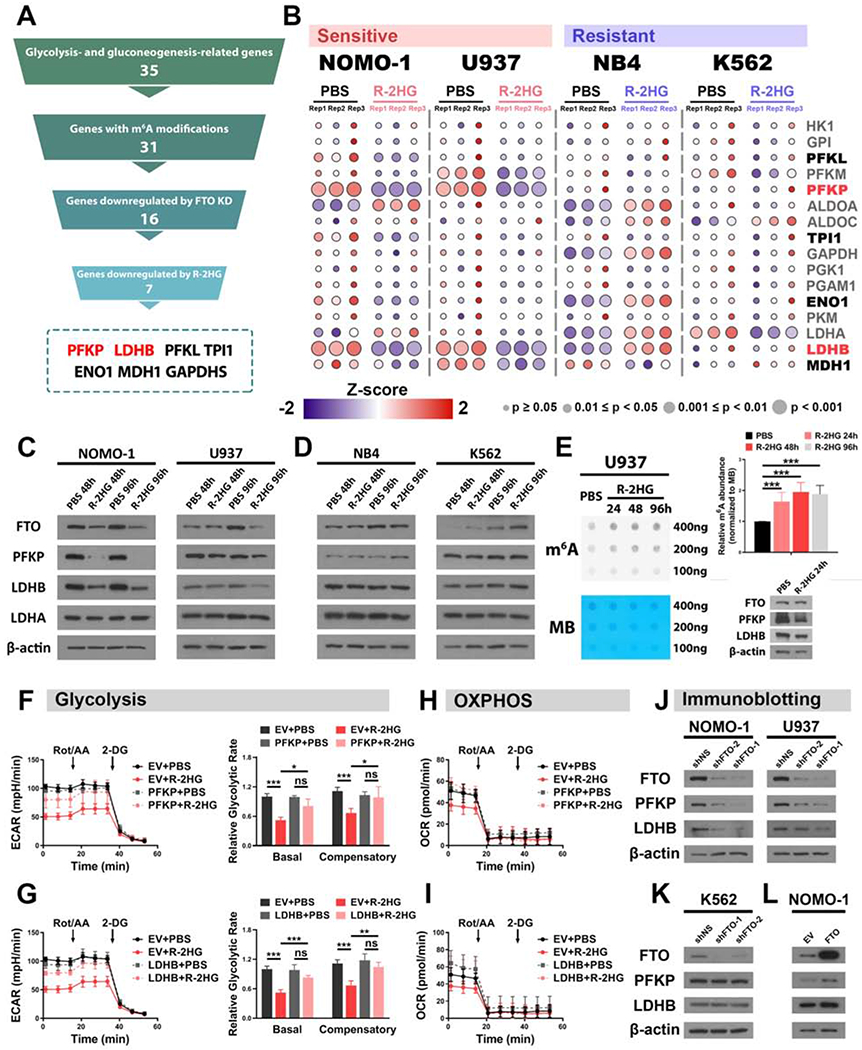
(A) Pyramid flowchart of the screening strategy to identify downstream metabolic target genes of the R-2HG/FTO axis.
(B) Heatmaps showing the relative expression of the screened out genes (marked in bold) along with other glycolytic enzymes in PBS- or R-2HG-treated sensitive and resistant cells. The color represents normalized expression level, and the diameter indicates significance of the difference between PBS- and R-2HG-treated samples. Among all tested genes, HK1, PFKL, PFKM, PFKP, PKM, LDHA, and LDHB are glycolysis-related, while MDH1 is gluconeogenesis-related; all other genes are related to both glycolysis and gluconeogenesis.
(C and D) Protein levels of FTO and the representative glycolytic enzymes in PBS- or 300 μM R-2HG-treated sensitive cells (C) and resistant cells (D).
(E) Effects of 300 μM R-2HG on global m6A modification and expression of target genes in U937 cells. Methylene blue (MB) staining is shown as a loading control. The quantification of normalized m6A signal is shown in the right upper panel.
(F and G) Rescue effects of PFKP (F) and LDHB (G) on glycolysis rates in R-2HG treated NOMO-1 cells.
(H and I) Mitochondrial respiration was measured in PBS- or R-2HG-treated NOMO-1 cells, with or without forced expression of PFKP (H) or LDHB (I).
(J and K) Effects of FTO KD on protein levels of PFKP and LDHB in sensitive (J) and resistant (K) cells.
(L) Effects of FTO overexpression on protein levels of PFKP and LDHB in NOMO-1 cells.
Data are represented as mean ± SD. ns, not significant (p ≥ 0.05); *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine the roles of PFKP and LDHB in R-2HG-mediated glycolytic inhibition and growth suppression, we ectopically expressed PFKP or LDHB in NOMO-1 and U937 cells (Figure S4F) to conduct rescue assays. As shown in Figures 4F and 4G, forced expression of PFKP and LDHB largely rescued R-2HG-induced glycolytic inhibition in NOMO-1 cells. Again, R-2HG treatment and/or forced expression of PFKP or LDHB showed minimal effects on mitochondrial respiration (Figures 4H and 4I). Moreover, forced expression of PFKP or LDHB also significantly rescued the inhibitory effects of R-2HG or FTO KD on cell proliferation/growth in sensitive cells (Figures S4H and S4I), demonstrating that R-2HG-induced metabolic reprogramming contributes substantially to its overall anti-tumor effects. Consistent with the qPCR results, FTO KD markedly decreased the protein levels of PFKP and LDHB in sensitive cells but not in resistant cells (Figures 4J and 4K), and the opposite is true when FTO is overexpressed in sensitive cells (Figure 4L). Thus, our data indicate that PFKP and LDHB are critical metabolic targets of the R-2HG/FTO axis in sensitive leukemia cells.
Another key downstream target of the R-2HG/FTO axis is MYC (Su et al., 2018), which is also heavily involved in cancer metabolism regulation (Dejure and Eilers, 2017; Stine et al., 2015). To determine whether MYC also contributes to the regulation of aerobic glycolysis by the R-2HG/FTO axis, we ectopically expressed MYC in sensitive cells (Figure S4G). While MYC overexpression significantly rescued the growth inhibition induced by R-2HG treatment (Su et al., 2018) or FTO KD (Figure S4J), it exhibited no significant rescue effect on R-2HG-induced glycolytic inhibition (Figure S4K), in contrast to the significant rescue effects of PFKP or LDHB overexpression (see Figures 4F and 4G). Therefore, our results suggest that MYC has considerable contribution to the anti-proliferation effects of R-2HG but plays a much less important role in R-2HG-mediated glycolysis inhibition, while PFKP and LDHB are the major mediators for the metabolic regulation of leukemia cells by the R-2HG/FTO axis.
The R-2HG/FTO axis regulates PFKP and LDHB expression in an m6A-dependent manner
Since our results showed that the m6A demethylase activity of FTO is important for the metabolic regulation by the R-2HG/FTO axis, we then sought to investigate how FTO/m6A regulates PFKP and LDHB expression. Multiple m6A peaks in both PFKP and LDHB transcripts were detected in an m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP) sequencing study (Figures S5A and S5B). Moreover, our gene-specific m6A RT-qPCR results showed that m6A abundance in both PFKP and LDHB mRNAs was increased upon R-2HG treatment or FTO KD (Figures 5A–5F), indicating that the R-2HG/FTO axis modulates m6A levels in PFKP and LDHB transcripts. Additionally, we conducted CLIP-RT-qPCR assays and demonstrated that FTO protein strongly binds with PFKP/LDHB transcripts (Figures 5G and S5C). Hence, these data demonstrate that PFKP and LDHB mRNAs contain m6A sites that are under regulation of the R-2HG/FTO axis.
Figure 5. Expression of PFKP and LDHB is downregulated by the R-2HG/FTO axis via an m6A-dependent mechanism.
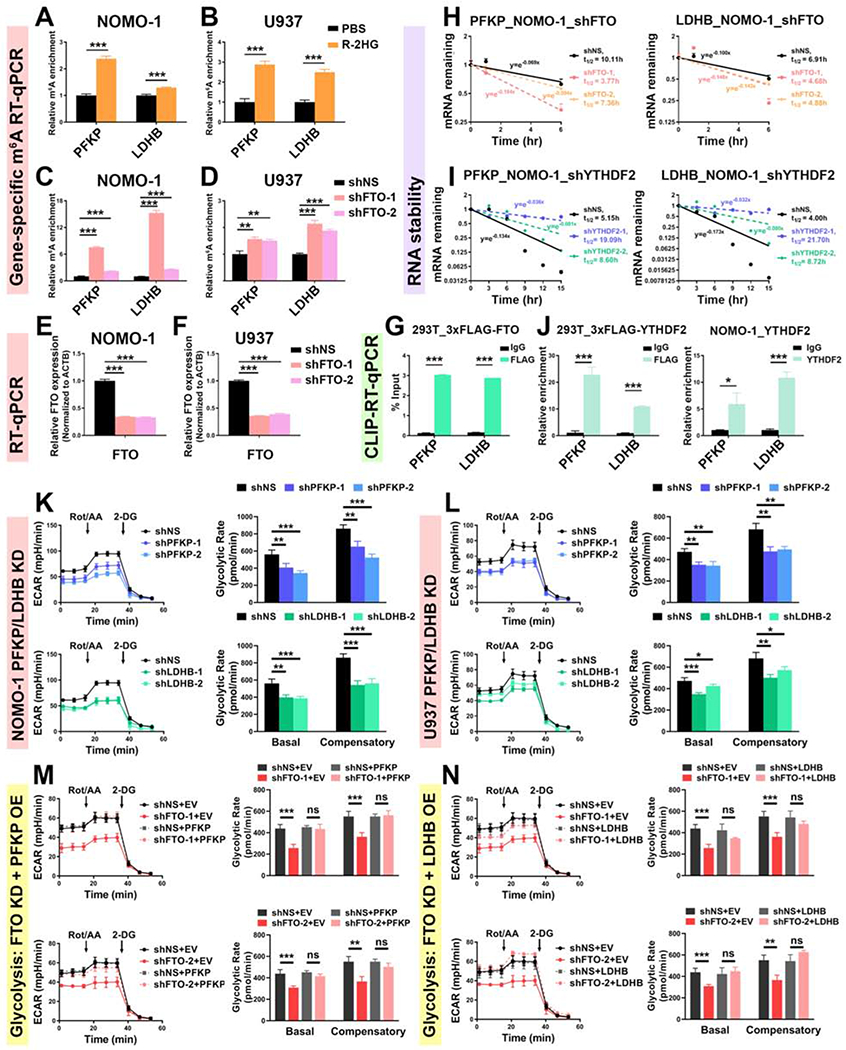
(A and B) Relative m6A abundance in PFKP and LDHB transcripts in NOMO-1 (A) and U937 (B) cells upon 300 μM R-2HG for 48 h.
(C and D) The m6A abundance changes in PFKP and LDHB transcripts in NOMO-1 (C) and U937 (D) cells upon FTO KD.
(E and F) Verification of FTO KD efficiency in NOMO-1 (E) and U937 (F) cells.
(G) Determination of the direct binding of FTO with PFKP and LDHB transcripts.
(H and I) Effects of FTO (H) and YTHDF2 (I) KD on the stability of PFKP (left panel) and LDHB (right panel) mRNAs.
(J) Determination of the direct binding of YTHDF2 with PFKP and LDHB transcripts in 293T (left panel) and NOMO-1 (right panel) cells.
(K and L) Effects of PFKP (upper panel) and LDHB (lower panel) KD on glycolytic rates in NOMO-1 (K) and U937 (L) cells. The same control shNS groups were used for the analysis.
(M and N) FTO KD-induced glycolytic inhibition by either shFTO-1 (upper panel) or shFTO-2 (lower panel) could be rescued by forced expression of PFKP (M) or LDHB (N). The same control groups (shNS+EV, shNS+PFKP, and shNS+LDHB) were used for the analysis.
Data are represented as mean ± SD. ns, not significant (p ≥ 0.05); *, p < 0.05; **, p < 0.01; ***, p < 0.001.
See also Figure S5.
As the most abundant internal modification in mRNA, m6A is known to exert its biological function by recruiting a series of proteins termed m6A readers to regulate mRNA stability and/or translation efficiency (Deng et al., 2018; Huang et al., 2020a; Huang et al., 2018; Huang et al., 2020b; Shi et al., 2017; Wang et al., 2014a; Wang et al., 2015b). Here, we first tested the half-time of PFKP and LDHB mRNA and showed that FTO KD decreased their mRNA stability in leukemia cells (Figures 5H and S5D). It is well known that YTHDF2 can recognize m6A modification and lead to mRNA decay (Wang et al., 2014a). CLIP-seq data indicated the direct binding of YTHDF2 on PFKP and LDHB transcripts (Figures S5A and S5B). Our RNA stability and CLIP-RT-qPCR data also demonstrated that YTHDF2 KD significantly increased the stability of PFKP and LDHB transcripts (Figure 5I and Figures S5E–S5G), which were bound directly by YTHDF2 (Figures 5J, S5H and S5I).
To determine whether PFKP and LDHB are functionally essential targets of FTO in regulating glycolysis, we conducted both loss-of-function and rescue assays. Similar to the effects induced by R-2HG treatment or FTO KD, genetic depletion of PFKP or LDHB significantly and consistently attenuated glycolysis (Figures 5K and 5L), whereas had a mild inhibitory effect on mitochondrial respiration (Figures S5J and S5K), in the two sensitive leukemia cell lines. Furthermore, glycolytic inhibition caused by FTO KD in NOMO-1 cells could be largely rescued by forced expression of PFKP (Figures 5M and S5L) or LDHB (Figures 5N and S5L). In contrast, mitochondrial respiration did not appear to be significantly and consistently affected by FTO KD and/or overexpression of PFKP or LDHB (Figures S5M and S5N). Taken together, our data indicate that PFKP and LDHB are bona fide and functionally essential metabolic targets of the R-2HG/FTO/m6A axis and are responsible for R-2HG- or FTO KD-induced glycolytic inhibition in sensitive leukemia cells.
PFKP and LDHB are essential for leukemia cells but are dispensable for normal cells
To investigate the biological function of PFKP and LDHB, we performed in vitro loss-of-function studies in leukemia cells and normal CD34+ hematopoietic stem/progenitor cells (HSPCs). KD of PFKP or LDHB significantly reduced cell proliferation/growth and promoted apoptosis and cell cycle arrest (at G0/G1 phase) in both NOMO-1 and U937 cells (Figures 6A–6H), consistent with the effects of R-2HG treatment or FTO KD (Li et al., 2017; Su et al., 2018). On the contrary, KD of FTO, PFKP, or LDHB did not show consistently significant inhibitory effects on glycolytic rates, mitochondrial respiration, or cell viability in normal CD34+ HSPCs (Figures 6I, 6J, and S6A–S6C), implying that FTO, PFKP, and LDHB are dispensable for normal HSPCs and thus represent potential safe therapeutic targets for leukemia treatment. We further conducted colony-forming/replating assays (CFAs) to determine the function of Pfkp and Ldhb in MLL-AF9-mediated mouse normal bone marrow (BM) progenitor cell transformation (Figure 6K). KD of Pfkp or Ldhb significantly decreased colony numbers compared to the control group in series of plating (Figure 6L), indicating that Pfkp and Ldhb are important for leukemic cell transformation (or immortalization).
Figure 6. Effects of PFKP or LDHB KD in leukemia cells and normal CD34+ HSPCs.
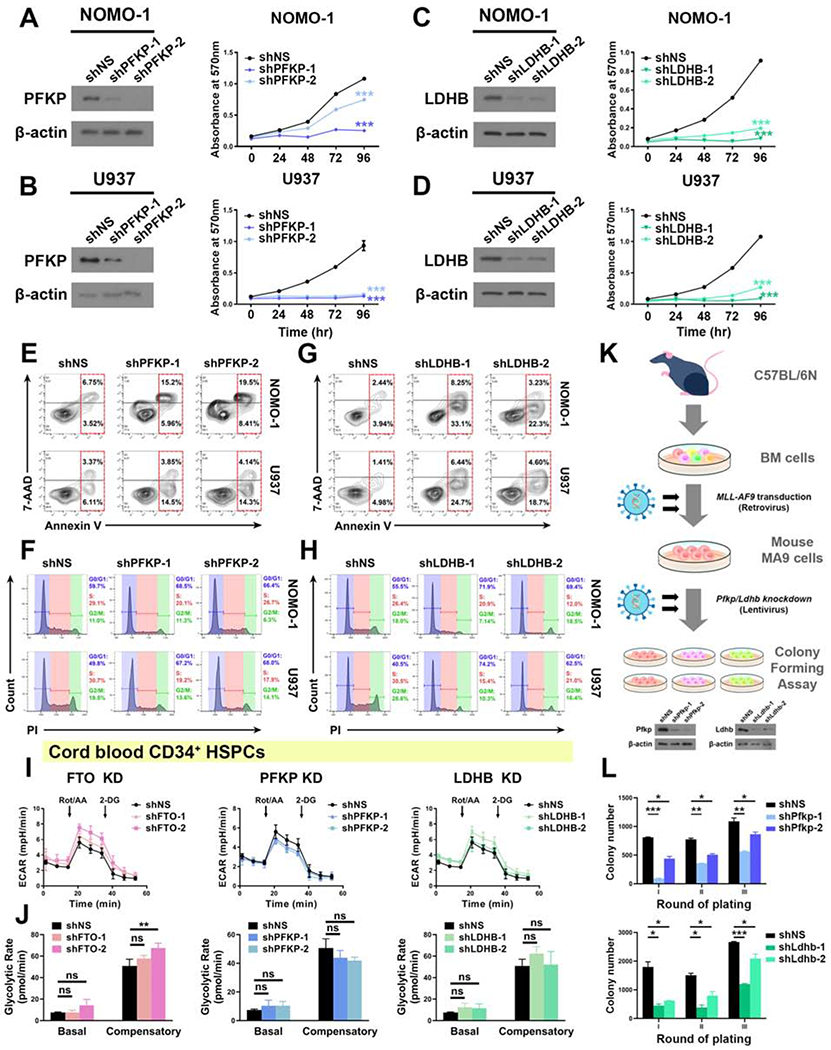
(A and B) Effects of KD of PFKP (left panel) on cell proliferation/growth (right panel) in NOMO-1 (A) and U937 (B) cells.
(C and D) Effects of KD of LDHB (left panel) on cell proliferation/growth (right panel) in NOMO-1 (C) and U937 (D) cells.
(E and F) Effects of PFKP KD on apoptosis (E) and cell cycle (F) in NOMO-1 and U937 cells.
(G and H) Effects of LDHB KD on apoptosis (G) and cell cycle (H) in NOMO-1 and U937 cells.
(I and J) Effects of KD of FTO (left panel), PFKP (middle panel), and LDHB (right panel) on glycolytic rates in normal CD34+ HSPCs. The same control shNS group was used for the analysis.
(K) Schematic illustration of mouse MA9 cell colony forming assay.
(L) Effects of Pfkp (upper panel) or Ldhb (lower panel) KD on colony forming/replating capacity of mouse MA9 cells.
Data are represented as mean ± SD. ns, not significant (p ≥ 0.05); *, p < 0.05; **, p < 0.01; ***, p < 0.001.
See also Figure S6.
LDHB is a critical component of LDH in leukemia
Lactate dehydrogenase (LDH) catalyzes the interconversion of lactate and pyruvate, with the net reaction direction depending on the concentrations of substrates and products (Doherty and Cleveland, 2013). LDH is a tetramer that comprises two different subunits, LDHA and LDHB (Dawson et al., 1964). These two subunits can assemble into five LDH isoenzymes: LDH-1 (4B), LDH-2 (3B1A), LDH-3 (2B2A), LDH-4 (1B3A), and LDH-5 (4A) (Figure S6D) (Markert and Appella, 1961). Because LDHA subunit catalyzes pyruvate reduction more efficiently than LDHB, it was thought that upregulation/activation of LDHA rather than LDHB promotes glycolysis and tumor progression in cancer (Dawson et al., 1964; Markert et al., 1975; Pathria et al., 2018; Wang et al., 2014b). However, patients with complete deficiency of LDHA subunits showed no symptoms under ordinary conditions and only manifested exertional myoglobinuria, indicating that LDHB is able to substitute for LDHA in most tissues except for in skeletal muscles (Kanno et al., 1988; Takashi et al., 1980). More importantly, LDHB has been recently reported to be phosphorylated at serine 162, which significantly improves its activity in pyruvate reduction and causes a boost in glycolytic flux (Cheng et al., 2019). Furthermore, increasing evidence suggests that LDHB is essential for the progression of many cancers (Brisson et al., 2016; Dennison et al., 2013; McCleland et al., 2013; Wang et al., 2020).
We analyzed the LDH isoenzyme distribution in leukemia cells by zymography (Li et al., 2019), and found that LDHB subunits compose 44.1% to 65.1% of the LDH subunits in the leukemia cell lines tested (Figure S6D). Since LDHA and LDHB together compose various LDH isoenzymes, we presumed that KD of either of them will reduce the overall LDH enzyme abundance and thereby inhibit glycolysis, leading to subsequent inhibition on leukemia cell growth and survival. To test this, we also conducted loss-of-function study of LDHA in leukemia cells. Indeed, similar to LDHB KD, LDHA KD also significantly inhibited glycolysis and cell proliferation/growth, and promoted apoptosis and cell cycle arrest (at G0/G1 phase) in both NOMO-1 and U937 cells (Figures S6E–S6J), while showing no consistently significant effects on mitochondrial respiration (Figures S6K and S6L). Taken together, our data indicate that suppression of either LDHB or LDHA leads to similar and significant effects on inhibition of glycolysis and leukemia cell growth/survival. Thus, although only LDHB seems to be a direct target of the R-2HG/FTO axis in leukemia cells, suppression of its expression can sufficiently reduce the overall abundance and function of LDH, leading to significant inhibitory effects on glycolysis and leukemia cell growth/survival.
PFKP and LDHB promote leukemogenesis in vivo and represent clinically relevant therapeutic targets in leukemia
To evaluate the in vivo function of PFKP and LDHB in leukemogenesis, we conducted loss-of-function studies with a “human-in-mouse” xenotransplantation leukemia model. We injected R-2HG-sensitive AML cells into NRGS immunodeficient mice (Wunderlich et al., 2014) and tracked leukemia burden with bioluminescence imaging. In line with our previous reports (Li et al., 2017; Su et al., 2018), FTO KD significantly inhibited leukemia progression in mice (Figure 7A). Consistently, KD of PFKP or LDHB also significantly delayed leukemogenesis in vivo (Figure 7B), although to a lesser extent than that caused by FTO KD. KD of FTO, PFKP, or LDHB substantially reduced the proportion of leukemic blast cells in BM and peripheral blood, and suppressed leukemia cell infiltration into spleen and liver (Figures S7A and S7B). Their KD significantly prolonged overall survival in xenotransplanted mice (Figures 7C and 7D). Overall, PFKP and LDHB, similar to FTO, play critical tumor-promoting roles in leukemia, indicating their basic functions in regulating glycolysis is essential for leukemogenesis.
Figure 7. The FTO/m6A/PFKP/LDHB axis regulates leukemogenesis in vivo and has clinical relevance.
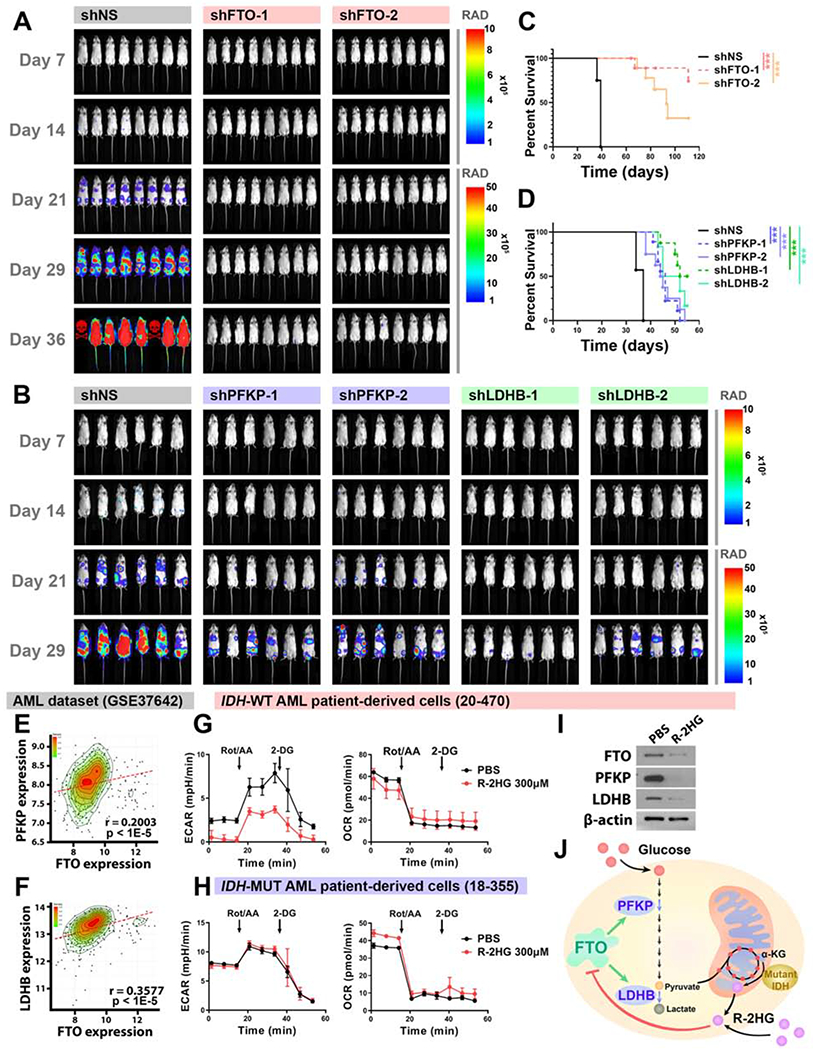
(A) KD of FTO decreased leukemia burden in vivo. Representative in vivo pseudocolor bioluminescence images of NRGS mice transplanted with control or FTO-KD MONO-MAC-6 cells. Unit of Radiance is “photons/second/cm2/steradian”.
(B) KD of PFKP or LDHB decreased leukemia burden in vivo.
(C and D) Kaplan-Meier survival curves showing the effects of FTO KD (C) and PFKP or LDHB KD (D) on progression of human AML cells in NRGS mice.
(E and F) Positive correlation between FTO and PFKP (E) or LDHB (F) in expression in AML.
(G and H) Effects of R-2HG treatment (24 h) on glycolytic rates (left panel) and mitochondrial respiration (right panel) in IDH-wildtype (G) and IDH-mutant (H) AML patient-derived cells.
(I) Effects of R-2HG treatment (48 h) on protein levels of FTO, PFKP, and LDHB in IDH-wildtype AML patient-derived cells.
(J) Cartoon illustration of the proposed model in this study.
Data are represented as mean ± SD. ***, p < 0.001.
To investigate the clinical relevance of the R-2HG/FTO/m6A/PFKP/LDHB axis, we first checked the correlation between FTO and PFKP or LDHB in expression in AML patient dataset (Herold et al., 2018; Li et al., 2013). We found that FTO expression is significantly positively correlated with the expression of both PFKP (Figure 7E) and LDHB (Figure 7F), suggesting the clinical relevance of the FTO/m6A/PFKP/LDHB axis. We further tested the effects of R-2HG on cell metabolism in both IDH-wildtype and IDH-mutant AML patient-derived cells (Figure S7C). In IDH-wildtype primary AML cells, R-2HG treatment significantly inhibited glycolytic flux without significantly altering mitochondrial respiration (Figure 7G), recapitulating its effects in sensitive leukemia cell lines. In contrast, R-2HG did not significantly suppress either glycolytic flux or mitochondrial respiration in IDH-mutant primary cells (Figure 7H), resembling its effects in resistant leukemia cell lines. Moreover, the protein levels of FTO, PFKP, and LDHB were markedly downregulated by R-2HG treatment in IDH-wildtype primary patient cells (Figure 7I), confirming the regulation of FTO/m6A/PFKP/LDHB axis by R-2HG in primary patient samples.
DISCUSSION
Proliferating tumor cells catabolize glucose at an unexpectedly high rate, and convert most of the glucose carbon to lactate rather than oxidizing it to CO2 even in the presence of excess oxygen, a phenomenon termed as the “Warburg effect” or aerobic glycolysis (Warburg, 1925, 1956). Recently, growing efforts have focused on developing advanced anticancer therapies exploiting this metabolic phenotypic difference between cancer cells and their normal counterparts, and it has already proved possible to safely target central metabolic enzymes in patients (Bonnet et al., 2007; Michelakis et al., 2010; Porporato et al., 2011; Vander Heiden, 2011). In the present study, we uncover that R-2HG primarily suppresses glycolysis in sensitive leukemia cells by targeting the FTO/m6A/PFKP/LDHB signaling pathway, which contributes to its anti-tumor activity (Figure 7J). The glycolytic inhibitory function of the R-2HG/FTO axis suggests that R-2HG, and especially specific FTO inhibitors, alone or in combination with other anticancer agents, hold great potential to treat IDH-wildtype leukemia by targeting tumor metabolism. Our findings also provide a crucial inspiration for treatment of IDH-mutant leukemia patients. For IDH-mutant AML patients treated with therapies targeting mutant IDH, less than half of the patients responded and only 20-30% attained complete remission (Pollyea et al., 2018; Roboz et al., 2019; Stein et al., 2017), which might be partially due to the release of R-2HG-caused glycolytic inhibition. Combinational therapy that combines mutant IDH inhibitors and glycolysis inhibitors (e.g., FTO inhibitors) might lead to a synergistic effect and confer benefits to non-responding patients.
Phosphofructokinase-1 (PFK-1) is a rate-limiting enzyme in glycolysis and catalyzes the irreversible conversion of fructose-6-phosphate to fructose-1,6-bisphosphate (Lang et al., 2019; Weber, 1977). Three PFK-1 isoforms exist in humans: PFKP (platelet), PFKM (muscle), and PFKL (liver). Notably, PFKP has been observed to predominate over PFKL and PFKM in various types of cancer (Kim et al., 2017; Lee et al., 2017; Moon et al., 2011), and elevated PFKP expression is a feature of malignant tissues (Sánchez-Martínez and J. Aragón, 1997). A recent work on PFKP in AML revealed that PFKP expression is increased in AML patients compared to healthy controls, especially in the subtypes with poor cytogenetic risk (Luo et al., 2018). Thus, PFKP appears to be an appealing target for cancer therapy. Nevertheless, very few small-molecule inhibitors of PFK-1 have been identified, which are also incapable of discriminating between different PFK-1 isoforms and cannot specifically inhibit PFKP (Riquelme et al., 1984; Spitz et al., 2009). We show here that PFKP is selectively downregulated by R-2HG treatment or FTO inhibition, affording an alternative strategy to selectively target PFKP in cancer treatment.
The other metabolic target of the R-2HG/FTO/m6A axis, LDHB, does not have a well-defined function in tumor biology: It is silenced by promoter hypermethylation in some cancer types (Leiblich et al., 2006; Maekawa et al., 2003), but is amplified or overexpressed in other cancers (McCleland et al., 2013; Rodriguez et al., 2003). Here, our functional studies show that both PFKP and LDHB promote glycolysis and play oncogenic roles in leukemia, clarifying the function of LDHB as oncogenic in leukemia. KD of either PFKP or LDHB is sufficient to retard leukemogenesis in vivo, but the delay in leukemogenesis is not as remarkable as that caused by FTO KD, implying the presence of a potential synergistic effect when both PFKP and LDHB are inhibited. Since inhibition of FTO effectively downregulates expression of both PFKP and LDHB, specific small-molecule FTO inhibitors are expected to exert a more potent anti-tumor effect than agents targeting only PFKP or LDHB in cancer therapy. Several FTO inhibitors have been discovered previously (Chen et al., 2012; Huang et al., 2019; Huang et al., 2015; Wang et al., 2015a), but are unlikely clinical applicable due to relatively low selectivity and/or low therapeutic efficacy. Very recently, we have identified two highly potent FTO inhibitor compounds (named CS1 and CS2), which showed high therapeutic efficacy and low side effects in our preclinical animal studies (Su et al., 2020). Our current findings revealed that CS1 treatment at a low concentration is sufficient to effectively attenuate glycolytic flux in leukemia cells, adding a new layer of mechanism for the anti-tumor activity of FTO inhibitors.
MYC, a pivotal transcription factor aberrantly overexpressed in many types of cancers, amplifies the expression of many genes controlling cell metabolism, proliferation, growth, and differentiation (Hsieh and Dang, 2016; Lin et al., 2012). MYC is documented to increase TCA cycle flux by 400% and glycolytic flux by roughly 20% in human B cells, and targets a set of glycolytic genes including LDHA (Dang et al., 2008; Murphy et al., 2013). Our data showed that LDHA expression is not significantly altered by R-2HG treatment, and that no consistent changes in the rate of TCA cycle/mitochondrial respiration are induced by R-2HG or FTO KD in sensitive leukemia cells. More importantly, our rescue experiments suggest that MYC is not a key player in the metabolic regulation of leukemia by R-2HG, though it is an intermediate mediator for the anti-proliferation effect of R-2HG.
In conclusion, our study revealed a previously unappreciated signaling involving the FTO/m6A/PFKP/LDHB axis, which controls aerobic glycolysis and plays an essential tumor-promoting role in leukemia; this axis can be suppressed by R-2HG or selective FTO inhibitors. Thus, our work provides novel insights into the mechanisms underlying the intrinsic anti-tumor activity of R-2HG and the tumor-promoting role of FTO in leukemia. Our study also provides encouraging results for leukemia therapy that target glycolysis, and inspires the clinical application of potent FTO inhibitors as both epigenetic-modulating and cancer metabolism-targeting agents for cancer therapy.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Jianjun Chen (jianchen@coh.org).
Materials Availability
All cell lines, plasmids, and other stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Metabolomic analysis results are available in Tables S1–S4. This study did not generate any unique code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
For leukemia cells, NOMO-1 and NB4 were obtained from DSMZ and cultured in endotoxin-free RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) (Gemini BioProducts); U937 and K562 were obtained from American Type Culture Collection (ATCC) and kept in RPMI 1640 supplemented with 10% FBS. MONO-MAC-6 (DSMZ) was maintained in 90% RPMI 1640 with 10% FBS plus 2 nM L-glutamine, 10 μg/mL human insulin, 1 mM sodium pyruvate, and 1× non-essential amino acids. The glioblastoma cell line U87 was originally maintained by Dr. David R. Plas from University of Cincinnati, and cultured in 90% RPMI 1640 with 10% FBS. HEK293T cells were grown in DMEM medium (Invitrogen) with 10% FBS. AML patient-derived primary cells were cultured in IMDM supplemented with 20% FBS, 10 ng/mL human cytokines SCF, IL-3, IL-6 and TPO. CD34+ cells isolated from cord blood were cultured in SFEM (09650, STEMCELL Technologies) with 10 ng/mL human cytokines SCF, IL-3, IL-6 and TPO. All cell lines are not among commonly misidentified cell lines, and were not authenticated by ourselves. All cell lines were cultured at 37°C in a humidified incubator with 5% CO2. All cell lines were tested for mycoplasma contamination yearly using a PCR Mycoplasma Detection Kit (G238, Applied Biological Materials Inc.). Penicillin-Streptomycin (15140122, Thermo Fisher Scientific) and Plasmocin prophylactic (ant-mpp, InvivoGen) were added to all cell culture media according to the manufacturer’s instruction to prevent potential contamination.
Care and Maintenance of Animals
NRG-SGM3 (NRGS) mice were originally purchased from the Jackson Laboratory (Stock No: 024099), and bred at core facilities of City of Hope. Both male and female mice at the age of 6-10 weeks were used for the xenotransplantation experiments and randomly distributed into each group. C57BL/6N mice were purchased from Charles River, and allowed for at least 1-week acclimation before being used for the generation of MLL-AF9 (MA9)-transformed mouse normal bone marrow (BM) progenitor cells. All laboratory mice were maintained under standard husbandry and housing conditions at the City of Hope Biological Resources Center. All experiments on mice in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of City of Hope.
Animal Procedures
For the “human-in-mouse” xenotransplantation leukemia model, MONO-MAC-6 cells were lentivirally infected with shNS, shRNAs against FTO (shFTO-1 and shFTO-2), or shRNAs against PFKP (shPFKP-1 and shPFKP-2) or LDHB (shLDHB-1 and shLDHB-2). Then 0.1 x 106 infected MONO-MAC-6 cells were transplanted into NRGS recipient mice intravenously. Leukemia development and progression were monitored by bioluminescence imaging over time. Leukemic mice were euthanized by CO2 inhalation if they showed typical symptoms of AML including hunched posture, paralysis, and decreased body weight. Peripheral blood (PB), bone marrow (BM), spleen, and liver samples were collected from euthanized mice for further analysis.
For the generation of mouse MA9 cells, 6- to 8-week-old C57BL/6N mice were treated with 5-fluorouracil (5-FU) for 5 days before euthanization by CO2 inhalation. BM cells were collected and enriched for progenitor cells (herein is lineage negative cells; Lin− cells) with Lineage Cell Depletion Kit (130-090-858, Miltenyi Biotec). The Lin− progenitor cells were then subject to retroviral transduction with MSCV-Neo-MA9 construct by two rounds of ‘spinoculation’, as described previously (Li et al., 2015). After being selected for 7 days with 0.5 mg/mL G418 Sulfate (10131027, Gibco) in ColonyGEL methylcellulose medium (1201, ReachBio Research Lab), the cells were regarded as mouse MA9 cells and were further knocked down for target genes lentivirally before in vitro colony-forming and replating assay.
METHODS DETAILS
Metabolite Extraction and Mass-Spectrometry-based Metabolomic Analysis
Cells were pre-treated with PBS, R-2HG, or Dox (doxycycline) for 48 hours, or directly used for metabolite extraction as indicated. For the metabolite extraction, 1 × 106 cells were washed with ice-cold 5% mannitol, and 1 mL cold 80% methanol (−80°C) was added to cell pellets. 5 nmol norvaline (Sigma-Aldrich) was added to each sample as internal standard. Samples were then vortexed for at least three times over 15 minutes on ice, and spun down at top speed for 5 minutes. The supernatant was transferred to a new Eppendorf tube, and the samples were dried down on Vacufuge Plus (Eppendorf) at 30°C. Extracted metabolite samples were stored at −80°C. The mass spectrometry-based analysis of extracted metabolites was conducted at UCLA Metabolomics Center. Metabolites were resuspended in 50% acetonitrile (ACN), and one tenth of the sample was analyzed with a Thermo Scientific Q Exactive mass spectrometer. The mass spectrometer was coupled to the UltiMate 3000 UPLC chromatography systems. The chromatographic separation was achieved on a Phenomenex Luna 3u NH2 100A (150×2.0 mm) column, and performed with 5 mM NH4AcO (pH 9.9, mobile phase A) and ACN (mobile phase B) at a flow rate of 200 μL/min. The linear gradient from 15% A to 95% A over 18 minutes was followed by an isocratic step at 95% A for 9 minutes and re-equilibration. The Q Exactive was run with polarity switching (+3.5 kV/−3.5 kV) in full scan mode with an m/z range of 65-975. TraceFinder 4.1 (Thermo Scientific) was used to quantify targeted metabolites by area under the curve using accurate mass measurements (±3 ppm) and prior established retention times. Data were normalized to the cell number. To plot the heatmap, the detection levels for all metabolites were further transformed by Z-score normalization. Pathways were enriched by hypergeometric test using KEGG metabolic pathway database (Kanehisa, 2019; Kanehisa and Goto, 2000). For enrichment of metabolic pathway classes, specific pathways that contain at least one metabolite from the overlapping metabolites were plotted as dots, and connections between pathways were plotted as lines.
Cell Viability and Proliferation Assay
Cells were seeded in 96-well plates at a concentration of 8,000-10,000 cells per well in triplicates, and cell proliferation/growth was determined with CellTiter 96 Non-Radioactive Cell Proliferation Assay (MTT, G400, Promega). Following the manufacturer’s instructions, 15 μL dye solution was added into the well at indicated time points. After incubation at 37°C for 2-4 hours, 100 μL solubilization/Stop Solution was added to quench the reaction. The absorbance at 570 nm of each well was recorded on the next day. To verify the effects of exogenous R-2HG, PBS or 300 μM R-2HG were added to cells at the time of cell seeding (time 0). To test the effects of endogenous R-2HG, IDH1R132H cells were pre-treated with PBS or 1 μg/mL doxycycline for 36 hours before being seeded into 96-well plates (time 0). To study the impacts of genetic depletion of FTO, PFKP, LDHB, and LDHA on cell viability and proliferation, cells were selected with 1 μg/mL puromycin for 96 hours after infection with lentivirus, and then seeded in 96-well plates. For cell counting assay, cells were counted by cell counter hemocytometer (Invitrogen Countess). Trypan blue (15-250-061, Thermo Fisher Scientific) staining and propidium iodide (PI, P4170, Sigma-Aldrich) staining was used to assess cell viability. For PI staining, 1× 106 cells were collected, washed once and suspended in 100 μL PBS. 5 μL PI staining solution was added to cells, mixed well and applied to flow cytometer directly.
Isolation of CD34+ cells
Fresh cord blood was purchased from StemCyte. CD34+ cells were isolated according to manufacturer’s instruction (130-046-702, Miltenyi Biotec). Mononuclear cells were isolated using Ficoll-Paque and suspended in 300 μL MACS buffer for up to 108 cells. 100 μL FcR blocking reagent and 100 μL CD34 MicroBeads were added to the cells and incubated for 30 minutes in refrigerator (4 °C). After washing with MACS buffer, the cells were isolated with MACS separation columns. CD34+ cells were cultured in SFEM medium with cytokines.
Metabolic Assays Using the XFe96 Extracellular Flux Analyzer
ECAR and OCR measurements were determined with a Seahorse XFe96 Extracellular Flux Analyzer and a Seahorse XF Glycolytic Rate Assay Kit (Agilent Technologies) according to the manufacturer’s instructions. All materials and compounds were obtained from Agilent Technologies. In brief, 1.6 × 105 leukemia cells per well were plated in XF96 Cell Culture Microplate coated with Cell-Tak (Corning). Then the cells were incubated in XF RPMI medium (without phenol red) supplemented with 2 mM glutamine, 10 mM glucose, 1 mM pyruvate, and 5 mM HEPES for 45 minutes at 37°C (non-CO2 incubator) prior to the assay. For measurement of U87 cells, 4 × 104 cells were seeded per well in XF96 Cell Culture Microplate in DMEM supplemented with 10% FBS, and incubated overnight at 37°C in 5% CO2. 1 hour prior to the assay, U87 cells were washed with XF RPMI medium (without phenol red) supplemented with 2 mM glutamine, 10 mM glucose, 1 mM pyruvate, and 5 mM HEPES, and maintained in this medium containing PBS of R-2HG at indicated concentrations. ECAR and OCR were measured at the basal stage (basal glycolysis + mitochondrial acidification), in response to Rot/AA (inhibitors of mitochondrial electron transport chain; compensatory glycolysis) and 2-deoxy-D-glucose (a glucose analog; post 2-DG acidification). The basal and compensatory glycolytic rates were calculated using the Seahorse Glycolytic Rate Assay Report Generator, and account for contribution of CO2 to extracellular acidification derived from mitochondrial respiration.
Radioactive Glycolysis Assay
This assay was conducted as described previously with some modifications (Ashcroft et al., 1972; Tandon et al., 2011). Duplicate wells containing 0.5 to 1.0×106 of the indicated cell lines were incubated with 5 μCi of 5-3H-glucose for 2 hours. The reaction was terminated by acid lysis, and triplicate samples drawn from each well were placed in a capless Eppendorf tube. Tubes were placed in a closed outer chamber containing non-radioactive water to undergo evaporative exchange for 1-3 days. Scintillation counting of samples from the outer chamber was referenced to scintillation counts from the inner chamber to determine the per cell rate of 3H-H2O production from 3H-glucose, an event that is largely dependent on the glycolytic conversion of 2-phosphoglycerate to phosphoenolpyruvate, correcting for background and evaporative exchange rate.
Lactate and ATP levels
Lactate levels were determined with the Lactate Colorimetric/Fluorometric Assay Kit (K607, BioVision), and the colorimetric assay was performed per the manufacturer’s recommendation with an additional deproteinization step to remove LDH from the cells cultured with FBS. Briefly, 1 × 106 cells from each group were washed once with PBS, lysed with 160 μL Lactate Assay Buffer (provided in the kit) and centrifuged to remove the insoluble materials. 150 μL of the soluble fraction was then transferred to a new Eppendorf tube for deproteinization with Deproteinizing Sample Preparation Kit (ab204708, Abcam). The deproteinized samples were then added to a 96-well plate in triplicates with appropriate dilution; a series of lactate standards with different dilution factors were also added to the plate for standard curve preparation. After incubation with the reaction mix (containing lactate enzyme mix and probe) for 30 minutes, absorbance of the samples at 570nm was measured and the lactate levels were calculated using the standard curve prepared. ATP levels were examined by the ApoSENSOR™ ATP Cell Viability Bioluminescence Assay Kit (K254, BioVision) in accordance with the supplier’s direction.
Cell Cycle and Apoptosis Analysis with Flow Cytometry
The percentage of cells located at G0/G1, S, and G2/M phases was assessed by Propidium Iodide (PI) DNA staining. 1 x 106 cells were collected, washed with PBS once, resuspended in 1 mL buffer containing 0.05 mg/mL PI, 0.02 mg/mL ribonuclease A, 0.1% trisodium citrate, 0.3% NP40, and incubated at 37°C for 30 minutes. The samples were then subjected to flow cytometry analysis directly. The evaluation of cell apoptosis was executed using the PE Annexin V Apoptosis Detection Kit I (559763, BD Biosciences) following the instructed protocol. The samples were analyzed on Fortessa X20 cell analyzer (BD Biosciences) and the data were analyzed with FlowJo V10 Software.
Lentivirus and Retrovirus Preparation and Infection
Lentiviral particles for pCDH-puro-FTO-WT (no GFP), pCDH-puro-FTO-Mut (no GFP) [carrying H231A and D233A mutations, which abolish the m6A demethylase activity of FTO (Jia et al., 2011; Li et al., 2017; Su et al., 2018)], pCDH-puro-PFKP (no GFP), pCDH-puro-LDHB-3xFLAG (no GFP), pCDH-puro (no GFP) (CD510B-1, System Biosciences), pMIRNA1-3×FLAG-FTO, pMIRNA1-3×FLAG-YTHDF2, pLenti-CMV-Puro-LUC (17477, Addgene), pLKO.1-shFTO-1 (TRCN0000246247, Sigma-Aldrich), pLKO.1-shFTO-2 (TRCN0000246249, Sigma-Aldrich), pLKO.1-shYTHDF2-1 (TRCN0000265510, Sigma-Aldrich), pLKO.1-shYTHDF2-2 (Table S7), pLKO.1-shPFKP-1 (TRCN0000199163, Sigma-Aldrich), pLKO.1-shPFKP-2 (TRCN0000199329, Sigma-Aldrich), pLKO.1-shLDHB-1 (TRCN0000028502, Sigma-Aldrich), pLKO.1-shLDHB-2 (TRCN0000279700, Sigma-Aldrich), pLKO.1-shLDHA-1 (TRCN0000159591, Sigma-Aldrich), pLKO.1-shLDHA-2 (TRCN0000026541, Sigma-Aldrich), pLKO.1-shPfkp-1 (TRCN0000274699, Sigma-Aldrich), pLKO.1-shPfkp-2 (TRCN0000274765, Sigma-Aldrich), pLKO.1-shLdhb-1 (TRCN0000041758, Sigma-Aldrich), pLKO.1-shLdhb-2 (TRCN0000041759, Sigma-Aldrich), and pLKO.1-shNS were packaged with pMD2.G (12259, Addgene), pMDLg/pRRE (12251, Addgene), and pRSV-Rev (12253, Addgene). The pCDH-puro-FTO (no GFP), pCDH-puro-PFKP (no GFP), and pCDH-puro-LDHB-3×FLAG (no GFP) were constructed by In-Fusion HD Cloning Plus CE kit (638916, Takara). Before In-Fusion, empty pCDH-puro (no GFP) vector was linearized by XbaI and NotI. The primers used in In-Fusion cloning were listed in Table S7. For packaging of lentiviral particles, 0.3 μg pMDLg/pRRE, 0.5 μg pMD2.G, 0.7 μg pRSV-Rev, and 1.8 μg construct for overexpression or knockdown of specific genes were co-transfected into Lenti-X 293T cells in 60mm cell culture dish with X-tremeGENE™ HP DNA Transfection Reagent (6366546001, Roche). The pTRIPZ-IDH1R132H was packaged with psPAX2 and pMG2.G. The lentiviral particles were harvested at 48 and 72 hours post transfection. To infect leukemia and HEK-293T cells, the lentiviruses were added into cells with existence of 4 μg/mL polybrene (H9268, Sigma-Aldrich). The leukemia cells were further subject to two rounds of ‘spinoculation’ at 1000 rpm, 32°C for 90 minutes. The infected cells were selected with puromycin (for pCDH-puro-FTO, pCDH-puro-PFKP, pCDH-puro-LDHB-3×FLAG, pCDH-puro, and all shRNAs) (P8833, Sigma-Aldrich) or GFP expression (for pMIRNA1-3×FLAG-FTO and pMIRNA1-3×FLAG-YTHDF2). 1 μg/mL Doxycycline (D9891, Sigma-Aldrich) was added to infected cells after selection to induce expression of IDH1R132H. Retrovirus production and infection were employed as reported previously (Li et al., 2015). Retroviral particles of MSCV-Neo-MA9 were packaged with pCL-ECO vector (IMGENEX, San Diego, CA), collected at 48 and 72 hours after transfection, and added with polybrene into mouse bone marrow progenitor cells for ‘spinoculation’.
RNA Extraction and Real-Time Quantitative PCR (RT-qPCR) Analysis
Total RNAs were extracted with miRNeasy Mini Kit (217004, Qiagen) and quantified by UV spectrophotometry. For cDNA synthesis, 200-1,000 ng of total RNA samples or immunoprecipitated RNA samples were reverse-transcribed into cDNA in 20 μL reaction volume with the QuantiTect Reverse Transcription Kit (205314, Qiagen) according to the manufacturer’s instruction. Quantitative PCR (qPCR) was then performed with 0.5 μL cDNA (with 2-10 fold dilution) using Maxima SYBR Green qPCR Master Mix (2×) (FEPK0253, Thermo Fisher) on the QuantStudio 7 Flex PCR system (Thermo Fisher Scientific). Each reaction was run in triplicates, and relative mRNA abundance was calculated by the comparative ΔΔCT method. The calculated mRNA abundance was further transformed by Z-score normalization to plot the heatmap. ACTB was used as endogenous control for gene expression evaluation, while 18S rRNA and 28S rRNA were selected as endogenous controls for RNA stability assay. All the primers were listed in Table S7.
Protein Extraction and Western Blot Analysis
For total protein extraction, cells were washed twice with ice-cold PBS, and lysed in RIPA buffer (R0278, Sigma-Aldrich) with Halt phosphatase inhibitor cocktail (78420, Thermo Fisher Scientific) and Halt protease inhibitor cocktail (78429, Thermo Fisher Scientific). Supernatants were collected after centrifugation and the protein concentration was measured with the Bio-Rad Protein Assay (5000006, Bio-Rad). Equal amounts of protein samples (10-30 μg) were loaded per well on 10% SDS-PAGE gel, and transferred onto methanol-activated polyvinylidene fluoride membranes (Thermo Fisher Scientific). Membranes were washed with 1× PBST, blocked with 5% non-fat milk, and incubated sequentially with indicated primary antibodies and secondary antibodies. Antibodies used for Western blot were as follows: FTO (ab124892, Abcam), β-actin (3700S, Cell Signaling Technology), PFKP (8164S, Cell Signaling Technology), LDHB/Ldhb (PA5-27505, Invitrogen), Pfkp (13389-1-AP, Proteintech), LDHA (2012S, Cell Signaling Technology), FLAG M2 (F1804, Sigma-Aldrich), YTHDF2 (24744-1-AP, Proteintech), MYC (13978S, Cell Signaling Technology), IDH1R132H (TA190113, Origene).
m6A dot blot assay
Dot blot assay was performed to determine the global m6A abundance. In brief, total RNA samples were mixed with RNA incubation buffer and denatured at 65 °C for 5 minutes. Then, the RNA samples were loaded on the Amersham Hybond-N+ membrane (RPN119B, GE Healthcare), washed with 10×SSC buffer (S6639, Sigma-Aldrich), and crosslinked to the membrane by 254 nm UV. The membrane was stained by 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2) for control. Then, the membrane was blocked with 5% nonfat milk and incubated with m6A antibody (202003, Synaptic Systems) overnight at 4°C. After washed with PBST, the membrane was incubated with HRP-conjugated secondary antibody (ab6721, Abcam) for 1 hour. The signal was detected by Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare).
Gene-Specific m6A RT-qPCR
To assess the relative m6A abundance in transcripts of individual genes, m6A RNA immunoprecipitation (MeRIP) was performed as described previously with some modifications (Dominissini et al., 2013), followed by RT-qPCR. Briefly, 100 μg of total RNA isolated with miRNeasy Mini Kit was sheared to 100 nucleotides or smaller fragments by metal-ion induced fragmentation. The fragmented RNA was purified and incubated with m6A antibody (202003, Synaptic Systems)- or normal rabbit IgG (NI01, Millipore)-conjugated Protein A/G Magnetic Beads (88803, Thermo Fisher Scientific) in 500 μL 1× IP buffer containing RNase inhibitors at 4°C for 2 hours. RNA fragments with m6A modifications were immunoprecipitated by m6A antibody-conjugated beads, eluted by competition with free m6A (23382, Cayman Chemical), and recovered with the RNeasy MinElute Cleanup Kit (74204, Qiagen). One tenth of fragmented RNA was saved as input control, and analyzed by RT-qPCR in parallel with the MeRIPed RNAs using primers listed in Table S7. Cycle threshold (Ct) values (ΔΔCt method) were used to determine the relative enrichment of m6A in each sample.
Data Analysis of miCLIP seq, YTHDF2 PAR-CLIP-seq, and RNA-seq data
The miCLIP seq, YTHDF2 PAR-CLIP seq, and RNA seq data were obtained from the public database Gene Expression Omnibus (GEO; for miCLIP seq: GSE63753; for YTHDF2 PAR-CLIP seq: GSE49339; for RNA seq in FTO-knockdown samples: GSE103494; for RNA-seq in R-2HG treated samples: GSE87187) (Huang et al., 2019; Linder et al., 2015; Su et al., 2018; Wang et al., 2014a). The peaks on PFKP and LDHB transcripts were displayed via IGV to show the presence of m6A modifications and YTHDF2 binding sites on the target mRNAs.
Cross-Linking Immunoprecipitation and RT-qPCR (CLIP-RT-qPCR)
To test the interactions between FTO/YTHDF2 proteins and their metabolic target transcripts (PFKP and LDHB), CLIP-qPCR was conducted. Briefly, one 150 mm plate of HEK293T cells at 80% confluency or NOMO-1 cells with a cell density of about 1 × 106 cells/mL were washed once with cold PBS, and cross-linked by ultraviolet (254nm) at 150 mJ/cm2. The cells were then harvested and lysed in RNA immunoprecipitation (RIP) buffer [150 mM KCl, 25 mM Tris (pH 7.4), 5 mM EDTA, 0.5 mM DTT, 0.5% NP40, with freshly added RNase inhibitor and protease inhibitor], followed by a sonication step to shear RNA. Each sonicated sample was pre-cleared by incubation with 50 μL Protein A/G magnetic beads at 4°C for 1 hour with end-over-end mixing. Flag antibody (F3165, Sigma-Aldrich) and normal mouse IgG (NI03, Millipore), or YTHDF2 antibody (24744-1-AP, Proteintech) and normal rabbit IgG (NI01, Millipore) were conjugated to Protein A/G magnetic Beads by incubation at 4°C for 4 hours, followed by 3× wash with RIP buffer. The pre-cleared samples were incubated with conjugated beads at 4°C overnight, washed three times with RIP buffer, and resuspended in 80 μL PBS for DNA digestion with RNase-free DNase I (EN0521, Thermo Scientific) and protein digestion with Proteinase K (EO0492, Thermo Scientific). The Protein A/G magnetic Beads were then removed from the samples, and the co-immunoprecipitated RNA were recovered from the supernatant by RNeasy MinElute Cleanup Kit along and dissolved in 12 μL RNase-free water. 4 μL co-immunoprecipitated RNA along with 4 μL input RNA from each group was used for reverse transcription and qPCR analysis using primers listed in Table S7.
Immunoprecipitation (IP) Assay
To verify the specificity of antibodies used in the CBIP-RT-qPCR assay, IP was conducted. Total protein samples were combined with 10 μg of the indicated antibody in 500 μL lysis buffer, and incubated at 4°C overnight. The sample/antibody mixture was subsequently added to a 1.5 mL Eppendorf tube containing Protein A/G Magnetic Beads pre-washed with Wash Buffer [Tris-buffered saline (TBS) containing 0.05% Tween-20 detergent], and incubated at room temperature for 1 hour with mixing. After washing the beads with Wash Buffer for three times and with purified water for one time, 100 μL SDS-PAGE reducing sample buffer was added to the beads and incubated for 10 minutes at room temperature with mixing. The beads were magnetically removed and the supernatant was analyzed by Western blot.
DNA isolation and IDH1/2 mutation status confirmation
Genomic DNA was extracted with DNeasy Blood & Tissue Kit (69506, Qiagen). The IDH1/2 mutation was detected as reported previously (Su et al., 2018). Briefly, primers containing M13 were used for IDH1/2 amplification. The PCR products were purified and sent for Sanger sequencing. The PCR primers were listed in Table S7.
LDH isoenzyme zymography assay
Native gel electrophoresis was performed based on a previously reported method with some modifications to determine the distribution of LDH isoenzymes in the AML cell lines used in this study (Lang et al., 2019). Cells were washed with PBS and lysed with 400 μL 1‰ Triton X-100 for 30 minutes at 37°C. After centrifugation at 4,000× g at 8°C for 30 minutes, the supernatant was transferred to a new Eppendorf tube, and the protein concentration was measured by the Bio-Rad Protein Assay. 50 μg protein samples were loaded (2× gel-loading buffer: 0.04g bromophenol blue and 10g saccharose dissolved in 1xTBE to a final volume of 20 mL) onto 0.5% agarose gel made with 1× TBE (pH 8.3) buffer. The gel was run at a constant voltage of 100 V and temperature of 4°C until the stained line was 95% of the way down the gel. After a brief wash with 1× TBE buffer, the gel was incubated with the staining solution [15 mg NAD+, 1.5 mL sodium lactate (1 M), 0.4 mL PMS (1 mg/mL), 4 mL NBT (1 mg/mL), 10.75 mL PBS] at 37°C for 40 minutes. The stained gel was rinsed with distilled water for three times and imaged by the gel imaging analysis system. Quantification of the bands were realized with the ImageJ software, and the percentage map was plotted with GraphPad Prism 8.
RNA Stability Assay
NOMO-1 and U937 cells upon FTO or YTHDF2 knockdown were treated with 5 μg/mL actinomycin D (A9415, Sigma-Aldrich) to assess RNA stability of PFKP and LDHB transcripts. Cells were harvested at indicated time points, and total RNA was extracted with miRNeasy Mini Kit for reverse transcription and qPCR analysis. The turnover rates and half-lives of mRNA were estimated according to previously published protocols (Chen et al., 2008; Liu et al., 2014). Since actinomycin D turns off mRNA transcription, the rate of disappearance of mRNA concentration at a given time (dC/dt) is proportional to both mRNA concentration (C) and the rate constant for mRNA decay (kdecay), as shown in the following equation:
Thus the rate constant for decay (kdecay) could be estimated by the derivation of the equation:
C0 is the concentration of mRNA at time 0 before transcription inhibition starts, t is the transcription inhibition time, and C is the mRNA concentration at time t. To determine the half-life (t1/2) of mRNA, which means C/C0 = 50%/100% = 1/2, the equation can be rearranged into the following equation:
from where:
In Vitro Colony-Forming and Replating assay
The serial colony-forming assay was employed as described previously with some modifications (Li et al., 2015). The mouse MA9 cells transformed by MSCV-Neo-MA9 from bone marrow progenitor cells were knocked down for Pfkp or Ldhb before being seeded into 35 mm culture dishes (20,000 cells/dish or 10,000 cells/dish) containing ColonyGEL methylcellulose medium supplemented with murine cytokines (10 ng/mL of IL-3, IL-6, GM-CSF, and 50 ng/mL of SCF), 1 mg/mL G418, and 2.5 mg/mL puromycin. Cultures were incubated at 37°C in a humidified atmosphere with 5% CO2 in air for 6 to 7 days. Serial replating was then conducted by collecting and replating colony cells in methylcellulose medium every 7 days. Colony numbers were counted and recorded for each passage.
In Vivo Bioluminescence Imaging
MONO-MAC-6 cells were lentivirally infected with pLenti CMV Puro LUC (17477, Addgene) by two rounds of ‘spinoculation’ and selected with puromycin for 7 days. Then, the luciferase-labeled MONO-MAC-6 cells were infected with lentivirus to knock down FTO, PFKP, or LDHB prior to xenograft transplantation. For in vivo bioluminescence imaging, NRGS mice were weighted, injected intraperitoneally with D-luciferin (LUCK-2G, Goldbio) at 150 mg/kg, and anesthetized with isoflurane. The mice were imaged using a Lago X (Spectral Instruments Imaging) 10 minutes post D-luciferin injection and Aura imaging software (Spectral Instruments Imaging) was used to quantify the bioluminescent signals. Radiance is in the unit of “photons/second/cm2/steradian”.
Histopathology Analysis
NRGS mice from shNS group were euthanized by CO2 inhalation when they developed leukemia and showed signs of systemic illness; some mice from the knockdown groups were euthanized at the same time points to collect specimens for histopathological examination. PB and BM cells isolated from the tibia and femur were smeared on slides and air dried. The slides were fixed with methanol for 30 seconds, followed by a 2-minute incubation with Wright-Giemsa Stain (24985, Polysciences), a 6-minute incubation with Wright-Giemsa Stain/Buffer (24984, Polysciences) mixture, and a 2-minute rinse in Wright-Giemsa Buffer. Portions of livers and spleens were collected from the euthanized mice and fixed in formalin. Paraffin embedding and H&E staining of the specimens was performed by the Pathology Core at City of Hope. The stained blood and bone marrow smear slides as well as the slides of liver and spleen sections were examined with a Widefield Zeiss Observer 7 microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were presented as mean ± SD and were analyzed with GraphPad Prism 8. Means between groups were compared by two-tailed Student’s t test as indicated; P < 0.05 was considered significant. Kaplan-Meier survival curves were plotted with GraphPad Prism 8 and the P values were calculated using the log-rank (Mantel-Cox) test. Mice in the knockdown groups that did not develop morbidity and were euthanized for sample collection were not included for the survival analysis. Pearson’s correlation test was used for the correlation analysis. For Western blot results, representative figures from three biological replicates were shown.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| m6A (N6-methyladenosine) antibody | Synaptic Systems | Cat# 202003; RRID: AB_2279214 |
| Anti-FTO antibody [EPR6895] | Abcam | Cat# ab124892; RRID: AB_10972698 |
| Goat anti-rabbit IgG H&L (HRP) | Abcam | Cat# ab6721; RRID: AB_955447 |
| Goat anti-mouse IgG H&L (HRP) | Abcam | Cat# ab6789; RRID: AB_955439 |
| β-Actin (8H10D10) Mouse mAb | Cell Signaling Technology | Cat# 3700S; RRID: AB_2242334 |
| Anti-Flag M2 antibody | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| Normal Mouse IgG control antibody | Millipore | Cat# 12-371; RRID: AB_145840 |
| PFKP antibody | Cell Signaling Technology | Cat# 8164S; RRID:AB_2713957 |
| LDHB/Ldhb antibody | Invitrogen | Cat# PA5-27505; RRID:AB_2544981 |
| Pfkp antibody | Proteintech | Cat# 13389-1-AP; RRID:AB_2252278 |
| LDHA antibody | Cell Signaling Technology | Cat# 2012S; RRID:AB_2137173 |
| YTHDF2 antibody | Proteintech | Cat# 24744-1-AP; RRID:AB_2687435 |
| MYC antibody | Cell Signaling Technology | Cat# 13987S; RRID:AB_2631168 |
| IDH1 R132H antibody | Origene | Cat# TA190113 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bisantrene (CS1) | MedChemExpress | Cat# HY-100875; CAS: 78186-34-2 |
| Puromycin dihydrochloride | Sigma-Aldrich | Cat# P8833; CAS: 58-58-2 |
| (2R)-2-Hydroxyglutaric Acid Octyl Ester Sodium Salt | Toronto Research Chemicals | Cat# H942595; CAS: 1391068-16-8 |
| 5-fluorouracil | Sigma-Aldrich | Cat# F6627; CAS: 51-21-8 |
| Propidium iodide | Sigma-Aldrich | Cat# P4170; CAS: 25535-16-4 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat# A2058; CAS: 9048-46-8 |
| Recombinant Human GM-CSF | PeproTech | Cat# 300-03 |
| Recombinant Human IL-6 | PeproTech | Cat# 200-06 |
| Recombinant Human IL-3 | PeproTech | Cat# 200-03 |
| Recombinant Human SCF | PeproTech | Cat# 300-07 |
| Recombinant Human TPO | PeproTech | Cat# 300-18 |
| D-Luciferin Firefly, potassium salt | Goldbio | Cat# LUCK; CAS: 115144-35-9 |
| L-Glutamine (200mM) | Thermo Fisher Scientific | Cat# 25030-081 |
| Ammonium Chloride Solution | STEMCELL Technologies | Cat# 07850 |
| MEM Non Essential Amino Acids Solution (100×) | Thermo Fisher Scientific | Cat# 11-140-050 |
| Sodium Pyruvate (100mM) | Thermo Fisher Scientific | Cat# 11360-070 |
| Insulin, human recombinant zinc solution | Thermo Fisher Scientific | Cat# 12585014 |
| Penicillin Streptomycin | Thermo Fisher Scientific | Cat# 15-140-122 |
| Plasmocin prophylactic | InvivoGene | Cat# ant-mpp |
| Corning Matrigel Membrane Matrix | Thermo Fisher Scientific | Cat# CB-40234A |
| Actinomycin D | Sigma-Aldrich | A9415; CAS: 50-76-0 |
| ColonyGEL-Mouse Base Medium | Reachbio | Cat# 1201 |
| RIPA Buffer | Sigma-Aldrich | Cat# R0278 |
| Halt Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | Cat# 78420 |
| Halt Protease Inhibitor Cocktail | Thermo Fisher Scientific | Cat# 78429 |
| Paraformaldehyde powder | Sigma-Aldrich | Cat# 158127 |
| 4× Laemmli Sample Buffer | Bio-Rad | Cat# 1610747 |
| Bio-Rad Protein Assay | Bio-Rad | Cat# 5000006 |
| RNaseOUT Recombinant Ribonuclease Inhibitor | Thermo Fisher Scientific | Cat# 10777019 |
| Amersham ECL Prime Western Blotting Detection Reagent | Fisher Scientific | Cat# 45-010-090 |
| Pierce Protein A/G Magnetic Beads | Thermo Fisher Scientific | Cat# 88803 |
| Ficoll Paque Plus | GE Healthcare | Cat# 17-1440-02 |
| CD34 MicroBead Kit, human | Miltenyi Biotec | Cat# 130-046-702 |
| FastDigest NotI | Thermo Fisher Scientific | Cat# FD0595 |
| FastDigest XbaI | Thermo Fisher Scientific | Cat# FD0684 |
| D-Mannitol (Powder/Certified ACS), Fisher Chemical | Fisher Scientific | Cat# M120500 |
| DL-Norvaline | Sigma-Aldrich | Cat# N7502-25G |
| Acetonitrile | Sigma-Aldrich | Cat# 271004-100ML |
| Wright-Giemsa Stain | Polysciences | Cat# 24985 |
| Wright-Giemsa Stain/Buffer | Polysciences | Cat# 24984 |
| Critical Commercial Assays | ||
| Seahorse XF Glycolytic Rate Assay Kit | Seahorse Bioscience | Cat# 103344-100 |
| miRNeasy Mini Kit | Qiagen | Cat# 217004 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Magna MeRIP m6A Kit | Millipore | Cat# 17-10499 |
| CellTiter 96 Non-Radioactive Cell Proliferation Assay | Promega | Cat# G4100 |
| Lineage Cell Depletion Kit | Miltenyi Biotec | Cat# 130-090-858 |
| PE Annexin V apoptosis Detection Kit | BD Biosciences | Cat# 559763 |
| QuantiTect Reverse Transcription Kit | Qiagen | Cat# 205314 |
| Deproteinizing Sample Preparation Kit | Abcam | Cat# ab204708 |
| Maxima SYBR Green qPCR Master Mix | Thermo Fisher Scientific | Cat# K0253 |
| Lactate Colorimetric/Fluorometric Assay Kit | BioVision | Cat# K607 |
| QIAGEN Plasmid Mini Kit | Qiagen | Cat# 12125 |
| Effectene Transfection Reagent | Qiagen | Cat# 301427 |
| ApoSENSOR™ ATP Cell Viability Bioluminescence Assay Kit | BioVision | Cat# K254 |
| PCR Mycoplasma Detection Kit | Applied Biological Materials | Cat# G238 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69506 |
| In-Fusion HD Cloning Plus CE | Takara | Cat# 638916 |
| Deposited Data | ||
| miCLIP seq | Linder et al., 2015 | GEO: GSE63753 |
| YTHDF2 PAR-CLIP seq | Wang et al., 2014a | GEO: GSE49339 |
| FTO knockdown RNA seq | Huang et al., 2019 | GEO: GSE103494 |
| R-2HG treated RNA seq | Su et al., 2018 | GEO: GSE87187 |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | (CRL-3216); RRID:CVCL_0063 |
| U937 | ATCC | (CRL-1593.2); RRID:CVCL_0007 |
| K562 | ATCC | (CCL-243); RRID:CVCL_0004 |
| NOMO-1 | DSMZ | (ACC-542); RRID:CVCL_1609 |
| MONO-MAC-6 | DSMZ | (ACC-124); RRID:CVCL_1426 |
| NB4 | DSMZ | (ACC-207); RRID:CVCL_0005 |
| U87 | Provided by Dr. David R. Plas | N/A |
| Experimental Models: Organisms/Strains | ||
| NRGS mouse | The Jackson Laboratory | Cat# JAX:024099; RRID: IMSR_JAX:024099 |
| NCI B6-Ly5.1/Cr mouse | Charles river | Cat# CRL:564; RRID: IMSR_CRL:564 |
| Oligonucleotides | ||
| DNA oligos listed in Table S7 | Integrated DNA Technologies (IDT) | N/A |
| Recombinant DNA | ||
| pCDH-puro (no GFP) | System Biosciences | Cat# CD510B-1 |
| pCDH-puro-FTO-WT (no GFP) | This paper | N/A |
| pCDH-puro-FTO-Mut (no GFP) | This paper | N/A |
| pCDH-puro-PFKP (no GFP) | This paper | N/A |
| pCDH-puro-LDHB-3xFLAG (no GFP) | This paper | N/A |
| pTRIPZ-IDH1R132H | A gift from Dr. Atsuo T. Sasaki | N/A |
| pLenti CMV Puro LUC (w168-1) | Campeau et al., 2009 | Addgene plasmid # 17477; RRID: Addgene_17477 |
| pMD2.G | A gift from Dr. Didier Trono | Addgene plasmid # 12259; RRID: Addgene_12259 |
| pMDLg/pRRE | Dull et al., 1998 | Addgene plasmid # 12251; RRID: Addgene_12251 |
| pRSV-Rev | Dull et al., 1998 | Addgene plasmid # 12253; RRID: Addgene_12253 |
| psPAX2 | A gift from Didier Trono | Addgene plasmid # 12260; RRID: Addgene_12260 |
| pLKO.1-shFTO-1 | Sigma-Aldrich | TRCN0000246247 |
| pLKO.1-shFTO-2 | Sigma-Aldrich | TRCN0000246249 |
| pLKO.1-shYTHDF2-1 | Sigma-Aldrich | Cat# TRCN0000265510 |
| pLKO.1-shYTHDF2-2 | This paper | Table S7 |
| pLKO.1-shNS | Stewart et al., 2003 | Addgene plasmid # 8453 |
| pLKO.1-shPFKP-1 | Sigma-Aldrich | TRCN0000199163 |
| pLKO.1-shPFKP-2 | Sigma-Aldrich | TRCN0000199329 |
| pLKO.1-shLDHB-1 | Sigma-Aldrich | TRCN0000028502 |
| pLKO.1-shLDHB-2 | Sigma-Aldrich | TRCN0000279700 |
| pLKO.1-shLDHA-1 | Sigma-Aldrich | TRCN0000159591 |
| pLKO.1-shLDHA-2 | Sigma-Aldrich | TRCN0000026541 |
| pLKO.1-shPfkp-1 | Sigma-Aldrich | TRCN0000274699 |
| pLKO.1-shPfkp-2 | Sigma-Aldrich | TRCN0000274765 |
| pLKO.1-shLdhb-1 | Sigma-Aldrich | TRCN0000041758 |
| pLKO.1-shLdhb-2 | Sigma-Aldrich | TRCN0000041759 |
| pMIRNA1-3×FLAG-FTO | This paper | N/A |
| pMIRNA1-3×FLAG-YTHDF2 | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| GelAnalyzer | GelAnalyzer | http://www.gelanalyzer.com/ |
| RSEM-1.2.31 | Li and Dewey, 2011 | https://deweylab.github.io/RSEM/ |
| STAR 2.7 | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| igv-2.3.72g | Thorvaldsdottir et al., 2013 | http://software.broadinstitute.org/software/igv/ |
| GSEA-2.2.3 | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
Highlights.
R-2HG attenuates glycolytic flux in R-2HG sensitive leukemia cells
Suppression of FTO contributes to R-2HG-mediated inhibition of glycolysis
R-2HG abrogates FTO/m6A/YTHDF2-mediated upregulation of PFKP and LDHB
PFKP and LDHB play critical roles in glycolysis and tumorigenesis in leukemia
ACKNOWLEDGEMENTS
We thank Jenna Minami for LC-MS data processing and quality control, and thank Atsuo T. Sasaki for providing plasmids. This work was supported in part by the National Institutes of Health (NIH) Grants R01 CA243386 (J.C.), R01 CA214965 (J.C.), R01 CA236399 (J.C.), R01 CA211614 (J.C.), R01 DK124116 (J.C.), The Margaret Early Medical Research Trust (R.S.), The Held Foundation Fellowship (Y.Q.), R35 CA197628 (M.M.), U10 CA180827 (M.M.), R01 CA137060 (M.M.), R01 CA157644 (M.M.), R01 CA172558 (M.M.), and R01 CA213138 (M.M.). J.C. is a Leukemia & Lymphoma Society (LLS) Scholar. Z.L. is an American Cancer Society (ACS) Research Scholar. M.M. is a Howard Hughes Medical Institute (HHMI) Faculty Scholar. B.T. is an American Association for Cancer Research (AACR) Breast Cancer Research Postdoc Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
J.C. is a scientific founder of Genovel Biotech Corp. and holds equities with the company.
REFERENCES
- Ashcroft SJ, Weerasinghe LC, Bassett JM, and Randle PJ (1972). The pentose cycle and insulin release in mouse pancreatic islets. The Biochemical journal 126, 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al. (2007). A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37–51. [DOI] [PubMed] [Google Scholar]
- Brisson L, Banski P, Sboarina M, Dethier C, Danhier P, Fontenille MJ, Van Hee VF, Vazeille T, Tardy M, Falces J, et al. (2016). Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 30, 418–431. [DOI] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, et al. (2015). Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med 372, 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BE, Ye F, Yu L, Jia GF, Huang XT, Zhang XJ, Peng SY, Chen K, Wang MN, Gong SZ, et al. (2012). Development of Cell-Active N-6-Methyladenosine RNA Demethylase FTO Inhibitor. Journal of the American Chemical Society 134, 17963–17971. [DOI] [PubMed] [Google Scholar]
- Chen CYA, Ezzeddine N, and Shyu AB (2008). Messenger RNA half - life measurements in mammalian cells. Methods in enzymology 448, 335–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Zhang P, Wang B, Yang D, Duan X, Jiang Y, Xu T, Jiang Y, Shi J, Ding C, et al. (2019). Aurora-A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate-inhibition effect. Nat Commun 10, 5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Kim JW, Gao P, and Yustein J (2008). The interplay between MYC and HIF in cancer. Nature reviews. Cancer 8, 51–56. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DM, Goodfriend TL, and Kaplan NO (1964). Lactic Dehydrogenases: Functions of the Two Types Rates of Synthesis of the Two Major Forms Can Be Correlated with Metabolic Differentiation. Science 143, 929–933. [DOI] [PubMed] [Google Scholar]
- Dejure FR, and Eilers M (2017). MYC and tumor metabolism: chicken and egg. EMBO J 36, 3409–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Su R, Weng H, Huang H, Li Z, and Chen J (2018). RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell research 28, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennison JB, Molina JR, Mitra S, Gonzalez-Angulo AM, Balko JM, Kuba MG, Sanders ME, Pinto JA, Gomez HL, Arteaga CL, et al. (2013). Lactate dehydrogenase B: a metabolic marker of response to neoadjuvant chemotherapy in breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 19, 3703–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, Swords R, Collins RH, Mannis GN, Pollyea DA, et al. (2018). Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med 378, 2386–2398. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty JR, and Cleveland JL (2013). Targeting lactate metabolism for cancer therapeutics. The Journal of clinical investigation 123, 3685–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, and Rechavi G (2013). Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc 8, 176–189. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol 72, 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Chin RM, Vergnes L, Hwang H, Deng G, Xing Y, Pai MY, Li S, Ta L, Fazlollahi F, et al. (2015). 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab 22, 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC, and Gale RE (2011). The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood 118, 409–412. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hay N (2016). Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nature reviews. Cancer 16, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold T, Jurinovic V, Batcha AMN, Bamopoulos SA, Rothenberg-Thurley M, Ksienzyk B, Hartmann L, Greif PA, Phillippou-Massier J, Krebs S, et al. (2018). A 29-gene and cytogenetic score for the prediction of resistance to induction treatment in acute myeloid leukemia. Haematologica 103, 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AL, and Dang CV (2016). MYC, Metabolic Synthetic Lethality, and Cancer. In Metabolism in Cancer. Cramer T, and Schmitt CA, eds. (Cham: Springer International Publishing; ), pp. 73–91. [DOI] [PubMed] [Google Scholar]
- Hsu PP, and Sabatini DM (2008). Cancer cell metabolism: Warburg and beyond. Cell 134, 703–707. [DOI] [PubMed] [Google Scholar]
- Huang H, Weng H, and Chen J (2020a). m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 37, 270–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al. (2018). Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HL, Weng HY, Deng XL, and Chen JJ (2020b). RNA Modifications in Cancer: Functions, Mechanisms, and Therapeutic Implications. Annu Rev Canc Biol 4, 221–240. [Google Scholar]
- Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, Ni T, Zhang ZS, Zhang T, Li C, et al. (2019). Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 35, 677–691 e610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, Gan J, Jiang H, Jia GF, Luo C, et al. (2015). Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res 43, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia GF, Fu Y, Zhao X, Dai Q, Zheng GQ, Yang Y, Yi CQ, Lindahl T, Pan T, Yang YG, et al. (2011). N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature Chemical Biology 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M (2019). Toward understanding the origin and evolution of cellular organisms. Protein science : a publication of the Protein Society 28, 1947–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Sudo K, Maekawa M, Nishimura Y, Ukita M, and Fukutake K (1988). Lactate dehydrogenase M-subunit deficiency: a new type of hereditary exertional myopathy. Clinica chimica acta; international journal of clinical chemistry 173, 89–98. [DOI] [PubMed] [Google Scholar]
- Kim NH, Cha YH, Lee J, Lee SH, Yang JH, Yun JS, Cho ES, Zhang X, Nam M, Kim N, et al. (2017). Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat Commun 8, 14374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang L, Chemmalakuzhy R, Shay C, and Teng Y (2019). PFKP Signaling at a Glance: An Emerging Mediator of Cancer Cell Metabolism. In Reviews on Biomarker Studies of Metabolic and Metabolism-Related Disorders. Guest PC, ed. (Cham: Springer International Publishing; ), pp. 243–258. [DOI] [PubMed] [Google Scholar]
- Lee JH, Liu R, Li J, Zhang C, Wang Y, Cai Q, Qian X, Xia Y, Zheng Y, Piao Y, et al. (2017). Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat Commun 8, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiblich A, Cross S, Catto J, Phillips J, Leung H, Hamdy F, and Rehman I (2006). Lactate dehydrogenase-B is silenced by promoter hypermethylation in human prostate cancer. Oncogene 25, 2953–2960. [DOI] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PJ, Qiu NH, Zhang L, Zhong GS, and Zeng FC (2019). A more universal and stable method for lactate dehydrogenase isoenzyme test. Analytical Methods 11, 4173–4183. [Google Scholar]
- Li Z, Chen P, Su R, Li Y, Hu C, Wang Y, Arnovitz S, He M, Gurbuxani S, Zuo Z, et al. (2015). Overexpression and knockout of miR-126 both promote leukemogenesis. Blood 126, 2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Herold T, He C, Valk PJ, Chen P, Jurinovic V, Mansmann U, Radmacher MD, Maharry KS, Sun M, et al. (2013). Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J Clin Oncol 31, 1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. (2017). FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 31, 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, and Jaffrey SR (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature methods 12, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, Yue YN, Han DL, Wang X, Fu Y, Zhang L, Jia GF, Yu M, Lu ZK, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N-6-adenosine methylation. Nature Chemical Biology 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Zheng D, Zheng R, Wang C, Xu L, and Tan H (2018). The Platelet Isoform of Phosphofructokinase in Acute Myeloid Leukemia: Clinical Relevance and Prognostic Implication. Blood 132, 5251–5251. [Google Scholar]
- Maekawa M, Taniguchi T, Ishikawa J, Sugimura H, Sugano K, and Kanno T (2003). Promoter hypermethylation in cancer silences LDHB, eliminating lactate dehydrogenase isoenzymes 1-4. Clin Chem 49, 1518–1520. [DOI] [PubMed] [Google Scholar]
- Markert CL, and Appella E (1961). Physicochemical nature of isozymes. Annals of the New York Academy of Sciences 94, 678–690. [DOI] [PubMed] [Google Scholar]
- Markert CL, Shaklee JB, and Whitt GS (1975). Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 189, 102–114. [DOI] [PubMed] [Google Scholar]
- McCleland ML, Adler AS, Deming L, Cosino E, Lee L, Blackwood EM, Solon M, Tao J, Li L, Shames D, et al. (2013). Lactate Dehydrogenase B Is Required for the Growth of KRAS-Dependent Lung Adenocarcinomas. Clinical Cancer Research 19, 773–784. [DOI] [PubMed] [Google Scholar]
- Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, and Swords R (2017). Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 31, 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al. (2010). Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2, 31ra34. [DOI] [PubMed] [Google Scholar]
- Moon JS, Kim HE, Koh E, Park SH, Jin WJ, Park BW, Park SW, and Kim KS (2011). Kruppel-like factor 4 (KLF4) activates the transcription of the gene for the platelet isoform of phosphofructokinase (PFKP) in breast cancer. J Biol Chem 286, 23808–23816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TA, Dang CV, and Young JD (2013). Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab Eng 15, 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. (2016). Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 374, 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, et al. (2012). Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 366, 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathria G, Scott DA, Feng Y, Sang Lee J, Fujita Y, Zhang G, Sahu AD, Ruppin E, Herlyn M, Osterman AL, et al. (2018). Targeting the Warburg effect via LDHA inhibition engages ATF4 signaling for cancer cell survival. EMBO J 37, e99735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan LM, Yeung SC, and Lee MH (2014). Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer biology & medicine 11, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollyea DA, Dinardo CD, de Botton S, Stein E, Roboz GJ, Mims AS, Swords RT, Altman JK, Collins R, Mannis GN, et al. (2018). Ivosidenib (IVO; AG-120) in mutant IDH1 relapsed/refractory acute myeloid leukemia (R/R AML): Results of a phase 1 study. Journal of Clinical Oncology 36, 7000–7000. [Google Scholar]
- Porporato PE, Dhup S, Dadhich RK, Copetti T, and Sonveaux P (2011). Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol 2, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme PT, Wernette-Hammond ME, Kneer NM, and Lardy HA (1984). Mechanism of action of 2,5-anhydro-D-mannitol in hepatocytes. Effects of phosphorylated metabolites on enzymes of carbohydrate metabolism. J Biol Chem 259, 5115–5123. [PubMed] [Google Scholar]
- Roboz GJ, Dinardo CD, Stein EM, de Botton S, Mims AS, Prince GT, Altman JK, Arellano ML, Erba HP, Pollyea DA, et al. (2019). Ivosidenib (IVO; AG-120) in IDH1-mutant newly-diagnosed acute myeloid leukemia (ND AML): Updated results from a phase 1 study. Journal of Clinical Oncology 37, 7028–7028. [Google Scholar]
- Rodriguez S, Jafer O, Goker H, Summersgill BM, Zafarana G, Gillis AJ, van Gurp RJ, Oosterhuis JW, Lu YJ, Huddart R, et al. (2003). Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2-p12.1. Oncogene 22, 1880–1891. [DOI] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. (2013). An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Martínez C, and J. Aragón J (1997). Analysis of phosphofructokinase subunits and isozymes in ascites tumor cells and its original tissue, murine mammary gland. FEBS letters 409, 86–90. [DOI] [PubMed] [Google Scholar]
- Shi HL, Wang X, Lu ZK, Zhao BXS, Ma HH, Hsu PJ, Liu C, and He C (2017). YTHDF3 facilitates translation and decay of N-6-methyladenosine-modified RNA. Cell research 27, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz GA, Furtado CM, Sola-Penna M, and Zancan P (2009). Acetylsalicylic acid and salicylic acid decrease tumor cell viability and glucose metabolism modulating 6-phosphofructo-1-kinase structure and activity. Biochem Pharmacol 77, 46–53. [DOI] [PubMed] [Google Scholar]
- Stein EM, DiNardo CD, Fathi AT, Pollyea DA, Stone RM, Altman JK, Roboz GJ, Patel MR, Collins R, Flinn IW, et al. (2019). Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 133, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, et al. (2017). Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130, 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, and Dang CV (2015). MYC, Metabolism, and Cancer. Cancer Discov 5, 1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al. (2018). R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 172, 90–105 e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, Deng X, Li H, Huang Y, Gao L, et al. (2020). Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 38, 79–96 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashi K, Kayoko S, Izumi T, Shinji K, Nishio H, Yoshiro N, and Kunio O (1980). Hereditary deficiency of lactate dehydrogenase M-subunit. Clinica Chimica Acta 108, 267–276. [DOI] [PubMed] [Google Scholar]
- Tandon P, Gallo CA, Khatri S, Barger JF, Yepiskoposyan H, and Plas DR (2011). Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency. Proceedings of the National Academy of Sciences of the United States of America 108, 2361–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG (2011). Targeting cancer metabolism: a therapeutic window opens. Nature reviews. Drug discovery 10, 671–684. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. (2013). Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626. [DOI] [PubMed] [Google Scholar]
- Wang J, Li Y, Wang P, Han G, Zhang T, Chang J, Yin R, Shan Y, Wen J, Xie X, et al. (2020). Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell 27, 81–97 e88. [DOI] [PubMed] [Google Scholar]
- Wang TL, Hong TT, Huang Y, Su HM, Wu F, Chen Y, Wei L, Huang W, Hua XL, Xia Y, et al. (2015a). Fluorescein Derivatives as Bifunctional Molecules for the Simultaneous Inhibiting and Labeling of FTO Protein. Journal of the American Chemical Society 137, 13736–13739. [DOI] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014a). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015b). N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG, and Scadden DT (2014b). Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158, 1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1925). über den Stoffwechsel der Carcinomzelle. Klinische Wochenschrift 4, 534–536. [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G (1977). Enzymology of cancer cells (second of two parts). N Engl J Med 296, 541–551. [DOI] [PubMed] [Google Scholar]
- Wunderlich M, Brooks RA, Panchal R, Rhyasen GW, Danet-Desnoyers G, and Mulloy JC (2014). OKT3 prevents xenogeneic GVHD and allows reliable xenograft initiation from unfractionated human hematopoietic tissues. Blood 123, e134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. (2009). IDH1 and IDH2 mutations in gliomas. N Engl J Med 360, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metabolomic analysis results are available in Tables S1–S4. This study did not generate any unique code.