

Abstract
Selumetinib, a highly specific MEK1/2 inhibitor, is approved for children older than 2 years of age with neurofibromatosis 1 (NF1) who have inoperable plexiform neurofibromas. By selectively binding to MEK1/2 proteins, selumetinib can arrest the mitogen-activated protein kinases/extracellular signal-regulated kinases signaling pathway that regulates critical cellular responses. Selumetinib has shown promising results as single-agent or in combination with conventional chemotherapy and other targeted therapies both preclinically and clinically, in multiple cancers including pediatric low-grade glioma (pLGG), non-small cell lung cancer, and melanoma, among others. The pharmacokinetic profiles of selumetinib and its active metabolite N-desmethyl selumetinib have been well characterized in both adults and children. Both compounds exhibited rapid absorption and mean terminal elimination half-lives about 7.5 hours, with minimal accumulation at steady state. Three population pharmacokinetic models have been developed in adults and children, characterizing large inter- and intra-patient variabilities, and identifying significant covariates including food intake on selumetinib absorption, weight metrics, age, co-administration of cytochrome modulators, and Asian ethnicity on selumetinib apparent oral clearance. The most common side effects associated with selumetinib are dermatologic, gastrointestinal toxicities and fatigue. Most toxicities are mild or moderate, generally tolerated and manageable. Cardiovascular and ocular toxicities remain less frequent but can be potentially more severe and require close monitoring. Overall, selumetinib exhibits favorable pharmacokinetic properties and safety profile, with promising activity in multiple solid tumors, supporting current and further evaluation in combination with conventional chemotherapy and other targeted agents.
1. Introduction
The mitogen-activated protein kinases/extracellular signal-regulated kinases (MAPK-ERK) signaling pathway regulates critical cellular responses including cell proliferation and survival, and is dysregulated in about 30% of cancers, which makes it an attractive therapeutic target [1, 2]. Inhibition of the MEK1/2 (mitogen-activated protein kinase kinase 1/2) proteins, a key component of MAPK-ERK pathway, has led to the development of promising and specific compounds currently being tested in clinical trials for various indications, such as trametinib, cobimetinib, and selumetinib [3, 4].
Selumetinib (AZD6244, ARRY-142886, Koselugo®) is an oral, small molecule, specific ATP noncompetitive inhibitor of MEK1/2 proteins, which has demonstrated cell proliferation arrest and increased apoptosis in various tumor cell lines in preclinical studies [5, 6]. Selumetinib has been evaluated as monotherapy or in combination in multiple adult trials with a maximum tolerated dose (MTD) of 75 mg/dose twice daily. Selumetinib has also been evaluated in children with solid tumors, including central nervous system (CNS) tumors. In April 2020, selumetinib received FDA-approval, at a dosage of 25 mg/m2/dose twice daily, for the treatment of children ≥ 2 years of age with neurofibromatosis type 1 (NF1) who have symptomatic and inoperable plexiform neurofibromas [7]. This is the first therapy approved for this often debilitating indication which is caused by a spontaneous or inherited mutation in the NF1 gene.
This review aims to provide a comprehensive summary of the clinical pharmacokinetic and pharmacodynamic properties of selumetinib. The mechanism of action, the pharmacokinetic characteristics in healthy volunteers and special populations, the clinical efficacy and toxicities observed in the main therapeutic indications are discussed.
2. Search strategy
A MEDLINE database search using PubMed was performed to identify relevant publications. The following keywords were used: “((selumetinib) AND ((pharmacokinetics) OR (pharmacodynamics) OR (activity) OR (population) OR (toxicity) OR (mechanism)))”. The search was limited to English articles. Additional studies were identified with the reference lists of selected articles. The American and European clinical trial databases were used to identify the clinical trials evaluating selumetinib mono- or combination therapy.
3. Structure and Mechanism of action
3.1. Chemical structure
Selumetinib is 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole- 5-carboxylic acid (2-hydroxy-ethoxy)-amide (C17H15BrClFN4O3) (Figure 1) [5]. Molecular weights of selumetinib free-base and its hydrogen sulfate salt are 457.7 and 555.8 g/mol. Selumetinib free-base has low permeability and aqueous pH-dependent solubility (274 μg/mL at pH 1; 3.4 μg/mL at pH 7.4) [8].
Figure 1:

Chemical structures of selumetinib (A) and its hydrogen-sulfate salt (B)
3.2. Mechanism of action
The MAPK-ERK (or Ras-Raf-MEK-ERK) pathway involves the specific series of serine/threonine protein kinases: Raf (MAPK), MEK1/2 (MAPKK), and ERK1/2 (MAPKKK) [1, 4]. The activation of the MAPK/ERK pathway has been well described previously and is depicted in Figure 2 [4, 9–11].
Figure 2:

Simplified representation of the MAPK-ERK1/2 signaling, NF1 pathway and selumetinib impact.
Activation of the pathway starts with extracellular growth and mitogenic factors binding to cell-membrane receptors tyrosine kinase (RTK), leading to their dimerization and autophosphorylation. Activated RTK recruit adaptor proteins (e.g., Grb2 and SOS) which activate Ras-GDP by inducing the GDP to GTP molecule change. Ras-GTP activates Raf which catalyzes the sequential activation of MEK1/2 and ERK1/2 through phosphorylation. Activated ERK1/2 proteins translocate into the nucleus to modulate cell proliferation, differentiation and survival, through the activation of transcription factors and the regulation of gene expression. ERK1/2 proteins also target cytosolic substrates to modulate cell migration and metabolism. The MAPK-ERK pathway is dependent on scaffold proteins and is tightly regulated by negative regulators (e.g., neurofibromin-mediated negative regulation of RAS), and feedback systems (e.g., ERK1/2-mediated feedback inhibition of Raf). Mutated NF1 leads to loss of neurofibromin function resulting in overactivation of MAPK-ERK pathway. MAPK(KK) mitogen-activated protein kinase, Grb2 growth factor receptor-bound protein 2, SOS Son of Sevenless, GDP/GTP guanosine diphosphate/triphosphate, ATP adenosine triphosphate, wt wild-type, mut mutated
Increased ERK1/2 signaling can be caused by an overexpression of growth factors and/or RTK, or by mutational activation or amplification of genes encoding Ras, Raf, and MEK1/2 [2, 12]. Inhibiting MEK1/2 kinases is of particular interest as they are the focal point of upstream activating mutations, only act on ERK1/2 kinases, and have a structure favoring the development of specific and selective inhibitors [4, 13]. Selumetinib binds to a unique and specific allosteric pocket on the N-terminal domain of MEK1/2, adjacent to a typical ATP binding site [4, 5, 13–15]. This leads to conformational changes preventing Raf-induced phosphorylation, and locking MEK1/2 into a catalytically inactive state, which thereby prevents the activation of ERK1/2 and arrests the MAPK signaling [13, 14].
In vitro enzymatic assays showed that selumetinib specifically inhibits the activity of MEK1/2 proteins (i.e., ERK phosphorylation) with an IC50 of 14.1 ± 0.79 nM, but fails to inhibit other kinases at concentrations up to 10 μM [5]. In preclinical studies of melanoma, colorectal, pancreatic, liver, breast and lung cancer cell lines, selumetinib arrested the MAPK-ERK signaling by inhibiting ERK1/2 phosphorylation (basal or induced through Ras/non-Ras processes), inhibiting cellular growth, arresting the cell-cycle in G1-S phase, and inducing apoptosis [5, 6]. However, the extent of cell growth inhibition and apoptosis induction largely differed between cell lines.
The impact on cell proliferation was strongly dependent on the cell Raf and Ras mutational state. Selumetinib was found more potent in inhibiting cell growth in cell lines containing activating B-Raf mutations and K- or N-Ras mutations with IC50 values <1 μM, while it had minimal effect in cell lines with wild-type B-Raf and Ras at concentrations up to 50 μM [5, 6]. A greater variability in sensitivity among Ras mutant cell lines is observed, explained by the implication of Ras in other Raf-independent signaling pathways. In vivo, selumetinib tumor growth inhibition was dose-dependent from 10-100 mg/kg oral bid dosing, correlated with inhibition of ERK1/2 phosphorylation, and required chronic dosing of selumetinib [5, 6]. Selumetinib-induced apoptosis observed in vitro and in vivo occurred regardless of the B-Raf mutational status [5, 6]. Selumetinib triggers apoptosis through various ERK-dependent substrates whom expression and/or mutational status may vary between cell lines. In vivo tumor micro-environments may also modulate the cellular response to MEK signaling and explain the differences between cell lines [6].
Lastly, by inhibiting MEK1/2, selumetinib can remove the ERK1/2-mediated feedback inhibition on Raf [15]. However, this potentially leads to a rebound in the MAPK pathway activity by increasing the interactions between Raf and MEK1/2 in some Ras mutant tumor cells [15].
4. Pharmacokinetic properties
4.1. Dosing and formulation
In the first-in-human phase 1 study, selumetinib was administered orally, fasted, as a free-base suspension, “mix and drink” formulation, presented as a powder for reconstitution in an aqueous sulfobutyl ether cyclodextrin vehicle [16]. The MTD was established at 100 mg bid [16], and subsequent phase 2 studies were conducted [17, 18]. However, the low solubility, its potential dose-limited absorption, and the inconvenience of the extemporaneous solution led to a change in the formulation.
In a new first-in-human phase I study, selumetinib was administered fasted as a 25-mg solid oral white hypromellose (“Phase II”) capsule incorporating a hydrogen sulfate salt in a D-α-tocopheryl polyethyleneglycol-1000 succinate [19]. The MTD was determined at 75 mg bid [19]. The capsule (75 mg) showed an improved bioavailability of 263% relative to the free-base suspension (100 mg).Similar time to maximum concentration (TMAX), apparent clearance (CL/F) and volume (V/F) were observed between the two formulations. The maximum concentration (CMAX) and exposure (area under the curve AUC0-24h) increased by 252% and 197%, respectively, with the capsule relative to the free-base suspension [19]. Before the start of pivotal trials, other types of capsule were investigated, leading to the Phase III blue capsules [20, 21]. The new capsule did not result in a change of selumetinib pharmacokinetic properties; thus, did not require dose adjustments [20]. Since, the clinical development of selumetinib has continued with the Phase III 10-mg and 25-mg capsules. The following sections will focus on the pharmacokinetic parameters reported with the capsule formulation.
4.2. Absorption
The first phase 1 studies with selumetinib administered fasted showed the drug was absorbed quickly, in a dose-proportional manner [8, 19, 22]. Following single 25-100 mg doses, mean selumetinib TMAX was between 1-2 hours, and mean selumetinib CMAX ranged from 1051-1726 ng/mL at the MTD 75 mg (Table 1). Typical pharmacokinetic profiles of selumetinib are depicted in Figures 3A–B.
Table 1.
Non-compartmental pharmacokinetic parameters estimated for selumetinib administered as oral capsule and N-desmethyl-selumetinib
| Study identifier | N patients (age range) Description | Selumetinib dosing | Selumetinib | N-desmethyl selumetinib | ||||
|---|---|---|---|---|---|---|---|---|
| CMAX (ng/mL) | AUC (h·ng/mL) | CL/F (L/h) | Half-life (h) | CMAX (ng/mL) | AUC (h·ng/mL) | |||
| Healthy volunteers (HV) | ||||||||
| NCT01931761 Phase I [22] | N=6 (52-61 yo) PK profile of [14C]-drug |
75 mg SD | 1520 (28.5) | 4510(19.2) | 15.7 ±2.87 | 13.7 ±5.04 | 109 (23.9) | 351 (23.1) |
| NCT01974349 Phase I [20] | N=34 (19-44 yo) Food effect |
Fasted 75 mg SD | 1430 (36.1) | 4160 (25.3) | 18 (25.3) | 8.3 (1.8) | ||
| Fed 75 mg SD | 711 (33.1) | 3490 (24.8) | 21.5 (24.7) | 8.0 (2.4) | ||||
| NCT01635023 Phase I [20] | N=27 (19-54 yo) Phase II and III capsules |
Phase II 75 mg SD | 1390 (40.4) | 4060 (23.9) | 19 (23.9) | 7.5 (21.8) | ||
| Phase III 75 mg SD | 1150 (47.8) | 3680 (32.5) | 21.5 (32.5) | 7.2 (19.8) | ||||
| NCT01960374 Phase I [30] | N=72 (28.5-41 yo) Effect of ethnicity (Asian vs Western) |
Japanese 35 mg SD | a1005 (31.0) | a2968 (21.0) | 12.0 ±2.56 | 11.5 ±3.96 | ||
| Non-Japanese 50 mg SD | a1215 (32.3) | a3945 (25.4) | 13.0 ±2.94 | 9.8 ±2.58 | ||||
| Indian 25 mg SD | a702.5 (16.0) | a1738 (16.7) | 14.6 ±2.32 | 9.5 ±3.31 | ||||
| NCT02093728 Phase I [31] | N=26 (18-44 yo) Effects of CYP3A4 and 2C19 inhibitors |
25 mg SD | 409 (36.8) | 1180 (27.2) | 21.9 ±5.76 | 8.24 ±2.07 | 35.7 (37.6) | 83.4 (30.0) |
| 25 mg SD + Itraconazole | 484 (30.8) | 1760 (31.8) | 14.9 ±4.92 | 14.0 ±7.77 | 27.0 (29.4) | 74.6 (29.2) | ||
| 25 mg SD +Fluconazole | 509 (34.0) | 1770 (33.4) | 14.8 ±4.39 | 9.75 ±3.14 | 37.5 (33.6) | 117 (30.4) | ||
| NCT02046850 Phase I [31] | N=22 (18-44 yo) Effect of CYP3A4 inducer |
75 mg SD | 1440 (36.5) | 4480 (19.9) | 17.0 ±3.33 | 9.27 ±2.57 | 106 (41.8) | 404 (27.1) |
| 75 mg SD + Rifampicin | 1070 (38.3) | 2200 (23.4) | 35.0 ±8.46 | 6.69 ±2.33 | 86.9 (40.4) | 170 (28.4) | ||
| NCT02056392 Phase I [25] | N = 50 (18-45 yo) Effect QT intervals |
75 mg SD | 1240 (33.1) | 3980 (24.6) | 19.0 ±4.66 | 9.18 ±2.64 | 90.4 (38.3) | 285 (29.3) |
| Adults patients | ||||||||
| NCT00463814 Phase I [19] | N = 59 (26-79 yo) FIH study with capsule formulation Advanced cancers |
25 mg bid SD 25 mg bid MD |
369 (268-533) 458 (326-678) |
1361 (1060-1910) 1515 (1310-1870) |
18.8 (13.1-24) 16.5 (13.3-19) |
6.9 (5.8-8.3) | ||
| 50 mg bid SD 50 mg bid MD |
1036 (570-1800) 987 (532-1440) |
3075 (1500-6430) 3381 (2040-6350) |
18 (7.8-33.3) 15.8 (7.9-24.5) |
7.2 (4.9-10) | ||||
| 75 mg bid SD 75 mg bid MD |
1207 (611-2000) 1439 (922-1770) |
6335 (5260-8510) 5448 (2940-7830) |
12.1 (8.8-14.2) 14.8 (9.6-25.5) |
5.3 (3.9-7.4) | 78 (36.8-173) 67 (20.2-115) |
820 (417-1520) 365 (324-412) |
||
| 100 mg bid SD 100 mg bid MD |
1483 (235-2690) 1365 (856-2630) |
7055 (4260-12500) 4758 (2710-9590) |
15.1 (8.0-23.4) 23.2 (10.4-37) |
6.8 (3.9-8.7) | 97 (14.3-245) 65 (43.2-187) |
506 (374-696) 321 (238-407) |
||
| NCT02063204 Phase I [27] | N=24 (50±6 yo) Effect of renal IP HV and ESRD patients |
HV 50 mg SD | 863 (41.5) | 2620 (22.3) | 19.1 (22.3) | 7.34 (2.90) | 53.4 (52.1) | 186 (35.0) |
| Pre-dialysis 50 mg SD | 507 (50.2) | 1560 (44.2) | 32.2 (44.2) | 5.81 (2.09) | 38.5 (57.7) | 153 (71.5) | ||
| Post-dialysis 50 mg SD | 725 (45.8) | 1880 (35.9) | 26.6 (36.0) | 6.55 (1.78) | 45.1 (40.4) | 171 (50.9) | ||
| NCT02063230 Phase I [27] | N=32 (56±6 yo) Effect of hepatic IP HV and subjects with hepatic IP |
HV 50 mg SD | a945 (37.0) | a2680 (37.3) | 18.6 (37.3) | 8.02 ±1.94 | a90.5 (57.0) | a242 (43.4) |
| Mild IP 50 mg SD | a740 (47.0) | a2295 (39.0) | 21.8 (38.9) | 7.72 ±2.28 | a40.2 (57.7) | a122.5 (41.1) | ||
| Moderate IP 50 mg SD | a1180 (37.4) | a4255 (38.3) | 11.7 (45.5) | 9.92 ±2.63 | a27.4 (102.8) | a91.5 (129.6) | ||
| Severe IP 20 mg SD | a370 (49.9) | a1686 (20.1) | 11.9 (19.9) | 9.02 ±1.28 | a3.46 (44.3) | ND | ||
| NCT01605916 Phase I [34] | N= 25 (32-88 yo) Selumetinib + docetaxel in Japanese Advanced solid tumors |
25 mg bid SD 25 mg bid MD |
630.3 (96.4) 642.6 (36.2) |
1570 (39.7) 2230 (30.7) |
15.9 ± 6.5 | 10.5 ± 5.7 | ||
| 50 mg bid SD 50 mg bid MD |
897.1 (69.3) 1012 (47.1) |
3556 (30.7) 4818 (20.1) |
14.4 ± 4.1 | 9.6 ± 2.3 | ||||
| 75 mg bid SD 75 mg bid MD |
2237 (46.2) 2261 (70.0) |
7362 (34.5) 9984 (49.0) |
10.6 ± 3.8 | 9.2 ± 2.7 | ||||
| NCT00600496 Phase I [32] | N=80 (59.1 ±9.9 yo) | 50 mg SD 75 mg SD |
92 (69.1) 1051 (43.6) |
b2699 (32.67) b3197 (39.80) |
41.7 (60.6) 66.8 (75.7) |
b173.4 (39.8) b223.9 (34.9) |
||
| Selumetinib + erlotinib or temsirolimus Advancer cancers |
50 mg SD + Erlotinib 75 mg SD + Erlotinib |
1334 (51.2) 1657 (55.7) |
b4011 (29.56) b4826 (36.70) |
43.4 (47.6) 72.9 (91.0) |
b153.3 (39.4) b262.7 (53.3) |
|||
| 50 mg SD 75 mg SD |
666 (108.9) 1237 (60.0) |
b2147 (59.92) b3368 (38.32) |
35.3 (109.0) 75.3 (88.6) |
b168.2 (54.2) b269.4 (62.6) |
||||
| 50 mg SD + Temsirolimus 75 mg SD + Temsirolimus |
675.5 (63.1) 887.4 (65.7) |
b2128 (48.74) b2938 (51.63) |
38.5 (81.6) 48.4 (65.6) |
b168.2 (57.9) b190.5 (55.1) |
||||
| NCT00600496 Phase 1 [33] | N=60 (58.9±9.5 yo) Selumetinib + docetaxel or dacarbazine Advancer cancers |
50 mg SD 75 mg SD |
382.5 (114.5) 1165 (62.3) |
b1479 (45.02) b2999 (42.33) |
31.8 (60.0) 69.6 (49.7) |
b151.2 (37.4) b236.5 (36.1) |
||
| 50 mg SD + Docetaxel 75 mg SD + Docetaxel |
576.6 (41.4) 1215 (70.1) |
b2056 (30.40) b5239 (31.04) |
29.4 (175.6) 40.9 (52.1) |
b258.5 (42.5) b172.7 (38.1) |
||||
| 50 mg SD 75 mg SD |
933.5 (43.4) 1537 (45.5) |
b2652 (22.38) b4210 (31.55) |
47.0 (77.3) 71.2 (69.7) |
b196.0 (70.1) b261.3 (40.6) |
||||
| 50 mg SD + Dacarbazine 75 mg SD + Dacarbazine |
692.7 (56.3) 1343 (74.4) |
b2822 (42.48) b5317 (43.00) |
14.9 (162) 24.9 (96.5) |
b277.9 (2.1) b146.4 (58.3) |
||||
| NCT02143466 Phase Ib [35] | N=36 (32-79 yo) Selumetinib + osimertinib EGFR-mutant lung cancer |
50 mg cont. SD 50 mg cont. MD 75 mg cont. SD 75 mg cont. MD |
1143 (54.9) 1308 (80.7) 1051 (41.4) 1750 (NA) |
c2792 (31.7) c3743 (40.0) c3933 (74.2) c5970 |
2.4 ±0.65 1.4 ±0.5 2.6 ±0.56 1.05 |
|||
| Asian 25 mg cont. SD 25 mg cont. MD 50 mg cont. SD 50 mg cont. MD |
379.5 (92.5) 485.3 (8.0) 1033 (67.3) 716.9 (58.6) |
c1450 (32.4) c1340, 1440 c2834 (56.5) c3260, 3660 |
2.2 ±0.35 0.7, 0.86 2.3 ±0.37 0.86, 2.6 |
|||||
| NCT02188264 Phase Ib [41] | N=39 (18-64 yo) +14 >65 yo Selumetinib + cisclosporin A Advanced Solid Tumors |
50 mg SD 75 mg SD |
- 1550 |
d2475 ±1129 d4396 ±2078 |
- 6.4 |
|||
| 50 mg SD + CsA 75 mg SD + CsA |
1250 |
d2269 ±872 d3865 ±1721 |
||||||
| NCT01809210 Phase I/Ib [42] | N=55 (39-76 yo) Selumetinib + platinum-doublet chemotherapy Advanced NSCLC |
50 mg bid MD | 1176 ±654 | 3761 ±1622 | 14.9 ±6.0 | |||
| 75 mg bid MD | 1481 ±707 | 4256 ±1470 | 19.3 ±6.7 | |||||
| 100 mg bid MD | 2408 ±911 | 5691 ±2754 | 20.9 ±9.8 | |||||
| NCT01029418 Phase Ib [44] | N=27 Asian (43-74 yo) Selumetinib+ sorafenib Advanced HCC |
50 mg bid MD | 500 (129) | c3081 (72) | 14.9 (50) | 5.1 (2.4-9.1) | 44 (112) | c342 (42) |
| 75 mg bid MD | 1270 (92) | c5933 (54) | 12.2 (78) | 4.5 (1.8-7.5) | 94 (99) | c543 (67) | ||
| 100 mg bid MD | 1407 (83) | c9173 (28) | 10.6 (28) | 5.0 (4.5-5.1) | 94 (52) | c852 (29) | ||
| NCT01287130 Phase I [24] | N=33 (27-89 yo) Selumetinib + cetuximab Solid tumors |
50 mg qd SD | 1324 (13) | 3213 (23) | 23.9 ± 5.48 | 2.28 ± 0.55 | ||
| 50 mg bid SD | 1143 (28) | 3614 (12) | 21.0 ± 2.53 | 2.31 ± 0.55 | ||||
| 75 mg bid SD | 1504 (40) | 5200 (58) | 16.4 ± 7.08 | 2.97 ± 0.89 | ||||
| NCT01242605 Phase Ib [43] | N=11 (45-81 yo) Selumetinib + CisGem Biliary tract cancer |
75 mg qd SD | 1331.3 | 5653.3 | 77.16 | 452.9 | ||
| 75 mg qd SD + CisGem | 1725.6 | 6286.3 | 48.41 | 277.6 | ||||
| NCT01061749 Phase I [45] | N=13 (37-82 yo) Selumetinib + cixutumumab Solid tumors |
50 mg bid SD | 1093 ±231 | d2822±694 | 73.5±37.6 | d210 ±77 | ||
| NCT01021748 Phase I [46] | N=62 (33-81 yo) Selumetinib + MK-2206 Solid tumors |
100 mg qd SD | 1140 (554-2540) | e4500 (2297-7875) | ||||
| NCT00710515 Phase I [8] | N=30 (32-77 yo) Food effect Advanced solid tumors |
Fasted 75 mg SD | 1450 | 5782 | 12.9 (29.9) | 9.4 (33) | 73.9 (45.7) | 418.4 (36.5) |
| Fed 75 mg SD | 557 | 4664 | 16.0 (35.8) | 8.6 (44.5) | 33.5 (63.8) | 379.1 (49) | ||
| Pediatric patients | ||||||||
| NCT01362803 Phase I [40] | N=18 (3-18.5 yo) NF1 and inoperable plexiform neurofibromas |
20 mg/m2 SD | 826 ±377 | 2343 ±449 | f9.4 ±1.0 | 9.3 ±4.7 | 68.6 ±38.5 | 216.3 ±60.3 |
| 25 mg/m2 SD | 940 ±180 | 2665 ±429 | f10.1 ±1.2 | 6.0 ±1.1 | 58.7 ±22.3 | 178 ±39.8 | ||
| 30 mg/m2 SD | 1059±517 | 3434 ±1494 | f10.0 ±3.2 | 7.0 ±1.2 | 63.3 ±36.6 | 206.6 ±72.8 | ||
| NCT01362803 Phase II [49] | N=50 (3.5-17.4 yo) Inoperable Plexiform Neurofibroma |
25 mg/m2 bid SD (25 mg) 25 mg/m2 bid MD (25 mg) |
a650 ± 622 a715 ± 565 |
a1883 ± 1200 a2120 ± 1278 |
f12.4 ± 4.8 f10.8 ± 4.1 |
2.55 ± 0.64 |
a50 ± 38 a30.4 ± 28 |
a156.3 ±92.8 a136 ± 79.5 |
| NCT01089101 Phase II [38] | N=23 (3.5-20.8 yo) LGG |
25 mg/m2 SD | 786.8 (420-2838) | d1812 (1585-2266) | 10.9 (9.0-16) | d150 (122-204) | ||
| NCT01089101 Phase I [37] | N=32 (5.6-20.8 yo) Recurrent or refractory LGG |
25 mg/m2 SD | 1400 (306-3570) | 3855 (1780-7250) | f6.5 (3.4-14.0) | 6.5 (4.7-15.8) | 82 (23-264) | 112 (276-750) |
| 33 mg/m2SD | 1750 (372-1860) | 7325(4747-14021) | f4.5 (2.4-7.0) | 10.4 (5.4-23) | 68 (10-92) | 286 (173-913) | ||
| 43 mg/m2 SD | 3430 (3400-3460) | 9109(7978-10231) | f4.8 (4.2-5.4) | 4.8 (4.3-5.3) | 152 (107-196) | 600 (509-691) | ||
Data are reported eitner as mean (CV%), mean + sd, or median (min-max).
CisGem cisplatin-gemcitabine, CsA cisclosporin A, ESRD end-stage renal disease, FIH first-in-human study, HCC hepatocellular carcinoma, HV healthy volunteers, IP impairment, LGG low-grade glioma, NF1 neurofibromatosis type 1, SD /MD single / multiple dose Unless stated otherwise:
Unless stated otherwise:
- Subjects received selumetinib dose(s) in a fasted state
- The majority of subjects included in the studies were Caucasians or Black
- The AUCs reported after single dose are the AUC0-∞, and the AUCs reported after multiple doses are AUC0-12h
Dose-normalized CMAX (ng/mL/mg) and AUC (h.ng/mL/mg) values were reported in original references.
AUC0-12h
AUC0-24h
AUC0-8h
AUC0-10h
L/h/m2
Figure 3:
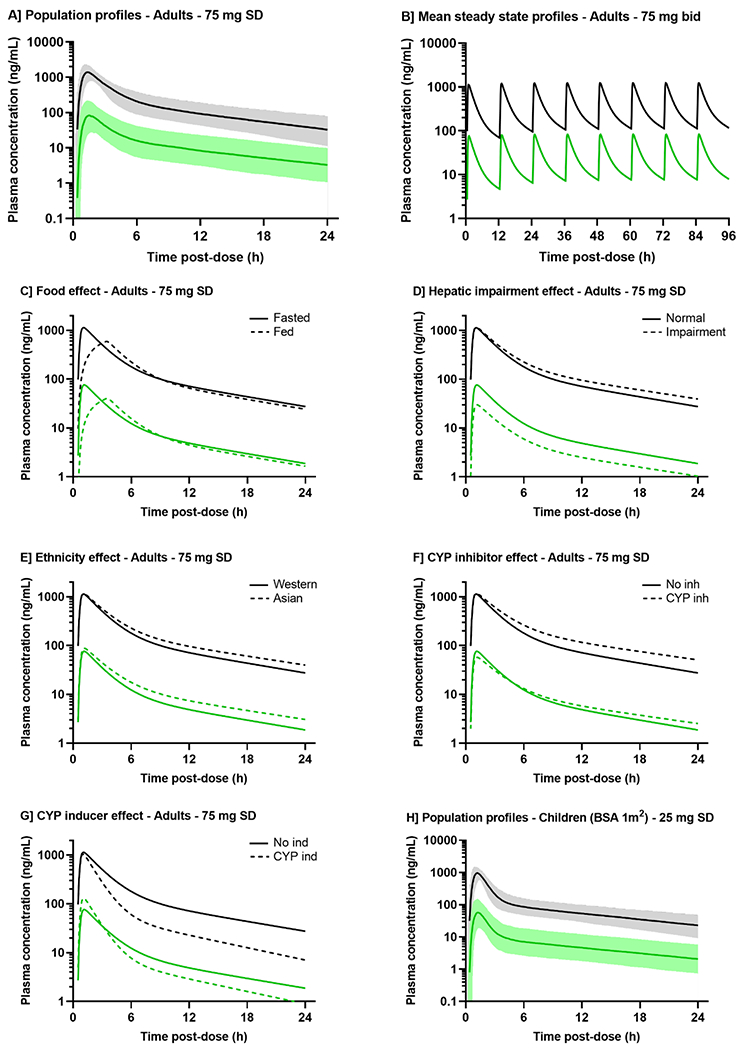
Simulated representative pharmacokinetic profiles of selumetinib (black) and N-desmethyl selumetinib (green).
A] Population predictions simulated based upon the model developed by Patel et al. (2017), for typical Caucasian adults of 30-year-old and 1.73 m2 BSA, receiving single dose of 75 mg selumetinib, fasted with no concomitant treatment. Solid line and shaded area are the mean and 90th prediction interval. For B-G] panels, the mean parameters estimated in the model by Patel et al. (2018) were used to simulate the pharmacokinetic profiles for typical adults (30-year-old, 70 kg) receiving 75 mg selumetinib: twice daily every 12 hours B] or once in fasted or fed state C], subjects with normal liver function or hepatic impairment D], Western or Asian subjects E], with or without a CYP inhibitor (itraconazole or fluconazole) F], with or without a CYP inducer rifampicin G]. H] Population predictions simulated based upon the model developed by Patel et al. (2017), for typical Caucasian children of 9-year-old and 1.0 m2 BSA, receiving single dose of 75 mg selumetinib, fasted with no concomitant treatment.
The absolute oral bioavailability was determined in two studies involving healthy volunteers receiving 75 mg oral and/or 80 μg intravenous [14C]-selumetinib [21, 22]. Mean absolute bioavailability ranged from 62%-71%. These data were also integrated in two population models, resulting in estimated bioavailability of 61% and 68% [21, 23].
Food intake effect on absorption was investigated in healthy volunteers and patients receiving one dose of 75 mg selumetinib in overnight fasted and fed (FDA-approved high-fat breakfast) states in two phase I studies [8, 20]. Both studies showed food intake significantly reduced the extent and rate of absorption with no impact on drug half-life (Figure 3C). After a high-fat meal, mean selumetinib CMAX and AUC0-∞ decreased by 49.8-62% and 16-19%, respectively, and median TMAX was delayed by 1.5-2.5 hours compared to in fasted state [8, 20]. The food impact is more limited on AUC vs CMAX, suggesting the fraction of drug available for absorption remain similar in both fed and fasted states [20]. Those data supported continuing administering selumetinib as fasted: no food/drink other than water for 2 hours prior and at least 1 hour after dosing.
4.3. Distribution
4.3.1. General distribution
After reaching CMAX, selumetinib concentrations rapidly decline overtime in a bi-exponential fashion (Figure 3A). After single dose, V/F was consistent across studies and tested doses, with mean values ranging from 65.2 to 175 L, suggesting a good distribution in tissues [8, 19, 20, 22, 24, 25]. In mice receiving oral doses of 5 and 10 mg/kg, selumetinib rapidly distributed to various tested tissues: liver, gut, muscle, lung, skin, and subcutaneous melanoma tumor cells, in a dose-proportional manner [26].
In two phase 1 studies, the extent of selumetinib plasma protein binding was determined using the plasma ultrafiltrate method [27]. After one 50 mg dose, median bound fraction ranged from 99.54-99.67%, and wasn’t concentration or time dependent. Other studies showed ~98.4% selumetinib bound to plasma proteins, and more largely to albumin [28]. With such small unbound fraction, any minor changes may have a large impact on selumetinib exposure.
Minimal distribution of selumetinib into red blood cells was observed, and concentration ratios of blood-to-plasma ranged from 60%-70% with no time dependency, confirming plasma to be the adequate matrix to analyze selumetinib [22].
4.3.2. CNS penetration
The brain penetration of selumetinib and other MEK inhibitors was investigated preclinically [29]. In vitro concentration equilibrium transport assays showed that all tested MEK inhibitors were substrates for human and murine P-glycoprotein (ABCB1/MDR1/P-gp) and Breast cancer resistance protein (ABCG2/BCRP) transporters. In vivo studies were conducted in wild-type, Abcg2−/−, Abcb1a/b−/−, and Abcg2;Abcb1a/b−/− FVB mice with analysis of drug concentrations in blood and homogenized brain tissues one hour after clinically relevant dosage administration. After 5 mg/kg IV, and 1 to 5 mg/kg oral dosing in wild-type mice, selumetinib exhibited mean total brain-to-plasma ratios of 0.02, and 0.014-0.018, respectively, suggesting poor brain penetration at clinically achievable selumetinib plasma concentrations. After 5 mg/kg IV dosing in Abcg2−/−, Abcb1a/b−/−, and Abcg2;Abcb1a/b−/− mice, selumetinib exhibited mean total brain-to-plasma ratios of 0.018, 0.028, and 0.048 respectively. Despite a marked increase in Abcg2;Abcb1a/b−/− mice, selumetinib CNS penetration remained largely restricted.
4.4. Elimination
4.4.1. Metabolism
Selumetinib is mainly metabolized (i.e., ~56%) by Phase I oxidation through cytochrome P450 enzymes, mainly CYP3A4 and to a lesser extent 2C19, 1A2, 2C09, 2E1 enzymes. It is also metabolized by Phase II glucuronidation (i.e., ~29%) through UDP-glucuronosyltransferase UGT1A1 and 1A3 enzymes [22, 30]. A total of 15 metabolites M1-M15 were identified with molecular weights ranging from 383-633 g/mol [22].
The distribution of selumetinib and metabolites in plasma, urine, and feces were evaluated in healthy volunteers receiving oral 75 mg [14C]-selumetinib [22]. In plasma, the major circulating compounds were selumetinib (40%) and the amide glucuronide M2 (22%). In urine, M2 was the most abundant compound (~10%) while selumetinib was minimally detected (≤ 1%). In feces, the parent drug was the major compound (6 to 34%).
Although M2 is the major circulating metabolite, it is ~50-fold less active than selumetinib [8]. However, N-desmethyl-selumetinib (M8), primarily generated by demethylation via CYP1A2 and 2C19 enzymes, was found 3 to 5-fold more potent in inhibiting ERK1/2 phosphorylation than selumetinib [22, 25]. This active metabolite was further investigated in several clinical studies (Table 1) [19, 25, 31–33]. N-desmethyl-selumetinib displayed a similar pharmacokinetic profile to that of selumetinib (Figure 3A). After 75 mg selumetinib, N-desmethyl-selumetinib reaches CMAX ranging from 40.9-109 ng/mL at a mean TMAX of ~1.5 h, suggesting rapid metabolism (Table 1). The metabolite-to-parent systemic exposure ratio ranged from 5-15%, relatively consistent across studies and doses. The fraction of selumetinib metabolized in N-desmethyl-selumetinib was also estimated at 10-11% in the population pharmacokinetic models (Table 2) [21, 23]. The plasma bound fraction of the metabolite was reported slightly higher than that of selumetinib, from 99.34-99.29% [27]. Similarly, food intake has an impact on metabolite pharmacokinetics [8]. After a meal, mean CMAX and AUC of N-desmethyl-selumetinib decreased by 55% and 9%, respectively, and TMAX was delayed by ~3 hours.
Table 2.
Population pharmacokinetic modeling: main parameter estimates and covariate relationships
| Patel et al. 2017 [36] | Patel et al. 2018 [21] | Tong et al. 2019 [23] | ||
|---|---|---|---|---|
| Study population | Patients with solid tumors Children with low grade glioma |
Healthy volunteers | Healthy volunteers | NSCLC patients |
| Dosing administration | 75 mg oral bid SD or MD 25-33-43 mg/m2 oral bid MD |
20-75 mg oral SD 80 μg IV |
80 μg IV, 25-75 mg oral SD | 75 mg oral bid MD |
| Significant covariates | Age, BSA, ALT, food, steady state | Food, comedication, bilirubin, age, WT, race, Child-Pugh score | Food, comedication, WT | WT |
| PK Parameters [IIV%] [[IOV%]] and covariate relationships Selumetinib | ||||
| Lag time ALAG1 (h) | 0.32 + 0.35 · Fed [41] [[56]] | 0.4 [24.8] | 0.38 [30] | 0.41 [62.3] |
| Bioavailability F | 1 – 0.117 · Fed | Fasted / Fed: 0.68 / 0.58 | Fasted / Fed: 0.61 / 0.51 [52.3] | 0.61 |
| Duration of absorption D1 (h) | 0.622 + 4.09 · Fed [41] [[50]] | Fasted / Fed: 0.37 / 3.0 [99.8] | Fasted / Fed: 0.34 / 2.1 [103] | 0.35 [169.2] |
| Absorption rate ka (/h) | 3.7 | 6.4 | Fasted / Fed: 5.22 / 1.28 | 5.34 [59.2] |
| Clearance CL (L/h) | [26.5] [[15.8]] | [24.4] | 12.0 · (1 – 0.24 · FLU) · (1 – 0.30 · ITR) · (1 + 0.93 · RIF) [14.8] | 11.9 [31.7] [[43.2]] |
| Central volume V2 (L) | [45] [[44]] | [36.9] | [26.8] | [57.1] |
| Peripheral clearance Q (L/h) | 8.2 [54] | 6.2 | 8.3 | 8.8 |
| Peripheral volume V3 (L) | 55 [62] | 47.4 [26.9] | 52.3 [18.5] | 54 [139.9] |
| N-desmethyl-selumetinib | ||||
| Fraction metabolized FM | [40] [[34]] | [99.1] | 0.11 [17.8] | 0.10 [80.4] |
| Central clearance CLM/F (L/h) | 240 [39] | 17 [94.7] | 16.6 | 16.6 |
| Central volume V4/F (L) | V2/F | - | 1 fixed | 1 fixed |
| Peripheral clearance QM/F (L/h) | 49.5 | - | - | - |
| Peripheral volume V5/F (L) | 413 | - | - | - |
NSCLC non-small cell lung cancer, SD single dose, MD multiple doses, BSA body surface area, ALT alanine amino-transferase, SS steady state, JAP Japanese, NJAP non-Japanese Asian, CP1/2 Child Pugh score 1 / 2 or 3, FLU fluconazole, ITR itraconazole, RIF rifampicin, BIL bilirubin, WT total body weight.
IIV inter-individual variability as coefficient of variation (CV%), IOV inter-occasion variability as coefficient of variation (CV%)
In the analysis by Patel et al. (2017), F was fixed to 1 and the relative impact of food on F was estimated. All the clearance and volume parameters are apparent.
In the analysis by Patel et al. (2018), the ratio FM/V4 was estimated instead of FM.
In the analysis by Tong et al. (2019), a population pharmacokinetic model was first developed based upon data from phase I studies in healthy volunteers and served as prior information for the development of the patient model based upon data from a phase II trial and a phase III study.
The model structure was similar across the studies: a two-compartment model with delayed zero-first order absorption and first-order elimination for selumetinib, a linear one-compartment model (Patel et al. 2018, Tong et al. 2019) or a two-compartment (Patel et al, 2017) for N-desmethyl-metabolite.
4.4.2. Excretion
Selumetinib excretion predominantly occurred through hepatic metabolism. In healthy volunteers, 48 hours after oral dosing of 75 mg [14C]-radiolabeled selumetinib, ~50% of the radioactive dose was found in feces primarily (59%) and in urine (33%) and feces, and a near total dose recovery (~93%) was observed within 9 days [22]. Most of the dose was recovered as metabolites. The amount of unchanged selumetinib in feces (~19%) suggest both unabsorbed selumetinib removed by first-pass effects and selumetinib back-transformed from the metabolites via gut microbial degradation [22].
Selumetinib oral CL/F estimates were largely consistent across all studied doses, ranging from ~11-24 L/h (Table 1). The estimated terminal elimination half-life was also consistent across studied doses, with a median of 7.3 hours (2.5-11 hours). Similar terminal half-life values were reported for N-desmethyl-selumetinib, ranging from 7.7-12.7 hours [8, 19, 22, 25]. Those values were consistent with the minimal accumulation in both selumetinib and N-desmethyl-selumetinib exposures observed after repeated doses (Figure 3B). After 25 to 100 mg bid dosing, the accumulation ratios calculated from selumetinib AUC0-12h after single dose and at steady state ranged from 0.9-1.8 [34, 35].
5. Population pharmacokinetics
5.1. Adult population
No significant difference was found in selumetinib and N-desmethyl-selumetinib pharmacokinetics between healthy volunteers and patients with solid tumors (Table 1). Large inter-patient variabilities were observed for both compounds and quantified in three population models [21, 23, 36]. Selumetinib profile was described with a linear two-compartment disposition model and a lag-time-delayed sequential zero-first order absorption model, while a linear one or two-compartment model was used for the metabolite (Figure 4). High variabilities were specifically observed among the absorption parameters with coefficients of variation exceeding 50% (Table 2). This can be related to inter-patient differences in the amount and activities of CYP3A4, P-gp and BCRP enzymes, implicated in intestinal absorption and hepatic metabolism [20].
Figure 4.
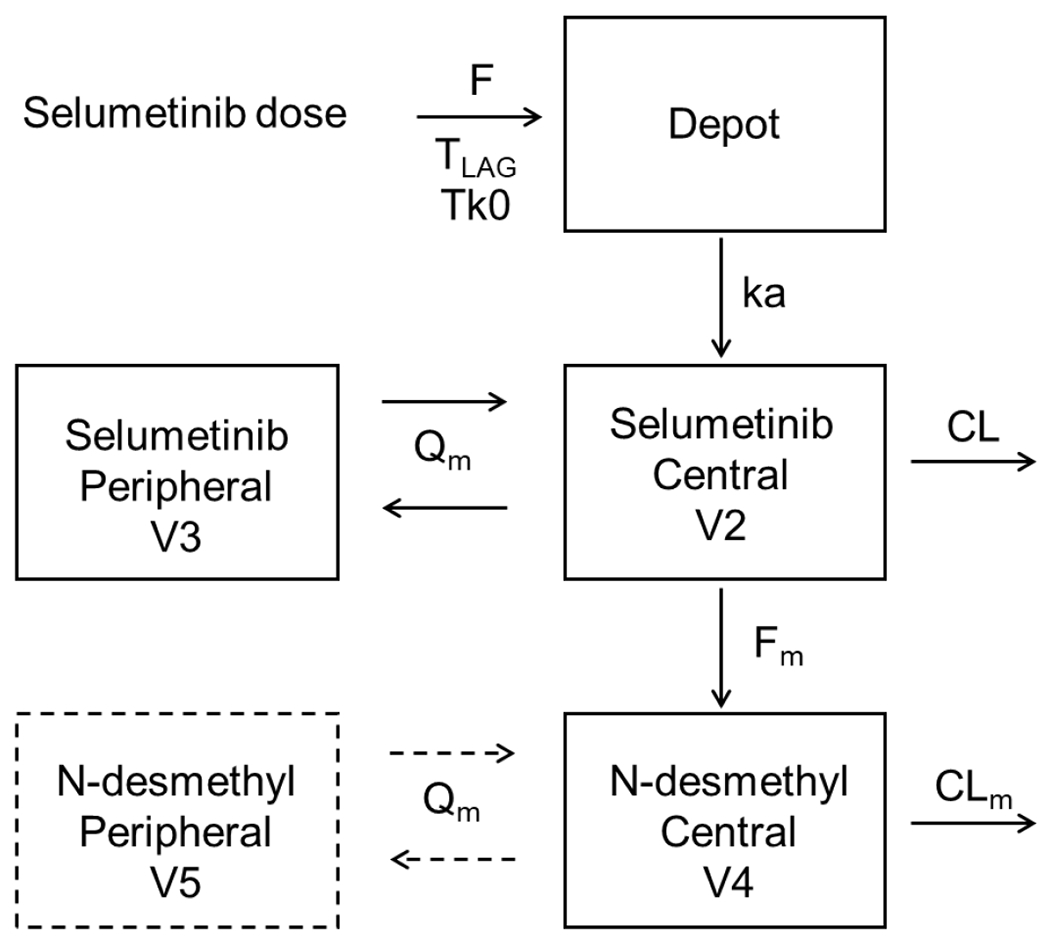
Selumetinib and N-desmethyl-selumetinib pharmacokinetic model structure.
Selumetinib was described by a two-compartment with delayed sequential zero-first order absorption and first-order elimination. N-desmethyl-selumetinib was described by a one- or two-compartment with first-order elimination. F bioavailability, TLAG absorption lag time, Tk0 duration of zero-order absorption, ka first-order absorption rate, CL/Q and CLm/Qm selumetinib and N-desmethyl-selumetinib apparent central/peripheral clearance, V2/V3 and V4/V5 selumetinib and N-desmethyl-selumetinib central/peripheral volume of distribution, Fm fraction metabolized in N-desmethyl-selumetinib.
In the adult population models, total bodyweight was included according to an allometric function on selumetinib CL/F, V/F, and/or on the metabolic fraction, with estimated or fixed (0.75 for CL/F and 1 for V/F) coefficients (Table 2). In those models, the subject ages (18-84 years old) correlated with selumetinib V/F [21, 23]. However, sensitivity analyses showed that total bodyweight and age only had minimal impact (<20%) on the overall exposure of selumetinib and its metabolite [21]. Therefore, no specific dose adjustments were suggested based on bodyweight or for elderly patients.
5.2. Pediatric population
Pharmacokinetic results were reported from two phase 1/2 studies conducted in children with recurrent/refractory or progressive low-grade glioma (LGG) or with inoperable plexiform neurofibromas [37–40]. Patients included in these studies ranged from 3 to 18.5 years of age. No significant difference in selumetinib and N-desmethyl-selumetinib pharmacokinetics was observed between children and adults, with superimposable plasma concentration-time profiles (Figures 3A and 3H). At the pediatric MTD 25 mg/m2 (~45 mg based on adult body surface area [BSA] of 1.8 m2), mean selumetinib AUC0-∞ and CMAX ranged from 1812-3855 h.ng/mL and 787-1400 ng/mL, respectively, comparable to exposures observed in adults at 50 mg (Table 1). Mean CL/F ranged from 4.5-12.4 L/h/m2 (~3.5-24 L/h) across dosages 20-43 mg/m2, in agreement with estimates in adults (Table 1). Pediatric terminal half-lives estimates were similar to adults (mean 2.55 to 10.4 hours). Selumetinib exposure proportionally increased with dose from 20-43 mg/m2, and minimal accumulation was observed after multiple doses. The metabolic-to-parent concentration ratio was also about 6-10% in pediatrics [39, 40]. In one study, BSA-adjusted CL/F positively correlated with age ranging from 3.5-17.4 years-old [39]. However, this association was not observed in the other studies with similar age range [37, 40].
Pharmacokinetic data collected in children with LGG were combined with data from adults in one population model [36]. Large inter-patient and intra-patient variabilities were quantified (Table 2). A sensitivity analysis showed that when children were dosed with BSA-based dosages, selumetinib steady state exposures varied between 95-104% of the median vs range of BSA values [36]. However, when dosed with flat dosages, steady state exposures were between 66-176% for the observed BSA range, highlighting the impact of BSA on drug clearance, and the importance of BSA-based dosages in pediatrics compared to flat-dosages in adults. The metabolic fraction was found to decrease at steady state in this population model (Table 2); however, the underlying mechanism was unclear.
5.3. Impact of food
As previously described, food intake significantly impacted selumetinib and N-desmethyl-selumetinib exposures. This effect was also quantified in the developed population models (Table 2). The food effect was implemented on the absorption parameters: lag time, bioavailability, zero-order and/or first-order rate, and led to delayed absorption and lower CMAX and AUC0-∞ (Figure 3C) [21, 23, 36].
5.4. Impact of renal and hepatic impairments
Renal impairment impact on selumetinib and N-desmethyl-selumetinib exposures was investigated in a phase 1 study with 12 healthy volunteers and 12 subjects with end-stage renal disease (ESRD) receiving 50 mg orally [27]. As expected, considering the minimal fractions of these drugs in urine, renal impairment did not significantly impact drug exposure (Table 1) [27]. Therefore, no dose adjustment is required for renal impaired patients.
The hepatic impairment phase 1 study included 8 healthy volunteers, 24 subjects with mild/moderate or severe hepatic impairment, dosed with 50 or 20 mg selumetinib [27]. A significant hepatic impairment effect was observed, as expected considering the large fraction of selumetinib excreted in feces. Mean dose-normalized selumetinib total AUC0-∞ was 59% and 57% higher in subjects with moderate and severe hepatic impairment compared with healthy subjects (Table 1, Figure 3D) [27]. Similar findings were observed with unbound plasma exposures. An increased Child-Pugh score, decreased serum albumin and increased prothrombin time were correlated with increasing unbound selumetinib exposure. Consistently, 49% and 62% lower N-desmethyl-selumetinib exposures were observed in patients with mild and moderate hepatic impairment, compared to healthy volunteers, while most concentrations were below the limit of quantification in patients with severe hepatic impairment (Table 1, Figure 3D) [27].
These data were included in a population model leading to the inclusion of two hepatic function-related covariates [21]. Bilirubin concentrations impacted selumetinib CL/F with an estimated 10% reduction in clearance for each unit rise in bilirubin levels (Table 2). The Child-Pugh score was associated with the fraction of selumetinib metabolized. A 34% and 54% reduction in N-desmethyl-selumetinib formation were estimated for patient with mild-to-moderate and severe hepatic impairment, respectively. In another population model, the alanine amino-transferase levels were associated with selumetinib clearance (Table 2) [36]. However, minimal changes in drug exposures were observed, as most of the population had normal liver function.
Therefore, caution should be used in patients with hepatic dysfunction. A dose reduction to 20 mg/m2 was suggested in children with at least moderate hepatic impairment [28].
5.5. Impact of ethnicity
The data from phase 1 studies conducted in Caucasian and Black subjects were pooled with one study including healthy Japanese, non-Japanese Asian, and Indian populations to explore the impact of race [30]. After 25-75 mg oral doses, no difference in exposure was found between Caucasian and Black subjects, but mean dose-normalized selumetinib AUC0-12h and CMAX were 35% and 39% higher in all Asian versus Western subjects. Among Asian subjects, minor differences (2-18%) were observed in selumetinib AUC0-12h and CMAX (Table 1) [30]. In another phase 1 study, Japanese patients with solid tumors receiving selumetinib with docetaxel, exhibited two-fold higher selumetinib exposures compared to those reported in Western patients (Table 1) [34]. Pharmacogenetic analyses were performed to further investigate associations between selumetinib pharmacokinetics and CYP2C19, UGT1A1 and ABCG2 variants [30]. However, none of the genetic polymorphisms showed any association with dose-normalized exposures.
Asian ethnicity was included as a categorical covariate in a population model on selumetinib CL/F and metabolic fraction (Table 2, Figure 3E) [21]. Typical clearance decreased by 17% and 12%, while the metabolic fraction increased by 36% and 23% in Japanese and non-Japanese Asian subjects, respectively. Despite these marked differences, no safety or tolerability concerns were observed in this population, and thus, no clinical dose adjustments in Asian subjects are currently recommended.
5.6. Impact of CYP inhibitors and inducers
The impact of potent CYP modulators was investigated in healthy volunteers dosed with 25, 35 or 75 mg selumetinib alone or combined with itraconazole/fluconazole (CYP3A4/2C19 inhibitors) or rifampicin (CYP3A4 inducer) [31]. The results were consistent with in vitro data identifying CYP3A4 and 2C19 responsible for selumetinib metabolism (Table 1) [31]. When co-dosed with itraconazole or fluconazole, selumetinib AUC0-∞ increased by ~49% and 53%, and CL/F decreased by one third. When co-dosed with rifampicin, selumetinib AUC0-∞ decreased by ~51%, and CL/F increased by two-fold. Different effects on N-desmethyl-selumetinib pharmacokinetics were observed: mean metabolite-to-parent exposure ratio decreased with itraconazole but remained similar with fluconazole and rifampicin co-administration [31].
The impact of CYP modulators was also quantified in two population modeling analyses [21, 23]. Administration of itraconazole, fluconazole, and rifampicin were associated with selumetinib CL/F only as categorical covariates (Table 2). Typical clearance decreased by 24-30% with co-treatment with CYP inhibitors but increased by 86-93% with rifampicin (Figure 3F–G). It is currently recommended to avoid concomitant administration of potent CYP3A4 or 2C9 modulators with selumetinib.
5.7. Drug-drug interactions
Unpublished in vitro data suggest selumetinib has low potential for any drug-drug interactions. Selumetinib has been evaluated in combination therapy for various solid tumors with multiple other agents. When combined with erlotinib, temsirolimus, docetaxel or dacarbazine [32, 33], osimertinib [35], ciclosporin A [41], gemcitabine, pemetrexed, carboplatin and/or cisplatin [42, 43], sorafenib [44], cetuximab, cixutumumab or MK-2206 [24, 45, 46], no clinically significant difference in selumetinib and/or co-administrated drug pharmacokinetic profiles was observed (Table 1).
6. Clinical efficacy
6.1. NF1-related tumors
NF1-related tumors such as plexiform neurofibromas and optic pathway gliomas (OPG) are caused by mutations in NF1 leading to the dysregulation of the MAPK-ERK pathway (Figure 2). In preclinical NF1-deficient mouse models of acute myeloid leukemia and neurofibroma, MEK inhibition resulted in significant reduction in tumor burden [40, 47, 48]. These results supported the evaluation of MEK inhibitors in NF1-related tumors, specifically, plexiform neurofibroma.
The first pediatric phase 1 trial enrolled 24 children (median age 10.9 years) with NF1 and inoperable plexiform neurofibroma [40]. The MTD was determined to be 25 mg/m2/dose twice daily, which is ~60% of the adult recommended phase 2 dose (RP2D) [19, 37, 40]. All patients showed tumor shrinkage, with a median reduction of 31%. Seventy-one percent of patients met criteria for partial response (PR), with durable PR sustained for a median of 23 cycles. No patients had disease progression while on trial [40].
These results led to a phase 2 trial (SPRINT study) enrolling 50 children (median age, 10.2 years) with inoperable NF1-plexiform neurofibroma at the MTD/RP2D of 25 mg/m2/dose twice daily [49]. Seventy-four percent of patients had radiographic PR. Twenty-eight patients had durable responses for at least one year with acceptable toxicities. Objective decreases in tumor size correlated with clinically meaningful benefit, evidenced by decrease in pain scores, improvement in daily functioning, improved health-related quality of life, and improved functional outcomes (strength and range of motion) [49]. These safety and efficacy data led to the regulatory approval of selumetinib by the FDA in 2020 for NF1-related inoperable plexiform neurofibromas.
6.2. Pediatric low-grade gliomas
Pediatric LGG (pLGG) are the most common CNS tumor in children, and despite excellent overall survival (OS), children with incompletely resected LGGs experience a high-rate of recurrence and significant tumor and treatment-related morbidities [50–52]. Recently, pLGG was shown as a specific MAPK-ERK pathway disease with the discovery of BRAF alterations in pilocytic astrocytoma and other alterations along this pathway [53]. Preclinical studies evaluating selumetinib in pediatric pilocytic astrocytoma xenograft models with BRAF mutation showed significant tumor regression and prolonged progression-free survival (PFS) while some adult early-phase clinical trials demonstrated excellent activity in patients with tumors harboring BRAF aberrations [54–56]. In the Pediatric Brain Tumor Consortium (PBTC) phase 1 study in recurrent/refractory pLGG, the two-year PFS of the 25 patients treated at the MTD/RP2D of 25 mg/m2/dose twice daily, was 69%; five patients sustained PR (20%), out of whom, four had known BRAF aberrations [37].
On the PBTC phase 2 study, patients were divided among six separate strata based on tumor location, presence of NF1 and BRAF aberration status. Data is currently published for stratum 1 (pilocytic astrocytoma with BRAF aberration) and stratum 3 (NF1-related LGG) [38]. In stratum 1, 36% (9/25) of patients had sustained PR with a 2-year PFS of 70%. Of the 25 patients in stratum 3, 40% of patients had a PR, with two-year PFS of 96%. Ten of the 13 patients with OPG had comparative visual testing at baseline and at one year of therapy, with 80% of patients showing stable visual acuity and 90% showing stable visual fields. Notably, two patients had improved visual acuity and one had improved visual fields [38]. These data led to two large prospective multi-institutional ongoing phase 3 trials, comparing selumetinib to standard chemotherapy (carboplatin and vincristine) in both NF1-related and sporadic pLGG (NCT03871257 & NCT04166409).
Relative to the restricted brain penetration of selumetinib observed in the murine studies (Section 4.3.2), the positive responses seen in patients with LGG may suggest that selumetinib has a potent anti-tumor effect even at concentrations reaching the CNS tumor or could also indicate a disruption of the blood-brain barrier in those patients leading to selumetinib tumor concentrations sufficiently high for adequate anti-tumor effect.
6.3. Non-small cell lung cancer
Approximately 30% of non-small cell lung cancers (NSCLC) harbor KRAS mutations resulting in constitutive activation of MAPK-ERK signaling. Preclinical studies with selumetinib showed great in vitro activity and in vivo tumor shrinkage in mouse models using RAS mutated cell lines/xenografts [5, 6]. Additionally, significant preclinical evidence of potential synergy has been reported when selumetinib was combined with cytotoxic chemotherapy and other targeted therapies, particularly with docetaxel [6, 57, 58]. An initial phase 2 trial of selumetinib monotherapy for recurrent advanced NSCLC (unselected for specific histology/genetic alteration) did not show a significant difference in PFS [59]. A subsequent phase 2 study of selumetinib combined with chemotherapy in KRAS wildtype or KRAS unknown tumors showed a higher response rate with the addition of selumetinib; but no statistical survival difference [60]. In the phase 2 study of docetaxel with selumetinib/placebo in patients with previously treated KRAS-mutant advanced NSCLC, the addition of selumetinib led to improved objective response (p<0.0001) and improved PFS (p=0.014); however, the trend towards improved OS was not significant (p=0.21) and more adverse events were observed [61]. In the phase 3 trial of docetaxel and selumetinib/placebo in patients with recurrent KRAS-mutant NSCLC, the addition of selumetinib to docetaxel did not improve PFS [62]. Some early data suggest variation in efficacy based on unique KRAS codon mutations or combinations; however, this is still being investigated [63].
6.4. Melanoma
6.4.1. Cutaneous melanoma
Up to 75% of melanoma have a dysregulated MAPK-ERK pathway through BRAF (50%) and NRAS (15-25%) mutations. Preclinical studies showed promising efficacy of selumetinib against melanoma cell-lines when used as monotherapy and combined with conventional chemotherapy or targeted therapies [6, 57, 64]. In the phase 1 study of selumetinib combined with dacarbazine, docetaxel, erlotinib or temsirolimus, 5/18 patients with metastatic melanoma (28%) had an objective response, and all five harbored BRAF mutation [65]. However, phase 2 trials evaluating selumetinib versus temozolomide in advanced melanoma, or docetaxel ± selumetinib in wild-type BRAF melanoma did not demonstrate significant improvement in PFS with selumetinib treatment [55, 66].
Two phase 2 trials have been completed in BRAF mutated melanoma, evaluating selumetinib plus dacarbazine, or selumetinib alone [56, 67]. In the first study, a small significant increase (5.6 months) in PFS was observed in the selumetinib group, versus placebo (p=0.021), but no improved OS [67]. The second study was stratified based on low or high phosphorylated-AKT expressions. The high-expressing cohort was closed after no responses seen in the first 10 patients enrolled. In the low-expressing cohort, tumor regressions were seen in 3/5 patients before premature closure due to poor accrual. These data confirm the need for comprehensive genetic testing to optimize therapy selection and support combinatory approaches in patients with high phosphorylated-AKT [56].
6.4.2. Uveal melanoma
Metastatic uveal melanoma doesn’t typically harbor BRAF mutations but is largely characterized by GNAQ and GNA11 mutations leading to downstream activation of MAPK and PI3K/AKT pathways. In a phase 2 study comparing selumetinib to conventional chemotherapy in advanced uveal melanoma, a significant improvement in the PFS was observed in the selumetinib group, but without any improvement in OS [68]. A subsequent phase 3 study evaluating selumetinib plus dacarbazine showed no statistical difference in survival [69].
6.5. Other solid tumors
Selumetinib has been evaluated in multiple phase 1 and 2 studies, alone or in combination to treat various solid tumors, based upon evidence of frequent RAS family or BRAF mutations in these diseases. Selumetinib was evaluated in patients with colorectal cancer versus capecitabine [18] or combined with irinotecan [70]; for patients with pancreatic cancers versus capecitabine [71] or combined with the PI3K/AKT inhibitor MK-2206 [72]; for patients with breast cancers alone or with fulvestrant [73]; for patients with hepatocellular carcinoma as monotherapy [17], in patients with soft-tissue sarcoma alone or combined with temsirolimus [74], in patients with papillary thyroid carcinoma as monotherapy [75], and in patients with relapsed/refractory diffuse large B-cell lymphomas or acute myelogenous leukemia as monotherapy [76, 77]. However, all these studies resulted in poor or limited clinical efficacy with no improvement in survival and variable tolerability.
Better outcomes were observed in a phase 2 study in women with recurrent low-grade serous carcinoma of the ovary or peritoneum receiving selumetinib as monotherapy. In this study, 15% of patients had objective responses and 65% of patients had stable disease [78]. Additionally, in a phase 2 trial of selumetinib in patients with advanced biliary cancer, 3 patients (12%) achieved PR, while 17 patients (68%) achieved stable disease [79].
6.6. Ongoing clinical trials
Selumetinib is currently under investigation in several notable clinical trials. In the NCI-COG Pediatric MATCH trial, selumetinib is one of several drugs tested specifically for children and adolescents with recurrent solid tumors with activating MAPK pathway mutations (NCT03213691). A phase 2 study of selumetinib monotherapy is also currently ongoing for advanced pancreatic cancer harboring KRAS G12R mutation (NCT03040986). Selumetinib is being investigated in combination with mTOR inhibitor sirolimus in patients with malignant peripheral nerve sheath tumors (MPNSTs) in a phase 2 trial (NCT03433183). Proofs of activation of both AKT/mTOR and MAPK pathways in MPNSTs, cross talk between these pathways, and promising preclinical results paved the way for this clinical study [80, 81]. Additionally, selumetinib is being trialed in combination with dexamethasone for the treatment of relapsed/refractory RAS-pathway mutated pediatric and adult acute lymphoblastic leukemia in an international phase 1/2 expansion trial (NCT03705507). This trial was developed based on preclinical studies showing that activating MAPK pathway mutations were highly prevalent in relapsed acute lymphoblastic leukemia, that mutated cells were specifically sensitive to selumetinib, and that the combination of selumetinib with dexamethasone was highly synergistic in this disease model [82].
6.7. Exposure-response associations
The inhibition of ERK phosphorylation (pERK) was considered as the primary proof-of-mechanism biomarker of selumetinib activity in preclinical studies [5, 6]. In a few clinical trials in adults and pediatrics, pERK inhibition after selumetinib treatment was evaluated as a pharmacodynamic marker, either in tumor biopsies or in peripheral blood mononuclear cells (PBMCs) ex-vivo. In the first-in-human phase I studies evaluating the free-base suspension and the capsule formulation, close to complete inhibition of pERK was observed in PBMCs at all selumetinib dosages (25 to 100 mg twice daily), with the greatest degree of inhibition seen 1-hour after selumetinib administration [16, 19]. Additionally, the magnitude of inhibition was related to selumetinib plasma concentrations, with a mean IC50 estimated at 0.77 μM [19]. Decreased expression of pERK was also observed in tumor biopsies after selumetinib administration [37, 41, 45, 68, 79] and was significantly correlated with selumetinib CMAX in one study [45]. However, no studies have yet established a significant association between pERK suppression and clinical benefit, such as improved PFS or OS [45, 68, 79].
Associations between model-derived selumetinib and N-desmethyl selumetinib exposures (i.e., steady state AUC0-12h, CMAX, 12-hour troughs) and direct clinical efficacy endpoints including PFS and OS were also evaluated in patients with NSCLC [23]. However, again, no significant relationships were identified.
7. Clinical toxicities
7.1. Dermatologic toxicity
Most patients treated with selumetinib develops dermatologic toxicity. In the first adult phase 1 trials, rash was both the most frequent and dose-limiting toxicity, occurring in 53-74% of all patients, within two weeks of therapy [8, 16, 19]. Rash mostly presented as acneiform dermatitis, erythematous and maculopapular rash, was dose dependent, and typically resolved with dosing interruption and/or reduction. At the adult RP2D, rash was mainly mild/moderate with most patient having grade 1-2 symptoms, while ~10% of patients reported grade 3/4 dermatitis [18, 19, 55, 71]. Other selumetinib-related dermatologic symptoms include xeroderma (60%), pruritis (46%), paronychia (48%), changes in hair color (32%) and eczema (28%). Severe palmar-plantar erythrodysesthesia syndrome has also been reported in adults receiving selumetinib [83–85]. In the pediatric SPRINT study, rash was reported in 91% of patients, most commonly as dermatitis acneiform (54%), maculopapular rash (39%) and eczema (28%). Grade 3 rash occurred in 8% of patients, resulting in dose interruption and reduction in 11% and 4% of patients [49]. The frequency and severity of these toxicities was similar in the phase 2 pLGG study [38].
The management these dermatologic toxicities should be individualized, based on type, severity, location and the necessity of continuation on treatment [84]. Co-management with a Dermatologist is typically recommended. As preventive measures, skin should be well moisturized routinely using fragrant- and alcohol-free emollient, sun exposure should be minimized and combined with broad-spectrum sunscreen. Low-potency topical steroid creams/ointment, antibiotic lotions, or oral antibiotics are usually recommended for patients with acneiform rash or eczema. For patients with dry/flaky scalp, ketoconazole 2% shampoo can be used once a day, along with topical antibiotics in the presence of pustules or acneiform lesion.
7.2. Gastro-intestinal toxicities
Most patients experience at least one gastro-intestinal toxicity. In the adult phase 1 trials, nausea, vomiting and diarrhea were the most common toxicities, with only small subsets of grade 3/4 toxicity [16, 19]. At the 75 mg RP2D, 48.6% of participants reported mild nausea, 54.3% had mild diarrhea, 25.7% had mild constipation, 17.1% had mild vomiting while 5.7% had grade 3/4 vomiting [19]. In the pediatric SPRINT study, grade 1/2 nausea and vomiting were reported in 66% and 82% of patients, respectively, while diarrhea occurred in 70% of patients including 16% reporting grade 3 symptoms [49]. Diarrhea resulted in dose interruption in 15% patients and in permanent drug discontinuation in 1.4% patients. Other gastro-intestinal toxicities reported in this study included mild abdominal pain (76%), mild stomatitis (50%) and constipation (34%) [49]. These results are similar to those reported in the phase 2 pLGG study [38].
The gastro-intestinal toxicities are generally well managed with supportive care measures [84]. For significant nausea and/or vomiting, anti-emetics can be given on an as-needed basis. For patients experiencing diarrhea, management plan should be tailored based on the severity of symptoms and usually include anti-diarrheal medications such as loperamide, temporary drug withholding, and dose reduction for patients with prolonged or recurrent symptoms.
7.3. Cardiovascular toxicities
Cardiovascular toxicities are potentially severe although less common, and include peripheral edema, hypertension, sinus tachycardia, and more rarely asymptomatic decrease in left ventricular ejection fraction (LVEF) [84, 86]. In both adult and pediatric early trials, mild-to-moderate peripheral edema (20-40%), hypertension (<20%) and sinus tachycardia (20%) were reported [8, 16, 19, 38, 49]. In the pediatric SPRINT and pLGG trials, 23% and 38% of patients exhibited mildly decreased LVEF, respectively [38, 49]. Rare severe LVEF decreases were reported, resulting in dose reduction or eventual permanent drug discontinuation [19, 38, 49]. A phase 1 study in healthy adult volunteers showed no relevant drug effect on cardiac repolarization and no QT/QTc interval prolongation [25].
Considering the potential severity and long-term impact of cardiovascular toxicities, close monitoring is recommended, especially for patients with a history of cardiac disease [84]. To assess LVEF, echocardiogram should be obtained prior to treatment initiation, and then monitored as clinically indicated. Based on the severity of toxicity, treatment with selumetinib may require dose reduction and/or discontinuation [87].
7.4. Ocular toxicities
Ocular toxicity can occur in a small proportion of patients; however, most are mild (grades 1/2) and reversible in nature. Most common ocular events reported include blurred vision, diplopia, photophobia, increased lacrimation, cataracts and ocular hypertension [88]. In the first adult trials at the RP2D, mild ocular toxicities were reported in 12-13% of patients [16, 19]. In the SPRINT study, ocular toxicity was reported in 15% of patients, and blurred vision resulted in dose interruption in 2.7% of patients [49]. Serious ocular toxicities including retinal vein occlusion and retinal pigment epithelial detachment remain rare but have been reported [84, 89].
Patients should undergo comprehensive ophthalmic assessment prior to selumetinib initiation and subsequently at regular intervals during treatment. In general, for grade 1/2 toxicities, treatment should be held until thorough ophthalmologic examination and resolution of symptoms. For more severe toxicities, dose interruption followed by dose reduction or cessation of therapy should be strongly considered.
7.5. Other toxicities
Several other toxicities are associated with selumetinib but are most of the time mild, asymptomatic and self-resolving without the need for dose reduction or cessation. Fatigue is usually experienced by more than half of patients, adults and children [84]. Reversible mild to moderate increases of serum alanine aminotransferase and serum alkaline phosphate were reported in 14-35% of adults and children treated with selumetinib [16, 19, 38, 49]. Creatine phosphokinase (CPK) elevation has also been frequently reported in those studies. In the SPRINT trial, increased CPK occurred in 76% of children, including grade 3/4 in 9% of patients [49]. Although usually asymptomatic, 7% of children required dose reduction, while 8% of patients reported myalgias [49]. These findings were consistent with the pLGG trial, where 78% of patients reported increased CPK, with only 10% of patients having grade 3/4 CPK increase [38]. Lastly, rhabdomyolysis is a rare complication that has been reported in the adult population, but it was not observed in either of the aforementioned pediatric clinical trials.
8. Conclusions
Selumetinib has been widely evaluated as monotherapy in a wide spectrum of tumors characterized by a dysregulation of the MAPK-ERK pathway, leading to the first FDA approval of selumetinib as monotherapy in children with NF1-associated plexiform neurofibromas. Combination of selumetinib with conventional chemotherapy and other targeted therapies is of specific interest as new therapeutic strategies to improve efficacy and overcome MEK inhibitor resistance. Combination of selumetinib with agents targeting other components of MAPK-ERK pathway such as BRAF or EGFR inhibitors, or with agents targeting other pathways in cross-talk with MAPK-ERK such as the AKT/PI3K pathway constitute promising approaches and are under investigations in various types of cancers.
Despite association with numerous side effects involving different organ systems, selumetinib is generally well tolerated by both adults and children at their respective RP2D and can be safely administered at home with supportive care and close monitoring throughout the duration of treatment. However, the ideal duration of therapy has not been standardized yet. However, the long-term toxicities of many targeted therapies remain unknown, and this is particularly important in pediatrics, as the effect of kinase inhibitors such as selumetinib on motor and cognitive development is unclear. Long-term longitudinal monitoring of patients on these agents are therefore necessary to better understand the late effects of these novel agents.
Key points:
Selumetinib is an oral, specific MEK1/2 inhibitor approved for the treatment of children ≥ 2 years with neurofibromatosis type 1-related plexiform neurofibromas at 25 mg/m2/dose twice daily, and currently being evaluated as single agent and in combination, in both adults and pediatrics, in a wide spectrum of solid tumors characterized by a dysregulation of MAPK-ERK pathway.
Selumetinib and its active N-desmethyl metabolite have a similar pharmacokinetic profile, with rapid absorption delayed by food intake and mean terminal half-life of 7.5 hours, leading to minimal accumulation after multiple dosing.
Selumetinib-related side effects most commonly include fatigue, dermatologic, and gastro-intestinal toxicities and are generally mild to moderate and manageable with supportive care and close monitoring.
Acknowledgments
Funding: The authors received no financial compensation for the preparation of this manuscript.
Footnotes
Conflicts of interest: The authors have no conflict of interests.
References
- 1.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 2.Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120(22):3446–56. doi: 10.1002/cncr.28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11(7):385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- 4.Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. MEK and the inhibitors: from bench to bedside. J Hematol Oncol. 2013;6:27. doi: 10.1186/1756-8722-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 6.Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6(8):2209–19. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 7.Ludden LK, Strong JM, Kohn EC, Collins JM. Similarity of metabolism for CAI (NSC 609974) in human liver tissue in vitro and in humans in vivo. Clinical Cancer Research. 1995;1:399–405. [PubMed] [Google Scholar]
- 8.Leijen S, Soetekouw PM, Jeffry Evans TR, Nicolson M, Schellens JH, Learoyd M et al. A phase I, open-label, randomized crossover study to assess the effect of dosing of the MEK 1/2 inhibitor Selumetinib (AZD6244; ARRY-142866) in the presence and absence of food in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;68(6):1619–28. doi: 10.1007/s00280-011-1732-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mandal R, Becker S, Strebhardt K. Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: an emerging threat to anticancer therapy. Oncogene. 2016;35(20):2547–61. doi: 10.1038/onc.2015.329. [DOI] [PubMed] [Google Scholar]
- 10.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kidger AM, Sipthorp J, Cook SJ. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol Ther. 2018;187:45–60. doi: 10.1016/j.pharmthera.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Luke JJ, Ott PA, Shapiro GI. The biology and clinical development of MEK inhibitors for cancer. Drugs. 2014;74(18):2111–28. doi: 10.1007/s40265-014-0315-4. [DOI] [PubMed] [Google Scholar]
- 13.Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11(12):1192–7. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 14.Roskoski R Jr. Allosteric MEK1/2 inhibitors including cobimetanib and trametinib in the treatment of cutaneous melanomas. Pharmacological research : the official journal of the Italian Pharmacological Society. 2017;117:20–31. doi: 10.1016/j.phrs.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 15.Wu PK, Park JI. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin Oncol. 2015;42(6):849–62. doi: 10.1053/j.seminoncol.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26(13):2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Neil BH, Goff LW, Kauh JS, Strosberg JR, Bekaii-Saab TS, Lee RM et al. Phase II study of the mitogen-activated protein kinase 1/2 inhibitor selumetinib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2011;29(17):2350–6. doi: 10.1200/JCO.2010.33.9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennouna J, Lang I, Valladares-Ayerbes M, Boer K, Adenis A, Escudero P et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs. 2011. ;29(5):1021–8. doi: 10.1007/s10637-010-9392-8. [DOI] [PubMed] [Google Scholar]
- 19.Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res. 2010;16(5):1613–23. doi: 10.1158/1078-0432.CCR-09-2483. [DOI] [PubMed] [Google Scholar]
- 20.Tomkinson H, McBride E, Martin P, Lisbon E, Dymond AW, Cantarini M et al. Comparison of the Pharmacokinetics of the Phase II and Phase III Capsule Formulations of Selumetinib and the Effects of Food on Exposure: Results From Two Randomized Crossover Trials in Healthy Male Subjects. Clin Ther. 2017;39(11):2260–75 e1. doi: 10.1016/j.clinthera.2017.08.022. [DOI] [PubMed] [Google Scholar]
- 21.Patel P, Howgate E, Martin P, Carlile DJ, Aarons L, Zhou D. Population pharmacokinetics of the MEK inhibitor selumetinib and its active N-desmethyl metabolite: data from 10 phase I trials. Br J Clin Pharmacol. 2018;84(1):52–63. doi: 10.1111/bcp.13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dymond AW, Howes C, Pattison C, So K, Mariani G, Savage M et al. Metabolism, Excretion, and Pharmacokinetics of Selumetinib, an MEK1/2 inhibitor, in Healthy Adult Male Subjects. Clin Ther. 2016;38(11):2447–58. doi: 10.1016/j.clinthera.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Tong X, Xu H, Carlile DJ, Tomkinson H, Al-Huniti N, Zhou D. Population Pharmacokinetic and Exposure-Response Analysis of Selumetinib and Its N-desmethyl Metabolite in Patients With Non-Small Cell Lung Cancer. J Clin Pharmacol. 2019;59(1):112–22. doi: 10.1002/jcph.1295. [DOI] [PubMed] [Google Scholar]
- 24.Deming DA, Cavalcante LL, Lubner SJ, Mulkerin DL, LoConte NK, Eickhoff JC et al. A phase I study of selumetinib (AZD6244/ARRY-142866), a MEK1/2 inhibitor, in combination with cetuximab in refractory solid tumors and KRAS mutant colorectal cancer. Invest New Drugs. 2016;34(2):168–75. doi: 10.1007/s10637-015-0314-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou D, So K, Dymond AW, Vik T, Al-Huniti N, Mariani G et al. Evaluation of the Effect of Selumetinib on Cardiac Repolarization: A Randomized, Placebo- and Positive-controlled Crossover QT/QTc Study in Healthy Subjects. Clin Ther. 2016;38(12):2555–66. doi: 10.1016/j.clinthera.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Denton CL, Gustafson DL. Pharmacokinetics and pharmacodynamics of AZD6244 (ARRY-142886) in tumor-bearing nude mice. Cancer Chemother Pharmacol. 2011;67(2):349–60. doi: 10.1007/s00280-010-1323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dymond AW, Martin P, So K, Huang Y, Severin P, Holmes V et al. Pharmacokinetics of a Single Oral Dose of the MEK1/2 Inhibitor Selumetinib in Subjects With End-Stage Renal Disease or Varying Degrees of Hepatic Impairment Compared With Healthy Subjects. J Clin Pharmacol. 2017;57(5):592–605. doi: 10.1002/jcph.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koselugo [Package insert]. AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850. May 2020. https://www.azpicentral.com/koselugo/koselugo.pdf#page=1. Accessed August 22, 2020. [Google Scholar]
- 29.de Gooijer MC, Zhang P, Weijer R, Buil LCM, Beijnen JH, van Tellingen O. The impact of P-glycoprotein and breast cancer resistance protein on the brain pharmacokinetics and pharmacodynamics of a panel of MEK inhibitors. Int J Cancer. 2018;142(2):381–91. doi: 10.1002/ijc.31052. [DOI] [PubMed] [Google Scholar]
- 30.Dymond AW, Elks C, Martin P, Carlile DJ, Mariani G, Lovick S et al. Pharmacokinetics and pharmacogenetics of the MEK1/2 inhibitor, selumetinib, in Asian and Western healthy subjects: a pooled analysis. Eur J Clin Pharmacol. 2017;73(6):717–26. doi: 10.1007/s00228-017-2217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dymond AW, So K, Martin P, Huang Y, Severin P, Mathews D et al. Effects of cytochrome P450 (CYP3A4 and CYP2C19) inhibition and induction on the exposure of selumetinib, a MEK1/2 inhibitor, in healthy subjects: results from two clinical trials. Eur J Clin Pharmacol. 2017;73(2):175–84. doi: 10.1007/s00228-016-2153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Infante JR, Cohen RB, Kim KB, Burris HA 3rd, Curt G, Emeribe U et al. A phase I dose-escalation study of Selumetinib in combination with Erlotinib or Temsirolimus in patients with advanced solid tumors. Invest New Drugs. 2017;35(5):576–88. doi: 10.1007/s10637-017-0459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LoRusso PM, Infante JR, Kim KB, Burris HA 3rd, Curt G, Emeribe U et al. A phase I dose-escalation study of selumetinib in combination with docetaxel or dacarbazine in patients with advanced solid tumors. BMC Cancer. 2017;17(1):173. doi: 10.1186/s12885-017-3143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seto T, Hirai F, Saka H, Kogure Y, Yoh K, Niho S et al. Safety and tolerability of selumetinib as a monotherapy, or in combination with docetaxel as second-line therapy, in Japanese patients with advanced solid malignancies or non-small cell lung cancer. Jpn J Clin Oncol. 2018;48(1):31–42. doi: 10.1093/jjco/hyx144. [DOI] [PubMed] [Google Scholar]
- 35.Oxnard GR, Yang JC, Yu H, Kim SW, Saka H, Horn L et al. TATTON: a multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann Oncol. 2020;31(4):507–16. doi: 10.1016/j.annonc.2020.01.013. [DOI] [PubMed] [Google Scholar]
- 36.Patel YT, Daryani VM, Patel P, Zhou D, Fangusaro J, Carlile DJ et al. Population Pharmacokinetics of Selumetinib and Its Metabolite N-desmethyl-selumetinib in Adult Patients With Advanced Solid Tumors and Children With Low-Grade Gliomas. CPT: pharmacometrics & systems pharmacology. 2017;6(5):305–14. doi: 10.1002/psp4.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol. 2017;19(8):1135–44. doi: 10.1093/neuonc/now282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. The lancet oncology. 2019;20(7):1011–22. doi: 10.1016/S1470-2045(19)30277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gross AM, Frone M, Gripp KW, Gelb BD, Schoyer L, Schill L et al. Advancing RAS/RASopathy therapies: An NCI-sponsored intramural and extramural collaboration for the study of RASopathies. Am J Med Genet A. 2020;182(4):866–76. doi: 10.1002/ajmg.a.61485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med. 2016;375(26):2550–60. doi: 10.1056/NEJMoa1605943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krishnamurthy A, Dasari A, Noonan AM, Mehnert JM, Lockhart AC, Leong S et al. Phase Ib Results of the Rational Combination of Selumetinib and Cyclosporin A in Advanced Solid Tumors with an Expansion Cohort in Metastatic Colorectal Cancer. Cancer Res. 2018;78(18):5398–407. doi: 10.1158/0008-5472.CAN-18-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greystoke A, Steele N, Arkenau HT, Blackhall F, Md Haris N, Lindsay CR et al. SELECT-3: a phase I study of selumetinib in combination with platinum-doublet chemotherapy for advanced NSCLC in the first-line setting. Br J Cancer. 2017;117(7):938–46. doi: 10.1038/bjc.2017.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bridgewater J, Lopes A, Beare S, Duggan M, Lee D, Ricamara M et al. A phase 1b study of Selumetinib in combination with Cisplatin and Gemcitabine in advanced or metastatic biliary tract cancer: the ABC-04 study. BMC Cancer. 2016;16:153. doi: 10.1186/s12885-016-2174-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tai WM, Yong WP, Lim C, Low LS, Tham CK, Koh TS et al. A phase Ib study of selumetinib (AZD6244, ARRY-142886) in combination with sorafenib in advanced hepatocellular carcinoma (HCC). Ann Oncol. 2016;27(12):2210–5. doi: 10.1093/annonc/mdw415. [DOI] [PubMed] [Google Scholar]
- 45.Wilky BA, Rudek MA, Ahmed S, Laheru DA, Cosgrove D, Donehower RC et al. A phase I trial of vertical inhibition of IGF signalling using cixutumumab, an anti-IGF-1R antibody, and selumetinib, an MEK 1/2 inhibitor, in advanced solid tumours. Br J Cancer. 2015;112(1):24–31. doi: 10.1038/bjc.2014.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tolcher AW, Khan K, Ong M, Banerji U, Papadimitrakopoulou V, Gandara DR et al. Antitumor activity in RAS-driven tumors by blocking AKT and MEK. Clin Cancer Res. 2015;21(4):739–48. doi: 10.1158/1078-0432.CCR-14-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lauchle JO, Kim D, Le DT, Akagi K, Crone M, Krisman K et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009;461(7262):411–4. doi: 10.1038/nature08279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123(1):340–7. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Engl J Med. 2020;382(15):1430–42. doi: 10.1056/NEJMoa1912735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouffet E, Jakacki R, Goldman S, Hargrave D, Hawkins C, Shroff M et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J Clin Oncol. 2012;30(12):1358–63. doi: 10.1200/JCO.2011.34.5843. [DOI] [PubMed] [Google Scholar]
- 51.Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641–7. doi: 10.1200/JCO.2011.36.6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019;21(Suppl 5):v1–v100. doi: 10.1093/neuonc/noz150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J et al. Integrated Molecular and Clinical Analysis of 1,000 Pediatric Low-Grade Gliomas. Cancer Cell. 2020;37(4):569–83 e5. doi: 10.1016/j.ccell.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kolb EA, Gorlick R, Houghton PJ, Morton CL, Neale G, Keir ST et al. Initial testing (stage 1) of AZD6244 (ARRY-142886) by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2010;55(4):668–77. doi: 10.1002/pbc.22576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18(2):555–67. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Catalanotti F, Solit DB, Pulitzer MP, Berger MF, Scott SN, lyriboz T et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res. 2013;19(8):2257–64. doi: 10.1158/1078-0432.CCR-12-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA, Nathanson KL et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14(1):230–9. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 58.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hainsworth JD, Cebotaru CL, Kanarev V, Ciuleanu TE, Damyanov D, Stella P et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5(10):1630–6. doi: 10.1097/JTO.0b013e3181e8b3a3. [DOI] [PubMed] [Google Scholar]
- 60.Melosky B, Bradbury P, Tu D, Florescu M, Reiman A, Nicholas G et al. Selumetinib in patients receiving standard pemetrexed and platinum-based chemotherapy for advanced or metastatic KRAS wildtype or unknown non-squamous non-small cell lung cancer: A randomized, multicenter, phase II study. Canadian Cancer Trials Group (CCTG) IND.219. Lung Cancer. 2019;133:48–55. doi: 10.1016/j.lungcan.2019.04.027. [DOI] [PubMed] [Google Scholar]
- 61.Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. The lancet oncology. 2013;14(1):38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 62.Janne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crino L et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA. 2017;317(18):1844–53. doi: 10.1001/jama.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Janne PA, Smith I, McWalter G, Mann H, Dougherty B, Walker J et al. Impact of KRAS codon subtypes from a randomised phase II trial of selumetinib plus docetaxel in KRAS mutant advanced non small-cell lung cancer. Br J Cancer. 2015;113(2):199–203. doi: 10.1038/bjc.2015.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70(21):8736–47. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patel SP, Lazar AJ, Papadopoulos NE, Liu P, Infante JR, Glass MR et al. Clinical responses to selumetinib (AZD6244; ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. Cancer. 2013;119(4):799–805. doi: 10.1002/cncr.27790. [DOI] [PubMed] [Google Scholar]
- 66.Gupta A, Love S, Schuh A, Shanyinde M, Larkin JM, Plummer R et al. DOC-MEK: a double-blind randomized phase II trial of docetaxel with or without selumetinib in wild-type BRAF advanced melanoma. Ann Oncol. 2014;25(5):968–74. doi: 10.1093/annonc/mdu054. [DOI] [PubMed] [Google Scholar]
- 67.Robert C, Dummer R, Gutzmer R, Lorigan P, Kim KB, Nyakas M et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. The lancet oncology. 2013;14(8):733–40. doi: 10.1016/S1470-2045(13)70237-7. [DOI] [PubMed] [Google Scholar]
- 68.Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311(23):2397–405. doi: 10.1001/jama.2014.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carvajal RD, Piperno-Neumann S, Kapiteijn E, Chapman PB, Frank S, Joshua AM et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J Clin Oncol. 2018;36(12):1232–9. doi: 10.1200/JCO.2017.74.1090. [DOI] [PubMed] [Google Scholar]
- 70.Hochster HS, Uboha N, Messersmith W, Gold PJ, BH ON, Cohen D et al. Phase II study of selumetinib (AZD6244, ARRY-142886) plus irinotecan as second-line therapy in patients with K-RAS mutated colorectal cancer. Cancer Chemother Pharmacol. 2015;75(1):17–23. doi: 10.1007/s00280-014-2609-3. [DOI] [PubMed] [Google Scholar]
- 71.Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30(3):1216–23. doi: 10.1007/s10637-011-9687-4. [DOI] [PubMed] [Google Scholar]
- 72.Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients With Metastatic Pancreatic Cancer After Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017;3(4):516–22. doi: 10.1001/jamaoncol.2016.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zaman K, Winterhalder R, Mamot C, Hasler-Strub U, Rochlitz C, Mueller A et al. Fulvestrant with or without selumetinib, a MEK 1/2 inhibitor, in breast cancer progressing after aromatase inhibitor therapy: a multicentre randomised placebo-controlled double-blind phase II trial, SAKK 21/08. Eur J Cancer. 2015;51(10):1212–20. doi: 10.1016/j.ejca.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 74.Eroglu Z, Tawbi HA, Hu J, Guan M, Frankel PH, Ruel NH et al. A randomised phase II trial of selumetinib vs selumetinib plus temsirolimus for soft-tissue sarcomas. Br J Cancer. 2015;112(10):1644–51. doi: 10.1038/bjc.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy BA et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18(7):2056–65. doi: 10.1158/1078-0432.CCR-11-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Galanina N, Smith SM, Liao C, Petrich A, Libao B, Gartenhaus R et al. University of Chicago phase II consortium trial of selumetinib (MEKi) demonstrates low tolerability and efficacy in relapsed DLBCL. Br J Haematol. 2018;181(2):264–7. doi: 10.1111/bjh.14544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jain N, Curran E, Iyengar NM, Diaz-Flores E, Kunnavakkam R, Popplewell L et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: a University of Chicago phase II consortium trial. Clin Cancer Res. 2014;20(2):490–8. doi: 10.1158/1078-0432.CCR-13-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farley J, Brady WE, Vathipadiekal V, Lankes HA, Coleman R, Morgan MA et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single arm, phase 2 study. The lancet oncology. 2013;14(2):134–40. doi: 10.1016/S1470-2045(12)70572-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bekaii-Saab T, Phelps MA, Li X, Saji M, Goff L, Kauh JS et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol. 2011;29(17):2357–63. doi: 10.1200/JCO.2010.33.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Endo M, Yamamoto H, Setsu N, Kohashi K, Takahashi Y, Ishii T et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin Cancer Res. 2013;19(2):450–61. doi: 10.1158/1078-0432.CCR-12-1067. [DOI] [PubMed] [Google Scholar]
- 81.Watson AL, Anderson LK, Greeley AD, Keng VW, Rahrmann EP, Halfond AL et al. Co-targeting the MAPK and PI3K/AKT/mTOR pathways in two genetically engineered mouse models of schwann cell tumors reduces tumor grade and multiplicity. Oncotarget. 2014;5(6):1502–14. doi: 10.18632/oncotarget.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matheson EC, Thomas H, Case M, Blair H, Jackson RK, Masic D et al. Glucocorticoids and selumetinib are highly synergistic in RAS pathway-mutated childhood acute lymphoblastic leukemia through upregulation of BIM. Haematologica. 2019;104(9):1804–11. doi: 10.3324/haematol.2017.185975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Balagula Y, Barth Huston K, Busam KJ, Lacouture ME, Chapman PB, Myskowski PL. Dermatologic side effects associated with the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886). Invest New Drugs. 2011. ;29(5):1114–21. doi: 10.1007/s10637-010-9567-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klesse LJ, Jordan JT, Radtke HB, Rosser T, Schorry E, Ullrich N et al. The Use of MEK Inhibitors in Neurofibromatosis Type 1-Associated Tumors and Management of Toxicities. Oncologist. 2020;25(7):e1109–e16. doi: 10.1634/theoncologist.2020-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dy GK, Adjei AA. Understanding, recognizing, and managing toxicities of targeted anticancer therapies. CA Cancer J Clin. 2013;63(4):249–79. doi: 10.3322/caac.21184. [DOI] [PubMed] [Google Scholar]
- 86.Abdel-Rahman O, ElHalawani H, Ahmed H. Risk of Selected Cardiovascular Toxicities in Patients With Cancer Treated With MEK Inhibitors: A Comparative Systematic Review and Meta-Analysis. J Glob Oncol. 2015;1(2):73–82. doi: 10.1200/JGO.2015.000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Totzeck M, Schuler M, Stuschke M, Heusch G, Rassaf T. Cardio-oncology - strategies for management of cancer-therapy related cardiovascular disease. Int J Cardiol. 2019;280:163–75. doi: 10.1016/j.ijcard.2019.01.038. [DOI] [PubMed] [Google Scholar]
- 88.Stjepanovic N, Velazquez-Martin JP, Bedard PL. Ocular toxicities of MEK inhibitors and other targeted therapies. Ann Oncol. 2016;27(6):998–1005. doi: 10.1093/annonc/mdw100. [DOI] [PubMed] [Google Scholar]
- 89.Avery RA, Trimboli-Heidler C, Kilburn LB. Separation of outer retinal layers secondary to selumetinib. J AAPOS. 2016;20(3):268–71. doi: 10.1016/j.jaapos.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]