

SUMMARY
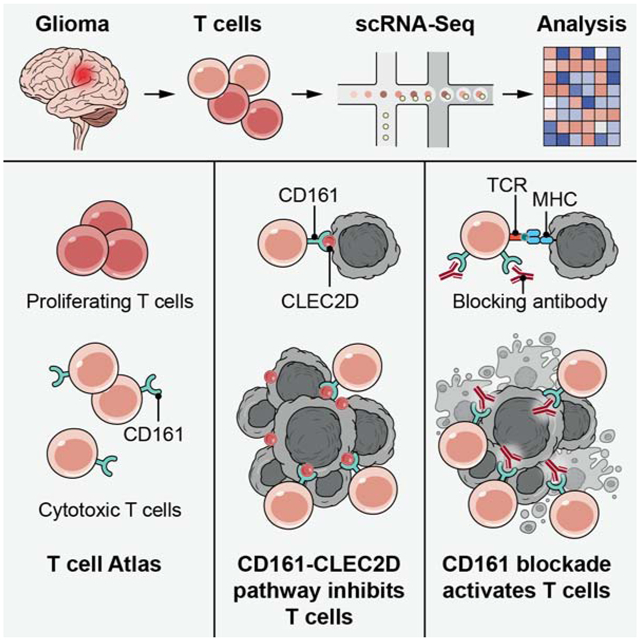
T cells are critical effectors of cancer immunotherapies, but little is known about their gene expression programs in diffuse gliomas. Here, we leverage single-cell RNA-seq to chart the gene expression and clonal landscape of tumor-infiltrating T cells across 31 patients with IDH-wildtype glioblastoma and IDH-mutant glioma. We identify potential effectors of anti-tumor immunity in subsets of T cells that co-express cytotoxic programs and several NK cell genes. Analysis of clonally expanded tumor-infiltrating T cells further identifies the NK gene KLRB1 (encoding CD161) as a candidate inhibitory receptor. Accordingly, genetic inactivation of KLRB1 or antibody-mediated CD161 blockade enhances T cell-mediated killing of glioma cells in vitro and their anti-tumor function in vivo. KLRB1 and its associated transcriptional program are also expressed by substantial T cell populations in other human cancers. Our work provides an atlas of T cells in gliomas and highlights CD161 and other NK cell receptors as immunotherapy targets.
Keywords: CD161, T cells, single-cell RNA-seq, glioblastoma, IDH-mutant gliomas
Graphical Abstract
In-brief
Single cell analysis of tumor-infiltrating T cells in glioma patients identifies T cell subsets coexpressing cytotoxic programs and NK cell receptors, highlighting the functional significance of NK cell receptors such as CD161 in mediating immunosuppression on T cells and their potential as immunotherapy targets.
INTRODUCTION
Diffuse gliomas are the most common type of primary human brain tumor and remain incurable. Current therapeutic approaches largely fail to prevent relapse in the major classes of diffuse gliomas –isocitrate dehydrogenase (IDH) mutant glioma (IDH-G) and IDH-wildtype glioblastoma (GBM) – likely due to striking genetic and cellular heterogeneity of malignant cells. Endogenous or therapy-induced T cell responses typically target diverse tumor antigens, offering the potential of treating heterogeneous tumors like diffuse gliomas through T cell-mediated immunity. While blockade of the inhibitory PD-1 and/or CTLA-4 receptors represent major therapeutic advances in several cancer types, a recent phase 3 clinical trial of anti-PD-1 therapy failed to demonstrate benefit in recurrent GBM patients (Filley et al., 2017; Reardon et al., 2020; Reardon, 2019). Several factors have been postulated to limit the efficacy of PD-1 monotherapy, including a highly immunosuppressive microenvironment with prominence of immunosuppressive myeloid cells, release of the onco-metabolite 2-hydroxyglutarate (2-HG) in IDH-G (Bunse et al., 2018), lymphopenia due to dexamethasone and chemo-radiation treatment, and sequestration of T cells in the bone marrow (Chongsathidkiet et al., 2018; Grossman et al., 2011; Gustafson et al., 2010).
Glioma-infiltrating T cells express several inhibitory receptors, including PD-1, CTLA-4 and LAG-3 (Woroniecka et al., 2018) and recent mass cytometry efforts have begun to unmask the immune landscape of brain tumors (Friebel et al., 2020; Klemm et al., 2020). However, little is known about the comprehensive expression programs and functional states of glioma-infiltrating T cell populations. Here, we profiled and characterized the types, programs, and states of glioma-infiltrating T cells isolated from fresh tumor samples using full-length or 5’end single-cell RNA-seq (scRNA-seq) in 31 adult patients with either GBM or IDH-G. We used this T cell atlas to identify new pathways regulating T cell function in diffuse gliomas. We focused on subsets of cytotoxic glioma-infiltrating T cells that co-expressed several NK cell genes and may serve as potential effectors of anti-tumor immunity. These analyses highlighted the NK gene KLRB1 (encoding CD161) as a potential inhibitory receptor. CLEC2D, the ligand for CD161, is a surface molecule expressed by myeloid cells and malignant cells, suggesting a ligand-receptor pathway for immunotherapy. In vitro and in vivo experiments demonstrated that the CD161 receptor inhibits key aspects of T cell function, including cytotoxicity and cytokine secretion. Generalizing our observations, we found that KLRB1, and the gene expression signature of a KLRB1 expressing T cell, was shared across diverse cancer types. Our atlas of T cell expression programs across the major classes of diffuse gliomas thus identified the CD161-CLEC2D pathway as a potential target for immunotherapy of diffuse gliomas and other human cancers.
RESULTS
A single cell atlas of glioma-infiltrating T cell populations
We profiled T cells from 16 GBM and 15 IDH-G patients, 18 of whom did not receive dexamethasone within four weeks prior to surgery (Figure 1A, Table S1). High-dose corticosteroids (dexamethasone) have a substantial negative impact on T cell function (Gustafson et al., 2010; Keskin et al., 2019). Indeed, flow cytometry analysis demonstrated that dexamethasone therapy was associated with substantially reduced numbers of infiltrating T cells (average reduction 4.14 and 7.72 fold for CD3+ and CD4+ T cells, respectively, Figure 1B). We sorted T cells from 26 freshly dissociated tumors (Figure S1A,B, STAR Methods) and profiled them by full-length scRNA-Seq (Picelli et al., 2014), retaining 8,252 T cells after quality control filtering for all subsequent analyses (Figure S1C, STAR Methods). Four major T cell clusters were identified across GBM and IDH-G (STAR Methods): CD8 T cells, CD4 conventional T cells (CD4 Tconv), CD4 regulatory T cells (Treg) and cycling T cells (Figure 1C). The overall representation of these clusters was similar in GBM and IDH-G and in patients receiving different prior therapies (Figure 1D, Figure S1D-G).
Fig. 1. Transcriptional features of glioma-infiltrating T cells.

(A) Experimental strategy. (B) Quantification of GBM infiltrating T cells from patients who did or did not receive prior dexamethasone. * P < 0.05, ** P < 0.01 (Mann-Whitney U test). Error bars denote SEM. (C) Uniform Manifold Approximation and Projection (UMAP) visualization of 8,252 T cells from 26 glioma patients colored by T cell sub-cluster. (D) UMAP representation from (C) colored based on patient ID (left) and tumor mutational status (right). (E) Sub-clustering and UMAP visualization of CD8 T cells and CD4 T cells, colored based on NMF clustering; ND – not determined. (F) Heat map showing relative expression of selected genes across glioma T cell subsets (clusters numbered at bottom as in E). Gene expression is shown separately for T cells from IDH-G and GBM based on subsets identified using NMF clustering (E). Gene expression is zero-centered and given in units of ln(TP100K+1).
Further clustering of CD8 and CD4 T cell subsets highlighted six sub-clusters for each population that were robustly identified using non-negative matrix factorization (NMF) (Figure 1E, STAR Methods). The sub-clusters spanned several distinct T cell states (Figure 1E,F, Table S2). Cytotoxicity-related genes were most highly expressed in one of the six CD8 T cell clusters which also showed increased expression of NK cell genes. Interestingly, expression of cytotoxicity genes was also detected in one cluster of CD4 T cells (Figure 1E, F). In both CD8 and CD4 T cells there were clusters with an interferon signature, an effector memory signature, or a stress signature (Figure 1E,F). We confirmed expression of genes of the stress signature by RNA in situ hybridization (RNA-ISH) for glioma-infiltrating CD3E+ T cells (Figure S1H), excluding a potential artifact of tissue dissociation. To further demonstrate the robustness of the clusters, we generated transcriptomic profiles by single-cell droplet-based RNA-Seq for 25,256 CD3+ single T cells from five additional GBM patients (3,424 to 6,026 cells/tumor) and found expression signatures to be highly consistent with the CD8 and CD4 T cell clusters identified in the initial 26 patients (Figure S2A-C, Table S3, STAR Methods).
Even though similar T cell clusters were identified in both types of gliomas, we performed further analyses to identify differences in T cell states. First, gene expression signatures reflecting T cell cytotoxicity, interferon, and cellular stress programs gave a stronger signal in the respective clusters for both CD8 and CD4 T cell populations in GBM than IDH-G (Figure 2A,B, Table S4). Second, interrogation of TCGA bulk RNA-seq profiles based on our scRNA-seq data revealed that T cell-specific genes were more highly expressed in GBM than IDH-G (Figure S2D-E), which is similar to results of prior studies (Bunse et al., 2018; Zhang et al., 2018a). When the expression of T cell-specific genes was normalized to the T cell signal per tumor, we observed higher expression of cytotoxicity genes (PRF1, GZMA) in GBM than IDH-G (Figure S2E), consistent with the higher cytotoxicity signature observed in the single cell data. Overall, these data suggest that GBM and IDH-G share similar composition in subsets of infiltrating T cells, but that the total abundance of T cells and their cytotoxicity program is higher in GBM, corresponding with an immunosuppressive role of 2-HG (Bunse et al., 2018).
Fig. 2. Expression of NK cell receptors by population of CD8 T cells.

(A, B) Expression of gene signatures for cytotoxicity (CD8: P < 2.2*1016; CD4: P = 1.52*1013), interferon (CD8: P < 2.2*1016; CD4: P < 2.2*1016) and stress (CD8: P < 2.2*1016; CD4: P < 2.2*1016) are elevated in their respective sub-clusters for GBM compared to IDH-G in both CD8 (A) and CD4 (B) T cells (Komolgorov-Smirnov two-sample test); cluster designation corresponds to Figure 1E, F. (C) Correlation of cytotoxicity signature (x-axis) and NK signature scores (y-axis) for CD8 T cells from cluster 1 (green) or other clusters (2-6, black); clusters as defined in Fig. 1E (STAR Methods). (D) CD8 T cells from all gliomas were split into those with high and low NK signature scores. Heatmap shows relative expression of the 20 most differentially expressed genes for each subset, separately for IDH-G and GBM. NK signature genes are bolded red, cytotoxicity signature genes are bolded black. (E) UMAP visualization of CD8 T cells colored by expression of cytotoxicity, NK receptor or inhibitory receptor signatures (STAR Methods). (F) Expression of three cytotoxicity markers in CD8 T cells. (G) UMAP visualization of CD4 T cells colored by expression of cytotoxicity or inhibitory receptor signatures. (H) Cytotoxic markers in CD4 T cells. (E-H) Displayed on UMAP visualizations as in Figure 1E. Gene expression is given in units of ln(TP100K+1).
High cytotoxicity in glioma-infiltrating CD8 T cells is associated with NK cell-like signature
We hypothesized that the increased expression of NK cell genes by cytotoxic CD8 T cells may be related to their functionality and may assist in identifying subsets of T cells with anti-tumor effector function. We scored each T cell across the 26 patients from the full-length scRNAseq dataset for a cytotoxicity signature (PRF1, GZMB, GZMA, GZMH, NKG7, GNLY) and a NK cell signature (KLRD1, FGFBP2, FCGR3A, S1PR5, KLRC1, KLRC3, KLRB1, KLRC2) (STAR Methods). Higher cytotoxicity signature scores correlated with higher NK cell signatures (Figure 2C-H). Key cytotoxicity genes (Figure 2D, bold black, Figure 2E, F) were more highly expressed in CD8 T cells with a high NK signature score (Figure 2D, bold red, Figure 2E, Table S2). Consistently, several NK cell receptors, including KLRC2 (NKG2C protein), KLRC3 (NKG2E protein), KLRC1 (NKG2A protein), KLRD1 (CD94 protein), and KLRB1 (CD161 protein) were expressed by CD8 T cells with high cytotoxicity (Figure 2E, Figure S2F, Table S2). High cytotoxicity and NK scores were also associated with lower expression of the PDCD1 gene (PD-1 protein) (Figure 2D, bold black underlined) and lower expression of a signature of well-known co-inhibitory receptors (PDCD1, CTLA4, HAVCR2, LAG3, TIGIT) (Figure 2E, Figure S2G). In CD4 T cells, we detected the highest level of PDCD1 in cytotoxic CD4 T cells, while TIGIT was most highly expressed by Tregs (Figure 2G,H, Figure S2H). We validated the expression of a subset of these NK cell receptors by GBM-infiltrating T cells at the protein level by flow cytometry (Figure 3A,B). The inhibitory CD161 receptor (KLRB1) and the activating NKG2C/CD94 receptor (KLRC2 and KLRD1) were expressed by substantial populations of CD8 T cells.
Fig. 3. Transcriptional signatures of clonally expanded T cells.

(A) Flow cytometric analysis of GBM-infiltrating CD8 T cells from one patient for protein levels of CD161 (x-axis), NKp80, NKG2C, and TIGIT (y-axis). (B) Summary of data from (A) for tumor-infiltrating T cells from two GBM patients. (C) TCR α and β chain sequences were reconstructed for each cell from scRNA-seq reads for the full-length scRNAseq dataset and displayed as mean percent of T cells with recurrent TCRs within each T cell subset. The mean percent is taken across all individual patient samples, and the error bars show the SEM. (D) Size of TCR clonotypes for five GBM samples based on 5’ scRNA-seq analysis. Clonotype sizes are grouped by colors, and the number of clonotypes for each size group is indicated for each tumor. (E) Comparison of expression for each gene in clonal versus non-clonal T cells from GBM tumors. Z-score of expression (y-axis) and Z-score of coefficient of variation (CV) of expression (x-axis) is shown for each gene separately for CD8 (left) and CD4 (right) T cells (STAR Methods). (F) Heatmap comparing gene expression of clonally expanded, KLRB1 positive and negative T cells from five GBM samples based on 5’ scRNA-seq analysis. Gene expression is zero-centered and given in units of ln(TP100K+1).
Distinct gene expression programs in clonal T cell populations
We reconstructed T cell receptor (TCR) sequences from the scRNA-seq reads to identify clonal T cells that may have proliferated following tumor antigen recognition (Stubbington et al., 2016) (STAR Methods). We detected full-length, productive TCR sequences (α and/or β chains) for 4,630 of 8,252 T cells (56%) across the 26 patients in the full-length scRNAseq dataset (Figure 3C, S3A-E). A substantial number (13±8) of distinct clonotypes (≥2 sequenced T cells with the same TCR sequences) were identified in most patients (Figure S3B,C). The degree of detected clonality is likely still an underestimate, due to the number of sampled T cells per patient. We therefore also examined the TCR repertoire for the five additional GBM cases using the V(D)J T cell profiling protocol (10X Genomics), which enabled analysis of a considerably larger number of T cells per patient (3,424 to 6,026 T cells per tumor). This analysis revealed the presence of highly expanded clonotypes in each patient (Figure 3D), thus providing important evidence for T cell activation and expansion in gliomas.
Analysis of the expression patterns of clonally expanded T cells identified four major clusters (Figure S3D), with no marked difference between GBM and IDH-G (Figure S3E, Table S2), but with distinct expression features compared to non-clonal cells. For each gene, we compared the difference in mean expression (Y-axis) as well as variance in expression (X-axis) between clonal and non-clonal T cells in CD8 T cells or CD4 Tconv cells (excluding Tregs), separately for GBM (Figure 3E) and IDH-G (Figure S3F, STAR Methods). Clonal CD8 T cells had higher expression of cytotoxicity genes (GZMB, NKG7) and NK receptors (KLRB1, KLRD1) than non-clonal cells in GBM (Figure 3E), supporting that these programs may identify tumor-reactive T cells. In particular, KLRB1 expression was both higher and less variable in clonal CD8 T cells compared to non-clonal cells in GBM (Figure 3E, STAR Methods) and less variable in clonal CD8 T cells in IDH-G (Figure S3F, p < 0.01, permutation test, STAR Methods). KLRB1 encodes the CD161 protein that was previously shown to inhibit NK cell-mediated cytotoxicity following binding to CLEC2D (LLT1) (Aldemir et al., 2005; Rosen et al., 2005). Clonal CD4 T cells also more highly expressed several cytotoxicity genes (PRF1, GZMA, GZMH) (Figure 3E, Figure S3F). These conclusions were confirmed by comparing the gene expression of clonally expanded, KLRB1 positive and negative T cell populations using the five additional GBM samples analyzed by 5’ scRNA-seq: in CD8 T cells, KLRB1+ cells expressed higher levels of cytotoxicity (GNLY, GZMB) and NK cell genes (NCR3, KLRD1) (Figure 3F, Figure S2C, Table S3).
Expression of CD161 receptor by T cells and its ligand CLEC2D by malignant and myeloid cells
We selected the KLRB1 gene for further investigation because (1) the gene was overexpressed by CD8 T cells with a cytotoxicity signature and CD4 effector T cells, but importantly not Tregs, (2) it encodes a surface protein that could be therapeutically modulated, (3) it was more highly expressed in clonally expanded T cells in human gliomas. Given that clonal expansion is induced by TCR signaling, this analysis enriched for genes over-expressed by tumor-reactive T cells. Because glioma cells express CLEC2D mRNA and protein (Roth et al., 2007), we hypothesized that the CD161 receptor could inhibit activation of T cell populations following CLEC2D binding. CLEC2D mRNA was expressed in both malignant cells and myeloid cells in GBM and IDH-G scRNA-seq datasets (Neftel et al., 2019; Tirosh et al., 2016b; Venteicher et al., 2017) (Figure 4A). KLRB1 was preferentially expressed by CD8 and CD4 Tconv cells in diffuse gliomas; expression was lower in CD4 Tregs (Figure S4A). Critically, these cells were polyclonal T cells and not NKT cells or mucosal associated invariant T cells (MAIT): they used diverse TCR V and J segments by both TCR α and β chains (Figure 4B and Figure S4B,C), whereas both NKT cells and MAIT cells have a highly restricted TCR repertoire (Mori et al., 2016). Furthermore, the polyclonal T cells expressed key T cell genes, including TCR-CD3 subunits (CD3G, CD3D, CD3E) and co-receptors (CD4 or CD8A/CD8B) (Figure S4D). We did not detect rearranged TCR δ chains, indicating that these cells were αβ rather than γδ T cells. KLRB1 expressing T cells were also not Th17 cells, based on very low expression of IL17A, IL17F, IL22, IL23R and RORC (Figure S4D). This is particularly relevant for KLRB1 expressing T cells because CD161 is used as a marker for MAIT cells that provide protection against intracellular bacterial infection (Keller et al., 2017). KLRB1 expressing T cells in glioma are thus distinct from CD161+ γδ T cells that produce IL-17, which were previously shown to be enriched in the cerebrospinal fluid of patients with multiple sclerosis (Schirmer et al., 2013).
Fig. 4. Investigation of CLEC2D – CD161 pathway in GBM.

(A) Quantification of transcript levels for CLEC2D and selected genes in malignant cells, myeloid cells, and oligodendrocytes from published glioma datasets. Gene expression is zero-centered and given in units of ln(TP100K+1). (B) Diversity of TCRα V and J gene segment usage by CD8 T cells expressing KLRB1. (C) Flow cytometric analysis of CD161 protein on the surface of CD8 (top) and CD4 (bottom) T cells in GBM (filled colors) versus blood T cells (blue line) from the same patient. Dotted line indicates staining with isotype control antibody; percentage of CD161+ tumor-infiltrating T cells is indicated. (D and E) Quantification of CD161 and PD-1 positive T cells in two GBM (E208 and E161), based on fluorescence minus one (FMO) and isotype controls, (D) and summary of data for five GBM (E). Percentage of CD161 versus PD-1 positive T cells, P < 0.006 for CD8 and P < 0.003 for CD4 T cells, Mann-Whitney U test. Error bars denote SEM. (F) In situ hybridization in two IDH-G (upper panel) and two GBM (lower panel) with probes for KLRB1 and CD3E (top) as well as CLEC2D and CD45 (bottom), counterstained with hematoxylin (light purple). Top row shows subsets of cells co-expressing CD3E and KLRB1 (black arrowheads); bottom row highlights CLEC2D expression in malignant cells (yellow arrowheads) and CD45 positive immune cells (black arrowheads); * lumen of blood vessels. (G, H) Quantification of infiltrating CD3+ T cells and CD3−CD56+ NK cells in five GBM.
We confirmed these expression patterns by flow cytometry and in situ hybridization (STAR Methods). Flow cytometry showed that a large fraction (>90%) of GBM-infiltrating CD8 T cells and most CD4 T cells were CD161 positive, while CD161 was detected in only a small subset of peripheral blood T cells from the same patients (Figure 4C). The proportion of CD161+ T cells was substantially larger (P < 0.005, Mann-Whitney U test) and less variable (P < 0.001, F-test) than that of PD-1+ T cells (Figure 4D,E and Figure S4E). Detection of CD161 with this antibody was specific based on isotype control staining (Figure 4C and Figure S4F), labeling of a cell line transduced with full-length KLRB1 cDNA (Figure S4G), and lack of detection for primary T cells in which the KLRB1 gene was inactivated by CRISPR/Cas9 (STAR Methods). RNA-ISH further confirmed that KLRB1 was expressed in populations of CD3E+ T cells and that CLEC2D mRNA was expressed primarily in malignant cells, but also in CD45+ immune cells (Figure 4F). CD161 is known to be expressed by NK cells, but flow cytometric analysis identified only small numbers of CD3− CD56+ NK cells in five GBM samples (Figure 4G,H), consistent with recent studies (Klemm et al, Cell 2020). We also identified CLEC2D protein expression on tumor-infiltrating dendritic cells in a murine glioma model (Figure S4H-J). Expression of CLEC2D by myeloid cells is consistent with published studies demonstrating transcription of this gene following activation of monocytes, dendritic cells, B cells, and T cells (Germain et al., 2011; Rosen et al., 2008).
CD161 acts as an inhibitory receptor for tumor-specific T cells
We interrogated the CD161 – CLEC2D pathway using a co-culture system with primary human T cells and patient-derived gliomasphere cultures (Figure 5A, STAR Methods). We focused on studying the interaction of human T cells and glioma cells because the human genome carries a single KLRB1 gene, whereas multiple Klrb1 homologs are present in the murine genome that encode both inhibitory and activating receptors (Zhang et al., 2012). These gliomasphere cultures expressed CLEC2D mRNA as shown by qPCR analysis (Figure 5B) and also expressed the HLA-A*02:01 protein (STAR methods), thus enabling co-culture assays with T cells that recognized the NY-ESO-1 tumor peptide presented by HLA-A*02:01. We isolated CD161+ T cells (negative for Vα7.2, a marker for MAIT cells) from peripheral blood samples of healthy donors. These cells were edited with high efficiency by electroporation of Cas9 protein with bound gRNAs (>90% editing efficiency for KLRB1) (Figure S5A-F). We introduced a NY-ESO-1 specific TCR (1G4) into KLRB1-inactivated or control T cells to generate a population of T cells with defined tumor antigen specificity (Figure S5G-K, STAR methods) (Zhao et al., 2005). This TCR recognized a NY-ESO-1 peptide presented by HLA-A*02:01, and previous clinical trials with TCR transduced T cells demonstrated therapeutic responses in multiple myeloma and synovial sarcoma (Rapoport et al., 2015; Robbins et al., 2011).
Fig. 5. Targeting of CD161 pathway enhances T cell activation and reduces PD-1 expression.

(A) Workflow of experimental strategy to interrogate the function of the KLRB1 gene in primary human T cells. (B) qPCR analysis of CLEC2D and GAPDH mRNA in four gliomasphere cultures and U-87 MG cell line. (C) T cell cytotoxicity assay. Gliomaspheres (MGG123, MGG75 or D270) were co-cultured with KLRB1 or control edited T cells at an effector to target (E:T) ratio of 1:1 or 0.25:1 for 8 hours. The percentage of killed (Zombie UV+) tumor cells was quantified by flow cytometry; cultures without T cells (0:1) were used to assess background levels of apoptosis. (D) Strategy for evaluation of the CD161 – CLEC2D pathway with a CD161 blocking monoclonal antibody. (E) T cell killing assay with CD161 blocking mAb (HP-3G10) or isotype control IgG. (F) Labeling of CD8 T cells after 24 hours for surface localization of CD107a degranulation marker. (G-I) Analysis of PD-1 expression by CD8 T cells. T cells were co-cultured for 72 hours with patient-derived gliomaspheres at the indicated E:T ratios and PD-1 surface expression was evaluated by flow cytometry. Pathway was targeted by editing of KLRB1 gene in T cells or addition of CD161 blocking mAb. (J, K) Cytokine release by T cells. T cells were co-cultured for 72 hours with gliomaspheres from GBM (MGG123, MGG75) (J) or IDH-G (BT142) (K) at the indicated E:T ratios. Experiments in (C) were performed three times, (E-K) were performed two times, (B) was performed once. * P < 0.05, ** P < 0.01, *** P < 0.001, error bars denote SEM. Mann-Whitney U test (C, E-F, H-K).
KLRB1-edited compared to control-edited T cells had higher cytotoxic activity (P < 0.005, Mann-Whitney U test) against gliomaspheres (Figure 5C; Figure S5L-N). Enhanced T cell-mediated cytotoxicity (Figure 5D,E) and cytokine secretion (Figure S5O) were also observed when a CD161 specific mAb (HP-3G10, Figure S5P, STAR Methods) was used to inhibit CLEC2D binding. Consistent with their higher cytotoxic activity, T cells edited for KLRB1 or blocked by HP-3G10 mAb had higher surface levels of the degranulation marker CD107a (P < 0.003, Mann-Whitney U test) (Figure 5F) and increased expression of T cell activation markers (CD25 and CD69) (Figure S6A,B). Inactivation of the KLRB1 gene with two different gRNAs resulted in a similar enhancement of T cell function (Figure S6C). Interestingly, a significantly smaller fraction of CD8 T cells co-cultured with patient-derived gliomaspheres or the U-87 MG cell line became positive for PD-1 protein (P < 0.0016, Mann-Whitney U test) when the KLRB1 pathway was inactivated (Figure 5G-I; Figure S6D-F). Expression of CTLA-4 and LAG-3 receptors was not affected by KLRB1 inactivation (Figure S6F). Editing of the KLRB1 gene also enhanced secretion of cytokines important in anti-tumor immunity (P < 0.0004, Mann-Whitney U test), including IL-2 and IFNγ, in co-cultures with two GBM and one IDH-G gliomaspheres (Figure 5J, K). Overall, these data demonstrated that targeting of the CD161 pathway enhanced T cell-mediated cytotoxicity against patient-derived gliomaspheres and other critical aspects of T cell function. Also, signaling through the inhibitory CD161 receptor may contribute to regulation of PD-1 expression. Previous studies had suggested that CD161 represents either an activating or inhibitory receptor (Fergusson et al., 2014; Le Bourhis et al., 2013), but had limitations due to the artificial stimulation conditions (CD3 + CD28 antibodies) that were used in the assays.
Inactivation of KLRB1 in tumor-infiltrating T cells enhances their anti-tumor activity in vivo
We generated two different humanized mouse models to assess the impact of the KLRB1 gene on in vivo T cell function. In the first model, we implanted 5x104 tumor cells from MGG123 gliomaspheres derived from a recurrent GBM (Nigim et al., 2015) (HLA-A*02:01 positive, transduced to express NY-ESO-1 antigen) into the CNS of immunodeficient mice (Figure 6A). GBM implantation was followed eight days later by injection of human T cells into the cerebrospinal fluid (CSF) via the contralateral ventricle (Figure 6A, Figure S6G), as performed for CAR T cell delivery in patients (Brown et al., 2016). Transferred T cells expressed the NY-ESO-1 TCR and were edited with either a KLRB1 or control gRNA (STAR Methods). Tumor cells from MGG123 formed highly aggressive tumors, with evidence of brain infiltration, mass effect and intratumoral hemorrhage (Figure 6B, S6H). We used bioluminescence imaging (BLI), starting on day −1 relative to T cell treatment, to assign mice with a similar BLI signals to the two experimental groups (Figure 6C); a second dose of T cells was administered 27 days later. Transfer of KLRB1 compared to control edited T cells resulted in significantly slower tumor growth at all time points starting approximately two weeks following initial T cell transfer based on BLI imaging, a finding that was reproducible in two independent experiments (Figure 6D, S6I). Importantly, transfer of KLRB1 compared to control edited T cells conferred a significant survival benefit in both experiments, despite the aggressive nature of this model (P = 0.0001, Log-rank (Mantel-Cox test) (Figure 6E, S6J).
Fig. 6. KLRB1 gene inactivation in T cells improves survival in two humanized GBM models.

(A) Schematic indicating injection sites of GBM cells into the striatum and T cells into the contralateral lateral ventricle. (B) Coronal section of the mouse brain at survival endpoint. (C) Bioluminescence imaging (BLI) on day −1 (relative to first T cell injection). (D) Kinetic analysis of tumor burden based on BLI signal from mice that received KLRB1 or control edited T cells; day 0 corresponds to first T cell injection; 2nd T cell injection is indicated. (E) Survival analysis for MGG123 model following transfer of KLRB1 or control edited T cells. (F-J) Flow cytometry analysis of KLRB1 or control edited T cells infiltrating MGG123 at a late disease stage (moribund). (K) Survival analysis for U87 model following transfer of KLRB1 or control edited T cells. (L-N) Flow cytometry analysis of T cells from U-87 MG tumors 8 days following T cell injection. Experiments in (C-E) & (K) were performed twice; (F-J) and (L-N) were performed once. * P < 0.05, ** P < 0.01, *** P < 0.001, error bars denote SEM. Mann-Whitney U test (D), (F-J), (L-N) and Log-rank (Mantel-Cox) test (E), (K).
Flow cytometric analysis of T cells at a late disease time point demonstrated a significantly larger number of infiltrating granzyme B+ CD8 T cells in MGG123 gliomas when transferred T cells were edited with KLRB1 rather than control gRNAs (Figure 6F) (P < 0.007, Mann-Whitney U test). Further investigation provided evidence that KLRB1 edited T cells had a less exhausted state than control edited T cells based on the following criteria: 1. a substantially larger fraction of granzyme B+ CD8 T cells were PD-1 negative; 2. a small percentage of CD8 T cells co-expressed the PD-1 and TIM-3 inhibitory receptors (Figure 6G-I) (P < 0.006, Mann-Whitney U test). The latter finding is relevant because exhausted T cells tend to co-express multiple inhibitory receptors (McLane et al., 2019). We also observed lower PD-1 expression by KLRB1-edited CD4 T cells (Figure 6J).
In the second humanized mouse model of T cell-mediated tumor immunity, U-87 MG cells were similarly engineered and implanted into the CNS of immunodeficient mice, followed by a single injection of KLRB1 or control edited human T cells expressing NY-ESO-1 TCR, 7 days later. Again, mice that received KLRB1 compared to control edited T cells showed significantly improved survival (Figure 6K P = 0.0059, Log-rank Mantel-Cox text) and lower BLI signal (Figure S6K). A limitation of the U-87 MG model was that tumors did not grow in an infiltrative manner as observed for the MGG123 model. Ex vivo analysis of tumor-infiltrating T cells from a separate experiment on day 8 following T cell transfer showed that PD-1 expression was substantially lower in KLRB1 edited CD8 T cells (Figure 6L) (P < 0.004, Mann-Whitney U test), consistent with the in vitro findings (Figure S6E-F). Furthermore, we identified increased numbers of granzyme B, IL-2, and TNFα positive CD8 T cells, as well as granzyme B and IL-2 positive CD4 T cells, in mice that had received KLRB1 edited T cells (Figure 6M,N). Thus, inactivation of the KLRB1 gene in transferred T cells provided a survival benefit in two humanized GBM mouse models. Together, ex vivo analysis demonstrated enhanced T cell function and reduced expression of the PD-1 inhibitory receptor.
The KLRB1 program is present in tumor-infiltrating T cells across human cancer types
We next examined whether KLRB1 may be associated with similar transcriptional programs in tumor-infiltrating T cells in other human tumor types using published scRNA-seq datasets from melanoma, non-small cell lung cancer (NSCLC), hepatocellular carcinoma, and colorectal cancer (Figure 7A, Figure S7A-C) (Guo et al., 2018; Jerby-Arnon et al., 2018; Zhang et al., 2018b; Zheng et al., 2017). KLRB1 was expressed by a subset of tumor-infiltrating CD4 and CD8 T cells in each of these cancers (Figure 7A, Figure S7A-C) and was particularly prominent in lung cancer (KLRB1 detected in 40% of CD4 T cells and 25% of CD8 T cells) (Figure 7A). KLRB1 expression was low in FOXP3 positive Tregs compared to CD4 Tconv and CD8 T cells, suggesting that it may primarily restrain the function of effector T cells (Figure 7A, Figure S7A-C) (t-test: P<2.2*10−16 for lung cancer; P<2.2*10−16 for hepatocellular carcinoma; P=2.1*10−9 for colorectal cancer; Treg annotation unavailable for melanoma). Most KLRB1 positive T cells in liver and colorectal cancers did not express Th17 cell markers (Figure S7D,E) and only few MAIT cells were detected in either cancer type (Figure S7F,G). Notably, while KLRB1 was expressed in both melanoma and diffuse gliomas, it was among the top 10 genes that were more highly expressed in diffuse gliomas versus melanoma-infiltrating CD8 and CD4 T cells (Figure 7B, Figure S7H, STAR Methods) (t-test: P=7.5*10−102).
Fig. 7. Identification of a T cell KLRB1 program across multiple cancer types.

(A) t-distributed stochastic neighbor embedding (t-SNE) visualization showing expression of key genes in T cells isolated from lung adenocarcinoma. Gene expression is given in units of ln(TP100K+1). (B) Averaged expression of genes in tumor-infiltrating T cells (TILs) in GBM + IDH-G (y-axis) versus melanoma (x-axis). The inset lists the genes with the greatest log2 fold change. (C and D) Overlap of KLRB1 transcriptional programs in CD8 T cells (left) and CD4 T cells (right) between the different pairs of cancer datasets. The p-values (C) and observed vs. expected ratio (D) express the significance (hypergeometric test) and magnitude of the overlap between genes in the KLRB1 transcriptional programs from the different cancers. The Melanoma1 and Melanoma2 datasets refer to two separate melanoma scRNA-seq studies (Jerby-Arnon et al., 2018, Sade-Feldman et al., 2018). (E) Heatmap showing the expression of all genes for the pan-cancer KLRB1 program. This pan-cancer KLRB1 program was defined based on overlap in the individual KLRB1 programs from six scRNA-seq cancer datasets. Gene expression of the pan-cancer program is shown for CD4 and CD8 T cells from the gliomas in this study (GBM and IDH-G), and T cells are further divided into KLRB1 expressing and KLRB1 nonexpressing cells. Gene expression is zero-centered, and given in units of ln(TP100K+1).
We next investigated whether the KLRB1 transcriptional program we originally detected in glioma-infiltrating T cells generalized to other cancer types. Each KLRB1 transcriptional program consisted of those genes whose expression was significantly greater (t-test, STAR Methods) in KLRB1+ T cells compared to KLRB1− T cells in a tumor type. There was extensive and significant overlap in the KLRB1 transcriptional programs from each of the five analyzed cancer types (Figure 7C,D, Table S5), and we used this overlap to define a pan-cancer KLRB1 program in both CD8 and CD4 T cells (STAR Methods, Figure 7E). The CD8 T cell pan-cancer KLRB1 program included several other NK cell genes (KLRD1, KLRF1, TYROBP, ZBTB16 and CD96, Figure 7E). CD96 was previously shown to act as an inhibitory receptor for NK cells, and its CD155 ligand (PVR gene) was expressed by malignant cells in GBM (Figure 4A) and other cancer types (Chan et al., 2014; Gao et al., 2017). IL-18 is a major pro-inflammatory cytokine that activates NK and CD8 T cells (Kaplanski, 2018). IL18R1 and IL18RAP, the two genes encoding the IL-18 receptor, were expressed in KLRB1 expressing T cells in diffuse gliomas (Figure S7I). The functional role of these KLRB1 signature genes will require further investigation. KLRB1 expressing T cells thus shared transcriptional features with ‘innate’ effector T cells, but did not have a restricted TCR repertoire (Figure 4B, Figure S4B,C) (Fergusson et al., 2014).
The KLRB1 signature included several genes that were previously shown to be upregulated in tissue-resident memory T cells (Trm), including ITGAE, ITGA1 and CXCR6, and Trm were previously identified in human brain (Smolders et al., 2018). However, KLRB1 expression did not correlate with a signature of genes upregulated in Trm (Kumar et al., 2017) for glioma-infiltrating CD8 or CD4 T cell clusters (correlation value of <0.1, Table S6). Our scRNA-seq data showed that KLRB1 is expressed in multiple CD8 and CD4 T cell clusters in diffuse gliomas, including clusters with effector, effector memory or memory signatures, and some of these cells may be Trm. Consistent with this interpretation, CD161+ CD8+ T cells in the blood (non-MAIT T cells) were previously shown to represent highly functional effector memory T cells with higher cytotoxic potential than their CD161 negative counterparts (Fergusson et al., 2016). Taken together, these analyses suggest that our observations based on the expression landscape of glioma-infiltrating T cells generalize to other cancer types and define a pan-cancer KLRB1 program with additional candidate targets for T cell immunity.
DISCUSSION
Here, we charted the gene expression landscape of infiltrating T cells in GBM and IDH-G, providing insights into their expression programs and suggesting strategies for enhancing T cell-mediated immunity in diffuse gliomas and other human cancers. The identification of largely similar subsets of T cells across different classes of diffuse gliomas suggests that common strategies could be leveraged for T cell-mediated immunity. The expression program of KLRB1-expressing T cells suggest that these cells represent effectors that share features with innate T cells, despite the fact that they possess a diverse TCR repertoire. CLEC2D, the CD161 ligand, is expressed by both tumor cells and immunosuppressive myeloid cells, thus the CD161 pathway shares certain similarities with the PD-1 pathway. PDCD1 or KLRB1 mRNAs identified distinct, but partially overlapping, T cell populations. Antibody-mediated inhibition of the CLEC2D–CD161 pathway may provide synergistic therapeutic benefit with PD-1 blockade by enhancing the anti-tumor function of distinct T cell populations. Potential side effects of antibody-mediated blockade of the CLEC2D–CD161 pathway remain unknown and will need to be investigated in a non-human primate model. The significance of these findings may extend beyond diffuse gliomas, as illustrated by expression of KLRB1 in tumor-infiltrating T cells from four other common human cancers. Also, several types of human lymphomas are characterized by high CLEC2D protein levels (Germain et al., 2015; Llibre et al., 2016), and inhibition of the CLEC2D–CD161 pathway may enhance T cell-mediated immunity against such hematological malignancies.
Our study highlights the significance of ‘NK cell’ receptors that are expressed by tumor-infiltrating T cells which possess diverse TCR repertoires. We hypothesize that expression of these NK cell receptors is induced in T cells by inflammatory mediators in the tumor microenvironment. In celiac disease, tissue-infiltrating T cells were previously shown to express several NK cell receptors (Meresse et al., 2006), including the NKG2C receptor that we also identified on glioma-infiltrating T cells. Inhibitory and activating NK cell receptors expressed by tumor-infiltrating T cells may offer opportunities for enhancing the efficacy of immunotherapy in diffuse gliomas and other malignancies.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for further information should be directed to Kai Wucherpfennig (Kai_Wucherpfennig@dfci.harvard.edu).
Materials availability
Requests for resources and reagents should be directed to and will be fulfilled by Kai Wucherpfennig (Kai_Wucherpfennig@dfci.harvard.edu).
Data and code availability
Processed data generated for this study are available at the Gene Expression Omnibus (GEO: GSE163108; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163108). Raw scRNA-seq data can be requested at Data Use Oversight System (https://duos.broadinstitute.org/). The code supporting the current study is available from M.L.S (Suva.Mario@mgh.harvard.edu) on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Procurement and processing of primary human glioma samples for scRNA-seq
Surgically resected glioma specimens were provided on ice by the Brigham and Women’s Hospital (BWH) and the Massachusetts General Hospital (MGH) Pathology Tissue Banks within 30 minutes of lesion excision. Prior to the procedure, all patients provided informed consent for the collection of glioma specimens. Samples were de-identified prior to receipt and acquired under a protocol approved by the Institutional Review Board (DF/HCC 10-417). Subjects included both females and males spanning a range of age from 25-77 years. Clinical characteristics and details of individual patients are provided in (Table S1). To determine sample size estimation, FACS purified T cells were isolated and sequenced, following removal of cells that did not meet quality criteria (73 – 531 cells analyzed per patient for the SmartSeq2 method; 3,424-6,026 cells analyzed per patient for the 10X genomics method). T cells were profiled at scale sufficient to enable statistical power. Tumor specimens from each subject were allocated to experimental groups through the determination of IDH1/2 mutational status and assigned to IDH mutant (IDH-G; 15 cases) or IDH wildtype (GBM; 16 cases) cohorts.
Experimental mouse models
Healthy, naïve female NOD.Cg-Prkdc<scid> IL2rg<tm1Wjl>/SzJ (NSG) mice aged 6-8 weeks were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were housed in specific pathogen free conditions in compliance with all guidelines of the Institutional Care and Use Committee (IACUC) and were subjected to procedures after approval of the Dana-Farber Cancer Institute (DFCI) Animal Care and Use Committee (Protocol: 08-049). Only treatment-naive mice (aged 10-12 weeks) were used that had not been part of any previous experimental procedures. Mice were housed together in indicated groups and monitored daily for neurological symptoms indicative of brain tumors (altered gait, seizure, loss of weight, lethargy). Mice were immediately euthanized upon reaching pre-determined clinical endpoints and when recommend by veterinary staff of the DFCI animal unit. All mice were purchased (no mice were bred) for this study.
Patient-derived and cell line tumor models
Patient derived glioblastoma neurosphere cells MGG123 (recurrent GBM) (Nigim et al., 2015), MGG75 (newly diagnosed GBM) (Higuchi et al., 2018), D270 (Gedeon et al., 2018) and BT142 (IDH-G) were maintained in Neurobasal Medium (Thermo Fisher Scientific) supplemented with 0.5% N-2 and 2% B-27 (Thermo Fisher Scientific), 1% Penicillin/Streptomycin (Thermo Fisher Scientific), 1.5% Glutamax (Thermo Fisher Scientific), 20 ng/mL of EGF and 20 ng/mL of FGF-2 (Shenandoah Biotechnology) in ultra-low attachment T-25 or T-75 flasks (Corning; Corning, NY). D270 cells were obtained from the laboratory of John Sampson at Duke University (Miao et al., 2014). BT142 cells and U-87 MG cells were procured from and authenticated by ATCC (Manassas, VA). All tumor lines were tested monthly for mycoplasma. U-87 MG cells were maintained in vitro with DMEM medium supplemented with 10% FBS, 2 mM Glutamax, and 100 units/ml penicillin/streptomycin. All tumor cells were HLA typed to confirm the HLA-A*02:01 genotype. Briefly, genomic DNA was extracted from MGG123, MGG75, BT142, and D270 using the Quick-DNA miniprep kit (Zymo Research; Tustin, CA) and subsequently PCR amplified using the forward primer: ACCGTCCAGAGGATGTATGG and reverse primer: CCAGGTAGGCTCTCAACTGC. The 202bp DNA band was extracted and gel purified using the MinElute gel extraction kit (Qiagen). Sanger sequencing was performed using the above forward primer and aligned to the HLA-A*02:01 sequence (GenBank: GQ996941.1). U-87 MG cells were HLA typed by the Tissue Typing Laboratory of Brigham and Women’s Hospital.
METHOD DETAILS
Single-cell isolation from tumor tissue
Tumor specimens were mechanically disrupted into small pieces with a disposable, sterile scalpel and further dissociated into single cell suspensions using the enzymatic brain dissociation kit (P) from Miltenyi Biotec (Bergisch Gladbach, Germany) following the manufacturer’s protocol. Fc receptor blocking was performed on the total cell suspension using Human TruStain FcX reagent (Biolegend; San Diego, CA). Cell suspensions were subsequently stained for flow cytometry using antibodies specific for CD45 [HI30]-BV605, CD3 [HIT3a]-BV510, PD-1 (CD279) [MIH4]-PE, CD161 [DX12]-BV421 from BD Bioscience (Franklin Lakes, NJ) (BWH specimens) and CD45 [REA747]-VioBlue, CD3 [BW264/56]-PE from Miltenyi Biotec (Bergisch Gladbach, Germany) (MGH specimens). The exclusion panel included the following APC-labeled mAbs: CD14 [63D3], CD64 [10.1], CD163 [GHI/61], CD15 [HI98] from Biolegend (San Diego, CA), and CD66b [G10F5] ThermoFisher Scientific (Waltham, MA). Tumor cell suspensions were next spiked with 0.5 μM Calcein AM (ThermoFisher) to enable gating of live cells and incubated in the dark at room temperature for 10 minutes. Live, single T cells (gating: Calcein, AM+, Exclusion−, CD45+, CD3+) from all IDHwt-GBM + IDHmut-gliomas were sorted into individual wells of a 96-well twin.tec PCR plate (Eppendorf; Hamburg, Germany) that contained 10μL/well of RLT buffer (Qiagen; Venlo, Netherlands), using a BD Biosciences Aria III fluorescence-activated cell sorter (FACS) with a 70μM nozzle. Plates were immediately centrifuged at 800 xg for 1 minute at 4°C and frozen on dry ice. For scRNA-seq library construction using the 10X Genomics protocol, 15,000 live, single T cells (gating: Calcein AM+, Exclusion−, CD45+, CD3+) were sorted in a 15ml Falcon tube containing 8ml media (RPMI).
Preparation of scRNA-seq libraries
Single cell cDNA and sequencing libraries were prepared using the SMART-seq2 protocol (Picelli et al., 2014) with several adaptations. Agencourt RNAClean beads were used to purify RNA prior to oligo (dT) hybridization. RNase inhibitor was added to the dT annealing step at 1U/μl. Trehalose (1M) was used instead of water to make up the reaction volume. For the reverse transcription step, Maxima RNaseH-minus RT (200 U/μl) was used as the RT enzyme at 2U/μl. MgCl2 was added at 0.01M and DTT was omitted from the reaction. Trehalose (1M) was used to stabilize the reaction rather than betaine. The RT step was performed at 50°C for 90 minutes followed by 85°C for 5 minutes. In the PCR pre-amplification step, an ISPCR primer was used at 0.2μM and PCR was performed for 21 cycles. PCR purification was performed using AMpure XP beads at 0.8X. Libraries were tagmented and enriched with dual indexes using Illumina Nextera XT Library Prep kits. Libraries from 384 or 768 cells with unique indexes were pooled together and sequenced at 2pM on an Illumina Nextseq 500 sequencer, yielding paired-end 38 base pair reads (Tirosh et al., 2016a).
Preparation and sequencing of scRNA-seq libraries using 10X Genomics Protocol
Gene expression and TCR enriched libraries were prepared as previously described (Luoma et al., 2020). Briefly, tumor samples were dissociated to generate single cell suspension and stained for FACS, as described above. 15,000 CD45+CD3+ cells were sorted and washed with PBS containing 0.05% RNase-free BSA (ThermoFisher Scientific #AM2616). The isolated cells were loaded into separate channels of a Single Cell Chip A with reverse transcriptase reagent mixture and 5’gel beads according to the manufacturer’s protocol (10X Genomics; Pleasanton, CA). Chips were next loaded into the 10X Genomics Chromium Controller for single-cell partitioning, immediately followed by emulsion recovery from the chip and incubated in a deep-well block Thermocycler (Bio-Rad Laboratories) for the reverse transcription reaction.
cDNA isolation and library preparation were completed as per the manufacturer’s protocol. Isolated cDNA was amplified (13 cycles). Consecutively, cDNA was allocated for preparation of a gene expression library or TCR enrichment/library preparation with the Chromium Single Cell V(D)J TCR kit (10X Genomics). The quality of the cDNA and library was evaluated using the D5000 and D1000 high-sensitivity kits on a 2200 TapeStation system (Agilent; Santa Clara, CA). T cell gene expression libraries and TCR V(D)J libraries were sequenced on the Illumina HiSeq platform with 150 bp paired-end read configuration. For gene expression libraries 20,000 reads per cell and for TCR enriched libraries 5,000 reads per cell were obtained, respectively.
RNA in situ hybridization
Paraffin-embedded tissue sections from human tumors from Massachusetts General Hospital were obtained according to Institutional Review Board-approved protocols (DF/HCC 10-417). Sections were mounted on glass slides and stored at −80°C. Slides were stained using the RNAscope 2.5 HD Duplex Detection Kit (Advanced Cell Diagnostics, Cat. No. 322430). Slides were baked for 1 h at 60°C, deparaffinized and dehydrated with xylene and ethanol. The tissue was pretreated with RNAscope Hydrogen Peroxide (Cat. No. 322335) for 10 min at room temperature and RNAscope Target Retrieval Reagent (Cat. No. 322000) for 15 min at 98°C. RNAscope Protease Plus (Cat. No. 322331) was then applied to the tissue for 30 min at 40°C. Hybridization probes were prepared by diluting the C2 probe (red) 1:40 into the C1 probe (green). Advanced Cell Technologies RNAscope Target Probes used included Hs-CD3E (Cat. No. 553971-C2), Hs-KLRB1 (Cat. No. 509031), Hs-Hs-PTPRC (Cat. No. 601991-C2), Hs-NPM1-X-CLEC2D (Cat. No. 419751). Probes were added to the tissue and hybridized for 2 h at 40°C. A series of 10 amplification steps were performed using instructions and reagents provided in the RNAscope 2.5 HD Duplex Detection Kit. Tissue was counterstained with Gill’s hematoxylin for 25 s at room temperature followed by mounting with VectaMount media (Vector Laboratories).
Analysis of NK cell receptors on T cells from primary human GBM specimens
GBM tissue was dissociated into a single cell suspension as described above. Next, Fc receptors were blocked with Human TruStain FcX (Biolegend), and cells were subsequently stained with CD45 [HI30]-PacificBlue, CD8ɑ [HIT8a]-AlexaFluor488, CD4 [OKT4]-AlexaFluor700, CD16 [3G8]-BV570, CD25 [BC96]-BV605, TIGIT [A15153G]-PE/Cy7, CD279 (PD-1) [EH12.2H7]-APC, Zombie NIR (obtained from biolegend), CD3 [HIT3a]-BV510, CD161 [DX12]-BV421 (obtained from BD), CD56 [CMSSB]-PerCP/eFluor710 (from Invitrogen), NKG2C [REA205]-PE from MiltenyiBiotec, and NKp80 [239127]-AlexaFluor750 from R&D systems. All reagents were titrated and confirmed to stain PBMCs from healthy individuals. Samples were analyzed on a Sony SP6800 spectral analyzer which captured fluorescence emission spectra from 500 nm to 800 nm using a 32-channel photomultiplier tube (PMT). The signals from each fluorophore were unmixed mathematically by the spectral shape using an algorithm based upon the least squares method. Cells were gated on live (ZombieNIR negative), CD45+, CD3+ and CD8+ or CD4+ populations. Cell populations were then analyzed for labeling with CD161 as well as PD-1, TIGIT, CD56, NKG2C and NKp80 antibodies. To assess the NK-cell to T-cell ratio in GBM samples, cells were gated on live (ZombieNIR negative) CD45+ population. The total cell numbers of CD3+ or CD3−CD56+ cells reported were determined based on the total number of cells in the dissociated single cell tumor suspensions and the percentage of gated ZombieNIR negative CD45+ cells that were either CD3+ or CD3−CD56+.
Analysis of murine dendritic cells in GL261 tumors
The study was performed following approval by the Dana-Farber Animal Care and Use Committee (Protocol: 08-049). GL261-luc2 cells (1 x 105) were resuspended in PBS and injected stereotactically into the right striatum of anesthetized, 7-week-old female albino C57BL/6 mice (The Jackson Laboratory, ME) using a Hamilton syringe and stereotactic frame. Mice with increasing bioluminescence signal between days 7 and 14 after tumor implantation were randomized into IgG control and PD-1 mAb (clone 8H3) treatment cohorts (6 mice per cohort). The antibodies were administered via intraperitoneal injection beginning on day 14 (500 μg/mouse) after tumor implantation with repeat injections every 3 days (250 μg/dose) for a total of three injections. Brain samples were collected on day 22 and analyzed as below.
Tumors were dissected from brain tissue under a stereo microscope. Tumors were minced thoroughly in petri dishes, and single cell suspensions were generated using the Brain Tumor Dissociation Kit (Miltenyi Biotec) according to the manufacture’s protocol with some modifications. Briefly, after dissociation enzymes had been added, samples were shaken and incubated for 15 min at 37°C, and then pipetted up and down (20 times). These incubation and pipetting steps were repeated once. The samples were passed through a 70 μm cell strainer before staining for cell surface markers. Single cell suspensions were incubated with Fc blocker (purified anti-mouse CD16/32), and cells were subsequently stained with anti-CD3 [17A2]-PE/Cy7, anti-CD45 [30-F11]-AlexaFluor 700, anti-NK1.1 [PK136]-Brilliant Violet 510, anti-CD4 [GK1.5]-Brilliant Violet 650, and anti-CD8a [53-6.7]-Brilliant Violet 785 (2 μg/ml). Cells were washed and incubated with phycoerythrin (PE)-conjugated streptavidin (Prozyme). CLEC2D positive cells were identified by staining with anti-I-A/I-E [M5/14.15.2]-AlexaFluor 647, anti-CD45 [30-F11]-AlexaFluor 700, anti-CD11c [N418]-Brilliant Violet 785 and anti-CLEC2D [166C1309]-AlexaFluor 488 (Novus biologicals), or AlexaFluor 488-conjugated isotype control. Cells were treated with Zombie UV (Biolegend) after surface marker staining, and analyzed using a LSR Fortessa (BD Biosciences). Data were analyzed using FlowJo software (Tree Star). Antibodies were purchased from Biolegend unless otherwise stated.
Engineering GBM and IDH-G cellular models
The MGG123, MGG75, and D270 cells were transduced with a lentivirus to express a NY-ESO-1 cDNA that encoded the relevant NY-ESO-1 protein (NYEP) containing the NY-ESO-1 epitope SLLMWITQC that is recognized by the 1G4 TCR when presented by HLA-A*02:01. This lentiviral vector also encoded the selection marker ZsGreen (for MGG123 and MGG75) or nonfunctional human nerve growth factor receptor (NGFR) (for D270 and BT142) to enable the detection and isolation of transduced cells. The BT142 cells were also transduced with a lentiviral construct that contained a cassette to induce the expression of HLA-A*02:01 which was followed by the NGFR extracellular domain. Gliomaspheres positive for the indicated selection marker (ZsGreen MGG123 and MGG75; or NGFR for D270 and BT142 cells) were purified by FACS to > 99% purity to select a population with uniform NY-ESO-1 expression. Gliomasphere tumor cells were utilized between 2 and 5 passages for all experiments, and the appropriate selection marker expression was periodically verified to be expressed by > 99% of the cells.
U-87 MG cells were transduced with a lentivirus to express the NY-ESO-1 cDNA and the NGFR (nonfunctional) extracellular domain. U87-NGFR+ cells were purified by FACS to > 99% purity and were utilized between 2 and 5 passages for all experiments. The NGFR marker expression was periodically verified.
Plasmid cloning and lentiviral vectors
To generate the NY-ESO-1+ tumor lines, cDNAs encoding NY-ESO-1 (accession number: NM_139250.2) and luciferase were connected through a 2A ribosomal skip sequence and cloned into the pHAGE-MCS lentiviral vector under the control of the EF-1ɑ promoter (Figure S5I). The cDNA encoding Zsgreen (for constructs transduced into MGG123 and MGG75) or the extracellular domain of NGFR (for constructs transduced into D270, BT142, and U-87 MG) was synthesized and cloned downstream of an IRES to serve as a marker for transduced cells; NGFR was non-functional as it lacked a cytoplasmic signaling domain. Recipient cells of NGFR were verified to be NGFR-negative prior to transduction. Following the amplification of the insert, the PCR product was run on an electrophoresis gel and the band of expected size was excised and purified using the MinElute gel extraction kit (Qiagen) following the manufacturer’s protocol. Next, the purified amplicon was ligated into the vector backbone using the NotI and ClaI restriction enzymes. Transformed bacteria were plated on agarose containing ampicillin (100 μg/ml) and a single colony was sequenced (Quintarabio; South San Francisco, CA).
For the gliomasphere cells, VSV-pseudotyped third-generation lentiviruses were produced by Lipofectamine 2000 (Thermo Fisher Scientific)-based transfection of 293T cells with a transfer plasmid, packaging plasmids (GAG/POL, RSV-Rev), and an envelope plasmid (VSV-G). Briefly, 293T cells were plated at a density of 6 x 104 viable cells/cm2 on a poly-L-lysine (Sigma) treated 100 mm dish. 24 hours after plating, a mixture of lentiviral plasmids (10 μg of transfer plasmid, 6.5 μg of GAG/POL, 2.5 μg of RSV-Rev and 3.5 μg of VSV-G) was transfected with Lipofectamine 2000, following the manufacturer’s protocol. After 12-15 hours incubation, the culture medium was changed, and the supernatant was collected 48 and 72 hours post-transfection. Lentiviruses were concentrated by ultracentrifugation or with Lentivirus Precipitation Solution (ALSTEM) and resuspended in PBS to remove serum that can cause irreversible differentiation of gliomasphere cells. Sphere-forming glioma cells were transferred to ultra-low attachment 6-well plates without disrupting the spheres and incubated with the lentiviruses in a serially diluted manner.
For the U-87 MG model, the plasmid was transfected with packaging plasmids pCMV-dR8.91 and pCMV-VSV-G (Addgene #8454) into HEK293FT cells. Transfection was performed using TransIT-293 (Mirus, MIR2700), following the manufacturer’s protocol. Viral supernatant was harvested at 24 hours and 48 hours post-transfection and concentrated using a Sorvall wX+ ultracentrifuge (ThermoFisher) at 25,000 x g for 1.5 hours at 4°C. Virus titer was determined after all viral aliquots were frozen on dry ice and stored at −80°C. Virus was applied to the indicated tumor cells in a serial dilution from 1:1 to 1:2048.
The transfected tumor cells were isolated by FACS to > 99% purity based on the expression of the selection marker ZsGreen (MGG123 and MGG75) or by staining for the human NGFR selfantigen (D270, BT142, and U-87 MG).
To generate the lentiviral transduced NY-ESO-1 [1G4] T cell receptor cells used in the U-87 MG experiments, HA and PC epitope tags were placed at the N-terminus of the mature TCR α and β chains, respectively (Figure S5H) of the NY-ESO-1 TCR cDNA (1G4 TCR) (Robbins, Paul F, JI 2008). cDNA was gel purified (as described above) and inserted into the pHAGE-MCS lentiviral vector backbone under the control of the EF-1ɑ promoter using NheI and ClaI restriction enzymes. The DNA sequence of the construct was confirmed for a single bacterial colony. Lentivirus was prepared as described above and stored at −80°C. NY-ESO-1 TCR lentivirus titration was performed on Jurkat J.RT3-T3.5 cells (ATCC) that lack the β chain of the T cell receptor. Briefly, a non-tissue culture treated 24-well plate was coated with retronectin (Takara; Kusatsu, Japan) at a final concentration of 15 μg/ml at 4°C overnight. The plate was then blocked with 2% bovine serum albumin (BSA) in phosphate buffered saline (PBS) for 15 minutes at room temperature. Virus was applied to the plate in a serial dilution from 1:1 to 1:8,192 in a volume of 300 μL tissue culture media per well. The virus was then attached to the plate in a prewarmed 32 °C centrifuge at 2,000 x g for 2.5 hours. Next, the viral supernatant was decanted, and wells were gently washed with PBS. Jurkat J.RT3-T3.5 cells (0.5 x 106) were then transferred to each well in the presence of protamine sulfate (10 μg/ml) in a total volume of 2 ml. The cells were next cultured for 3 days and subsequently stained with a primary antibody that detects the HA-tag [3F10] (SigmaAldrich; St. Louis, MO) on the transfected NY-ESO-1 TCRɑ chain and a secondary anti-rat-FITC conjugated antibody (Life Technologies; Carlsbad, CA). HA-positive cells were quantified by flow cytometry using a Fortessa X-20 (BD Bioscience) flow cytometer.
Virus titer (viral particles/μL) was calculated by the equation: (percentage of HA-tag positive cells x the number of cells infected / volume in well) x dilution factor. The multiplicity of infection (MOI) of 15 was subsequently calculated by the equation: (number of cells infected x 15) / virus titer.
Generation of bivalent CD161 and CLEC2D fusion proteins
To generate bivalent CD161- and CLEC2D-fusion proteins, the cDNAs of KLRB1 (Accession number: NM_002258.3) and CLEC2D (Accession number: NM_013269.6) were synthesized as gBlocks (IDT). Both CD161 and CLEC2D are expressed on the cell surface as homodimers. To generate bivalent fusion proteins, two copies of each gene were connected by a flexible linker followed by a mutated human IgG1 Fc region (to prevent binding to activating Fc receptors). Constructs were expressed via transient transfection in Expi293F cells using the ExpiFectamine 293 Transfection Kit (Gibco). Transfected cells were grown in Optimum Growth flasks (Thomson Instrument) for 4-6 days at 37°C, 8% CO2, 125-150 rpm. Supernatants were spun, filtered, and passed over a Protein G Sepharose 4 Fast Flow column (GE). Fusion proteins were eluted at pH 11.5, neutralized, and concentrated in Amicon Ultra spin concentrators (Millipore Sigma), then further purified by gel filtration using a Superose 6 HPLC column (GE). Final yields for bivalent CLEC2D-Ig and CD161-Ig fusion proteins were 13 mg/L and 0.6 mg/L, respectively.
Isolation of primary human T cells for co-culture experiments
Primary human T cells were isolated from fresh leukophoresis blood collars provided by the Brigham and Women’s Hospital blood bank. Briefly, PBMCs were isolated by density gradient centrifugation using Premium Ficoll-Paque 1.078 g/ml (GE Healthcare). T cells were isolated using the Human T cell isolation kit (EasySep, cat#17951) following the manufacturer’s instructions. Human T cells were maintained in vitro in RPMI-1640 medium supplemented with 9% fetal bovine serum (FBS), 1% human serum, 50 units/ml penicillin/streptomycin (Pen/Strep), 5 mM HEPES, 2 mM Glutamax, 5 mM non-essential amino acids, 5 mM sodium pyruvate, 50 μM β-mercaptoethanol, and 30 units/ml of recombinant human IL-2 (Peprotech; Rocky Hill, NJ). CD161+Vα7.2- T cells were next purified by FACS after blocking of Fc receptors with Human TruStain FCX (Biolegend) followed by staining of cells with pre-conjugated flow cytometry antibodies specific for CD3 [HIT3a]-APC, CD161 [HP-3G10]-PerCP_Cy5.5, Vα7.2 [3C10]-BV786. Following sorting using an Aria III (BD Biosciences), and where indicated, polyclonal CD161+Vα7.2- T cells were expanded using ɑCD3/ɑCD28 coated human Dynabeads (Thermo Fisher Scientific; Waltham, MA) at a bead to cell ratio of 1:1 in the presence of recombinant human IL-2 (30 IU/ml) (Peprotech; Rocky Hill, NJ) for three days, followed by electroporation with RNPs containing gRNAs targeting TRAC and either KLRB1 or control. T cells used in U-87 MG experiments were next expanded using fresh Dynabeads and IL-2 for an additional 3 days followed by transduction with the NY-ESO-1 TCR lentivirus. Transduced T cells were labeled using anti-HA [3F10] primary antibody (SigmaAldrich) and a secondary anti-rat FITC antibody (Invitrogen) to enable isolation of NY-ESO-1 TCR+ T cells (HA positive) using an Aria III flow cytometer.
Gene editing of primary human T cells
Guide RNA (gRNA) sequences were designed using the ChopChop Harvard software suite to identify five sequences that specifically targeted each gene (Labun et al., 2016). Briefly, crisprRNAs (crRNA) targeting the human CD4, CD2, KLRB1 or TRAC genes as well as two “non-targeting” control crRNAs (targeting LacZ or an intergenic region) were synthesized by Integrated DNA Technologies (IDT; Coralville, IA). Guide RNAs (gRNAs) were generated by incubating equimolar concentrations of crRNAs targeting KLRB1 region#1 (AATTAAAGCCACTTACCCCG), KLRB1 region#2 (TTACCCCGAGGAAGAGATGA), TRAC (TCAGGGTTCTGGATATCTGT), CD4 (GGCAAGGCCACAATGAACCG) or controls with tracrRNA (IDT) in nuclease free buffer (IDT), according to the manufacturer’s instructions, by incubation at 95°C for 5 minutes; this yielded gRNAs at a final concentration of 60 μM. Reactions were cooled to room temperature, followed by addition of an equal volume of Cas9 protein (20 μM) modified to carry two nuclear localization signals (Cas9-NLS provided by QB3 Macrolab of the University of California Berkeley; Berkeley, CA). The Cas9 protein and gRNA mixture was next incubated at 37°C for 15 minutes to form ribonucleoprotein complexes (RNP). To edit target genes, freshly isolated human T cells were stimulated with human ɑCD3/ɑCD28 Dynabeads with hIL-2 (30 U/mL) for one to three days, as indicated. The expanded T cells were collected and resuspended in P3 buffer (Lonza; Basel, Switzerland) according to the manufacturer’s protocol. Next, 1 x 106 T cells (in 20 μL P3 buffer) were electroporated with 3 μL RNP per well of a nucleocuvette strip using an Amaxa 4D nucleofector with program EH-100. The final concentration of Cas9 in each nucleocuvette well was 10 μM. Electroporated T cells were then transferred to individual wells of a 48 well recovery plate containing human αCD3/αCD28 Dynabeads and hIL-2 (30 U/ml) in fully supplemented RPMI-1640 media (as described above). Dual gene editing was achieved by resuspending 1 x 106 primary human T cells in 17 μL of P3 buffer and electroporation with 3 uL each of the two gRNAs, as indicated.
Generation of NY-ESO-1 TCR T cells
For experiments with the gliomasphere tumor cells, the NY-ESO-1 (1G4) TCR was induced using a single stranded DNA homology directed repair template (HDR) with an RNP (TRAC gRNA + Cas9 protein) directed against the human TRAC locus. The HDR template was designed as previously described (Roth et al., 2018) with several modifications. Briefly, a T2A self-cleaving peptide was placed upstream of a PC tag, which was immediately followed by the NY-ESO-1 β-variable region, the β-constant region, and a P2A peptide. These regions were followed with an HA tag, the NY-ESO-1 α-variable region, and a partial (45 nt) α-constant region that resulted in the TRAC gRNA binding site to be inactivated thus preventing multiple rounds of editing. Upon incorporation of the HDR template, the partial α-constant region of the template inserted in-line with the remaining endogenous α-constant region resulting in a functional α-chain upon protein translation (Figure S5G). The 5’ and 3’ ends of the described template are surrounded by ~300nt homology arms.
Purified (described above) CD161+Vα7.2- T cells (1 x 106 per well) were electroporated with 3 μL of TRAC RNP + 4 μg of single stranded HDR template in 16 μL of fully supplemented P3 buffer. Following electroporation, 80 μL of prewarmed RPMI supplemented media was added to each well, and the T cells were incubated in the nucleovette strips for 10 mins at 37 °C. T cells were transferred to pre-acclimated recovery plates containing IL-2 (30 U/ml) and a 1:1 ratio of CD3/CD28 dynabeads (Invitrogen) and expanded for 10 days.
The experiments for the U-87 MG model utilized CD161+Vα7.2- T cells that were transduced with lentivirus by spin infection (as described above) to introduce the NY-ESO-1 TCR. Briefly, a non-tissue culture treated 24 well plate was coated with 0.8 ml of 15 μg/ml Retronectin (Takara; Kyoto, Japan) overnight at 4°C. Wells were blocked with sterile 2% BSA for 15 minutes at room temperature and gently washed once with PBS. Next, lentivirus was added to wells of the retronectin-coated plate at a multiplicity of infection (MOI) of 15, and plates were spun for 2.5 hours at 2,000 x g at 32°C. The supernatant in the wells was then carefully decanted, and wells were gently washed with 0.5 ml of PBS. 0.5 x 106 control or KLRB1 edited T cells were transferred to wells containing 10 μg/ml protamine sulfate (SigmaAldrich) in RPMI-1640 media containing 30 U/ml IL-2 and cultured for three days. NY-ESO-1+ T cells were isolated to >90% purity by FACS and immediately expanded with Dynabeads and IL-2 (30 U/ml) for 5 days.
Co-culture of edited human T cells with GBM cells
Patient-derived human gliomasphere cells (MGG123, MGG75, or BT142) were dissociated with Accutase (Millipore Sigma) for 3 minutes, filtered with a 100 μM cell strainer to generate a single cell suspension, and seeded into ultra-low attachment 96-well plates at a density of 2.5 x 104 cells per well. The tumor cells were allowed to reform neurospheres for 4 hours prior to the addition of T cells. U87-NYEP+ tumor cells were seeded into 96-well plates at a density of 3 x 104 cells per well and allowed to attach to the plate for 8 hours. NY-ESO-1 TCR+ T cells, that had been edited with KLRB1 (or control) gRNAs as well as a TRAC gRNA, were seeded into wells in triplicate at the indicated effector T cell to tumor cell ratios (E:T). For co-culture experiments with CD161 blocking mAb, NY-ESO-1 TCR+ T cells that expressed wildtype KLRB1 (but that had the endogenous TCR inactivated by editing the TRAC locus) were incubated with indicated target tumor cells in the presence of 10 μg/ml of either CD161 [HP-3G10] mAb or mouse IgGκ isotype control Ab. Co-cultures were incubated for 24 or 72 hours. The supernatants from the cocultures that contained KLRB1 or control edited T cells or CD161 mAb were collected for detection of IL-2 and IFNγ by enzyme linked immunosorbent assays (ELISA) according to the manufacturer’s instructions (Biolegend). ELISAs were performed in triplicate in DELFIA high-binding 96-well assay plates (Perkin-Elmer; Waltham, MA). Each well was coated with capture antibody (based on lot specific antibody concentration) that was diluted in carbonate buffer with pH of 9.5 and incubated overnight at 4°C. Plates were washed with PBS + 0.05% Tween-20 and subsequently blocked with 1% BSA in PBS at room temperature for 1 hour. Following a two hour incubation with experimental cell culture supernatants and the manufacturer provided standard, plates were washed and incubated with the manufacturer’s biotinylated detection antibody for 1 hour at room temperature. The secondary antibody was next detected with streptavidin-Europium (Perkin-Elmer; Waltham, MA) and DELFIA Enhancement Solution (Perkin-Elmer). The Europium fluorescence was analyzed using an EnVision multimode plate reader (Perkin-Elmer).
Gliomasphere co-culture experiments that were analyzed by flow cytometry were collected at the indicated time points and stained with the viability dye Zombie UV (Biolegend) in PBS followed by monoclonal antibodies specific for extracellular markers dependent upon the assay. The eight hour cytotoxicity killing experiments were stained with: CD3 [HIT3a]-APC and CD45 [HI30]-APC/Cy7. The co-cultures examining T cell activation were stained with: CD3 [HIT3a]-BV510, CD8 [RPA-T8]-BV650, CD69 [FN50]-BV421, CD25 [BC96]- BV785, CD107a [H4A3]-PE, PD-1 [EH12.2H7]-PE/Dazzle594, and CD161 [HP-3G10]-PerCP/Cy5.5 in 2% FBS in PBS. The cells were next fixed (Cytofix/Cytoperm, BD bioscience) for analysis.
For U-87MG co-culture assays analyzed by flow cytometry, cells were co-cultured in the presence of protein transport inhibitor cocktail (ThermoFisher) for the final six hours of culture in the absence of PMA/ionomycin; cells were then stained with Zombie UV (viability) in PBS and then the following monoclonal antibodies specific for the extracellular markers CD3 [HIT3a]-BV510, CD8 [RPA-T8]-BV650, CD4 [OKT4]-APC/Cy7, CD69 [FN50]-BV421, CD161 [HP-3G10]-PerCP/Cy5.5 in 2% FBS in PBS. The cells were fixed (Cytofix/Cytoperm, BD bioscience), permeabilized (Permeabilization buffer, Thermofisher), and stained for the intracellular markers Perforin [B-D48]-PE, GranzymeB [GB11]-AlexaFluor647, IL-2 [MQ1-17H12]-FITC, IFNγ [4S.B3]-BV711, TNFɑ [MAb11]-PE/Cy7. To further examine exhaustion markers, a separate overlapping panel was used that contained monoclonal antibodies that detected CD3 [HIT3a]-BV510, CD8 [RPA-T8]-BV650, CD4 [OKT4]-APC/Cy7, LAG-3 [11C3C65]-FITC, CTLA-4 [L3D10]-PE/Cy7, TIM-3 [F38-2E2]-BV785, TIGIT [A15153G]-PE, PD-1 [EH12.2H7]-PE/Dazzle594.
Flow cytometry acquisition was performed using an LSR Fortessa X-20 (BD Bioscience). Flow cytometry files were analyzed using FlowJo v10.7.1 (BD; Franklin Lakes, NJ). The CD3 [HIT3a]-BV510 was purchased from BD Bioscience, CD161 [HP-3G10]-PerCP/Cy5.5 from eBioscience, and all other flow cytometry antibodies were purchased from Biolegend.
Humanized mouse model to evaluate impact of KLRB1 gene on T cell function
The following procedures were performed following approval by the Dana-Farber Animal Care and Use Committee (Protocol: 08-049). Tumor implantation into the left striatum of the mouse cortex and the subsequent administration of T cells into the right lateral ventricle were achieved by stereotactic surgical injections. NOD.Cg-Prkdc<scid> IL2rg<tm1Wjl>/SzJ (NSG) mice were shaved on the crown of the head from midbrow to ears, weighed, and tagged one day prior to surgery. On the day of surgery, anesthesia was induced using a precision isoflurane vaporizer (VetEquip; Livermore, CA) at an input air flow rate of 3.0 liters/min and isoflurane of 4% for 3 minutes. The anesthetized mouse was next transferred into a small animal stereotactic instrument (Kopf Instruments; Tujunga, CA) that was equipped with continuous adjustable flow of isoflurane to maintain anesthesia with input air flow of 1.5 liters/min and between 0.75% and 1.5% isoflurane. The eyes of the animal were protected with Puralube ophthalmic ointment (Dechra Veterinary; Overland Park, KS) and the crown was disinfected with povidone-iodine (PDI Healthcare; Woodcliff Lake, NJ). A 1 cm midline incision was made above the sagittal suture allowing the underlying fascia to be retracted by blunt dissection. The calvaria was next cleaned with sterile gauze. The skull was adjusted to a level plane by ensuring that the dorsal/ventral coordinates of bregma and lambda were within +/− 50 μm. Using Bregma as coordinate zero for all mice, the needle was adjusted to the following coordinates: medial/lateral: +0.2 mm; anterior/posterior: +0.1 mm; dorsal/ventral: −0.3 mm. A burr hole was drilled and the needle inserted for 3 minutes to form a void for the injected cells to aggregate. The needle was then partially withdrawn to the final injection depth of −0.25 mm. Next, 5 x 104 MGG123 or 1.5 x 104 U87 MG tumor cells that expressed the NY-ESO-1 protein were injected in a volume of 2 μL at a rate of 0.5 μL/min using a programmable Pump 11 Elite Nanomite micro-syringe pump (Harvard Apparatus; Holliston, MA). The needle was fully withdrawn three minutes following completion of the cell infusion and the trepanation was sealed with bone wax (Medline; Northfield, IL). The midline incision was closed using 9mm wound clips (MikRon Precision; Gardena, CA) and 4-0 Ethilon sutures (Ethicon; Somerville, NJ), followed by the application of triple antibiotic ointment to the incision daily for 2 days. All animals were administered buprenorphine 75 mg/kg subcutaneously immediately following surgery and twice daily thereafter for 2 days, as needed if animal exhibited signs of distress.
The T cells were administered into the contralateral ventricle seven or eight days following tumor implantation in a second stereotactic surgery using the methods described above, at the following coordinates: medial/lateral: +0.03 mm; anterior/posterior: −0.1 mm; and dorsal/ventral: −0.285mm. KLRB1 or control edited NY-ESO-1 TCR+ T cells were injected in a volume of 7.5 μL at a rate of 1.5 μL/min. The MGG123 gliomasphere model received two injections of 0.8 x106 NY-ESO-1 TCR+ T cells for survival experiments and flow cytometry analysis. The U-87 MG model received a single injection of 0.4 x 106 T cells for survival experiments or 2 x 106 T cells for flow cytometry analysis. The needle was removed 2 minutes after cell infusion was completed and closing was performed as described above. Buprenorphine was given for analgesia at the same dose and administration schedule used following tumor implantation. All instruments were sterilized between each mouse using a dry bead sterilizer (CellPoint Scientific; Gaithersburg, MD) at 350 °C for three minutes.
Bioluminescence imaging of tumor bearing mice
Following tumor implantation, mice where analyzed by bioluminescence imaging (BLI) starting on day −1, relative to T cell administration via the lateral ventricle. Briefly, mice were anesthetized using a precision isoflurane vaporizer (VetEquip; Livermore, CA) at an input air flow rate of 3.0 liters/min and isoflurane of 4% for 5 minutes followed by the intraperitoneal administration of luciferin (150 mg/kg). Eight minutes after the luciferin injection, the mice were imaged using a Xenogen Imaging System (PerkinElmer) with an automatic exposure time. BLI was performed twice weekly, as indicated, and analyzed using living image software package (PerkinElmer) to quantify signal flux (photons / second).
Histological analysis of intracranial MGG123 growth
MGG123-bearing mouse was euthanized at survival endpoint and perfused via the left cardiac ventricle with 10 ml of PBS, followed by 10 ml of buffered formalin (MilliporeSigma). The intact brain was removed from the cranium, and three gross sections were made: the first containing the frontal lobe anterior to bregma, the second encompassing the region intermediate of bregma and lambda, the third posterior of lambda. The gross sections were placed into a conical tube containing 50 ml of buffered formalin for 36 hours. The sections were then dehydrated in 70% ethanol for at least 24 hours prior to embedding in paraffin. 6 μM paraffin-embedded sections were stained with H&E by the Rodent Histopathology Core of Dana-Farber/Harvard Cancer Center and scanned using an Aperio VERSA 200 (Leica Biosystems; Wetzlar, Germany). Images were captured with Aperio Image Scope software v.12.4.0.5043 (Leica).
Ex vivo analysis
Upon reaching the protocol endpoint (moribund), MGG123 gliomasphere-bearing mice were euthanized, and the intact brain was extracted from the cranium. The whole brain was grossly sectioned into three regions (as described above) and the tumor was carefully isolated from the surrounding parenchyma. The lesion was dissociated, filtered into a single cell suspension, and total cells were counted. The cells were then incubated with the viability dye Zombie UV per the manufacturer instructions in PBS and next stained in 2% FBS in PBS with monoclonal antibodies to detect the extracellular markers: CD3 [HIT3a]-BV510, CD8 [RPA-T8]-BV650, CD4 [OKT4]-APC/Cy7, TIM3 [F38-2E2]-BV421, PD-1 [EH12.2H7]-PE/Dazzle594, and CD161 [HP-3G10]-PerCP/Cy5.5. The cells were fixed (BD bioscience), permeabilized (Thermofisher), and stained for the intracellular marker GranzymeB [GB11]-AlexaFluor647.
The mice in the U-87 MG model were euthanized on day 8 following T cell administration and the whole brain was removed from the cranium. The brain was grossly dissected, the tumor mass was removed, dissociated into a single cell suspension, total cells were counted, and incubated with Zombie UV viability dye. The cells were then stained in 2% FBS in PBS with monoclonal antibodies to detect: CD3 [HIT3a]-BV510, CD8 [RPA-T8]-BV650, CD4 [OKT4]-APC/Cy7, TIGIT [A15153G]-PE, PD-1 [EH12.2H7]-PE/Dazzle594, and CD161 [HP-3G10]-PerCP/Cy5.5. The cells were then fixed, permeabilized, and stained for GranzymeB [GB11]-AlexaFluor647, IL-2 [MQ1-17H12]-FITC, IFNγ [4S.B3]-BV711, TNFɑ [MAb11]-PE/Cy7.
Flow cytometry acquisition for all ex vivo experiments was performed using an LSR Fortessa X-20 (BD Bioscience) and analyzed using FlowJo v10.7.1 (BD; Franklin Lakes, NJ).
qPCR analysis of CLEC2D mRNA
mRNA was isolated from the indicated cell lines using a RNeasy kit according to the manufacturer’s instructions (Qiagen, Venlo, the Netherlands). mRNA (1 μg) was used to synthesize cDNA using SuperScript VILO (Invitrogen; Carlsbad, CA). The following qPCR primers were designed for the detection of the following human cDNAs: GAPDH-Forward: GGAGCGAGATCCCTCCAAAAT, GAPDH-Reverse: GGCTGTTGTCATACTTCTCATGG; CLEC2D-Forward: CTGCCATCAAGAGCCATCAG, CLEC2D-Reverse: TCAGCCCAATCCAGTGATCA. Power SYBR Green master mix (Thermo Fisher) was used following the manufacturer’s instructions. All primers were verified for the production of a single specific PCR product with a melting curve program.
QUANTIFICATION AND STATISTICAL ANALYSIS
All computational analyses were performed in R (v4.0.0).
scRNA-seq data processing (SMART-Seq2)
We processed sequencing data from raw reads to gene expression matrices as previously described (Tirosh et al., 2016a). Briefly, we used bcl2fastq to generate demultiplexed FASTQ files, and aligned the resulting paired-end scRNA-seq reads to the human transcriptome (hg19) using Bowtie (v0.12.7) (Langmead et al., 2009). We quantified gene expression levels from these alignments as transcripts-per-million (TPM) values by running RSEM (v1.2.19) in paired-end mode (Li and Dewey, 2011). We normalized the total transcripts per cell to one-hundred thousand (TP100K), as this is our estimated complexity of single-cell libraries prepared by SMART-Seq2 (Tirosh et al., 2016a), and then log-transformed these values to report gene expression as E = ln(TP100K+1).
Quality control of scRNA-seq (SMART-Seq2)
For each cell, we considered a gene to be detected if its TP100K > 0. We excluded all cells with either <500 or >7,500 unique genes detected, or with fewer than 20 housekeeping genes detected based on a previously curated gene set (Tirosh et al., 2016a). Three patient samples (E88, E119, and MGH142), where the majority of T cells failed to meet these criteria, were completely omitted from further analysis.
To ensure our studied cell population consisted only of T cells, we carried out an initial round of graph-based clustering (Blondel et al., 2008), described below, to identify cell types that might have been flow-sorted with the T cells. We first identified and removed B cells, myeloid cells, and astrocytes. We next identified NK cells and cell doublets by carrying out clustering using non-negative matrix factorization (NMF) with the Brunet algorithm and 7 factors in the factorization. This was implemented in the R package NMF (v0.21.0) (Gaujoux and Seoighe, 2010). We identified doublets as a distinct cluster of cells expressing both T cell markers (CD3D, CD3E, CD3G) and either astrocyte markers (GFAP) or myeloid markers (C1QB, C1QC). We searched for potential NK cells by examining the cluster of CD8 T cells, as NK cells and CD8 T cells share transcriptional programs. We identified a small cell population (127 cells) within the cluster of CD8 T cells with low mean expression (ln(TP100K+1) < 1) of T cell markers (CD3D, CD3E, CD3G, CD247). We identified differentially expressed genes, as described below, between this cell population and the remaining CD8 T cells and found that this population had lower expression of other T cell markers (CD5, IL7R, CD8A, CD8B) and higher expression of NK markers (KIR2DL1, KIR2DL3, KIR2DS4). To ensure our results were not confounded by NK cells, we removed this population. The above filtering steps removed 8% of the cells. Finally, we also excluded a small percentage of cells (2%) that were either cycling T cells that expressed both CD4 and CD8, cycling T cells that lacked expression of CD4 and CD8, or Tregs that expressed CD8. Positive gene expression was defined as ln(TP100K+1) > 1 and lack of gene expression was defined as ln(TP100K+1) < 1. After these quality control steps, overall we retained 8,252 T cells from 27 glioma samples (from 26 patients), with a total of 22,448 genes detected in at least one cell. These T cells are used in all analyses reported in this work (clustering, cell type and cell state identification, clonal expansion, pan-cancer metaanalysis).
Cell type and cell state identification (SMART-Seq2)
We carried out analyses of all T cells to identify cell types, states, and their corresponding gene signatures using the R package Seurat (v2.3.4) (https://github.com/satijalab/seurat) (Butler et al., 2018). We chose highly variable genes for clustering analyses using FindVariableGenes, which controls for the inherent relationship between the mean and the variance of gene expression. We used the following thresholds for the mean expression (x) and the variance to mean ratio (y): x.low.cutoff = 0.1, x.high.cutoff = 7, y.cutoff = 0.5. We identified variable genes within each patient sample and selected the 1,500 variable genes that were most commonly shared across all patient samples. Next, we centered the expression of each gene to have a mean of zero using ScaleData and performed Principal Components Analysis (PCA) with RunPCA. We performed graph-based Louvain clustering on the top 20 principal components using FindClusters, with the resolution parameter set to 0.7 and k for the k-nearest neighbor algorithm set to 30. We identified differentially expressed genes for each cluster of cells using the t-test implemented in FindMarkers while adjusting p-values for multiple hypothesis testing with the Bonferroni correction. Finally, we visualized gene expression and clustering results on a Uniform Manifold Approximation and Projection embedding (UMAP) of the top twenty PCs using RunUMAP with the following settings: min_dist = 0.5, number of neighbors = 30, and distance metric = Euclidean.
To sub-cluster CD4 T cells (4,369 cells) or CD8 T cells (3,883 cells), we carried out clustering using NMF (Figure 1E). When clustering CD4 T cells and CD8 T cells, we used 6 factors in the factorization, and when clustering all glioma T cells found in clonotypes, we used 4 factors. As an alternative to NMF clustering, we also carried out graph-based Louvain clustering using resolution parameters of 0.5 and 0.7 for CD4 and CD8 T cells respectively. We calculated UMAP visualizations for the CD4 T cells and CD8 T cells using the top twenty PCs as input features, and the same settings as above.
We annotated the cell type or state represented by each cluster by considering the cluster’s differentially expressed genes together with the known cell type markers CD3D, CD3E, CD3G, CD4, CD8A, CD8B, and FOXP3. To identify a cell’s state in the cell cycle, we used CellCycleScoring, which scores the levels of programs for S phase and G2/M phase using previously defined gene sets for DNA replication and mitosis (Tirosh et al., 2016a).
To examine gene expression signatures within single cells, we scored the levels of specific signatures using AddModuleScore, which calculates the average expression levels of the genes in a signature and then subtracts off the average expression levels of control gene sets (Tirosh et al., 2016a). Control gene sets were chosen to have similar expression values to the genes in the signature. All genes were binned into 25 bins based on their average expression across all cells, and for each gene in a signature, a random set of 20 genes was chosen from the same average expression bin as that gene. This procedure controls a signature’s expression level for the differences in cell quality and library complexity across single cells. We defined cytotoxicity, NK, and inhibitory signature scores based on established marker genes. For the cytotoxicity signature, we included the genes PRF1, GZMB, GZMA, GZMH, NKG7, and GNLY, for the NK signature, we included the genes KLRD1, FGFBP2, FCGR3A, S1PR5, KLRC1, KLRC3, KLRB1, and KLRC2, and for the inhibitory signature, we included the genes PDCD1, CTLA4, HAVCR2, LAG3, and TIGIT. We defined a CD8 T cell as having a high NK signature score if it had an NK signature score > 0.5; otherwise we defined the cell as having a low NK signature score (Figure 2C). We then identified the differentially expressed genes between the high NK and low NK signature score CD8 T cells using FindMarkers as described above. We confirmed that the top differentially expressed genes were not sensitive to the threshold used to define cells as having a high NK vs. a low NK signature score by repeating the analyses with thresholds of 0 and of 1.
We identified significant changes in cell type proportions under different treatment regimes (prior corticosteroids or immune checkpoint inhibitors) or in different glioma classes (GBM or IDH-G) using a Dirichlet-multinomial regression model (Smillie et al., 2019).
Differential gene expression in IDH-G and GBM T-cells (SMART-Seq2)
To look for variability between the expression profiles of CD4 and CD8 T cells isolated from IDH-G and GBM, we used the FindMarkers function to identify differentially expressed genes between cells of the two tumor classes of a given subset. For three subsets, Interferon, Cytotoxicity, and Stress, we calculated the signature scores for the list of DE genes using AddModuleScore and compared the signature scores between the two classes by a two-sample Komolgorov-Smirnov test.
KLRB1 and tissue resident memory T cell signature comparison (SMART-Seq2 and 5’-end scRNA-Seq)
In order to determine if any of the subsets of T cells we identified were Tissue Resident Memory T Cells (Trms), we first calculated a signature score using AddModuleScore for a Trm signature (CA10, ITGA1, ITGAE, IL2, IL10, CXCR6, CXCL13, KCNK5, RGS1, CRTAM, DUSP6, PDCD1) and the expression of KLRB1. We then calculated the Pearson correlation coefficients between the Trm signature and KLRB1 expression in all CD4 T cells, all CD8 T cells, and their respective subsets of cells.
Reconstructing TCR chain sequences (SMART-Seq2)
For each single T cell, we computationally reconstructed its TCR α and β chain sequences from the corresponding scRNA-seq data using TraCeR (v0.6.0, https://github.com/Teichlab/tracer) (Stubbington et al., 2016). We ran TraCeR assembly with the following Trinity (Grabherr et al., 2011) settings in our configuration file: inchworm_only = True and trinity_kmer_length = 15. We classified a T cell as having a reconstructed TCR if we could reconstruct productive sequences for α and/or β chains. We defined clonotypes based on grouping T cells sharing an identical α and/or β chain, and we reported the size of the clonotype as the number of cells in this grouping. We defined a T cell as a singleton if it had a reconstructed TCR that did not share an identical α and/or β chain with any other T cell from that same tumor.
Differential gene expression in clonal cells (SMART-Seq2)
To compare expression levels between clonal and non-clonal T cells, we first calculated the mean expression of each gene in these respective populations. These populations differ in cell composition and cell quality across different patient tumors. To control for this, we matched every clonotype in each patient tumor with ten sets of non-clonal T cells randomly drawn from that same patient tumor. Each randomly selected set of non-clonal T cells had the same number of cells as the corresponding clonotype, and each selected T cell was a singleton. For each gene, we calculated its mean expression value across all cells in clonotypes from all tumors, and across all random sets of non-clonal cells from all tumors, and subtracted its mean expression in all non-clonal cells from its mean expression in clonal cells. Because we take the mean expression value across all cells from all clonotypes, this gives greater weight to clonotypes with greater size.
To compare expression variability between clonal and non-clonal T cells we followed a similar approach. To quantify variability of each gene for clonal cells, we calculated the L1 distance between the expression of a gene in cell x and in cell y in the same clonotype. (We used L1 and not L2, because squaring the expression difference will emphasize larger differences, which are often driven by technical noise in scRNA-seq.) For each clonotype from each patient tumor, we calculated the L1 distances for every possible pair of cells within the clonotype, and then calculated the mean distance across all these distances. We divided this mean L1 distance by the gene’s mean expression across all cells from all clonotypes (as above), to get a coefficient of variation (CV). To quantify variability of each gene for non-clonal cells, we use the same approach but instead calculated L1 distances within the random sets of non-clonal cells, with non-clonal cells defined as above. We finally calculated the difference in CV values between clonal and non-clonal cells. We excluded genes with low mean expression in both the clonal and nonclonal cells (ln(TP100K+1) < 0.1) as these genes would have greatly inflated CV values and not be reliably quantified.
To test for each gene whether the differences in mean expression or CV are significantly different between clonal and non-clonal cells, we empirically generated a background distribution. We shuffled the expression values between clonal and non-clonal cells for each gene and calculated the differences in mean expression and CV for this shuffled expression data. We repeated this 100 times for each gene and then calculated a z-score of the observed difference relative to this empirical null distribution. If the magnitude of the z-score was greater than 3, the observed difference in the actual data was more extreme than any of the values in the empirical null distribution.
scRNA-seq data processing (5’-end scRNA-seq)
We processed raw sequencing reads from both library types (gene expression and V(D)J) to generate gene expression matrices and to reconstruct TCR alpha and beta chain sequences. For both library types, we demultiplexed FASTQ files from the raw sequencing reads using Cell Ranger mkfastq (v3.1) (10x Genomics). For the gene expression libraries, we then aligned these reads to the human GRCh38 genome (refdata-cellranger-GRCh38-3.0.0) using Cell Ranger count (v3.1) (10x Genomics). We normalized the total number of unique molecular identifiers (UMIs) per cell to ten thousand (TP10K) and then log-transformed these values to report gene expression as E = ln(TP10K+1). For the V(D)J libraries, we assembled the sequencing reads into contigs and annotated the segments and CDR3 regions using Cell Ranger vdj (v3.1) (10x Genomics) and a V(D)J reference (refdata-cellranger-vdj-GRCh38-alts-ensembl-3.1.0). We used the clonotype groups as determined by Cell Ranger vdj, where cells in a clonotype share the same productive CDR3 nucleotide sequence.
Quality control of scRNA-seq (5’-end scRNA-seq)
For each cell, we considered a gene to be detected if its TP10K > 0. We excluded all cells with <400 UMIs detected, either <200 or >5,000 unique genes detected, or with >10% of UMIs coming from mitochondrial genes. As with our SMART-Seq2 analysis, we carried out an initial round of clustering, described below, to identify and remove cells that were not T cells or droplets that contained cell doublets. We first identified and removed myeloid cells and astrocytes. We then identified and removed two types of cell doublets: T cells expressing CD4, CD8A, and CD8B, and a cluster of Tregs expressing CD8. After these quality control steps, overall we retained 25,256 T cells from 5 glioma samples (from 5 patients). These T cells are used in all analyses reported in this work.
Cell type and cell state identification (5’-end scRNA-seq)
To identify the cell states present in CD4 T cells (3,665 cells) and CD8 T cells (21,502 cell), we used Seurat (v3.2.0). In order to perform batch correction and identify highly variable genes in the two populations of T cells, we split the data by sample, used NormalizeData to normalize gene expression, and identified variable genes using FindVariableFeatures. In order to integrate the samples, we used FindIntegrationAnchors to identify shared variable genes across samples and used SelectIntegrationFeatures to integrate the datasets. We then centered mean gene expression across all samples to zero using ScaleData and performed Principle Components Analysis (PCA) using RunPCA. We then used Louvain clustering on the top 20 principal components (PCs) using FindNeighbors, with the k-nearest neighbor algorithm set to 20, and FindClusters, with the resolution parameter set to 1. For two-dimensional visualization, we employed Uniform Manifold Approximation and Projection embedding (UMAP) using RunUMAP (min.dist = 0.5, number of neighbors = 15, and distance metric = Euclidean). Lastly, to identify differentially expressed genes we implemented FindAllMarkers.
In order to identify the different cell states in the CD4 T cells and CD8 T cells, we clustered these subpopulations using NMF. For clustering of the CD4 T cells, we used 8 factors; when clustering the CD8 T cells we used 9 factors. We then generated UMAP visualizations of the CD4 T cells and CD8 T cells using the top 20 PCs as described above. We used the differentially expressed genes and known cell type markers to annotate the cell type or cell state of each cluster identified using NMF.
Differential gene expression in clonal cells (5’-end scRNA-seq)
To compare expression levels in clonally expanded CD4 and CD8 T cells, we first binned cells by expression of KLRB1, those cells with KLRB1 expression greater than zero were deemed KLRB1-positive and those with expression less than or equal to zero were deemed KLRB1-negative. We then used FindMarkers to identify differentially expressed genes between the KLRB1-positive and KLRB1-negative populations.
Analysis of gene expression in additional scRNA-seq glioma datasets
To examine gene expression from malignant cells, myeloid cells and glial cells in diffuse gliomas, we analyzed our published, quality-controlled, scRNA-seq datasets for IDH-mutant oligodendroglioma, IDH-mutant astrocytoma, and IDH-wildtype GBM . Gene expression was given in units of ln(TP100K+1), and malignant cells, myeloid cells, and oligodendrocytes were identified based on the cell-type annotations provided in our published studies.
Analysis of gene expression by tumor-infiltrating T cells in additional scRNA-seq cancer datasets
We collected processed scRNA-seq datasets for the following human cancers: melanoma (Jerby-Arnon et al., 2018; Sade-Feldman et al., 2019), colorectal cancer (Zhang et al., 2018b), http://crc.cancer-pku.cn/), non-small cell lung cancer (Lambrechts et al., 2018), http://scope.aertslab.org/#/Bernard_Thienpont/), and liver cancer (Zheng et al., 2017), http://hcc.cancer-pku.cn/). In some studies, samples were collected from both malignant tissue and normal, adjacent tissues. We used the sample and cell-type annotations provided by the authors to select only T cells from the malignant tissue samples. In liver cancer and colorectal cancer, 4% and 1% of the T cells, respectively, were MAIT cells (as defined by the dataset authors). For melanoma, colorectal cancer, and liver cancer, we constructed t-distributed stochastic neighbor embedding (t-SNE) visualizations using RunTSNE with perplexity=30. For lung cancer, we used the t-SNE visualization provided by the authors.
Pan-cancer metanalysis of KLRB1 transcriptional programs
We identified KLRB1 programs by comparing between cells in which KLRB1 was detected (KLRB1+) and cells in which KLRB1 was not detected (KLRB1−). Each KLRB1 program consisted of the genes whose expression was significantly greater in KLRB1+ T cells relative to KLRB1− T cells, and as above, differential expression analysis was performed using a t-test on the log-transformed TPM100K values. We identified the KLRB1 programs separately in CD4 and CD8 T cells, using the glioma IDH-G + GBM cohort. We then repeated this analysis in the five other scRNA-Seq T cell datasets listed above. We evaluated the significance of overlap in genes between the programs that were identified in each T cell cohort with a hypergeometric test (Figure 7C). For each pair of programs we also report the observed vs. expected ratio (Figure 7D), that is, the number of genes in the overlap between the two programs, divided by the number of genes that are expected to be in the overlap if the programs were randomly sampled. Lastly, to integrate the results that were obtained across the six cohorts in this metanalysis and derive the final pan-cancer program, we used Fisher's combined probability test and corrected the resulting p-values for multiple hypotheses testing using the Benjamini-Hochberg correction. The different programs and the final pan-cancer p-values are provided in Table S5. To define the pan-cancer KLRB1 programs in CD4 T cells and in CD8 T cells (Figure 7E), we used those significant genes that appeared in at least 3 of 6 of the KLRB1 programs from the six cancer cohorts, including the glioma IDH-G + GBM cohort.
T cell differences between IDH-G and GBM in bulk RNA-seq profiles from TCGA
To evaluate the differences in T cell expression in a larger dataset, we leveraged the GBM and IDH-G bulk RNA-seq expression profiles from the TCGA. First, we combined the scRNA-seq data with our previously published glioblastoma scRNA-seq data, which were generated using the same protocol, to compare the average expression of each gene across different cell types and identify genes that were at least 8-fold higher expressed in T cells than in any other cell-type. Next, we found the log-transformed expression of each of these T cell specific genes in the bulk RNA seq dataset. Next, we calculated the total T cell signal for each tumor, defined by the average expression of the genes CD2, CD3D, CD3E, and CD3G. Since the state of T cells cannot be properly evaluated if the total T cell signal is low, we restricted further analysis to 129 out of 148 tumors from GBM dataset and 161 out of 428 tumors for the IDH-G dataset that had a total T cell signal above 2. Next, to normalize each gene’s expression in the bulk RNA-seq to its total T cell expression, we generated a relative expression profile for each T cell specific gene, by subtracting from its expression the median expression of all T cell specific genes. Next, to visualize the difference in expression between the GBM and IDH-G tumors, we performed a Student’s t test comparison between the normalized expression (as well as the non-normalized log2-transformed expression) of each T cell specific gene in GBM tumors compared to the IDH-G tumors and plotted the −log10(p value) from this comparison against the log2(fold change) value in a volcano plot.
Supplementary Material
Figure S1. Analysis of glioma-infiltrating T cells, Related to Figure 1. (A) Sorting of T cells from surgical glioma specimens. Single cell suspensions were generated for isolation of T cells by FACS. Individual live T cells were sorted into 96 well plates for scRNA-seq using the SMART-seq2 chemistry. T cells were sorted at two different facilities: T cells from GBM that were processed at DFCI were sorted as CD3+CD4+ and CD3+CD8+ cells; T cells from IDH-G that were processed at MGH were sorted as CD3+ cells. (B) Impact of tissue dissociation procedure on CD8 surface protein levels. Peripheral blood T cells were treated with PBS (left) or tumor dissociation buffer (right) and then analyzed for expression of CD4 and CD8 proteins following gating on live CD45+CD3+ cells. Incubation with the tumor dissociation buffer reduced the staining intensity with the CD8 mAb. (C) Quality-control analysis of scRNA-seq (SMART-seq2) data based on the number of detected genes (top) and housekeeping genes (bottom; dotted line indicates interrogated number of genes) for which transcripts were detected per cell. T cells from patients E88, E119, and MGH142 were excluded from further analysis. (D) UMAP plot with cell annotation based on prior dexamethasone (left) or immune checkpoint inhibitor (ICI) treatment (right). (E) Fraction of T cells assigned to the indicated T cell clusters based on glioma subtype (GBM or IDH-G) and prior treatment (dexamethasone, left; immune checkpoint inhibitor, right). (F) Fraction of T cells assigned to CD8 T cell, CD4 T cell, Treg, and cycling T cell clusters based on tumor type (IDH-G, GBM) and disease state (new onset versus recurrent). (G) Quantification of each tumor for CD8 T cell, CD4 T cell, regulatory T cell (Treg), and cycling T cell clusters; higher frequency of CD8 T cells in IDH-G versus GBM may be related to differences in details of cell isolation procedures as described in (A). (H) RNA-ISH for mRNA expression of three different cellular stress genes (HSPA1A, HSPH1, HSPA6) by CD3E+ T cells; labeling is shown for IDH-G (top) and GBM (bottom).
Figure S2. Transcriptional signatures of glioma-infiltrating T cells, Related to Figure 2 & 3. (A-C) scRNA-seq analysis of T cells from five GBM samples by 5’ sequencing using the 10X Genomics platform. (A) UMAP visualization of T cells color-coded based on tumor specimen. (B) UMAP visualization of major T cell subpopulations. (C) Heatmap showing top differentially expressed genes for clusters of CD8 (left) and CD4 (right) T cells. Similar cluster designation is shown as in Figure 1F for T cells from the SMART-seq2 dataset. (D-E) Analysis of TCGA datasets from GBM and IDH-G for expression of T cell specific genes. T cell-specific genes were identified using T cell scRNA-seq data from this study and our previously published GBM scRNA-seq data (Neftel et al., 2019). These genes were used to interrogate bulk RNA-seq data from TCGA, limiting the analysis to those gliomas with a sufficient T cell signal (129 of 148 GBM and 161 of 428 IDH-G samples). Expression of these genes is shown without (D) and with (E) normalization of the total T cell signal (average expression of CD2, CD3D, CD3E, and CD3G). Volcano plots compare expression of these genes in IDH-G (left) versus GBM (right); genes are color coded with respect to T cell cluster in scRNA-seq data (Figure 1E). (F) Heat map showing relative expression of selected genes across CD8 T cells in cytotoxicity-NK (1) cluster (Figure 1E), with emphasis on key cytotoxicity, NK cell receptor, and T cell markers. Gene expression is zero-centered and given in units of ln(TP100K+1). (G, H) Expression of inhibitory receptor genes (PDCD1, HAVCR2 and TIGIT) in CD8 (G) and CD4 (H) T cells.
Figure S3. Transcriptional programs of clonally expanded T cells, Related to Figure 3. (A-F) Analysis of gene expression in clonally expanded T cells using the SMART-seq2 dataset. (A) Number of T cells from each patient passing quality control and number of those cells with reconstructed TCR α and/or β chains. (B) Quantification of clonally expanded T cells in each patient’s tumor. TCR sequences were used to identify T cells with identical TCR sequences (clonotypes). For each patient tumor, the number of T cells belonging to each clonotype (clonotype size) is shown; the number of clonotypes composed of two T cells is also indicated. (C) Percentage of T cells from each patient that shared a TCR chain with another T cell (recurrent TCRs) (y-axis) and the number of distinct identified T cell clonotypes per patient (x-axis). GBM shown in dark blue and IDH-G shown in light green. (D) UMAP visualization of T cells with recurrent TCRs colored by subsets. (E) Heatmap showing relative expression of selected cluster-related marker genes across subsets of clonally expanded T cells defined in (D). The gliomas are separated into IDH-G and GBM. Gene expression is zero-centered and given in units of ln(TP100K+1). (F) Comparison of expression for each gene in clonal versus non-clonal T cells in IDH-G. Z-score of gene expression (y-axis) and Z-score of the coefficient of variation (CV; x-axis) of gene expression is shown separately for each gene in CD8 (left) and CD4 (right) T cells.
Figure S4. Expression of KLRB1 in glioma-infiltrating T cells, Related to Figure 4. (A-D) Analysis of KLRB1 expressing T cells in SMART-seq2 dataset. (A) Violin plots showing KLRB1 expression across major T cell subpopulations in GBM (left) and IDH-G (right). Gene expression is given in units of ln(TP100K+1). (B-C) Diversity of TCR V-segment (left) and J-segment (right) rearrangements of KLRB1 expressing T cells shown for TCRβ chains of KLRB1 expressing CD8 T cells (B) and TCRα and β chains of KLRB1 expressing CD4 T cells (C). (D) Heat map showing absolute expression of selected genes across KLRB1 expressing CD4 and CD8 T cells with emphasis on key T cell, Th17, and MAIT cell markers. Gene expression is given in units of ln(TP100K+1). (E, F) Quantification of GBM-infiltrating T cell populations for CD161 and PD-1 protein surface levels in three additional patients (E). CD161 gating is based on labeling with an isotype control mAb for each patient, shown for one patient in (F); live CD45+ CD3+ GBM T cells were stained with isotype control mAb (isotype matching CD161 mAb). (G) Specific detection of CD161 protein by flow cytometry with mAb used to study GBM-infiltrating T cells. Jurkat [E6-1] cells were transduced with the human KLRB1 cDNA using a lentiviral vector and labeling with a CD161 mAb was compared between WT control Jurkat cells (left) and KLRB1 cDNA transduced Jurkat cells (right). (H-J) Analysis of dendritic cells from murine GL261 tumors for expression of CLEC2D. (H) Gating strategy for analysis of murine dendritic cells (DCs). (I) Representative examples of CLEC2D mAb labeling (filled red) compared to mIgG1 isotype staining control (grey dashed line) on dendritic cells in GL261 tumors from mice treated with isotype control antibody (left) or PD-1 blocking antibody (right). (J) Summary of CLEC2D protein levels on dendritic cells in GL261 tumors from mice treated with isotype control antibody (left) or PD-1 blocking antibody (right). ** P < 0.01 (Mann-Whitney U test), error bars denote SEM. MFI, mean fluorescent intensity. n = 6 tumors/group. (J) was performed twice.
Figure S5. Engineering of human T cells for functional studies, Related to Figure 5. (A) Efficiency of gene editing procedure. Editing of primary human CD4+ T cells by electroporation of Cas9 protein with bound control (LacZ, left) or CD4 (right) gRNAs. Loss of CD4 protein expression (ExCD4) was quantified three days following editing. (B) CD161+ T cells negative for Vα7.2 (MAIT cell marker) were sorted from peripheral blood mononuclear cells (PBMCs) of a healthy donor. (C) CD161+ T cells were edited by electroporation of Cas9 protein with a bound control or KLRB1 gRNA and CD161 surface protein levels were quantified by flow cytometry. (D) CD161+ T cells were sorted from PBMCs of a healthy donor. T cells were either not edited (no electroporation) or edited by electroporation with Cas9 protein with bound gRNAs, using either control (Cntrl) + TRAC gRNAs, Cntrl + KLRB1 gRNAs, or KLRB1 + TRAC gRNAs. The TRAC gRNA targeted an exon encoding the constant domain of the TCR α chain to inactivate endogenous TCR α chains. Surface levels of TCR and CD161 proteins were quantified by flow cytometry three days following editing. (E) Quantification of editing efficiency for KLRB1 gene by Sanger sequencing using TIDE software, illustrating the frequency of nucleotide deletions/insertions. (F) CD161+ T cells were edited by electroporation of Cas9 protein with a bound control (intergenic) gRNA or with one of two KLRB1 gRNAs. CD161 surface protein levels were quantified by flow cytometry; both KLRB1 gRNAs yielded similar editing efficiencies. (G) Schema of endogenous TCR replacement with NY-ESO-1 (1G4) TCR by homology directed repair (HDR) template (2214 nt), used for all experiments utilizing gliomasphere models. The NY-ESO-1 β-chain and α-chain were preceded by a PC tag and HA tag, respectively, to enable identification of the individual NY-ESO-1 TCR chains by flow cytometry. Each was preceded by a self-cleaving peptide enabling translation of separate TCR chains. An ~300nt homology arm was placed on either side of the construct which allowed site directed insertion into the TRAC gRNA cut site (Figure S5D), enabling the 3’ portion of the endogenous α-constant region to be utilized, thereby decreasing the size of the required HDR template. T cells generated with this approach were used for all in vitro and in vivo experiments with patient-derived gliomasphere models (Figure 5, Figure S5M-O, Figure 6B-J, Figure S6A-C, H-J). (H) Construct design for expression of NY-ESO-1 specific (1G4) TCR by lentivirus in primary human T cells, used for U-87 model experiments (Figure 6K-N, Figure S6D-F). HA and PC epitope tags were placed at the N-terminus of the mature TCR α and β chains, respectively, to enable identification of both TCR chains on the cell surface. (I) Map of lentiviral construct with EF-1ɑ promoter regulated expression of the NY-ESO-1 protein containing the 1G4 epitope. A T2A skip sequence was used for co-expression of luciferase and an IRES was used for expression of the extracellular (EC), non-functional, domain of NGFR as a marker for transduced cells. (J) Labeling of transduced, unsorted T cells with PC and HA tag antibodies to identify 1G4 TCR α and β chains, respectively. (K) Characterization of T cells used for functional experiments shown in Fig. 5. Edited T cells that expressed the NY-ESO-1 TCR were labeled with antibodies specific for CD161 and HA tag (NY-ESO-1 TCRα chain). T cells had been edited with Cas9 plus either Cntrl + TRAC or KLRB1 + TRAC gRNAs. The TRAC gRNA targeted the endogenous TCRα chain but not the introduced NY-ESO-1 TCR. Editing of the KLRB1 gene resulted in loss of CD161 protein at the cell surface (which also demonstrated the antigen specificity of the mAb used to quantify CD161 surface levels in Fig. 4C-E). (L-N) T cell cytotoxicity assay with patient-derived gliomaspheres. After an 8 hour co-culture period, the percentage of Zombie UV+ (dead) glioma cells was quantified by gating first on single, CD3− cells (L) and then identifying the percentage of Zombie UV+ tumors cells (M, N). (M, N) Example flow cytometry plots for quantification of Zombie UV+ MGG123 (M) and MGG75 (N) glioma cells co-cultured either with control or KLRB1 edited T cells; data correspond to Figure 5C. (O) Cytokine release by T cells co-cultured with MGG75 glioma cells in the presence of CD161 blocking mAb or control IgG Ab for 72 hours at the indicated E:T ratios. Secretion of IFNγ and IL-2 was quantified by ELISA. (P) Validation of HP-3G10 as a mAb that blocks CLEC2D ligand binding by the CD161 receptor. Detection of CD161 on the surface of WT Jurkat [E6-1] or Jurkat cells transduced with KLRB1 cDNA. Transfectants were stained with a biotinylated CLEC2D-Ig fusion protein (CLEC2D, green histogram) or biotinylated CD161 mAb [HP-3G10-biotin] (blue histogram) and bound proteins were detected with streptavidin-PE (SA 2°). Preincubation of CD161+ Jurkat cells with non-biotinylated HP-3G10 Ab (10 μg/ml) fully blocked binding of the biotinylated CLEC2D fusion protein (red histogram); bound CLEC2D fusion protein was detected with SA 2°-PE. Experiments in (A-E) & (K) were performed more than three times; (F), (L-O) were performed two times, (J) and (P) were performed once. * P < 0.05, ** P < 0.01, *** P < 0.001, error bars denote SEM. Mann-Whitney U test (O).
Figure S6. Enhanced function of T cells against glioma cells following inactivation of KLRB1 gene, Related to Figures 5 and 6. (A-B) Analysis of T cell activation markers (CD25 and CD69) following co-culture with the patient-derived gliomasphere MGG75 for 72 hours. T cells that expressed the NY-ESO-1 TCR were co-cultured with glioma cells at the indicated effector to target ratio (E:T). The CD161 pathway was investigated by editing T cells with a KLRB1 or control gRNA (A); alternatively, a CD161 blocking mAb or control IgG was added to co-cultures (B). (C) Cytokine release by T cells edited with two different KLRB1 gRNAs. KLRB1 gRNA#1 or KLRB1 gRNA#2 or control edited T cells were co-cultured for 72 hours with GBM (MGG75) gliomaspheres at the indicated E:T ratios. Secretion of IFNγ and IL-2 was quantified by ELISA. (D-F) Co-culture experiments with T cells and U-87 MG tumor cells. (D) KLRB1 or control edited primary T cells that expressed a NY-ESO-1 specific TCR were co-cultured with U-87 MG cells that expressed NY-ESO-1 protein, and secreted cytokines (IFNg and IL-2) were quantified by ELISA after 72 hours for the indicated T cell to tumor cell ratios. (E, F) Quantification of surface levels of PD-1 (E) and a larger panel of inhibitory receptors (F) on CD8 T cells edited with control or KLRB1 gRNAs. PD-1 expression is shown for different T cell to tumor ratios (E); analysis of a panel of inhibitory receptors is shown for 2:1 ratio of T cells to tumors (F), gated on CD161 positive or negative cells. (G) Proof of concept surgical injection of 1.75% Brilliant blue R into the lateral ventricle of an anesthetized mouse. Mice were immediately euthanized following injection and the intact brain was removed at necropsy. Gross examination revealed the needle track lateral of the sinus artery (top). A coronal section intersecting the injection site visualized the dye in both lateral ventricles (which are connected), even though the needle was inserted to predetermined coordinates (right) of only one ventricle. No staining of the lateral ventricles was observed when the needle was withheld at the corpus callosum (control injection, left). (H) H&E stained coronal section of the mouse brain shows MGG123 GBM in the left hemisphere, accompanied by mass effect with midline shift and hemorrhage. Insets 1 (white matter) and 2 (cortex) highlight infiltration of brain parenchyma by pleomorphic GBM cells (arrowheads) (corresponds to Figure 6B). (I-J) Second in vivo experiment with GBM formed by patient-derived MGG123 gliomaspheres implanted into NSG mice. Mice received T cells that expressed a NY-ESO-1 TCR and had been edited with a KLRB1 or control gRNA. Tracking of tumor burden by BLI (I) and analysis of survival (J) for the experimental and control groups; corresponds to similar experiment in Figure 6C-E. (K) BLI tracking of tumor burden for U-87 MG model; mice received a single transfer of KLRB1 or control edited T cells that expressed a NY-ESO-1 TCR; experiment corresponds to survival curve shown in Figure 6K. Experiments in (D) were performed three times; (A-C), (E-F) & (I-K) were performed two times; (H) was performed once. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.001, error bars denote SEM. Mann-Whitney U test (A-F), (I) and Log-rank (Mantel-Cox) test (J).
Figure S7. T cell KLRB1 program across multiple cancer types, Related to Figure 7. (A-C) t-SNE visualization of gene expression by T cells from three cancer types, colored by expression of T cell marker genes (CD3D, CD4, CD8A, and FOXP3) as well as KLRB1 and PDCD1 genes. Cancers include melanoma (A), liver cancer (B), and colorectal cancer (C). (D-E) t-SNE visualizations of tumor-infiltrating T cells showing low expression of signature genes for Th17 cells (RORC, IL17A, and IL17F) in liver cancer (D) and colorectal cancer (E). Gene expression is given in units of ln(TP100K+1) for (A-E). (F-G) t-SNE visualization of T cells from liver cancer (F) and colorectal cancer (G) colored by the authors’ annotations of T cell subsets (Zheng et al., 2017, Zhang et al., 2018). MAIT cells represented 4% of T cells in liver cancer and 1% in colorectal cancer. The ‘unknown’ T cell subset in liver cancer is based on the author’s annotation. (H) Averaged expression of genes in tumor-infiltrating T cells in GBM + IDH-G (y-axis) versus melanoma (x-axis) shown for CD8 T cells (left) and CD4 T cells (right). The insets list selected genes that were expressed at a higher level in gliomas (up) compared to melanoma; expression is log2 fold change. (I) Correlation of expression between KLRB1 and genes encoding IL-18 receptor subunits (IL18R1, IL18RAP). The correlation between KLRB1 and IL18R1 was 0.12 (P < 1e-15) and the correlation between KLRB1 and IL18RAP was 0.19 (P < 1e-15). Gene-gene correlations in single cell RNA-seq data are often lower due to technical dropout effects.
Supplemental Table 1 (Table S1): Clinical cohort.
Supplemental Table 2 (Table S2) Gene expression data for SMART-seq2 dataset, Related to Figure 1, 2, & S3.
Sheet 1. Marker genes in GBM and IDH-G CD8 T cells (Fig1F_CD8_DE_genes) All differentially expressed genes in IDHwt-GBM and IDHmut-glioma CD8 T cells are listed. This corresponds to the left heatmap in Fig. 1F. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (following the cluster numbering in Fig. 1E), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 2. Marker genes in GBM and IDH-G CD4 T cells (Fig1F_CD4_DE_genes) All differentially expressed genes in GBM and IDH-glioma CD4 T cells are listed. This corresponds to the right heatmap in Fig. 1F. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (following the cluster numbering in Fig. 1E), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 3. Marker genes in GBM and IDH-G CD8 T cells with high versus low NK signature scores (Fig2D_HighNK_CD8_DE_genes) All differentially expressed genes between CD8 T cells with high NK signature scores compared to low NK signature scores are listed. The CD8 T cells are from GBM and IDH-G. This corresponds to Fig. 2D. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 4. Marker genes in clonally expanded GBM and IDH-G T cells (FigS3E_ClonalCells_DE_genes) All differentially expressed genes in clonally expanded IDHwt-GBM and IDHmut-glioma T cells are listed. This corresponds to Fig. S3E. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (following the cluster annotations in Fig. S3D), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Supplemental Table 3 (Table S3): Gene expression data for 10X Genomics dataset, Related to Figure 3, & S2.
Sheet 1. Marker genes in GBM CD8 T cells (FigS2C_CD8_DE_genes) All differentially expressed genes in GBM CD8 T cells profiled using the 10X Genomics platform are listed. This corresponds to the left heatmap in Fig. S2C. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed, the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 2. Marker genes in IDH-G CD4 T cells (FigS2C_CD4_DE_genes) All differentially expressed genes in GBM CD4 T cells profiled using the 10X Genomics platform are listed. This corresponds to the right heatmap in Fig. S2C. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed, the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 3. Marker genes in clonally expanded KLRB1+ vs KLRB1− cells in GBM CD8 T cells (Fig3F_DEgenes_CD8_KLRB1+_KLRB1−) All differentially expressed genes in GBM KLRB1+ versus KLRB1− CD8 T cells profiled using the 10X Genomics platform are listed. This corresponds to the columns labelled CD8+ in the heatmap in Fig. 3F. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed, the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 4. Marker genes in clonally expanded KLRB1+ vs KLRB1− cells in GBM CD4 T cells (Fig3F_DEgenes_CD4_KLRB1+_KLRB1−) All differentially expressed genes in GBM KLRB1+ versus KLRB1− CD4 T cells profiled using the 10X Genomics platform are listed. This corresponds to the columns labelled CD4+ in the heatmap in Fig. 3F. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed, the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Supplemental Table 4 (Table S4): Marker genes in cellular signatures from the SMART-seq2 dataset, Related to Figure 2.
Sheet 1. Marker genes between Cytotoxicity-NK cells CD8 T between GBM and IDH-G (Fig2A_CD8_Cyto-NK_topDEgenes) All differentially expressed genes between Cytotoxicity-NK CD8 T cells between GBM and IDH-G. This corresponds to the left panel of Fig. 2A. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 2. Marker genes between Interferon CD8 T cells between GBM and IDH-G (Fig2A_CD8_Interferon_topDEgenes) All differentially expressed genes between interferon CD8 T cells between GBM and IDH-G. This corresponds to the center panel of Fig. 2A. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 3. Marker genes between Stress CD8 T cells between GBM and IDH-G (Fig2A_CD8_Stress_topDEgenes) All differentially expressed genes between stress CD8 T cells between GBM and IDH-G. This corresponds to the right panel of Fig. 2A. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 4. Marker genes between Cytotoxicity-Chemokine CD4 T cells between GBM and IDH-G (Fig2B_CD4_Cyto_Chemo_topDEgenes) All differentially expressed genes between cytotoxicity-chemokine CD4 T cells between GBM and IDH-G. This corresponds to the left panel of Fig. 2B. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 5. Marker genes between Interferon CD4 T cells between GBM and IDH-G (Fig2B_CD4_Interferon_topDEgenes) All differentially expressed genes between interferon CD4 T cells between GBM and IDH-G. This corresponds to the center panel of Fig. 2B. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 6. Marker genes between Stress CD4 T cells between GBM and IDH-G (Fig2B_CD4_Stress_topDEgenes) All differentially expressed genes between stress CD4 T cells between IDHwt-GBM and IDHmut gliomas. This corresponds to the right panel of Fig. 2B. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Supplemental Table 5 (Table S5). Pan-cancer KLRB1 program, Related to Figure 7.
Sheet 1. Pan-cancer KLRB1 program for GBM and IDH-G CD8 T cells (Fig7E_CD8_KLRB1program)
Each row lists a gene identified in a KLRB1 transcriptional program in CD8 T cells from at least one of the analyzed cancer datasets (glioma, lung, melanoma, colon, liver). There are two melanoma datasets: Melanoma1 and Melanoma2 (Jerby-Arnon et al., 2018, Sade-Feldman et al., 2018). The columns indicate for each cancer dataset whether the gene is detected (1) or not detected (0) in the KLRB1 program for that cancer. The genes are sorted based on the number of cancer datasets that support their association with KLRB1 (N). The p-value is calculated using Fisher's combined probability test with Benjamini-Hochberg correction. From these genes, we derive the pan-cancer KLRB1 program for CD8 T cells shown in the left heatmap of Fig. 7E (STAR Methods).
Sheet 2. Pan-cancer KLRB1 program for GBM and IDH-G CD4 T cells (Fig7E_CD4_KLRB1program)
Each row lists a gene identified in a KLRB1 transcriptional program in CD4 T cells from at least one of the analyzed cancer datasets (glioma, lung, melanoma, colon, liver). There are two melanoma datasets: Melanoma1 and Melanoma2 (Jerby-Arnon et al., 2018, Sade-Feldman et al., 2018). The columns indicate for each datasets dataset whether the gene is detected (1) or not detected (0) in the KLRB1 program for that cancer. The genes are sorted based on the number of cancer datasets that support their association with KLRB1 (N). The p-value is calculated using Fisher's combined probability test with Benjamini-Hochberg correction. From these genes, we derive the pan-cancer KLRB1 program for CD4 T cells shown in the right heatmap of Fig. 7E (STAR Methods).
Supplemental Table 6 (Table S6). Correlation of KLRB1 expression with Trm signature, Related to Figure 7.
Sheet 1. Correlation between KLRB1 expression and Trm signature in each subset of CD8 and CD4 T cells (KLRB1_Trm_Correlation) Correlation between expression of KLRB1 and the Trm gene signature for each subset of CD8 and CD4 T cells from both data sets (SMART-seq2 and 10x Genomics). For each subset, we list the Pearson correlation coefficient and p value.
Highlights.
Single cell analysis charts clonal landscape of tumor-infiltrating T cells in glioma
T cells with a cytotoxicity program express multiple NK cell receptors
The NK cell receptor CD161 inhibits killing of glioma cells by T cells
Tumor cells and immunosuppressive myeloid cells express the CLEC2D ligand
ACKNOWLEDGEMENTS:
We thank Ania Hupalowska for help with graphics and Dr. John Sampson (Duke University) for generously providing cells from the D270 PDX model. This work was supported by a grant from the Ben and Catherine Ivy Foundation (to D.A.R., M.L.S., and K.W.W.) and the Bridge Project, a partnership between the Koch Institute for Integrative Cancer Research at MIT and the Dana-Farber/Harvard Cancer Center (to K.W.W, M.L.S. and A.R). The work was supported by grants from the NIH (R01 CA238039 to K.W.W., P01 CA236749 to K.W.W., M.L.S., D.A.R. and E.A.C., R37CA245523 to M.L.S.), a gift from the Ambrose Monell Foundation (to K.W.W.), the MGH Research Scholars (to M.L.S.), the Sontag Foundation (to M.L.S.), R01 CA234018 to K.W.W. and X.S.L., the Zuckerman STEM Leadership Program (to I.T.), the Human Frontier Science Program (to I.T.), the Mexican Friends New Generation (to I.T.) and the Benoziyo Endowment Fund for the Advancement of Science (to I.T.). K.W.W. is a member of the Parker Institute for Cancer Immunotherapy (PICI). N.D.M. was supported by a postdoctoral fellowship from the American Cancer Society (PF-17-042-01-LIB) and the NIH education loan repayment program funded by the NCI (L30 CA231679-01). S.M. was supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (DFG, MA-8489/1-1). A.M. is an investigator at the Chan Zuckerberg Biohub, holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and the Cancer Research Institute (CRI) Lloyd J. Old STAR award, has received funding from the Innovative Genomics Institute (IGI) and is a member of the Parker Institute for Cancer Immunotherapy (PICI). A.R. was supported by funds from the Howard Hughes Medical Institute, the Klarman Cell Observatory, NCI grant 1U24CA180922, NCI grant R33CA202820, by the Koch Institute Support (core) grant P30-CA14051 from the National Cancer Institute, and the Ludwig Center at MIT. E.M.P is supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE1745303. S.G. was supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (DFG, GR 5252/1-1). L.J.A. is a Chan Zuckerberg Biohub investigator and holds a Career Award at the Scientific Interface from Burroughs Wellcome Fund (BWF). L.J.A. was a fellow of the Eric and Wendy Schmidt postdoctoral program and a CRI Irvington Fellow supported by the CRI. L.N.G.C was supported by a K12 award (CA090354). D.P.C. was supported by NIH R01CA227821, P50CA165962, and the Tawingo Fund and Loglio Foundation. T.H. was supported by Grant-in-Aid for JSPS Fellows from the Japan Society for the Promotion of Science, SENSHIN Medical Research Foundation, and Kanae foundation for the promotion of medical science.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: N.D.M., O.A., I.T., A.R., M.L.S and K.W.W. are co-inventors of a patent application on the CLEC2D–CD161 pathway for the treatment of cancer. K.W.W., M.L.S. and A.R. are co-founders of Immunitas Therapeutics. K.W.W., M.L.S. and I.T. are advisory board members of Immunitas Therapeutics. K.W.W. serves on the scientific advisory board of TCR2 Therapeutics, T-Scan Therapeutics, SQZ Biotech and Nextechinvest and received sponsored research funding from Bristol-Myers Squibb and Novartis. A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and until August 31, 2020 was a SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and Thermo Fisher Scientific. From August 1, 2020, A.R. is an employee of Genentech. N.D.M. serves as a scientific advisor to Immunitas Therapeutics. A.M. is cofounder, member of the Boards of Directors and member of Scientific Advisory Boards of Spotlight Therapeutics and Arsenal Biosciences. A.M. has served as an advisor to Juno Therapeutics, was a member of the scientific advisory board at PACT Pharma and an advisor to Trizell. A.M. has received an honorarium from Merck, a consulting fee from AlphaSights, and is an investor in and informal advisor to Offline Ventures. A.M. owns stock in Arsenal Biosciences, Spotlight Therapeutics and PACT Pharma. The Marson lab has received research support from Juno Therapeutics, Epinomics, Sanofi, GlaxoSmithKline, Gilead and Anthem. E.A.C. is currently an advisor to Advantagene Inc., Alcyone Biosciences, Insightec, Inc., Sigilon Therepeutics and DNAtrix Inc. and has equity interest in DNAtrix; A.M. has advised Oncorus, Merck, Tocagen, Ziopharm, Stemgen, NanoTx., Ziopharm Oncology, Cerebral Therapeutics, Genenta, Merck, Janssen, Karcinolysis, and Shanaghai Biotech and has received research support from Advantagene, NewLink Genetics and Amgen. A.M. is a named inventor on patents related to oncolytic HSV1. D.A.R has received research support from Acerta Phamaceuticals; Agenus; Celldex; EMD Serono; Incyte; Inovio; Midatech; Omniox; Tragara and he has served as paid consultant for: Abbvie; Advantagene; Agenus; Amgen; Bayer; Bristol-Myers Squibb; Celldex; DelMar; EMD Serono; Genentech/Roche; Inovio; Merck; Merck KGaA; Monteris; Novocure; Oncorus; Oxigene; Regeneron; Stemline; Taiho Oncology, Inc. P.K.B, outside the scope of this work, has consulted for Angiochem, Genentech-Roche, Lilly, Tesaro, ElevateBio, Pfizer (Array), SK Life Sciences and Dantari, and is supported by the Breast Cancer Research Foundation, Damon Runyon Cancer Research Foundation, Ben and Catherine Ivy Foundation, and the National Cancer Institute (5R01CA244975-02, 5R21CA220253-02, 5R01CA227156-03), BMS, Lilly, and honoraria from Merck, Genentech-Roche and Lilly. D.P.C. has consulted for Lilly and Boston Pharmaceuticals, and has received honoraria and travel reimbursement from Merck. O.R.R is an employee of Genentech since October 2020. O.R.R. is a co-inventor on patent applications filed by the Broad Institute for inventions relating to work in single cell genomics, such as in PCT/US2018/060860 and US provisional application no. 62/745,259. E.A.C. is currently an advisor to Advantagene Inc., Alcyone Biosciences, Insightec, Inc., DNAtrix Inc, Immunomic Therapeutics, Sangamo Therapeutics, Seneca Therapeutics and has equity interest in DNAtrix, Immunomic Therapeutics, Seneca Therapeutics; and has also advised Oncorus, Merck, Tocagen, Ziopharm, Stemgen, NanoTx., Ziopharm Oncology, Cerebral Therapeutics, Genenta. Merck, Janssen, Karcinolysis, Shanghai Biotech. E.A.C has received research support from NIH, US Department of Defense, American Brain Tumor Association, National Brain Tumor Society, Alliance for Cancer Gene Therapy, Neurosurgical Research Education Foundation, Advantagene, NewLink Genetics and Amgen and also is a named inventor on patents related to oncolytic HSV1 and noncoding RNAs. X.S.L. is a cofounder, board member, and consultant of GV20 Oncotherapy and its subsidiaries, SAB of 3DMedCare, consultant for Genentech, and stockholder of BMY, TMO, WBA, ABT, ABBV, and JNJ, and receives research funding from Takeda and Sanofi.
REFERENCES
- Aldemir H, Prod'homme V, Dumaurier MJ, Retiere C, Poupon G, Cazareth J, Bihl F, and Braud VM (2005). Cutting edge: lectin-like transcript 1 is a ligand for the CD161 receptor. J Immunol 175, 7791–7795. [DOI] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment. [Google Scholar]
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. (2016). Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 375, 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, Alansary D, Sonner JK, Green E, Deumelandt K, et al. (2018). Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med 24, 1192–1203. [DOI] [PubMed] [Google Scholar]
- Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, and Smyth MJ (2014). The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol 15, 431–438. [DOI] [PubMed] [Google Scholar]
- Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, Woroniecka K, Elsamadicy AA, Dechant CA, Kemeny HR, et al. (2018). Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 24, 1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergusson JR, Huhn MH, Swadling L, Walker LJ, Kurioka A, Llibre A, Bertoletti A, Hollander G, Newell EW, Davis MM, et al. (2016). CD161(int)CD8+ T cells: a novel population of highly functional, memory CD8+ T cells enriched within the gut. Mucosal Immunol 9, 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergusson JR, Smith KE, Fleming VM, Rajoriya N, Newell EW, Simmons R, Marchi E, Bjorkander S, Kang YH, Swadling L, et al. (2014). CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell reports 9, 1075–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filley AC, Henriquez M, and Dey M (2017). Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget 8, 91779–91794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebel E, Kapolou K, Unger S, Nunez NG, Utz S, Rushing EJ, Regli L, Weller M, Greter M, Tugues S, et al. (2020). Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 181, 1626–1642 e1620. [DOI] [PubMed] [Google Scholar]
- Gao J, Zheng Q, Xin N, Wang W, and Zhao C (2017). CD155, an onco-immunologic molecule in human tumors. Cancer Sci 108, 1934–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R, and Seoighe C (2010). A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedeon PC, Schaller TH, Chitneni SK, Choi BD, Kuan CT, Suryadevara CM, Snyder DJ, Schmittling RJ, Szafranski SE, Cui X, et al. (2018). A Rationally Designed Fully Human EGFRvIII:CD3-Targeted Bispecific Antibody Redirects Human T Cells to Treat Patient-derived Intracerebral Malignant Glioma. Clin Cancer Res 24, 3611–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain C, Guillaudeux T, Galsgaard ED, Hervouet C, Tekaya N, Gallouet AS, Fassy J, Bihl F, Poupon G, Lazzari A, et al. (2015). Lectin-like transcript 1 is a marker of germinal center-derived B-cell non-Hodgkin's lymphomas dampening natural killer cell functions. Oncoimmunology 4, e1026503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain C, Meier A, Jensen T, Knapnougel P, Poupon G, Lazzari A, Neisig A, Hakansson K, Dong T, Wagtmann N, et al. (2011). Induction of lectin-like transcript 1 (LLT1) protein cell surface expression by pathogens and interferon-gamma contributes to modulate immune responses. The Journal of biological chemistry 286, 37964–37975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SA, Ye X, Lesser G, Sloan A, Carraway H, Desideri S, Piantadosi S, and Consortium NC (2011). Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin Cancer Res 17, 5473–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, et al. (2018). Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 24, 978–985. [DOI] [PubMed] [Google Scholar]
- Gustafson MP, Lin Y, New KC, Bulur PA, O'Neill BP, Gastineau DA, and Dietz AB (2010). Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro-oncology 12, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi F, Fink AL, Kiyokawa J, Miller JJ, Koerner MVA, Cahill DP, and Wakimoto H (2018). PLK1 Inhibition Targets Myc-Activated Malignant Glioma Cells Irrespective of Mismatch Repair Deficiency-Mediated Acquired Resistance to Temozolomide. Molecular cancer therapeutics 17, 2551–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller AN, Corbett AJ, Wubben JM, McCluskey J, and Rossjohn J (2017). MAIT cells and MR1-antigen recognition. Curr Opin Immunol 46, 66–74. [DOI] [PubMed] [Google Scholar]
- Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. (2019). Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, Brouland JP, Iacobuzio-Donahue CA, Brennan C, Tabar V, et al. (2020). Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 181, 1643–1660 e1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, et al. (2017). Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell reports 20, 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Gagnon JA, Thyme SB, and Valen E (2016). CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic acids research 44, W272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwe H, Pircher A, Van den Eynde K, et al. (2018). Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 24, 1277–1289. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bourhis L, Dusseaux M, Bohineust A, Bessoles S, Martin E, Premel V, Core M, Sleurs D, Serriari NE, Treiner E, et al. (2013). MAIT cells detect and efficiently lyse bacterially-infected epithelial cells. PLoS Pathog 9, e1003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llibre A, Lopez-Macias C, Marafioti T, Mehta H, Partridge A, Kanzig C, Rivellese F, Galson JD, Walker LJ, Milne P, et al. (2016). LLT1 and CD161 Expression in Human Germinal Centers Promotes B Cell Activation and CXCR4 Downregulation. J Immunol 196, 2085–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoma AM, Suo S, Williams HL, Sharova T, Sullivan K, Manos M, Bowling P, Hodi FS, Rahma O, Sullivan RJ, et al. (2020). Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 182, 655–671 e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLane LM, Abdel-Hakeem MS, and Wherry EJ (2019). CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 37, 457–495. [DOI] [PubMed] [Google Scholar]
- Meresse B, Curran SA, Ciszewski C, Orbelyan G, Setty M, Bhagat G, Lee L, Tretiakova M, Semrad C, Kistner E, et al. (2006). Reprogramming of CTLs into natural killer-like cells in celiac disease. J Exp Med 203, 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G, Sayour EJ, McLendon R, Herndon JE 2nd, Healy P, et al. (2014). EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One 9, e94281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori L, Lepore M, and De Libero G (2016). The Immunology of CD1- and MR1-Restricted T Cells. Annu Rev Immunol 34, 479–510. [DOI] [PubMed] [Google Scholar]
- Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. (2019). An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849 e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigim F, Cavanaugh J, Patel AP, Curry WT Jr., Esaki S, Kasper EM, Chi AS, Louis DN, Martuza RL, Rabkin SD, et al. (2015). Targeting Hypoxia-Inducible Factor 1alpha in a New Orthotopic Model of Glioblastoma Recapitulating the Hypoxic Tumor Microenvironment. J Neuropathol Exp Neurol 74, 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J, et al. (2015). NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 21, 914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Bahr O, et al. (2020). Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol 6, 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon DAe.a. (2019). Randomized phase 3 study evaluating the efficacy and safety of nivolumab versus bevacizumab in patients with recurrent glioblastoma: Checkmate 143. 5th Quadrennial Meeting of the World Federation of Neuro-Oncology Societies (WFNOS) Neuro-Oncol 19 (suppl 3), abstract OS103. [Google Scholar]
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, et al. (2011). Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29, 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DB, Bettadapura J, Alsharifi M, Mathew PA, Warren HS, and Lanier LL (2005). Cutting edge: lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J Immunol 175, 7796–7799. [DOI] [PubMed] [Google Scholar]
- Rosen DB, Cao W, Avery DT, Tangye SG, Liu YJ, Houchins JP, and Lanier LL (2008). Functional consequences of interactions between human NKR-P1A and its ligand LLT1 expressed on activated dendritic cells and B cells. J Immunol 180, 6508–6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth P, Mittelbronn M, Wick W, Meyermann R, Tatagiba M, and Weller M (2007). Malignant glioma cells counteract antitumor immune responses through expression of lectin-like transcript-1. Cancer Res 67, 3540–3544. [DOI] [PubMed] [Google Scholar]
- Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, et al. (2018). Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2019). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 176, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer L, Rothhammer V, Hemmer B, and Korn T (2013). Enriched CD161high CCR6+ gammadelta T cells in the cerebrospinal fluid of patients with multiple sclerosis. JAMA Neurol 70, 345–351. [DOI] [PubMed] [Google Scholar]
- Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, et al. (2019). Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730 e722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolders J, Heutinck KM, Fransen NL, Remmerswaal EBM, Hombrink P, Ten Berge IJM, van Lier RAW, Huitinga I, and Hamann J (2018). Tissue-resident memory T cells populate the human brain. Nature communications 9, 4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbington MJT, Lonnberg T, Proserpio V, Clare S, Speak AO, Dougan G, and Teichmann SA (2016). T cell fate and clonality inference from single-cell transcriptomes. Nat Methods 13, 329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016a). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. (2016b). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, et al. (2017). Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, Elsamadicy AA, Cui X, Koyama S, Jackson C, et al. (2018). T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res 24, 4175–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Sorensen MD, Kristensen BW, Reifenberger G, McIntyre TM, and Lin F (2018a). D-2-Hydroxyglutarate Is an Intercellular Mediator in IDH-Mutant Gliomas Inhibiting Complement and T Cells. Clin Cancer Res 24, 5381–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, Gao R, Kang B, Zhang Q, Huang JY, et al. (2018b). Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Rahim MM, Allan DS, Tu MM, Belanger S, Abou-Samra E, Ma J, Sekhon HS, Fairhead T, Zein HS, et al. (2012). Mouse Nkrp1-Clr gene cluster sequence and expression analyses reveal conservation of tissue-specific MHC-independent immunosurveillance. PLoS One 7, e50561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, and Morgan RA (2005). Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol 174, 4415–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, et al. (2017). Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356 e1316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of glioma-infiltrating T cells, Related to Figure 1. (A) Sorting of T cells from surgical glioma specimens. Single cell suspensions were generated for isolation of T cells by FACS. Individual live T cells were sorted into 96 well plates for scRNA-seq using the SMART-seq2 chemistry. T cells were sorted at two different facilities: T cells from GBM that were processed at DFCI were sorted as CD3+CD4+ and CD3+CD8+ cells; T cells from IDH-G that were processed at MGH were sorted as CD3+ cells. (B) Impact of tissue dissociation procedure on CD8 surface protein levels. Peripheral blood T cells were treated with PBS (left) or tumor dissociation buffer (right) and then analyzed for expression of CD4 and CD8 proteins following gating on live CD45+CD3+ cells. Incubation with the tumor dissociation buffer reduced the staining intensity with the CD8 mAb. (C) Quality-control analysis of scRNA-seq (SMART-seq2) data based on the number of detected genes (top) and housekeeping genes (bottom; dotted line indicates interrogated number of genes) for which transcripts were detected per cell. T cells from patients E88, E119, and MGH142 were excluded from further analysis. (D) UMAP plot with cell annotation based on prior dexamethasone (left) or immune checkpoint inhibitor (ICI) treatment (right). (E) Fraction of T cells assigned to the indicated T cell clusters based on glioma subtype (GBM or IDH-G) and prior treatment (dexamethasone, left; immune checkpoint inhibitor, right). (F) Fraction of T cells assigned to CD8 T cell, CD4 T cell, Treg, and cycling T cell clusters based on tumor type (IDH-G, GBM) and disease state (new onset versus recurrent). (G) Quantification of each tumor for CD8 T cell, CD4 T cell, regulatory T cell (Treg), and cycling T cell clusters; higher frequency of CD8 T cells in IDH-G versus GBM may be related to differences in details of cell isolation procedures as described in (A). (H) RNA-ISH for mRNA expression of three different cellular stress genes (HSPA1A, HSPH1, HSPA6) by CD3E+ T cells; labeling is shown for IDH-G (top) and GBM (bottom).
Figure S2. Transcriptional signatures of glioma-infiltrating T cells, Related to Figure 2 & 3. (A-C) scRNA-seq analysis of T cells from five GBM samples by 5’ sequencing using the 10X Genomics platform. (A) UMAP visualization of T cells color-coded based on tumor specimen. (B) UMAP visualization of major T cell subpopulations. (C) Heatmap showing top differentially expressed genes for clusters of CD8 (left) and CD4 (right) T cells. Similar cluster designation is shown as in Figure 1F for T cells from the SMART-seq2 dataset. (D-E) Analysis of TCGA datasets from GBM and IDH-G for expression of T cell specific genes. T cell-specific genes were identified using T cell scRNA-seq data from this study and our previously published GBM scRNA-seq data (Neftel et al., 2019). These genes were used to interrogate bulk RNA-seq data from TCGA, limiting the analysis to those gliomas with a sufficient T cell signal (129 of 148 GBM and 161 of 428 IDH-G samples). Expression of these genes is shown without (D) and with (E) normalization of the total T cell signal (average expression of CD2, CD3D, CD3E, and CD3G). Volcano plots compare expression of these genes in IDH-G (left) versus GBM (right); genes are color coded with respect to T cell cluster in scRNA-seq data (Figure 1E). (F) Heat map showing relative expression of selected genes across CD8 T cells in cytotoxicity-NK (1) cluster (Figure 1E), with emphasis on key cytotoxicity, NK cell receptor, and T cell markers. Gene expression is zero-centered and given in units of ln(TP100K+1). (G, H) Expression of inhibitory receptor genes (PDCD1, HAVCR2 and TIGIT) in CD8 (G) and CD4 (H) T cells.
Figure S3. Transcriptional programs of clonally expanded T cells, Related to Figure 3. (A-F) Analysis of gene expression in clonally expanded T cells using the SMART-seq2 dataset. (A) Number of T cells from each patient passing quality control and number of those cells with reconstructed TCR α and/or β chains. (B) Quantification of clonally expanded T cells in each patient’s tumor. TCR sequences were used to identify T cells with identical TCR sequences (clonotypes). For each patient tumor, the number of T cells belonging to each clonotype (clonotype size) is shown; the number of clonotypes composed of two T cells is also indicated. (C) Percentage of T cells from each patient that shared a TCR chain with another T cell (recurrent TCRs) (y-axis) and the number of distinct identified T cell clonotypes per patient (x-axis). GBM shown in dark blue and IDH-G shown in light green. (D) UMAP visualization of T cells with recurrent TCRs colored by subsets. (E) Heatmap showing relative expression of selected cluster-related marker genes across subsets of clonally expanded T cells defined in (D). The gliomas are separated into IDH-G and GBM. Gene expression is zero-centered and given in units of ln(TP100K+1). (F) Comparison of expression for each gene in clonal versus non-clonal T cells in IDH-G. Z-score of gene expression (y-axis) and Z-score of the coefficient of variation (CV; x-axis) of gene expression is shown separately for each gene in CD8 (left) and CD4 (right) T cells.
Figure S4. Expression of KLRB1 in glioma-infiltrating T cells, Related to Figure 4. (A-D) Analysis of KLRB1 expressing T cells in SMART-seq2 dataset. (A) Violin plots showing KLRB1 expression across major T cell subpopulations in GBM (left) and IDH-G (right). Gene expression is given in units of ln(TP100K+1). (B-C) Diversity of TCR V-segment (left) and J-segment (right) rearrangements of KLRB1 expressing T cells shown for TCRβ chains of KLRB1 expressing CD8 T cells (B) and TCRα and β chains of KLRB1 expressing CD4 T cells (C). (D) Heat map showing absolute expression of selected genes across KLRB1 expressing CD4 and CD8 T cells with emphasis on key T cell, Th17, and MAIT cell markers. Gene expression is given in units of ln(TP100K+1). (E, F) Quantification of GBM-infiltrating T cell populations for CD161 and PD-1 protein surface levels in three additional patients (E). CD161 gating is based on labeling with an isotype control mAb for each patient, shown for one patient in (F); live CD45+ CD3+ GBM T cells were stained with isotype control mAb (isotype matching CD161 mAb). (G) Specific detection of CD161 protein by flow cytometry with mAb used to study GBM-infiltrating T cells. Jurkat [E6-1] cells were transduced with the human KLRB1 cDNA using a lentiviral vector and labeling with a CD161 mAb was compared between WT control Jurkat cells (left) and KLRB1 cDNA transduced Jurkat cells (right). (H-J) Analysis of dendritic cells from murine GL261 tumors for expression of CLEC2D. (H) Gating strategy for analysis of murine dendritic cells (DCs). (I) Representative examples of CLEC2D mAb labeling (filled red) compared to mIgG1 isotype staining control (grey dashed line) on dendritic cells in GL261 tumors from mice treated with isotype control antibody (left) or PD-1 blocking antibody (right). (J) Summary of CLEC2D protein levels on dendritic cells in GL261 tumors from mice treated with isotype control antibody (left) or PD-1 blocking antibody (right). ** P < 0.01 (Mann-Whitney U test), error bars denote SEM. MFI, mean fluorescent intensity. n = 6 tumors/group. (J) was performed twice.
Figure S5. Engineering of human T cells for functional studies, Related to Figure 5. (A) Efficiency of gene editing procedure. Editing of primary human CD4+ T cells by electroporation of Cas9 protein with bound control (LacZ, left) or CD4 (right) gRNAs. Loss of CD4 protein expression (ExCD4) was quantified three days following editing. (B) CD161+ T cells negative for Vα7.2 (MAIT cell marker) were sorted from peripheral blood mononuclear cells (PBMCs) of a healthy donor. (C) CD161+ T cells were edited by electroporation of Cas9 protein with a bound control or KLRB1 gRNA and CD161 surface protein levels were quantified by flow cytometry. (D) CD161+ T cells were sorted from PBMCs of a healthy donor. T cells were either not edited (no electroporation) or edited by electroporation with Cas9 protein with bound gRNAs, using either control (Cntrl) + TRAC gRNAs, Cntrl + KLRB1 gRNAs, or KLRB1 + TRAC gRNAs. The TRAC gRNA targeted an exon encoding the constant domain of the TCR α chain to inactivate endogenous TCR α chains. Surface levels of TCR and CD161 proteins were quantified by flow cytometry three days following editing. (E) Quantification of editing efficiency for KLRB1 gene by Sanger sequencing using TIDE software, illustrating the frequency of nucleotide deletions/insertions. (F) CD161+ T cells were edited by electroporation of Cas9 protein with a bound control (intergenic) gRNA or with one of two KLRB1 gRNAs. CD161 surface protein levels were quantified by flow cytometry; both KLRB1 gRNAs yielded similar editing efficiencies. (G) Schema of endogenous TCR replacement with NY-ESO-1 (1G4) TCR by homology directed repair (HDR) template (2214 nt), used for all experiments utilizing gliomasphere models. The NY-ESO-1 β-chain and α-chain were preceded by a PC tag and HA tag, respectively, to enable identification of the individual NY-ESO-1 TCR chains by flow cytometry. Each was preceded by a self-cleaving peptide enabling translation of separate TCR chains. An ~300nt homology arm was placed on either side of the construct which allowed site directed insertion into the TRAC gRNA cut site (Figure S5D), enabling the 3’ portion of the endogenous α-constant region to be utilized, thereby decreasing the size of the required HDR template. T cells generated with this approach were used for all in vitro and in vivo experiments with patient-derived gliomasphere models (Figure 5, Figure S5M-O, Figure 6B-J, Figure S6A-C, H-J). (H) Construct design for expression of NY-ESO-1 specific (1G4) TCR by lentivirus in primary human T cells, used for U-87 model experiments (Figure 6K-N, Figure S6D-F). HA and PC epitope tags were placed at the N-terminus of the mature TCR α and β chains, respectively, to enable identification of both TCR chains on the cell surface. (I) Map of lentiviral construct with EF-1ɑ promoter regulated expression of the NY-ESO-1 protein containing the 1G4 epitope. A T2A skip sequence was used for co-expression of luciferase and an IRES was used for expression of the extracellular (EC), non-functional, domain of NGFR as a marker for transduced cells. (J) Labeling of transduced, unsorted T cells with PC and HA tag antibodies to identify 1G4 TCR α and β chains, respectively. (K) Characterization of T cells used for functional experiments shown in Fig. 5. Edited T cells that expressed the NY-ESO-1 TCR were labeled with antibodies specific for CD161 and HA tag (NY-ESO-1 TCRα chain). T cells had been edited with Cas9 plus either Cntrl + TRAC or KLRB1 + TRAC gRNAs. The TRAC gRNA targeted the endogenous TCRα chain but not the introduced NY-ESO-1 TCR. Editing of the KLRB1 gene resulted in loss of CD161 protein at the cell surface (which also demonstrated the antigen specificity of the mAb used to quantify CD161 surface levels in Fig. 4C-E). (L-N) T cell cytotoxicity assay with patient-derived gliomaspheres. After an 8 hour co-culture period, the percentage of Zombie UV+ (dead) glioma cells was quantified by gating first on single, CD3− cells (L) and then identifying the percentage of Zombie UV+ tumors cells (M, N). (M, N) Example flow cytometry plots for quantification of Zombie UV+ MGG123 (M) and MGG75 (N) glioma cells co-cultured either with control or KLRB1 edited T cells; data correspond to Figure 5C. (O) Cytokine release by T cells co-cultured with MGG75 glioma cells in the presence of CD161 blocking mAb or control IgG Ab for 72 hours at the indicated E:T ratios. Secretion of IFNγ and IL-2 was quantified by ELISA. (P) Validation of HP-3G10 as a mAb that blocks CLEC2D ligand binding by the CD161 receptor. Detection of CD161 on the surface of WT Jurkat [E6-1] or Jurkat cells transduced with KLRB1 cDNA. Transfectants were stained with a biotinylated CLEC2D-Ig fusion protein (CLEC2D, green histogram) or biotinylated CD161 mAb [HP-3G10-biotin] (blue histogram) and bound proteins were detected with streptavidin-PE (SA 2°). Preincubation of CD161+ Jurkat cells with non-biotinylated HP-3G10 Ab (10 μg/ml) fully blocked binding of the biotinylated CLEC2D fusion protein (red histogram); bound CLEC2D fusion protein was detected with SA 2°-PE. Experiments in (A-E) & (K) were performed more than three times; (F), (L-O) were performed two times, (J) and (P) were performed once. * P < 0.05, ** P < 0.01, *** P < 0.001, error bars denote SEM. Mann-Whitney U test (O).
Figure S6. Enhanced function of T cells against glioma cells following inactivation of KLRB1 gene, Related to Figures 5 and 6. (A-B) Analysis of T cell activation markers (CD25 and CD69) following co-culture with the patient-derived gliomasphere MGG75 for 72 hours. T cells that expressed the NY-ESO-1 TCR were co-cultured with glioma cells at the indicated effector to target ratio (E:T). The CD161 pathway was investigated by editing T cells with a KLRB1 or control gRNA (A); alternatively, a CD161 blocking mAb or control IgG was added to co-cultures (B). (C) Cytokine release by T cells edited with two different KLRB1 gRNAs. KLRB1 gRNA#1 or KLRB1 gRNA#2 or control edited T cells were co-cultured for 72 hours with GBM (MGG75) gliomaspheres at the indicated E:T ratios. Secretion of IFNγ and IL-2 was quantified by ELISA. (D-F) Co-culture experiments with T cells and U-87 MG tumor cells. (D) KLRB1 or control edited primary T cells that expressed a NY-ESO-1 specific TCR were co-cultured with U-87 MG cells that expressed NY-ESO-1 protein, and secreted cytokines (IFNg and IL-2) were quantified by ELISA after 72 hours for the indicated T cell to tumor cell ratios. (E, F) Quantification of surface levels of PD-1 (E) and a larger panel of inhibitory receptors (F) on CD8 T cells edited with control or KLRB1 gRNAs. PD-1 expression is shown for different T cell to tumor ratios (E); analysis of a panel of inhibitory receptors is shown for 2:1 ratio of T cells to tumors (F), gated on CD161 positive or negative cells. (G) Proof of concept surgical injection of 1.75% Brilliant blue R into the lateral ventricle of an anesthetized mouse. Mice were immediately euthanized following injection and the intact brain was removed at necropsy. Gross examination revealed the needle track lateral of the sinus artery (top). A coronal section intersecting the injection site visualized the dye in both lateral ventricles (which are connected), even though the needle was inserted to predetermined coordinates (right) of only one ventricle. No staining of the lateral ventricles was observed when the needle was withheld at the corpus callosum (control injection, left). (H) H&E stained coronal section of the mouse brain shows MGG123 GBM in the left hemisphere, accompanied by mass effect with midline shift and hemorrhage. Insets 1 (white matter) and 2 (cortex) highlight infiltration of brain parenchyma by pleomorphic GBM cells (arrowheads) (corresponds to Figure 6B). (I-J) Second in vivo experiment with GBM formed by patient-derived MGG123 gliomaspheres implanted into NSG mice. Mice received T cells that expressed a NY-ESO-1 TCR and had been edited with a KLRB1 or control gRNA. Tracking of tumor burden by BLI (I) and analysis of survival (J) for the experimental and control groups; corresponds to similar experiment in Figure 6C-E. (K) BLI tracking of tumor burden for U-87 MG model; mice received a single transfer of KLRB1 or control edited T cells that expressed a NY-ESO-1 TCR; experiment corresponds to survival curve shown in Figure 6K. Experiments in (D) were performed three times; (A-C), (E-F) & (I-K) were performed two times; (H) was performed once. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.001, error bars denote SEM. Mann-Whitney U test (A-F), (I) and Log-rank (Mantel-Cox) test (J).
Figure S7. T cell KLRB1 program across multiple cancer types, Related to Figure 7. (A-C) t-SNE visualization of gene expression by T cells from three cancer types, colored by expression of T cell marker genes (CD3D, CD4, CD8A, and FOXP3) as well as KLRB1 and PDCD1 genes. Cancers include melanoma (A), liver cancer (B), and colorectal cancer (C). (D-E) t-SNE visualizations of tumor-infiltrating T cells showing low expression of signature genes for Th17 cells (RORC, IL17A, and IL17F) in liver cancer (D) and colorectal cancer (E). Gene expression is given in units of ln(TP100K+1) for (A-E). (F-G) t-SNE visualization of T cells from liver cancer (F) and colorectal cancer (G) colored by the authors’ annotations of T cell subsets (Zheng et al., 2017, Zhang et al., 2018). MAIT cells represented 4% of T cells in liver cancer and 1% in colorectal cancer. The ‘unknown’ T cell subset in liver cancer is based on the author’s annotation. (H) Averaged expression of genes in tumor-infiltrating T cells in GBM + IDH-G (y-axis) versus melanoma (x-axis) shown for CD8 T cells (left) and CD4 T cells (right). The insets list selected genes that were expressed at a higher level in gliomas (up) compared to melanoma; expression is log2 fold change. (I) Correlation of expression between KLRB1 and genes encoding IL-18 receptor subunits (IL18R1, IL18RAP). The correlation between KLRB1 and IL18R1 was 0.12 (P < 1e-15) and the correlation between KLRB1 and IL18RAP was 0.19 (P < 1e-15). Gene-gene correlations in single cell RNA-seq data are often lower due to technical dropout effects.
Supplemental Table 1 (Table S1): Clinical cohort.
Supplemental Table 2 (Table S2) Gene expression data for SMART-seq2 dataset, Related to Figure 1, 2, & S3.
Sheet 1. Marker genes in GBM and IDH-G CD8 T cells (Fig1F_CD8_DE_genes) All differentially expressed genes in IDHwt-GBM and IDHmut-glioma CD8 T cells are listed. This corresponds to the left heatmap in Fig. 1F. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (following the cluster numbering in Fig. 1E), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 2. Marker genes in GBM and IDH-G CD4 T cells (Fig1F_CD4_DE_genes) All differentially expressed genes in GBM and IDH-glioma CD4 T cells are listed. This corresponds to the right heatmap in Fig. 1F. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (following the cluster numbering in Fig. 1E), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 3. Marker genes in GBM and IDH-G CD8 T cells with high versus low NK signature scores (Fig2D_HighNK_CD8_DE_genes) All differentially expressed genes between CD8 T cells with high NK signature scores compared to low NK signature scores are listed. The CD8 T cells are from GBM and IDH-G. This corresponds to Fig. 2D. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 4. Marker genes in clonally expanded GBM and IDH-G T cells (FigS3E_ClonalCells_DE_genes) All differentially expressed genes in clonally expanded IDHwt-GBM and IDHmut-glioma T cells are listed. This corresponds to Fig. S3E. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (following the cluster annotations in Fig. S3D), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Supplemental Table 3 (Table S3): Gene expression data for 10X Genomics dataset, Related to Figure 3, & S2.
Sheet 1. Marker genes in GBM CD8 T cells (FigS2C_CD8_DE_genes) All differentially expressed genes in GBM CD8 T cells profiled using the 10X Genomics platform are listed. This corresponds to the left heatmap in Fig. S2C. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed, the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 2. Marker genes in IDH-G CD4 T cells (FigS2C_CD4_DE_genes) All differentially expressed genes in GBM CD4 T cells profiled using the 10X Genomics platform are listed. This corresponds to the right heatmap in Fig. S2C. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed, the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 3. Marker genes in clonally expanded KLRB1+ vs KLRB1− cells in GBM CD8 T cells (Fig3F_DEgenes_CD8_KLRB1+_KLRB1−) All differentially expressed genes in GBM KLRB1+ versus KLRB1− CD8 T cells profiled using the 10X Genomics platform are listed. This corresponds to the columns labelled CD8+ in the heatmap in Fig. 3F. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed, the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 4. Marker genes in clonally expanded KLRB1+ vs KLRB1− cells in GBM CD4 T cells (Fig3F_DEgenes_CD4_KLRB1+_KLRB1−) All differentially expressed genes in GBM KLRB1+ versus KLRB1− CD4 T cells profiled using the 10X Genomics platform are listed. This corresponds to the columns labelled CD4+ in the heatmap in Fig. 3F. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed, the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Supplemental Table 4 (Table S4): Marker genes in cellular signatures from the SMART-seq2 dataset, Related to Figure 2.
Sheet 1. Marker genes between Cytotoxicity-NK cells CD8 T between GBM and IDH-G (Fig2A_CD8_Cyto-NK_topDEgenes) All differentially expressed genes between Cytotoxicity-NK CD8 T cells between GBM and IDH-G. This corresponds to the left panel of Fig. 2A. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 2. Marker genes between Interferon CD8 T cells between GBM and IDH-G (Fig2A_CD8_Interferon_topDEgenes) All differentially expressed genes between interferon CD8 T cells between GBM and IDH-G. This corresponds to the center panel of Fig. 2A. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 3. Marker genes between Stress CD8 T cells between GBM and IDH-G (Fig2A_CD8_Stress_topDEgenes) All differentially expressed genes between stress CD8 T cells between GBM and IDH-G. This corresponds to the right panel of Fig. 2A. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 4. Marker genes between Cytotoxicity-Chemokine CD4 T cells between GBM and IDH-G (Fig2B_CD4_Cyto_Chemo_topDEgenes) All differentially expressed genes between cytotoxicity-chemokine CD4 T cells between GBM and IDH-G. This corresponds to the left panel of Fig. 2B. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 5. Marker genes between Interferon CD4 T cells between GBM and IDH-G (Fig2B_CD4_Interferon_topDEgenes) All differentially expressed genes between interferon CD4 T cells between GBM and IDH-G. This corresponds to the center panel of Fig. 2B. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Sheet 6. Marker genes between Stress CD4 T cells between GBM and IDH-G (Fig2B_CD4_Stress_topDEgenes) All differentially expressed genes between stress CD4 T cells between IDHwt-GBM and IDHmut gliomas. This corresponds to the right panel of Fig. 2B. For each gene we list: its p value from the t-test and its Bonferroni-adjusted p value (p_val_adj), the cluster in which the gene appears as differentially expressed (high NK signature or low NK signature), the frequency at which the gene is detected in the listed cluster (pct.1) and in all other clusters (pct.2), and the average log-fold change (avg_lnFC, base e) in expression between the gene in the listed cluster and all other clusters.
Supplemental Table 5 (Table S5). Pan-cancer KLRB1 program, Related to Figure 7.
Sheet 1. Pan-cancer KLRB1 program for GBM and IDH-G CD8 T cells (Fig7E_CD8_KLRB1program)
Each row lists a gene identified in a KLRB1 transcriptional program in CD8 T cells from at least one of the analyzed cancer datasets (glioma, lung, melanoma, colon, liver). There are two melanoma datasets: Melanoma1 and Melanoma2 (Jerby-Arnon et al., 2018, Sade-Feldman et al., 2018). The columns indicate for each cancer dataset whether the gene is detected (1) or not detected (0) in the KLRB1 program for that cancer. The genes are sorted based on the number of cancer datasets that support their association with KLRB1 (N). The p-value is calculated using Fisher's combined probability test with Benjamini-Hochberg correction. From these genes, we derive the pan-cancer KLRB1 program for CD8 T cells shown in the left heatmap of Fig. 7E (STAR Methods).
Sheet 2. Pan-cancer KLRB1 program for GBM and IDH-G CD4 T cells (Fig7E_CD4_KLRB1program)
Each row lists a gene identified in a KLRB1 transcriptional program in CD4 T cells from at least one of the analyzed cancer datasets (glioma, lung, melanoma, colon, liver). There are two melanoma datasets: Melanoma1 and Melanoma2 (Jerby-Arnon et al., 2018, Sade-Feldman et al., 2018). The columns indicate for each datasets dataset whether the gene is detected (1) or not detected (0) in the KLRB1 program for that cancer. The genes are sorted based on the number of cancer datasets that support their association with KLRB1 (N). The p-value is calculated using Fisher's combined probability test with Benjamini-Hochberg correction. From these genes, we derive the pan-cancer KLRB1 program for CD4 T cells shown in the right heatmap of Fig. 7E (STAR Methods).
Supplemental Table 6 (Table S6). Correlation of KLRB1 expression with Trm signature, Related to Figure 7.
Sheet 1. Correlation between KLRB1 expression and Trm signature in each subset of CD8 and CD4 T cells (KLRB1_Trm_Correlation) Correlation between expression of KLRB1 and the Trm gene signature for each subset of CD8 and CD4 T cells from both data sets (SMART-seq2 and 10x Genomics). For each subset, we list the Pearson correlation coefficient and p value.
Data Availability Statement
Processed data generated for this study are available at the Gene Expression Omnibus (GEO: GSE163108; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163108). Raw scRNA-seq data can be requested at Data Use Oversight System (https://duos.broadinstitute.org/). The code supporting the current study is available from M.L.S (Suva.Mario@mgh.harvard.edu) on request.