

Abstract
Cancer stem‐like cells (CSCs) induce drug resistance and recurrence of tumors when they experience DNA replication stress. However, the mechanisms underlying DNA replication stress in CSCs and its compensation remain unclear. Here, we demonstrate that upregulated c‐Myc expression induces stronger DNA replication stress in patient‐derived breast CSCs than in differentiated cancer cells. Our results suggest critical roles for mini‐chromosome maintenance protein 10 (MCM10), a firing (activating) factor of DNA replication origins, to compensate for DNA replication stress in CSCs. MCM10 expression is upregulated in CSCs and is maintained by c‐Myc. c‐Myc‐dependent collisions between RNA transcription and DNA replication machinery may occur in nuclei, thereby causing DNA replication stress. MCM10 may activate dormant replication origins close to these collisions to ensure the progression of replication. Moreover, patient‐derived breast CSCs were found to be dependent on MCM10 for their maintenance, even after enrichment for CSCs that were resistant to paclitaxel, the standard chemotherapeutic agent. Further, MCM10 depletion decreased the growth of cancer cells, but not of normal cells. Therefore, MCM10 may robustly compensate for DNA replication stress and facilitate genome duplication in cancer cells in the S‐phase, which is more pronounced in CSCs. Overall, we provide a preclinical rationale to target the c‐Myc‐MCM10 axis for preventing drug resistance and recurrence of tumors.
Keywords: anticancer drug resistance, breast cancer, cancer stem cell, c‐Myc, DNA replication stress, drug sensitivity/drug resistance‐relating factors/gene expression analysis, MCM, oncogenes and tumor‐suppressor genes, others, tumor spheroids
We provide evidence that upregulated c‐Myc expression induces stronger DNA replication stress in patient‐derived breast cancer stem‐like cells than in differentiated cancer cells. Our results suggest critical roles for mini‐chromosome maintenance protein 10 (MCM10), which is a firing (activating) factor of the DNA replication origins, to compensate for the DNA replication stress.

1. INTRODUCTION
Breast cancer is the most frequently observed tumor type among women worldwide. Some patients with breast cancer show poor prognosis due to resistance to therapy and tumor recurrence. 1 Over the past few decades, studies have shown that a subset of cancer cells possess the capacity to initiate tumors. 2 , 3 These tumor‐initiating cells or cancer stem‐like cells (CSCs) are resistant to conventional chemotherapeutic agents, resulting in tumor recurrence. Thus, CSC‐targeted therapeutic strategies are urgently required to improve the prognosis of breast cancer patients. Clarification of the features of CSCs is important for developing CSC‐targeting therapies to remove these cells and prevent recurrence. In vitro tumor spheroid formation in serum‐free floating culture conditions has been established to enrich CSCs. 4 , 5 Researchers, including our group, have used this method to elucidate the features of CSCs. 6 , 7 , 8 , 9 , 10 , 11 Although these features have been studied extensively, the roles of DNA replication initiation factors in CSCs remain unclear.
In preparation for cell division, the whole genome must be replicated during the S‐phase of the cell cycle. To rapidly generate a complete copy of the entire genome, eukaryotic genome replication is initiated from thousands of origins. 12 , 13 The inactive MCM2‐7 helicases (composed of MCM family proteins MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7) bind numerous sites of DNA replication origins in the genome to form pre‐replicative complexes (pre‐RCs) in the late M and G1‐phases. Following the activation of S‐phase cyclin‐dependent kinase (S‐CDK) in the S‐phase, MCM2‐7 in pre‐RCs are activated to form the CDC45/MCM2‐7/GINS (CMG) helicase, and only about 1/10 of the chromatin‐bound MCM2‐7 are converted to the CMG helicase in normal cells. Subsequent recruitment of firing (activating) factors, including MCM10, activates the CMG helicase to form the replisome, which includes DNA polymerases, followed by initiation of bidirectional DNA replication. 14 , 15 , 16 , 17 , 18 , 19 MCM10 opens the MCM2‐7 ring within the CMG, creating a gate for passage of a single DNA strand when the CMG helicase engages in fork progression. 20 However, most origins remain dormant, and pre‐RCs are passively removed from DNA when the replisomes approach these dormant origins during replication progression.
DNA replication stress is defined as the stalling or slowing of replication progression due to interference with the normal replication process by a variety of mechanisms including DNA strand breaks, lack of nucleotides, and so on. 21 , 22 Recently, repair processes responding to DNA strand breaks have received much attention as potential therapeutic targets. Inhibitors of poly(ADP‐ribose) polymerase (PARP), a repair enzyme for single‐stranded breaks, are clinically used to treat breast cancer with BRCA mutations. 23 However, in most patients without BRCA mutations, PARP inhibitors are not obviously effective. In cancer cells, constitutive activation of oncogenes is a primary cause of replication stress. 24 , 25 , 26 DNA replication stress is reported to be upregulated in glioblastoma stem cells 27 , however the level of DNA replication stress in other CSC types, including breast CSCs, remains unknown. When cells suffer from replication stress, checkpoint pathways are activated. 25 , 26 , 28 Ataxia telangiectasia‐ and Rad 3‐related protein (ATR) kinase, and subsequently checkpoint kinase 1 (Chk1), are phosphorylated and activated. Activated Chk1 slows down cell cycle progression in the S‐phase and allows dormant origins to be activated for the completion of DNA replication. Many proteins included in the aforementioned DNA replication initiation machinery work together to activate the dormant origins.
c‐myc is a typical oncogene that is frequently overexpressed in numerous cancer types. The transcription factor c‐Myc can induce the transcription of c. 15% of all genes in the genome. 29 Transcription in the G1‐phase is sequentially followed by DNA replication in the S‐phase in normal cells, however, in cancer cells, c‐Myc overexpression in cancer cells disrupts the cooperation between the transcription and replication machineries. 30 As a result, they collide with DNA strands, leading to DNA replication stress.
In this study, we examined breast cancer patient‐derived primary samples to clarify the specific features of CSCs. We compared whole transcriptomes of CSC‐enriched spheroid cells and cultured cells under regular adherent conditions. We found that pathways contributing to c‐Myc activation and DNA replication stress were upregulated in CSC‐enriched spheroid cells. Our results suggest that c‐Myc causes frequent collisions between the transcription and replication machinery in the nuclei. These collisions may be one of the major causes of higher DNA replication stress levels in CSCs compared with that in differentiated cancer cells. Furthermore, we showed that the expression of MCM10 was increased in CSCs and in differentiated cancer cells, and that its expression was higher in the former than in the latter, compared with that in normal cells. MCM10 may compensate for replication stress by activating dormant origins. Moreover, we demonstrated that MCM10 plays critical roles in the maintenance of CSCs, including those resistant to paclitaxel, a commonly used chemotherapeutic agent for breast cancer. Furthermore, by analyzing patient‐derived cancer cells (PDCs), we found that MCM10 is also essential for the growth of differentiated cancer cells. Thus, inhibition of MCM10 may be a novel therapeutic strategy to target replication initiation in CSCs.
This study is the first to demonstrate that increased c‐Myc activity is a major cause of DNA replication stress in breast CSCs. Furthermore, MCM10, a c‐Myc‐dependent firing factor, which is required for replisome activation, is essential for CSCs, probably compensating for DNA replication stress.
2. MATERIALS AND METHODS
2.1. Primary cell culture
To isolate lineage‐negative (Lin−) breast cancer cells, cells obtained from breast tumor specimens were incubated with a mixture of biotin‐conjugated antibodies against Lin+ cells, as previously described. 10 The antibody mixture included a magnetic cell separation (MACS) lineage kit for depletion of hematopoietic and erythrocyte precursor cells (CD2, CD3, CD11b, CD14, CD15, CD16, CD19, CD56, CD123, and CD235a; Miltenyi Biotec, Birgisch Gladbach, Germany), endothelial cells (CD31, eBioscience, San Diego, CA), and stromal cells (CD140b, Biolegend, San Diego, CA). After incubation, cells were separated using the MACS system (Miltenyi Biotec). Isolated Lin− breast cancer cells were cultured in Human EpiCult™‐B Medium Kit medium (Stem Cell Technologies, Vancouver, Canada) containing a supplement mix, freshly prepared 0.48 μg/mL hydrocortisone (Stem Cell Technologies), 2 mmol/L l‐glutamine (Nacalai Tesque), 100 units/mL penicillin (Nakarai tesque, Inc), and 100 µg/mL streptomycin (Nakarai tesque, Inc). Isolated single cells were cultured in a humidified atmosphere at 37°C in 5% CO2, and the culture medium was changed every 2 d.
Tumor spheres were cultured as follows. Single‐cell suspensions were cultured in ultra‐low attachment plates and cells were grown in sphere culture medium (SCM), which consists of DMEM/F‐12 (GIBCO), 20 ng/mL epidermal growth factor (Millipore, Burlington, MA), 20 ng/mL basic fibroblast growth factor (PeproTech, Cranbury, NJ), B27 supplement (GIBCO), and 2 µg/mL heparin (Stem Cell Technologies), as previously described. 10 Adherent cells attached to regular cell culture plates were cultured in RPMI1640 (GIBCO) supplemented with 10% FBS (GIBCO) and 1% penicillin/streptomycin (P/S) (Nacalai Tesque Inc). Patient‐derived ovarian cancer spheroid cells (OVN62) were established from clinical specimens using previously reported procedures. 31
2.2. Tumor sphere formation assay
We have confirmed previously that patient‐derived breast cancer cells plated at 5000 cells/mL yield tumor spheres clonally derived from single cells. 11 Hence, cells were plated as single‐cell suspensions on ultra‐low attachment 24‐well plates (1000‐5000 cells/well) to obtain single‐cell–derived tumor spheres. The cells were grown in SCM. Spheres with diameter >75 µm were counted after 4‐7 d.
2.3. Study approval
All human breast carcinoma specimens were obtained from the University of Tokyo Hospital, Minami Machida Hospital, and Kanazawa University Hospital. Human ovarian cancer specimens were obtained from Niigata University Medical & Dental Hospital. This study was approved by the institutional review boards of the Institute of Medical Science, The University of Tokyo; The University of Tokyo Hospital, Minami Machida Hospital, National Cancer Center, Niigata University, and Kanazawa University. Written informed consent was received from all participants before inclusion in the study.
3. RESULTS
3.1. CSCs are enriched in spheroid culture conditions and show distinct features
To identify the specific features of CSCs, we first cultured patient‐derived breast cancer cells in spheroid conditions; cells were cultured in SCM on ultra‐low attachment dishes and under normal adherent conditions (Figure 1A). Spheroid cells retained stem cell features such as high tumor‐initiating ability and expression of stemness marker proteins. 4 , 9 , 32 Patient‐derived spheroid cells showed significantly higher tumor‐initiating ability in vivo compared with their counterparts under adherent conditions (Figure 1B and Figure S1A). In addition, western blotting revealed that the expression of the stemness marker proteins Nanog and Oct‐4 was higher in spheroid cells than in adherent cells (Figure 1C). Sox2, another stemness marker protein, also showed slightly higher expression in spheroid cells than in adherent cells (Figure S1B). Immunocytochemistry (ICC) showed strong Nanog staining in the nucleus and that more spheroid cells were strongly positive for Nanog (Figure S1C). A subpopulation of breast CSCs is known to be enriched in the CD24low/−/CD44high cell population 33 , thus we investigated this population using flow cytometry. The proportion of cells in the CD24low/−/CD44high cell fraction was higher in spheroid cells than in adherent cells (Figure 1D). These results indicated that breast CSCs were more abundant among spheroid cells, whereas differentiated cancer cells were more abundant among adherent cells.
FIGURE 1.
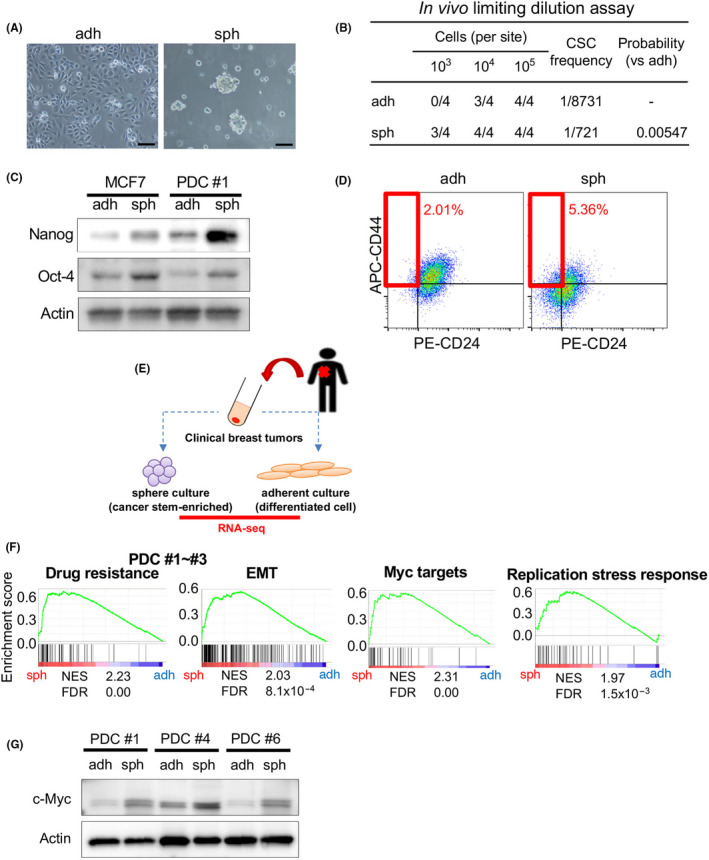
CSCs are enriched in the sphere culture population and show activation of distinct pathways; c‐Myc expression is upregulated in CSC‐enriched spheroid cells. A, Images of PDC #1 cultured under adherent conditions (adh; left), and in sphere culture conditions (sph; right) are shown. Scale bars = 100 μm. B, Results of limiting dilution assays of PDC#1 obtained under adherent and sphere culture conditions were compared. CSC frequency and P‐value were determined using ELDA software (http://bioinf.wehi.edu.au/software/elda/index.html). C, Expression levels of Nanog and Oct‐4 in MCF7 and PDC #1 cells were compared between cells cultured under adherent and sphere conditions. Actin was used for the loading control. D, PDC #1 cells obtained by adherent and sphere culture conditions were stained with CD44 and CD24 antibodies, and then subjected to flow cytometry analysis. E, Schematic of the experimental procedure. Cancer cells were separated from clinical breast tumor samples, and then were cultured under adherent and sphere conditions. RNA was collected from both conditions for RNA‐seq transcriptome analysis. F, Gene set enrichment analysis (GSEA) was used to compare gene expression profiles of PDC #1‐#3. FDR, false discovery rate; NES, normalized enrichment score. G, Expression levels of c‐Myc in PDC #1, #4 and #6 determined by immunoblotting were compared between cells cultured under the adherent and sphere conditions. Actin was used for the loading control
Next, we performed RNA sequencing (RNA‐seq) to compare the transcriptomes of spheroid cells and adherent cells (Figure 1E). All samples were derived from breast tumor tissues of 3 individual patients (PDCs #1, #2, and #3; the clinical sample information is summarized in Table S1). Gene set enrichment analysis (GSEA) (http://software.broadinstitute.org/gsea/index.jsp) based on the RNA‐seq data revealed that genes associated with drug resistance and epithelial‐mesenchymal transition (EMT) were upregulated in spheroid cells compared with adherent cells (Figure 1F). Because these are well known features of CSCs, we confirmed that CSCs were enriched in spheroid cells. 34 We also noted that other gene sets related to Myc targets and DNA replication stress responses were highly upregulated in spheroid cells.
3.2. c‐Myc expression is increased in CSC‐enriched spheroid cells and may lead to strong DNA replication stress
We then investigated c‐Myc protein levels. Western blotting revealed that c‐Myc expression was higher in spheroid cells than in adherent cells among all 3 PDC samples irrespective of the different subtypes (Figure 1G, triple negative [#1], HER2 [#4], and luminal‐like [#6]). ICC showed strong accumulation of c‐Myc in the nucleus, and more spheroid cells were strongly positive for c‐Myc compared with adherent cells (Figure S1D), suggesting that a subpopulation of CSCs strongly expresses c‐Myc.
Because gene sets related to DNA replication stress responses were upregulated in spheroid cells, we examined the proteins involved in the checkpoint pathways. 25 , 26 We found that the phosphorylation levels of ATR and Chk1 were higher in spheroid cells than in adherent cells (Figure 2A). These results suggested that spheroid cells experience more DNA replication stress, which activates the checkpoint pathways. To directly monitor DNA replication fork stalling caused by DNA replication stress, we performed DNA fiber assays. 35 We labeled cells with iodo‐deoxyuridine (IdU) for 30 min and then with chloro‐deoxyuridine (CldU) for 30 min (Figure 2B). Using this approach, bidirectional forks can be observed when a single replication origin is activated in the first 30 min (IdU: green) and then proceeds in 2 opposite directions (Figure 2C, left and middle panels). If the 2 forks proceed normally, a CIdU‐labeled (red) fork of the same length should be observed. In contrast, when 1 fork stalls because of DNA replication stress, the 2 forks have different lengths. We observed fork stalling more frequently in spheroid cells, as reflected in the higher frequency of asymmetric CIdU‐containing forks (Figure 2C, right panel, and Figure S2A,C). If forks stalled during the first 30 min, the stalled forks would be labeled only with IdU (green) (Figure 2D, left panel). We observed these stalled forks more frequently in spheroid cells (Figure 2D, right panel, and Figure S2B,D). Taken together, we concluded that DNA replication stress is upregulated in CSCs compared with that in differentiated cancer cells.
FIGURE 2.
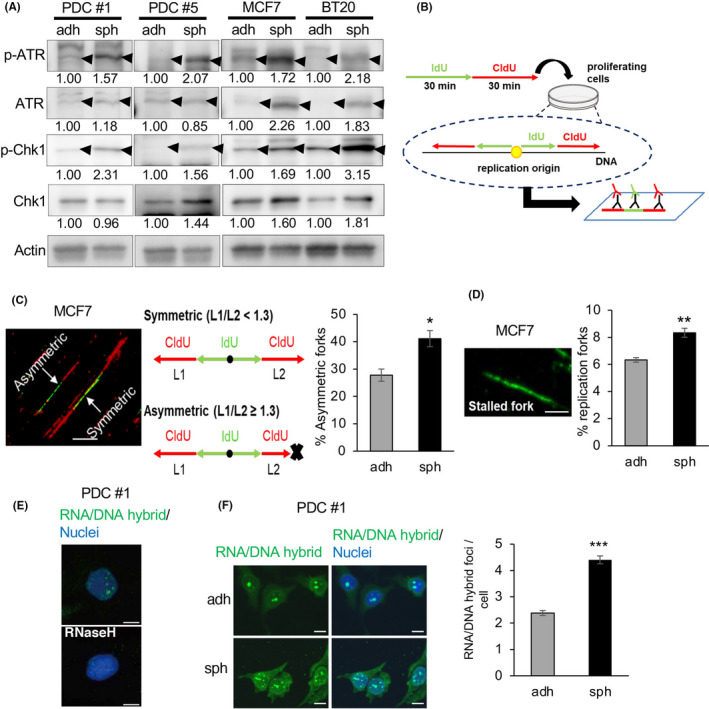
c‐Myc expression and DNA replication stress are upregulated in CSC‐enriched spheroid cells. A, Expression levels of ATR, p‐ATR, Chk1, and p‐Chk1, determined by immunoblotting, were compared between cells cultured under the adherent and sphere conditions. Expression levels were quantified using ImageJ software and normalized to Actin. B, Schematic of the experimental procedure. Cells were incubated sequentially with iodo‐deoxyuridine (IdU) then chloro‐deoxyuridine (CldU). Labeled DNA was spread onto glass slides, and then stained with antibodies for IdU (green) and CldU (red). If replication started in the first 30 min, bidirectional forks stained with green and red could be observed. C, Proportion of asymmetric forks, representative of replication stress. The ratio of longer CldU tracks (L1) to shorter tracks (L2) was calculated; forks with L1/L2 ≥ 1.3 were regarded as asymmetric. Thirty bidirectional forks in each slide were counted. Three slides for each population were prepared (mean ± SEM, n = 3; *P < .05). Scale bar = 10 μm. D, Proportion of stalled forks, labeled only with green was calculated. In total, 200 labeled forks in each slide were counted. Three slides for each population were prepared (mean ± SEM, n = 3; **P < .01). Scale bar = 5 μm. E, Immunofluorescence images of RNA/DNA hybrid staining in PDCs are shown. Cells were cultured under the sphere condition with or without RNase H treatment. Nuclei were counterstained with DAPI. Scale bars = 10 μm. F, (Left) Immunofluorescence images of RNA/DNA hybrid staining in PDCs cultured under adherent and sphere conditions are shown. (Right) Number of RNA/DNA hybrid foci in each cell was counted and compared between the 2 conditions. In total, 100 cells in each slide were counted. Three slides for each population were prepared (mean ± SEM, n = 3; ***P < .001). Scale bars = 10 μm
Upregulation of c‐Myc induces collisions between the transcription and replication machinery in the nucleus, leading to DNA replication stress. 30 We hypothesized that upregulated c‐Myc in CSCs causes collisions between the transcription and replication machinery more frequently than in differentiated cancer cells. These collisions are associated with stabilized R‐loops, that is an RNA/DNA hybrid and the displaced single‐stranded DNA behind the elongation of RNA polymerases. 22 , 36 R‐loops can be detected by ICC staining using the monoclonal antibody S9.6, which is a tool used widely to recognize RNA/DNA hybrids. 37 RNA/DNA hybrid foci detected with the S9.6 antibody were localized in the nucleus (Figure 2E). The staining disappeared following treatment with ribonuclease H (RNase H), which cleaves the RNA strands in RNA/DNA hybrids, confirming the specificity of the antibody (Figure 2E). We found that spheroid cells showed a higher number of RNA/DNA foci than adherent cells (Figure 2F and Figure S3A,B). These results suggest that collisions between transcription and replication machinery occur more frequently in CSCs than in differentiated cancer cells.
We then depleted c‐Myc expression using small interfering RNAs (siRNAs) for c‐myc (Figure 3A and Figure S4A). Depletion of c‐Myc led to a decreased number of RNA/DNA hybrid foci and decreased the phosphorylation levels of ATR and ChK1 (Figure 3B,C and Figure S4B,C). Therefore, c‐Myc‐induced collisions between transcription and replication machinery in the nuclei are likely to lead to DNA replication stress, which is more frequent in CSCs than in differentiated cancer cells.
FIGURE 3.
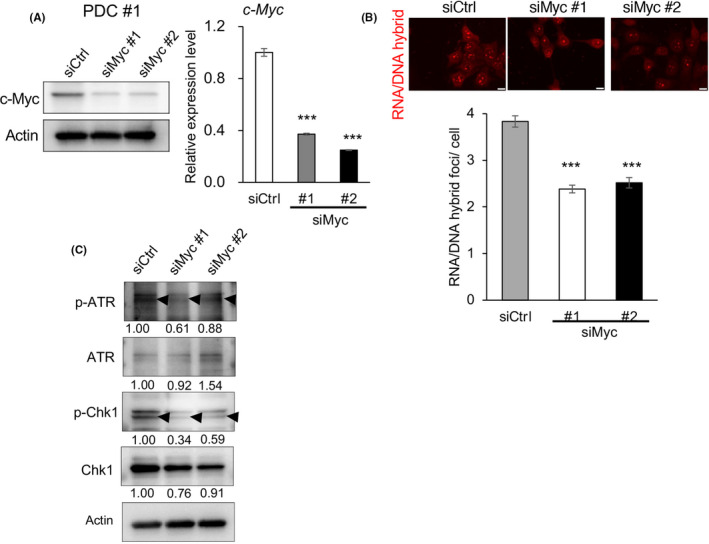
c‐Myc expression contributes to replication stress. A, Knockdown efficiencies of siRNAs targeting c‐myc (siMyc #1 and #2) or control siRNA (siCtrl) in PDCs was compared by immunoblotting (left) and qPCR (right) (mean ± SEM, n = 3; ***P < .001). B, The number of RNA/DNA hybrid foci in each cell was counted and compared among spheroid cells treated with siCtrl, siMyc #1, and siMyc #2. Scale bars = 10 μm. In total, 100 cells in each slide were counted. Three slides for each population were prepared (mean ± SEM, n = 3; ***P < .001). C, Expression levels of ATR, p‐ATR, Chk1, and p‐Chk1 as determined by immunoblotting, were compared among cells treated with siCtrl, siMyc #1, and siMyc #2. Expression was quantified by ImageJ software and normalized to Actin
3.3. MCM10 expression is upregulated in CSC‐enriched spheroid cells and co‐localizes with the RNA/DNA hybrid foci in nuclei
Based on the results described above, we expected that CSCs possess mechanisms to manage higher levels of DNA replication stress. Gene Ontology (GO) enrichment analysis based on the RNA‐seq data showed that several gene sets related to DNA replication were upregulated in the spheroid cells (Figure S5). We focused our attention on MCM10, which was the fifth most highly upregulated gene (Table S2) considering that MCM10 can activate dormant origins to compensate for DNA replication stress. 38 Western blotting showed that MCM10 expression levels were higher in several breast cancer cell lines than in MCF10A cells, a normal mammary epithelial cell line (Figure 4A). We found that MCM10 mRNA and protein levels were reduced in c‐myc‐depleted cancer cells both under adherent conditions (Figure 4A,B) and spheroid conditions (Figure S6A), indicating that MCM10 expression is associated with c‐Myc expression. qPCR and western blotting analyses showed that MCM10 expression levels were higher in spheroid cells than in adherent cells in several PDCs and breast cancer cell lines (Figure 4C,D). In addition, the expression of MCM10 was higher in the CD24low/−/CD44high cell population, a subpopulation of CSCs, compared with in the control population (Figure S6B,C).
FIGURE 4.
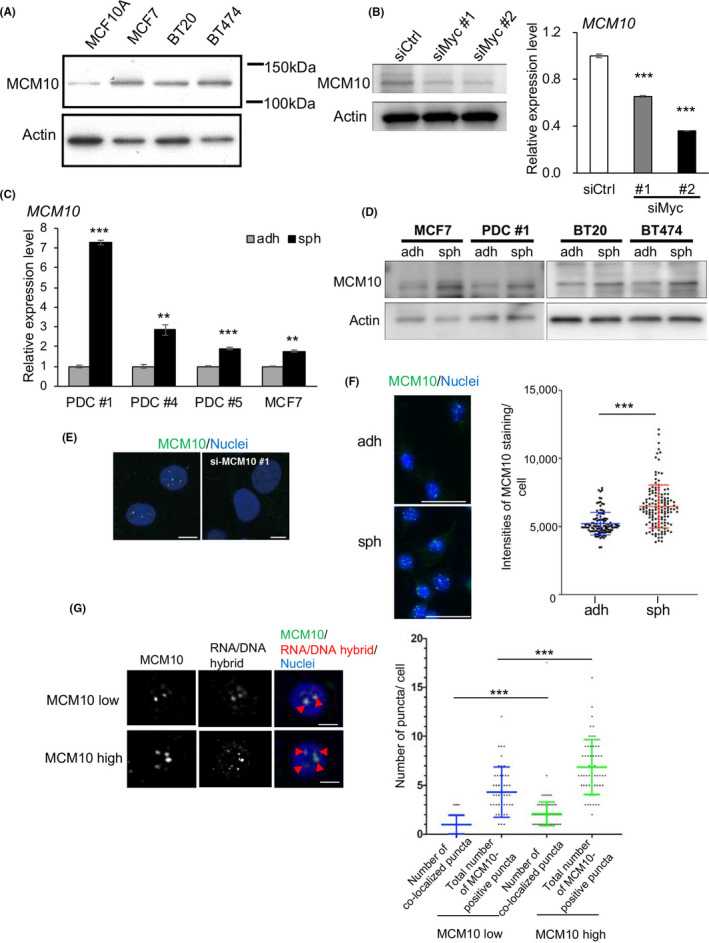
MCM10 expression is upregulated in CSC‐enriched spheroid cells and MCM10 is co‐localized with RNA/DNA hybrid foci. A, Expression levels of MCM10 in MCF10A, MCF7, BT20, and BT474 were compared by immunoblotting. Actin was used for the loading control. B, Expression levels of MCM10 in PDCs treated with siCtrl, siMyc #1 and siMyc #2 were compared by immunoblotting (left) and qPCR (right) (mean ± SEM, n = 3; ***P < .001). Actin was used for the loading control. C, Expression levels of MCM10 in PDC #1, #4, #5 and MCF7 cells were compared between cells cultured under adherent and sphere conditions, by qPCR (mean ± SEM, n = 3; ***P < .001, **P < .01). D, Expression levels of MCM10 were compared in cells cultured under the adherent and sphere conditions, by immunoblotting. Actin was used for the loading control. E, Immunofluorescence images of MCM10 staining in PDCs after transfection with control siRNA (left) or siMCM10 #1 (right) are shown. Nuclei were counterstained with DAPI. Scale bars = 5 μm. F, (left) Immunofluorescence images of MCM10 staining in PDCs cultured under adherent and sphere conditions are shown. Nuclei were counterstained with DAPI. Scale bars = 50 μm. (Right) The intensities of MCM10 staining were quantified using ImageJ software. In total, 100 cells in each slide were counted (mean ± SD, n = 3; ***P < .001). G, (Left) Immunofluorescence images of MCM10 and S9.6 antibody staining in PDCs cultured in the sphere conditions are shown. The median values of the intensities of MCM10 staining (F) were used as the cut‐off to determine MCM10‐low cells and MCM10‐high cells. Nuclei were counterstained with DAPI. Arrowheads indicate double‐positive puncta. Scale bars = 5 μm. (Right) Scatter plot showing the total number of MCM10‐positive puncta and double‐positive puncta in each cell. MCM10‐positive puncta and double‐positive puncta were quantified using ImageJ software. Fifty cells were counted for each group. (mean ± SD; ***P < .001)
Further, ICC staining showed that MCM10‐positive puncta were present in the nucleus (Figure 4E). The staining disappeared after depletion of MCM10 using siRNAs for MCM10, confirming the specificity of the antibodies (Figure 4E). The number of MCM10‐positive puncta was significantly higher in spheroid cells than in adherent cells (Figure 4F). These results suggested that MCM10 expression is maintained by c‐Myc and is upregulated in CSCs.
To test whether MCM10 is recruited close to the collisions between the transcription and replication machinery, we stained PDCs using antibodies against MCM10 and with S9.6. We found significant co‐localization of MCM10‐positive puncta and RNA/DNA hybrid foci (Figure 4G). This result supports the notion that MCM10 is recruited to stalled forks due to collisions in nuclei. Together, our findings suggested that MCM10 expression is upregulated in both CSCs and in differentiated cancer cells, and that its expression is higher in the former than in the latter, compared to normal cells. MCM10 expression may thus be partly induced by upregulated c‐Myc expression. Furthermore, MCM10 may be recruited to stalled forks in the nuclei.
3.4. MCM10 expression levels are prognostic and MCM10 plays important roles in the proliferation of adherent cells
To examine the clinical relevance of MCM10, we analyzed the data obtained from publicly available gene expression profiles of breast cancer tissues. 39 , 40 We found that patients with breast cancer showing high levels of MCM10 expression had poor prognosis (Figure 5A), supporting the possibility that MCM10 plays an important role in tumorigenesis. Based on the Oncomine database (https://www.oncomine.org), MCM10 expression was higher in various cancer tissues including breast and colon cancer, than in their normal counterparts (Figure S6C).
FIGURE 5.
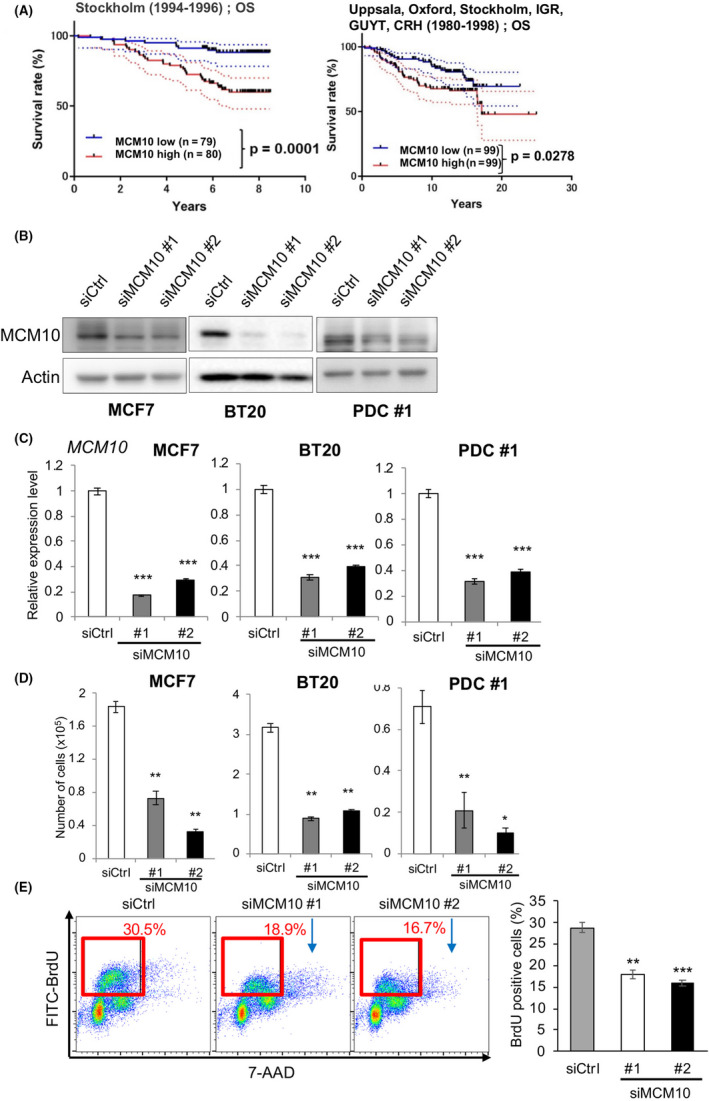
MCM10 plays important roles for proliferation of cancer cells. A, Kaplan‐Meier survival curves were drawn using the Stockholm cohort (GSE1456; overall survival) and the Uppsala, Oxford, Stockholm, IGR, GUYT, and CRH cohorts (GSE7390; overall survival). The median values were used as the cut‐off. P‐values were obtained by log‐rank test. B and C, Knockdown efficiencies of siRNAs targeting MCM10 (siMCM10 #1 and #2) in MCF7, BT20 and PDC #1 were compared by immunoblotting (B) and qPCR (C) (mean ± SEM, n = 3; ***P < .001). D, Cells were seeded in 12‐well plates (10 000 cells/well) and cultured. Then they were harvested and counted after 4 d (mean ± SEM, n = 3; **P < .01, *P < .05). E, (Left) PDCs treated with siCtrl, siMCM10 #1, or siMCM10 #2 were incubated with BrdU for 30 min. DNA and incorporated BrdU content were analyzed by flow cytometry. (Right) The proportion of BrdU‐positive cells was averaged from 3 biological replicates (mean ± SEM, n = 3; ***P < .001, **P < .01)
To examine the functions of MCM10 in cancer cells, we depleted MCM10 by using siRNAs. We confirmed that 2 types of siRNAs efficiently suppressed MCM10 expression compared with the control siRNA (siCtrl) (Figure 5B,C). We evaluated the effect of these siRNAs on the proliferation of adherent cells. Knockdown of MCM10 greatly decreased the proliferation of MCF7 (luminal), BT20 (triple negative), and PDC #1 (triple negative) cells relative to a nonspecific control siRNA (siCtrl) (Figure 5D), indicating that MCM10 is essential for the proliferation of differentiated cancer cells regardless of the breast cancer subtype. To verify the requirement of high MCM10 expression for the proliferation in ovarian cancer cells, another type of gynecological cancer cell, we utilized the CRISPR‐caspase 9 (CRISPR/Cas9)‐mediated conditional knockout system to deplete MCM10 in PDC #8) (Figure S7A,B). We found that doxycycline (Dox)‐induced depletion of MCM10 significantly reduced the proliferation of ovarian cancer cells (Figure S7C). In contrast, knockdown of MCM10 in normal MCF10A cells did not significantly alter their proliferation (Figure S7D). These results suggested that MCM10 plays important roles in the proliferation of differentiated cancer cells, but not in normal cells.
We next measured DNA replication activity by examining BrdU incorporation. When MCM10 was depleted in adherent cells, BrdU incorporation was decreased, indicating that DNA replication activity was significantly decreased in MCM10‐depleted cells (Figure 5E). Together, these results are consistent with the notion that MCM10 depletion increases DNA replication stress, slows S‐phase progression, and decreases cell proliferation. MCM10 thus appears to be a limiting factor for dealing with DNA replication stress to complete the S‐phase, leading to cell proliferation.
We next examined whether MCM5, a component of pre‐RCs, is a limiting factor for cancer cell proliferation to the same extent as MCM10. We then depleted MCM5 in MCF7 cells with siRNAs. However, we found that cell proliferation was not significantly altered in these cells (Figure S8A,B). These results indicated that MCM5 is not a limiting factor for the cancer cell proliferation under these conditions. Consistently, a previous report has shown that MCM2‐7 is highly abundant and that MCM5‐depleted cells do not show a significant growth defect under normal culture conditions. 41 We then asked whether MCM10 overexpression contributes to the replication stress response and thus promotes cell proliferation. To test this, we first overexpressed MCM10 and found that cell proliferation was not significantly altered by MCM10 overexpression alone (Figure S8C,D). We further treated cells with hydroxyurea (HU), an inhibitor of ribonucleotide reductase, to induce replication stress. Treatment with HU leads to a shortage of deoxyribonucleotides that are used for DNA synthesis in the S‐phase. 42 As expected, treatment with HU decreased cell proliferation because the cells experienced stronger replication stress (Figure S8E). We found that decreased cell proliferation was partly restored by MCM10 overexpression when the cells were treated with 500 µmol/L HU. Under these conditions, we found that MCM5 depletion blocked the restored effects on cell proliferation by MCM10 overexpression (Figure S8F). These results suggested that MCM10 overexpression counteracts the strong replication stress induced by HU in cooperation with MCM5.
3.5. MCM10 plays important roles in CSC properties
Next, we focused on the association between MCM10 upregulation and CSCs. To this end, we first examined its effects on tumor sphere‐forming capacity. MCM10 depletion greatly decreased the sphere‐forming ability of all tested cancer cells (Figure 6A and Figure S9A). We subsequently examined the proportion of CD24low/−/CD44high cells, a subpopulation of breast CSCs and found that they were decreased in MCM10‐depleted cells than in control cells (Figure 6B and Figure S9B). Furthermore, the expression levels of Nanog and Oct‐4 were lower in MCM10‐depleted cells than in control cells (Figure 6C). These results suggested that MCM10 plays an important role in CSC maintenance.
FIGURE 6.
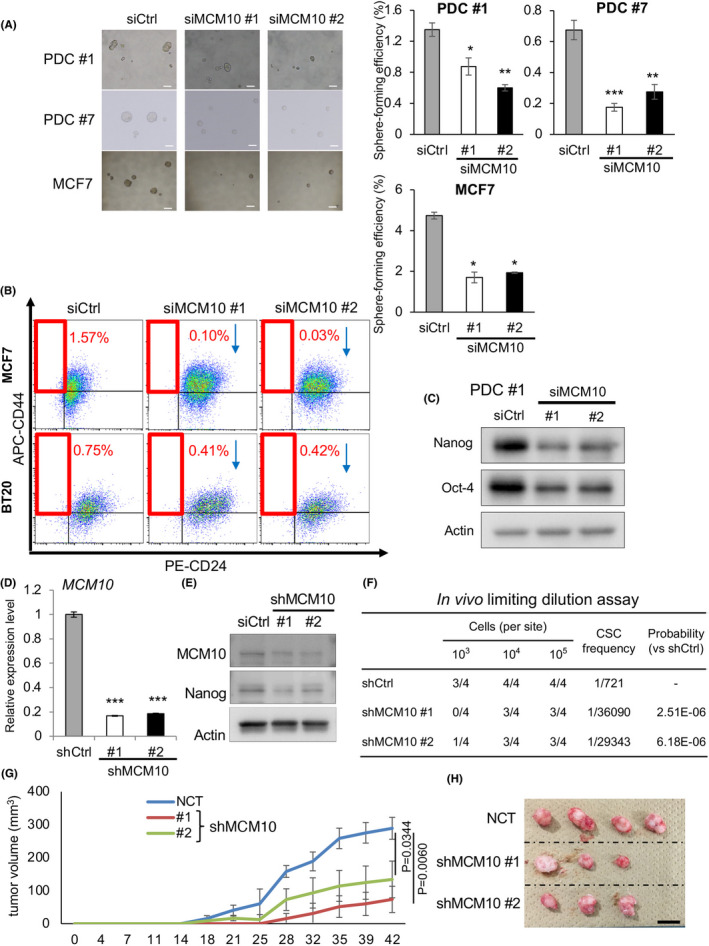
MCM10 plays important roles for CSC properties. A, (Left) Representative images of tumor spheres. PDC #1, #7 and MCF7 cells treated with siRNA targeting MCM10 were cultured under sphere conditions. Scale bars = 100 μm. (Right) Quantification of tumor sphere formation efficiency. Spheres were formed for 6 d (mean ± SEM, n = 4; ***P < .001, **P < .01, *P < .05). B, MCF7 and BT20 cells treated with siCtrl, siMCM10 #1 or siMCM10 #2 were stained with CD44 and CD24 antibodies, and then subjected to flow cytometry analysis. C, Expression levels of Nanog and Oct‐4, as determined by immunoblotting, were compared between PDCs treated with siCtrl, siMCM10 #1 and siMCM10 #2. D, Expression levels of MCM10 were analyzed by qPCR in PDCs introduced with shCtrl, shMCM10 #1, or shMCM10 #2 (mean ± SEM, n = 3; ***P < .001). E, Expression levels of MCM10 and Nanog were compared by immunoblotting in PDCs introduced with shCtrl, shMCM10 #1, or shMCM10 #2. F, Results of limiting dilution assay of shRNA‐introduced PDCs #1 are shown. Tumors larger than 50 mm3 were counted. CSC frequency and P‐values were determined using ELDA software. G,H, Growth curves (G) and representative images (H) of tumors are shown (1 × 105 cells/site) (mean ± SEM, n = 4). Scale bar = 10 mm
We next constructed short hairpin RNAs (shRNAs) against MCM10 (shMCM10 #1 and #2) and confirmed that these constructs significantly decreased the levels of MCM10 at both the RNA and protein levels relative to the control (shCtrl) (Figure 6D,E). An in vitro limiting dilution assay revealed that MCM10 depletion using shRNA decreased tumor sphere‐forming ability and estimated CSC frequency (Figure S8G). We then examined the tumor‐initiating capacity using a patient‐derived xenograft model. Using the in vivo limiting dilution assay, we found that MCM10‐depleted cells had a greatly reduced tumor‐initiating ability and estimated CSC frequency (Figure 6F). The tumor growth rate following inoculation of 105 cells was also decreased by MCM10 depletion (Figure 6G,H). Together, these results illustrate the importance of MCM10 in the maintenance of CSCs both in vitro and in vivo.
3.6. Expression of c‐Myc and MCM10 is enriched in paclitaxel‐resistant CSCs that are dependent on MCM10 for their maintenance
Finally, we examined the possibility that MCM10 depletion can eradicate CSCs. CSCs are enriched after paclitaxel treatment, as only differentiated cancer cells are efficiently killed by this chemotherapeutic drug. 43 , 44 Indeed, when cancer cells were treated with paclitaxel, the remaining resistant cells showed significantly higher sphere‐forming abilities (Figure 7A,B). We also found that the expression levels of MCM10, c‐Myc, and Nanog were enriched after treatment (Figure 7C). However, when MCM10 was depleted, the paclitaxel‐resistant cells displayed a significant reduction in sphere‐forming abilities, and enrichment of CSCs with high Nanog and c‐Myc expression was abrogated (Figure 7C,D). These results are consistent with the notion that CSCs that are resistant to paclitaxel can be eradicated by MCM10 depletion.
FIGURE 7.
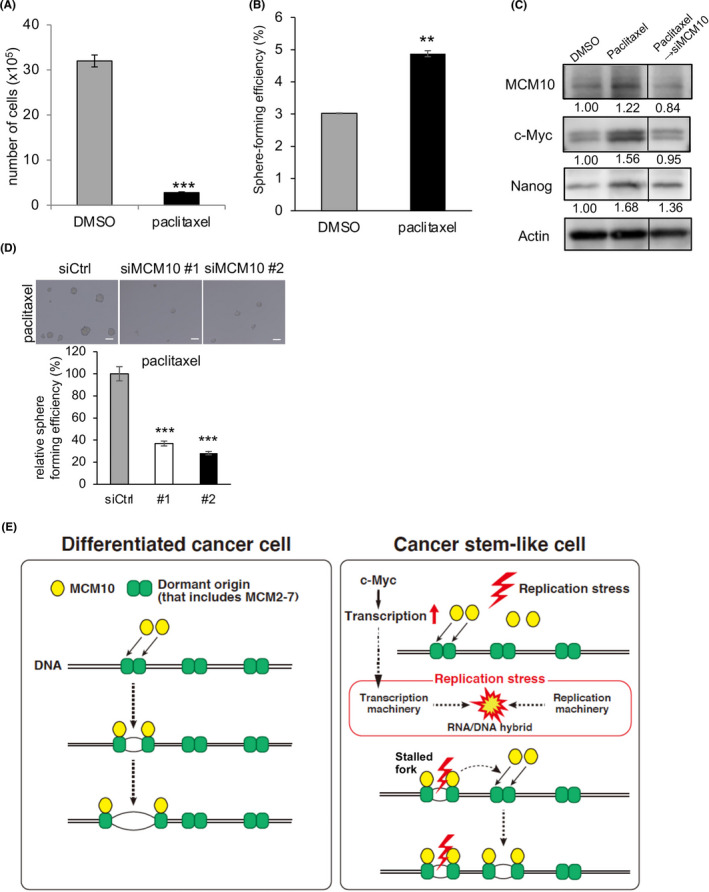
Paclitaxel‐resistant cancer cells are dependent on MCM10 for their maintenance. A, PDCs were seeded into 12‐well plates (10 000 cells/well) and cultured with 10 nmol/L paclitaxel or control DMSO for 3 d. Cells were harvested and counted (mean ± SEM, n = 3; ***P < .001). B, Cells that survived after the treatment with paclitaxel or DMSO were used to analyze sphere‐forming ability. Spheres were counted after 6 d (mean ± SEM, n = 4; **P < .01). C and D, The cells that survived after the treatment with paclitaxel or DMSO were treated with siRNA for MCM10 or control siRNA (Ctrl), and then cultured under the sphere conditions. Expression levels of Nanog, c‐Myc, and MCM10, as determined by immunoblotting, were compared (C). Expression was quantified using ImageJ software and normalized to Actin. D, Representative images of tumor spheres are shown (upper panels). Scale bars = 100 μm. Spheres were counted after 4 d (mean ± SEM, n = 4; ***P < .001, **P < .01) (lower panel). E, Models of MCM10 function in CSCs and differentiated cancer cells are illustrated. In CSCs, upregulation of c‐Myc leads to higher levels of replication stress due to collisions between transcription machinery and replication machinery. MCM10 is necessary to deal with such DNA replication stress. MCM10 promotes the completion of DNA replication by activating dormant origins near the stalled forks
Taken together, our findings suggested that upregulated c‐Myc increases RNA transcription in CSCs (Figure 7E). This increased RNA transcription may result in collisions between the transcription and replication machinery, thereby causing DNA replication stress, which is more frequent in CSCs than in differentiated cancer cells. MCM10 may then activate the dormant origins near the stalled replication forks. Upregulated MCM10 by c‐Myc may robustly compensate for DNA replication stress by activating the dormant origins. Therefore, MCM10 is essential for the proliferation of cancer cells and maintenance of CSCs that are resistant to paclitaxel.
4. DISCUSSION
In this study, we provide evidence that MCM10, a component of the DNA replication initiation machinery, is essential for maintaining CSCs, probably by helping them compensate for DNA replication stress. MCM10 expression is upregulated in many types of cancer cells. 45 , 46 , 47 Although previous studies have reported that MCM10 upregulation is correlated with tumor malignancy, the underlying molecular mechanisms remain largely unclear. In this study, we have provided mechanistic insights into how MCM10 is required in cancer cells, including CSCs. MCM10 may efficiently activate dormant origins to compensate for DNA replication stress, which is more frequent in CSCs than in differentiated cancer cells. In contrast, depletion of MCM10 did not significantly alter cell proliferation in normal cells, indicating that a small amount of MCM10 is sufficient for DNA replication in normal cells. In cancer cells, an increased amount of MCM10 appears to be required to compensate for the increased DNA replication stress. Thus, targeting MCM10 is likely to be effective for eliminating cancer cells, including CSCs, without adverse effects on normal cells.
We also provide evidence that the replication stress in breast cancer cells and CSCs may be caused by collisions between the transcription and replication machinery. In neural progenitor cells and glioblastoma CSCs, transcription of long neural genes is reported to increases the frequency of collisions between the transcription and replication machinery, leading to a higher level of DNA replication stress. 27 In contrast, in breast CSCs, it appears that upregulated c‐Myc induces collisions between the transcription and replication machinery. Emerging evidence indicates that CSCs are maintained by unique mechanisms that may endow them with a stress‐resistant phenotype relative to differentiated cancer cells. CSCs may need to produce a higher amount of proteins compared with differentiated cancer cells to maintain their properties, possibly explaining why CSCs upregulate c‐Myc expression. The molecular mechanisms that link c‐Myc and stemness genes are still unclear in our study. c‐Myc is reported to directly interact with Nanog and Sox2, leading to self‐renewal in T‐cell leukemia cells. 48 c‐Myc activation in hepatoma cells is also reported to cause upregulation of Nanog, Oct4, and Sox2 in a p53‐dependent manner. 49 It would thus be intriguing to study the detailed molecular mechanisms by which c‐Myc regulates stemness in breast CSCs. A more detailed understanding of the mechanisms of replication stress in CSCs will facilitate the development of therapeutic strategies targeting CSCs.
MCM10 was found to be the most highly upregulated gene among DNA replication initiation factors in spheroid cells compared with adherent cells. Our finding that reduction of MCM10 expression in c‐myc‐depleted cells raises the intriguing possibility that c‐Myc induces MCM10 transcription. However, further studies are required to clarify the detailed molecular mechanisms for this. We found that other MCM proteins in the pre‐RCs were also among the top 100 upregulated genes. MCM5 was the 43rd most highly upregulated gene and the second most highly upregulated MCM family gene. However, MCM5 depletion alone did not affect cell proliferation significantly (Figure S3B). 50 Thus, MCM10, but not MCM5, is likely to be essential for cancer cell proliferation. In fact, MCM2‐7 helicase complexes are expressed abundantly, and cancer cells can survive for some time after depletion of these molecules. 41 MCM5 remains in the pre‐RCs in the licensed or dormant origins, whereas MCM10 is recruited as a firing factor to activate these origins. This functional difference between these MCMs may explain the respective phenotypes in cancer cells after the knockdown of each molecule. GINS and CDC45, other firing factors, were the 12th and 19th most highly upregulated genes, respectively, but their roles in CSCs remain unknown. We are interested in analyzing their roles in future projects.
Overall, we provide a proof‐of‐principle that molecules inhibiting the functions of MCM10 will be useful for targeting cancer cells as well as CSCs in breast cancer. We also provide evidence that MCM10 can be an appropriate target for ovarian cancer. It is possible that MCM10 could be targeted for other cancer types as well. Our future analysis will clarify these points. Suramin, an anti‐parasitic agent, is known to inhibit the functions of MCM10. 51 It may also be possible to develop MCM10 inhibitors without enzymatic activity using Proteolysis Targeting Chimera (PROTAC) technology. 52 The combination of such MCM10 inhibitors with chemotherapeutic reagents that induce DNA replication stress is expected to synergistically target cancer cells.
DISCLOSURE
The authors declare no potential conflicts of interest.
Supporting information
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Nakauchi, Y. Ishii, and A. Fujita for their help with flow cytometry. This work was supported in part by an Extramural Collaborative Research Grant from the Cancer Research Institute, Kanazawa University, a Research Grant from Princess Takamatsu Cancer Research Fund (17‐2924), a Grant‐in‐Aid for Scientific Research from JSPS (16H06279[PAGS], 17K19587 and 18H02679), and a research grant from AMED Project for Development of Innovative Research on Cancer Therapeutics, Project for Cancer Research and Therapeutic Evolution (16cm0106120h0001) and Practical Research for Innovative Cancer Control (16ck0106194h0001) to NG This work was also supported by MEXT KAKENHI (No. 221S0002) to NG. This work was supported by the Graduate Program for Leaders in Life Innovation (GPLLI) at The University of Tokyo Life Innovation Leading Graduate School (to TM, http://square.umin.ac.jp/gplli/en/index.html) and a Grant‐in‐Aid for a Japan Society for Promotion of Science (JSPS) fellowship (to TM, http://www.jsps.go.jp/english/e‐pd/index.html).
Murayama T, Takeuchi Y, Yamawaki K, et al. MCM10 compensates for Myc‐induced DNA replication stress in breast cancer stem‐like cells. Cancer Sci. 2021;112:1209–1224. 10.1111/cas.14776
Takahiko Murayama and Yasuto Takeuchi equally to this work.
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA: Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 2. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. 2019;24:25‐40. [DOI] [PubMed] [Google Scholar]
- 3. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124‐1134. [DOI] [PubMed] [Google Scholar]
- 4. Ablett MP, Singh JK, Clarke RB. Stem cells in breast tumours: are they ready for the clinic? Eur J Cancer. 2012;48:2104‐2116. [DOI] [PubMed] [Google Scholar]
- 5. Ponti D, Costa A, Zaffaroni N, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506‐5511. [DOI] [PubMed] [Google Scholar]
- 6. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG‐GLI1 signaling regulates human glioma growth, cancer stem cell self‐renewal, and tumorigenicity. Curr Biol. 2007;17:165‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(−) glioblastoma‐derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010‐4015. [DOI] [PubMed] [Google Scholar]
- 8. Sansone P, Storci G, Tavolari S, et al. IL‐6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988‐4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murayama T, Nakaoku T, Enari M, et al. Oncogenic fusion gene CD74‐NRG1 confers cancer stem cell‐like properties in lung cancer through a IGF2 autocrine/paracrine circuit. Cancer Res. 2016;76:974‐983. [DOI] [PubMed] [Google Scholar]
- 10. Tominaga K, Minato H, Murayama T, et al. Semaphorin signaling via MICAL3 induces symmetric cell division to expand breast cancer stem‐like cells. Proc Natl Acad Sci U S A. 2019;116:625‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hinohara K, Kobayashi S, Kanauchi H, et al. ErbB receptor tyrosine kinase/NF‐kappaB signaling controls mammosphere formation in human breast cancer. Proc Natl Acad Sci U S A. 2012;109:6584‐6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fragkos M, Ganier O, Coulombe P, Mechali M. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol. 2015;16:360‐374. [DOI] [PubMed] [Google Scholar]
- 13. Masai H, Matsumoto S, You Z, Yoshizawa‐Sugata N, Oda M. Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem. 2010;79:89‐130. [DOI] [PubMed] [Google Scholar]
- 14. Watase G, Takisawa H, Kanemaki MT. Mcm10 plays a role in functioning of the eukaryotic replicative DNA helicase, Cdc45‐Mcm‐GINS. Curr Biol. 2012;22:343‐349. [DOI] [PubMed] [Google Scholar]
- 15. Kanke M, Kodama Y, Takahashi TS, Nakagawa T, Masukata H. Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO J. 2012;31:2182‐2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Deursen F, Sengupta S, De Piccoli G, Sanchez‐Diaz A, Labib K. Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO J. 2012;31:2195‐2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Looke M, Maloney MF, Bell SP. Mcm10 regulates DNA replication elongation by stimulating the CMG replicative helicase. Genes Dev. 2017;31:291‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gambus A, Jones RC, Sanchez‐Diaz A, et al. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol. 2006;8:358‐366. [DOI] [PubMed] [Google Scholar]
- 19. Douglas ME, Ali FA, Costa A, Diffley JFX. The mechanism of eukaryotic CMG helicase activation. Nature. 2018;555:265‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wasserman MR, Schauer GD, O'Donnell ME, Liu S. Replication fork activation is enabled by a single‐stranded DNA gate in CMG helicase. Cell. 2019;178:600‐611.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Techer H, Koundrioukoff S, Nicolas A, Debatisse M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet. 2017;18:535‐550. [DOI] [PubMed] [Google Scholar]
- 23. Pettitt SJ, Lord CJ. Dissecting PARP inhibitor resistance with functional genomics. Curr Opin Genet Dev. 2019;54:55‐63. [DOI] [PubMed] [Google Scholar]
- 24. Gaillard H, Garcia‐Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15:276‐289. [DOI] [PubMed] [Google Scholar]
- 25. Kotsantis P, Petermann E, Boulton SJ. Mechanisms of oncogene‐induced replication stress: jigsaw falling into place. Cancer Discov. 2018;8:537‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petropoulos M, Champeris Tsaniras S, Taraviras S, Lygerou Z. Replication licensing aberrations, replication stress, and genomic instability. Trends Biochem Sci. 2019;44:752‐764. [DOI] [PubMed] [Google Scholar]
- 27. Carruthers RD, Ahmed SU, Ramachandran S, et al. Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem‐like cells. Cancer Res. 2018;78:5060‐5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blow JJ, Ge XQ. A model for DNA replication showing how dormant origins safeguard against replication fork failure. EMBO Rep. 2009;10:406‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976‐990. [DOI] [PubMed] [Google Scholar]
- 30. Macheret M, Halazonetis TD. Intragenic origins due to short G1 phases underlie oncogene‐induced DNA replication stress. Nature. 2018;555:112‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ishiguro T, Sato A, Ohata H, et al. Establishment and characterization of an in vitro model of ovarian cancer stem‐like cells with an enhanced proliferative capacity. Cancer Res. 2016;76:150‐160. [DOI] [PubMed] [Google Scholar]
- 32. Dontu G, Al‐Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36(Suppl 1):59‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983‐3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Y, Weinberg RA. Epithelial‐to‐mesenchymal transition in cancer: complexity and opportunities. Front Med. 2018;12:361‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwab RA, Niedzwiedz W. Visualization of DNA replication in the vertebrate model system DT40 using the DNA fiber technique. J Vis Exp. 2011;56:e3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gan W, Guan Z, Liu J, et al. R‐loop‐mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011;25:2041‐2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vijayraghavan S, Tsai FL, Schwacha A. A checkpoint‐related function of the MCM replicative helicase is required to avert accumulation of RNA:DNA hybrids during S‐phase and ensuing DSBs during G2/M. PLoS Genet. 2016;12:e1006277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baxley RM, Bielinsky AK. Mcm10: a dynamic scaffold at eukaryotic replication forks. Genes (Basel). 2017;8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pawitan Y, Bjohle J, Amler L, et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population‐based cohorts. Breast Cancer Res. 2005;7:R953‐R964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Desmedt C, Piette F, Loi S, et al. Strong time dependence of the 76‐gene prognostic signature for node‐negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007;13:3207‐3214. [DOI] [PubMed] [Google Scholar]
- 41. Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2‐7 are required for human cells to survive replicative stress. Genes Dev. 2007;21:3331‐3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vesela E, Chroma K, Turi Z, Mistrik M. Common chemical inductors of replication stress: focus on cell‐based studies. Biomolecules. 2017;7(4):19. [Google Scholar]
- 43. Li Y, Atkinson K, Zhang T. Combination of chemotherapy and cancer stem cell targeting agents: preclinical and clinical studies. Cancer Lett. 2017;396:103‐109. [DOI] [PubMed] [Google Scholar]
- 44. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia‐inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci U S A. 2014;111:E5429‐E5438. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Li WM, Huang CN, Ke HL, et al. MCM10 overexpression implicates adverse prognosis in urothelial carcinoma. Oncotarget. 2016;7:77777‐77792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cui F, Hu J, Ning S, Tan J, Tang H. Overexpression of MCM10 promotes cell proliferation and predicts poor prognosis in prostate cancer. Prostate. 2018;78(16):1299‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahadevappa R, Neves H, Yuen SM, et al. DNA replication licensing protein MCM10 promotes tumor progression and is a novel prognostic biomarker and potential therapeutic target in breast cancer. Cancers (Basel). 2018;10(9):282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Das B, Pal B, Bhuyan R, et al. MYC regulates the HIF2alpha stemness pathway via Nanog and Sox2 to maintain self‐renewal in cancer stem cells versus non‐stem cancer cells. Cancer Res. 2019;79:4015‐4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Akita H, Marquardt JU, Durkin ME, et al. MYC activates stem‐like cell potential in hepatocarcinoma by a p53‐dependent mechanism. Cancer Res. 2014;74:5903‐5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Woodward AM, Gohler T, Luciani MG, et al. Excess Mcm2‐7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol. 2006;173:673‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Paulson CN, John K, Baxley RM, et al. The anti‐parasitic agent suramin and several of its analogues are inhibitors of the DNA binding protein Mcm10. Open Biol. 2019;9:190117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. An S, Fu L. Small‐molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine. 2018;36:553‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material