

Abstract
Cancer is characterized by an accumulation of somatic mutations that represent a source of neoantigens for targeting by antigen‐specific T cells. Head and neck squamous cell carcinoma (HNSCC) has a relatively high mutation burden across all cancer types, and cellular immunity to neoantigens likely plays a key role in HNSCC clinical outcomes. Immune checkpoint inhibitors (CPIs) have brought new treatment options and hopes to patients with recurrent and/or metastatic HNSCC. However, many patients do not benefit from CPI therapies, highlighting the need for novel immunotherapy or combinatorial strategies. One such approach is personalized cancer vaccination targeting tumor‐associated antigens and tumor‐specific antigens, either as single agents or in combination with other therapies. Recent advances in next‐generation genomic sequencing technologies and computational algorithms have enabled efficient identification of somatic mutation‐derived neoantigens and are anticipated to facilitate the development of cancer vaccine strategies. Here, we review cancer vaccine approaches against HNSCC, including fundamental mechanisms of a cancer vaccine, considerations for selecting appropriate antigens, and combination therapies.
Keywords: cancer vaccine therapy, clinical trial, head and neck squamous cell carcinoma, tumor‐associated antigen, tumor‐specific antigen
Many patients with head and neck squamous cell carcinoma (HNSCC) do not benefit from checkpoint inhibitor therapies, which highlights the need for novel immunotherapy or combinatorial strategies. One such approach is personalized cancer vaccination targeting tumor‐associated antigens and tumor‐specific antigens, either as single agents or in combination with other therapies. Here, we review cancer vaccine approaches against HNSCC, including fundamental mechanisms of cancer vaccine, considerations for selecting appropriate antigens, and combination therapies.
Abbreviations
- APC
antigen‐presenting cell
- cDC1
conventional type 1 dendritic cell
- cDC2
conventional type 2 dendritic cell
- CPI
immune checkpoint inhibitor
- CR
complete response
- CRT
chemoradiotherapy
- CTA
cancer testis antigen
- DC
dendritic cell
- EBNA1
Epstein‐Barr nuclear antigen 1
- EZH2
enhancer of zeste homolog 2
- HNSCC
head and neck squamous cell carcinoma
- HPV
human papillomavirus
- IFN‐γ
interferon gamma
- IL
interleukin
- indel
insertion/deletion
- LMP2
latent membrane protein 2
- LN
lymph node
- MS
mass spectrometry
- NGS
next‐generation sequencing
- OS
overall survival
- PD‐1
programmed cell death‐1
- PD‐L1
PD‐1 ligand
- PFS
progression‐free survival
- Rag
recombination activating gene
- RNA‐seq
RNA sequencing
- SLP
synthetic long peptide
- SNV
single nucleotide variant
- TAA
tumor‐associated antigen
- TAP
transporter associated with antigen processing
- TCR
T‐cell receptor
- Th1
type 1 helper T cell
- TMB
tumor mutational burden
- TSA
tumor‐specific antigen
1. INTRODUCTION
Head and neck squamous cell carcinoma, which originates in the oral cavity, pharynx, and larynx, is the most common malignant histology arising in the head and neck region. Head and neck squamous cell carcinoma is the sixth most common cancer type in the world, with 800,000 new diagnoses and 400,000 patient deaths per year. 1 Alcohol and tobacco use are the main etiologies of conventional HNSCC, and HPV is an independent risk factor. Thus, HPV‐related HNSCC is more common among patients without significant alcohol and tobacco use histories, and these patients tend to be younger. Human papillomavirus‐related HNSCC has significantly increased in recent years and is generally more responsive to CRT, with a better prognosis than HPV‐negative conventional HNSCC. 1 , 2
Head and neck squamous cell carcinoma occurs in regions essential for swallowing, breathing, and speaking and has a significant functional impact on afflicted patients. Balancing clinical outcomes and preservation of these functions through optimization of multimodal treatments, including surgery, chemotherapy, and radiotherapy, has thus been a major focus in the last 50 years. With these extended efforts, the 5‐year survival rate of patients with conventional HNSCC has improved modestly over several decades, but remains at 40%‐50% for locally advanced disease. 1 , 2
The success of cancer immunotherapy has dramatically altered the landscape of cancer treatment introducing a fourth pillar of therapy for patients. There are various kinds of immunotherapeutic treatments for cancer, such as CPIs, small molecules, vaccines, cell‐based, and cytokine therapies. Notably, in these immunotherapeutic treatment options, the revolution in cancer treatment has been through the application of CPIs, including anti‐CTLA4, anti‐PD‐1, and anti‐PD‐L1. 3 The promising effect of CPIs on various kinds of cancers also validated the existence of immunity against self‐generated cancer cells. A key feature of immunotherapy is its durability that, when successful, represses cancer relapse.
Pivotal phase III trials (CheckMate 141 and KEYNOTE 048) resulted in the approval of anti‐PD‐1 in the recurrent and/or metastatic HNSCC setting. 4 Despite the excitement surrounding this approach, only 15%‐20% of HNSCC patients benefit from CPIs, thus highlighting the development of new immunotherapeutic methods or novel combinatorial strategies as an urgent task. Across cancer types, response rates to CPIs have, at least in part, been linked to TMB. 5 The genomics analyses of HNSCC has not identified widely shared oncogenic driver mutations but did show relatively high TMB. 6 , 7 In addition, HNSCC is among the most highly immune‐infiltrated cancers. 8 These data suggest that HNSCC is potentially immunotherapy‐responsive. Despite this feature of HNSCC and early success, negative findings from two recent highly anticipated phase III trials highlight the need for additional approaches. The EAGLE trial combining anti‐CTLA4 (tremelimumab) and anti‐PD‐L1 (durvalumab) (NCT02369874) failed to meet endpoints vs standard of care. 9 The phase III JAVELIN Head and Neck 100 study (NCT02952586) asked whether the addition of anti‐PD‐L1 Ab (avelumab) to standard HNSCC CRT improved outcomes compared with CRT alone. 10 This trial was terminated early because interim analysis found it was unlikely to show a difference compared with standard of care. These data indicate the urgent need for approaches to enhance antitumor immunity in HNSCC treatment. Cancer vaccination therapy represents an important approach. In this review, we aim to discuss the mechanism of cancer vaccination therapy, recent advances in antigen selection, and the current status of clinical cancer vaccination therapy for HNSCC.
2. CANCER IMMUNOEDITING CONCEPT
Prior to discussing cancer vaccination, we briefly review the long and sometimes controversial history of defining host immune response to cancers. For several decades there was an ongoing debate about whether a cancer immune‐surveillance mechanism exists. In the 1950s, Thomas and Burnet conceived of an idea for a host immune‐surveillance system that served to prevent the widespread development of cancers. 11 , 12 Specifically, they hypothesized that the immune system would recognize and eliminate cancers that continually developed from normal tissues. However, multiple lines of experimentation embroiled this idea in controversy until experiments completed two decades ago by Schreiber, Smyth and others. 13 , 14 In particular, Schreiber definitively determined the existence of immune‐surveillance for cancer using immunodeficient genetically engineered mouse models. 15 This concept was further developed, and a new hypothesis of cancer immunoediting was proposed. 16 In this framework, cancer development can be divided into three phases: elimination, equilibrium, and escape phases. Nascent tumors undergo immunoediting before manifesting as clinically recognizable disease. In the first elimination phase, cancer cells with high‐affinity neoantigens are eliminated by the immune system. In the subsequent equilibrium phase, cancer cells that have lost strong neoantigens or have developed resistance mechanisms remain dormant in balance with the immune system. Clinically evident cancers are seen in the escape phase, having lost potent antigens. This latter concept was first illustrated by Matsushita et al, who analyzed an unedited methylcholanthrene‐induced sarcoma from Rag KO mice and found the tumor retained a highly immunogenic neoantigen. 17 Notably, when this Rag‐derived tumor was transplanted into immunocompetent WT mice, many of the tumors were rejected, but some tumors grew and formed escape tumors. Escape tumors lost a specific neoantigen present in parental tumors, consistent with the cancer immunoediting concept. Cancer vaccination therapy targets immunoedited tumors and as highly immunogenic antigens could be lost in these tumors, which antigens to use for vaccination is an important consideration.
3. MECHANISMS OF CANCER VACCINATION
The goal of cancer vaccination therapy is to increase antigen‐specific CD4+ and CD8+ T cells that recognize and eradicate tumor cells. Thus, understanding of endogenous antigen‐specific T cell responses to vaccination therapies and developing efficient methods to increase antigen‐specific effector T cells have been primary goals in current approaches.
The APCs are the central players to induce robust antigen‐specific T cell responses by vaccination therapies. Antigen‐presenting cells are specialized immune cells that are capable of taking up antigen and presenting it to naïve CD4+ and CD8+ T cells to enhance activation (Figure 1). CD8α+/CD103+ (mouse) and CD141+ (human) cDC1s are the critical APCs for cancer cell‐derived antigen presentation. 18 CD4+ naïve T cells are activated by TCR recognition of MHC‐II of cDC1s or cDC2s. 19 , 20 Interferon‐gamma and IL‐12 produced by APCs polarize the CD4+ naïve T cells into Th1 cells. The Th1 cells help prime CD8+ T cells to differentiate into effector and memory CTLs by secreting cytokines such as IFN‐γ and IL‐2, which support cellular immunity in conjunction with APCs. In addition, CD4+ T cell help induces increased expression of CD80/86 costimulatory signals and the secretion of various cytokines, including type 1 IFN, IL‐15, and IL‐12 in LN‐resident cDC1s, promoting CD8+ T cell expansion and differentiation. 19 Further details of the essential role of the CD4+ T cells in antitumor immunity have been described elsewhere. 19 , 21
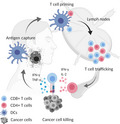
FIGURE 1.
Cancer immunity cycle. The cancer immunity cycle is composed of several phases. Tumor antigens from dying/necrotic tumor cells are captured by tissue migrating antigen‐presenting cells (APCs), in particular dendritic cells (DCs) (conventional type 1 and type 2 DCs) through phagocytosis or endocytosis. Subsequently, APCs traffic into the lymph nodes where antigen‐specific T cells are primed and T cell trafficking to the tumor is induced. Tumor‐specific CD4+/CD8+ T cells cooperatively eradicate cancer cells by recognition of cancer‐specific antigens. IFN‐γ, interferon‐gamma; IL‐2, interleukin‐2; TNF‐α, tumor necrosis factor‐α
Antigen‐presenting cells additionally present tumor antigens on MHC‐I molecules and activate CD8+ T cells through cross‐presentation (Figure 2). Costimulatory signals such as CD80/86 from DCs are essential for T cell priming. 18 CD8+ TCR engagement with MHC‐I bound peptide results in activation and differentiation into CTLs. Thus, these CTLs are the ultimate effectors in recognizing MHC‐I/peptide on cancer cells with resultant release of cytokines such as IFN‐γ/tumor necrosis factor‐α, perforin, and granzyme to eradicate cancer cells.
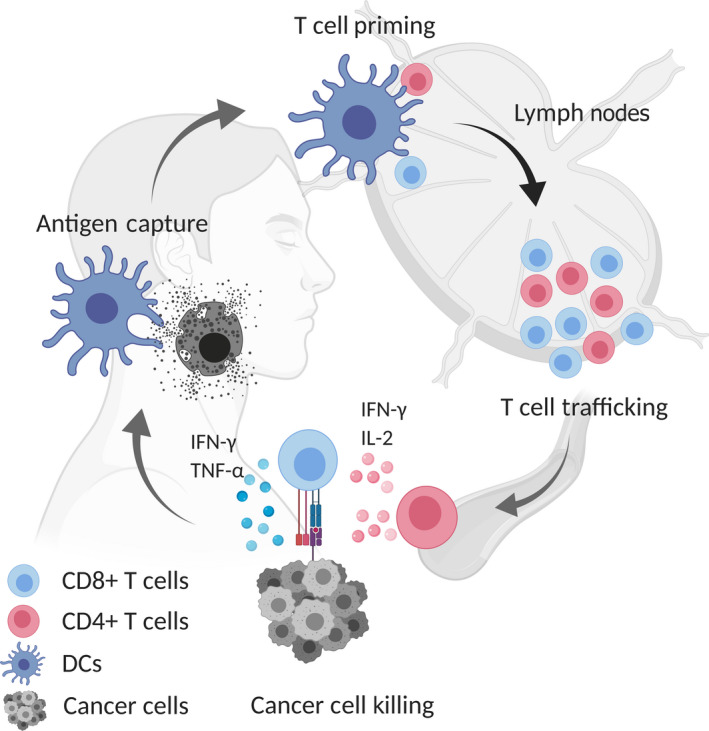
FIGURE 2.
Antigen presentation process. Tumor antigen‐derived proteins are taken up in antigen‐presenting cells by phagocytosis or endocytosis. In the MHC‐I pathway, proteins are degraded by the proteasome and enter into the endoplasmic reticulum (ER) through transporter associated with antigen processing (TAP). Subsequently, 8‐11 residues peptides are loaded onto the MHC‐I molecule and translocate to the cell surface where they could activate CD8+ T cells through cross‐presentation. In the MHC‐II pathway, endosomes or lysosomes take up tumor antigen‐derived proteins and digest them to 10‐30 (optimal 12‐16) residues peptides, which bind with MHC‐II for CD4+ T cell activation. Antigen‐specific CD4+/CD8+ T cells are activated by conventional type 1 dendritic cells (cDC1s) through T cell receptor (TCR) and costimulatory molecules. Various cytokines, including interleukin (IL)‐2 and IL‐21 from CD4+ T cells, especially type 1 helper cells, and IL‐15, IL‐12, and type I interferons from dendritic cells are produced. In part, these cytokines support differentiation and proliferation of CD8+ T cells
Recent data have highlighted dynamic epigenetically driven CD8+ T cell differentiation using viral and antigen‐specific murine tumor models. Notably, cancer vaccination therapy using lymphocytic choriomeningitis virus gp33 expressing B16 melanoma showed induction of Tcf1+PD‐1+CD8+ stem‐like T cells that were critical for tumor control. 22 These data show that cancer vaccination therapy could have an impact on T cell character in addition to increasing antigen‐specific T cells. Moreover, clinical trials of RNA‐based neoantigen vaccination for melanoma patients showed one‐third of selected neoepitopes for vaccination strengthened existing T cell responses, while two‐thirds of neoepitopes induced de novo responses, 23 indicating cancer vaccination expands the TCR clonality and provokes novel T cell responses. In agreement with these studies, a recent paper reported a phase Ib clinical trial in which combination therapy of anti‐PD‐1 and neoantigen vaccination was used in advanced melanoma, non‐small‐cell lung cancer, and bladder cancer patients. Intriguingly, patients with prolonged PFS showed increased frequency of effector memory T cells, expanded TCR clonality, and increased infiltration of TCF7+CD8+ stem‐like T cells in tumors. Notably, novel T cell responses generated after neoantigen vaccination suggested the existence of antigen spreading. Patients with antigen spreading showed prolonged PFS, indicating that this phenomenon could be involved to induce robust anticancer effect using cancer vaccination therapy. 24
4. VACCINATION ANTIGENS
The goal of cancer vaccination is to obtain anticancer effects by activating or increasing an effective CD4+/CD8+ antigen‐specific T cell response. Most commonly, vaccination can be accomplished by injecting peptides or antigen‐encoding DNA or RNA. 25 , 26 The ideal properties for vaccine peptides include cancer cell‐specific expression, high immunogenicity and, ideally, a cancer cell‐specific functional dependency. In general, the types of antigens used for treatment can be divided into TAAs and TSAs. 27 In the following section, we describe the characteristics of each antigen subtype, including those relevant for HNSCC cancer vaccination therapy.
4.1. Tumor‐associated antigens
Tumor‐associated antigens are strongly expressed in cancer cells but are retained, often weakly, in normal tissues. 28 Tumor‐associated antigens are nonmutated antigens and are thought to be applicable for a variety of cancers. However, a major concern with TAAs as vaccine targets is the possibility of inducing autoimmune toxicity in normal tissues, such as colitis, hepatitis, or rapid respiratory failure. 29 To date, many vaccination studies using TAAs have been completed without clinical efficacy, likely due to TAAs being normal tissue‐derived antigens, which are subject to central or peripheral tolerance. 30 , 31 Although some T cells that specifically bind to TAAs could remain, most TAA‐specific T cells with high‐affinity TCRs are removed during development by negative selection. 32 Cytotoxic ability and activation of T cells are thought to be associated with TCR binding affinity, 33 which may be one of the reasons why TAA vaccination is not sufficiently effective. For example, TP53 is the most frequently mutated gene in HNSCC. 34 , 35 As a result, mutant and WT TP53 tetramer proteins tend to accumulate in most HNSCC cancer tissue harboring TP53 mutations. 36 The TP53 tetramer reagents identify specific endogenous antigen‐specific T cells in HNSCCs. In a phase I clinical study, patient‐derived DCs were loaded with WT peptides then injected into inguinal LNs of HNSCC patients (NCT00404339). The results showed increased TP53‐specific T cells in 11/16 patients but IFN‐γ secretion in only 4/16 patients. 37
Cancer testis antigens are TAAs that are expressed in a variety of tumors and also show limited expression in germline tissues, including ovary or testis. As CTAs are only present in cancer cells in peripheral tissue, 38 they frequently show immunogenicity. 39 In general, germline tissues do not express MHC‐I molecules, 31 and CTAs are thought to be less likely to cause autoimmune side‐effects in normal tissues. Cancer testis antigens are known to be expressed in only a limited number of cancer types, potentially reducing the applicability of CTA‐targeted immunotherapy. 25 In the HNSCC setting, Zandberg et al completed a phase I trial involving seven patients with recurrent and/or metastatic HNSCC. They used a MAGE‐A3 vaccine and found four of seven patients with T cell and Ab response without serious side‐effects. 40 Another phase I trial (UMIN000008379) has been reported in which three kinds of CTAs were combined and injected s.c. They injected short peptides restricted to HLA‐A24 and showed that peptide‐specific CD8+ T cell responses were observed only when these peptides were injected into HLA‐A24 patients. 41 Notably, patients who responded to all three CTAs showed improved OS, suggesting that the combination of several epitopes could be useful to prolong OS.
4.2. Tumor‐specific antigens
Tumor‐specific antigens are theoretically more attractive vaccination targets due to the specificity of tumor cell expression. Tumor‐specific antigens are recognized as nonself by the immune system and are less likely to induce autoimmunity compared with TAAs. 28 , 42 Tumor‐specific antigens are classified as foreign antigens associated with viral infection or neoantigens that arise from cancer‐specific SNVs and indels.
Human papillomavirus‐related HNSCCs have dramatically increased in the last two decades, and viral‐specific proteins have been explored as targets. T cells can recognize viral antigens in HPV‐related cancers, including HNSCC and cervical cancer, 43 and HPV‐related oncoproteins such as E6 and E7, can be targeted in these cancers. 44 A pilot study (NCT00257738) that treated recurrent HNSCC patients with HPV‐16‐derived peptides in conjunction with HIV‐derived “Penetrin” peptide showed T cell responses in PBMCs from vaccinated patients. 45 Penetrin peptide is thought to allow the entire peptide to translocate through the cell membrane and into the endoplasmic reticulum and Golgi apparatus, which promotes TAP independent antigen presentation. 46 A recent phase II clinical trial (NCT02426892) combined nivolumab and HPV‐16 vaccine ISA101, which encodes nine overlapping E6 peptides and four overlapping E7 peptides. 47 In this study, ISA101 long peptide was tested in 24 patients with incurable HPV‐related cancer, of whom 22 patients had oropharyngeal cancer. The results showed that the overall response rate was 33% with the median OS of 17.5 months. 47 These promising data from combination therapy suggest that ISA101 long peptides might increase antigen‐specific CD4+/CD8+ T cells and augment the efficacy of anti‐PD‐1 treatment. Furthermore, nasopharyngeal cancer is often an Epstein‐Barr virus‐related malignancy 48 with EBNA1 and LMP2 as virus‐specific antigens. 49 In a clinical trial (NCT01147991) using modified vaccinia Ankara encoding a functionally inactive EBNA1 and LMP2 as a therapeutic vaccine, CD4+/CD8+ T cell responses were also observed. 50 , 51
Cancer‐specific somatic mutations can be classified as driver mutations that mainly contribute to cancer development and progression, and passenger mutations, which are bystander mutations that accumulate in the process of cancer development. 52 In addition, fusion‐genes, which are sometimes present and expressed in the head and neck cancers, including adenoid cystic carcinoma, have been shown to stimulate cancer immunity. 53 Despite the potential for these genomic alterations to be immune targets, clinically evident cancers will either not express or will have lost many of the potential neoantigens. The rare neoantigens that remain to be recognized by the immune system likely have either low immunogenicity or “survived” due to other escape mechanisms. Considering the diversity of HLA types in each patient, although targeting driver mutations represents an attractive approach that can also target a cancer cell dependency, only limited patients might benefit. 54 However, there is another potential benefit that shared driver mutation vaccines can be used across patients. Similar to small molecule therapies, this approach can also lead to the escape‐tumor formation. 55 In parallel, other groups have focused on using combinations of neoantigens from passenger mutations for vaccinations and showed promising results. 23 , 56 , 57 The process of selecting neoantigens from passenger mutations that are unique to individual patients is an evolving field.
5. TUMOR‐SPECIFIC ANTIGEN SELECTION ALGORITHMS
Although many groups are trying to use TSAs as neoepitope candidates, it is still challenging to predict them accurately. The classical cDNA library screening approach is labor‐intensive and ineffective in detecting some TSAs from GC‐rich or low expression transcripts. 58 The revolution in cancer genomics has made it possible to predict actionable neoepitopes with high probability by combining NGS data (whole exome and RNA‐seq) with in silico analysis. 59 However, many false‐positive candidates are reported using only in silico prediction. 60 A few reasons for this include: (i) less than accurate consideration of the numerous transcriptional and posttranscriptional events that regulate antigen processing and presentation; 61 (ii) the position of certain mutations within the epitope that could function as an anchor for MHC molecules; 62 and (iii) some mutations in neoepitopes alter TCR structural interaction. 63 Together, these and other factors highlight bona fide neoepitope prediction from in silico analysis as still an imperfect approach.
Analysis of presented peptides on cancer cell MHC molecules with MS is considered by many to be the most reliable technique to identify neoepitopes. A typical schema to detect neoepitopes is shown in Figure 3. Ebrahimi‐Nik et al 63 suggested that the combination of genomics, unbiased discovery MS immune‐peptidomics, and targeted MS was useful to detect neoepitopes that elicit actual tumor rejection. Notably, they found that some of these neoepitopes showed low‐affinity binding to MHC‐I and could be missed by in silico analysis, suggesting the high specificity and robustness of MS. 63 However, MS neoantigen identification is not widely accessible, requires synthesis of candidate peptides, and is time‐intensive. Thus, new methods of narrowing down potential neoepitopes and reducing the time of the identification process are essential, especially in the context of clinical vaccine development where initiation of therapy is critical. To address this latter issue, Chen et al 54 proposed two strategies to accelerate neoantigen identification: (i) targeted sequencing of cancer‐related gene mutations; and (ii) building an inventory of shared neoantigen peptide libraries of common solid tumors. They showed that these methods were effective in narrowing down the number of neoepitope candidates and also contributed to reducing the time from prediction to patient vaccination. Importantly, current approaches primarily focus on SNVs to detect TSAs, but there are numerous indel events in cancer development that also yield TSAs. Recently, MS data identified neoantigen candidates from noncanonical reading frames, including introns, 5′‐UTRs, 3′‐UTRs, and noncoding RNAs. 64 Considering that exons occupy only approximately 2% of the genome and 75% of the remaining region can be potentially translated, current in silico algorithms might miss the detection of many useful neoepitopes. 60 However, harvesting a sufficient amount of high‐quality tumor tissues to perform MS is often difficult in the clinical setting. In addition, little is known about the long‐time harvesting effect on proteomics, especially in surgery‐derived clinical samples. 65 Needle biopsies can overcome these complications but might not be able to process sufficient tissues. The establishment of an efficient procedure to carry out MS should accelerate identification of neoantigens.
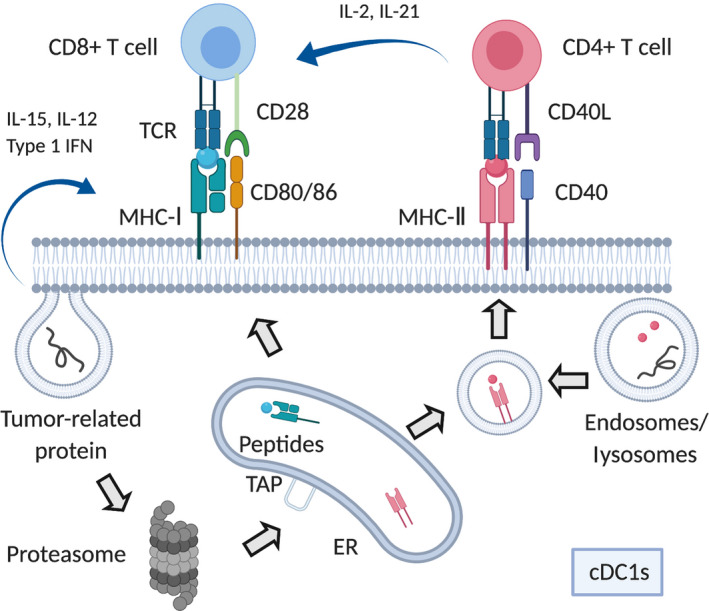
FIGURE 3.
Pipeline for neoantigen identification. The sequence data (whole exome and RNA sequencing data) identifies neoantigen candidates by single nucleotide variants or insertions/deletions. Gene expression data allows narrowing down of expressed candidates. In silico analysis is then carried out with prediction software. In most cases, high‐affinity (IC50 < 500 nmol/L) candidates are selected. Mass spectrometry is an approach to validate candidates based on MHC bound peptides from tumor/cell lysis extraction. Functional analysis including FACS, ELISA, and ELISPOT further validate neoantigen candidates. After the integration of information, peptides/DNA/RNA vaccines are synthesized
6. VACCINATION PLATFORMS
6.1. Peptide vaccine
Peptide vaccination is generally thought to be safe and represents the most common technology. However, the major limitation is the time required for vaccine generation (2‐3 months), which is relevant in the context of a patient with metastatic disease and limited lifespan. Many clinical trials using peptide vaccination have been completed. According to these trials, it is becoming clear that single epitope vaccination only could lead to escape‐tumor formation due to the tumor heterogeneity. 66 Short peptides under 11 amino acids do not require processing by APCs and can efficiently bind to MHC‐I of nucleated cells as epitopes. However, when T cells are stimulated by nucleated cells other than professional APCs without costimulatory factors, anergy or a dysfunctional state could be induced. 67 Synthetic long peptides, ranging from 20 to 35 amino acids, are efficiently processed by DCs and can be presented on both MHC‐I and MHC‐II molecules with costimulatory factors. Thus, SLPs are suggested to be able to activate both CD4+ and CD8+ T cells in a balanced manner of they include strong MHC‐I and MHC‐II epitopes. As a result, robust T cell priming and induction of memory T cells lead to a robust therapeutic effect. 68 , 69 , 70
A clinical trial identified neoantigens from six patients with advanced melanoma, and vaccinated these patients with up to 20 distinct neoantigen peptides. As a result, an increase of neoantigen‐specific CD4+/CD8+ T cells in the patients’ PBMCs was detected, and CR were observed in four cases. Two cases, who showed progressive disease and were treated with anti‐PD‐1 therapy after vaccination therapy, finally achieved CR. 57 These results indicate that multiple neoantigens could prevent the development of escape tumors. Notably, the neoantigen‐specific T cells, which were increased by vaccination therapy, could be augmented with subsequent anti‐PD‐1 treatment. Although preliminary and requiring trial validation, this finding is promising for combining cancer vaccination and CPIs. In an HNSCC preclinical study using the mouse oral carcinoma syngeneic mouse cancer cell lines, our group detected several neoantigen candidates by combining NGS data with in silico analysis. Validation of candidates indicated prophylactic vaccination with mutant ICAM1 neoantigen‐derived SLP induced significant T cell response and robust tumor suppression. 71 Given that HNSCC has relatively high TMB, 6 , 7 cancer vaccination therapy using neoantigen‐derived SLPs represents a promising method to treat HNSCC. 72
6.2. DNA vaccine
One of the advantages of using nucleic acid‐derived vaccines is that DNA and RNA vaccines can be synthesized easier and faster than peptide vaccines. DNA vaccines, made from bacterial plasmids encoding antigens, are often used with immune‐stimulatory molecules such as IL‐2 and granulocyte/macrophage colony‐stimulating factor. The advantages of DNA vaccines are that they are relatively stable, can be presented by MHC‐I and MHC‐II of APCs, and could activate CD4+ and CD8+ T cells. 73 In addition, plasmid DNA can also activate innate immune responses by double‐stranded DNA structure recognition. 74 DNA vaccines have to penetrate not only the cell membrane but also the nuclear membrane to be effective. Therefore, many methods have attempted to improve the efficiency of nuclear delivery, such as electroporation and a “gene gun” technique. 75 Although there were some early concerns about genomic integration of DNA vaccines, no evidence supporting this has emerged. 76
6.3. RNA vaccine
The benefits of RNA vaccines is that they do not need to penetrate the nuclear membrane and can function when they are delivered into the cytoplasm of APCs. 26 Although RNA is thought to be unstable compared to DNA, manufactured RNA vaccines are relatively stable. 77 In a phase I trial in advanced melanoma patients, RNA epitopes that encoded four different endogenous self‐antigens were given i.v. in liposomal complex formation. The RNA‐liposomal complex was effectively taken up by DCs, and antigen‐specific T cell responses were observed. 78 Another recent study reported that RNA‐based polyneoepitope vaccination induced a significant reduction in the cumulative event of metastasis and sustained PFS. This group undertook neoantigen‐derived RNA‐based vaccination on melanoma patients and obtained CRs in some patients. 23
7. CLINICAL VACCINATION STUDIES IN HNSCC
A systematic search was carried out to identify clinical trials related to the keywords “head and neck cancer” and “vaccine” from ClinicalTrials.gov or PubMed. We included clinical trials from 1995 to the present. As shown in Table 1, there are 17 completed trials, some of which were discussed above. There are also 12 pre/active trials, which include six HPV‐antigen related vaccine trials (NCT03821272, NCT02002182, NCT02865135, NCT03260023, NCT03418480, and NCT04369937), three TAA‐related vaccine trials (NCT02544880, NCT04247282, and NCT0368919), one cellular vaccine trial (NCT02999646), and two neoantigen vaccine trials (NCT03568058 and NCT04266730).
TABLE 1.
Cancer vaccination clinical trials for head and neck squamous cell carcinoma
| Vaccine type/target antigens | Adjuvants/other therapy | Type | Phase | Identifier | Period | Reference |
|---|---|---|---|---|---|---|
| Pre/active trials | ||||||
| ADXS11‐001 (HPV 16‐E6/E7) | Viral Ag | II | NCT02002182 | Dec 2013‐Aug 2022 | 82 | |
| MUC1 peptide | PDE5 inhibitor | TAA | I/II | NCT02544880 | Apr 2016‐Jun 2020 | 83 |
| DPX‐E7 (HPV 16‐E7 11‐19 nanomer) | Cyclophosphamide | Viral Ag | I | NCT02865135 | Dec 2016‐Dec 2020 | |
| MVX‐ONCO1 (irradiated tumor cell lysate) | GM‐CSF | Cellular | II | NCT02999646 | Jun 2017‐Dec 2024 | |
| TG4001 (HPV 16‐E6/E7 coded vector) | Avelumab | Viral Ag | I/II | NCT03260023 | Sep 2017‐May 2021 | |
| E6/E7 RNA | Anti‐CD40 | Viral Ag | I/II | NCT03418480 | Apr 2017‐Dec 2020 | |
| Tumor‐derived neoantigens | Anti‐PD‐1 | TSA | I | NCT03568058 | Jul 2018‐Aug 2023 | |
| Arginase1 peptide | Montanide ISA‐51 | TAA | I | NCT03689192 | Dec 2018‐Jun 2021 | |
| PepCan (HPV‐16 E6 peptides) | Viral Ag | I/II | NCT03821272 | Nov 2019‐Sep 2021 | ||
| Autologous tumor antigen (TriAD) | Anti‐PD‐L1, TGF‐β, GM‐CSF | TAA | I/II | NCT04247282 | Apr 2020‐Dec 2021 | |
| PANDA‐VAC (neoantigen peptides) | PolyICLC, pembrolizumab | TSA | I | NCT04266730 | Sep 2020 | |
| HPV‐16 E6/E7 peptides | Cisplatin‐IMRT, pembrolizumab | Viral Ag | II | NCT04369937 | May 2020‐Jun 2022 | |
| Completed | ||||||
| HPV‐16 E6/E7 peptides | Viral Ag | I | NCT00019110 | Nov 1995 | ||
| Ras protein | IL‐2, sargramostim | TAA | II | NCT00019331 | Oct 1997‐May 2007 | |
| B7‐1, ICAM1, LFA‐1 transgenes (TRICOM) | TAA | I | NCT00021424 | Jun 2001 | ||
| TRICOM‐CEA peptide | TAA | I | NCT00027534 | January 2002‐October 2007 | ||
| MAGE‐A3/ HPV‐16 peptides | GM‐CSF, IFA | TAA/viral Ag | I | NCT00257738 | Nov 2005‐Oct 2012 | 40 |
| Modified p53 pulsed dendritic cells | Th tetanus toxoid | TAA | I | NCT00404339 | Mar 2011‐Mar 2014 | 37 |
| MVA‐EL (EBV‐EBNA/LMP2) | Viral Ag | I | NCT01147991 | Mar 2005‐Apr 2011 | 50, 51 | |
| VicOryx trial (p16 peptides) | Montanide ISA‐51 | TAA | I/II | NCT01462838 | Aug 2011‐May 2015 | |
| AlloVAX: chaperone rich cell lysate | AlloStim (adjuvant) | Cellular | II | NCT01998542 | Jan 2016‐Nov 2017 | |
| HPV‐E7 | Surgery (TOVS) | Viral Ag | II | NCT02002182 | Dec 2013‐Aug 2019 | |
| MEDI‐0457 (HPV16/18‐E6/E7) | Cisplatin or surgery | Viral Ag | I/II | NCT02163057 | Jun 2014‐Nov 2017 | |
| HPV‐E7 | Durvalumab | Viral Ag | I/II | NCT02291055 | Apr 2015‐Dec 2019 | |
| ISA 101 (HPV16‐E6/E7) | Nivolumab | Viral Ag | II | NCT02426892 | Dec 2015‐Dec 2016 | 47 |
| VicOryx‐2 trial (p16 peptides) | Montanide ISA‐51, cisplatin | TAA | I | NCT02526316 | Jun 2015‐May 2017 | |
| MEDI‐0457 (HPV16/18‐E6/E7) | Durvalumab | Viral Ag | I/II | NCT03162224 | Jun 2017‐Aug 2019 | |
| Survivin‐2B peptide | TAA | I | UMIN000000976 | Sep 2003‐Jul 2006 | 84 | |
| LY6K, CDCA1, IMP3 peptides | IFA | TAA | I/II | UMIN000008379 | Dec 2008 | 41 |
Abbreviations: Ag, antigen; GM‐CSF, granulocyte/macrophage colony‐stimulating factor; HPV, human papillomavirus; IL, interleukin; IMRT, intensity‐modulated radiation therapy; PD‐1, programmed cell death‐1; PD‐L1, PD‐1 ligand; PDE5, phosphodiesterase‐5; polyICLC, polyinosinic‐polycytidylic acid mixed with the stabilizers carboxymethylcellulose and polylysine; TAA, tumor‐associated antigen; TGF‐β, transforming growth factor‐β; TOVS, trans oval videolaryngoscopic surgery; TSA, tumor‐specific antigen.
NCT03568058 is a phase Ib trial examining the combination therapy of personalized cancer vaccination and anti‐PD‐1 (pembrolizumab) for adult patients with advanced cancers, including HNSCC. Vaccine candidates will be selected from tumor‐derived neoantigens. The NCT04266730 phase I trial includes patients with squamous lung cancer, non‐small‐cell lung cancer, or HNSCC, who showed stable disease or nonthreatening progressive disease after anti‐PD‐1 or anti‐PD‐L1 treatment. Patients in this trial, termed “personalized and adjusted neoantigen peptide vaccine” (PANDA‐VAC), will be vaccinated with six neoantigens concurrently with pembrolizumab. Neoepitopes for each patient will be selected with in silico analysis using NGS data (Exome, RNA‐seq).
8. COMBINATION CONSIDERATIONS: CANCER VACCINATION WITH OTHER THERAPIES
As discussed above, combination therapies of cancer vaccination with CPIs are promising because cancer vaccination could augment the effects of CPIs by increasing antigen‐specific T cells. 24 , 47 , 57 Notably, Zhou et al showed that the inhibitor of EZH2 promoted antigen presentation by epigenetically repressing H3K27me3, especially in the β2‐microglobulin promoter region in HNSCC. 79 These data show EZH2 might promote cancer cell recognition and killing by antigen‐specific T cells and suggest that combination therapy with EZH2 and cancer vaccination represents a promising approach. In addition, several lines of evidence show that CD4+ T cells are important to eliciting a strong vaccination effect. 21 , 23 , 57 , 80 Peptide vaccination in conjunction with Toll‐like receptor agonist and OX40/CD40 stimulation effectively induces robust antitumor CD4+ T cell response. 81 Thus, EZH2 targeting and Toll‐like receptor/OX40/CD40 combinations are some of several approaches to strengthen vaccination efficacy.
9. CLOSING COMMENTS
Recent remarkable advances in genomics and cancer immunology have highlighted that cancer vaccination could represent a personalized and effective cancer treatment approach. As HNSCC is a cancer type with relatively high TMB, cancer vaccination therapy might be applicable for difficult‐to‐treat HNSCCs. Notably, recent progress in various techniques including NGS and MS has enabled the detection of strong neoepitopes more efficiently than before. Both fundamental understanding of cancer vaccines using preclinical models and studies of outcomes in clinical settings both contribute to further progress in this field. Appropriately designed neoantigen vaccine treatments are highly personalized therapy and might be able to address tumor heterogeneity in each patient. However, obstacles remain, including efficiently detecting neoepitopes and delivering the vaccine. The rapid pace of developments in this field is addressing these challenges and will ultimately change the landscape of current HNSCC therapy.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
ACKNOWLEDGMENTS
Figures were created with BioRender.com. RU is funded by NIH/NIDCR R01DE024403, R01DE027736, and NIH/NCI/NIDCR U01DE029188. HS received funding from the Uehara Foundation.
Shibata H, Zhou L, Xu N, Egloff AM, Uppaluri R. Personalized cancer vaccination in head and neck cancer. Cancer Sci. 2021;112:978–988. 10.1111/cas.14784
Funding information
RU is funded by NIH/NIDCR R01DE024403, R01DE027736, and NIH/NCI/NIDCR U01DE029188. HS received funding from the Uehara Foundation.
Contributor Information
Hirofumi Shibata, Email: hirofumi_shibata@dfci.harvard.edu.
Ravindra Uppaluri, Email: ravindra_uppaluri@dfci.harvard.edu.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Chaturvedi AK, Anderson WF, Lortet‐Tieulent J, et al. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J Clin Oncol. 2013;31:4550‐4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56‐61. [DOI] [PubMed] [Google Scholar]
- 4. Ferris RL, Blumenschein G, Fayette J, et al. Nivolumab for recurrent squamous‐cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856‐1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mandal R, Şenbabaoğlu Y, Desrichard A, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1:e89829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferris RL, Haddad R, Even C, et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: EAGLE, a randomized, open‐label phase III study. Ann Oncol. 2020;31:942‐950. [DOI] [PubMed] [Google Scholar]
- 10. Yu Y, Lee NY. JAVELIN head and neck 100: a phase III trial of avelumab and chemoradiation for locally advanced head and neck cancer. Future Oncol. 2019;15:687‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1‐27. [DOI] [PubMed] [Google Scholar]
- 12. Thomas L. On immunosurveillance in human cancer. Yale J Biol Med. 1982;55:329‐333. [PMC free article] [PubMed] [Google Scholar]
- 13. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137‐148. [DOI] [PubMed] [Google Scholar]
- 14. Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1‐50. [DOI] [PubMed] [Google Scholar]
- 15. Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107‐1111. [DOI] [PubMed] [Google Scholar]
- 16. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991‐998. [DOI] [PubMed] [Google Scholar]
- 17. Matsushita H, Vesely MD, Koboldt DC, et al. Cancer exome analysis reveals a T‐cell‐dependent mechanism of cancer immunoediting. Nature. 2012;482:400‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7‐24. [DOI] [PubMed] [Google Scholar]
- 19. Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635‐647. [DOI] [PubMed] [Google Scholar]
- 20. Ferris ST, Durai V, Wu R, et al. cDC1 prime and are licensed by CD4(+) T cells to induce anti‐tumour immunity. Nature. 2020;584:624‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alspach E, Lussier DM, Miceli AP, et al. MHC‐II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siddiqui I, Schaeuble K, Chennupati V, et al. Intratumoral Tcf1(+)PD‐1(+)CD8(+) T cells with stem‐like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50:195‐211.e10. [DOI] [PubMed] [Google Scholar]
- 23. Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly‐specific therapeutic immunity against cancer. Nature. 2017;547:222‐226. [DOI] [PubMed] [Google Scholar]
- 24. Ott PA, Hu‐Lieskovan S, Chmielowski B, et al. A Phase Ib trial of personalized neoantigen therapy plus anti‐PD‐1 in patients with advanced melanoma, non‐small cell lung cancer, or bladder cancer. Cell. 2020;183:347‐362.e24. [DOI] [PubMed] [Google Scholar]
- 25. Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17:569. [DOI] [PubMed] [Google Scholar]
- 26. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aldous AR, Dong JZ. Personalized neoantigen vaccines: a new approach to cancer immunotherapy. Bioorg Med Chem. 2018;26:2842‐2849. [DOI] [PubMed] [Google Scholar]
- 28. Ward JP, Gubin MM, Schreiber RD. The role of neoantigens in naturally occurring and therapeutically induced immune responses to cancer. Adv Immunol. 2016;130:25‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pan R‐Y, Chung W‐H, Chu M‐T, et al. Recent development and clinical application of cancer vaccine: targeting neoantigens. J Immunol Res. 2018;2018:4325874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Melero I, Gaudernack G, Gerritsen W, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11:509‐524. [DOI] [PubMed] [Google Scholar]
- 31. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14:135‐146. [DOI] [PubMed] [Google Scholar]
- 32. Stone JD, Harris DT, Kranz DM. TCR affinity for p/MHC formed by tumor antigens that are self‐proteins: impact on efficacy and toxicity. Curr Opin Immunol. 2015;33:16‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tian S, Maile R, Collins EJ, Frelinger JA. CD8+ T cell activation is governed by TCR‐peptide/MHC affinity, not dissociation rate. J Immunol. 2007;179:2952‐2960. [DOI] [PubMed] [Google Scholar]
- 34. Cancer Genome Atlas Network . Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sirianni N, Ha PK, Oelke M, et al. Effect of human papillomavirus‐16 infection on CD8+ T‐cell recognition of a wild‐type sequence p53264–272 peptide in patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2004;10:6929‐6937. [DOI] [PubMed] [Google Scholar]
- 37. Schuler PJ, Harasymczuk M, Visus C, et al. Phase I dendritic cell p53 peptide vaccine for head and neck cancer. Clin Cancer Res. 2014;20:2433‐2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Old LJ. Cancer is a somatic cell pregnancy. Cancer Immun. 2007;7:19. [PMC free article] [PubMed] [Google Scholar]
- 39. Andersen RS, Thrue CA, Junker N, et al. Dissection of T‐cell antigen specificity in human melanoma. Cancer Res. 2012;72:1642‐1650. [DOI] [PubMed] [Google Scholar]
- 40. Zandberg DP, Rollins S, Goloubeva O, et al. A phase I dose escalation trial of MAGE‐A3‐ and HPV16‐specific peptide immunomodulatory vaccines in patients with recurrent/metastatic (RM) squamous cell carcinoma of the head and neck (SCCHN). Cancer Immunol Immunother. 2015;64:367‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoshitake Y, Fukuma D, Yuno A, et al. Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clin Cancer Res. 2015;21:312‐321. [DOI] [PubMed] [Google Scholar]
- 42. Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125:3413‐3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lorenz FKM, Ellinger C, Kieback E, et al. Unbiased identification of T‐cell receptors targeting immunodominant peptide‐MHC complexes for T‐cell receptor immunotherapy. Hum Gene Ther. 2017;28:1158‐1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92:709‐720. [DOI] [PubMed] [Google Scholar]
- 45. Voskens CJ, Sewell D, Hertzano R, et al. Induction of MAGE‐A3 and HPV‐16 immunity by Trojan vaccines in patients with head and neck carcinoma. Head Neck. 2012;34:1734‐1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu J, Wettstein PJ, Higashimoto Y, Appella E, Celis E. TAP‐independent presentation of CTL epitopes by Trojan antigens. J Immunol. 2001;166:7063‐7071. [DOI] [PubMed] [Google Scholar]
- 47. Massarelli E, William W, Johnson F, et al. Combining immune checkpoint blockade and tumor‐specific vaccine for patients with incurable human papillomavirus 16‐related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5:67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chan KCA, Woo JKS, King A, et al. Analysis of plasma Epstein‐Barr virus DNA to screen for nasopharyngeal cancer. N Engl J Med. 2018;378:973. [DOI] [PubMed] [Google Scholar]
- 49. Fox CP, Haigh TA, Taylor GS, et al. A novel latent membrane 2 transcript expressed in Epstein‐Barr virus‐positive NK‐ and T‐cell lymphoproliferative disease encodes a target for cellular immunotherapy. Blood. 2010;116:3695‐3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hui EP, Taylor GS, Jia H, et al. Phase I trial of recombinant modified vaccinia ankara encoding Epstein‐Barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013;73:1676‐1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taylor GS, Jia H, Harrington K, et al. A recombinant modified vaccinia ankara vaccine encoding Epstein‐Barr Virus (EBV) target antigens: a phase I trial in UK patients with EBV‐positive cancer. Clin Cancer Res. 2014;20:5009‐5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pon JR, Marra MA. Driver and passenger mutations in cancer. Annu Rev Pathol. 2015;10:25‐50. [DOI] [PubMed] [Google Scholar]
- 53. Yang W, Lee K‐W, Srivastava RM, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25:767‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen F, Zou Z, Du J, et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J Clin Invest. 2019;129:2056‐2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tran E, Robbins PF, Lu Y‐C, et al. T‐cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375:2255‐2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu YC, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol. 2016;28:22‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan‐4.0: improved peptide‐MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. 2017;199:3360‐3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ehx G, Perreault C. Discovery and characterization of actionable tumor antigens. Genome Med. 2019;11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pearson H, Daouda T, Granados DP, et al. MHC class I‐associated peptides derive from selective regions of the human genome. J Clin Invest. 2016;126:4690‐4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Capietto A‐H, Jhunjhunwala S, Pollock SB, et al. Mutation position is an important determinant for predicting cancer neoantigens. J Exp Med. 2020;4:e20190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ebrahimi‐Nik H, Michaux J, Corwin WL, et al. Mass spectrometry driven exploration reveals nuances of neoepitope‐driven tumor rejection. JCI Insight. 2019;5:e129152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Laumont CM, Daouda T, Laverdure J‐P, et al. Global proteogenomic analysis of human MHC class I‐associated peptides derived from non‐canonical reading frames. Nat Commun. 2016;7:10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Macklin A, Khan S, Kislinger T. Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clin Proteomics. 2020;17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cecco S, Muraro E, Giacomin E, et al. Cancer vaccines in phase II/III clinical trials: state of the art and future perspectives. Curr Cancer Drug Targets. 2011;11:85‐102. [DOI] [PubMed] [Google Scholar]
- 67. Hailemichael Y, Dai Z, Jaffarzad N, et al. Persistent antigen at vaccination sites induces tumor‐specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med. 2013;19:465‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang H, Hong H, Li D, et al. Comparing pooled peptides with intact protein for accessing cross‐presentation pathways for protective CD8+ and CD4+ T cells. J Biol Chem. 2009;284:9184‐9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Janssen EM, Droin NM, Lemmens EE, et al. CD4+ T‐cell help controls CD8+ T‐cell memory via TRAIL‐mediated activation‐induced cell death. Nature. 2005;434:88‐93. [DOI] [PubMed] [Google Scholar]
- 70. Rosalia RA, Quakkelaar ED, Redeker A, et al. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T‐cell activation. Eur J Immunol. 2013;43:2554‐2565. [DOI] [PubMed] [Google Scholar]
- 71. Zolkind P, Przybylski D, Marjanovic N, et al. Cancer immunogenomic approach to neoantigen discovery in a checkpoint blockade responsive murine model of oral cavity squamous cell carcinoma. Oncotarget. 2018;9:4109‐4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hanna GJ, Adkins DR, Zolkind P, Uppaluri R. Rationale for neoadjuvant immunotherapy in head and neck squamous cell carcinoma. Oral Oncol. 2017;73:65‐69. [DOI] [PubMed] [Google Scholar]
- 73. Lopes A, Vandermeulen G, Preat V. Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res. 2019;38:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ori D, Murase M, Kawai T. Cytosolic nucleic acid sensors and innate immune regulation. Int Rev Immunol. 2017;36:74‐88. [DOI] [PubMed] [Google Scholar]
- 75. Lambricht L, Lopes A, Kos S, Sersa G, Preat V, Vandermeulen G. Clinical potential of electroporation for gene therapy and DNA vaccine delivery. Expert Opin Drug Deliv. 2016;13:295‐310. [DOI] [PubMed] [Google Scholar]
- 76. Liu MA. A comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines. 2019;7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Stitz L, Vogel A, Schnee M, et al. A thermostable messenger RNA based vaccine against rabies. PLoS Negl Trop Dis. 2017;11:e0006108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kranz LM, Diken M, Haas H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396‐401. [DOI] [PubMed] [Google Scholar]
- 79. Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents anti‐PD‐1 resistance in head and neck cancer. Clin Cancer Res. 2020;26:290‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sahin U, Oehm P, Derhovanessian E, et al. An RNA vaccine drives immunity in checkpoint‐inhibitor‐treated melanoma. Nature. 2020;585:107‐112. [DOI] [PubMed] [Google Scholar]
- 81. Kumai T, Lee S, Cho H‐I, et al. Optimization of peptide vaccines to induce robust antitumor CD4 T‐cell responses. Cancer Immunol Res. 2017;5:72‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus‐associated cancers ‐ novel immunologic vaccines: ADXS11–001. Gynecol Oncol Res Pract. 2017;4. 10.1186/s40661-017-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Weed DT, Zilio S, Reis IM, et al. The reversal of immune exclusion mediated by Tadalafil and an anti‐tumor vaccine also induces pdl1 upregulation in recurrent head and neck squamous cell carcinoma: interim analysis of a phase I clinical trial. Frontiers in Immunology. 2019;10. 10.3389/fimmu.2019.01206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Miyazaki A, Kobayashi J, Torigoe T, et al. Phase I clinical trial of survivin‐derived peptide vaccine therapy for patients with advanced or recurrent oral cancer. Cancer Sci. 2011;102:324‐329. [DOI] [PubMed] [Google Scholar]