

Abstract
Genetic alterations in adult T‐cell leukemia/lymphoma (ATLL), a T‐cell malignancy associated with HTLV‐1, and their clinical impacts, especially from the perspective of viral strains, are not fully elucidated. We employed targeted next‐generation sequencing and single nucleotide polymorphism array for 89 patients with ATLL in Okinawa, the southernmost islands in Japan, where the frequency of HTLV‐1 tax subgroup‐A (HTLV‐1‐taxA) is notably higher than that in mainland Japan, where most ATLL cases have HTLV‐1‐taxB, and compared the results with previously reported genomic landscapes of ATLL in mainland Japan and the USA. Okinawan patients exhibited similar mutation profiles to mainland Japanese patients, with frequent alterations in TCR/NF‐ĸB (eg, PRKCB, PLCG1, and CARD11) and T‐cell trafficking pathways (CCR4 and CCR7), in contrast with North American patients who exhibited a predominance of epigenome‐associated gene mutations. Some mutations, especially GATA3 and RHOA, were detected more frequently in Okinawan patients than in mainland Japanese patients. Compared to HTLV‐1‐taxB, HTLV‐1‐taxA was significantly dominant in Okinawan patients with these mutations (GATA3, 34.1% vs 14.6%, P = .044; RHOA, 24.4% vs 6.3%, P = .032), suggesting the contribution of viral strains to these mutation frequencies. From a clinical viewpoint, we identified a significant negative impact of biallelic inactivation of PRDM1 (P = .027) in addition to the previously reported PRKCB mutations, indicating the importance of integrated genetic analysis. This study suggests that heterogeneous genetic abnormalities in ATLL depend on the viral strain as well as on the ethnic background. This warrants the need to develop therapeutic interventions considering regional characteristics.
Keywords: adult T‐cell leukemia/lymphoma, geographical mutation heterogeneity, human T‐cell leukemia virus type I, integrated clinico‐genetic analysis, tax subgroup
Targeted next‐generation sequencing and single nucleotide polymorphism array were applied to analyze aggressive adult T‐cell leukemia/lymphoma in Okinawa, which were not included in prior genomic studies. Our results showed that HTLV‐1 tax subgroup‐A was associated with high alteration frequencies in GATA3 and RHOA. Clinically, biallelic alterations, not heterozygous deletions or mutations, of PRDM1 were significantly associated with poor prognosis.
![]()
Abbreviations
- ATLL
adult T‐cell leukemia/lymphoma
- CNA
copy number alternation
- HAM
HTLV‐1‐associated myelopathy
- HBZ
HTLV‐1 bZIP
- HTLV‐1
human T‐cell leukemia virus type 1
- HTLV‐1‐taxA
HTLV‐1 tax subgroup‐A
- HTLV‐1‐taxB
HTLV‐1 tax subgroup‐B
- MST
median survival time
- OS
overall survival
- SNP
single nucleotide polymorphism
1. INTRODUCTION
Human T‐cell leukemia virus type I (HTLV‐1) is a retrovirus associated with adult T‐cell leukemia/lymphoma (ATLL), 1 , 2 , 3 , 4 a distinct peripheral T‐cell malignancy. ATLL is classified into four clinical subtypes, namely acute, lymphoma, chronic, and smoldering. 5 Patients with the former two subtypes, which are referred to as aggressive ATLLs, show very poor prognosis, with a median survival time (MST) of approximately 1 year, even if they receive combined chemotherapy. 6 On the contrary, patients with the latter two types, referred to as indolent ATLLs, show relatively long‐term survival without treatment. 7
HTLV‐1 carriers are endemically distributed worldwide, mainly in Central Africa, South America, the Caribbean coast, Melanesia, and Japan. 8 Phylogenetic variations in HTLV‐1 are related to geography and/or ethnicity. 8 The polymorphism of the HTLV‐1 tax gene, encoding the viral oncoprotein, Tax, 9 is closely associated with subgrouping. HTLV‐1 tax subgroup‐A (HTLV‐1‐taxA) and tax subgroup‐B (HTLV‐1‐taxB) are prevalent in Japan. 10 These two viral strains differ in two amino acids in Tax and one amino acid in HTLV‐1 bZIP factor (HBZ), 10 another viral oncoprotein on the antisense strand. 11 Infection with HTLV‐1‐taxA increases the risk of HTLV‐1‐associated myelopathy (HAM), an inflammatory disease caused by HTLV‐1, compared with HTLV‐1‐taxB infection. 10 These differences in viral strains have been described as affecting the biological characteristics of infected T‐cells, such as different expression levels of several genes, including HBZ and FOXP3. 12 , 13 However, the effect of variations in the virus on the biological characteristics of ATLL tumor cells remains to be elucidated.
The Okinawa islands, located in the southernmost area off mainland Japan, are one of the largest endemic areas for HTLV‐1. The germline genomic background of Okinawa residents has been reported to differ from that of the mainland Japanese population. 14 , 15 We previously reported that HTLV‐1‐taxA and HTLV‐1‐taxB had similar frequencies in Okinawa (taxA = 44%, taxB = 56%) 16 in contrast to the extremely higher frequency of HTLV‐1‐taxB in mainland Japan (taxA = 11%, taxB = 89%). 10
Although previous studies have identified numerous genetic aberrations in ATLL, 17 , 18 , 19 , 20 , 21 there have been few studies that analyzed their clinical significance. 20 , 22 Recent studies conducted in North America and Japan suggest regional or racial heterogeneity in the genetic profiles of patients with ATLL and thereby imply that the outcome of a therapeutic intervention may vary among different regions or races. 19 , 20 Based on these reports, we hypothesized that phylogenetic variations in HTLV‐1 contribute to the geographically heterogeneous genetic profiles of ATLL and their clinical impacts. In this study, we employed targeted next‐generation sequencing and a single nucleotide polymorphism (SNP) array to unravel the genetic alteration profile of aggressive ATLL in Okinawa, which was not included in prior studies, 17 , 18 , 19 , 20 , 21 , 22 and to determine how it is associated with the tax subgroup and clinical outcome. In addition, we compared the frequency of genetic alterations among patients in our cohort with those in prior studies conducted in mainland Japan and North America to identify geographical variations of the genetic landscape of ATLL. The results of this study indicate that viral strains might be associated with the heterogeneity of genetic alterations in ATLL.
2. MATERIALS AND METHODS
2.1. Materials and patients
We used genomic DNA samples from 89 patients with aggressive ATLL (64 acute type, 12 lymphoma type, 2 chronic type with poor prognosis, and 11 indolent form with disease progression) in Okinawa, diagnosed between 2013 and 2017 (Table S1). Monoclonal integrations of HTLV‐1 provirus were observed in all DNA samples analyzed in this study. ATLL subtypes were assessed based on the Shimoyama classification. 5 Genomic DNA was extracted from 76 peripheral blood mononuclear cell samples, 11 lymph nodes, and 2 tissue samples from the mammary glands and subcutaneous tissue using the QIAamp Blood/DNA Mini Kit (QIAGEN, Hilden, Germany). We also analyzed the tax subgroups as described in our prior research. 16 Viral genotyping identified HTLV‐1‐taxA and HTLV‐1‐taxB in 41 and 48 cases, respectively (Table S1).
This study was approved by the ethics review board of the University of the Ryukyus and each participating institution, adhering to the Helsinki Declaration revised in 2008 or 2013. Before sampling, all the patients provided their physicians at the hospitals with informed consent for participation in the study.
2.2. Targeted sequencing
We performed targeted next‐generation sequencing for 89 genomic DNA samples using the HaloPlex HS target enrichment system (Agilent Technology) and the Miseq platform (Illumina). The SureCall software (Agilent Technologies) was used for identifying the variants. We designed probes for 51 cancer‐related genes, especially focusing on the genes frequently mutated in ATLL patients (Table S2). 19 , 20
Variants were filtered out under the following exclusion criteria: (a) synonymous mutations and variants in introns except for splice sites and a promoter region (TERT) or (b) variants with ≥1% allele frequency in online SNP databases (details in Document S1). Subsequently, putative driver mutations were selected when they satisfied at least one of the following conditions: (a) frameshift, in‐frame, or splice‐site mutations, (b) variants considered to be recurrent mutations in cancers by the Catalogue of Somatic Mutations in Cancer (https://cancer.sanger.ac.uk/cosmic) or a previous report, 19 or (c) nonsynonymous mutations with a SIFT score < 0.05. 23
2.3. SNP array
The OncoScan® CNV FFPE Assay Kit (Thermo Fisher Scientific) was used to analyze copy number alterations (CNAs) in the DNA samples from 87 cases. Preparation of the PCR amplicon library, array hybridization, and array scanning were performed in accordance with the protocol recommended by Affymetrix. The Chromosome Analysis Suite v4.0 software (Thermo Fisher Scientific) was used for detecting CNAs. The probes on chromosomes X and Y were excluded from the analysis because each experiment was not sex matched.
2.4. Statistical analysis
Fisher's exact test was used for assessing the significance of coexistence or mutual exclusiveness of mutations, alteration frequency, and/or clinical characteristics between tax subgroups or clinical subtypes, and mutation frequencies between patients in Okinawa and those in other regions. 19 , 20 Altered genes selected for statistical analysis are shown in Table S3. Overall survival (OS) was calculated from the day of diagnosis or disease progression to the last follow‐up day or death by any cause. Univariate analysis was performed using the Kaplan‐Meier method and log‐rank test. We adjusted the effect of variables with statistical significance by multivariate analysis with the Cox proportional hazard model. ATLL‐Prognostic Index (high/intermediate risk and low risk groups) 24 and corrected calcium, 25 a previously reported prognostic predictor, were selected as clinical covariates. P < .05 were considered significant in all statistical analyses. All analyses were performed using the Stata software version 14 (Stata Corporation).
Detailed methods for each section are provided in the online Supporting Information.
3. RESULTS
3.1. Genetic alteration profile of aggressive ATLL in Okinawa prefecture
Eighty‐eight of the 89 (98.9%) patients carried two or more mutations. A total of 470 mutations were identified in 44 out of 51 genes (Figure 1A and Table S4). The most frequently mutated gene was PRKCB (39/89, 43.8%), followed by PLCG1 (36/89, 40.4%), TP53 (31/89, 34.8%), TBL1XR1 (22/89, 24.7%), GATA3 (21/89, 23.6%), CCR4 (21/89, 23.6%), IRF4 (18/89, 20.2%), and CARD11 (18/89, 20.2%). The mutations included components of TCR/NF‐ĸB signaling (PRKCB, PLCG1, IRF4, CARD11, RHOA, VAV1, FYN, and CD28), G protein‐coupled receptors (CCR4 and CCR7), and other T‐cell related genes (TBL1XR1, GATA3, NOTCH1, and STAT3), in agreement with those identified in a prior study. 19 We analyzed the coexistence and mutual exclusiveness of the mutated genes (Table 1). In particular, strong coexistence‐association was observed in the combinations of PRKCB/CARD11, EP300/CARD11, CD28/VAV1, and PRKCB/IRF4 (P < .01).
FIGURE 1.
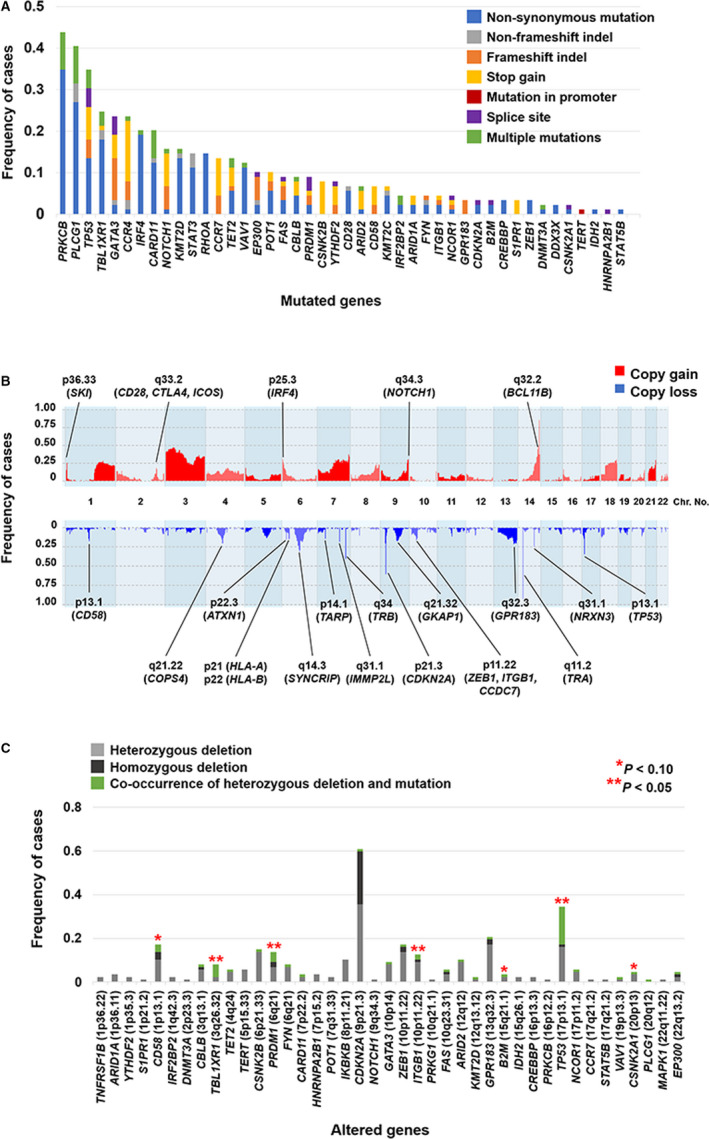
(A) The mutational profile of 44 mutated genes out of 51 genes in 89 patients with ATLL, analyzed by targeted next‐generation sequencing. (B) The frequency histograms of copy number alterations in 87 patients. Genes frequently altered (>10%) and previously reported in ATLL are annotated. (C) Analysis of genetic status via combinations of mutations and copy number losses in 87 patients. The association between mutations and deletions was assessed by the two‐sided Fisher's exact test
TABLE 1.
Mutual association of mutated genes
| Coexistence | Number of cases with mutations in both genes, n (%) | P | |
|---|---|---|---|
| Left gene | Right gene | ||
| PRKCB/CARD11 | 15/39 (39) | 15/18 (83) | <.001 |
| EP300/CARD11 | 6/9 (67) | 6/18 (33) | .002 |
| CD28/VAV1 | 4/6 (67) | 4/11 (36) | .002 |
| PRKCB/IRF4 | 13/39 (33) | 13/18 (72) | .008 |
| CSNK2B/PLCG1 | 6/7 (86) | 6/36 (17) | .016 |
| NOTCH1/FAS | 4/14 (29) | 4/8 (50) | .019 |
| CBLB/TBL1XR1 | 5/8 (63) | 5/22 (23) | .020 |
| PLCG1/ITGB1 | 4/36 (11) | 4/4 (100) | .024 |
| PRKCB/NOTCH1 | 10/39 (26) | 10/14 (71) | .038 |
| CCR7/CARD11 | 5/11 (45) | 5/18 (28) | .041 |
| POT1/CBLB | 3/10 (30) | 3/8 (38) | .044 |
| CCR7/PLCG1 | 8/11 (73) | 8/36 (22) | .045 |
| FYN/TBL1XR1 | 3/4 (75) | 3/22 (14) | .045 |
| Mutual exclusiveness | |||
| TP53/PLCG1 | 7/31 (23) | 7/36 (19) | .014 |
| CARD11/TP53 | 2/18 (11) | 2/31 (6) | .025 |
| CCR4/STAT3 | 0/21 (0) | 0/13 (0) | .033 |
| CCR4/RHOA | 0/21 (0) | 0/13 (0) | .033 |
The CNAs in 87 cases are shown in Figure 1B and Table S5. Several reported alterations in aggressive ATLL 17 , 18 , 19 were also frequently observed (>20%) in our cohort: gains on chromosome 3, the long‐arm of chromosomes 1, 7, 18, and 21, 1p36.33 (SKI), 2q33.2 (CD28, CTLA4, and ICOS), 6p25.3 (IRF4), 9q34.3 (NOTCH1), and 14q31.1‐32.33, and loss of 6q14.3 (SYNCRIP), 7q31.1 (IMMP2L), 7q34 (TRB), 9p21.3 (CDKN2A), 13q32.2‐34, 14q11.2 (TRA), and 17p13.1 (TP53). We investigated the frequency of biallelic alterations, namely, homozygous deletions or co‐occurrences of heterozygous deletions and mutations. Co‐occurrences of deletions and mutations were significantly frequent in TBL1XR1, PRDM1, ITGB1, and TP53 (Figure 1C). Homozygous deletions were frequently identified in CDKN2A (21/87, 24.1%), whereas no homozygous deletions of the other analyzed genes were identified in more than 5% of the patients. In particular, more than 50% of the mutations in CD58, PRDM1, and TP53 were truncating mutations, probably leading to loss of function (Figure 1A). This suggests that biallelic inactivation is induced by co‐occurrences of deletions and mutations of these genes.
3.2. Association of the HTLV‐1 tax subgroup with genetic alterations
We examined the association between each tax subgroup (HTLV‐1‐taxA, 41 cases; HTLV‐1‐taxB, 48 cases) and genetic alterations. Regarding mutations (Figure 2A and Table S6), GATA3 mutations were detected among 34.1% (14/41) and 14.6% (7/48) in patients with HTLV‐1‐taxA and HTLV‐1‐taxB, respectively (P = .044). RHOA mutations were also more frequent among patients with HTLV‐1‐taxA (10/41, 24.4%) compared with those with HTLV‐1‐taxB (3/48, 6.3%) (P = .032). Patients with HTLV‐1‐taxB had more frequently mutated EP300 (8/48, 16.7%) than those with HTLV‐1‐taxA (1/41, 2.4%) (P = .035). Copy number gains of RHOA (3p21.31) were more frequently observed in patients with HTLV‐1‐taxA (22/41, 53.7%) than in patients with HTLV‐1‐taxB (13/46, 28.3%), showing the same tendency for this RHOA mutation (P = .018) (Figure 2B and Table S7). The association between types of RHOA mutations, copy number gains, and viral strains was further analyzed. However, no significant relationship was identified among them.
FIGURE 2.
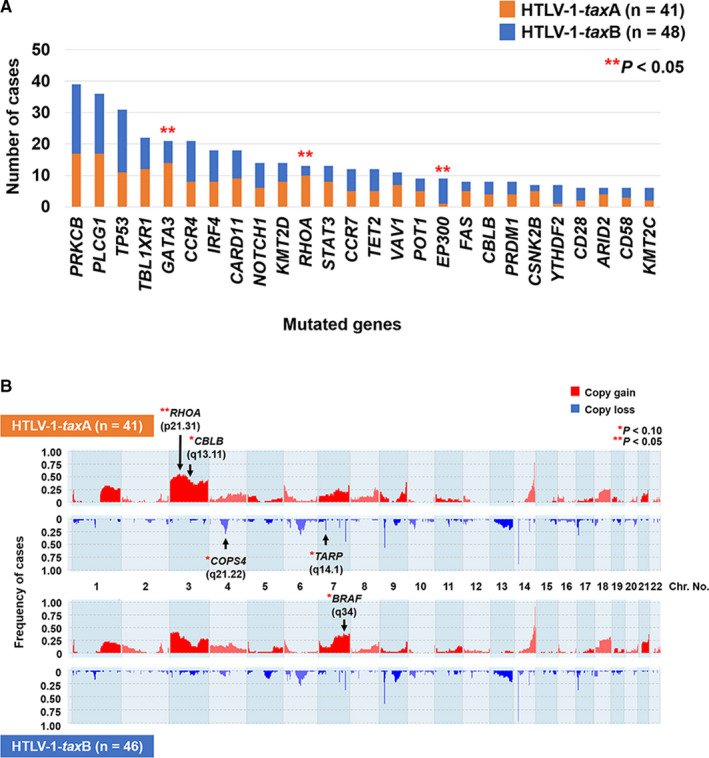
Association between genetic alterations and tax subgroups as assessed by the two‐sided Fisher's exact test. (A) The association between each mutated gene and tax subgroup. Genes mutated in more than 5% of patients were analyzed. (B) The frequency histograms of copy number alterations by the tax subgroup. Altered genes found in more than 10% of patients, including genes reported as strongly significant in ATLL, 22 were analyzed. Arrows indicate altered regions more frequently detected at a statistically different level in HTLV‐1‐taxA or HTLV‐1‐taxB
3.3. Comparison of genetic alteration profiles between patents in Okinawa, mainland Japan, and North America
We compared the frequency of each mutation between patients included in studies in Okinawa, mainland Japan, 19 and North America. 20 STAT3 mutations were significantly more frequent among patients in Okinawa (13/89, 14.6%) than in patients in North America (0/30, 0%) (Figure 3A). CARD11 and GATA3 mutations tended to be more frequently detected among patients in Okinawa (CARD11, 18/89, 20.2%; GATA3, 21/89, 23.6%) than among patients in North America (CARD11, 2/30, 6.7%; GATA3, 2/30, 6.7%). Although EP300 mutations were reported to be significantly more frequent in North American patients with ATLL than in the Japanese counterparts, 20 no significant difference was identified in the frequency between patients in Okinawa (9/89, 10.1%) and North America (6/30, 20.0%) in the present study.
FIGURE 3.
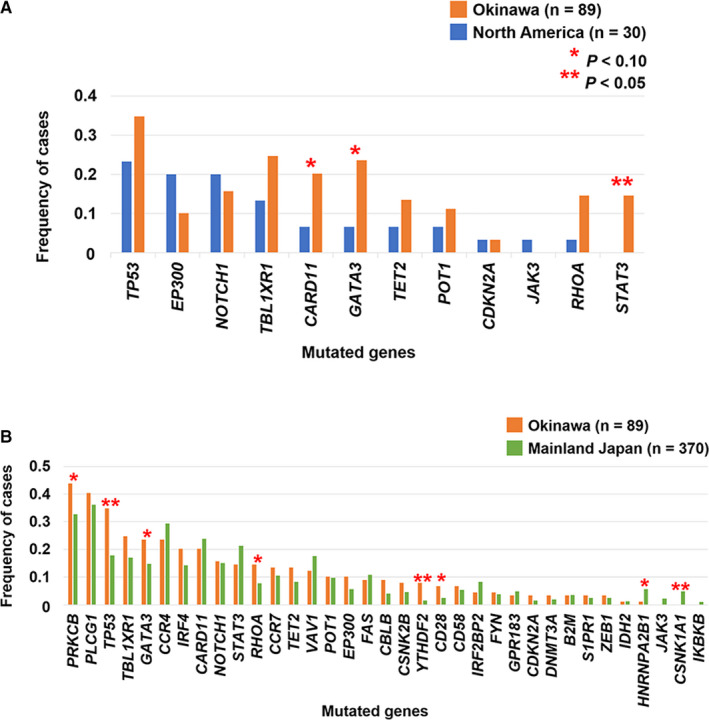
Comparison of mutation frequencies between patients in Okinawa and North America (A), and Okinawa and mainland Japan (B). The two‐sided Fisher's exact test was used for calculating the P values
TP53 and YTHDF2 showed significantly higher mutation rates among patients in Okinawa than among patients in mainland Japan (Figure 3B). Patients with mutated PRKCB, GATA3, RHOA, and CD28 were also more frequently observed in Okinawa, but with marginal significance (P values 0.051‐0.063). Although the number of patients was small, CSNK1A1 and HNRNPA2B1 mutations, which were reported in 5‐6% of mainland Japanese patients with ATLL, 19 were rarely detected among patients in Okinawa (CSNK1A1, P = .031; HNRNPA2B1, P = .095).
3.4. Association between genetic alteration and prognosis in patients with aggressive ATLL in Okinawa
Detailed clinical findings for 87 cases with available follow‐up data were analyzed as described in Tables 2 and S8. The MST of patients with the acute and lymphoma types in this cohort was 7.5 and 10.8 months, respectively (Figure S1A). These were equivalent to 8.3 months for the acute type and 10.6 months for the lymphoma type in the largest retrospective study of ATLL. 7 Mutated PRKCB, reported as an independent prognostic predictor in aggressive ATLL, 22 and loss of CD58 (1p13.1), a characteristic genetic alteration in the aggressive type, 18 were significantly associated with poor OS (Figure 4A,B, and Tables S9 and S10). We further analyzed prognosis by focusing on frequent biallelic alterations: TBL1XR1 (3q26.32), PRDM1 (6q21), CDKN2A (9p21.3), ITGB1 (10p11.22), and TP53 (17p13.1). Among them, only the biallelic alteration of PRDM1 (6q21) was significantly associated with inferior OS, whereas heterozygous deletion or mutation of PRDM1 did not affect the prognosis (Figure 4C,D and Table S11). We subsequently performed multivariate analysis for 73 patients with complete clinical data to adjust the prognostic impact of PRKCB mutations (31 cases), CD58 (1p13.1) losses (11 cases), and biallelic alterations of PRDM1 (6q21) (6 cases). Multivariate analysis showed independent negative impacts of all these three alterations on OS (Table 3).
TABLE 2.
Clinical characteristics of patients
| Clinical characteristics | Number of patients | P | ||
|---|---|---|---|---|
| Total (n = 87) | taxA (n = 40) | taxB (n = 47) | ||
| Sex | ||||
| M | 38 | 19 | 19 | .328 |
| F | 49 | 21 | 28 | |
| Age | ||||
| <70 | 46 | 21 | 25 | .560 |
| ≥70 | 41 | 19 | 22 | |
| Median (range) | 69 (35‐90) | 69 (53‐90) | 69 (35‐90) | |
| Clinical subtype | ||||
| Acute | 62 | 27 | 35 | .477 |
| Lymphoma | 12 | 7 | 5 | |
| Unfavorable chronic | 2 | 0 | 2 | |
| Disease progression from indolent form | 11 | 6 | 5 | |
| PS | ||||
| 0‐1 | 40 | 17 | 23 | .667 |
| 2‐4 | 47 | 23 | 24 | |
| Ann Arbor stage | ||||
| I‐II | 5 | 1 | 4 | .227 |
| III‐IV | 81 | 39 | 42 | |
| Unevaluable | 1 | 0 | 1 | |
| LDH (U/L) | ||||
| <230 | 6 | 2 | 4 | .683 |
| ≥230 | 81 | 38 | 43 | |
| Serum albumin (g/dL) | ||||
| <3.5 | 38 | 18 | 20 | .832 |
| ≥3.5 | 49 | 22 | 27 | |
| Corrected Ca (mmol/L) | ||||
| <2.75 | 58 | 26 | 32 | .469 |
| ≥2.75 | 29 | 14 | 15 | |
| sIL‐2R (U/mL) | ||||
| ≤20 000 | 29 | 13 | 16 | .536 |
| >20 000 | 56 | 26 | 30 | |
| Not tested | 2 | 1 | 1 | |
| Regimen of chemotherapy | ||||
| VCAP‐AMP‐VECP | 17 | 5 | 12 | .223 |
| CHOP/CHOP‐like | 42 | 20 | 22 | |
| Etoposide | 11 | 5 | 6 | |
| Others | 9 | 7 | 2 | |
| Untreated | 8 | 3 | 5 | |
| Mogamulizumab | ||||
| − | 24 | 10 | 14 | .400 |
| + | 63 | 30 | 33 | |
| Stem cell transplantation | ||||
| − | 79 | 38 | 41 | .192 |
| + | 8 | 2 | 6 | |
| ATLL‐PI | ||||
| Low | 11 | 4 | 7 | .934 |
| Intermediate | 37 | 17 | 20 | |
| High | 27 | 13 | 14 | |
| Unevaluable | 12 | 6 | 6 | |
| JCOG‐PI | ||||
| Moderate | 30 | 12 | 18 | .280 |
| High | 57 | 28 | 29 | |
| Follow‐up period (month) | ||||
| Median (range) | 7.7 (0.2‐64) | 7.6 (0.2‐50.7) | 10.0 (0.2‐64.0) | |
P values are calculated by two‐sided Fisher's exact test.
Abbreviations: ATLL‐PI, ATLL Prognostic Index; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; JCOG‐PI, Japan Clinical Oncology Group‐Prognostic Index; LDH, lactate dehydrogenase; PS, performance status; sIL‐2R, soluble interleukin 2 receptor; VCAP‐AMP‐VECP, vincristine, cyclophosphamide, doxorubicin, ranimustine, vindesine, carboplatin, etoposide, and prednisone; WBC, white blood cell.
FIGURE 4.
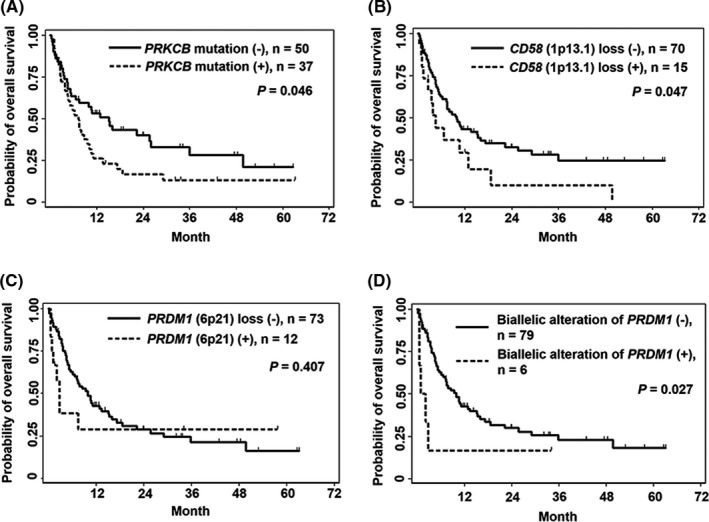
Survival analysis by the Kaplan‐Meier method for PRKCB mutation (A), CD58 (1p13.1) loss (B), PRDM1 (6q21) loss (C), and biallelic alteration of PRDM1 (D) are shown. Survival curves were evaluated by the log‐rank test
TABLE 3.
Results of multivariate analysis
| Variable | Hazard ratio | 95% confidence interval | P |
|---|---|---|---|
| ATLL‐prognostic index | |||
| High/intermediate | 3.31 | 1.22‐8.98 | 0.019 |
| Low | 1.00 | ||
| Corrected Ca (mmol/L) | |||
| ≥2.75 | 0.72 | 0.38‐1.36 | 0.306 |
| <2.75 | 1.00 | ||
| PRKCB mutation | |||
| Positive | 1.87 | 1.01‐3.46 | 0.047 |
| Negative | 1.00 | ||
| CD58 copy number loss | |||
| Positive | 3.62 | 1.68‐7.82 | 0.001 |
| Negative | 1.00 | ||
| PRDM1 biallelic alteration | |||
| Positive | 2.86 | 1.07‐7.64 | 0.036 |
| Negative | 1.00 | ||
Seventy‐three patients with evaluable ATLL‐Prognostic Index scores, data of mutation, and copy number alteration were analyzed using Cox's proportional hazard model.
We did not find statistically significant differences in clinical characteristics and prognosis between HTLV‐1‐taxA and HTLV‐1‐taxB patients (Table 2 and Figure S1B). A recently proposed prognostic model for ATLL based on PRKCB mutations, 9p24.1 (PD‐L1) amplifications, and older age (≥70) significantly distinguished cases with poorer prognosis among aggressive ATLL patients. 22 We applied this clinico‐genetic prognostic index to our cohort. The worst prognoses with statistical significance were observed in patients with more risk factors (Figure S1C).
4. DISCUSSION
In this study, we confirmed the importance of genetic alterations involved in several molecular pathways, such as TCR/NF‐ĸB signaling, G protein‐coupled receptors, and other T‐cell related genes, in ATLL cases in Okinawa as well as in mainland Japan through comprehensive genetic analysis. 19 On the contrary, genetic alterations in RHOA and GATA3, which were frequent in patients with HTLV‐1‐taxA, were significantly more frequent among patients in Okinawa compared with cases in other areas. 19 , 20 These results suggest that the different tax subgroups contribute to the geographical heterogeneity of genetic alterations in ATLL. Furthermore, we identified the negative prognostic impact of biallelic alterations in PRDM1 by examining combinations of mutations and copy number alterations.
Genetic alterations of GATA3 and RHOA, characterized by ATLL associated with HTLV‐1‐taxA, are related to Tax protein as with PRKCB, CARD11, CD28, TP53, and CDKN2A, 19 , 26 although Tax is downregulated in ATLL cells to escape from immune surveillance. 19 , 27 These alterations seem to replace the functions of Tax to promote proliferation and survival of ATLL cells. The promoter of GATA3, a transcription factor associated with Th2 cell differentiation, is regulated by Tax via the repressor ZEB protein, resulting in reduction of gene expression activity. 26 The sites of mutations inducing protein truncation were widely distributed in the entire coding region of GATA3 in the present and prior studies, 19 suggesting the essential role of the loss of function in this gene to ATLL pathogenesis. These interactions support the possibility that Tax variants affect the context of genetic alterations in ATLL cells. Moreover, RHOA, one of the Tax‐binding proteins, 28 belongs to the small GTPase Rho family, which is involved in cell cycle progression, cell survival, and cytoskeletal rearrangement. 29 Interestingly, not only mutations but also copy number gains of RHOA were more frequently identified in the HTLV‐1‐taxA subgroup (Figure 2A,B). However, no significant association was identified among each type of mutation, copy number status, and viral strain in the present cohort. Expanded studies in other HTLV‐1 endemic areas, especially where different HTLV‐1 subgroups are prevalent, will clarify the relationship between the viral strain and tumor biology of ATLL.
Differences in HTLV‐1 strains do not completely explain the geographical heterogeneity of mutation rates. A recent genomic study reported a higher frequency of mutations in genes associated with the epigenome and a lower frequency of mutations in the components of TCR/NF‐ĸB and JAK/STAT signaling in North American patients (Caribbean descent) than in Japanese patients. 19 , 20 In this study, STAT3 and CARD11 mutations were more frequent in Okinawan patients than in North American patients. However, there was no statistically significant difference in these mutations between ATLL cases in Okinawa and those in mainland Japan. These results suggest that TCR/NF‐ĸB and JAK/STAT3 signaling are important pathways for ATLL tumor formation in Japanese patients, unlike in North American patients. Moreover, mutation rates in PRKCB, TP53, YTHDF2, CD28, HNRNPA2B1, and CSNK1A1 differed among patients in mainland Japan and Okinawa. It was previously reported that mutation rates of PRKCB and TP53 were significantly higher in aggressive ATLL compared with indolent ATLL. 22 Higher rates of PRKCB and TP53 mutations among patients in Okinawa might be ascribed to the fact that all patients in this cohort had aggressive ATLL. Regarding other genes showing geographical variations in mutation frequency, it is possible that the genetic background contributes to the incidence of somatic mutations as people from Okinawa showed different genetic polymorphism compared with those from mainland Japan in studies using SNP genotyping. 14 , 15 Several cancers show heterogeneous mutation profiles in patients between different geographical regions. For example, trans‐ethnic analysis of genetic alterations identified race‐dependent differences in the frequency of BRAF/KRAS mutations in colon cancer 30 and ancestry‐dependent diversity of mutational signatures in hepatocellular carcinoma. 31 Some genetic polymorphisms in HLA class II locus were reported to influence the susceptibility to EGFR mutation‐positive lung adenocarcinoma in Japanese. 32 These reports suggest that some germline genetic variations might contribute to the different incidence of some somatic mutations in tumor cells among patients in Okinawa and in mainland Japan. 14 , 15
PRKCB mutations, CD58 (1p13.1) losses, and biallelic alterations in PRDM1 had a negative impact on survival. In a prior large cohort, PRKCB mutation was identified as an independent factor of poor prognosis. 22 The loss of PRDM1 (6q21) was also reported to be significantly associated with an unfavorable OS in activated B‐cell‐like DLBCL. 33 In DLBCL, homozygous deletions, not heterozygous deletions, were significantly associated with poor prognosis and induced upregulation of several transcription factors compared to the cases without homozygous deletions. In the prior study, 22 although multivariate analysis did not show statistical significance, the loss of PRDM1 (6q21) was a candidate for poor prognosis predictor in patients with ATLL in univariate analysis. Our observations suggest that the biallelic inactivation of PRDM1 affects the malignant potential of ATLL tumor cells more than the heterozygous alteration.
We found a high frequency of coexistence between PRKCB and CARD11 mutations, which is consistent with a previous report. 19 These two genes were some of the most frequently mutated genes in aggressive ATLL. In vitro experiments in a prior study demonstrated that co‐expression of the p.D427N PKCβ and the p.E626K CARD11 mutants enhanced NF‐ĸB activation more than either of the mutants alone. 19 Reproducible results in the two independent cohorts in Okinawa and mainland Japan indicated the crucial cooperative roles of PRKCB and CARD11 mutations in the pathogenesis of ATLL. Previous studies also described the importance of intragenic focal deletions in the inhibitory domain of CARD11, which cause CARD11 to be constitutively active. 19 , 34 However, these small deletions were not detected in the present study owing to the resolution of the SNP array, which is one of the limitations in this study. Detailed examination of the intragenic focal deletions in CARD11 will accurately determine the significance of the cooperation between CARD11 and PRKCB alterations.
Another limitation of this study is that mutational signature has not been understood because we performed targeted sequencing and not whole genome sequencing. Furthermore, the driver mutations were defined based on several databases as described in the Materials and Methods section, and possibly somatic passenger mutations as well as germline polymorphisms were excluded. Therefore, it was difficult to completely distinguish somatic mutations, including driver and passenger ones from germline polymorphisms due to the lack of normal control samples.
In summary, this study uncovers genetic alterations in aggressive ATLL in Okinawa and suggests an association between HTLV‐1 strains and the regional heterogeneity of genetic alterations. In addition, we discovered adverse outcomes in patients with CD58 (1p13.1) losses and PRDM1 biallelic alterations and confirmed the clinical impact of PRKCB mutations and the importance of several molecular pathways, such as TCR/NF‐ĸB signaling. Our results will contribute to the identification of optimal therapeutic targets for different geographical regions. Our integrated clinico‐genetic data, along with those of prior genomic studies, 19 , 20 , 22 form the foundation for future studies on ATLL from biological, clinical, virologic, and epidemiological viewpoints.
DISCLOSURE
The authors have no conflict of interest to declare.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Prof. Tetsuya Mitsudomi for organizing the research collaboration with the Department of Genome Biology, Kindai University Faculty of Medicine. The authors would also like to thank Mutsumi Isa and Dr Ryosuke Kimura for providing technical support. This work was supported by grants from Okinawa Prefecture, JSPS KAKEN Grant Number JP19K17835 (to S. S.) and JP19K07438 (to K. K.), the Ichiro Kanehara Foundation (to K. K.), the Yasuda Medical Foundation (to K. K.), the Japan Leukemia Research Fund (to K.K.), the Okinawa Internal Medicine Research Promotion Society (to K. K.), a Japanese Society of Hematology research grant (to K. K.), and the Life Medicine Research Promotion Foundation (to K. K.). The authors would like to thank Editage for English language editing.
Sakihama S, Morichika K, Saito R, et al. Genetic profile of adult T‐cell leukemia/lymphoma in Okinawa: Association with prognosis, ethnicity, and HTLV‐1 strains. Cancer Sci. 2021;112:1300–1309. 10.1111/cas.14806
Contributor Information
Shugo Sakihama, Email: sakihama@med.u-ryukyu.ac.jp.
Kazuho Morichika, Email: karube@med.u-ryukyu.ac.jp.
REFERENCES
- 1. Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. Adult T‐cell leukemia: clinical and hematologic features of 16 cases. Blood. 1977;50(3):481; ‐492. [PubMed] [Google Scholar]
- 2. Hinuma Y, Nagata K, Hanaoka M, et al. Adult T‐cell leukemia: antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc Natl Acad Sci USA. 1981;78(10):6476‐6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T‐cell lymphoma. Proc Natl Acad Sci USA. 1980;77(12):7415‐7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yoshida M, Seiki M, Yamaguchi K, Takatsuki K. Monoclonal integration of human T‐cell leukemia provirus in all primary tumors of adult T‐cell leukemia suggests causative role of human T‐cell leukemia virus in the disease. Proc Natl Acad Sci USA. 1984;81(8):2534‐2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimoyama M,Members of the Lymphoma Study Group . Diagnostic criteria and classification of clinical subtypes of adult T‐cell leukaemia‐lymphoma. Br J Haematol. 1991;79(3):428‐437. [DOI] [PubMed] [Google Scholar]
- 6. Tsukasaki K, Utsunomiya A, Fukuda H, et al. VCAP‐AMP‐VECP compared with biweekly CHOP for adult T‐cell leukemia‐lymphoma: Japan Clinical Oncology Group study JCOG9801. J Clin Oncol. 2007;25(34):5458‐5464. [DOI] [PubMed] [Google Scholar]
- 7. Katsuya H, Ishitsuka K, Utsunomiya A, et al. Treatment and survival among 1594 patients with ATL. Blood. 2015;126(24):2570‐2577. [DOI] [PubMed] [Google Scholar]
- 8. Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV‐1 infection. Front Microbiol. 2012;3:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanaka A, Takahashi C, Yamaoka S, Nosaka T, Maki M, Hatanaka M. Oncogenic transformation by the tax gene of human T‐cell leukemia virus type I in vitro. Proc Natl Acad Sci USA. 1990;87(3):1071‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Furukawa Y, Yamashita M, Usuku K, Izumo S, Nakagawa M, Osame M. Phylogenetic subgroups of human T cell lymphotropic virus (HTLV) type I in the tax gene and their association with different risks for HTLV‐I‐associated myelopathy/tropical spastic paraparesis. J Infect Dis. 2000;182(5):1343‐1349. [DOI] [PubMed] [Google Scholar]
- 11. Satou Y, Yasunaga J‐I, Zhao T, et al. HTLV‐1 bZIP factor induces T‐cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 2011;7(2):e1001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yasuma K, Matsuzaki T, Yamano Y, Takashima H, Matsuoka M, Saito M. HTLV‐1 subgroups associated with the risk of HAM/TSP are related to viral and host gene expression in peripheral blood mononuclear cells, independent of the transactivation functions of the viral factors. J Neurovirol. 2016;22(4):416‐430. [DOI] [PubMed] [Google Scholar]
- 13. Naito T, Yasunaga J‐I, Mitobe Y, et al. Distinct gene expression signatures induced by viral transactivators of different HTLV‐1 subgroups that confer a different risk of HAM/TSP. Retrovirology. 2018;15:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamaguchi‐Kabata Y, Nakazono K, Takahashi A, et al. Japanese population structure, based on SNP genotypes from 7003 individuals compared to other ethnic groups: effects on population‐based association studies. Am J Hum Genet. 2008;83(4):445‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sato T, Nakagome S, Watanabe C, et al. Genome‐wide SNP analysis reveals population structure and demographic history of the Ryukyu islanders in the southern part of the Japanese archipelago. Mol Biol Evol. 2014;31(11):2929‐2940. [DOI] [PubMed] [Google Scholar]
- 16. Sakihama S, Saito M, Kuba‐Miyara M, et al. Human T‐cell leukemia virus type I Tax genotype analysis in Okinawa, the southernmost and remotest islands of Japan: different distributions compared with mainland Japan and the potential value for the prognosis of aggressive adult T‐cell leukemia/. Leuk Res. 2017;61:18‐24. [DOI] [PubMed] [Google Scholar]
- 17. Oshiro A, Tagawa H, Ohshima K, et al. Identification of subtype‐specific genomic alterations in aggressive adult T‐cell leukemia/lymphoma. Blood. 2006;107(11):4500‐4507. [DOI] [PubMed] [Google Scholar]
- 18. Yoshida N, Karube K, Utsunomiya A, et al. Molecular characterization of chronic‐type adult T‐cell leukemia/lymphoma. Cancer Res. 2014;74(21):6129‐6138. [DOI] [PubMed] [Google Scholar]
- 19. Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47(11):1304‐1315. [DOI] [PubMed] [Google Scholar]
- 20. Shah UA, Chung EY, Giricz O, et al. North American ATLL has a distinct mutational and transcriptional profile and responds to epigenetic therapies. Blood. 2018;132(14):1507‐1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamagishi M, Nakano K, Miyake A, et al. Polycomb‐mediated loss of miR‐31 activates NIK‐dependent NF‐κB pathway in adult T cell leukemia and other cancers. Cancer Cell. 2012;21(1):121‐135. [DOI] [PubMed] [Google Scholar]
- 22. Kataoka K, Iwanaga M, Yasunaga JI, et al. Prognostic relevance of integrated genetic profiling in adult T‐cell leukemia/lymphoma. Blood. 2018;131(2):215‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073‐1082. [DOI] [PubMed] [Google Scholar]
- 24. Katsuya H, Yamanaka T, Ishitsuka K, et al. Prognostic index for acute‐ and lymphoma‐type adult T‐cell leukemia/lymphoma. J Clin Oncol. 2012;30(14):1635‐1640. [DOI] [PubMed] [Google Scholar]
- 25. Fukushima T, Nomura S, Shimoyama M, et al. Japan Clinical Oncology Group (JCOG) prognostic index and characterization of long‐term survivors of aggressive adult T‐cell leukemia‐lymphoma (JCOG0902A). Br J Haematol. 2014;166:739‐748. [DOI] [PubMed] [Google Scholar]
- 26. Gilli SCO, Salles TSI, Saad STO. Regulation of the GATA3 promoter by human T‐cell lymphotropic virus type I Tax protein. J Cell Biochem. 2004;93(6):1178‐1187. [DOI] [PubMed] [Google Scholar]
- 27. Takeda S, Maeda M, Morikawa S, et al. Genetic and epigenetic inactivation of tax gene in adult T‐cell leukemia cells. Int J Cancer. 2004;109(4):559‐567. [DOI] [PubMed] [Google Scholar]
- 28. Wu K, Bottazzi ME, De La Fuente C, et al. Protein profile of tax‐associated complexes. J Biol Chem. 2004;279(1):495‐508. [DOI] [PubMed] [Google Scholar]
- 29. Orgazy JL, Herraizy C, Sanz‐Moreno V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases. 2014;5(4):e983867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoon HH, Shi Q, Alberts SR, et al. Racial differences in BRAF/KRAS mutation rates and survival in stage III colon cancer patients. J Natl Cancer Inst. 2015;107(10):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Totoki Y, Tatsuno K, Covington KR, et al. Trans‐ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46(12):1267‐1273. [DOI] [PubMed] [Google Scholar]
- 32. Shiraishi K, Okada Y, Takahashi A, et al. Association of variations in HLA class II and other loci with susceptibility to EGFR‐mutated lung adenocarcinoma. Nat Commun. 2016;7:12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xia Y, Xu‐Monette ZY, Tzankov A, et al. Loss of PRDM1/BLIMP‐1 function contributes to poor prognosis of activated B‐cell‐like diffuse large B‐cell lymphoma. Leukemia. 2017;31(3):625‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Z, Hutcherson SM, Yang C, et al. Coordinated regulation of scaffold opening and enzymatic activity during CARD11 signaling. J Biol Chem. 2019;294(40):14648‐14660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material