

Abstract
α‐Methyl‐l‐tyrosine (AMT) has a high affinity for the cancer‐specific l‐type amino acid transporter 1 (LAT1). Therefore, we established an anti‐cancer therapy, with 211At‐labeled α‐methyl‐l‐tyrosine (211At‐AAMT) as a carrier of 211At into tumors. 211At‐AAMT had high affinity for LAT1, inhibited tumor cell growth, and induced DNA double‐stranded breaks in vitro. We evaluated the accumulation of 211At‐AAMT in vivo and the role of LAT1. Treatment with 0.4 MBq/mouse 211At‐AAMT inhibited tumor growth in the PANC‐1 tumor model and 1 MBq/mouse 211At‐AAMT inhibited metastasis in the lung of the B16F10 metastasis model. Our results suggested that 211At would be useful for anti‐cancer therapy and that LAT1 is suitable as a target for radionuclide therapy.
Keywords: amino acid, anti‐cancer drug, astatine‐211, large neutral amino acid transporter 1, nuclear medicine
α‐Methyl‐l‐tyrosine (AMT) has a high affinity for the cancer‐specific l‐type amino acid transporter 1 (LAT1). Therefore, we establish an anti‐cancer therapy using 211At‐labeled α‐methyl‐l‐tyrosine (211At‐AAMT) as a carrier of 211At into tumors. 211At is useful for anti‐cancer therapy and LAT1 was suitable as a target for nuclear medicine.

1. INTRODUCTION
In recent years, the survival rate of cancer patients has increased due to the development of various treatments. However, some cancers, such as pancreatic cancer, are very resistant to treatment. 1 One approach is to use antibodies or chemicals conjugated to radionuclides, which provide specific radiotherapy. However, targeted β‐therapy may not be suitable for micro‐metastasis. Targeted α‐therapy (TAT) is the selective delivery of α‐emitters to tumors using high linear energy transfer (LET) α‐particles, while minimizing damage to surrounding tissues. 2 Radium‐223 (223Ra) dichloride has been used as a treatment for bone metastasis (ie, Xofigo®), as well as for pain management. 3 , 4 Actinium‐225 (225Ac)‐PSMA‐617 has been shown to have significant benefits for patients with advanced‐stage or chemo‐naïve prostate cancer. 5 , 6 , 7 , 8 , 9 , 10 Radiopharmaceuticals are practical and effective treatment, albeit with their restrictions.
Astatine‐211 (211At) is one of the most promising radionuclides for TAT. 211At is a radioisotope of a halogen element that decays via α‐emission to 207Bi (42%), subsequent electron capture to 211Po (58%), and subsequent α‐emission to 207Pb, resulting in 100% α‐particle emission. 211At is generally produced in the 209Bi (α, 2n)211At reaction with a cyclotron capable of 28‐29 MeV α‐beam irradiation. 211At compounds can be synthesized using general halogenation methods. Adding 211At to a substrate with the ability to accumulate within tumors is a good strategy for developing novel TAT treatments. The most applicable method for labeling astatine to a substrate could involve the use of tin precursors. 11 , 12 , 13 , 14 , 15
l‐Type amino acid transporter 1 (LAT1) is an isoform of system L, a Na+‐independent neutral amino acid transport agency. LAT1 is expressed in primary human cancers that have originated in various organs such as brain, lung, thymus, and skin, 16 and it is a well known specific cancer marker. 17 Amino acid tracers containing radioactive halogen have attracted attention for use as probes for single photon emission computed tomography (SPECT) and positron emission tomography (PET). For example, iodine‐123 labeled 3‐[123I]iodo‐α‐methyl‐l‐tyrosine (123I‐IAMT) was developed as a probe for SPECT. 18 , 19 In particular, l‐3‐[18F]‐α‐methyl‐tyrosine (18F‐FAMT) has higher potential for tumor specificity compared with 2‐deoxy‐2‐[18F]fluoro‐glucose (18F‐FDG), which is widely employed as a PET probe for cancer staging. FDG has the potential for false‐positive accumulation within inflammation related to high glucose metabolism in macrophages or neutrophils, whereas 18F‐FAMT accumulates in tumors via LAT1, which is expressed only in cancer cells. 20 In contrast, 18F‐FAMT is not transported by other isoforms of the system L (eg, LAT2, LAT3, and LAT4), which are expressed in normal tissues. 21 , 22 Therefore, our l‐3‐[211At]‐α‐methyl‐tyrosine (211At‐AAMT) reagent is also expected to have LAT1 specificity and potential as a TAT treatment (Figure S1).
In an earlier study, 211At‐AAMT was studied as a treatment for melanoma because α‐methyl‐l‐tyrosine (AMT) exhibited positive results for melanoma cells in in vitro uptake. 23 However, this study did not use animal models. Furthermore, understanding the amino acid uptake mechanism through LAT1 had not yet been advanced, and the 211At‐AAMT applicability was underestimated.
This study aimed to evaluate the specificity of 211At‐AAMT for LAT1 and to confirm the anti‐tumor effect of 211At‐AAMT in vitro and in vivo. We also evaluated the induction of DNA double‐stranded breaks (DSB) to understand the cytotoxic effect of alpha radiation in tumors.
2. MATERIALS AND METHODS
2.1. Astatine‐211 (211At) production
A 209Bi(α,2n)211At reaction using the AVF Cyclotron at the Research Center for Nuclear Physics, Osaka University (Ibaraki, Japan) was used to produce 211At. 14 211 At was also produced with the same nuclear reaction at the Nishina Center for Accelerator‐Based Science, RIKEN, and was then transported to the Osaka University.
2.2. Synthesis of 211At‐AAMT
211At‐AAMT was synthesized via mercuration of AMT, as previously reported. 23 , 24 AMT was purchased from Watanabe Chemical Industries, Ltd., and other chemicals were purchased from the FUJIFILM Wako Pure Chemical Corporation. In total, 22 μmol of α‐methyl‐tyrosine and 20 μmol of HgSO4 were dissolved in 0.5 mL of H2SO4 (0.2 mol/L). The reaction mixture was stirred at 25℃ for 2 hours. Then, 45 μmol of NaCl was added, followed by stirring for 5 min. Next, 100 μL of 211At water solution (10 MBq) and 3 μL of KI3 (1 mol/L) were added, with subsequent stirring for 30 min at room temperature. After that, 0.2 mL KI (1 mol/L) was added until the solution became clear. The crude solution was passed into a column filled with cation exchange resin (Dowex™ 50W × 8, H + form). The reaction vessel was washed with 0.5 mL of H2SO4 (0.2 mol/L) and then a rinse solution was passed through the column. Anions (eg, SO4 2−, I−) were removed in this step. Next, 211At‐AAMT was eluted from the cation exchange column. We added 1 mol/L NH4OH to the column and collected 10 fractions of 0.5 mL each, all measured with a curie‐meter (IGC‐7, HITACHI, Ltd.). The first 4 fractions of 1 mol/L NH4OH eluted nearly all (about. 90%) 211At‐AAMT. We then passed the 3 fractions through a column filled with anion exchange resin (DowexTM 1 × 8, OH– form). Cations (eg, Hg2+, K+) were removed in this step. Prior to elution of 211At‐AAMT, 6 mL of 0.02% AcOH was passed through the column to elute the remaining AMT and 0.2% AcOH was passed through the column to elute 211At‐AAMT. The 211At‐AAMT solution at a final concentration of 5 MBq/mL was mixed with ascorbic acid as a reducing agent 14 , 25 to a final concentration of 1.0 w/v% at pH 6.0. DowexTM 50W × 8 and DowexTM 1 × 8 were purchased from the FUJIFILM Wako Pure Chemical Corporation. An S‐size Muromac™ mini‐column (Φ8 × 50 mm) (Muromachi Chemical Inc) was used to prepare the ion‐exchange column. 211At‐AAMT was analyzed by thin‐layer chromatography (TLC) (silica gel G60, Merck Millipore; solvent: n‐butanol/acetic acid/water (4/1/1)), and the radioactivity on the plate was measured using a Typhoon FLA7000 biomolecular imager (GE Healthcare). 15 To confirm the stability of 211At‐AAMT, we compared it with or without ascorbic acid by TLC. We also investigated the effect of ascorbic acid on stabilized 211At‐AAMT.
2.3. Cells and in vitro cytotoxicity
The human pancreatic cancer cell line PANC‐1 was purchased from the American Type Culture Collection (ATCC). The mouse melanoma cell line B16F10 was purchased from the RIKEN cell bank. These cells were maintained in D‐MEM (Sigma‐Aldrich, Merck KGaA) supplemented with 10% heat‐inactivated FBS (GIBCO, Thermo Fisher Scientific, Inc) and 1% penicillin‐streptomycin (FUJIFILM Wako Pure Chemical Corporation). Mock‐HEK293 (HEK293 cells transfected with Mock vector), hLAT1‐HEK293 (HEK293 cells overexpressing hLAT1), and hLAT2‐HEK293 (HEK293 cells overexpressing hLAT2) were established and provided by Dr. Kanai. 26 These cells were maintained in E‐MEM (FUJIFILM Wako Pure Chemical Corporation) as previously detailed. 26
For the cytotoxicity assay, PANC‐1 cells were seeded at 1 × 105 cells/mL in 96‐well plates. After 2 d of incubation, cells were treated with 211At‐AAMT. 211At‐AAMT was added at 10 kBq to each well. After 10 min of 211At‐AAMT treatment, cells were washed twice with phosphate‐buffered saline without calcium and magnesium (PBS(−)), and fresh medium was added. After 18 h of incubation, cell viability was measured using a cell counting kit‐8 (DOJINDO LABORATORIES, Kumamoto, Japan). Absorbance was measured using the microplate reader MultiSkan MC (Thermo Fisher Scientific, Inc).
2.4. In vitro cellular uptake assay
To confirm the uptake ability of 211At‐AAMT by PANC‐1 cells via LAT1, cells were seeded into 24‐well plates (1 × 105 cells/mL) and cultured for 2 d. Following incubation in Hanks’ balanced salt solution for 30 min, cells were treated with 211At‐AAMT and other reagents. 211At‐AAMT was added 1 kBq to each well. After treatment, cells were washed twice with PBS(−), lysed with 0.1 N NaOH and radioactivity was measured with a gamma counter 2480 Wizard2 (PerkinElmer, Inc). Protein levels were measured on a plate reader using a BCA protein assay kit (FUJIFILM Wako Pure Chemical Corporation). We used 2‐amino‐2‐norbornanecarboxylic acid (BCH, Sigma‐Aldrich) as the LAT inhibitor. Uptake was measured after 0.5, 1, 2, 5, 10, and 30 min. The uptake assay was also performed using 3 cell lines: Mock‐HEK293, hLAT1‐HEK293, and hLAT2‐HEK293. The procedure implemented was the same as that used for the cancer cell lines. The incubation time was 10 min. BCH was used at 200 mmol/L and AMT at 1 mmol/L. Mock treatment was represented by cells expressing an empty vector. We evaluated LATs affinity by the uptake inhibitory effects of 211At‐AAMT for LAT1 and LAT2 in the presence or absence of BCH, and non‐labeled AMT. 15 , 26
2.5. Observation of double‐stranded breaks in cell lines
We used the HCS DNA Damage Kit (Thermo Fisher Scientific, Inc) for detection of DNA DSB. We seeded PANC‐1 or B16F10 cells and, after 2 d, cells were treated with 211At‐AAMT for 5 min. Following incubation with 211At‐AAMT medium, the cells were incubated for 18 h. The cells were stained with the HCS DNA Damage Kit and fluorescence was measured with the fluorescence microscope BZ‐X810 (Keyence Corporation).
Imaging of 211At‐AAMT genotoxicity and cytotoxicity in PANC1 or B16F10 cells was carried out as follows. Cells were treated with 0.3‐10 kBq of 211At‐AAMT for 5 min. After washing with PBS(−), cells were incubated for 18 h and toxicity was assayed. Red fluorescence indicated DSB, green fluorescence was a signal of cell death, and blue fluorescence represented the cell number. Imaging and analyses were performed using a 10× objective and Image‐J software.
2.6. Distribution, imaging, and therapy of 211At‐AAMT in mice
The experimental protocol was approved by the Animal Care and Use Committee of the Osaka University Graduate School of Science. All animals were housed under a 12 h : 12 h, dark : light cycle (light from 08:00 to 20:00) at 25 ± 1°C with ad libitum food (CRF‐1: Oriental Yeas Co., Ltd.) and water in Multi‐chamber Animal Housing System (Nippon Medical & Chemical Instruments. Co., Ltd.). BALB/c nu/nu mice, C57BL/6 mice, and ICR mice were purchased from Japan SLC Inc.
Normal ICR mice (n = 3; 6 wk old; male body weight = 25.5 ± 1.7 g, female body weight = 24.9 ± 1.8 g) were used for evaluation of distribution at 1 h following administration of the 211At ‐AAMT solution (4.0 ± 0.4 MBq/mL). PANC‐1 xenograft mice (n = 3) were also sacrificed after 1 h following 211At‐AAMT administration. Details of tumor‐bearing mice was described later. All organs were packed into a zippered polyethylene bag and measured with a gamma counter.
We also generated PANC‐1 xenograft mice for imaging separately from distribution and therapeutic experiments. For imaging, 211At‐AAMT was injected at 1 MBq; groups included 211At‐AAMT only and 211At‐AAMT with BCH. Each group consisted of 2 mice (n = 2). Planar images were acquired using a gamma camera system (E‐cam, Siemens, Munich, Germany) with a low‐energy all‐purpose collimator. 14 The energy window was set with X‐rays emitted from the daughter nuclide of 211Po. The acquisition time of scintigram was 10 min. Tumorous uptake was estimated from the planar images at 10 min post‐injection.
PANC‐1 cells were cultured at 37°C in D‐MEM containing 10% fetal bovine serum and 1% antibiotics in a humidified incubator with 5% CO2. Cultured cells were washed in PBS(−) and harvested with trypsin. Tumor xenograft models were established by subcutaneous injection of 1 × 107 cells in 0.2 mL of serum‐free medium and Matrigel (1:1; BD Bioscience) into female BALB/c nu/nu mice. PANC‐1 xenograft mice (10 wk old; body weight = 19.3 ± 1.4 g) were used when tumor size reached approximately 50 mm3 on average. Mice were divided into 2 groups based on the injected dose (0.4 MBq [n = 4, 4.0 ± 0.2 MBq/mL]; control [n = 4]). The control group only received solvents. Tumor sizes and body weights were measured 3 times per week. Mice were sacrificed when the tumor size reached more than 10% of the total weight. Mice were followed for 40 d. Uptakes were normalized by the injected dose (MBq) and body weight (g).
B16F10 cells were cultured, washed, and harvested as described above. Tumor metastasis models were established by intravenous injection, via the tail vein, of 2 × 105 cells in 0.1 mL of serum‐free medium into C57BL/6 mice. Mice were divided into 3 groups based on the injected dose (1 MBq [n = 3, 10.0 ± 1.50 MBq/mL], 0.1 MBq [n = 3, 1.0 ± 0.1 MBq/mL]). The control group only received solvents. 211At‐AAMT was administered by intravenous injection via tail vein. At 2 wk after 211At‐AAMT injection, all experimental mice were euthanized and dissected. Nodules in the lungs were counted in each mouse. A pulmonary metastatic model of murine melanoma was selected to verify the cytotoxic properties reported in a previous study. 23
2.7. Statistical analysis
Comparisons between 2 groups were performed using Student's t test using Statcel3 for Excel software (OMS Publishing). P‐values of less than .05 were considered statistically significant.
3. RESULTS
3.1. 211At‐AAMT preparation
α‐Methyl‐l‐tyrosine was successfully radiolabeled with 211At by the replacement reaction of Hg2+ for 211At+ with a radiochemical yield of 60%‐80%. 211At‐AAMT was obtained with a high radiochemical purity of 98%, after purification by consecutive cation and anion exchange columns. Rf ratio of 211At‐AAMT was kept at 0.7 to 0.8 at least for 7 h in the presence of 1% ascorbic acid, although the product was decomposed (Rf ratio = 0) without ascorbic acid (Figure S2).
3.2. LAT1 specificity of 211At‐AAMT in vitro
We first compared uptake via LAT1 or LAT2 using HEK293 cells and showed that LAT1 contributed to the specificity of 211At‐AAMT. In hLAT2‐HEK293 cells, no significant uptake of 211At‐AAMT was observed compared to that in Mock‐HEK293 cells (Figure 1).
FIGURE 1.
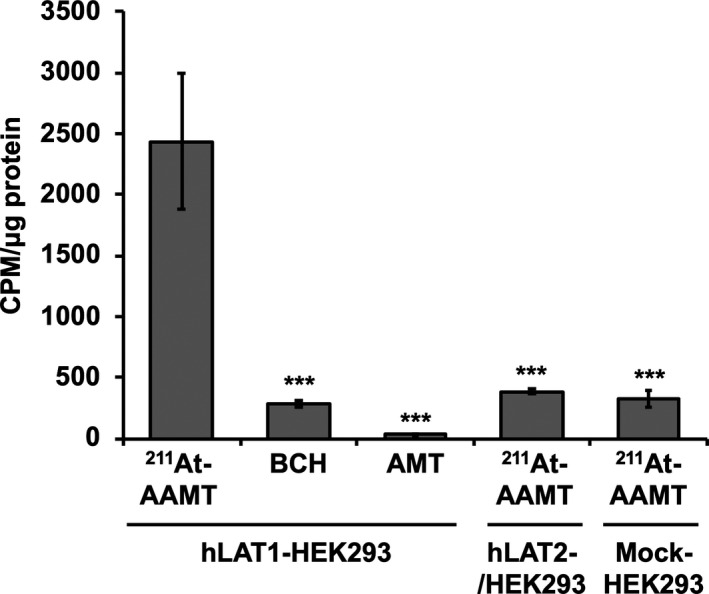
Uptake of 211At‐AAMT in HEK293 cells. Uptake of 211At‐AAMT measured using LAT1 or LAT2 overexpressing HEK293 cell lines. LAT1 represented the uptake results of hLAT1‐HEK293 cells, LAT2 represented the uptake results of hLAT2‐HEK293, and Mock represented the uptake results of Mock‐HEK293 cells. BCH: 2‐aminobicyclo [2.2.1] heptane‐2‐carboxylic acid, AMT: α‐methyl‐l‐tyrosine. ***P < .001
Second, we evaluated cellular uptake (Figure 2) and viability (Figure 3) in the PANC‐1 cell line by comparing it with LAT1 inhibitor groups. AMT was used for competitive inhibition at high concentrations. 211At‐AAMT was incorporated into the cells in a time‐dependent manner (Figure 2). Inhibitors reduced the amount of incorporated 211At‐AAMT, with BCH inhibiting 211At‐AAMT more efficiently compared with AMT. Figure 3 shows the LAT1 specificity of 211At‐AAMT, as well as its cytotoxicity. After 10 min of treatment with 211At‐AAMT, 94.1% of PANC‐1 cells were killed. LAT1 inhibitors prevented the incorporation of 211At‐AAMT as well as intracellular alpha‐ray exposure; thus, viability was the same as that of the control group (Figure 3). In summary, 211At‐AAMT exhibited LAT1 specificity and BCH and AMT inhibited its incorporation into PANC‐1 cells.
FIGURE 2.
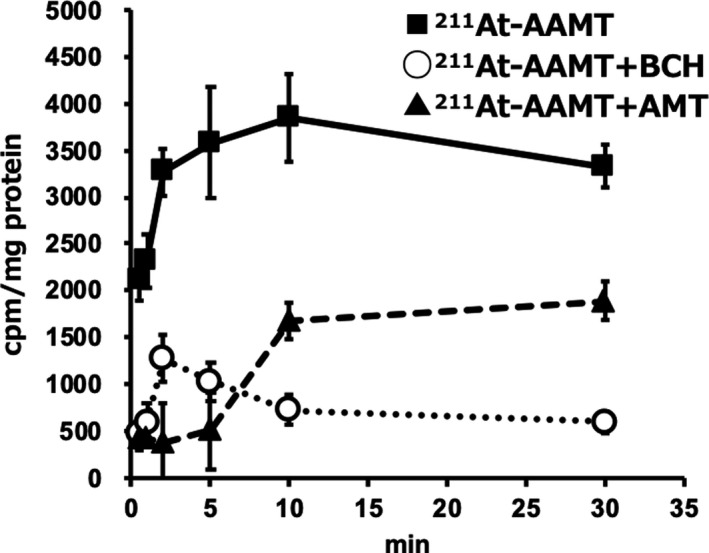
Uptake of 211At‐AAMT in PANC‐1 cells. Black squares represent samples lacking inhibitor, white circles represent LAT‐specific inhibitor treatment BCH (200 mmol/L). Black triangles represent competitive inhibitor treatment non‐labeled α‐methyl‐l‐tyrosine (1 mmol/L)
FIGURE 3.
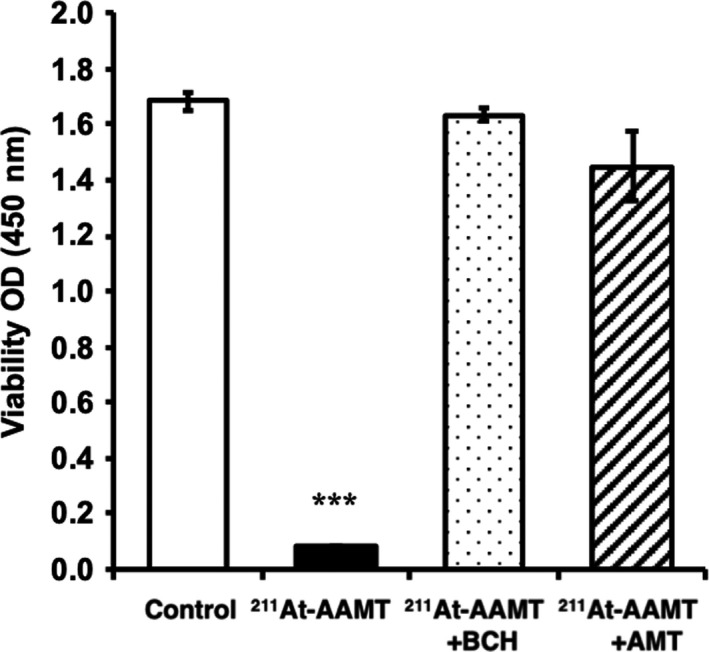
Cell toxicity of 211At‐AAMT in cancer cell lines. White bar is control, black bar is treatment by 211At‐AAMT, dot bar is treatment by 211At‐AAMT and BCH, and diagonal line bar is treatment 211At‐AAMT and AMT. ***P < .001
211At‐AAMT also induced both DNA and membrane damage in PANC‐1 and B16F10 cell lines. Figure 4A shows a fluorescence photograph of PANC‐1 (a) and B16F10 (b) cells treated with 0, 0.3, and 10 kBq 211At‐AAMT for 5 min. Figure 4B shows the quantitative representation of 211At‐AAMT‐treated cells. The cell death signal and DSB were observed after treatment with 0.3 kBq 211At‐AAMT. At 10 kBq 211At‐AAMT, cells showed genotoxic and cytotoxic effects, as demonstrated by the positive pH2AX and Image‐iT® DEAD GreenTM viability stain fluorescence. In addition, the number of viable cells was obviously reduced by treatment with 10 kBq 211At‐AAMT.
FIGURE 4.
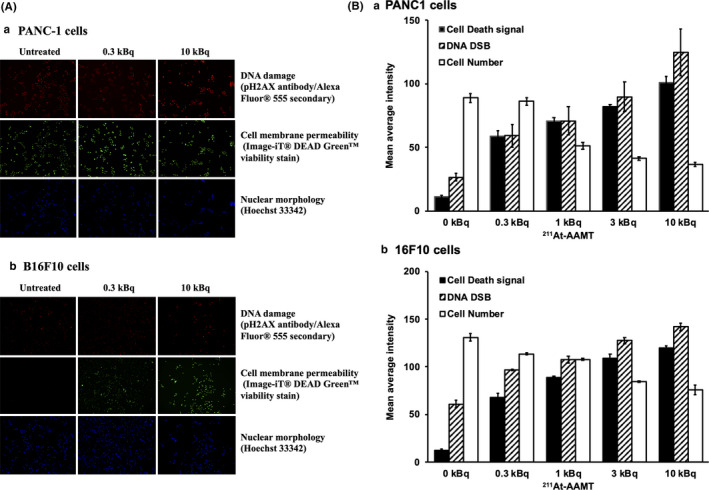
Induction of DNA double‐stranded breaks by 211At‐AAMT in PANC‐1 cells. Imaging of 211At‐AAMT genotoxicity and cytotoxicity in PANC1 or B16F10 cells. Cells were treated with 0.3‐10 kBq of 211At‐AAMT for 5 min. Even at 0.3 kBq 211At‐AAMT, cells were positive for pH2AX, and the Image‐iT® DEAD GreenTM viability stain indicated DNA damage and compromise in plasma membrane integrity. Hoechst 33342 stain was used as a nuclear segmentation tool (A). The bar graph (B) shows the quantitative representation of 211At‐AAMT‐treated cells
3.3. Administration of 211At‐AAMT, distribution, and imaging in mice
At 10 min after the injection, 211At‐AAMT was highly accumulated in the tumors and accumulated in the thyroid gland, kidney, and pancreas. BCH intake clearly inhibited the uptake of 211At‐AAMT into the tumors (Figure 5).
FIGURE 5.
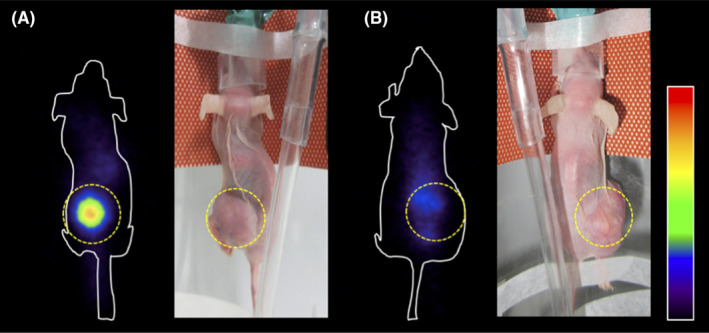
Coronal images of 211At‐AAMT in tumor‐bearing model. PANC‐1 cells tumor‐bearing mice were intravenously injected with 211At‐AAMT at 1 MBq/mouse. At 10 min after injection gamma camera imaging was performed. A, Mice were treated only with 211At‐AAMT. B, Mice were treated with 200 mg/kg BCH prior to 211At‐AAMT injection. Tumor locations were indicated by the yellow dotted line. BCH, 2‐aminobicyclo [2.2.1] heptane‐2‐carboxylic acid; inhibitor of l‐type amino acid transporter LAT
At 1 h after injection, 211At‐AAMT accumulated in the kidneys of both normal (Table 1) and tumor‐bearing mice (Table 2). The organ with the highest rate of accumulation in both sexes was the thyroid gland, followed by the kidneys highly related with urinary excretion. 211At‐AAMT was excreted in the urine (Table S1). The Rf ratio of urine was the same as that of 211At‐AAMT injected into mice (Figure S3). These results confirmed the in vivo stability of 211At‐AAMT.
TABLE 1.
Biodistribution of 211At‐AAMT in normal mice
| % ID/g | Male | Female |
|---|---|---|
| Thyroid | 13.67 ± 2.32 | 8.30 ± 2.72 |
| Salivary gland | 2.62 ± 0.66 | 3.02 ± 1.07 |
| Liver | 0.43 ± 0.12 | 1.65 ± 0.75 |
| Stomach | 7.01 ± 1.87 | 2.60 ± 0.53 |
| Kidney | 7.19 ± 2.88 | 11.09 ± 4.40 |
| Pancreas | 1.16 ± 0.40 | 6.50 ± 3.19 |
| Spleen | 2.32 ± 0.27 | 4.22 ± 0.93 |
| Blood | 0.68 ± 0.28 | 2.37 ± 1.10 |
Each value represents the mean percentage of injected dose per gram (% ID) of organ ± SE (n = 3).
TABLE 2.
Distribution of 211At‐AAMT in tumor‐bearing mice
| %ID/g | Mean ± SE |
|---|---|
| Thyroid | 11.33 ± 4.47 |
| Salivary gland | 6.02 ± 1.26 |
| Liver | 3.40 ± 0.50 |
| Stomach | 5.50 ± 1.40 |
| Kidney | 22.32 ± 7.68 |
| Pancreas | 21.02 ± 3.60 |
| Spleen | 8.40 ± 2.51 |
| Blood | 5.28 ± 0.39 |
| Tumor | 24.07 ± 11.95 |
Whole‐body distribution following intravenous administration of 211At‐AAMT solution in the mouse PANC‐1 xenograft model (n = 3). Mice were dissected 1 h after injection. Each value represents the mean of % ID of organ ± SE (n = 3).
3.4. Tumor growth
In the PANC‐1 model, control mice were injected only with solvents (0.2 w/v% AcOH and 1 w/v% ascorbic acid solution) and the 211At‐AAMT treatment group received intravenous injections of the 0.4 MBq/mouse 211At‐AAMT solution. No inflammation or abnormalities were observed around the injection site. In the 211At‐AAMT treatment group, tumor growth was clearly inhibited and body weight was not significantly decreased compared to the control group (Figure 6).
FIGURE 6.
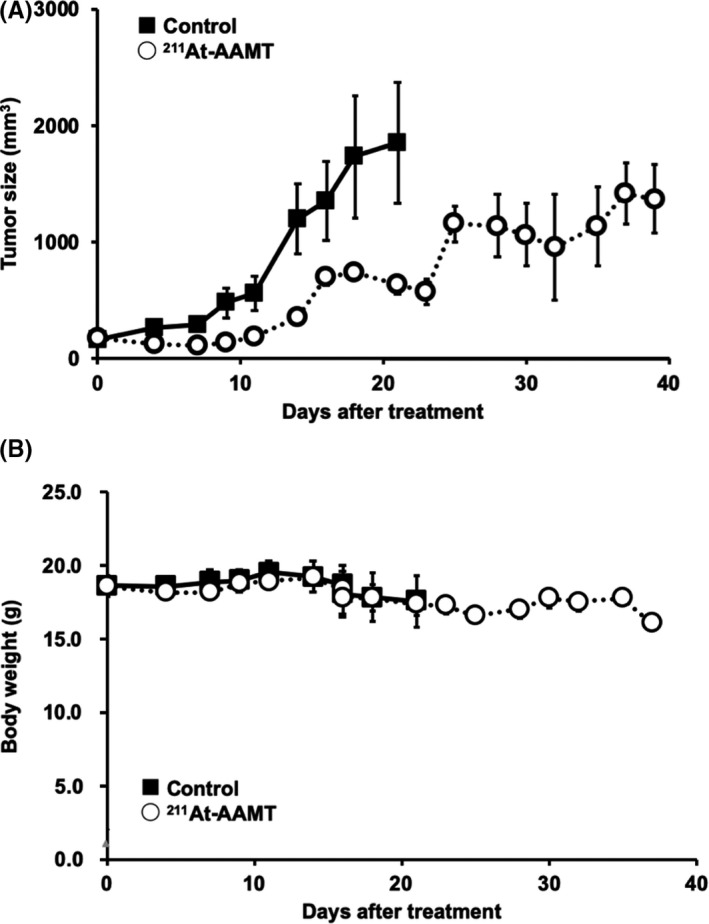
Tumor growth inhibition by 211At‐AAMT. Using the PANC‐1 xenograft model, we evaluated the efficacy of 211At‐AAMT. At 2 wk following tumor inoculation 211At‐AAMT (0.4 MBq/mouse) was injected. Tumor size (A) and weight (B) were measured 3 times a week
After B16F10 cell transplantation, mice were treated with either solvent only or 0.1 or 1 MBq/mouse of 211At‐AAMT. At 2 wk after cell transplantation, all mice were sacrificed and lung nodules were counted. Significant differences were observed between the groups (Figure 7A,B). During the experimental period, the body weight of mice was not significantly different between the groups (Figure 7C). 211At‐AAMT resulted in inhibition of nodule formation in the lung.
FIGURE 7.
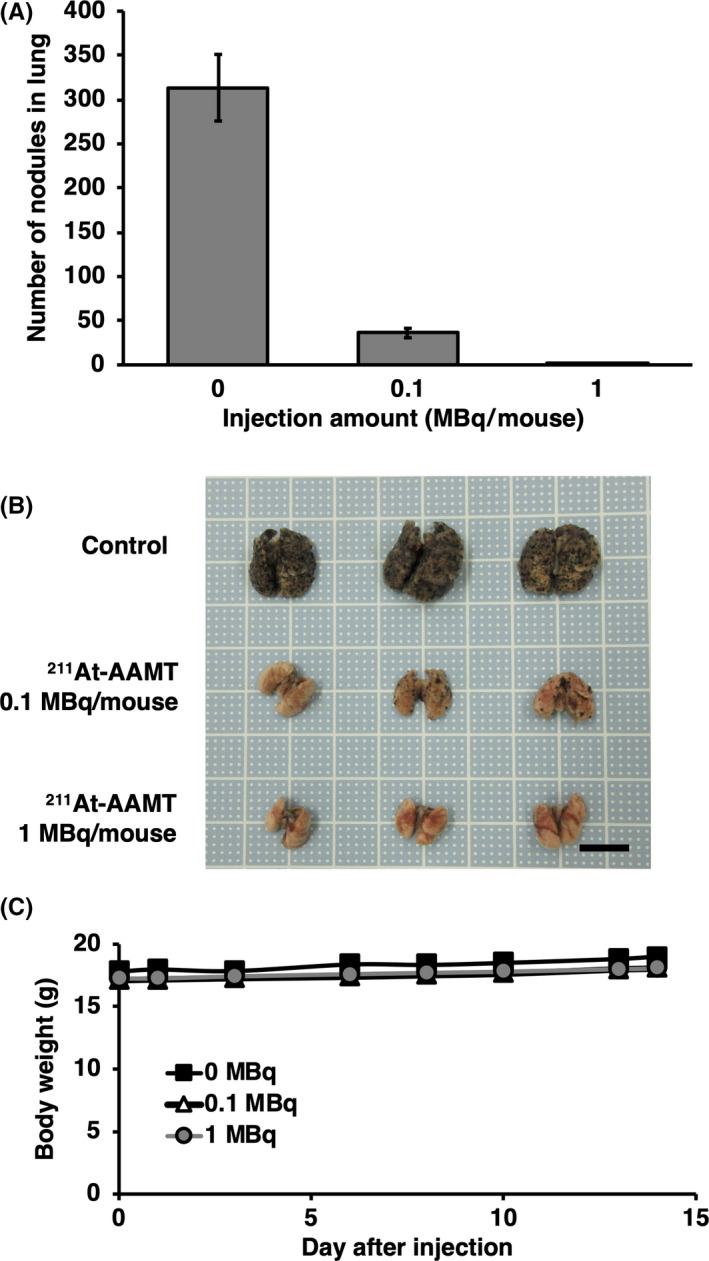
Tumor metastasis inhibition via 211At‐AAMT. Using a B16F10 model, we evaluated the efficacy of 211At‐AAMT. At 1 d after tumor inoculation, a single dose of 211At‐AAMT was injected. After 2 wk, mice were sacrificed and lung nodules were counted. A, Number of nodules in the lungs. B, Photographs of experimental mice lung. Scale bar is 10 mm. C, Weight of mice during the 2 wk period. ***P < .001
4. DISCUSSION
Uptake of 211At‐AAMT was shown in hLAT2‐HEK293, but as it was not different from that of Mock‐HEK293, it was considered the result of facilitated diffusion by a mechanism other than LAT1. In addition, as uptake was suppressed by specific inhibitors (BCH) and competitive inhibition (AMT), it was considered that the affinity of 211At‐AAMT for LAT1 was high (Figure 1). The uptake of 211At‐AAMT was also measured using PANC‐1 cells (Figure 2). BCH was used for specific inhibition and AMT was used for competitive inhibition. Uptake was assumed to be saturated at 10 min. Inhibitors reduced the amount of incorporated 211At‐AAMT, with BCH inhibiting 211At‐AAMT more efficiently compared with AMT. This may be because BCH blocked LAT1 and markedly decreased the incorporation of 211At‐AAMT, whereas under excessive AMT conditions, amino acid exchange still occurred and a small amount of 211At‐AAMT was incorporated. Specific uptake of 211At‐AAMT induced cell death and uptake for 10 min was sufficient for almost all cells to be killed (Figure 3).
211At‐AAMT had high cellular toxicity and induced DNA DSB after short exposure (Figure 4). In total, 5 min of 211At‐AAMT treatment may have been sufficient to induce DSB and enough DNA damage to induce cell death. This strong cytotoxicity was useful to establish the clear anti‐tumor effects of 211At‐AAMT.
We also confirmed the specificity of 211At‐AAMT in vivo (Figure 5) and we observed the accumulation of 211At‐AAMT in the tumor tissues as early as 10 min after injection (Figure 5A). Pretreatment with BCH clearly inhibited the accumulation of 211At‐AAMT in the tumor but it did appear to accumulate in the kidney and bladder (Figure 5B). It was thought that 211At‐AAMT also had high specificity to LAT1 like 18F‐FAMT in vivo.
We confirmed the LAT1 specificity and anti‐tumor effect of 211At‐AAMT in vitro and in vivo. To inhibit the oxidative decomposition of 211At‐AAMT, we also added ascorbic acid to 211At‐AAMT as a reducing agent. As previously observed with Na211At, 14 ascorbic acid also excelled as a reducing agent, stabilizing 211At‐AAMT in a safe manner (Figure S2). As such, we used 211At‐AAMT treated with ascorbic acid in the animal experiments. Using the urine collected from 211At‐AAMT‐injected mice, we performed the TLC analysis and did not detect the free 211At spot (Rf ratio = 1). 211At‐AAMT in urine was stable (Figure S3). There was also accumulation of 211At‐AAMT in the kidney. For evaluating the side effects of 211At‐AAMT, induction of renal tubular disorder by 211At‐AAMT should be confirmed in future. Accumulation in the thyroid and stomach was not of 211At‐AAMT itself, it is thought to be 211At from dehalogenation of 211At‐AAMT. Even if 211At is released in the body, if thyroid glands are protected with iodine block, side effects have not been reported with these doses. 27 , 28 The difference in accumulation of PANC‐1 model was recognized in comparison with the normal mice (Tables 1 and 2). It is known that liver metastasis in PANC‐1 model was infrequently, however there were some mice with nodules in a part of the liver. In pancreas, metastatic tissues were not so clear, but weights of pancreas were heavier compared with that of no tumor animal. Thus, the reason for strong accumulation of 211At‐AAMT in xenograft models was thought to be metastasized to pancreas and liver. Pathological analysis might be necessary to verify the relationship between accumulation and metastases. The accumulation of 211At‐AAMT in the kidney was also different between normal and tumor‐bearing mice. This is because there were no LAT1‐expressing tissues in normal mice, therefore injected 211At‐AAMT might be released faster in the urine of normal mice (Table S1). Fast accumulation is a suitable feature for short‐lived nuclides, such as 211At.
Amount of accumulation of 211At‐AAMT was higher in females compared with males. It was considered that the speed of excretion was slow in female. This might be because the amount of urine from females was smaller compared with that of males (examples of urine volume, 6‐wk‐old ICR mice; male: 2.39 ± 0.30 g/mouse for 24 h; female: 1.73 ± 0.29 g/mouse for 24 h). In males, only the stomach had more accumulation. Food consumption of male mice is more compared with that of female mice. It was thought that NIS (SLC5A5, sodium (Na)‐iodide symporter), which transports 211At expressed in the stomach wall, plays a role in accumulation of 211At released from AMT rather than 211At‐AAMT. We already confirmed that the administered 211At is transferred to the contents of the stomach, but the mechanism is not clear. As shown in this study, there were certainly differences between distribution of males and females 1 h after 211At‐AAMT injection. However, although no data are shown, most of them were excreted when the amount accumulated in the body was measured 1 d later. From these facts, it is considered that the difference in the accumulation of males and females at 1 h after administration does not have a significant effect. We also confirmed the anti‐tumor effect of 211At‐AAMT in tumor‐bearing models (Figures 6 and 7). 211At‐AAMT could also inhibit tumor metastasis with a single treatment (Figure 7). The lungs in the control group were larger compared with in the experimental group. It was because the proliferation of melanoma cells in the lung inhibited by 211At‐AAMT. In future research, we should calculate the absorbed dose of tumor tissues to estimate the accurate effectiveness of 211At‐AAMT.
Compared with the distribution results of 18F‐FAMT and 77Br‐BAMT, 29 although the accumulation in the kidney was similar, it was found that 211At‐AAMT efficiently accumulated in the tumor. As the results of excretion into urine were similar (Table S1), it could be inferred that the high anti‐tumor effect of 211At‐AAMT was due to the high amount of accumulation in the tumor. Approximately 70% of the administered 211At‐AAMT was excreted within 1 h without disassembly (Figure S3). 211At‐AAMT was hardly excreted from exhaled breath (Table S1). The excretion of 211At‐AAMT in feces was thought to be due to accumulation in the stomach. Excretion levels of 211At‐AAMT might be the same as other radiolabeled chemicals. The clearance of AMT derivatives was very fast. Fast excretion from normal organs is needed for lower side effects. The effects of 211At in normal organs for 2 mo after administration were already reported. 28 A high dose of 211At‐AAMT might also induce unexpected side effects. As we did not observe a rapid decrease in body weight or changes in the animals’ condition, the toxicity of 211At‐AAMT may be low in our experiments. However, side effects and dose–response induced by 211At‐AAMT should be assessed in future studies.
We established an alpha therapy targeting LAT1 using 211At‐AAMT. The 211At‐AAMT has 2 additional benefits compared with other TAT candidates. First, 211At‐AAMT could have the ability to accumulate within various cancer types. Other 211At‐labeled candidates that have advanced into clinical research target specific types of cancer. 211At‐AAMT would have similar accumulation ability as 18F‐FAMT, 76Br‐BAMT, and 77Br‐BAMT, which accumulate within various tumors where LAT1 is highly expressed. Second, 211At‐AAMT has strong cytotoxic effects. Its rapid effects might reduce the burden on patients, and its rapid elimination might reduce the side effects. High affinity to LAT1 and high cytotoxicity of alpha rays worked synergistically for anti‐tumor effects.
211At‐AAMT may be a novel anti‐cancer drug. Although 211At‐AAMT could inhibit tumor growth with a single treatment (Figure 6), the tumor was not completely abolished, so a single injection was not enough to continuously decrease tumor size. Multiple dose administration might be necessary to take full advantage of the high anti‐tumor effect of 211At‐AAMT. In conclusion, 211At‐AAMT might be an effective anti‐cancer drug when administered multiple times or in combination with existing anti‐cancer drugs.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
We thank Honoka Obata, Soichiro Ichimura, Takumi Ikeda, and Akimitsu Kanda for their excellent technical assistance. This study was funded by KAKENHI (C) (18K07323), JSPS KAKENHI in Middle Molecular Strategy (15H05836), from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), and the QiSS program of the OPERA (JPMJOP1721) from the Japan Science and Technology Agency. 211At was partly supplied through the Supply Platform of Short‐lived Radioisotopes, supported by a JSPS Grant‐in‐Aid for Scientific Research on Innovative Areas, Grant Number 16H06278. We would like to thank Editage (www.editage.com) for English language editing.
Kaneda‐Nakashima K, Zhang Z, Manabe Y, et al. α‐Emitting cancer therapy using 211At‐AAMT targeting LAT1. Cancer Sci. 2021;112:1132–1140. 10.1111/cas.14761
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin. 2020;70:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Makvandi M, Dupis E, Engle JW, et al. Alpha‐emitters and targeted alpha therapy in oncology: from basic science to clinical investigations. Target Oncol. 2018;13:189‐203. [DOI] [PubMed] [Google Scholar]
- 3. Hosono M, Ikebuchi H, Nakamura Y, et al. Introduction of the targeted α therapy (with radium‐223) into clinical practice in Japan: learnings and implementation. Ann Nucl Med. 2019;33:211‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gallicchio R, Mastrangelo PA, Nardelli A, et al. Radium‐223 for the treatment of bone metastases in castration‐resistant prostate cancer: when and why. Tumori J. 2019;105:367‐377. [DOI] [PubMed] [Google Scholar]
- 5. Sathekge M, Bruchertseifer F, Lawal I, et al. Treatment of brain metastases of castration‐resistant prostate cancer with 225 Ac‐PSMA‐617. Eur J Nucl Med Mol Imaging. 2019;46:1756‐1757. [DOI] [PubMed] [Google Scholar]
- 6. Sathekge M, Bruchertseifer F, Vorster M, et al. Predictors of overall and disease‐free survival in metastatic castration‐resistant prostate cancer patients receiving 225Ac‐PSMA‐617 Radioligand Therapy. J Nucl Med. 2020;61:62‐69. [DOI] [PubMed] [Google Scholar]
- 7. Sathekge M, Bruchertseifer F, Knoesen O, et al. 225Ac‐PSMA‐617 in chemotherapy‐naive patients with advanced prostate cancer: a pilot study. Eur J Nucl Med Mol Imaging. 2019;46:129‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rathke H, Kratochwil C, Hohenberger R, et al. Initial clinical experience performing sialendoscopy for salivary gland protection in patients undergoing 225Ac‐PSMA‐617 RLT. Eur J Nucl Med Mol Imaging. 2019;46:139‐147. [DOI] [PubMed] [Google Scholar]
- 9. Kratochwil C, Bruchertseifer F, Rathke H, et al. Targeted α‐therapy of metastatic castration‐resistant prostate cancer with 225Ac‐PSMA‐617: swimmer‐plot analysis suggests efficacy regarding duration of tumor control. J Nucl Med. 2018;59:795‐802. [DOI] [PubMed] [Google Scholar]
- 10. Kratochwil C, Bruchertseifer F, Rathke H, et al. Targeted α‐therapy of metastatic castration‐resistant prostate cancer with 225Ac‐PSMA‐617: dosimetry estimate and empiric dose finding. J Nucl Med. 2017;58:1624‐1631. [DOI] [PubMed] [Google Scholar]
- 11. Meyer GJ. Astatine. J Label Compd Radiopharm. 2018;61:154‐164. [DOI] [PubMed] [Google Scholar]
- 12. Guerard F, Gestin JF, Brechbiel MW. Production of [211At]‐Astatinated radiopharmaceuticals and applications in targeted α‐particle therapy. Cancer Biother Radiopharm. 2013;28:1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zalutsky MR, Pruszynski M. Astatine‐211: production and availability. Curr Radiopharm. 2011;4:177‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watabe T, Kaneda‐Nakashima K, Liu Y, et al. Enhancement of astatine‐211 uptake via the sodium iodide symporter by the addition of ascorbic acid in targeted α therapy of thyroid cancer. J Nucl Med. 2019;60:1301‐1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Watabe T, Kaneda‐Nakashima K, Shirakami Y, et al. Targeted alpha therapy using astatine (211At)‐labeled phenylalanine: A preclinical study in glioma bearing mice. Oncotarget. 2020;11:1388‐1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hafliger P, Charles RP. The L‐type amino acid transporter LAT1 – An emerging target in cancer. Int J Mol Aci. 2019;20:2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nobusawa A, Kim M, Kaira K, et al. Diagnostic usefulness of 18F‐FAMT PET and L‐type amino acid transporter 1 (LAT1) expression in oral squamous cell carcinoma. Eur J Nucl Med Mol Imaging. 2013;40:1692‐1700. [DOI] [PubMed] [Google Scholar]
- 18. Kawai K, Fujibayashi Y, Yonekura Y, et al. An artificial amino acid radiopharmaceutical for single photon emission computed tomographic study of pancreatic amino acid transports 123I‐3‐iodo‐α‐methyl‐l‐tyrosine. Ann Nucl Med. 1992;6:169‐175. [DOI] [PubMed] [Google Scholar]
- 19. Kawai K, Fujibayashi Y, Yonekura Y, et al. Canine SPECT studies for cerebral amino acid transport by means of 123I‐3‐iodo‐α‐methyl‐l‐tyrosine and preliminary kinetic analysis. Ann Nucl Med. 1995;9:47‐50. [DOI] [PubMed] [Google Scholar]
- 20. Achmad A, Bhattarai A, Yudistiro R, et al. The diagnostic performance of 18F‐FAMT PET and 18F‐FDG PET for malignancy detection: a meta‐analysis. BMC Med Imaging. 2017;17:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei L, Tominaga H, Ohgaki R, et al. Transport of 3‐fluoro‐l‐α‐methyl‐tyrosine (FAMT) by organic ion transporters explains renal background in [18F]FAMT positron emission tomography. J Pharmacol Sci. 2016;130:101‐109. [DOI] [PubMed] [Google Scholar]
- 22. Wei L, Tominaga H, Ohgaki R, et al. Specific transport of 3‐fluoro‐l‐α‐methyl‐tyrosine by LAT1 explains its specificity to malignant tumors in imaging. Cancer Sci. 2016;107:347‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McLaughlin WH, Thramann WM Jr, Lambrecht RM, et al. Preliminary observations of malignant melanoma therapy using radiolabeled α‐methyltyrosine. J Surg Oncol. 1988;37:192‐197. [DOI] [PubMed] [Google Scholar]
- 24. Bloomer WD, McLaughlin WH. Experimental therapeutics using α particles. Radiochim Acta. 1989;47:149‐152. [Google Scholar]
- 25. Visser GWM, Diemer EL, Kaspersen FM. The preparation and stability of astatotyrosine and astato‐iodotyrosine. Int J Appl Radiat Isot. 1979;30:749‐752. [Google Scholar]
- 26. Khunweeraphong N, Nagamori S, Wiriyasermkul P, et al. Establishment of stable cell lines with high expression of heterodimers of human 4F2hc and human amino acid transporter LAT1 or LAT2 and delineation of their differential interaction withα‐alkyl moieties. J Pharmacol Sci. 2012;119:368‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ikeda H, Hayashi Y, Takahashi N, et al. Application of astatine‐210: Evaluation of astatine distribution and effect of pre‐injected iodide in whole body of normal rats. Appl Radiat Isot. 2018;139:251‐255. [DOI] [PubMed] [Google Scholar]
- 28. Liu Y, Watabe T, Kaneda‐Nakashima K, et al. Preclinical evaluation of radiation‐induced toxicity in targeted α therapy using [(211)At] NaAt in mice: a revisit. Transl Oncol. 2020;13:100757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohshima Y, Hanaoka H, Watanabe S, et al. Preparation and biological evaluation of 3‐[(76)Br]bromo‐α‐methyl‐l‐tyrosine, a novel tyrosine analog for positron emission tomography imaging of tumors. Nucl Med Biol. 2011;38:857‐865. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1