

Abstract
We have previously identified receptor tyrosine kinase‐like orphan receptor 1 (ROR1) as a direct transcriptional target of TTF‐1/NKX2‐1, a lineage‐survival oncogene in lung adenocarcinoma. ROR1 sustains prosurvival signaling from multiple receptor tyrosine kinases including epidermal growth factor receptor, MET, and insulin‐like growth factor 1 receptor in part by maintaining the caveolae structure as a scaffold protein of cavin‐1 and caveolin‐1. In this study, a high throughput screening of the natural product library containing 2560 compounds was undertaken using a cell‐based FluoPPI assay detecting ROR1‐cavin‐1 interaction. As a result, geldanamycin (GA), a known inhibitor of heat shock protein 90 (HSP90), was identified as a potential inhibitor of ROR1. Geldanamycin, as well as two GA derivatives tested in the clinic, 17‐allylamino‐17‐demethoxygeldanamycin (17‐AAG) and 17‐dimethylaminoethylamino‐17‐demethoxygeldanamycin (17‐DMAG), decreased ROR1 protein expression. We found that ROR1 physically interacted with HSP90α, but not with other HSP90 paralogs, HSP90β or GRP94. Geldanamycin in turn destabilized and degraded ROR1 protein in a dose‐ and time‐dependent manner through the ubiquitin/proteasome pathway, resulting in a significant suppression of cell proliferation in lung adenocarcinoma cell lines, for which the kinase domain of ROR1, but not its kinase activity or N‐glycosylation, was required. Our findings indicate that HSP90 is required to sustain expression of ROR1 crucial for lung adenosarcoma survival, suggesting that inhibition of HSP90 could be a promising therapeutic strategy in ROR1‐positive lung adenocarcinoma.
Keywords: geldanamycin, HSP90, lung adenocarcinoma, proteasomal degradation, ROR1
High throughput screening of the natural product library was carried out using a cell‐based FluoPPI assay detecting ROR1‐cavin‐1 interaction. As a result, geldanamycin, a known inhibitor of heat shock protein 90 (HSP90), was identified as a potential inhibitor of ROR1. Our findings indicate that HSP90 is required to sustain expression of ROR1 crucial for lung adenosarcoma survival, suggesting that inhibition of HSP90 could be a promising therapeutic strategy in ROR1‐positive lung adenocarcinoma.
1. INTRODUCTION
Lung cancer is the leading cause of cancer death in both men and women. 1 Adenocarcinoma is the most common histological subtype, arising in the distal lung and accounting for ~40% and more than 50% of all lung cancer cases in the United States and in Japan, respectively. 2 Substantial progress in genomic profiling during the past decade enabled us to identify a subset of lung adenocarcinoma patients who harbor key oncogenic driver mutations and benefit from molecular targeted therapies, but also revealed lung adenocarcinoma as a group of distinct diseases with widespread and complex molecular heterogeneity, 3 , 4 suggesting the need for novel therapeutic targets and further personalized therapy based on the molecular features of the tumors.
The TTF‐1/NKX2‐1 homeodomain transcription factor is essential for lung morphogenesis and epithelial cell differentiation. 5 It is amplified in 10%‐15% of lung adenocarcinoma and plays a crucial role as a lineage‐survival oncogene in lung adenocarcinoma. 6 , 7 , 8 , 9 We previously reported that receptor tyrosine kinase‐like orphan receptor 1 (ROR1) is a direct transcriptional target of TTF‐1/NKX2‐1 that sustains epidermal growth factor receptor (EGFR)‐mediated PI3K‐AKT prosurvival signaling in kinase‐dependent and independent manners. 10 , 11 Intriguingly, ROR1 also possesses a kinase‐independent function as a scaffold protein of cavin‐1 and caveolin‐1 to maintain caveolae structure, which is crucially involved in sustaining prosurvival signaling of multiple receptor tyrosine kinases (RTKs) including EGFR, MET, and insulin‐like growth factor 1 receptor (IGF1R). 12 In addition, ROR1 is involved in caveolae‐dependent endocytosis through binding to cavin‐3, which in turn contributes to RTK‐mediated prosurvival signaling. 13 Expression levels of ROR1 in normal tissues are attenuated after birth, 14 , 15 but ROR1 is significantly upregulated in a variety of cancers. 14 , 15 , 16 , 17 , 18 The oncofetal expression pattern as well as multiple roles crucial to sustaining survival signaling in cancer point to a notion that ROR1 might be an effective drug target for cancer therapy development. In this regard, the involvement of ROR1 in sustaining signaling pathways activated by multiple tyrosine kinases, which can bypass signaling, conferring resistance to tyrosine kinase inhibitors, makes it an attractive therapeutic target to inhibit. 19
In the present study, we aimed at identifying compounds that potentially target ROR1‐cavin‐1 interaction to inhibit the scaffold function of ROR1. A high throughput screening of the natural product library containing 2560 compounds using a cell‐based fluorescent‐based technology detecting protein–protein interactions (FluoPPI) assay resulted in the identification of geldanamycin (GA) as a potential inhibitor of ROR1‐cavin‐1 interaction. We report here that ROR1 interacts with heat shock protein 90 (HSP90) and GA destabilizes and degrades ROR1 protein through the ubiquitin/proteasome pathway, suggesting that HSP90 inhibition could be a promising therapeutic strategy in ROR1‐positive lung adenocarcinoma.
2. MATERIALS AND METHODS
2.1. Reagents
Geldanamycin (G0334) was purchased from Tokyo Chemical Industry, and 17‐AAG (100068) and 17‐DMAG (100069) were purchased from Calbiochem. Cycloheximide (01810), MG132 (M7449), and gefitinib (SML1657) were purchased from Sigma‐Aldrich. Recombinant HGF (100‐39) was purchased from Peprotech. Chloroquine (catalog no. tlrl‐chq) was purchased from InvivoGen. Tunicamycin (202‐08241) was purchased from Fujifilm Wako Pure Chemical Corporation.
2.2. DNA constructs and transfection
Constructions of expression vectors of full‐length human ROR1 cDNA (pCMVpuro‐ROR1) and its various deletion mutants were carried out as previously described. 10 , 12 pCMV‐puro‐HA‐Ub plasmid was constructed as previously described. 20 Construction of hAG‐ROR1‐ICD and cavin‐1‐Ash plasmids was carried out as described for the FluoPPI assay. For transfection, 5 × 104 COS‐7 cells were seeded in 6‐well plates. After 24 hours, cells were transfected with plasmids at a concentration of 0.5 μg/mL using FuGENE 6 (Promega) for 24 hours according to the manufacturer’s instructions. For the ubiquitination assay, 24 hours after transfection with pCMV‐puro‐ROR1 or pCMV‐puro‐HA‐Ub, COS‐7 cells were treated with 10 μmol/L MG132 with or without 0.5 μmol/L GA for 3 hours.
2.3. Cell culture
NCI‐H1975, NCI‐H441, NCI‐H1299, NCI‐H2228, HCC4006, and HeLa cells were purchased from ATCC, and PC‐9 cells were obtained from Riken Cell Bank. All cell lines were cultured in RPMI‐1640 medium with 10% FBS. Cell lines were authenticated by short tandem repeat DNA profiling and were free from mycoplasma contamination.
For treatment with the proteasomal inhibitor MG132 and the lysosomal inhibitor chloroquine, a total of 1 × 105 cells per well were seeded in 6‐well plates and treated 24 hours later with GA, MG132, chloroquine, or DMSO at the indicated concentrations. The cells were harvested 8 hours after treatment for western blot analysis.
2.4. Fluorescent‐based technology detecting protein–protein interactions assay
The intracellular domains of ROR1 and full‐length cavin‐1 were subcloned into phAG‐MCL and pAsh‐MCL plasmids, respectively. A FluoPPI assay was used by cointroducing expression constructs of the ROR1 intracellular domain fused with an N‐terminal fluorescent protein tag (hAG‐ROR1‐ICD) and cavin‐1 with a C‐terminal assembly helper tag (cavin‐1‐Ash) in HeLa cells. For high throughput screening of a natural product library (~260 000 samples) containing 2560 compounds, HeLa clones stably expressing hAG‐ROR1‐ICD and cavin‐1‐Ash were seeded into 384‐well plates at a density of 5 × 103 cells/well. After 4 hours, screening samples were added at 0.5% and incubation was continued for 24 hours. Cells were then fixed with a 10% formalin solution and stained with 1 µg/mL Hoechst33342. Fluorescent images were acquired using an Opera Phenix High‐Content Screening System (PerkinElmer) and analyzed with the Harmony software package (PerkinElmer). Formation of foci was quantified by calculating the mean fluorescent intensity in foci divided by that intensity in cells. Hit criteria were set to under 3.0 of that value without inducing irregular cell shape or cytotoxicity.
2.5. Western blot analysis, immunoprecipitation‐western blot analysis, and protein ubiquitination assay
Western blot and immunoprecipitation‐western blot analyses were carried out according to standard procedures using Immobilon‐P filters (Millipore) and an Amersham ECL western blotting detection reagent (GE Healthcare). Ubquitination assay was carried out as previously described. 21 Antibodies against ROR1 (cat. no. 4102; Cell Signaling Technology), HSP90α (cat. no. ab2928; Abcam), HSP90β (cat. no. ab53497, Abcam), GRP94 (60012‐1‐Ig; Proteintech), p21 (cat. no. 2947; Cell Signaling Technology), LC3B (cat. no. 2775; Cell Signaling Technology), hemagglutinin (HA) (cat. no. M180‐3; MBL), Azami‐Green (hAG) (cat. no. PM011M; MBL), and cavin‐1 (cat. no. ab135655, Abcam; cat. no. A301‐271 A, Bethyl Laboratories) were used for western blot analysis. β‐Actin (cat. no. A5441; Sigma) was used as a loading control. For immunoprecipitation, anti‐goat IgG and immunoprecipitation‐specific anti‐ROR1 were used. Signal intensities of ROR1 protein bands were quantified using ImageJ (https://imagej.nih.gov/ij/) and normalized to β‐actin signal intensities.
2.6. RNA interference
Cells were transfected at a final concentration of 20 nmol/L siRNA using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer’s instructions using the following siRNAs: siControl (AllStars Negative Control siRNA, Qiagen), siHSP90α #1 (Hs‐HSPCA‐4; Qiagen), siHSP90α #2 (Hs‐HSPCA‐5; Qiagen), siHSP90β #1 (Hs‐HSPCB‐1; Qiagen), siHSP90β #2 (Hs‐HSPCB‐5; Qiagen), and siGRP94 (M‐006417‐02‐0005; Dharmacon). Cells were harvested 72 hours after transfection and cell lysates were subjected to western blot analysis. For double knockdown experiments, 10 nmol/L of each siRNA was mixed and transfected.
2.7. Real‐time quantitative PCR analysis
Total RNA was extracted using an miRNeasy mini kit (Qiagen). cDNA was prepared using a High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems). A quantitative PCR assay was carried out with a 7500 Fast Real‐Time PCR System using Power SYBR Green PCR Master Mix (Applied Biosystems). The following primers were used: ROR1 (forward, 5′‐TTCTTCATTTGCGTCTGTCG‐3′; reverse, 5′‐GGCACACTCACCCAATTCTT‐3′) and GAPDH (forward, 5′‐TGGTATCGTGGAAGGACTCATGAC‐3′; reverse, 5′‐ATGCCAGTGAGCTTCCCGTTCAGC‐3′). Each sample was run in triplicate. The Ct values for each gene were calculated and normalized to Ct values for GAPDH (ΔCt). The expression was estimated by 2−ΔCt, and the ratio was calculated relative to control.
2.8. Cell proliferation assay
For GA treatment, NCI‐H1975, PC‐9, NCI‐H441, and HCC4006 cells were seeded at 1 × 105 cells/well, and NCI‐H1299 and NCI‐H2228 cells were seeded at 5 × 104 cells/well in 6‐well plates. After 24 hours, cells were treated with different concentrations of GA for 96 hours. For treatment with gefitinib and hepatocyte growth factor (HGF), PC‐9 cells were seeded at 1 × 105 cells/well in 6‐well plates. After 24 hours, cells were treated with 0.5 μmol/L GA, 1 μmol/L gefitinib, and/or 50 ng/mL HGF for 96 hours. Cell proliferation was determined by colorimetric assay using CCK‐8 (Dojindo Laboratories).
3. RESULTS
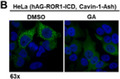
To identify compounds that potentially inhibit ROR1‐cavin‐1 interaction, we used the cell‐based FluoPPI assay, in which protein‐protein interaction can be evaluated as the formation of fluorescent foci in living cells (Figure 1A). A high throughput screening of an isolated natural product library containing 2560 compounds identified GA as a potential inhibitor of ROR1‐cavin‐1 interaction (Figure 1B). Geldanamycin is known as an HSP90 inhibitor that binds to ATP‐binding site of HSP90, 22 which is a molecular chaperone that plays a pivotal role in determining the stability and activity of client proteins. 23 However, ROR1 and cavin‐1 are not known as HSP90 clients. To determine whether GA targets ROR1 or cavin‐1 proteins themselves or physical interaction between ROR1 and cavin‐1, we undertook immunoprecipitation‐western blot analysis using ROR1 and cavin‐1 antibodies. As shown in Figure 1C, GA decreased ROR1 protein expression but not cavin‐1 protein expression. Interaction between ROR1 and cavin‐1 remained 6 hours after GA treatment in NCI‐H1975 cells, at which time point the total amount of ROR1 protein had already decreased markedly. Consistent with the findings observed in the FluoPPI assay, treatment with GA for 24 hours almost completely diminished ROR1 protein as well as ROR1‐cavin‐1 interaction (Figure 1D), suggesting that ROR1 might be a client of HSP90 and degraded by GA treatment.
FIGURE 1.
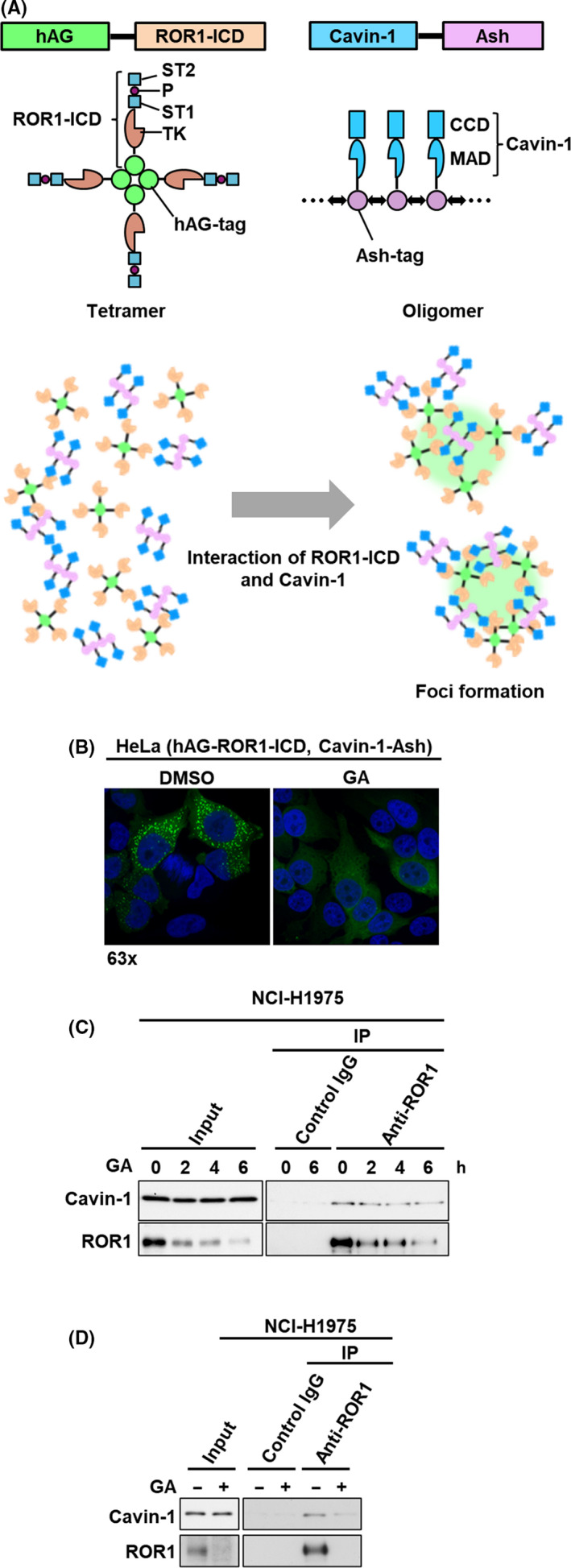
Identification of geldanamycin (GA) as a potential inhibitor of ROR1‐Cavin‐1 interaction. A, Schematic diagrams for constructs of humanized fluorescent Azami‐Green (hAG)‐ROR1‐intracellular domain (ICD), cavin‐1 assembly helper (Ash), and the fluorescent‐based technology detecting protein–protein interactions assay. ROR1‐ICD and cavin‐1 were genetically fused with tetramerizing hAG‐tag and oligomerizing Ash‐tag, respectively. A tetramer of hAG‐ROR1‐ICD and an oligomer of cavin‐1 can interact with multiple copies of each other, allowing the fluorescent proteins to form bright foci in cells. B, Fluorescent images of ROR1‐ICD and cavin‐1 interaction. Formation of fluorescent foci was suppressed after 24 h with 1 μmol/L GA (right panel) compared to control (left panel) in HeLa cells stably expressing hAG‐ROR1‐ICD and cavin‐1‐Ash. C, D, Immunoprecipitation‐western blot analyses using ROR1 Ab in NCI‐H1975 cells treated with 0.5 μmol/L GA for up to 6 h (C) and for 24 h (D). CCD, coiled‐coil domain; IP, immunoprecipitation; MAD, membrane association domain; P, proline‐rich domain; ST, serine/threonine‐rich domain; TK, tyrosine kinase domain
There are four major HSP90 paralogs, including HSP90α and HSP90β in cytoplasm, GRP94 in endoplasmic reticulum, and TRAP1 in mitochondria. 24 As endogenous ROR1 can be localized in cytoplasm or endoplasmic reticulum, we examined whether ROR1 can bind to HSP90α, HSP90β, or GRP94. Immunoprecipitation‐western blot analysis showed interaction between ROR1 and HSP90α, but not HSP90β or GRP94 (Figure 2A). To determine whether HSP90α is involved in ROR1 protein expression, we knocked down HSP90α, HSP90β, and GRP94 in two lung adenocarcinoma cell lines, PC‐9 and NCI‐H1975 cells. As shown in Figure 2B,C, knockdown of only HSP90α reduced levels of ROR1 protein expression, suggesting ROR1 as a novel client protein of HSP90α. mRNA expression levels of HSP90α in lung adenocarcinoma was significantly increased in tumor tissues compared to adjacent normal lung but was not significantly associated with ROR1 mRNA expression levels in 574 lung adenocarcinoma RNA sequencing data from The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/) and in our previous microarray data (GSE13213) 4 , 25 (Figure 2D,E).
FIGURE 2.
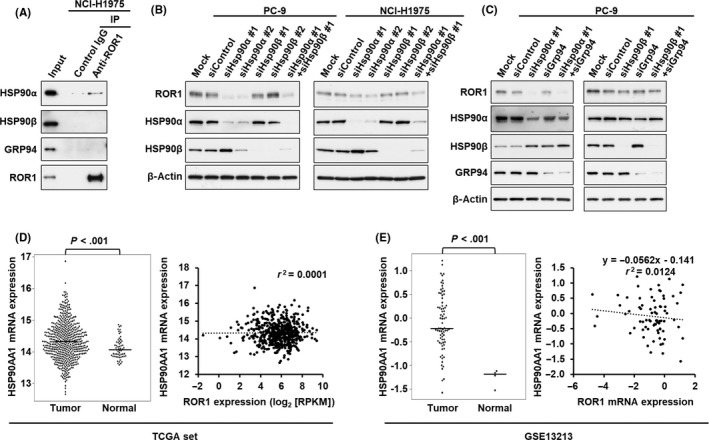
ROR1 interacts with heat shock protein (HSP)90α. A, Immunoprecipitation (IP)‐western blot analysis using ROR1 Ab in NCI‐H1975 cells, showing physical interaction between ROR1 and HSP90α, but not HSP90β or GRP94. B, C, Levels of ROR1 protein expression were decreased in PC‐9 and NCI‐H1975 cells treated with siRNAs against HSP90α, but not siRNAs against HSP90β (B) or GRP94 (C). D, E, mRNA expression levels of HSP90AA1, which encodes HSP90α protein, in lung adenocarcinoma tumors and adjacent normal tissues in The Cancer Genome Atlas (TCGA) dataset (tumor, N = 515; normal, N = 59) (D, left panel) and the GSE13213 dataset (tumor, N = 75; normal lung mixture, N = 4) (E, left panel). P values were calculated by unpaired t test. Correlation of mRNA expression levels of HSP90AA1 and ROR1 in tumor tissue in the TCGA dataset (D, right panel) and the GSE13213 dataset (E, right panel). Pearson correlation coefficient and P values were calculated
We further examined the effects of GA on ROR1 expression. Levels of ROR1 protein decreased in both NCI‐H1975 and PC‐9 cells in a time‐dependent manner, whereas mRNA expression levels of ROR1 were not obviously altered with GA treatment (Figure 3A), suggesting that GA regulated ROR1 at the protein level. Treatment with two GA derivatives, 17‐AAG and 17‐DMAG, also decreased ROR1 protein expression in NCI‐H1975 and PC‐9 cells (Figure 3B). As some other RTKs, including EGFR, MET, and IGF1R, have been known as HSP90 clients, 26 , 27 , 28 we examined whether GA would reduce expression levels of these proteins in lung adenocarcinoma cell lines as well. Protein levels of EGFR, MET, and IGF1R, as well as ROR1, were decreased after 8 hours of treatment with GA in a dose‐dependent manner (Figure 3C). Sensitivity of EGFR, MET, and IGF1R to GA was widely different among lung adenocarcinoma cell lines, but ROR1 was highly sensitive to GA in all six lung adenocarcinoma cell lines tested in this study. Interestingly, GA significantly decreased cell proliferation in these six cell lines (Figure 3D), suggesting the potential of HSP90 inhibitors in ROR1‐positive lung adenocarcinoma. To determine whether GA can suppress activation of bypass signaling that potentially drives resistance to tyrosine kinase inhibitors, we treated gefitinib‐sensitive PC‐9 cells with HGF to activate MET signaling, which can compensate the inhibitory effect of gefitinib on EGFR. 29 Treatment with HGF restored cell proliferation reduced by gefitinib, and GA negated the effect of HGF (Figure 3E), suggesting that GA overcame HGF‐mediated resistance to gefitinib in PC‐9 cells.
FIGURE 3.
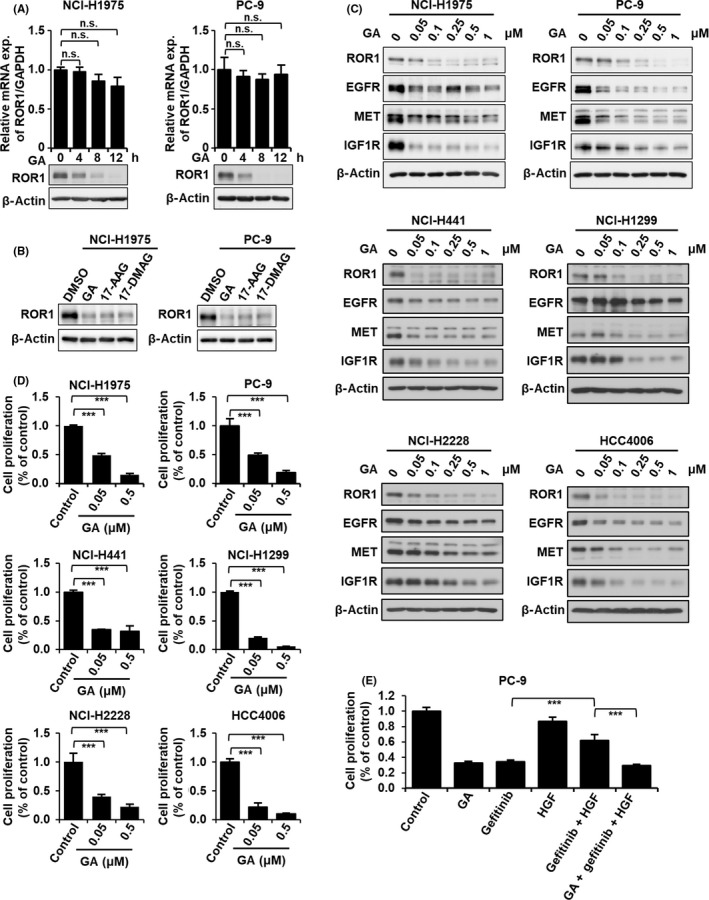
Geldanamycin (GA) decreases ROR1 protein and inhibits cell growth. A, Quantitative RT‐PCR and western blot analyses of ROR1 in NCI‐H1975 (left panel) and PC‐9 (right panel) cells treated with 0.5 μmol/L GA for the indicated time points. B, Western blot analysis of ROR1 protein in NCI‐H1975 and PC‐9 cells with 0.5 μmol/L GA, 17‐allylamino‐17‐demethoxygeldanamycin (17‐AAG), and 17‐dimethylaminoethylamino‐17‐demethoxygeldanamycin (17‐DMAG). C, Western blot analysis of ROR1, epidermal growth factor receptor (EGFR), MET, and insulin‐like growth factor 1 receptor (IGF1R) in NCI‐H1975, PC‐9, NCI‐H441, NCI‐H1299, NCI‐H2228, and HCC4006 cells treated with indicated concentrations of GA. β‐Actin served as a loading control. D, Cell proliferation analysis of NCI‐H1975, PC‐9, NCI‐H441, NCI‐H1299, NCI‐H2228, and HCC4006 cells treated with 0.05 or 0.5 μmol/L GA using the colorimetric method. ***P < .01. E, Cell proliferation analysis of PC‐9 cells treated with 0.5 μmol/L GA, 1 μmol/L gefitinib, and/or 50 ng/mL hepatocyte growth factor (HGF) using the colorimetric method. In (A), (D), and (E), columns indicate the average of triplicate samples from a representative experiment and bars indicate SD. P values were calculated by unpaired t test. ***P < .01. n.s., not significant
To determine the molecular mechanisms for GA‐mediated regulation of ROR1 protein, we examined whether GA suppressed ROR1 protein expression by attenuating protein stability or inhibiting protein synthesis. Although treatment with cycloheximide (CHX), an inhibitor of protein biosynthesis, did not largely affect expression levels of ROR1 protein, treatment with GA combined with CHX more rapidly decreased ROR1 protein than treatment with GA alone (Figures 3A and 4A, B), suggesting that GA reduced the stability of ROR1 protein. Next, we determined whether ROR1 protein is degraded through the ubiquitin/proteasome pathway or lysosomal degradation pathway. 30 NCI‐H1975 and PC‐9 cells were treated with GA by combining a proteasomal inhibitor MG132 or a lysosomal degradation inhibitor chloroquine (Figures 5A,B and S1). Protein expressions of p21 and LC3B were used as positive controls of MG132 and Chloroquine treatment, respectively. As shown in Figures 5A,B and S1, MG132, but not chloroquine, partially restored GA‐mediated reduction of ROR1 protein levels. Degradation of ROR1 intracellular domain following GA treatment was also almost completely rescued by MG132 in HeLa cells expressing phAG‐ROR1‐ICD (Figure 5C). In addition, we showed that ROR1 protein was polyubiquitinated following GA treatment (Figure 5D), suggesting that ROR1 was degraded by GA through the ubiquitin/proteasome pathway.
FIGURE 4.
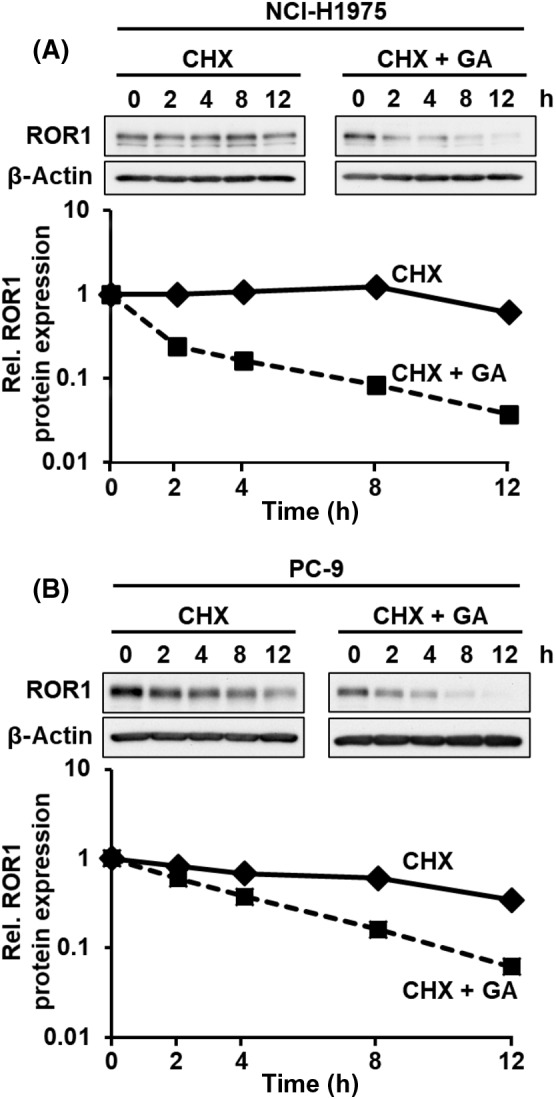
Geldanamycin (GA) destabilizes ROR1 protein. NCI‐H1975 (A) and PC‐9 (B) cells treated with 50 μg/mL cycloheximide (CHX) in the presence or absence of 0.5 μmol/L GA. Signal intensities of ROR1 protein bands were normalized to β‐actin signal intensities
FIGURE 5.
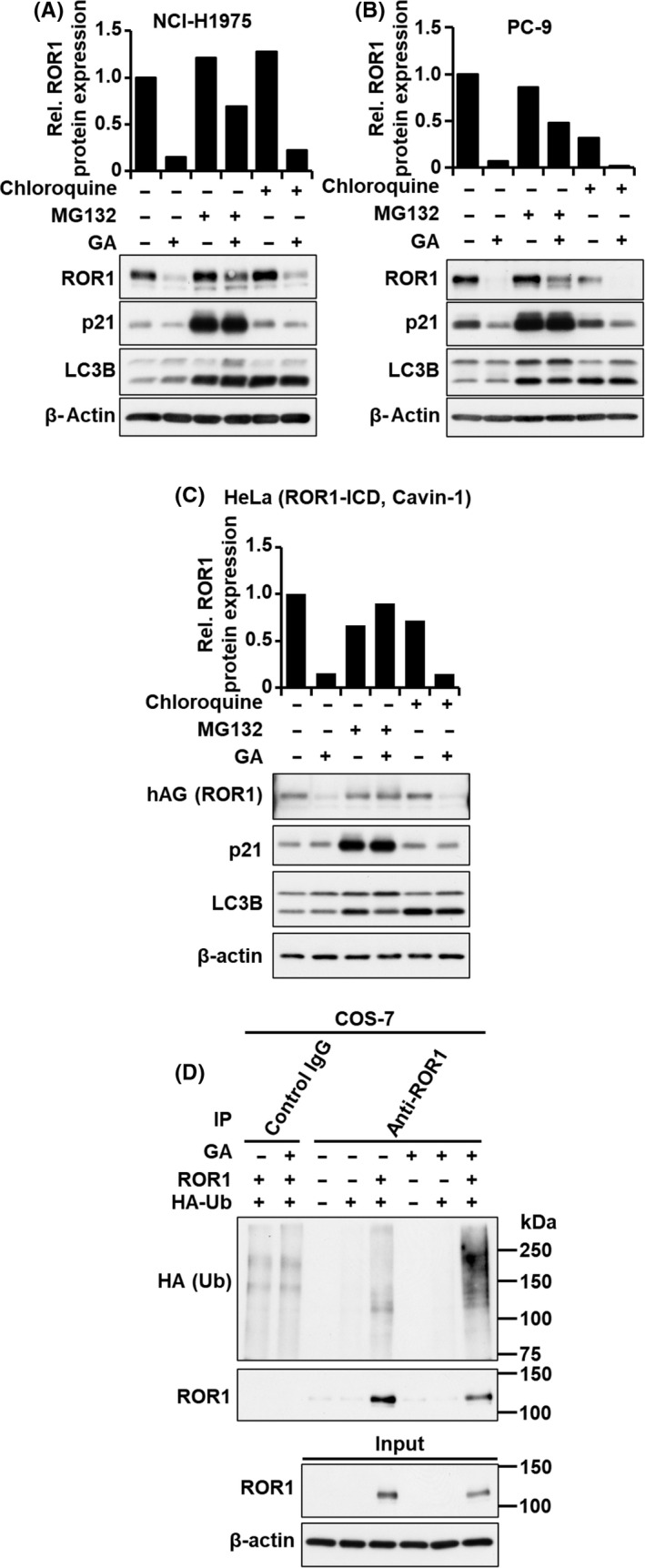
Geldanamycin (GA) degrades ROR1 protein through the ubiquitin/proteasome pathway. Western blot analysis of ROR1, p21, and LC3B in NCI‐H1975 (A), PC‐9 (B), and HeLa cells expressing humanized fluorescent Azami‐Green‐ROR1‐intracellular domain (hAG‐ROR1‐ICD) (C), treated with proteasomal inhibitor MG132 (10 μmol/L) or lysosomal inhibitor chloroquine (10 μmol/L) for 8 h in the presence or absence of 0.5 μmol/L GA. β‐Actin served as a loading control. Columns indicate relative signal intensities of ROR1 protein. D, Immunoprecipitation (IP)‐western blot analysis using ROR1 Ab in COS‐7 cells transfected with HA‐tagged ubiquitin (HA‐Ub) and/or ROR1 in the presence or absence of 0.5 μmol/L GA for 3 h
We next determined the region of ROR1 protein responsible for GA‐mediated proteasome degradation. The ROR1 WT and a deletion mutant of the C‐terminal region (ΔS/T1 + P+S/T2), but not a deletion mutant of the kinase domain (△TK), were degraded by GA (Figures 6A and S2). In addition, immunoprecipitation of ROR1 revealed that HSP90α did not bind to △TK (Figure 6B). Our findings indicate that the kinase domain of ROR1 is responsible for binding with HSP90α and GA‐mediated proteasome degradation, which is consistent with a recent study that identified the amino acid sequence ELHHPNIV in the kinase domain of ROR1 as a binding motif of HSP90. 31 Kinase activity was not involved in the interaction of ROR1 with HSP90α, or proteasome degradation (Figure 6C,D). Although N‐linked glycosylation of ROR1 could be associated with its subcellular localization and function, 32 inhibition of N‐linked glycosylation of ROR1 with tunicamycin did not clearly interfere with GA‐mediated proteasome degradation (Figure 6E).
FIGURE 6.
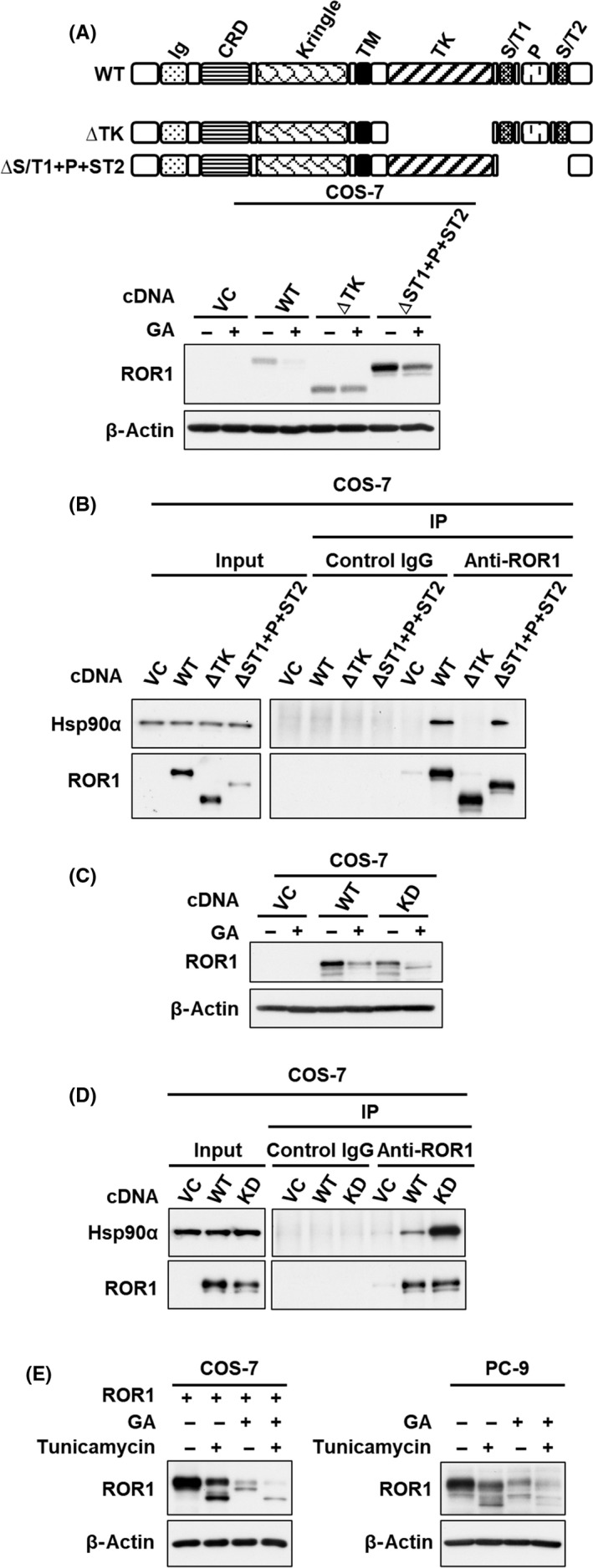
Kinase domain of ROR1 is required for geldanamycin (GA)‐induced proteasome degradation. Western blot analysis of ROR1 in COS‐7 cells transfected with ROR1 WT, (A) a deletion mutant of entire kinase domain (ROR1‐△TK) and the C‐terminal region (ROR1‐△ST1 + P+ST2), and (C) a kinase‐dead mutant (ROR1 K558R), in the presence or absence of 0.5 μmol/L GA for 24 h. Immunoprecipitation‐western blot analysis using ROR1 Ab in COS‐7 cells transfected with ROR1 WT, (B) ROR1‐△TK and ROR1‐△ST1 + P+ST2, and (D) a kinase‐dead mutant (ROR1 K558R). E, Western blot analysis of ROR1 in COS‐7 cells with overexpressing ROR1 (left panel) and PC‐9 cells (right panel), treated with 0.5 μg/mL tunicamycin for 12 h in the presence or absence of 0.5 μmol/L GA. In (A) to (E), β‐actin served as a loading control. CRD, cysteine‐rich domain; Ig, Ig‐like domain; P, proline‐rich domain; ST, serine/threonine‐rich domain; TK, tyrosine kinase domain; VC, vector control
4. DISCUSSION
We and others have reported that cell survival of a subset of lung adenocarcinoma depends on TTF‐1/NKX2‐1 signaling, but TTF‐1/NKX2‐1 cannot be considered as a molecular target because of its crucial role for maintaining physiological functions of normal lung. 33 In this connection, we previously found that ROR1 is a transcriptional target for TTF‐1/NKX2‐1 and sustains EGFR‐mediated prosurvival signaling in TTF‐1/NKX2‐1‐positive lung adenocarcinoma cell lines. 10 Furthermore, ROR1 was shown to act as a scaffold protein of cavin‐1 and caveolin‐1, two essential structural components of caveolae, sustaining caveolae formation and prosurvival signaling through multiple additional RTKs, such as MET and IGF1R. 12 As bypass signaling through diverse RTKs confers EGFR‐TKI resistance, 29 , 34 , 35 , 36 the scaffold function of ROR1 appears to be an attractive target for overcoming EGFR‐TKI resistance due to bypass signaling. It is also of note that, given its broad expression in common solid tumors and hematologic malignancies with minimal expression in healthy adult tissues, 14 , 15 , 16 , 17 , 18 ROR1‐targeted therapies using CAR‐T cells, 37 mAbs, 38 and small‐molecule inhibitors 39 are also being developed in a variety of cancer.
In the present study, we showed direct interaction between ROR1 and HSP90α, but not with its paralog, HSP90β; both are localized in the cytoplasm and their functions are mostly overlapping, but some paralog‐specific functions have been reported. 22 Heat shock protein 90α is induced by cellular stimuli such as heat shock, whereas HSP90β is constitutively expressed. It is also of note that HSP90α and HSP90β have variable affinities for client proteins as well as small‐molecule inhibitors including GA 40 ; expression of Hsp90β alone could not rescue yeast cells from the HSP90 inhibitor radicicol, in contrast to the conferral of marked resistance by comparable expression of Hsp90α alone. 41 Geldanamycin, a benzoquinone ansamycin derived from Streptomyces hygroscopicus, is the first established inhibitor of HSP90 that interferes with the ATP‐binding site of HSP90. 24 Despite its potent cytotoxic effects, its phase I clinical trial was suspended due to liver toxicity, metabolic instability, and poor solubility. 42 Geldanamycin derivatives with improved efficacy and side‐effects, including 17‐AAG and 17‐DMAG, were then developed. In addition to IPI‐504, a reduced form of 17‐AAG, derivatives of another natural HSP90 inhibitor, radicicol (such as ganetespib), purine analogues, and some other compounds have been tested in multiple clinical trials for advanced solid tumors and hematologic malignancies, including lung cancer. 43 , 44
Epidermal growth factor receptor, MET, and IGF1R are known clients of HSP90, 26 , 27 , 28 and GA indeed reduced those RTK proteins in the six lung adenocarcinoma cell lines examined in the present study. Furthermore, it is interesting to note that the concentrations of GA required to decrease each RTK protein were noticeably different among the cell lines. In contrast, ROR1 protein was consistently decreased in all of the lung adenocarcinoma cell lines treated with GA at a concentration lower than that required for significant reduction of other RTK proteins, which was accompanied by significant cell growth inhibition (Figure 3C,D). ROR1 itself has been shown to sustain signaling of multiple RTKs in both kinase‐dependent and independent manners, 10 , 12 , 13 thus it is difficult to determine the contributions of ROR1 or other RTKs to the inhibitory effects of GA on cell growth. Nevertheless, these findings suggest a critical role for ROR1 in lung adenocarcinoma cell growth.
In conclusion, we have shown that ROR1 is a novel client protein of HSP90 with specific binding to HSP90α isoform. Our present findings also suggest promising potential of HSP90 inhibitors targeting ROR1 and its scaffold function to overcome resistance caused by bypass signaling through activation of RTKs.
CONFLICT OF INTEREST
The authors declare no competing interests to disclose.
Supporting information
Fig S1‐S2
ACKNOWLEDGMENTS
We thank Ms Arisa Tamura for her technical assistance. This work was supported in part by a Grant‐in‐Aid for Scientific Research (A) (grant number: 16H02468) from the Japan Society for the Promotion of Science and the Project for Development of Innovative Research on Cancer Therapeutics (P‐Direct) (project number: 18cm0106107h9903) from the Japan Agency for Medical Research and Development.
Khaledian B, Taguchi A, Shin‐ya K, et al. Inhibition of heat shock protein 90 destabilizes receptor tyrosine kinase ROR1 in lung adenocarcinoma. Cancer Sci. 2021;112:1225–1234. 10.1111/cas.14786
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Cheng TY, Cramb SM, Baade PD, Youlden DR, Nwogu C, Reid ME. The International Epidemiology of Lung Cancer: Latest Trends, Disparities, and Tumor Characteristics. J Thorac Oncol. 2016;11:1653‐1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015;16:e342‐e351. [DOI] [PubMed] [Google Scholar]
- 4. Takeuchi T, Tomida S, Yatabe Y, et al. Expression profile‐defined classification of lung adenocarcinoma shows close relationship with underlying major genetic changes and clinicopathologic behaviors. J Clin Oncol. 2006;24:1679‐1688. [DOI] [PubMed] [Google Scholar]
- 5. Maeda Y, Dave V, Whitsett JA. Transcriptional control of lung morphogenesis. Physiol Rev. 2007;87:219‐244. [DOI] [PubMed] [Google Scholar]
- 6. Tanaka H, Yanagisawa K, Shinjo K, et al. Lineage‐specific dependency of lung adenocarcinomas on the lung development regulator TTF‐1. Cancer Res. 2007;67:6007‐6011. [DOI] [PubMed] [Google Scholar]
- 7. Weir BA, Woo MS, Getz G, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893‐898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kendall J, Liu Q, Bakleh A, et al. Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci U S A. 2007;104:16663‐16668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kwei KA, Kim YH, Girard L, et al. Genomic profiling identifies TITF1 as a lineage‐specific oncogene amplified in lung cancer. Oncogene. 2008;27:3635‐3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamaguchi T, Yanagisawa K, Sugiyama R, et al. NKX2‐1/TITF1/TTF‐1‐Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell. 2012;21:348‐361. [DOI] [PubMed] [Google Scholar]
- 11. Ida L, Yamaguchi T, Yanagisawa K, et al. Receptor tyrosine kinase‐like orphan receptor 1, a target of NKX2‐1/TTF‐1 lineage‐survival oncogene, inhibits apoptosis signal‐regulating kinase 1‐mediated pro‐apoptotic signaling in lung adenocarcinoma. Cancer Sci. 2016;107:155‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamaguchi T, Lu C, Ida L, et al. ROR1 sustains caveolae and survival signalling as a scaffold of cavin‐1 and caveolin‐1. Nat Commun. 2016;7:10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamaguchi T, Hayashi M, Ida L, et al. ROR1‐CAVIN3 interaction required for caveolae‐dependent endocytosis and pro‐survival signaling in lung adenocarcinoma. Oncogene. 2019;38:5142–5157. 10.1038/s41388-019-0785-7. [DOI] [PubMed] [Google Scholar]
- 14. Fukuda T, Chen L, Endo T, et al. Antisera induced by infusions of autologous Ad‐CD154‐leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc Natl Acad Sci USA. 2008;105:3047‐3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang S, Chen L, Wang‐Rodriguez J, et al. The onco‐embryonic antigen ROR1 is expressed by a variety of human cancers. Am J Pathol. 2012;181:1903‐1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bicocca VT, Chang BH, Masouleh BK, et al. Crosstalk between ROR1 and the Pre‐B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell. 2012;22:656‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Daneshmanesh AH, Porwit A, Hojjat‐Farsangi M, et al. Orphan receptor tyrosine kinases ROR1 and ROR2 in hematological malignancies. Leuk Lymphoma. 2013;54:843‐850. [DOI] [PubMed] [Google Scholar]
- 18. Balakrishnan A, Goodpaster T, Randolph‐Habecker J, et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin Cancer Res. 2017;23:3061‐3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17:637‐658. [DOI] [PubMed] [Google Scholar]
- 20. Liu Z, Yanagisawa K, Griesing S, et al. TTF‐1/NKX2‐1 binds to DDB1 and confers replication stress resistance to lung adenocarcinomas. Oncogene. 2017;36:3740‐3748. [DOI] [PubMed] [Google Scholar]
- 21. Xiong H, Wang D, Chen L, et al. Parkin, PINK1, and DJ‐1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Investig. 2009;119:650‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515‐528. [DOI] [PubMed] [Google Scholar]
- 23. Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18:345‐360. [DOI] [PubMed] [Google Scholar]
- 24. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761‐772. [DOI] [PubMed] [Google Scholar]
- 25. Tomida S, Takeuchi T, Shimada Y, et al. Relapse‐related molecular signature in lung adenocarcinomas identifies patients with dismal prognosis. J Clin Oncol. 2009;27:2793‐2799. [DOI] [PubMed] [Google Scholar]
- 26. Shimamura T, Lowell AM, Engelman JA, Shapiro GI. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005;65:6401‐6408. [DOI] [PubMed] [Google Scholar]
- 27. Webb CP, Hose CD, Koochekpour S, et al. The geldanamycins are potent inhibitors of the hepatocyte growth factor/scatter factor‐met‐urokinase plasminogen activator‐plasmin proteolytic network. Cancer Res. 2000;60:342‐349. [PubMed] [Google Scholar]
- 28. Martins AS, Ordonez JL, Garcia‐Sanchez A, et al. A pivotal role for heat shock protein 90 in Ewing sarcoma resistance to anti‐insulin‐like growth factor 1 receptor treatment: in vitro and in vivo study. Cancer Res. 2008;68:6260‐6270. [DOI] [PubMed] [Google Scholar]
- 29. Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res. 2008;68:9479‐9487. [DOI] [PubMed] [Google Scholar]
- 30. Pedersen NM, Madshus IH, Haslekas C, Stang E. Geldanamycin‐induced down‐regulation of ErbB2 from the plasma membrane is clathrin dependent but proteasomal activity independent. Mol Cancer Res. 2008;6:491‐500. [DOI] [PubMed] [Google Scholar]
- 31. Liu Z, Liu J, Zhang T, et al. Destabilization of ROR1 enhances activity of Ibrutinib against chronic lymphocytic leukemia in vivo. Pharmacol Res. 2020;151:104512. [DOI] [PubMed] [Google Scholar]
- 32. Kaucka M, Krejci P, Plevova K, et al. Post‐translational modifications regulate signalling by Ror1. Acta Physiol (Oxf). 2011;203:351‐362. [DOI] [PubMed] [Google Scholar]
- 33. Yamaguchi T, Hosono Y, Yanagisawa K, Takahashi T. NKX2‐1/TTF‐1: an enigmatic oncogene that functions as a double‐edged sword for cancer cell survival and progression. Cancer Cell. 2013;23:718‐723. [DOI] [PubMed] [Google Scholar]
- 34. Guix M, Faber AC, Wang SE, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF‐binding proteins. J Clin Invest. 2008;118:2609‐2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth‐factor‐driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505‐509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hudecek M, Lupo‐Stanghellini MT, Kosasih PL, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1‐specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19:3153‐3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Choi MY, Widhopf GF 2nd, Ghia EM, et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell. 2018;22(951–9):e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hojjat‐Farsangi M, Daneshmanesh AH, Khan AS, et al. First‐in‐class oral small molecule inhibitor of the tyrosine kinase ROR1 (KAN0439834) induced significant apoptosis of chronic lymphocytic leukemia cells. Leukemia. 2018;32:2291‐2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prince TL, Kijima T, Tatokoro M, et al. Client Proteins and Small Molecule Inhibitors Display Distinct Binding Preferences for Constitutive and Stress‐Induced HSP90 Isoforms and Their Conformationally Restricted Mutants. PLoS One. 2015;10:e0141786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Millson SH, Truman AW, Racz A, et al. Expressed as the sole Hsp90 of yeast, the alpha and beta isoforms of human Hsp90 differ with regard to their capacities for activation of certain client proteins, whereas only Hsp90beta generates sensitivity to the Hsp90 inhibitor radicicol. FEBS J. 2007;274:4453‐4463. [DOI] [PubMed] [Google Scholar]
- 42. Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol. 1995;36:305‐315. [DOI] [PubMed] [Google Scholar]
- 43. Garcia‐Carbonero R, Carnero A, Paz‐Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol. 2013;14:e358‐e369. [DOI] [PubMed] [Google Scholar]
- 44. Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S2