

Abstract
Background and Objectives
Use of parenteral amikacin to treat refractory nontuberculous mycobacterial (NTM) lung disease is limited by systemic toxicity. A population pharmacokinetic model was developed using data pooled from two randomized trials to evaluate the pharmacokinetic properties of once-daily amikacin liposome inhalation suspension (ALIS) in patients with treatment-refractory NTM lung disease.
Methods
In phase 2 (TR02-112) and phase 3 (CONVERT) studies, patients with sputum cultures positive for Mycobacterium avium complex (both studies) or M. abscessus (TR02-112) despite ≥ 6 months of guideline-based therapy were treated with once-daily ALIS 590 mg.
Results
Fifty-three patients (28 Japanese; 25 White) were assessed. At baseline and ≈ 6 months after daily dosing, median maximum concentration (Cmax) was < 2 mg/L and median area under the concentration-time curve (AUC0–24) was < 20 mg·h/L, suggesting low systemic exposure at both time points. Exposure estimates were similar between Japanese and White patients. The median unchanged amikacin fraction excreted in urine was < 10% of inhaled dose throughout the TR02-112 study, indicating that relatively small amounts reached systemic circulation. Median t1/2 was 5.5 h. Amikacin concentrations were much higher in sputum than in serum, demonstrating the ability to achieve higher drug concentration at the site of infection. Median sputum amikacin concentrations in the CONVERT study were high at 1–4 h postdose (range 242–426 μg/g) and decreased by 8 h (median 7 μg/g).
Conclusions
Systemic exposure to amikacin in serum and urine following once-daily ALIS administration in patients with treatment-refractory NTM lung disease was notably lower than that previously reported for parenteral amikacin.
Trial registration
ClinicalTrials.gov NCT01315236 (registered March 15, 2011) and NCT02344004 (registered January 22, 2015)
Supplementary Information
The online version contains supplementary material available at 10.1007/s13318-020-00669-7.
Key Points
| Amikacin liposome inhalation suspension (ALIS) was developed as a treatment for refractory nontuberculous mycobacterium (NTM) lung disease caused by Mycobacterium avium complex in order to limit systemic exposure and increase amikacin uptake into alveolar macrophages at the site of infection. |
| In patients treated with once-daily ALIS, amikacin concentrations in sputum were much higher than in serum; systemic exposure was low at baseline and after 6 months of treatment. |
| Serum and urine amikacin concentrations in patients treated with once-daily ALIS were much lower than previously reported for parenteral amikacin. |
Introduction
Nontuberculous mycobacteria (NTM) are ubiquitous environmental organisms that are generally innocuous but can cause progressive lung disease, predominantly in people with underlying chronic lung diseases such as bronchiectasis or chronic obstructive pulmonary disease [1, 2]. The prevalence of NTM lung disease is increasing in many countries and can vary by patient demographics and clinical characteristics (e.g., age, sex, race/ethnicity, geography, pre-existing comorbidities) [1–3]. Treatment generally includes at least three active antibiotics for ≥ 12 months depending on the species and disease severity [4]. Amikacin is an aminoglycoside antibiotic with potent activity against NTM species and has a role in the treatment of NTM lung disease. However, the clinical use of parenteral aminoglycosides in NTM lung disease is generally limited to severe or macrolide-resistant disease, and administration is of limited duration due to the potential for ototoxicity and nephrotoxicity [1, 4]. Amikacin liposome inhalation suspension (ALIS) was developed to limit the systemic exposure of amikacin and is recommended to be added to treatment regimens for patients with Mycobacterium avium complex (MAC) lung disease who do not have a microbiologic response (from MAC positive sputum to MAC negative) following 6 months of antibiotic treatment [5, 6]. This recommendation was based on phase 2 (TR02-112) and phase 3 (CONVERT) clinical trials in which the addition of ALIS to guideline-based multidrug therapy in patients with treatment-refractory NTM lung disease significantly increased sputum culture conversion rates versus treatment with guideline-based therapy alone [7, 8].
Amikacin is rapidly absorbed following intramuscular injection, with peak serum concentrations (Cmax) obtained within 1 h following a single dose and a serum half-life (t1/2) of slightly over 2 h; 91.9% and 98.2% of the dose is excreted unchanged in urine within 8 and 24 h, respectively, in adults with normal renal function [9]. When administered intravenously (IV) in normal adults, Cmax is obtained by the end of a 30-min infusion with a single dose (7.5 mg/kg); 84% and 94% of the dose is excreted in the urine within 9 and 24 h, respectively [9]. In patients with MAC treated with IV amikacin (15 mg/kg mean daily dose), mean (SD) Cmax was 55.3 (16.9) µg/mL and mean (SD) area under the concentration-time curve (AUC0–6/7) was 152.8 (50.8) µg·h/mL [10]. Following 3 months of daily ALIS 590 mg in patients with MAC, mean Cmax was 2.8 µg/mL and mean AUC0–24 was 23.5 µg·h/mL; apparent serum t1/2 ranged from 5.9 to 19.5 h, and 7.42% of the dose was excreted unchanged in the urine [11].
Population pharmacokinetic data for ALIS (up to 28 days administration) have been reported in children and young adults with cystic fibrosis and chronic Pseudomonas aeruginosa infection [12, 13] but not in adults with MAC lung disease. Since pharmacokinetic parameters of aminoglycosides, including amikacin, can vary depending on the disease for which they are being used as well as the age of the patient [14, 15], and ethnic differences in pharmacokinetic parameters may contribute to variable responses to different drug types [16, 17], it is important to characterize ALIS pharmacokinetic data in adults with NTM lung disease to assess amikacin exposure with nebulized administration in this specific population.
In this pharmacokinetic analysis, data from the TR02-112 and CONVERT clinical trials were used to characterize systemic exposure to amikacin in adults with NTM lung disease with up to 6 months of ALIS administration, compare systemic exposure in Japanese versus White patients, and evaluate sputum amikacin concentrations before and after dosing.
Methods
Study Designs and Patients
Pharmacokinetic data from the two randomized, multicenter, prospective studies—TR02-112 and CONVERT—were pooled and analyzed. TR02-112 was a double-blind, placebo-controlled phase 2 study [8], and CONVERT was an open-label phase 3 study [7]. Both studies assessed the efficacy and safety of ALIS in adult patients with treatment-refractory NTM lung disease. Patients in both studies met 2007 American Thoracic Society/Infectious Diseases Society of America diagnostic criteria [18] for NTM lung disease and had sputum cultures that were persistently positive for MAC (both studies) or M. abscessus (TR02-112) despite receiving guideline-based multidrug therapy for ≥ 6 months. Key exclusion criteria in TR02-112 were forced expiratory volume in 1 sec (FEV1) predicted < 30%; systemic immune deficiency; clinically significant cardiac, pulmonary, hepatic, or renal disease; current smoking; and active malignancy. Key exclusion criteria in CONVERT were cystic fibrosis, active pulmonary tuberculosis, immunodeficiency syndromes, MAC isolates with amikacin resistance (minimum inhibitory concentration [MIC] > 64 µg/mL) on culture screening, and active malignancy.
In both studies, ALIS was administered once daily by an investigational eFlow® Technology nebulizer (PARI Pharma GmbH, Munich, Germany). In CONVERT [7], patients were randomly assigned 2:1 to receive ALIS 590 mg added to their ongoing guideline-based multidrug therapy or continued multidrug therapy alone; patients could receive ALIS for up to 16 months (open-label) followed by a 12-month off-treatment period. In TR02-112 [8], patients were randomly assigned 1:1 to receive ALIS 590 mg or placebo (empty liposomes) once daily; patients could receive ALIS for up to 168 days (84-day initial double-blind period followed by 84-day open-label administration) followed by a 28-day off-treatment period.
The pharmacokinetic substudies were conducted at 12 CONVERT sites (5 in the USA; 7 in Japan) and 19 TR02-112 sites (18 in the USA; 1 in Canada).
Pharmacokinetic Samples
The sampling scheme for each trial is shown in Table S1 of the Electronic Supplementary Material. In TR02-112, systemic exposure to amikacin was assessed in blood, urine, and sputum in a subset of White patients. Blood samples were drawn at 0–4 h predose and at 0–4 h postdose on days 1, 2, 28, 56, 84, 112, and 168. Sputum samples (minimum 3 mL expectorated or induced) were collected at 0–4 h predose and at least two samples were collected at 0–1 h postdose on days 1, 2, 28, 56, 84, 112, and 168. Urine samples were collected at 0–24 h postdose on days 1, 84, and 168.
In CONVERT, blood and sputum samples were collected in a population pharmacokinetic subset comprising patients who agreed to participate and adhere to the sampling schedule (select sites in the USA and Japan only) and in a separate comprehensive pharmacokinetic substudy conducted only in Japanese patients participating at trial sites in Japan. In the population pharmacokinetic subset, blood and sputum samples were collected at 0–1 h predose and at 1–4 h postdose at months 1, 3, and 6. In addition, sputum samples were collected in a sputum-only subset of patients at 8 and 24 h postdose on day 1; 72 h postdose at months 4, 5, and 6; and the 28-day and 3-month follow-up visits. The predose and 72-h postdose sputum samples (at least two samples; minimum 3 mL expectorated or induced) were collected after 2 days of dose interruption. In the comprehensive pharmacokinetic substudy in Japanese patients, blood and sputum samples were collected at 0–1 h predose on day 1 and at months 1, 3, and 6. Postdose blood samples were collected at 1, 2, 4, 6, 8, 12, and 24 h postdose on day 1 and at month 3, and at 1–4 h postdose at months 1 and 6. Postdose sputum samples were collected at 8 (optional) and 24 h postdose on day 1 and at 1–4 h postdose at months 1, 3, and 6.
Amikacin concentration was measured in serum and sputum samples using liquid chromatography with tandem mass spectrometry with a lower limit of quantification (LLOQ) of 0.15 mg/L for serum and 0.6 μg/g for sputum. Additional details regarding the measurement of amikacin concentration are provided in the Electronic Supplementary Material.
Population Pharmacokinetic Analysis
The population pharmacokinetic dataset was constructed using SAS software v9.4 (SAS Institute, Inc; Cary, NC, USA). When available, dosing start and end times were used to calculate the duration of inhalation; if unavailable, the duration was imputed based on the previous dose for that patient. No data points met the criteria for an outlier (defined as an aberrant observation that substantially deviated from the rest of the observations for an individual subject) and therefore all data points were used in the analysis. Serum concentration values reported as below the limit of quantification (BLQ) were included in the population pharmacokinetic analysis with the exception of those that were obtained before the first dose, which were excluded. All concentrations reported as BLQ were handled by employing the M3 method of Beal [19].
The original population pharmacokinetic model that was developed using data from cystic fibrosis patients [13] was used as the basis for the present analyses, with plans to refine the model as necessary to obtain a reasonable fit to the data from patients with NTM. In the 3-compartment model with absorption (lungs), central, and urine compartments, zero-order drug administration to the lungs via nebulization, a first-order process, carried the drug from the lungs to the central compartment, and the drug was removed from the body by linear elimination (Fig. 1). Amikacin in urine was modeled as accumulating in the urine compartment after clearance from the central compartment in a first-order process in order to predict bioavailability. Candidate models were simultaneously fit to serum concentration and urine amount data using Monte Carlo parametric expectation maximization in open-source population analysis software S-ADAPT v1.57 [20, 21]. Since serum sampling in the CONVERT pharmacokinetic substudy (White patients) was relatively sparse, the pharmacokinetic model that was applied to the data from TR02-112 [13] was used to analyze the pooled data from the two studies.
Fig. 1.
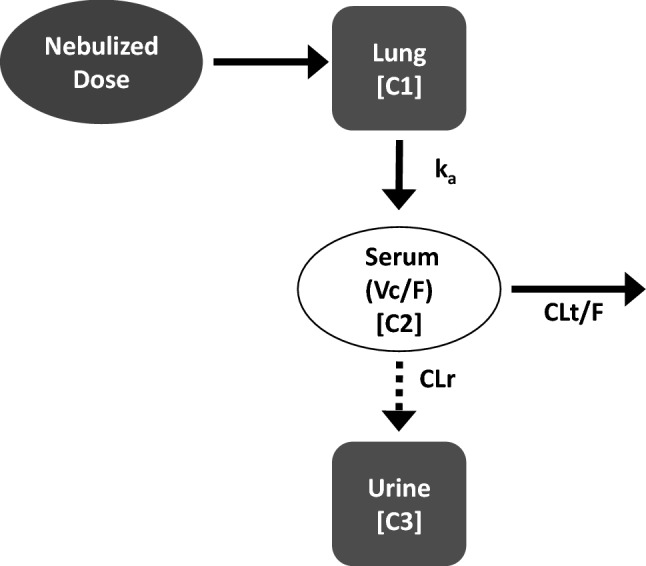
3-compartment pharmacokinetic model. Absorption (lungs), central, and urine compartments, zero-order drug administration to the lungs via nebulization occurred in the lungs, a first-order process carried the drug from the lungs to the central compartment, and the drug was removed from the body by linear elimination. C1, C2, C3 compartments 1, 2, 3; CLr renal clearance; CLt/F apparent clearance; ka absorption rate constant; Vc/F apparent volume of the central compartment
Where possible, inter-individual variability (IIV) was modeled for each pharmacokinetic parameter using an exponential variability model assuming log-normal distributions. For individual population pharmacokinetic parameter estimates, the presence of significant inter-occasion variability was also assessed; however, since samples in individual patients were drawn on up to six different occasions with only one sample being drawn on some occasions, random inter-occassion variability was not estimated for any of the pharmacokinetic parameters. Residual variability was initially described using an additive error model, with the option of using other error models if necessary; separate error models were used for serum and urine data.
Amikacin exposure estimates (AUC0–24 and Cmax) at baseline (day 1) and on day 168 (≈ 6 months) were generated for each patient using their individual pharmacokinetic parameter estimates from the fit of the population pharmacokinetic model. The model-derived AUC0–24 and Cmax estimates were compared between Japanese and White patients using box-and-whisker plots. In addition to the model-derived values, which were generated for all patients, estimates for AUC0–24 and Cmax for the Japanese patients who participated in the comprehensive pharmacokinetic substudy in CONVERT were also calculated using non-compartmental methods. The non-compartmental estimates were used as a form of model qualification by comparing them to the model-derived estimates of AUC0–24 and Cmax for these patients.
Sputum Pharmacokinetic Analyses
Sputum amikacin concentrations were not included in the population pharmacokinetic analysis. However, summary statistics of the sputum concentrations at nominal sampling times were calculated separately for the entire pharmacokinetic population and for the comprehensive pharmacokinetic substudy (Japanese patients) in CONVERT. Given the relatively high number of sputum concentrations that were reported as BLQ, three methods of handling BLQs for the summary statistic calculations were employed—(1) assigning each BLQ concentration a value of 0.3 μg/g (0.5 × LLOQ), (2) assigning each BLQ concentration a value of 0.6 μg/g (1 × LLOQ), and (3) assigning each BLQ concentration a value of 0 μg/g. The resultant summary statistics were then compared to test the sensitivity of the results to the imputation strategy. Power and sample size calculations for this study were based on clinical outcomes as pharmacokinetic endpoints were considered secondary.
Results
Population Demographics and Baseline Clinical Characteristics
The pharmacokinetic analysis population (n = 53) included 14 patients from TR02-112 and 39 patients from CONVERT (Table 1). The median age of the patients was 63 years, with 43% aged ≥ 65 years and the majority being female (85%). Table 1 shows selected clinical characteristics at baseline. Weight, body mass index (BMI), and body surface area were significantly greater among White patients, while the estimated glomerular filtration rate (eGFR) was significantly lower in Japanese patients.
Table 1.
Demographics and baseline clinical characteristics (population pharmacokinetic analysis population)
| Characteristic | TR02-112 (N = 14) |
CONVERT (N = 39) |
Total (N = 53) |
|---|---|---|---|
| Age, median (range), years | 57 (20–77) | 66 (47–84) | 63 (20–84) |
| ≥ 65 years, n (%) | 3 (21.4) | 20 (51.3) | 23 (43.4) |
| Female, n (%) | 14 (100) | 31 (79.5)a | 45 (84.9) |
| Race/ethnicity, n (%) | |||
| Japanese | 0 (0) | 28 (71.8)a | 28 (52.8) |
| White | 14 (100) | 11 (28.2)a | 25 (47.2) |
| Weight, median (range), kg | 65.2 (43.9–80) | 50 (33.8–77.1) | 52.6 (33.8–80) |
| BMI, median (range), kg/m2 | 21.6 (18.6–29.3) | 20.1 (13.5–24.9) | 20.6 (13.5–29.3) |
| BSA, median (range), m2 | 1.75 (1.35–1.91) | 1.48 (1.25–1.97) | 1.54 (1.25–1.97) |
| eGFR, median (range), mL/min/1.73 m2 | 86.3 (63.3–140) | 88.4 (57.4–124) | 88.3 (57.4–140) |
BSA body surface area; BMI body mass index; eGFR estimated glomerular filtration rate
aData previously reported in Griffith et al. [7]
Data Disposition
In TR02-112, 111 serum and 23 urine samples from 14 patients were available for pharmacokinetic analysis; serum samples included 16 concentrations that were BLQ. In CONVERT, 380 serum samples (including 73 with BLQ concentrations) from 39 patients were available for pharmacokinetic analysis. Because all observations were consistent with the expected concentration-time profiles following ALIS administration, all serum and urine data were included in the analyses. Despite the number of samples collected from each patient (average 7.6 samples), the overall sampling scheme was relatively sparse, as the samples were spread out over ≤ 6 occasions. This, combined with the variability inherent in the absorption process, precluded formal identification of outliers.
Sample time points were consistent with the sampling scheme described in each study (see Table S1 in the Electronic Supplementary Material for the sampling scheme). The serum samples in TR02-112 were clustered in two primary windows—0–4 h and 12–24 h postdose (Fig. 2). Some samples were drawn > 24 h after the previous dose, likely reflecting the timing of at-home doses relative to the timing of clinic visits when pharmacokinetic samples were drawn. The serum samples in CONVERT were less clustered throughout the first 24 h postdose, with an additional cluster at approximately 72 h postdose (Fig. 2).
Fig. 2.
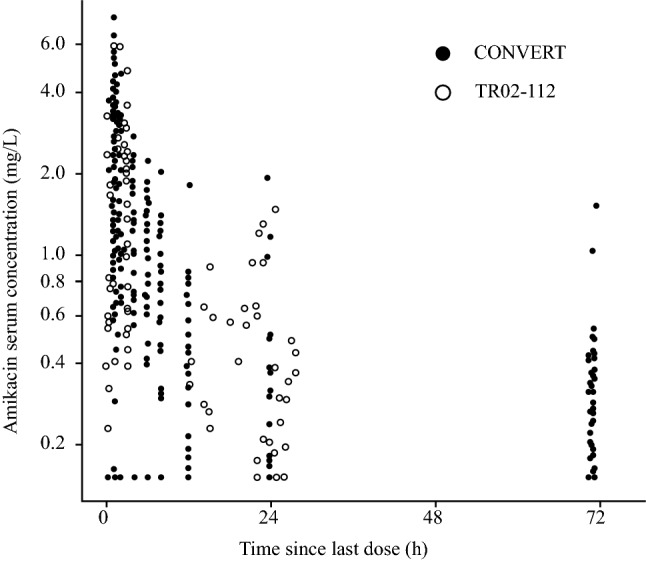
Observed serum amikacin concentration versus time since last dose. Closed and open circles represent individual values for patients in CONVERT and TR02-112 studies, respectively. Amikacin concentrations that were BLQ are shown at LLOQ (i.e., 0.15 mg/L). One BLQ concentration in the TR02-112 study, observed at > 500 h after the previous dose, was excluded; it was retained in the population pharmacokinetic dataset but did not impact the model fit. BLQ below the limit of quantification; LLOQ lower limit of quantification
Population Pharmacokinetic Model
The population pharmacokinetic model derived from previous studies in patients with CF [13] provided a robust fit to the data from patients with NTM from studies TR02-112 and CONVERT. No changes to the model structure or the covariate relationships were deemed necessary given the relatively small sample size and sparse pharmacokinetics, as well as previous analyses demonstrating that additional covariate relationships were unlikely to be clinically relevant due to the use of inhaled administration, which results in substantial random variability in drug concentrations. Population pharmacokinetic parameter estimates and associated precision from the model fit to the data are shown in Table 2. Substantial IIV was observed in the pharmacokinetic parameters, likely due to the variability in systemic absorption after inhaled administration. The IIV estimates for apparent clearance (CLt/F) and apparent volume of the central compartment (Vc/F) were 71.82% and 65.09%, respectively; those for absorption rate constant (ka) and renal clearance (CLr) were more moderate (40.30% and 30.24%, respectively). Overall, the population pharmacokinetic parameters were estimated with acceptable precision, with the exception of IIV in ka and CLr (standard error of mean, 122.1% and 142.3%, respectively). Inter-occasion variability was not required.
Table 2.
Population pharmacokinetic parameter estimates for amikacin
| Parameter | Population mean | Inter-individual variability (% CV) | ||
|---|---|---|---|---|
| Final estimate | %SEM | Final estimate | %SEM | |
| CLt/F, L/h | 34.29 | 16.36 | 71.82 | 45.25 |
| Vc/F, L | 272.6 | 11.41 | 65.09 | 31.2 |
| ka, h−1 | 1.866 | 24.86 | 40.30 | 122.1 |
| CLr, L/h | 1.990 | – | 30.24 | 142.3 |
| CLr coefficient, L/h | 1.931 | 14.29 | – | – |
| CLr WTKG power | 0.75 | – | – | – |
| Residual error (serum) | 0.615 | 3.941 | – | – |
| Residual error (urine) | 14.0 | 27.30 | – | – |
CLr coefficient is the population mean value for CLr in a patient weighing 51 kg (population mean CLr = 1.931 × (WTKG/51)0.75)
CLr renal clearance; CLt/F apparent clearance; CV coefficient of variation; ka absorption rate constant; SEM standard error of mean; Vc/F apparent volume of the central compartment; WTKG body weight in kg
The goodness-of-fit plots suggest that the population pharmacokinetic model is providing a robust fit to the data. The coefficient of determination (r2) for the observed versus individual fitted values was 0.802 for serum (pooled data from both studies) and 0.891 for urine (TR02-112 only) (Fig. 3). The individual data points were scattered appropriately around the line of identity, suggesting the absence of systematic bias.
Fig. 3.

Goodness-of-fit plots for the pooled population pharmacokinetic model (individual fitted values) versus observed data. Panels a and b show the observed versus individual fitted concentrations for serum and urine, respectively. Closed circles represent individual values for patients in CONVERT and open circles represent individual values for patients in TR02-112; the solid line indicates the line of best fit, while the dashed line represents the line of identity. Panels c and d show conditional weighted residuals versus population fitted concentrations for serum and urine, respectively; the dashed line represents a conditional weighted residual of zero. r2 coefficient of determination
Amikacin Pharmacokinetic Exposure Estimates
Model-derived serum amikacin exposure and t1/2 estimates on day 1 and at approximately 6 months are shown in Table 3. The median Cmax values in CONVERT were slightly higher than those in TR02-112; values for AUC0–24 were similar between the two studies. The overall median Cmax and AUC0–24 values at approximately 6 months were slightly higher than those on day 1; the differences were < 10%. The median t1/2 of amikacin was approximately 5.5 h. In TR02-112, the proportion of amikacin dose excreted in urine was consistently low on day 1 (median 3.25%; range 2.71–8.95%) through to approximately 6 months (median 8.42%; range 0.72–22.6%) (Electronic Supplementary Material Table S2).
Table 3.
Summary statistics of serum amikacin exposure and t1/2 at day 1 and following approximately 6 months of active ALIS treatment (population pharmacokinetic analysis population)
| Parameter | TR02-112 (N = 14) |
CONVERT (N = 39) |
Total (N = 53) |
|---|---|---|---|
| Cmax, median (range), mg/L | |||
| Day 1 | 1.31 (0.621–5.48) | 1.75 (0.465–6.61)b | 1.70 (0.465–6.61) |
| ≈ 6 months | 1.65 (0.636–5.65) | 1.85 (0.482–6.87)b | 1.81 (0.482–6.87) |
| AUC0–24, median (range), mg·h/L | |||
| Day 1 | 15.1 (4.99–44.3) | 15.8 (4.16–53.5)b | 15.8 (4.16–53.5) |
| ≈ 6 months | 16.1 (5.11–51.7) | 16.7 (4.31–55.6)b | 16.7 (4.31–55.6) |
| t1/2, median (range), ha | 5.48 (4.32–14.0) | 5.48 (3.29–7.37) | 5.48 (3.29–14.0) |
Note: Since BLQ concentrations were derived from the fit of the model to the dataset in which they were retained, the pharmacokinetic parameters reported here take these concentrations into account
ALIS amikacin liposome inhalation suspension; AUC0–24 area under the plasma concentration time curve over a 24-h dosing interval; BLQ below the limit of quantification; Cmax maximum concentration; CLt/F apparent clearance; t1/2 elimination half-life; Vc/F apparent volume of the central compartment
at1/2 estimates were derived from post hoc CLt/F and Vc/F and are independent of study day
bData previously reported in Griffith et al. [7]
Exposure estimates derived from the population pharmacokinetic model in Japanese patients (CONVERT) were compared with those in White patients (TR02-112 and CONVERT) (Electronic Supplementary Material Figure S2). AUC0–24 and Cmax distributions were similar between Japanese and White patients (P= 0.895 and 0.411, respectively; Mann–Whitney U test). The potential for two key patient factors—lung and renal function—to affect systemic exposure to amikacin after ALIS administration was also explored. No apparent relationship was observed between serum amikacin AUC0–24 and lung function (represented by baseline FEV1% predicted) or renal function (represented by eGFR) (see Electronic Supplementary Material Figure S3). Exposure estimates derived from the population pharmacokinetic model were compared with those from non-compartmental methods for Japanese patients who participated in the comprehensive pharmacokinetic substudy in CONVERT (Electronic Supplementary Material Figure S1). The two estimates for AUC0–24 were reasonably congruent, although the population pharmacokinetic estimates were generally higher than the non-compartmental estimates; the Cmax estimates were similar for the two methods.
Sputum Amikacin Concentrations
In TR02-112, 12 patients provided 143 sputum samples (73 predose; 70 postdose). Overall, 52.1% of the predose samples and 37.1% of the postdose samples had BLQ concentrations (i.e., < 0.6 μg/g), and sputum amikacin concentrations were highly variable (Fig. 4). The median postdose sputum amikacin concentration (3,185 μg/g; range 1.1–16,799 μg/g) was much higher than the median predose concentration (38.2 μg/g; range 0.8–7,080 μg/g).
Fig. 4.

Sputum amikacin concentrations (CONVERT). Each circle represents individual patient data. The solid line represents a smoother through the data, assuming a 3-compartment pharmacokinetic model; the shaded region is the 90% prediction interval from a simulation assuming a % coefficient of variation of 95 for all parameters
In CONVERT, sputum samples from 59 patients (sputum concentration analysis population) were analyzed. As in TR02-112, the sputum amikacin concentrations in patients from CONVERT were highly variable (Fig. 4). As expected, the median sputum amikacin concentrations at 1–4 h postdose (426, 242, and 414 μg/g at 1, 3, and 6 months, respectively) were consistently much higher than the median predose concentrations (3.6, 6.1, and 1.3 μg/g at 1, 3, and 6 months, respectively). The concentrations markedly decreased by 8 h postdose (median for samples collected on day 1, 7.0 μg/g) and were maintained for up to 72 h postdose (range for samples collected at 24 and 72 h postdose on day 1 and at months 4, 5, and 6, 7.1–17.4 μg/g). As expected, the median sputum amikacin concentrations at the 28-day and 3-month follow-up visits were very low and BLQ.
The sensitivity analyses, in which the BLQ observations were assigned a value of 0 μg/g, 0.5 × LLOQ, or 1 × LLOQ, did not change the above conclusions regarding the sputum concentrations over time after ALIS administration.
Discussion
In the present analyses, systemic exposure to amikacin following once-daily nebulization of ALIS 590 mg in patients with treatment-refractory NTM lung disease was relatively low, with a median Cmax < 2 mg/L and median AUC0–24 < 20 mg·h/L. In CONVERT, the relatively small difference (< 10%) in median AUC0–24 between day 1 and after approximately 6 months of daily dosing suggests that systemic exposure is similar regardless of treatment duration. In TR02-112, < 10% of amikacin (mean or median) was excreted in urine, suggesting that once-daily nebulized ALIS leads to low systemic circulation of amikacin; there also was little accumulation after 168 days, as previously reported [22]. Additionally, ALIS dose adjustments may not be needed for Japanese patients based on similar serum amikacin concentrations found in Japanese and White patients.
The amikacin exposure reported here was similar to that found in ALIS-treated patients with cystic fibrosis and chronic P. aeruginosa infection [12, 13] but much lower than that with parenteral amikacin. Commonly administered as short intravenous infusions of 7.5–30 mg/kg, parenteral amikacin produces a mean AUC0–6/7 of approximately 150 mg·h/L [10] and a mean or median AUC0–24 ranging from 67 to 548 mg·h/L [23–25]. Similarly, the mean Cmax values observed here (range 0.47–6.87 mg/L) are markedly lower than those previously reported for parenteral amikacin (35–77 mg/L) [10, 24, 26]. It is important to note that no pharmacokinetic–pharmacodynamic targets for ALIS have been established to guide therapeutic drug monitoring. Furthermore, no target drug exposure needed for efficacy has been established. However, NTM isolate susceptibility to amikacin has been described by breakpoint resistance, with a minimum inhibitory concentration of ≥ 64 μg/mL for parenteral amikacin and ≥ 128 μg/mL for ALIS, defining resistance in patients with MAC lung disease [27].
The challenge in achieving high drug concentrations in the lungs in order to maximize antimicrobial activity is highlighted by a hollow fiber infection model study of M. abscessus lung disease, which showed that the standard dose of parenteral amikacin (75–1,500 mg/day) did not achieve desirable efficacy (80% of maximal kill [EC80]), and that a considerably higher dose (4 g/day) was required to achieve EC80 in 70% of patients [28]. Local administration of amikacin directly to the lung is one strategy that can be used to maximize pulmonary concentrations. In the overall population of patients in CONVERT [7], culture conversion was achieved by 6 months in 29% of those receiving ALIS plus a multidrug regimen versus 9% of those treated with a multidrug regimen alone (P < 0.0001). Similarly, 32% of ALIS-treated patients in TR02-112 had negative sputum cultures at day 84 versus 9% of those treated with placebo (P = 0.006) [8]. In this population pharmacokinetic analysis, ALIS inhalation resulted in median sputum amikacin concentrations (a proxy for lung concentrations) that were consistently higher than amikacin concentrations in serum. Liposomal drug delivery targeting macrophage-mediated disease is another strategy that can be used to increase the intracellular concentration of drugs in target cells [29]. Alveolar macrophages are the primary reservoir for NTM in humans, where the bacteria persist intracellularly and lead to chronic lung disease in susceptible individuals. Compared with non-liposomal amikacin, ALIS increased amikacin uptake into cultured human macrophages and increased drug retention in the lungs of rats [6]. Both the inhalation route to deliver amikacin directly to the lungs and the liposomal delivery mechanism to increase the concentration of amikacin in target cells are hypothesized to contribute to the efficacy of ALIS in NTM lung disease.
As previously shown with the hollow fiber infection model, having high concentrations at the target site is essential to achieving exposures sufficient for killing of NTM; however, the parenteral amikacin dose needed to achieve efficacy was predicted to result in high rates of ototoxicity within 1–2 months of treatment [28]. The risk for developing ototoxicity or nephrotoxicity is associated with genetic susceptibility [30] and treatment variables, such as dosing schedule (once vs twice daily), exposure (AUC), concomitant medications, and overall treatment duration [25, 31]. Although a direct comparison of previously published data with the data in this report is limited by differences in patient populations, pathogens, treatment duration, and use of concomitant medications, amikacin exposure in ALIS-treated patients in the present analyses was considerably lower than those reported to correlate with the risk of ototoxicity (cumulative AUC 87,232–120,000 days·mg·h/L) [25] or nephrotoxicity (AUC ≈ 750 mg·h/L) [31]. In the overall population in CONVERT, ototoxicity- and nephrotoxicity-related adverse events were non-serious, infrequent, and generally similar between patients treated with ALIS plus a multidrug regimen and those treated with a multidrug regimen alone, with the exception of tinnitus (7.6% and 0.9% of patients, respectively) [7].
The pharmacokinetic data were best described with a 3-compartment population model. The model fit to the observed data resulted in relatively precise parameter estimates with the exception of IIV in ka and CLr, which is a likely consequence of the incomplete and variable transfer of ALIS from lung to serum. An allometric relationship between body weight and CLr was the only covariate relationship included in the model. A formal covariate evaluation was not conducted because the sample size was relatively small and the inherent random variability in amikacin pharmacokinetic properties after ALIS administration did not allow identification of significant covariate effects (data on file). The AUC0–24 estimates from the population pharmacokinetic model were higher than the non-compartmental estimates, likely due to the relatively sparse sampling scheme, which tends to result in lower non-compartmental estimates [32]. Regardless, the low systemic exposure and bioavailability observed in these analyses suggest that once-daily nebulized ALIS may be associated with reduced risk of ototoxicity and nephrotoxicity compared with parenteral amikacin, although direct comparative data are not available.
Limitations to our analyses included the small sample size and the sparse sampling scheme. The population pharmacokinetic substudy of the CONVERT trial was an optional analysis limited to select sites in the USA and Japan. As participation required additional visits and separate consent, not all eligible patients elected to participate. As urine samples were not collected in CONVERT, it was not possible to assess amikacin bioavailability in the pooled dataset. However, as the systemic exposure in TR02-112 and CONVERT was comparable, the bioavailability is likely to be similarly low in both studies. Additionally, sputum amikacin concentrations were variable, which is likely an inherent limitation of these measurements and is neither unexpected nor unprecedented. Such variability has been previously reported [12, 13] and may represent the difficulty in sample collection, variability in the time period elapsed since dosing, or perhaps a property inherent to sputum drug concentration measurements. It is unclear whether sputum concentrations directly reflect drug concentration at the site of NTM infection in the lung. However, the measurement of drug concentrations in the lung is not routine, and currently there is no standard in the sampling and measurement of lung pharmacokinetics [33]. Lastly, because of the highly variable nature of amikacin concentrations measured in sputum, simultaneously modeling sputum and serum concentrations has not been feasible, although we recognize the usefulness that such an analysis could provide.
In conclusion, the systemic exposure to amikacin in serum and urine of patients with NTM lung disease receiving ALIS once daily was notably lower than that previously reported for parenteral amikacin. A comparison of AUC at day 1 and after approximately 6 months of active treatment suggests that systemic exposure is similar at both time points; < 10% of amikacin reached the systemic circulation. Furthermore, systemic exposure to amikacin was similar in both Japanese and White patients with NTM lung disease. Amikacin concentrations in sputum were considerably higher than those in serum, demonstrating that once-daily ALIS administration in patients with NTM lung disease results in relatively high concentrations of amikacin in the lungs.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Kristen A. Andersen, PhD, of MediTech Media, Ltd, provided medical writing assistance; editorial and creative support were also provided by MediTech Media, Ltd. Insmed Incorporated provided funding to Meditech Media, Ltd, for these services. We acknowledge Gina Eagle, MD, who was an employee of Insmed Incorporated and oversaw the conduct of the TR02-112 and CONVERT clinical trials.
Declarations
Funding
This work and the Open Access fee for the published article were supported by Insmed Incorporated, Bridgewater, NJ, USA.
Conflicts of Interest/Competing Interests
ICPD Inc received funding from Insmed Incorporated to conduct the analysis and provide general consulting to Insmed. DEG has received consulting fees and research grants from Insmed. JvI is a member of the Insmed Advisory Board. DWY and KCM are employed by Insmed. KLW has received consulting fees and research grants from Insmed.
Ethics Approval
The clinical studies TR02-112 (NCT01315236) and CONVERT (NCT02344004) were conducted in accordance with the principles of Good Clinical Practice and Declaration of Helsinki. The study protocols were approved by an independent ethics committee or institutional review board for each site.
Consent to Participate
All patients provided written informed consent.
Consent for Publication
Not applicable.
Availability of Data and Material (data transparency)
Not applicable.
Code Availability (software application or custom code)
Not applicable.
Authors’ Contributions
The authors made the following contributions: authors from the Institute for Clinical Pharmacodynamics, Inc (ICPD, Schenectady, NY, USA), and Insmed Incorporated (Bridgewater, NJ, USA) designed the study; KLW and DEG were trial investigators. CMR, NJO, and SMB conducted the analyses. CMR, NJO, JvI, KLW, DWY, and KCM interpreted the data. CMR, NJO, JvI, KLW, and DWY drafted the content. All authors provided critical review and approval of the manuscript.
Footnotes
The original online version of this article was revised: Text correction and revised version of figure 2 updated. Revised ESM file updated.
Change history
4/19/2021
A Correction to this paper has been published: 10.1007/s13318-021-00687-z
References
- 1.Larsson LO, Polverino E, Hoefsloot W, Codecasa LR, Diel R, Jenkins SG, et al. Pulmonary disease by non-tuberculous mycobacteria – clinical management, unmet needs and future perspectives. Expert Rev Respir Med. 2017;11(12):977–989. doi: 10.1080/17476348.2017.1386563. [DOI] [PubMed] [Google Scholar]
- 2.Adjemian J, Olivier KN, Seitz AE, Holland SM, Prevots DR. Prevalence of nontuberculous mycobacterial lung disease in U.S. Medicare beneficiaries. Am J Respir Crit Care Med. 2012;185(8):881–886. doi: 10.1164/rccm.201111-2016OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prevots DR, Marras TK. Epidemiology of human pulmonary infection with nontuberculous mycobacteria: a review. Clin Chest Med. 2015;36(1):13–34. doi: 10.1016/j.ccm.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daley CL, Iaccarino JM, Lange C, Cambau E, Wallace RJ, Andrejak C, et al. Treatment of nontuberculous mycobacterial pulmonary disease: an official ATS/ERS/ESCMID/IDSA clinical practice guideline: executive summary. Clin Infect Dis. 2020;71:905–913. doi: 10.1093/cid/ciaa1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malinin V, Neville M, Eagle G, Gupta R, Perkins WR. Pulmonary deposition and elimination of liposomal amikacin for inhalation and effect on macrophage function after administration in rats. Antimicrob Agents Chemother. 2016;60(11):6540–6549. doi: 10.1128/AAC.00700-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Leifer F, Rose S, Chun DY, Thaisz J, Herr T, et al. Amikacin liposome inhalation suspension (ALIS) penetrates non-tuberculous mycobacterial biofilms and enhances amikacin uptake into macrophages. Front Microbiol. 2018;9:915. doi: 10.3389/fmicb.2018.00915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffith DE, Eagle G, Thomson R, Aksamit TR, Hasegawa N, Morimoto K, et al. Amikacin liposome inhalation suspension for treatment-refractory lung disease caused by Mycobacterium avium complex (CONVERT). a prospective, open-label, randomized study. Am J Respir Crit Care Med. 2018;198(12):1559–1569. doi: 10.1164/rccm.201807-1318OC. [DOI] [PubMed] [Google Scholar]
- 8.Olivier KN, Griffith DE, Eagle G, McGinnis JP, 2nd, Micioni L, Liu K, et al. Randomized trial of liposomal amikacin for inhalation in nontuberculous mycobacterial lung disease. Am J Respir Crit Care Med. 2017;195(6):814–823. doi: 10.1164/rccm.201604-0700OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amikacin sulfate injection . USP (For intramuscular or intravenous use) [package insert] Schaumburg: Sagent Pharmaceuticals; 2018. [Google Scholar]
- 10.van Ingen J, Egelund EF, Levin A, Totten SE, Boeree MJ, Mouton JW, et al. The pharmacokinetics and pharmacodynamics of pulmonary Mycobacterium avium complex disease treatment. Am J Respir Crit Care Med. 2012;186(6):559–565. doi: 10.1164/rccm.201204-0682OC. [DOI] [PubMed] [Google Scholar]
- 11.ARIKAYCE® [package insert]. Bridgewater, NJ: Insmed Incorporated; 2018
- 12.Okusanya OO, Bhavnani SM, Hammel J, Minic P, Dupont LJ, Forrest A, et al. Pharmacokinetic and pharmacodynamic evaluation of liposomal amikacin for inhalation in cystic fibrosis patients with chronic pseudomonal infection. Antimicrob Agents Chemother. 2009;53(9):3847–3854. doi: 10.1128/AAC.00872-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okusanya OO, Bhavnani SM, Hammel JP, Forrest A, Bulik CC, Ambrose PG, et al. Evaluation of the pharmacokinetics and pharmacodynamics of liposomal amikacin for inhalation in cystic fibrosis patients with chronic pseudomonal infections using data from two phase 2 clinical studies. Antimicrob Agents Chemother. 2014;58(9):5005–5015. doi: 10.1128/AAC.02421-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Begg EJ, Barclay ML, Kirkpatrick CM. The therapeutic monitoring of antimicrobial agents. Br J Clin Pharmacol. 2001;52(Suppl 1):35S–43S. doi: 10.1046/j.1365-2125.2001.0520s1035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez E, Perez R, Hernandez A, Tejada P, Arteta M, Ramos JT. Factors and mechanisms for pharmacokinetic differences between pediatric population and adults. Pharmaceutics. 2011;3(1):53–72. doi: 10.3390/pharmaceutics3010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasunuma T, Tohkin M, Kaniwa N, Jang IJ, Yimin C, Kaneko M, et al. Absence of ethnic differences in the pharmacokinetics of moxifloxacin, simvastatin, and meloxicam among three East Asian populations and Caucasians. Br J Clin Pharmacol. 2016;81(6):1078–1090. doi: 10.1111/bcp.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasuda SU, Zhang L, Huang SM. The role of ethnicity in variability in response to drugs: focus on clinical pharmacology studies. Clin Pharmacol Ther. 2008;84(3):417–423. doi: 10.1038/clpt.2008.141. [DOI] [PubMed] [Google Scholar]
- 18.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367–416. doi: 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 19.Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481–504. doi: 10.1023/A:1012299115260. [DOI] [PubMed] [Google Scholar]
- 20.Bauer RJ. S-ADAPT Software. Biomedical Simulations Resource. University of Southern California. [cited 2020 January 15]; Available from: https://bmsr.usc.edu/downloads/s-adapt/
- 21.D'Argenio DZ, Schumitzky A. A program package for simulation and parameter estimation in pharmacokinetic systems. Comput Programs Biomed. 1979;9(2):115–134. doi: 10.1016/0010-468X(79)90025-4. [DOI] [PubMed] [Google Scholar]
- 22.Olivier KN, Maas-Moreno R, Whatley M, Cheng K, Lee JH, Fiorentino C, et al. Airway deposition and retention of liposomal amikacin for inhalation in patients with pulmonary nontuberculous mycobacterial disease. San Francisco: Am J Respir Crit Care Med. American Thoracic Society; 2016. [Google Scholar]
- 23.Byl B, Baran D, Jacobs F, Herschuelz A, Thys JP. Serum pharmacokinetics and sputum penetration of amikacin 30 mg/kg once daily and of ceftazidime 200 mg/kg/day as a continuous infusion in cystic fibrosis patients. J Antimicrob Chemother. 2001;48(2):325–327. doi: 10.1093/jac/48.2.325. [DOI] [PubMed] [Google Scholar]
- 24.Garraffo R, Drugeon HB, Dellamonica P, Bernard E, Lapalus P. Determination of optimal dosage regimen for amikacin in healthy volunteers by study of pharmacokinetics and bactericidal activity. Antimicrob Agents Chemother. 1990;34(4):614–621. doi: 10.1128/AAC.34.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Modongo C, Pasipanodya JG, Zetola NM, Williams SM, Sirugo G, Gumbo T. Amikacin concentrations predictive of ototoxicity in multidrug-resistant tuberculosis patients. Antimicrob Agents Chemother. 2015;59(10):6337–6343. doi: 10.1128/AAC.01050-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peloquin CA. Mycobacterium avium complex infection. Pharmacokinetic and pharmacodynamic considerations that may improve clinical outcomes. Clin Pharmacokinet. 1997;32(2):132–144. doi: 10.2165/00003088-199732020-00004. [DOI] [PubMed] [Google Scholar]
- 27.Clinical and Laboratory Standards Institute . Performance standards for susceptibility testing of mycobacteria, Nocardia spp., and other aerobic actinomycetes. CLSI Guideline M62. 1. Wayne, PA: Clinical and Laboratory Standards Institute; 2018. [PubMed] [Google Scholar]
- 28.Ferro BE, Srivastava S, Deshpande D, Sherman CM, Pasipanodya JG, van Soolingen D, et al. Amikacin pharmacokinetics/pharmacodynamics in a novel hollow-fiber Mycobacterium abscessus disease model. Antimicrob Agents Chemother. 2015;60(3):1242–1248. doi: 10.1128/AAC.02282-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly C, Jefferies C, Cryan SA. Targeted liposomal drug delivery to monocytes and macrophages. J Drug Deliv. 2011;2011:727241. doi: 10.1155/2011/727241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanvers-Kaminsky C, Ciarimboli G. Pharmacogenetics of drug-induced ototoxicity caused by aminoglycosides and cisplatin. Pharmacogenomics. 2017;18(18):1683–1695. doi: 10.2217/pgs-2017-0125. [DOI] [PubMed] [Google Scholar]
- 31.Rybak MJ, Abate BJ, Kang SL, Ruffing MJ, Lerner SA, Drusano GL. Prospective evaluation of the effect of an aminoglycoside dosing regimen on rates of observed nephrotoxicity and ototoxicity. Antimicrob Agents Chemother. 1999;43(7):1549–1555. doi: 10.1128/AAC.43.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DiStefano JJ., 3rd Noncompartmental vs. compartmental analysis: some bases for choice. Am J Physiol. 1982;243(1):R1–6. doi: 10.1152/ajpcell.1982.243.1.C1. [DOI] [PubMed] [Google Scholar]
- 33.Sou T, Kukavica-Ibrulj I, Soukarieh F, Halliday N, Levesque RC, Williams P, et al. Model-based drug development in pulmonary delivery: pharmacokinetic analysis of novel drug candidates for treatment of Pseudomonas aeruginosa lung infection. J Pharm Sci. 2019;108(1):630–640. doi: 10.1016/j.xphs.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.