

Abstract
Passive NOx adsorbers (PNA) using Pd/zeolites have emerged as a promising solution for the reduction of cold-start emissions from vehicle exhaust. However, the nature of the active sites and the mechanisms underlying NOx adsorption in Pd/zeolites remain a subject of ongoing investigation. In this study, we employ quantum chemical simulations to investigate the structure of Pd species in cation-exchange sites at isolated Al and Al pairs in the 6-ring and 8-ring of the CHA framework, before the introduction of NOx. Our calculations show that the speciation of Pd in these exchange sites strongly depends on the precise Al arrangement within the framework, as well as the operating conditions. Ionically dispersed Pd is found to be the most favorable species over a wide range of oxidizing and reducing conditions. Small oligomers of PdO and metallic Pd do not appear to be competitive at either isolated Al or Al pairs. Notably, our calculations show that ion exchange sites other than next-next-nearest neighbor Al pairs in the 6-ring will be preferentially occupied by Pd+ instead of Pd2+. The stability of Pd+ in the zeolite environment is an interesting contrast with its rareness in molecular Pd compounds. Nonetheless, a detailed analysis of the electronic structure shows that predicted Pd oxidation states are consistent with chemical intuition for all complexes investigated in this study. We also discuss the potential ambiguity in Pd characterization provided by typical experimental techniques such as XANES, EXAFS and UV-VIS, and highlight the need for additional EPR spectroscopy studies to further elucidate the initial Pd speciation in zeolites for PNA applications.
Keywords: zeolites, passive NOx adsorbers, emission control, QM/MM, thermodynamics
Graphical Abstract

Quantum chemical simulations show that the Al configuration determines whether Pd+ or Pd2+ is thermodynamically most favorable in Pd/H-CHA.
1. Introduction
In recent years, growing concerns over the detrimental effects of air pollution from the operation of internal combustion engines have led to regulations imposing increasingly stringent limits on the emission of NOx and hydrocarbons. Further reduction of cold-start NOx emissions is critical to meeting these new standards. Unfortunately, standard abatement technologies require temperatures above 200°C to function efficiently.1–3 Passive NOx adsorbers (PNA) using Pd/zeolites have emerged as a promising solution to the cold-start problem.4–8 These materials can trap NO at low temperatures, and subsequently release it once the main emission control system has reached its optimal working temperature. The use of Pd-doped MFI, CHA and BEA zeolites as passive NOx adsorbers was first reported by Johnson Matthey.4 Pd/zeolites have been found to offer key advantages over related materials such as Pd dispersed on alumina or ceria, including a higher NO trapping efficiency and greater resistance to deactivation by sulfur and hydrocarbon poisoning.4
However, the nature of the active sites, and the precise mechanisms underlying NO adsorption in Pd/zeolites and their eventual degradation, remain a subject of ongoing investigation. Such molecular-level insights are key to the selection and design of Pd/zeolites with the required efficiency and durability for implementation in practical PNA applications.
A variety of experimental techniques has been employed in several studies to examine the nature of Pd species exchanged into zeolites either directly by X-ray spectroscopy9–11, EXAFS11, or indirectly by H2 temperature programmed reduction12–14 and infra-red spectroscopy with CO and NO as probe molecules11,15–17. The results of these studies have helped define the species present in Pd/zeolites and their interconversion as a function of temperature and gas composition. When prepared using typical ion-exchange methods, MFI, CHA and BEA zeolites loaded with ~1 wt% of Pd were found to contain a variety of species, ranging from isolated Pd cations compensating the charges associated with isolated Al atoms (−1) or proximate Al pairs (−2) in the zeolite framework, to differently sized PdO clusters enclosed inside the zeolite pores or deposited on the external surface.5 More recently, Khivantsev et al. have reported the synthesis of Pd/H-CHA with fully atomic dispersion at Pd loadings of up to 2 wt%.11 This material was shown to be highly efficient at removing NOx and CO from a simulated exhaust flow, surpassing the performance and Pd utilization of conventionally prepared Pd/zeolites.11
Atomically dispersed Pd species in zeolites are commonly designated as Pd2+ cations.5,11 However, IR spectra of NO adsorbed on Pd/CHA typically show two peaks, around 1848 cm−1 and 1808 cm−1, which have been assigned in the literature to “NO on Pd2+” and “NO on Pd+”, respectively. The assignment of the higher-wave number band dates back to an early study of Pd/Y zeolites by Che et al., in which a band at 1865 cm−1 was assigned to NO on Pd2+ species.15 The assignment of the second band is less definitive, but several studies have reported similar features upon NO adsorption and assigned them to NO adsorption on Pd+ and Pd2+.11,16,17 Khivantsev et al. have also performed DFT calculations to support these peak assignments.17 These studies suggest that the “NO on Pd+” peak emerges as the oxidation state of some Pd sites changes upon NO adsorption.5,11,16,17 However, the question remains whether Pd+ species might already be present in the as-synthesized Pd/H-CHA. Similarly, FTIR spectroscopy using CO as a probe molecule has been used to probe the speciation of Pd in zeolites, but there is no consensus in the literature on the assignment of the various absorption peaks.17–19 Furthermore, studies of the performance of Pd zeolites as PNA materials have found that CO improves the NOx storage below 100°C, but it remains unclear whether this improved performance is due to reaction of NO with the reductant enabling the formation of more stable chemisorbed species, or whether it is due to partial reduction of CO with the Pd itself.20
The +2 and +4 oxidation states are most common for Pd compounds,21 including typical precursors used for the preparation of Pd/zeolites by aqueous ion exchange such as Pd(NH3)4(NO3)2.12–14 While higher oxidation states (Pd6+) have been reported in organometallic complexes,22 Pd+ and Pd3+ compounds are exceedingly rare. Pd+ had only been observed in a small number of binuclear Pd complexes,23 until a family of mononuclear Pd+ species was very recently characterized by Luo et al.24 In the zeolite literature, Jacquemin et al. have reported the presence of Pd+ cations based on observation of EPR signals consistent with the presence of paramagnetic species in their study of the catalytic properties of Pd and Fe-exchanged beta zeolites for toluene oxidation.25
In a recent study, Paolucci et al. assessed the speciation of Pd in zeolites under PNA conditions using both experiments and periodic DFT calculations.26 For Pd/H-CHA, these authors conclude that at typical loadings, Pd will occur as Pd2+ at specific proximate Al pairs (next-next-nearest neighbors, separated by two Si atoms) in the 6-ring on the basis of the favorable energetics of Pd2+ at this ion-exchange site, combined with a model for the Al distribution in CHA introduced in previous work.26 It is notable, though, that there is still no general consensus in the literature about the Al distribution in CHA, or the abundance of specific proximate Al pairs, which are determined by the Si/Al ratio and the energetics of the interaction between the framework and the structure-directing agent during the zeolite synthesis process.27 While Al pairs in the 6-ring are often recognized as the most stable sites for cation exchange in CHA, Al pairs in the 8-ring may also stabilize divalent cations. Göltl et al. have reported that the stability of Cu2+ and Co2+ at Al pairs in the 6-ring and 8-ring depends on the temperature and the partial pressure of water.28–31 Mlekodaj et al. have shown that in CHA with a Si/Al ratio of 12, Al pairs in the 8-ring separated by up to three Si atoms can also host Co2+ cations, especially when solvated with water molecules.32
In this study, we have employed quantum chemical simulations to investigate the structure and relative stability of various previously proposed Pd species in cation-exchange sites in CHA in order to elucidate the nature of the adsorption sites available before the introduction of NO or other redox-non-innocent molecules that may be present under PNA conditions. Both univalent and divalent species were considered at isolated Al atoms as well as different proximate Al pairs in the 6-ring and 8-ring of the CHA framework (Table 1). Pd/H-CHA is ideally suited for a fundamental study because all T-atoms in the CHA framework are crystallographically identical,33 allowing a systematic exploration of all possible configurations for the various sites. We find that the speciation of Pd in ion exchange sites, and especially the relative stability of Pd2+ vs. Pd+ cations depends strongly on the operating conditions, as well as the specific arrangement of the framework Al atoms. We also consider how NH3 introduced along with Pd2+ by aqueous ion exchange might facilitate the formation of more stable Pd+ species at specific ion exchange sites. Finally, we compare our findings with various experimental characterization studies not relying on probe molecules that have been reported in the literature and discuss how Pd+ species occurring in addition to Pd2+ might be overlooked by XANES, EXAFS and UV-VIS spectroscopy.
Table 1.
Possible Pd species (Pd oxidation state = 0, 1, 2) present at cation-exchange sites associated with isolated Al atoms and proximate Al pairs in CHA
| Pd ox. state | isolated Al | proximate Al pair |
|---|---|---|
| - | HZ | HZ HZ |
| 0 | Pdn0 HZ ↔ PdH+ Z− (n=1–4) | HZ Pdn0 HZ (n=1–4) |
| +1 | Pd+(H2O)n Z− (n=0–2) | HZ Pd+(H2O)n Z− (n=0–2) Z−Pd+ Pd+Z− |
| +2 | (PdO) HZ ↔ PdOH+ Z− | Z− Pd2+(H2O)n Z− (n=0–4) HZ (PdO) HZ ↔ HZ (PdOH)+ Z− HZ (PdO)2 HZ ↔ Z− (PdOH)22+ Z− Z− (Pd-O-Pd)2+ Z− |
2. Models and Methods
2.1. Zeolite model
A large cluster model containing 696 tetrahedral atoms (T696) is used to represent the zeolite topology. The crystallographic structure of CHA was taken from the database maintained by the International Zeolite Association (IZA).33 The cluster model was formed by selecting all framework atoms within a 25 Å radius of a central cha cage containing the active site and trimming the edges of the resulting fragment to obtain a cluster terminated by double six-ring units. This large model avoids any boundary effects and prevents the introduction of unintended anisotropy in the description of long-range interactions.34
To model univalent species, a single Al atom was placed in the double six-membered ring at the bottom of the central cha cage. Divalent cation-exchange species associated with pairs of framework Al atoms were investigated. In the 6-ring, two cases with different Al-Al distances were examined: a next-nearest neighbor (NNN) pair, in which the two framework Al atoms are separated by one framework Si atom and a next-next nearest neighbor (NNNN) pair, in which the two framework Al atoms are separated by two framework Si atoms. To create the pair sites in the 6-ring, an additional Al atom was inserted in the 6-ring in an NNN or NNNN position relative to the first Al. Additionally, three Al pairs in the 8-ring were created by inserting the second Al atom in the adjacent 8-ring, separated from the first by up to three Si atoms (next-next-next-nearest neighbors or NNNNN) The location of the 6-ring and 8-ring in the central cha cage is highlighted in Figure 1.
Figure 1.
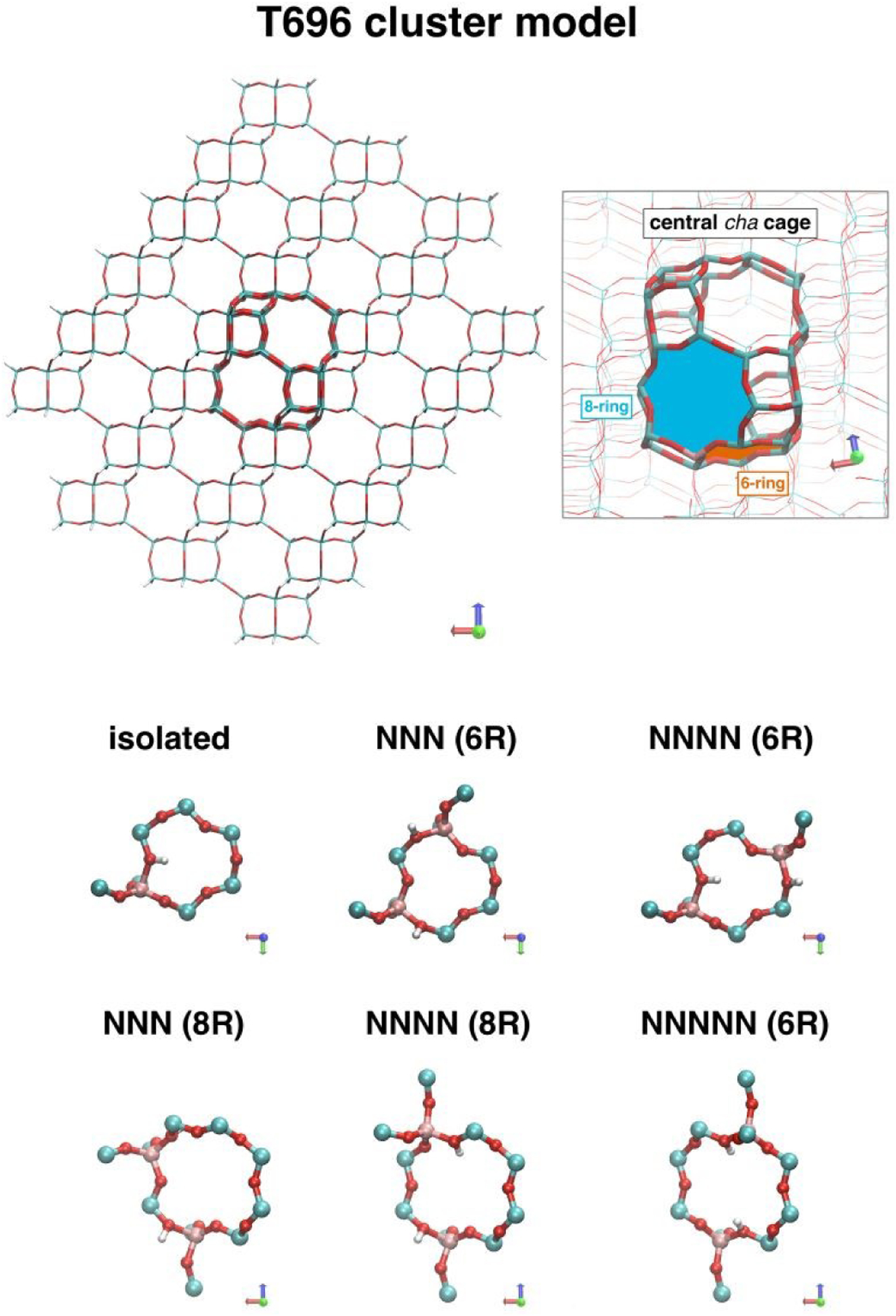
Top left: T696 cluster model for CHA used in QM/MM calculations. Heavier lines highlight the central cha cage in which the active sites are placed. Top right: Close up view of the central cha cage showing the location of the 6-ring and 8-ring. Bottom: QM regions (8–12 T-atoms) used to model active sites at isolated Al and different proximate Al pairs in the 6-ring (NNN and NNNN) or 8-ring (NNN, NNNN and NNNNN). Si atoms are shown in cyan, Al in pink, O in red, H in white.
2.2. QM/MM calculations
A hybrid quantum mechanics/molecular mechanics (QM/MM) approach is applied to limit the computational cost. A smaller cluster containing the active site is treated quantum mechanically (QM), and the remaining framework atoms are treated using molecular mechanics (MM).35,36 The Pd species and (where applicable) their hydration shells are always treated at the QM level. For the zeolite support, the complete 6-ring or 8-ring in which the Al atom(s) are located is included, as well as any Si atoms outside the ring that also neighbor an Al atom, are included in the QM region. This leads to QM regions comprising 8 T-atoms for the isolated Al in the 6-ring, 10 T-atoms for Al pairs sharing a 6-ring, and 12 T-atoms for Al pairs sharing an 8-ring. The QM regions used to model the isolated Al and Al pairs are shown using a ball-and-stick representation in Figure 1. The selection of the QM regions used in our study is mainly motivated by the need to describe all specific interactions (other than long-range electrostatics and dispersive interactions) between Pd species, adsorbates and the zeolite at the QM level. In their study of metal dimer sites in Cu-MFI, Arvidsson et al. compared adsorption energies obtained with gas-phase cluster models of increasing size to those calculated with a fully periodic model, and found that a “ring model”, which includes the entire 10-ring in which the Al atoms are located, provides a good compromise between accuracy and computational cost.37 Shor et al. explored the effect of QM region size in a QM/MM model for on the energetics of adsorption complexes of water and Rh6 in H-FAU.38 These authors reported that a small QM region consisting of 5 T-atoms was sufficient to capture the structure of Brønsted sites and adsorbed Rh6 species, while describing the structure and energetics of hydrated complexes required larger QM regions (8 to 14 T-atoms) to account for additional hydrogen bonding interactions with the framework.38 The QM regions selected in the current study fulfill both of these criteria. As an a posteriori validation of the choice of QM region, all optimized geometries were carefully inspected to ensure that all crucial chemical interactions are represented within the QM subsystem. XYZ coordinates of all optimized geometries are included in the ESI. Finally, additional test calculations were performed in which the 6-ring and 8-ring are both included in the QM region to ascertain that our final conclusions are unaffected by our choice of the size of our QM region (see Section S3 in the ESI).
The active sites and adsorbates are described using the ωB97X-D functional39,40, combined with def2 basis sets41, which include an effective core potential for Pd. The ωB97X-D functional is a dispersion-corrected, range-separated hybrid functional, which was found to be one of the best performers out of a test set of 53 hybrid GGA functionals in an extensive benchmarking study by Mardirossian and Head-Gordon.42 Geometry optimizations and frequency calculations are performed at the ωB97X-D/def2-SV(P) level of theory, followed by single-point energy refinements at the ωB97X-D/def2-TZVPD level of theory. Additional test calculations were performed using the range-separated hybrid meta GGA functional ωB97M-V43, which has emerged as the overall best performer out of the 200 density functionals evaluated by Mardirossian and Head-Gordon42 to ensure the robustness of our findings with regard to density functional choice. (see Section S4 in the ESI).
The MM region is described with a CHARMM-type force field using the P2 parameters for the zeolite Si and O atoms.36 The P2 forcefield parameters used to describe the bulk of the zeolite at the MM level were calibrated on the basis of both QM calculations and experimental adsorption data, and have been tested extensively in previous work.36 The zeolite atoms included in the MM region are fixed at their crystallographic positions, and the cluster is terminated by hydrogen atoms to saturate all dangling bonds.
Initial geometries were constructed with ZEOBUILDER.44 All QM/MM calculations were performed with a developmental version of Q-Chem.45 Enthalpy and entropy corrections were derived from a normal mode analysis on the various stationary points obtained from QM/MM to calculate free energy values under operating conditions. The quasi-rigid rotor/harmonic oscillator approximation (RRHO) was used, which applies an interpolation between a one-dimensional free rotor to capture the thermochemical contributions from the low-frequency modes more accurately.36,46 Single point TD-DFT calculations are performed using the ωB97X-D/def2-TZVPD level of theory for the QM region to calculate UV-VIS spectra for select Pd sites.47,48
3. Results and Discussion
3.1. Model structures for Pd exchange sites in CHA
For each of the possible site topologies for Pd exchange sites listed in Table 1, several chemically equivalent yet structurally different configurations can occur within the zeolite, which may vary significantly in energy. Therefore, an extensive configurational search (see Section S1 in the ESI) was performed in order to identify the lowest free energy structure for each candidate species at isolated Al and NNN and NNNN proximate Al pairs in the 6-ring. Figures 2 and 3 depict these lowest free energy structures of Pd0—H+, Pd+ and (PdOH)+ at the isolated Al, and Pd0—H+/H+, Pd+/H+, Pd2+, (PdOH)+/H+, (PdOH)22+ and (Pd-O-Pd)2+ at the NNN and NNNN Al pairs. Relative free energies for alternative structures explored in the configurational search are included in Table S1 of the Supplementary Information, highlighting the significant variations (up to ~50 kJ/mol) in free energy between the most and least stable configurations of the various sites. Khivantsev et al. have reported characteristic Pd-O distances in Pd2+, Pd+H+ and (Pd-O-Pd)2+ structures at the NNNN Al pair in the 6-ring obtained using periodic DFT calculations with the PW91-D2 functional.17 In their recent study, Paolucci et al. have also reported Pd-O distances in Pd2+ and Pd+ complexes in CHA.26 Comparing the Pd-O distances in our model structures with those reported by Khivantsev et al.17 and Paolucci et al.26 shows excellent agreement despite the use of different computational protocols (see Table S2 in the ESI).
Figure 2.

Lowest free energy configurations for Pd0—H+ and Pd+ at the isolated Al, and Pd0—H+/H+, Pd+/H+ and Pd2+ at the NNN and NNNN Al pairs in the 6-ring in CHA. Si atoms are represented in cyan, Al in pink, O in red, H in white. For clarity, only the zeolite atoms included in the QM region are shown.
Figure 3.

Lowest free energy configurations for (PdOH)+ at the isolated Al, and (PdOH)+/H+, (PdOH)22+ and (Pd-O-Pd)2+ at the NNN and NNNN Al pairs in the 6-ring in CHA. Si atoms are represented in cyan, Al in pink, O in red, H in white. For clarity, only the zeolite atoms included in the QM region are shown.
The overall geometric characteristics of the Pd0, Pd+ and Pd2+ and complexes are relatively independent of their specific environment - isolated Al, NNN or NNNN Al pairs. Pd2+ and Pd+ cations have four framework oxygen atoms in their first coordination shell. The Pd-O distances for Pd+ are slightly larger (~ 0.2 Å) than for Pd2+. Pd0 atoms hover above the Brønsted acid protons, and are therefore in less close contact with the framework oxygen atoms. Both PdO monomers (at isolated Al and Al pairs) and dimers (at Al pairs) (n=1,2) undergo protonation upon interaction with the Brønsted acid protons, forming PdOH+ Z− at isolated Al, and PdOH+Z− H+Z− or (PdOH)22+Z− Z− at Al pairs.
3.2. Relative stability under oxidizing conditions
To assess the relative stability of the various Pd species exchanged in CHA, the free energy of formation per Pd atom for each candidate structure was calculated relative to gas phase Pd0 atoms and a single Brønsted site (at isolated Al) or pair of Brønsted sites (at Al pairs).
Equations 1 and 2 were used to determine the free energy of formation of specific Pd structures under oxidizing conditions:
| (1) |
| (2) |
| (3) |
While not directly comparable to any experimental observable, these free energies of formation allow a direct comparison of the stability of different Pd species both within the same Al environment, as well as across isolated Al and different Al pairs. By using the originally present Brønsted sites as a baseline, this definition of the free energy of formation explicitly excludes energy differences originating from the positioning of the Al atoms. Because the Al siting itself is determined by the energetics of the interaction between the framework and the structure-directing agent during the zeolite synthesis process,27 this definition is more suitable for assessing the thermodynamic driving force for the formation of various exchange species.
Figure 4 shows the free energies of formation for all Pd species at isolated Al and proximate Al pairs in the 6-ring at room temperature (300 K, PO2 = 20 kPa and PH2O = 5 kPa) and at an elevated temperature corresponding to a typical oxidative pretreatment (1050 K, PO2 = 20 kPa and PH2O = 5 kPa), which Ryou et al. have shown to be key to maximize the NO adsorption performance of Pd/H-CHA materials.6,49
Figure 4:

Relative stability of Pd species at isolated Al and proximate Al pairs (NNN and NNNN) in the 6-ring in CHA under oxidizing conditions at 300 K (left) and 1050 K (right). pO2 = 20 kPa; pH2O = 5 kPa.
For both sets of conditions, ionically dispersed Pd is more thermodynamically favorable than PdO monomers and dimers at either isolated Al or proximate Al pairs. At room temperature, Pd+ and Pd2+ are expected to be completely hydrated. Under these conditions, hydrated Pd+ or Pd2+ species are 60–120 kJ/mol lower in free energy than the most stable PdO monomer or dimer at the same Al configuration. After a high-temperature oxidative treatment, only the bare cations remain, with free energies 100–150 kJ/mol below the most stable PdO species in our dataset. These results are in agreement with XPS measurements on highly-dispersed Pd/H-CHA reported by Khivantsev et al.17 The side-by-side comparison of the free energies of formation in Figure 4 highlights the exceptional stability of Pd2+ at the NNNN Al pair in the 6-ring, which allows for a near-perfect square planar coordination of the Pd2+ cation with four framework oxygen atoms (see Figure 2). For the NNN Al pair, the orientation of the zeolite oxygens is less optimal, and Pd+ H+ emerges as more stable than Pd2+. Paolucci et al. also observed that Pd2+ at the NNNN Al pair exhibits a square planar coordination with four Al-connected zeolite oxygens, similar to the common configuration observed in molecular Pd2+ complexes, while Pd2+ at the NNN Al pair features a less optimal coordination to three Al-connected and one Si-connected oxygens.26 These authors assessed the preference of Pd2+ for different exchange sites by directly comparing the total energies of Pd2+ complexes in 25 Al-Al arrangements in CHA, obtained from periodic calculations using the PBE functional.26 Even though this approach does not exclude energy differences originating from the positioning in the framework of the Al atoms themselves, the authors also found that Pd2+ strongly prefers the NNNN Al pair in the 6-ring. This Pd2+ complex is 65 kJ/mol lower in energy than next-lowest configuration at the NNN Al pair in the 6-ring.26 Referring to a previously introduced model of the Al distribution in CHA,50 Paolucci et al. then argue that CHA materials with typical Si/Al ratios (Si/Al = 5–12) contain sufficient NNNN Al pair sites in the 6-ring to accommodate all Pd introduced at typical loadings, and therefore other Al pairs need not be considered in CHA. We note, however, there is still no general consensus in the literature about the Al distribution in CHA (and therefore the prevalence of specific Al pairs) and consequently our study examines the relative stability of the candidate species listed in Table 1 at different proximate Al pairs in the 6-rings as well as the 8-rings (see Section 3.4).
To elucidate the relative stability of the various Pd species under a wider range of oxidizing conditions, phase diagrams are constructed by plotting the regions in which specific structures exhibit the lowest free energy of formation as a function of temperature and H2O partial pressure at a given O2 partial pressure of 20 kPa, i.e. flowing air (Figure 5).
Figure 5.

[PH2O,T] phase diagrams showing the thermodynamically preferred Pd species at isolated Al and Al pairs - next-nearest neighbors (NNN) and next-next-nearest neighbors (NNNN) - in the 6-ring of H-CHA under flowing air (pO2 = 20 kPa).
The phase diagrams shown in Figure 5 reveal that at the NNNN Al pair, Pd2+ in its near-perfect square planar coordination with the four Al-connected framework oxygen atoms is more stable than the next most favorable species under most conditions. Figure S6 in the ESI shows the difference in free energy between the most favorable and the next most favorable species, illustrating the uncertainty in the exact delineation between different areas of the phase diagrams. At the NNNN Al pair, Pd2+ is more stable than the next most favorable species by >30 kJ/mol over a wide range of conditions, such that it can be considered unchangeable throughout repeated adsorption-desorption-regeneration cycles on the Pd-CHA samples. On the other hand, at the NNN Al pair, Pd2+ or Pd+ is preferred depending on the temperature and the partial pressure of water. Since the boundaries between the different areas in the phase diagram are less well-defined, these species may be more easily interconverted (Figure S6).
3.3. Relative stability under reducing conditions
H2-TPR (temperature-programmed reaction) is frequently employed to characterize the speciation of Pd in zeolites. Homeyer et al. have shown that ionically dispersed Pd in Pd/zeolites can be completely reduced to Pd0 by exposing the material to H2 at 250 C.12–14 To assess the driving force for reduction of Pd2+ under these conditions, the free energies of reaction (ΔGR) for the subsequent reduction steps were calculated at T = 523 K, pH2 = 100 kPa and pH2O = 5 kPa:
These calculations show that while reduction of Pd2+ to Pd+ appears to be favorable, further reduction of Pd+ cations to isolated Pd0 atoms would not occur. One possible explanation for this discrepancy is that metallic Pd0 only occurs as small nanoparticles inside the zeolite pores, with the cohesive energy gained in the formation of such nanoparticles tipping the balance of the second reduction reaction in favor of Pd0. The cohesive energy in bulk Pd amounts to ~31 kJ/mol per Pd-Pd bond, since each Pd atom has 12 neighbors in the fcc structure.21 To examine this possibility further, additional structures in which the isolated Pd0 atom is replaced by small nanoparticles of up to four Pd atoms were included in our study. The structure and relative stability of Pd nanoparticles is a complex problem in its own right, and a comprehensive discussion is beyond the scope of the present study. Several authors have studied the properties of metallic Pd clusters in the gas phase.51–53 Inside the zeolite framework, the relative stability of these particles was found to be additionally influenced by their specific interactions with the Brønsted sites. Hafner et al. reported that Pdn (n=1–6) clusters inside mordenite undergo geometric distortions relative to their gas phase structures due both strong interactions with the framework oxygens close to the Al sites and weaker interactions with more distant framework oxygens.54 In this study, we have limited ourselves to neutral Pd particles of up to four atoms physisorbed at the Brønsted sites in the different Al configurations. The optimized structures for these complexes at the isolated Al are shown in Figure 6.
Figure 6.

Optimized structures for Pdn0 nanoparticles (n = 2–4) interacting with a Brønsted acid proton at an isolated Al in CHA. Si atoms are represented in cyan, Al in pink, O in red, H in white. For clarity, only the zeolite atoms included in the QM region are shown.
Upon interaction with a Brønsted site, Pd particles containing two or more Pd atoms may also protonate to form several variations of [PdnH]+ Z−. In an effort to estimate the additional stabilization that might be gained from protonation, we have evaluated the reaction energy of Pdn0 HZ ↔ PdH+ Z− for n=2 and n=4 at the isolated Al. We find that protonation might stabilize Pd2 by an additional 63 kJ/mol per Pd atom, and Pd4 by an additional 22 kJ/mol. From Figure 4 it is clear that additional stabilization through protonation by this amount is well short of the relative stability difference between any of the Pd particles and Pd+/Pd2+ cations regardless of the Al arrangement.
Subsequently, phase diagrams under reducing conditions were constructed by plotting the regions in which specific structures exhibit the lowest free energy of formation as a function of temperature, and O2 or H2 partial pressure at a given H2O partial pressure, as defined by Equations 4 and 5:
| (4) |
| (5) |
| (6) |
The resulting phase diagrams under dry reducing conditions are shown in Figure 7. Heat maps illustrating the uncertainties in the boundary between different areas in the diagrams are shown in Figure S8 in the ESI. The phase diagrams show that Pd+ Z− HZ is still the thermodynamically most favorable state under a wide range of conditions. Reduction of Pd2+ at the NNNN Al pair is possible, but requires increasing hydrogen partial pressure at above 600 K. While Pd04 particles are barely competitive at sub-ambient temperatures, and only at very high hydrogen partial pressures. Their appearance in this region of the phase diagram for the NNNN Al pair indicates the existence of a critical size beyond which Pd nanoparticles would resist oxidative redistribution.
Figure 7.

[PH2,T] phase diagrams showing the thermodynamically preferred Pd species at isolated Al and Al pairs - next-nearest neighbors (NNN) and next-next-nearest neighbors (NNNN) - in the 6-ring of H-CHA under dry reducing conditions (pH2O = 0.01 Pa).
3.4. Effect of Al-Al distance on stability of Pd species
As mentioned above, while Al pairs in the 6-ring have received the most attention as cation exchange sites in CHA, several authors have shown that Al pairs in the 8-ring could also host divalent cations, depending on the material properties and operating conditions such as temperature and water partial pressure.28–31 To assess whether such Al pairs may also provide potential NO adsorption sites in Pd/H-CHA, the relative stability of Pd+, Pd2+, and PdO monomers and dimers at three Al pairs in the 8-ring (NNN, NNNN and NNNNN; see Figure 1) was examined, using the same approach as that described in Section 3.2. Figure 8 shows the formation free energies under oxidizing conditions for these species at Al pairs in the 8-ring under ambient conditions (300 K, PO2 = 20 kPa and PH2O = 5 kPa) and oxidative pretreatment conditions (1050 K, PO2 = 20 kPa and PH2O = 5 kPa). Phase diagrams showing the thermodynamically most stable species as a function of temperature and H2O partial pressure under flowing air (PO2 = 20 kPa) are presented in Figure 9.
Figure 8:

Relative stability of Pd species at proximate Al pairs (NNN, NNNN and NNNNN) in the 8-ring in. CHA under oxidizing conditions at 300 K (left) and 1050 K (right). pO2 = 20 kPa; pH2O = 5 kPa.
Figure 9.

[PH2O,T] phase diagrams showing the thermodynamically preferred Pd species at next-nearest neighbor (NNN), next-next-nearest neighbor (NNNN) and next-next-next-nearest neighbor (NNNNN) Al pairs in the 8-ring of H-CHA under flowing air (pO2 = 20 kPa).
Heat maps illustrating the uncertainties in the boundary between different areas are shown in Figure S9 in the ESI. While the stability of Pd+ seems to be relatively insensitive to the exact location of the Al or Al pair in either the 6-ring or the 8-ring, the stability of Pd2+ in the 8-ring rapidly declines as the distance between the Al atoms increases. Although solvation with water molecules can potentially stabilize the Pd2+ at NNNN and NNNNN Al pairs in the 8-ring at 300 K (Figure 8, left), solvation is thermodynamically unfavorable at 1050 K (Figure 8, right), indicating that it is unlikely that (solvated) Pd2+ cations remain present in Pd/H-CHA at the end of the high temperature re-oxidation treatment. Both the formation free energies (Figure 8) and the phase diagrams (Figure 9) show that Pd cations are also favored thermodynamically at Al pairs in the 8-ring. At room temperature, Pd+ and Pd2+ are again expected to be hydrated. Hydrated Pd+ and Pd2+exhibit free energies of formation that are up to 120 kJ/mol lower in free energy than the most stable PdO monomer or dimer at the same Al configuration. While fully hydrated Pd2+ (H2O)4 was found to be most favorable at the 6-ring Al pairs under ambient conditions, at the 8-ring Al pairs Pd2+ (H2O)3 is lower in (free) energy. Comparing the minimum energy structures of Pd2+ (H2O)3 and Pd2+ (H2O)4 reveals that the latter complexes are too bulky to fit in the plane of the 8-ring and are therefore unable to maximize hydrogen bonding interactions with the zeolite framework. Comparing the phase diagrams for the 8-ring Al pairs with those for the 6-ring pairs in Figure 5 shows that both Pd+ and Pd2+ retain at least part of their hydration shell at higher temperatures in the 8-ring compared to the 6-ring, because the larger open space mitigates the entropy losses. Nevertheless, oxidative treatment up to 1050 K will again remove the hydration shells entirely such that only the bare cations remain at the 8-ring Al pairs (Figure 8). As in the 6-ring, Pd2+ and Pd+ at the NNN Al pair in the 8-ring may be interconverted, depending on the temperature and the water partial pressure. At both the NNNN and NNNN Al pairs in the 8-ring, Pd+ H+ is preferred once the hydration shell is driven off. Because of the larger Al-Al distances in the 8-ring (7.20 Å for the NNNN Al pair; 7.84 Å for the NNNNN Al pair) than in the 6-ring (6.20 Å for the NNNN Al pair), the Pd2+ cation is unable to efficiently compensate the negative charges associated with both Al atoms at the same time. These findings hint at a critical Al-Al distance (between 6.20 and 7.20 Å) beyond which Al atoms can be expected to behave as isolated for the purposes of stabilizing Pd species.
3.5. Oxidation state of Pd in cation exchange complexes
The oxidation state of the metal center is often used to characterize the catalytic behavior of transition metal complexes. Unlike for simple complexes, the assignment of oxidation state for metal centers in complex environments such as zeolites is not straightforward. Furthermore, from a quantum mechanical perspective, the concept of oxidation states is not well-defined. While atomic charges derived from global population analysis schemes (e.g., Natural Bonding Orbital analysis or NBO55–57) can give an indication of the oxidation state of the metal center, these charges are usually lower than the formal oxidation states and can be difficult to interpret. The localized orbital bonding analysis (LOBA) developed by Thom et al.58 has proven to be a robust and reliable alternative for determining oxidation states consistent with chemical intuition.58,59
To verify whether the oxidation state of Pd in the cation exchange complexes identified in this study corresponds to the formal oxidation state expected for these species, we have employed LOBA using Pipek-Mezey (PM) localized orbitals,60,61 combined with a Löwdin population analysis62,63and a localization threshold of 60%. Table 2 shows the natural charges on Pd obtained from NBO alongside the oxidation states obtained using LOBA. The results demonstrate that, while the natural charges do vary between the different Pd complexes, they are lower than expected and the correlation to the oxidation state of the Pd is not immediately clear. On the other hand, the oxidation states obtained with LOBA are in line with chemical intuition: 0 for Pd0H+H+, +1 for Pd+H+ and +2 for Pd2+. For Pd0H+H+, Pd+H+ and Pd2+, all electrons are localized on Pd resulting in d10, d9 and d8 electron configurations, respectively. For PdOH+, (PdOH)22+ and (Pd-O-Pd)2+, the populations of the bonding orbitals corresponding to the Pd-O bonds show that these bonds are strongly polarized, such that the electrons involved are no longer assigned to Pd, resulting in +2 oxidation state in each of these complexes.
Table 2.
Natural charges obtained from NBO analysis and oxidation states determined with LOBA for Pd in various cationic exchange complexes at isolated Al and proximate Al pairs (NNN and NNNN) in the 6-ring in CHA.
| isolated | NNN Al pair | NNNN Al pair | ||
|---|---|---|---|---|
| Pd natural charge (NBO) | Pd natural charge (NBO) | Pd natural charge (NBO) | Pd oxidation state (LOBA)a | |
| Pd2+ Z− Z− | n/a | 1.13251 | 1.10955 | +2 |
| Pd+ Z− H+Z− | 0.82720 | 0.82084 | 0.81724 | +1 |
| Pd0 H+Z− H+Z− | 0.18343 | 0.17001 | 0.16788 | 0 |
| PdOH+ Z− | 1.15024 | 1.12887 | 1.10567 | +2 |
| (PdOH)22+ Z− Z− | n/a | 1.03444, 1.04167 | 1.04556, 1.04547 | +2, +2 |
| Pd+ Z− Pd+Z− | n/a | 0.77625, 0.84798 | 0.86166, 0.80567 | +1, +1 |
| [Pd-O-Pd]2+ Z− Z− | n/a | 1.05567, 1.09424 | 1.03178 | +2, +2 |
This oxidation state is attained for each of the applicable environments for which NBO charges are shown.
3.6. Validation of Pd oxidation state
The theoretical prediction that Pd+ would be thermodynamically preferred at isolated Al and next-nearest neighbor Al pairs over a wide range of oxidizing and reducing conditions may be surprising given the relative scarcity of Pd+ compounds discussed in Section 1, and the nature of Pd in typical precursors used for aqueous ion exchange, such as Pd(NH3)4(NO3)2.12–14 Pd introduced by aqueous ion exchange needs to undergo partial reduction to enable the formation of Pd+. To assess whether the NH3 that is introduced along with the Pd might drive the reduction of Pd2+ to Pd+ upon insertion in H-CHA, the free energies of reaction (ΔGR) for the equilibria were calculated (T = 300 K, p = patm):
These results indicate that the presence of NH3 could drive the reduction of Pd2+ to the thermodynamically preferred Pd+ at isolated Al and NNN Al pairs, even if no additional reducing agents are applied. Note that the complete reduction to atomically dispersed Pd0 remains unfavorable and would presumably require the agglomeration of Pd into small particles, as discussed above.
In summary, our calculations strongly indicate that Pd can be present as Pd+ in cation exchange sites other than NNNN sites in the 6-ring. In the following sections, we explore how this finding might be corroborated with experimental evidence from various spectroscopic techniques that have been used for the characterization of metal-modified zeolites.
3.6.1. XAS spectroscopy
X-ray absorption spectroscopy has been used to characterize the nature and oxidation state of Pd in Pd catalysts, including Pd/zeolites. Ali et al. collected X-ray absorption spectra of Pd/H-ZSM-5 and Pd/SiO2 samples before and after complete oxidation.9 While different features emerge in the EXAFS region between the PdO standard and the oxidized Pd/H-ZSM-5 samples, these authors conclude that Pd in H-ZSM-5 is in the metallic state prior to oxidation, and in the Pd(II) state after complete oxidation, on the basis of the apparently very strong agreement between the shape of the XANES edges for the Pd/H-ZSM-5 samples before and after oxidation and those for a Pd foil and for PdO powder, respectively.9 Okumura et al. performed similar experiments on Pd/H-ZSM-5 before and after interaction with NO.10 The agreement between the XANES edges for Pd/H-ZSM-5 and PdO in their study is less strong, and these authors conclude that Pd in H-ZSM-5 is distinguishable from Pd in PdO. Additionally, there is only a slight shift in the spectrum for Pd/HZSM-5 after NO adsorption, which Okumura et al. characterize as “NO on Pd(I)”. An overlay of the spectra obtained before and after oxidation by Ali et al.9 is shown in the left-hand panel of Figure 10; an overlay of the spectra obtained before and after interaction with NO by Okumura et al.10 is shown in the right-hand panel of Figure 10. These composite images highlight the subtlety of the differences in the XANES and explain the difficulty in assigning oxidation states to Pd in zeolites on the basis of similarity of the XANES to known standards.
Figure 10:

(left) K-edge XANES of Pd/H-ZSM-5 before and after complete oxidation compared to Pd foil as a standard for Pd(0) and PdO as a standard for Pd(II). Adapted from ref.9 (right) K-edge XANES of Pd/H-ZSM-5 before and after interaction with NO compared to Pd foil as a standard for Pd(0) and PdO as a standard for Pd(II). Adapted from ref.10
While the oxidation state of the Pd is inferred from the XANES, the distance between Pd and the zeolite oxygen atoms can be estimated from the EXAFS region of the XAS spectrum, which depends on the coordination of the Pd. Theoretical estimates for both the Pd-O distances and the coordination number of Pd can also be directly determined from the optimized geometries. Figure 11 shows in ascending order, all Pd-O distances in the lowest free energy configurations of Pd+ and Pd2+ in the 6-ring identified in Section 5.2. This analysis reveals that both Pd+ and Pd2+ are coordinated to four zeolite oxygens in their first coordination shell. The Pd-O distance in this first shell appears to be fairly independent of the exact Al placement. While this Pd-O distance is found to be slightly longer for Pd+ (~2.3Å) than for Pd2+ (~2.1Å), the difference is likely too subtle to resolve in the interpretation of experimental EXAFS signals, and is therefore not helpful for distinguishing Pd+ from Pd2+.
Figure 11.

Pd-O interaction distances (in ascending order) in lowest free energy configurations of Pd+ sites at isolated Al, and Pd+ and Pd2+ sites at proximate Al pairs (NNN and NNNN) in the 6-ring in CHA.
3.6.2. UV-VIS spectroscopy
The use of DRUV-VIS spectroscopy to elucidate the nature of the metal species exchanged in zeolites has also been investigated in the literature. While variations in d-d transitions and ligandto-metal charge transfer among different species can be expected to result in distinct contributions to measured UV-VIS absorption spectra, an unambiguous identification of these features is not straightforward.64 Li et al. reported a combined experimental and theoretical UV-VIS study of Cu-SSZ-13.64 These authors found that even for zeolite samples specifically prepared to favor the presence of certain types of Cu species (CuOH+ or Cu2+), the experimental UV-VIS spectra contain features that are difficult to assign by comparison with calculated spectra, even when thermal dynamics and configurational variations are taken into account.64 To assess whether UV-VIS measurements could offer corroboration for our prediction based on thermodynamics that Pd2+ species are strongly favored only at NNNN Al pairs in the 6-ring, while any other Al arrangement in CHA is likely to favor Pd+ species, we have calculated UV-VIS spectra for Pd+ species at isolated Al and NNN and NNNN Al pairs, and Pd2+ species at NNN and NNNN Al pairs in the 6-ring using TD-DFT calculations on the respective optimized geometries. The resulting spectra are shown in Figure 12. The calculated UV-VIS spectra for Pd+ and Pd2+ are found to be relatively independent of the precise location of the Al atoms. The spectrum for Pd+ does not appear to contain any contributions from d-d transitions at wavelengths between 290 and 750 nm. On the other hand, the wavelengths of the characteristic absorption peaks for Pd+ and Pd2+ in the ligand-to-metal charge transfer region are very close. As a result, differentiating between these species on the basis of experimental UV-VIS measurements is likely not possible; the presence of Pd+ cannot be excluded if a sample also contains Pd2+.
Figure 12.

TD-DFT calculated UV-VIS spectra for (left) Pd+ at isolated Al and Al pairs (NNN and NNNN) and (right) Pd2+ species at Al pairs (NNN and NNNN) in the 6-ring in CHA.
3.6.3. Electron Paramagnetic Resonance spectroscopy
Electron paramagnetic resonance spectroscopy (EPR) has been used to identify and quantify Cu+ and Cu2+ in Cu/zeolites.65–67 Diamagnetic Cu+ (3d10) sites are EPR silent, while paramagnetic Cu2+ (3d9) sites give rise to characteristic EPR signals, depending on their structural environment (octahedral, square planar, square pyramidal).65 In Pd/zeolites, Pd+ sites (4d9) are paramagnetic, and should therefore be observable by EPR spectroscopy. Unfortunately, this technique has not been widely used in studies aiming to characterize Pd/zeolites. A rare exception is found in study of the catalytic properties of Pd and Fe-exchanged beta zeolites for toluene oxidation, in which Jacquemin et al. reported the presence of Pd+ species after observing EPR signals consistent with the presence of paramagnetic species.25 As discussed in the preceding sections, more commonly used characterization methods are likely unable to differentiate conclusively between Pd+ and Pd2+. We therefore believe that additional EPR studies of Pd/zeolites are warranted to verify whether at least a fraction of ionically dispersed Pd occurs as Pd+, as predicted by our calculations.
4. Conclusions
We have employed computational methods to examine the relative stability of a variety of monovalent and divalent Pd species at isolated Al and different Al pairs in the 6-ring and 8-ring of the CHA framework. Our calculations show that the speciation of Pd in these exchange sites strongly depends on the precise Al arrangement within the framework, as well as the temperature and gas composition at which Pd/H-CHA is held. Ionically dispersed Pd is found to be most favorable over a wide range of oxidizing and reducing conditions. Pd cations are expected to be fully hydrated under ambient conditions. After treating the material at higher temperatures, the hydration shell is removed resulting in the formation of bare cation sites. Small oligomers of PdO and metallic Pd do not appear to be competitive with Pd cations at either isolated Al or Al pairs.
While NNNN Al pairs in the 6-ring offer exceptional stabilization to Pd2+ cations due to a favorable orientation of the surrounding zeolite oxygen atoms, other Al pairs, especially those located in the 8-ring, are unable to provide Pd2+ cations with the ideal square planar coordination. Our calculations show that these exchange sites will be preferentially occupied by Pd+ and H+. While this finding may be surprising given the relative elusiveness of Pd+ in molecular compounds, a detailed analysis of the electronic structure has shown that the oxidation state of Pd in all of the structures investigated in this study is consistent with chemical intuition.
Careful examination of our results in the context of various experimental characterization studies (without probe molecules) reported in the literature shows how Pd+ cations occurring in addition to Pd2+ cations might be overlooked by XANES, EXAFS and UV-VIS spectroscopy. Assigning oxidation states to Pd in zeolites on the basis of similarity of the XANES to PdO and Pd0 standards is not straightforward. While our calculations show that the distance between Pd and the adjacent zeolite oxygens is slightly longer for Pd+ than for Pd2+, the difference is likely too subtle to resolve in EXAFS signals. UV-VIS spectra for Pd+ and Pd2+ species calculated using TD-DFT were also found to be relatively independent of the precise location of the Al atoms. However, the wavelengths of the characteristic absorption peaks in the ligand to metal charge transfer region for Pd+ and Pd2+, respectively, were found to be very close in wavelength, so that unambiguous identification of these species on the basis of experimental UV-VIS measurements is likely not possible. In light of the potential ambiguity in Pd characterization provided by XANES, EXAFS and UV-VIS, EPR spectroscopy may be the only experimental technique to conclusively prove the presence of paramagnetic Pd+, which highlights the need for additional EPR studies of Pd/zeolites to gain detailed insights into the nature of the initially available potential NO adsorption sites.
The predictions from this work showing that Pd+ and Pd2+ are both present in Pd/H-CHA is interesting and has important implications for understanding the mechanisms underlying NOx adsorption in Pd/zeolites and the eventual degradation of these materials, which will in turn enable the selection and design of suitable Pd/zeolites for practical PNA applications. Our results set the stage for a careful computational examination of the interaction of NOx species with the array of Pd+ and Pd2+ sites that we have predicted to exist in Pd/H-CHA; we hope to report progress on this complex issue in due course.
Supplementary Material
Acknowledgements
This material is based upon work supported by the U.S. Department of Energy’s Office of Energy Efficiency and Renewable Energy (EERE) under the Vehicle Technologies Program Award Number DE-EE0008213. Computational resources were provided by UC Berkeley’s Molecular Graphics and Computation Facility (supported by NIH S10OD023532).
Footnotes
Conflicts of interest
There are no conflicts to declare.
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
Electronic supplementary information (ESI)
Relative free energies of all structures considered in the configurational search performed to identify the lowest free energy structure for each candidate species at isolated Al and NNN and NNNN proximate Al pairs in the 6-ring; characteristic Pd-O distances for select model structures; validation of the QM region selections used in this work; corresponding electronic energies of formation for the Pd species in the 6-ring and the 8-ring; additional phase diagrams with sensitivity analyses; XYZ coordinates of all stationary points (lowest free energy structures).
5 References
- 1.Forzatti P, Nova I and Tronconi E, Ind. Eng. Chem. Res, 2010, 49, 10386–10391. [Google Scholar]
- 2.Beale AM, Gao F, Lezcano-Gonzalez I, Peden CHF and Szanyi J, Chem. Soc. Rev, 2015, 44, 7371–7405. [DOI] [PubMed] [Google Scholar]
- 3.Lambert C and Cavataio G, Development of the 2010 Ford Diesel Truck Catalyst System BT - Urea-SCR Technology for DeNOx After Treatment of Diesel Exhausts; Nova I and Tronconi E, Eds.; Springer New York: New York, NY, 2014; pp 659–689. [Google Scholar]
- 4.Chen H-Y, Collier JE, Liu D, Mantarosie L, Durán-Martín D, Novák V, Rajaram RR and Thompsett D, Catal. Letters, 2016, 146, 1706–1711. [Google Scholar]
- 5.Zheng Y, Kovarik L, Engelhard MH, Wang YY, Wang YY, Gao F and Szanyi J, J. Phys. Chem. C, 2017, 121, 15793–15803. [Google Scholar]
- 6.Ryou Y, Lee J, Cho SJ, Lee H, Kim CH and Kim DH, Appl. Catal. B Environ, 2017, 212, 140–149. [Google Scholar]
- 7.Khivantsev K, Gao F, Kovarik L, Wang Y and Szanyi J, J. Phys. Chem. C, 2018, 122, 10820–10827. [Google Scholar]
- 8.Mihai O, Trandafilović L, Wentworth T, Torres FF and Olsson L, Top. Catal, 2018, 61, 2007–2020. [Google Scholar]
- 9.Ali A, Alvarez W, Loughran CJ and Resasco DE, Appl. Catal. B Environ, 1997, 14, 13–22. [Google Scholar]
- 10.Okumura K, Amano J, Yasunobu N and Niwa M, J. Phys. Chem. B, 2000, 104, 1050–057. [Google Scholar]
- 11.Khivantsev K, Jaegers NR, Kovarik L, Hanson JC, Tao F, Tang Y, Zhang X, Koleva IZ, Aleksandrov HA, Vayssilov GN, Wang Y, Gao F and Szanyi J, Angew. Chemie Int. Ed, 2018, 57, 16672–16677. [DOI] [PubMed] [Google Scholar]
- 12.Homeyer ST, J. Catal, 1989, 118, 266–274. [Google Scholar]
- 13.Homeyer ST, J. Catal,1989, 117, 91–101. [Google Scholar]
- 14.Homeyer ST and Sachtler WMH, Appl. Catal, 1989, 54, 189–202. [Google Scholar]
- 15.Che M, Dutel JF, Gallezot P and Primet M, J. Phys. Chem, 1976, 80, 2371–2381. [Google Scholar]
- 16.Descorme C, Gelin P, Primet M, Lecuyer C, Catal. Letters, 1996, 41, 133–138. [Google Scholar]
- 17.Khivantsev K, Jaegers NR, Koleva IZ, Aleksandrov HA, Kovarik L, Engelhard M, Gao F, Wang Y, Vayssilov GN and Szanyi J, J. Phys. Chem. C, 2020, 124, 309–321. [Google Scholar]
- 18.Aylor AW, Lobree LJ, Reimer JA and Bell AT, J. Catal, 1997, 172, 453–462. [Google Scholar]
- 19.Chakarova K, Ivanova E, Hadjiivanov K, Klissurski D and Knözinger H, Phys. Chem. Chem. Phys, 2004, 006, 3702–3709. [Google Scholar]
- 20.Theis JR and Lambert CK, Catal. Today, 2015, 258, 367–377. [Google Scholar]
- 21.The Elements Handbook at KnowledgeDoor, http://www.knowledgedoor.com/2/elements_handbook/cohesive_energy.html.
- 22.Crabtree RH, Science, 2002, 295, 288–289. [DOI] [PubMed] [Google Scholar]
- 23.Goggin PL and Mink J, J. Chem. Soc. Dalt. Trans, 1974, 0, 534–540. [Google Scholar]
- 24.Luo J, Tran GN, Rath NP and Mirica LM, Inorg. Chem, 2020, 59, 15659–15669. [DOI] [PubMed] [Google Scholar]
- 25.Jacquemin J, Siffert S, Lamonier J-F, Zhilinskaya E and Aboukaïs A, Stud. Surf. Sci. Catal, 2002, 142, 699–706. [Google Scholar]
- 26.Mandal K, Gu Y, Westendorff KS, Li S, Pihl JA, Grabow LC, Epling WS and Paolucci C, ACS Catal., 2020, 10, 12801–12818. [Google Scholar]
- 27.Sastre G, Fornes V and Corma A, J. Phys. Chem. B, 2002, 106, 701–708. [Google Scholar]
- 28.Göltl F and Hafner J, J. Chem. Phys, 2012, 136, 64501. [DOI] [PubMed] [Google Scholar]
- 29.Göltl F, Bulo RE, Hafner J and Sautet P, J. Phys. Chem. Lett, 2013, 4, 2244 2249. [Google Scholar]
- 30.Göltl F, Love AM and Hermans I, J. Phys. Chem. C, 2017, 121, 6160–6169. [Google Scholar]
- 31.Göltl F, Müller P, Uchupalanun P, Sautet P and Hermans I, Chem. Mater, 2017, 29, 6434–6444. [Google Scholar]
- 32.Mlekodaj K, Dedecek J, Pashkova V, Tabor E, Klein P, Urbanova M, Karcz R, Sazama P, Whittleton SR, Thomas HM, Fishchuk AV and Sklenak S, J. Phys. Chem. C, 2019, 123, 7968–7987. [Google Scholar]
- 33.Structure Commission of the International Zeolite Association, Database of Zeolite Structures. http://www.iza-structure.org/databases/
- 34.Mansoor E, Van der Mynsbrugge J, Head-Gordon M and Bell AT, Catal. Today, 2018, 312, 51–65. [Google Scholar]
- 35.Zimmerman PM, Head-Gordon M and Bell AT, J. Chem. Theory Comput, 2011, 7, 1695–1703. [DOI] [PubMed] [Google Scholar]
- 36.Li Y-P, Gomes J, Mallikarjun Sharada S, Bell AT and Head-Gordon M, J. Phys. Chem. C, 2015, 119, 1840–1850. [Google Scholar]
- 37.Arvidsson AA, Zhdanov VP, Carlsson P-A, Grönbeck H and Hellman A, Catal. Sci. Technol, 2017, 7, 1470–1477. [Google Scholar]
- 38.Shor AM, Shor EAI, Laletina S, Nasluzov VA, Vayssilov GN and Rösch N, Chem. Phys, 2009, 363, 33–41. [Google Scholar]
- 39.Chai J-D and Head-Gordon M, Phys. Chem. Chem. Phys, 2008, 10, 6615. [DOI] [PubMed] [Google Scholar]
- 40.Chai J-D and Head-Gordon M, J. Chem. Phys, 2009, 131, 174105. [DOI] [PubMed] [Google Scholar]
- 41.Weigend F and Ahlrichs R, Phys. Chem. Chem. Phys, 2005, 7, 3297. [DOI] [PubMed] [Google Scholar]
- 42.Mardirossian N and Head-Gordon M, Mol. Phys, 2017, 115, 2315–2372. [Google Scholar]
- 43.Mardirossian N and Head-Gordon M, J. Chem. Phys, 2016, 144, 0–23. [DOI] [PubMed] [Google Scholar]
- 44.Verstraelen T, Van Speybroeck V and Waroquier M, J. Chem. Inf. Model, 2008, 48, 1530–1541. [DOI] [PubMed] [Google Scholar]
- 45.Shao Y, Gan Z, Epifanovsky E, Gilbert ATB, Wormit M, Kussmann J, Lange AW, Behn A, Deng J, Feng X, Ghosh D, Goldey M, Horn PR, Jacobson LD, Kaliman I, Khaliullin RZ, Kuś T, Landau A, Liu J, Proynov EI, Rhee YM, Richard RM, Rohrdanz MA, Steele RP, Sundstrom EJ, Woodcock HL, Zimmerman PM, Zuev D, Albrecht B, Alguire E, Austin B, Beran GJO, Bernard YA, Berquist E, Brandhorst K, Bravaya KB, Brown ST, Casanova D, Chang C-M, Chen Y, Chien SH, Closser KD, Crittenden DL, Diedenhofen M, DiStasio RA, Do H, Dutoi AD, Edgar RG, Fatehi S, Fusti-Molnar L, Ghysels A, Golubeva-Zadorozhnaya A, Gomes J, Hanson-Heine MWD, Harbach PHP, Hauser AW, Hohenstein EG, Holden ZC, Jagau T-C, Ji H, Kaduk B, Khistyaev K, Kim J, Kim J, King RA, Klunzinger P, Kosenkov D, Kowalczyk T, Krauter CM, Lao KU, Laurent AD, V Lawler K, V Levchenko S, Lin CY, Liu F, Livshits E, Lochan RC, Luenser A, Manohar P, Manzer SF, Mao S-P, Mardirossian N, V Marenich A, Maurer SA, Mayhall NJ, Neuscamman E, Oana CM, Olivares-Amaya R, O’Neill DP, Parkhill JA, Perrine TM, Peverati R, Prociuk A, Rehn DR, Rosta E, Russ NJ, Sharada SM, Sharma S, Small DW, Sodt A, Stein T, Stück D, Su Y-C, Thom AJW, Tsuchimochi T, Vanovschi V, Vogt L, Vydrov O, Wang T, Watson MA, Wenzel J, White A, Williams CF, Yang J, Yeganeh S, Yost SR, You Z-Q, Zhang IY, Zhang X, Zhao Y, Brooks BR, Chan GKL, Chipman DM, Cramer CJ, Goddard WA, Gordon MS, Hehre WJ, Klamt A, Schaefer HF, Schmidt MW, Sherrill CD, Truhlar DG, Warshel A, Xu X, Aspuru-Guzik A, Baer R, Bell AT, Besley NA, Chai J-D, Dreuw A, Dunietz BD, Furlani TR, Gwaltney SR, Hsu C-P, Jung Y, Kong J, Lambrecht DS, Liang W, Ochsenfeld C, Rassolov VA, V Slipchenko L, Subotnik JE, Van Voorhis T, Herbert JM, Krylov AI, Gill PMW and Head-Gordon M, Mol. Phys, 2015, 113, 184–215. [Google Scholar]
- 46.Grimme S, Chemistry, 2012, 18, 9955–9964. [DOI] [PubMed] [Google Scholar]
- 47.Runge E and Gross EKU, Phys. Rev. Lett, 1984, 52, 997–1000. [Google Scholar]
- 48.Petersilka M, Gossmann UJ and Gross EKU, Phys. Rev. Lett, 1996, 76, 1212–1215. [DOI] [PubMed] [Google Scholar]
- 49.Ryou YS, Lee J, Lee H, Kim CH and Kim DH, Catal. Today, 2017, 320, 175–180. [Google Scholar]
- 50.Paolucci C, Parekh AA, Khurana I, Di Iorio JR, Li H, Albarracin Caballero JD, Shih AJ, Anggara T, Delgass WN, Miller JT, Ribeiro FH, Gounder R and Schneider WF, J. Am. Chem. Soc, 2016, 138, 6028–6048. [DOI] [PubMed] [Google Scholar]
- 51.Krüger S, Vent S and Rösch N, Berichte der Bunsengesellschaft für Phys. Chemie, 1997, 101, 1640–1643. [Google Scholar]
- 52.Estiu GL and Zerner MC, J. Phys. Chem, 1994, 98, 4793–4799. [Google Scholar]
- 53.Morse MD, Chem. Rev, 1986, 86, 1049–1109. [Google Scholar]
- 54.Grybos R, Benco L, Bučko T and Hafner J, J. Chem. Phys, 2009, 130, 104503. [DOI] [PubMed] [Google Scholar]
- 55.Reed AE and Weinhold F, J. Chem. Phys, 1983, 78, 4066–4073. [Google Scholar]
- 56.Reed AE, Weinstock B and Weinhold F, J. Chem. Phys, 1985, 83, 735–746. [Google Scholar]
- 57.Reed AE, Curtiss LA and Weinhold F, Chem. Rev, 1988, 88, 899–926. [Google Scholar]
- 58.Thom AJW, Sundstrom EJ and Head-Gordon M, Phys. Chem. Chem. Phys, 2009, 11, 11297. [DOI] [PubMed] [Google Scholar]
- 59.Panetier JA, Letko CS, Tilley TD and Head-Gordon M, J. Chem. Theory Comput, 2016, 12, 223–230. [DOI] [PubMed] [Google Scholar]
- 60.Pipek J and Mezey P, J. Chem. Phys, 1989, 90, 4916–4926. [Google Scholar]
- 61.Pipek J and Mezey P, Int. J. Quantum Chem, 1988, 34, 1–13. [Google Scholar]
- 62.Löwdin P-O, J. Chem. Phys, 1950, 18, 365–375. [Google Scholar]
- 63.Löwdin P-O, Phys. Rev, 1955, 97, 1474–1489. [Google Scholar]
- 64.Li H, Paolucci C, Khurana I, Wilcox L, Göltl F, Albarracin-Caballero J, Shih A, Ribeiro F, Gounder R, Schneider W, Chem. Sci, 2018, 10, 2373–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Larsen SC, Aylor A, Bell AT and Reimer JA, J. Phys. Chem, 1994, 98, 11533–11540. [Google Scholar]
- 66.Godiksen A, Vennestrøm PNR, Rasmussen SB and Mossin S, Top. Catal, 2017, 60, 13–29. [Google Scholar]
- 67.Godiksen A, Stappen FN, Vennestrøm PNR, Giordanino F, Rasmussen SB, Lundegaard LF and Mossin S, J. Phys. Chem. C, 2014, 118, 23126–23138. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.