

Abstract
Tissue engineered vascular grafts (TEVGs) are a promising technology, but are hindered by occlusion. Seeding with bone-marrow derived mononuclear cells (BM-MNCs) mitigates occlusion, yet the precise mechanism remains unclear. Seeded cells disappear quickly and potentially mediate an anti-inflammatory effect through paracrine signaling. Here, we report a series of reciprocal genetic TEVG implantations plus recombinant protein treatment to investigate what role interleukin-10, an anti-inflammatory cytokine, plays from both host and seeded cells. TEVGs seeded with BM-MNCs from wild-type and IL-10 KO mice, plus unseeded grafts, were implanted into wild-type and IL-10 KO mice. Wild-type mice with unseeded grafts also received recombinant IL-10. Serial ultrasound evaluated occlusion and TEVGs were harvested at 14 days for immunohistochemical analysis. TEVGs in IL-10 KO mice had significantly higher occlusion incidence compared to wild-type mice attributed to acute (<3day) thrombosis. Cell seeding rescued TEVGs in IL-10 KO mice comparable to wild-type patency. IL-10 from the host and seeded cells did not significantly influence graft inflammation and macrophage phenotype, yet IL-10 treatment showed interesting biologic effects including decreasing cell proliferation and increasing M2 macrophage polarization. IL-10 from the host is critical for preventing TEVG thrombosis and seeded BM-MNCs exert a significant anti-thrombotic effect in IL-10 KO mice.
Keywords: interleukin-10, tissue engineered vascular grafts, thrombosis, macrophages, tissue factor
1. Introduction
Tissue engineered vascular grafts (TEVGs) pose a unique solution in vascular surgery due to their ability to grow and remodel into an autologous vessel over time. However, TEVGs still face translational hurdles primarily from graft stenosis in our clinical trial.[1–3] In our murine models with small diameter TEVGs (<1mm), occlusion occurs due to thrombosis, stenosis, or a dynamic interplay between the two.[4] Efforts to improve TEVG outcomes include incubation in a bioreactor, scaffold coatings, and cell seeding such as endothelial progenitor cells, fibroblasts, smooth muscle cells, and bone-marrow derived mononuclear cells (BM-MNCs).[5–10] Currently, our group utilizes a cell seeding approach with BM-MNCs to mediate neotissue formation in situ.[11,12] This cell population has the capacity to prevent both stenosis and thrombosis in our TEVG model.[10,13,14]
The precise mechanism of how the seeded cells assert their protective effects remains unclear, as does the host-response that induces graft occlusion. Infiltrating host macrophages appear to play a paradoxical role that is influenced by cell seeding. Decreasing the degree of host macrophage infiltration into the graft via cell seeding decreases occlusion, yet total ablation of the host macrophage response leads to a failure in graft development.[14–16] Seeded cells disappear approximately one day after graft implantation, suggesting their effects may be due to a paracrine mechanism rather than through direct contribution to neotissue formation.[10,17,18] Identifying the source of seeded BM-MNC’s protective effects offers the potential to bypass cell seeding all together and optimize a next generation “off-the-shelf” TEVG for use in the clinic.
Preliminary work demonstrated that seeded BM-MNCs in our model have the potential to secrete a wide range of cytokines including interleukin-10 (IL-10)[19]. IL-10 is classically described as an anti-inflammatory cytokine known for its involvement in monocyte differentiation, macrophage phenotype switching/polarization, venous thrombosis formation, coagulation, and monocyte tissue factor expression.[20–24] The extent, phenotype, and chronology of host macrophages responding to implanted TEVGs is an area of active research. The alternatively activated macrophage, commonly referred to as “M2”, has properties that may lend to improving TEVG development and patency, such as secretion of anti-inflammatory cytokines, improved scaffold vascularization, and down-regulation of the host immune response to the graft.[25–27] We hypothesized that IL-10 secreted from seeded cells could modify the phenotype of host infiltrating macrophages to an alternative phenotype “M2” thus decreasing TEVG inflammation and improving patency. However, utilizing an IL-10 knockout (KO) mouse model to investigate the role of secreted IL-10 from seeded cells as well as the reciprocal experiment of IL-10 KO in the graft recipient, we uncovered that recipient IL-10 is critical for TEVG patency whereas donor IL-10 from seeded cells appears to play little role. This work furthers the understanding of the graft-host interface, highlighting the importance of host IL-10 signaling for reduction of thrombosis in TEVGs.
2. Results
2.1. Graft Occlusion
TEVGs seeded with BM-MNCs (Figure 1a) from wild-type mice, IL-10 KO mice, or unseeded TEVGs were implanted into wild-type recipient and IL-10 KO recipient mice (Figure 1b). An additional group of wild-type recipient mice with unseeded grafts received recombinant IL-10 treatment (Figure 1b). Serial ultrasound at days 3, 7, and 10 with H&E cross-sectional histology was used to determine patency (Figure 1c). Ultrasound demonstrated that if graft occlusion occurred during the study’s course, it occurred by day 3 and did not reverse (Figure 2a,b). Histological analysis of explanted graft lumens at day 14 confirmed ultrasound results (Figure 2c,d). Unseeded grafts had 22.2% (4/18) occlusion for wild-type recipients versus 100% (12/12) for IL-10 KO recipients. Wild-type seeded grafts had 16.7% (3/18) occlusion for wild-type recipients versus 33.3% (4/12) for IL-10 KO seeded recipients. IL-10 KO seeded grafts had 22.2% (4/18) occlusion for wild-type recipient versus 54.5% (6/11) for IL-10 KO recipient mice. Wild-type seeding in IL-10 KO recipient mice significantly rescued patency compared to unseeded grafts in IL-10 KO recipient mice (pA = 0.001 and pB = 0.0009). IL-10 treatment of wild-type recipient mice that had unseeded grafts did not significantly influence patency on H&E examination (29.4% occluded) or on ultrasound (29.4% occluded by day 3) (Figure 3a, b). H&E staining of graft sections showed occluded lumens to be fibrin rich with trapped platelets and signs of re-vascularization (Figure 2c). These results indicate a highly pro-thrombotic state for IL-10 KO mice in the setting of TEVG implantation that can be rescued by wild-type BM-MNC seeding.
Figure 1. Schematic overiew of study.
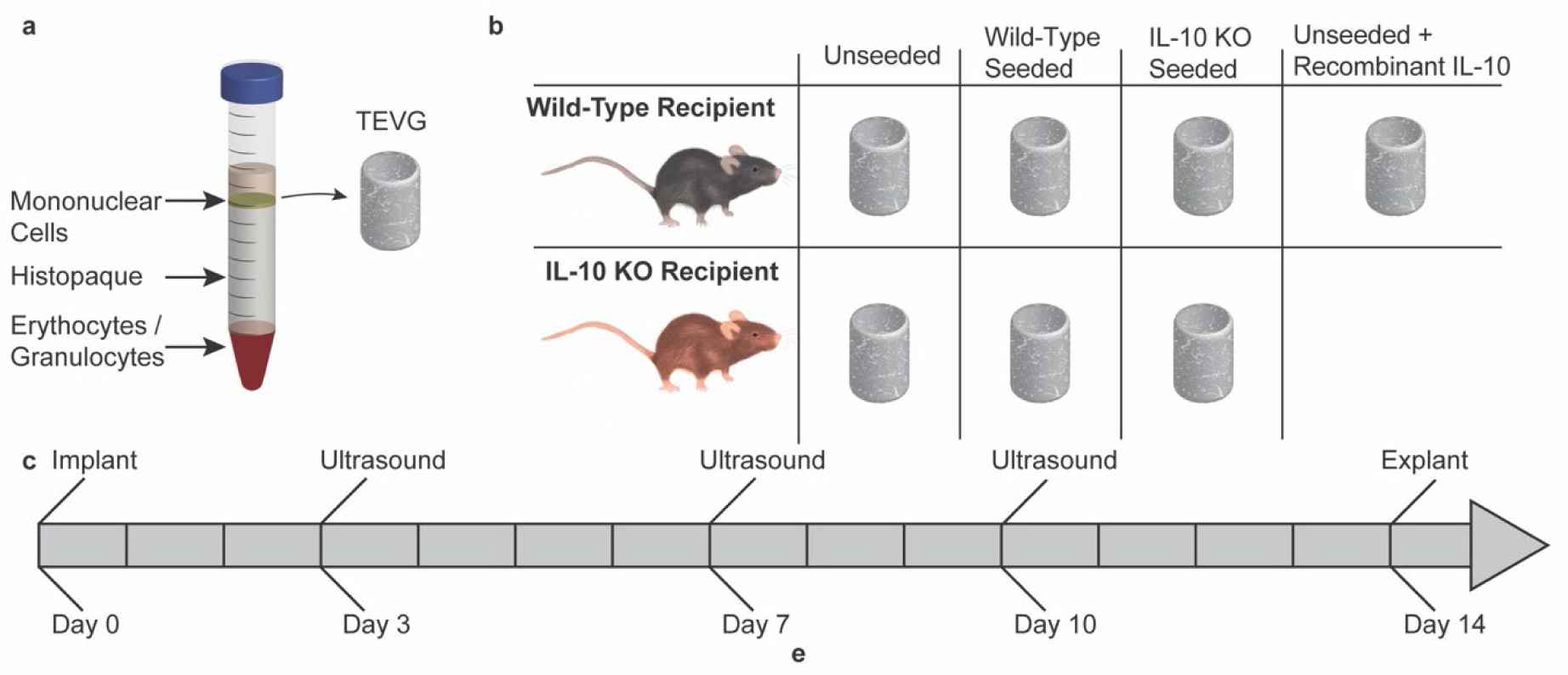
a) Graphic depicting collection of the mononuclear cell layer from harvested bone-marrow utilizing denisty based centrifugation. The collected mononuclear layer is statically seeded onto tissue engineered vascular grafts (TEVGs) and incubated overnight prior to implantation. b) Table depicting implant scheme. Grafts were either unseeded, seeded with bone-marrow derived monunclear cells (BM-MNCs) from wild-type mice, or seeded with BM-MNCs from IL-10 mice. Grafts were then implanted into either wild-type recipient mice (n = 18 per group), or into IL-10 KO recipient mice (n = 12 for unseeded and wild-type seeded, n = 11 for IL-10 KO seeded). One group of wild-type recipient mice with unseeded grafts was treated with recombinant IL-10 (n = 17). c) Timeline showing course of study.
Figure 2. Ultrasound and graft patency.
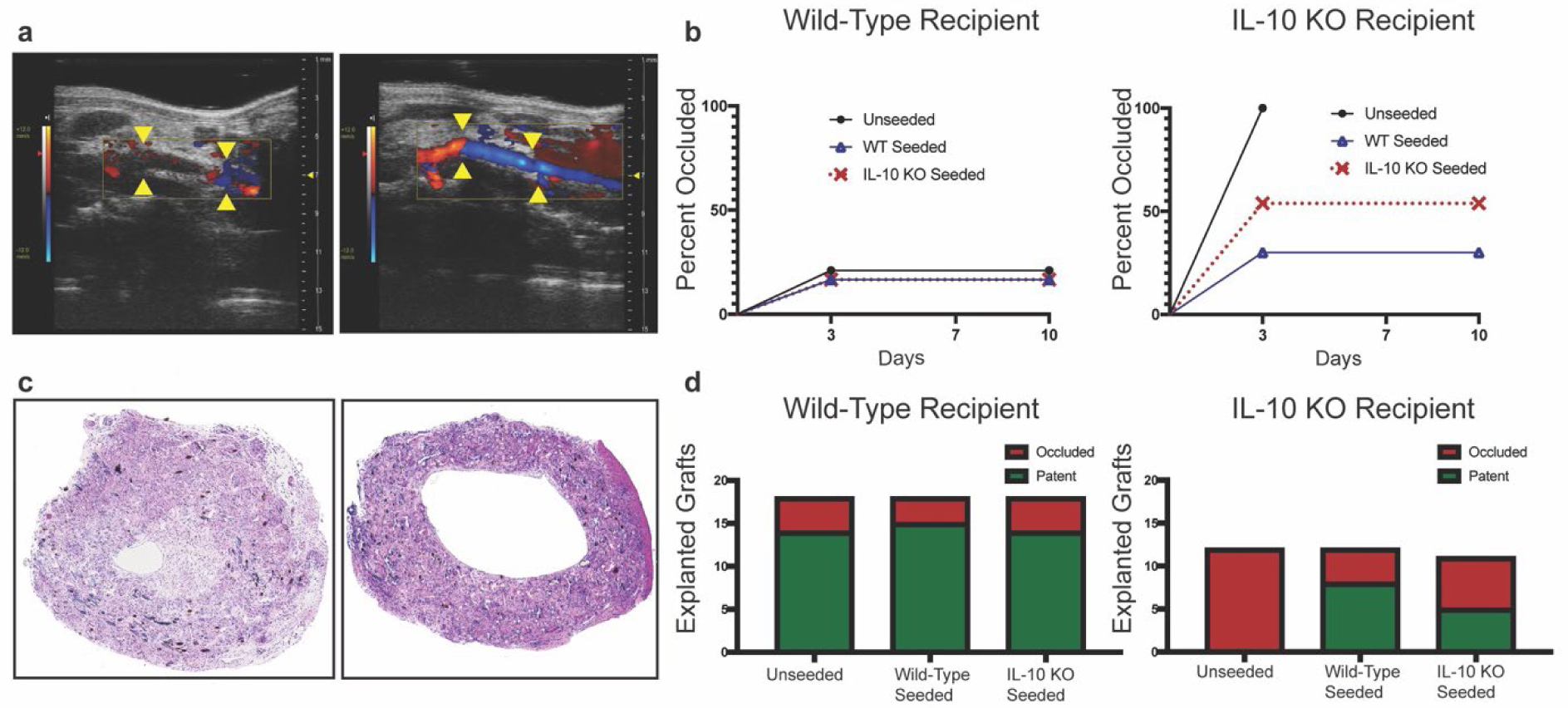
a) Representative ultrasound images showing an occluded TEVG (left) versus a patent TEVG (right). The anastomic sights of the graft at the native inferior vena cava are marked with yellow arrows. b) Percent occlusion of implanted TEVGs for both wild-type recipient (n = 18 per seeding group) and IL-10 KO recipient mice (n = 12 for unseeded and wild-type seeded, n = 11 for IL-10 KO seeded) based on ultrasound analysis at day 3, 7, and 10 post-surgery. c) Representative H&E images of an occluded TEVG (left) versus a patent TEVG (right). d) TEVG patency based on histological analysis of H&E stained TEVGs at day 14 post-surgery for wild-type recipient and IL-10 KO recipient mice. Wild-type seeding in IL-10 KO recipient mice significantly improved patency compared to unseeded in IL-10 KO recipient mice (Modified Fisher’s Exact test with Freeman-Halton Extension, pA = 0.001 and pB = 0.0009).
Figure 3. Patency results and cellular infiltrate analysis of unseeded grafts in IL-10 treated mice versus unseeded and wild-type seeded grafts in wild-type mice.
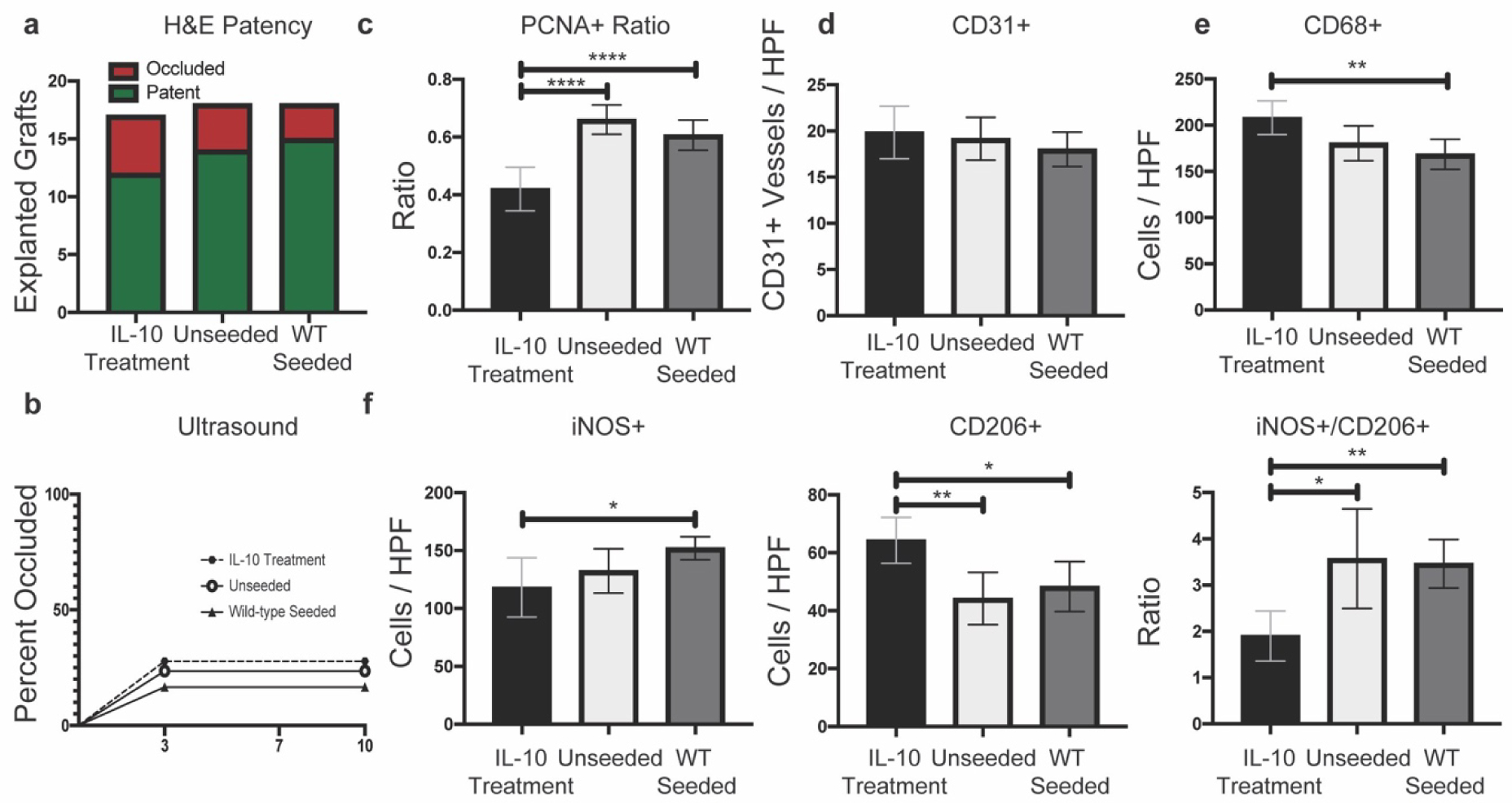
a-b) H&E patency results and ultrasound results (n= 17 / IL-10 treatment, n = 18 per unseeded and 18 per wild-type seeded in wild-type recipient mice) showed no significant differences between either groups. c) IL-10 treatment mice had significantly reduced PCNA+ cellular ratio (p < 0.0001) compared to unseeded and wild-type seeded. d) No significant differences in CD31+ vessel quantification. e) IL-10 treatment mice had significantly increased CD68+ cellular infiltration (p = 0.0048) compared to wild-type seeded only. f) Macrophage phenotype quantification. IL-10 treatment significantly decreased iNOS+ cellular infiltration compared to wild-type seeded grafts only (p = 0.0287). IL-10 treatment increased the CD206+ cellular infiltration and decreased the iNOS+/CD206+ ratio compared to unseeded (p = 0.0042 and 0.0176) as well as wild-type seeded (p =0.0246 and 0.0013) grafts. For histological quantification, n size = 16–18 per group with Ordinary one-way ANOVA for normally distributed data or Kruskal-Wallis for non-normally distributed data. Presented as means with 95% confidence intervals. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****).
2.2. Cellular Proliferation and Vascularization
Anti-Proliferating Cell Nuclear Antigen (PCNA) antibody was used to assess for cell proliferation (Figure 4a) and normalized to the total number of cells (Figure 4b).[28] Seeding grafts with wild-type BM-MNCs reduced the ratio of proliferating cells in wild-type recipient mice versus IL-10 KO recipient mice (mean ratio 0.6066 vs 0.7602, p = 0.0075). Wild-type seeding of grafts in wild-type recipient mice also reduced the ratio of proliferating cells compared to IL-10 KO seeded grafts in wild-type recipient mice (mean ratio 0.6066 vs 0.7264, p = 0.0230). IL-10 treatment (Figure 3c) significantly reduced the ratio of proliferating cells compared to unseeded and wild-type seeded grafts in wild-type recipient mice (mean ratio 0.4200 vs 0.6604 and 0.6066, respectively, p <0.0001). Anti-CD31 staining for endothelial cell junctions revealed 2 distinction observations: 1) grafts had prominent neovessel formation in the body of the graft and 2) based on a cross-sectional view of the middle of the graft, the luminal surface was endothelialized (Figure 4a). Quantification of neo-capillary formation within the graft showed a significant difference between wild-type recipient and IL-10 KO recipient mice (predicted mean difference 3.814 vessels/high powered field (HPF), p <0.0001) (Figure 4c). Multiple comparisons showed unseeded grafts in IL-10 KO recipient mice had reduced neo-capillary formation compared to unseeded grafts in wild-type mice (mean 13.46 vessels/HPF vs 19.16, p = 0.0015). IL-10 treatment did not significantly affect neo-capillary formation as assessed by CD31+ staining (Figure 3d). PCNA+ staining results indicate that neither host IL-10 and nor seeded cell IL-10 plays a large role in modulating the ratio of proliferating cells in a TEVG at 2 weeks. Reduced CD31+ staining within the graft wall in IL-10 KO recipient mice highlights a potential impairment for neovessel development owing to the importance of neoangiogenesis for neotissue formation.
Figure 4. PCNA and CD31 analysis.
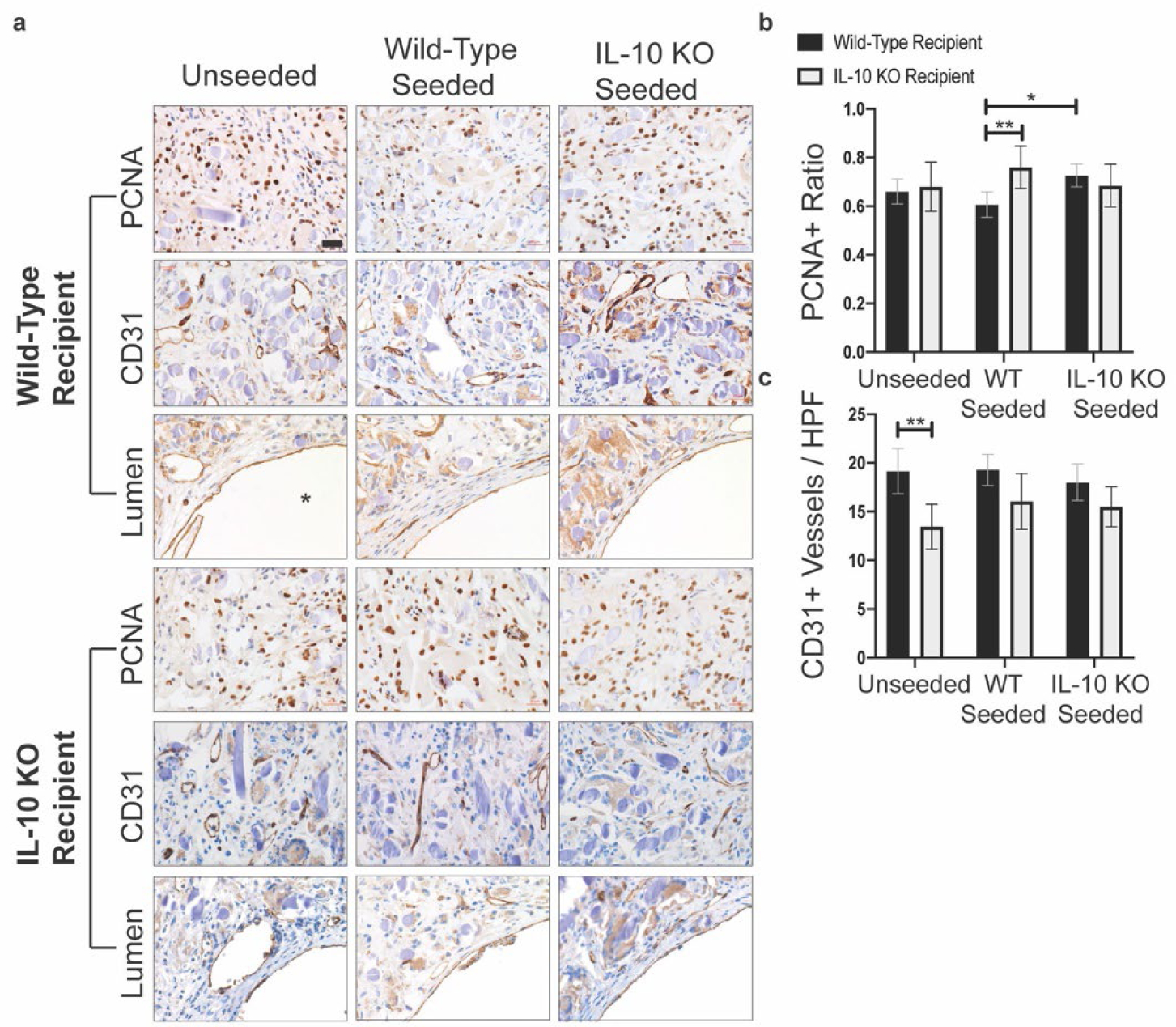
a) Representative images of PCNA and CD31 immunohistochemical staining. CD31 representative images were taken at two distinct locations: in the graft wall and at the luminal surface. For the images showing luminal endothelialization, the lumen is marked by a “*” in the bottom right hand corner. Images taken with a 40x objective. Scale bar = 20μm. b) Quantification of PCNA+ cells. PCNA+ cells presented as a ratio of total cells / high power field (HPF). Wild-type seeded TEVGs in wild-type recipient mice had significantly reduced PCNA+ ratio compared to wild-type seeded TEVGs in IL-10 KO recipient mice and to IL-10 KO seeded grafts in wild-type recipient mice (p = 0.0075 and 0.0230). c) Quantification of CD31+ vessels. Unseeded grafts in IL-10 KO recipient mice had significantly reduced CD31+ vessels compared to unseeded grafts in wild-type recipient mice (p = 0.0015). For CD31+ quantification, n = 18 per seeding group for wild-type recipient mice and n = 11–12 per seeding group for IL-10 KO recipient mice. For PCNA+ quantification, n = 18 per seeding group for wild-type recipient mice and n = 11 per seeding group for IL-10 KO recipient mice. Analysis with Two-Way ANOVA adjusted for multiple comparisons and data presented as means with 95% confidence interval. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****).
2.3. Macrophage Infiltration and Phenotype
Total macrophage infiltration was assessed with anti-CD68 antibody staining (Figure 5a). Neither graft seeding status nor host recipient IL-10 status influenced the total macrophage infiltration (Figure 5b). IL-10 treatment significantly increased the degree of macrophage infiltration compared to wild-type seeded grafts in wild-type recipient mice (mean 208.1 positive cells / HPF vs 168.5, p = 0.0048) (Figure 3e). Macrophages were further assessed for phenotype polarization with anti-iNOS staining for inflammatory (M1) phenotype or with anti-CD206 (mannose receptor) for anti-inflammatory (M2) phenotype (Figure 5a). Recipient mouse phenotype was significant for iNOS+ staining with IL-10 KO mice having overall reduced total iNOS+ cellular staining (Figure 5c) (predicted mean difference 21.14 positive cells / HPF, p = 0.0009). In wild-type seeded grafts, IL-10 KO recipient mice had reduced iNOS+ cellular staining (mean 116.6 positive cells / HPF vs 152.1, p = 0.0130). IL-10 treatment significantly reduced the number of iNOS+ cells compared to wild-type seeded grafts (Figure 3f), but not unseeded grafts (mean 118.2 positive cells / HPF vs 152.1, p = 0.0287). Unseeded grafts in wild-type recipient mice had significantly reduced CD206+ cell staining compared to unseeded grafts in IL-10 KO recipient mice (Figure 5d) (mean 44.18 cells/HPF vs 65.73, p = 0.0081). In IL-10 treated mice, the degree of CD206+ cell staining (Figure 3f) was significantly increased compared to unseeded grafts and wild-type seeded grafts in wild-type recipient mice (mean 64.31 positive cells / HPF vs 44.18 and 48.33, respectively, p = 0.0042 and 0.0246). The ratio of iNOS+ cells / CD206+ cells was significantly influenced by host phenotype (mean predicted difference 1.083, p = 0.0003). On multiple comparison analysis, unseeded grafts in IL-10 KO recipient mice had a significantly reduced iNOS+/CD206+ ratio compared to unseeded grafts in wild-type mice (Figure 5e) (mean rank 20.33 vs 51.12, p = 0.0110). IL-10 treatment significantly reduced the ratio of iNOS+ to CD206+ cells (Figure 3f) compared to unseeded and wild-type seeded grafts (mean rank 13.50 vs 27.71 and 31.39, respectively, p = 0.0176, 0.0013). Treatment with IL-10 recombinant protein does appear to play a significant role in polarizing infiltrating macrophages to an M2 phenotype.
Figure 5. Macrophage infiltration and phenotype analysis.
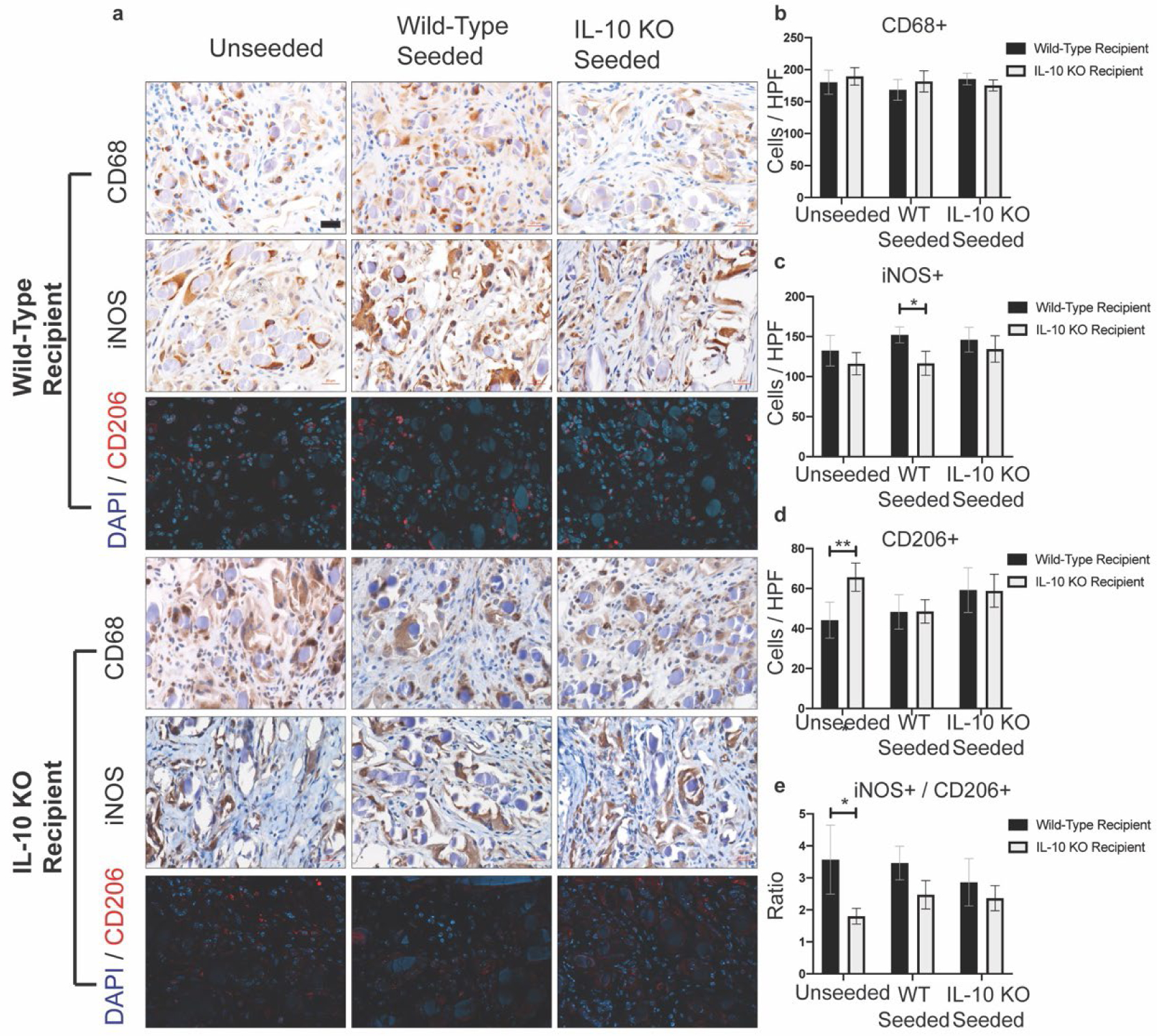
a) Representative immunohistochemical (CD68 and iNOS) and immunofluorescence (CD206) images of TEVG samples. Images taken with a 40x objective. Scale bar = 20μm. b) Quantification of CD68+ cellular infiltration. No significant differences. c) Quantification of iNOS+ cellular infiltration. Wild-type seeded TEVGs in wild-type recipient mice had significantly increased iNOS+ staining compared to wild-type seeded TEVGs in IL-10 KO mice (p = 0.0130). d) Quantification of CD206+ cellular infiltration. Unseeded TEVGs in IL-10 KO recipient mice had significantly increased CD206+ cellular infiltration compared to unseeded TEVGs in wild-type recipient mice (p = 0.0081). e) Ratio of iNOS+ cells / CD206+ cells. Unseeded TEVGs in IL-10 KO recipient mice had a significantly reduced ratio compared to unseeded TEVGs in wild-type recipient mice (p = 0.0110). For CD68+ and iNOS+ quantification, n = 18 per seeding group for wild-type recipient mice and n = 11–12 per seeding group for IL-10 KO recipient mice. For CD206+ quantification, n = 17–18 per seeding group for wild-type recipient mice and n = 11–12 for IL-10 KO recipient mice. Analysis with Two-Way ANOVA adjusted for multiple comparisons and data presented as means with 95% confidence interval. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****).
2.4. Graft Morphology and Collagen Content
H&E stained TEVG sections were divided into sections demarcating luminal area, neotissue area, and graft area and quantified (Figure 6a,b). Recipient mouse genotype significantly influenced luminal area (mean predicted difference, 181,660 μm2 p <0.0001). Unseeded grafts in IL-10 KO recipient mice had significantly reduced luminal area compared to unseeded grafts in wild-type recipient (mean rank 14.70 vs 58.72, p <0.0001). Luminal neotissue analysis showed graft seeding and recipient mouse genotype significantly influenced total area of neotissue development along the luminal surface (predicted mean difference for genotype 99,732 μm2, p = 0.0075 and p <0.0001, respectively). Unseeded grafts in IL-10 KO recipients had significantly increased neotissue development compared to unseeded grafts in wild-type recipient (mean rank 71.80 vs 39.17, p = 0.0062) as well as compared to wild-type seeded grafts in IL-10 KO recipient (mean rank 71.80 vs 42.09, p = 0.0478). Finally, total graft area quantification showed no significant differences. IL-10 treatment did not significantly influence luminal area or neotissue area (Figure 7a), but did significantly increase graft area compared to wild-type seeded grafts (1,650,630 μm2 vs 1,253,436 μm2, p < 0.0001). These results indicate that unseeded grafts in IL-10 KO recipient mice experienced occlusion from decreased luminal area due to acute thrombosis that partially remodeled into an increased deposition of neotissue compared to unseeded grafts in wild-type recipient mice.
Figure 6. Graft H&E, PSR, and PGA fiber analysis.
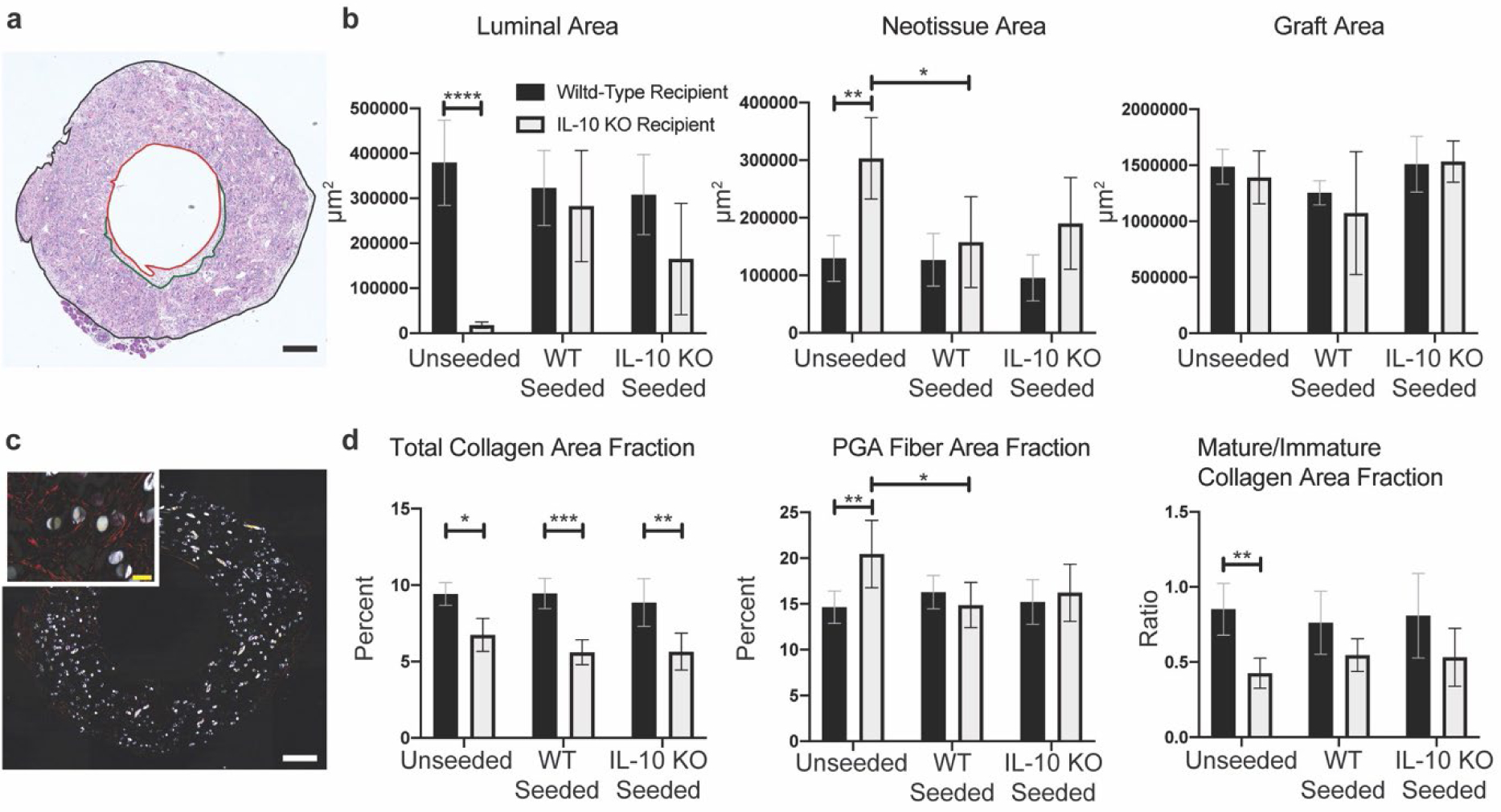
a) Representative tiled image of an H&E stained graft section. Luminal area demarcated in red outline; neotissue area demarcated in green outline; graft area demarcated in black outline. Image taken with a 10x objective. Scale bar = 200μm. b) Quantification of luminal area only, neotissue area only, and graft wall area (not including lumen and neotissue) from 10x H&E tiled images. Unseeded TEVGs in IL-10 KO recipient mice had significantly reduced luminal area compared to unseeded TEVGs in wild-type recipient mice (p < 0.0001). Unseeded TEVGs in IL-10 KO recipient mice had significantly increased neotissue area compared to unseeded TEVGs in wild-type recipient mice and wild-type seeded TEVGs in IL-10 KO recipient mice (p = 0.0062 and 0.0478, respectively). c) Representative tiled image of a tiled PSR stained graft section under polarized light. Image taken with 10x objective. Scale bar = 200μm. High magnification 63x inset in upper left-hand corner. Scale bar = 20μm. d) Total collagen, PGA fiber, and mature/immature quantifications presented as percent area fractions normalized to graft area as quantified in Figure 5b. All TEVGs in IL-10 KO recipient mice had significantly reduced total collagen area fractions compared to wild-type recipient counterparts (p = 0.0170, 0.0001, and 0.0065, respectively) and an overall trend in decreased ratio of mature/immature collagen. Unseeded TEVGs in IL-10 KO recipient mice had significantly increased PGA fiber area fraction compared to unseeded in wild-type recipient mice and wild-type seeded in IL-10 KO recipient mice (p = 0.0059 and p = 0.0202, respectively). For H&E histological quantification, n = 16–18 / seeding group for wild-type recipient mice and n = 11–12 per seeding group for IL-10 KO recipient mice. For PSR histological quantification, n = 18 per seeding group wild-type recipient mice and n = 11–12 per seeding group for IL-10 KO recipient mice. Two-way ANOVA utilized with multiple comparisons and data presented as means with 95% confidence interval. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****).
Figure 7. H&E and PSR staining quantification of unseeded grafts in IL-10 treated mice versus unseeded grafts in wild-type mice.
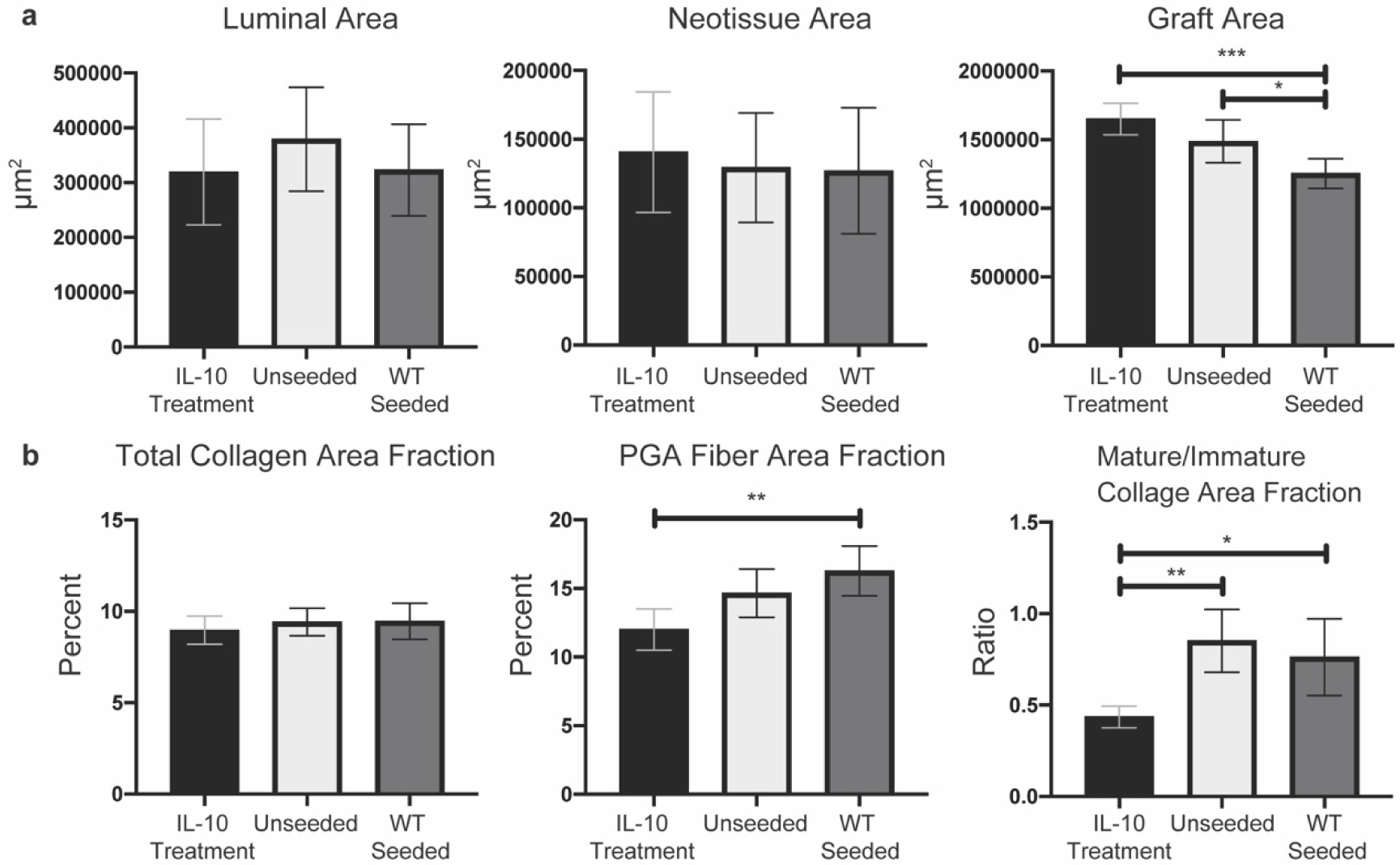
a) Quantification of luminal area, neotissue area, and graft area from H&E stained sections. No significant differences for luminal area and neotissue area. IL-10 treatment significantly increased graft fiber area compared to wild-type seeded grafts (p = <0.0001). b) Quantification of total collagen, PGA fiber, and mature/immature ratio from PSR stained sections under polarized light. TEVGs from IL-10 treated mice had significantly increased PGA fiber area fraction only compared to wild-type seeded grafts (p = 0.0014). TEVGs from IL-10 treated mice had a significantly decreased ratio of mature / immature collagen area fraction compared to unseeded grafts (p = 0.0011) and wild-type seeded grafts (p = 0.0122). For histological quantification, n size = 17–18 per group with Ordinary one-way ANOVA for normally distributed data or Kruskal-Wallis for non-normally distributed data. Presented as means with 95% confidence interval. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****).
Picrosirius red/fast green (PSR) staining was imaged under polarized light to determine total collagen, mature collagen (red-orange fibers), immature collagen (green-yellow fibers), and polyglycolic acid (PGA) fiber area (Figure 6c). Quantification was relative to total graft area to normalize for variances in explanted graft sizes (Figure 6d). Total collagen area fraction quantification showed that IL-10 KO recipient mice had significantly reduced collagen deposition compared to wild-type recipients for unseeded, wild-type seeded, and IL-10 KO seeded grafts (mean rank 31.58 vs 61.50, 19.50 vs 61.72, 19.73 vs 53.17, p = 0.0170, 0.0001, and 0.0065, respectively). Graft fiber area fraction quantification showed unseeded grafts in wild-type recipient mice had significantly less area fraction of PGA fibers at 2 weeks compared to IL-10 KO recipient mice (mean 14.66 vs 20.44, p = 0.0059). Additionally, unseeded grafts in IL-10 KO recipient mice had significantly greater fiber area fraction compared to wild-type seeded grafts in IL-10 KO recipient mice (mean 20.44 vs 14.87, p = 0.0202). Mature and immature collagen area fractions were calculated and then converted to a ratio of mature/immature. Two-way ANOVA showed this ratio to be significantly influenced by host recipient genotype (predicted mean difference 0.3060, p = 0.0004). Multiple comparison analysis showed a significant decrease in mature to immature collagen for unseeded grafts in IL-10 KO recipient mice compared to wild-type recipient mice (mean rank 26.33 vs 58.83, p = 0.0066). IL-10 treatment did not significantly influence total collagen area fraction (Figure 7b), but did significantly decrease fiber area fraction compared to wild-type seeded grafts (mean 12.01 vs 16.28, p = 0.0014), and significantly decreased mature/immature collagen ratio compared to unseeded and wild-type seeded grafts (mean 0.4353 vs 0.8513 and 0.7618, respectively, p = 0.0011, 0.0122). These results indicate that host IL-10 plays a role in collagen deposition and maturation in TEVGs.
2.5. Serology
Mouse whole blood platelet analysis revealed no significant differences between male/female wild-type mice or male/female IL-10 KO mice (Figure 8a). Mouse whole blood white blood cell (WBC) analysis (Figure 8b) showed that female IL-10 KO mice had significantly increased numbers of white blood cells (WBC) compared to wild-type male/female mice as well as male IL-10 KO mice (mean 6.292 million cells / mm3 vs 2.942, 6.292 vs 2.242, 6.292 vs 4.142, p = 0.0119, 0.0002, and 0.0239, respectively). IL-10 KO male mice had significantly increased WBC compared to wild-type female mice only (mean 4.142 million cells / mm3 vs 2.242, p = 0.0321).
Figure 8. Mouse platelet and white blood cell count analysis.
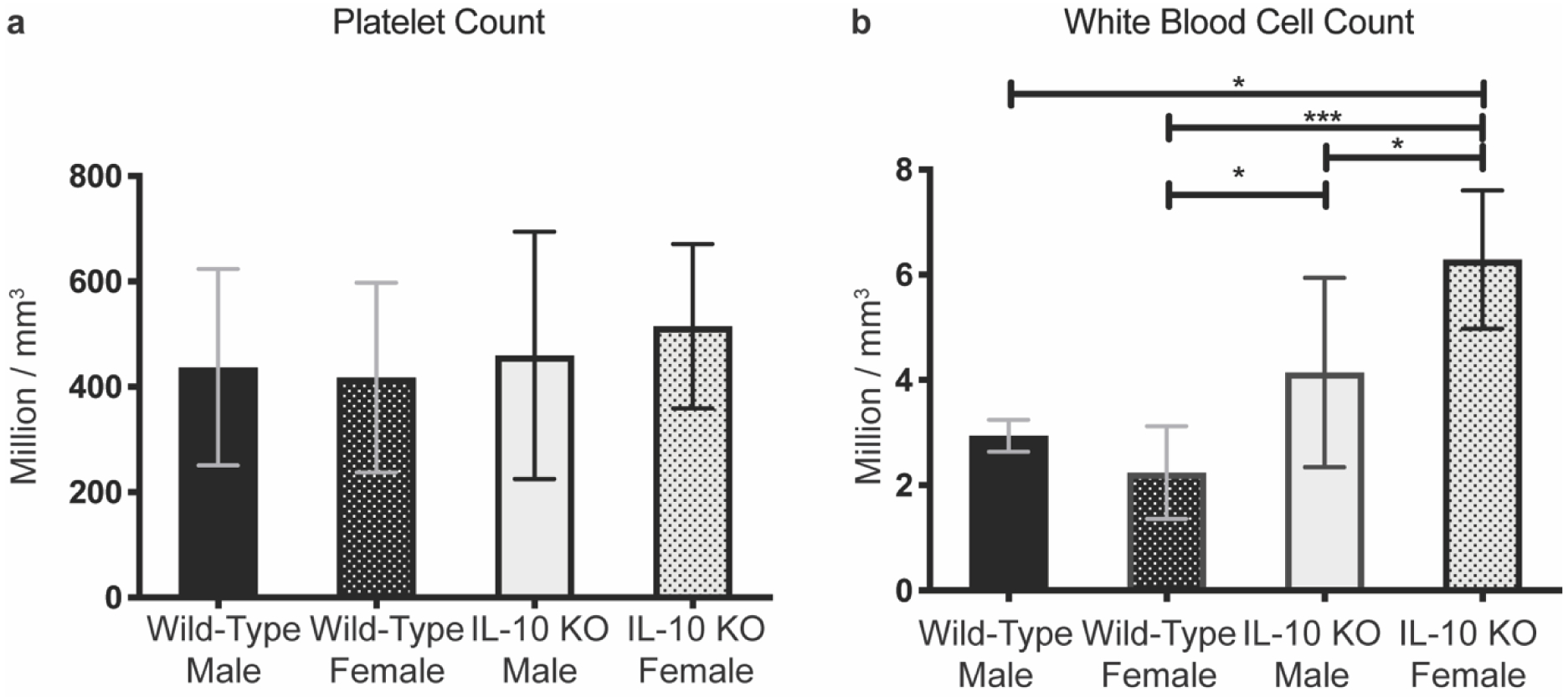
Following IVC blood draw (n= 6 per group), blood was mixed with EDTA and read on an ABX Micros 60 hematology analyzer. a) Platelet count analysis showed no significant differences. b) White blood cell count analysis showed IL-10 KO female mice had significantly increased counts compared to IL-10 male, wild-type female, and wild-type male mice (p = 0.0239, 0.0002, 0.0119, respectively). IL-10 KO male mice had significantly increased counts compared to wild-type female mice (p =0.0321). Ordinary one-way ANOVA was utilized and data presented as means with 95% confidence interval. p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***), p ≤ 0.0001 (****).
3. Discussion
To investigate the role of IL-10 in TEVG growth and remodeling, we performed a series of reciprocal genetic experiments by placing TEVG implants into both wild-type and IL-10 KO recipient mice. TEVGs were unseeded, wild-type BM-MNC seeded, or IL-10 KO BM-MNC seeded. Our results demonstrate that IL-10 released from seeded cells had little effect on graft inflammatory status, remodeling, and overall patency. However, recipient IL-10 status was critical for graft patency and collagen deposition / maturation. Cell seeding with wild-type BM-MNCs rescued TEVGs from complete thrombosis in IL-10 KO mice.
If a graft occluded, it occurred by day 3 post-operatively due to thrombosis and did not reverse course. Ultrasound results were confirmed with visual inspection of H&E graft sections. The ultrasound and histology results revealed a clear predisposition for 100% of unseeded grafts to thrombose in IL-10 KO mice. While no known studies directly show that IL-10 KO mice have an increased risk of venous thrombosis, several other works provide strong support for a critical role in IL-10 reducing the risk of venous thrombosis.[21,29,30] When seeded with wild-type BM-MNCs, TEVGs were rescued from acute thrombosis with patency levels similar to TEVGs in wild-type recipient mice. This suggests in a pro-thrombotic environment, the greatest protective effect afforded by seeded BM-MNCs is their anti-thrombotic properties. The precise cause of why IL-10 deficiency led to an enhanced pro-thrombotic state and how the seeded cells mitigated it remains unclear. Previous work has shown that seeded BM-MNCs in TEVGs potentially inhibit TEVG thrombosis by preventing adhesion of platelets to the graft luminal surface.[13,19] We performed a platelet count on whole collected blood and observed no quantitative platelet differences between IL-10 KO and wild-type mice. IL-10 KO mice are known to have leukocytosis and we confirmed this finding, indicating an upregulated inflammatory state. IL-10 is also known to play a prominent role in modulating monocyte tissue factor expression, a pro-thrombotic protein responsible for initiating the coagulation cascade leading to a fibrin clot.[31,32] Monocytes expressing tissue factor represent that greatest source for this protein’s expression in circulating blood.[33] Specifically, IL-10 is understood to strongly down-regulate the surface level expression of tissue factor on circulating monocytes.[23,24] IL-10 KO mice are relatively understudied in the context of the clotting cascade, but work by Caligiuri, et al has previously shown upregulation in tissue factor production in IL-10 KO mice.[29] We posit that the pro-thrombotic state we observe for TEVGs in IL-10 KO may be due to upregulated tissue factor expression. Given the evidence of cardiac surgical patients to have upregulated circulating tissue factor and the nature of our repeat congenital heart disease patients this represents an interesting area of future research and investigation.[11,34]
We performed an in-depth histological analysis to characterize graft inflammation and development. No impact of cell seeding or host IL-10 status was observed on the ratio of PCNA+ cells. Our prior work characterized proliferating cells within TEVGs and found two predominate populations, F4/80+ (macrophage specific) staining and COL1A1+ staining cells in accordance with classically described foreign body response.[4,35] Presently, we found that the degree of macrophage infiltration and ratio of PCNA+ cells in the graft at 14 days was independent of both seeding scheme and recipient genotype. This is in contrast to previous work showing TEVGs seeded with BM-MNCs have decreased macrophage infiltration compared to unseeded TEVGs at 14 days post implantation.[14] The macrophage discrepancies may be attributable to the quantification method. The previous study utilized anti-F4/80 staining to quantify macrophage infiltration as opposed to the anti-CD68 staining described herein. Both F4/80 and CD68 suffer from off-target staining of leukocytes other than macrophages in addition to expression intensity differences in TEVGs at different time points. [4,36–38]
Phenotype characterization of infiltrating macrophages focused on separating them into either a pro-inflammatory “M1” or an alternative “M2” category. For M1 macrophages, iNOS+ staining showed significant differences only between wild-type seeded grafts in wild-type recipient and IL-10 KO recipient mice. For M2 macrophages, CD206+ staining was only significantly influenced by recipient host genotype for unseeded grafts. As an M1/M2 ratio, IL-10 KO mice had significantly decreased M1/M2 indicating a greater portion of M2 macrophages compared to its wild-type counterpart. The decreased M1/M2 ratio appears counter-intuitive in a host lacking IL-10, a cytokine known for its ability to polarize macrophages along an M2 phenotype. However, macrophage phenotype characterization in implantable materials requires a high degree of scrutiny. While both iNOS+ and CD206+ staining have been specifically used for classifying M1 and M2 macrophages in the foreign body response, macrophage polarization is a continuous spectrum rather than a dichotomous classification with M2 macrophages being further broken down into M2a, M2b, M2c, and M2d phenotypes.[39–41] In an in vivo, foreign body reaction setting, the classification becomes even more complicated. At 2 weeks, foreign body giant cells compose a significant portion of the macrophage lineage in the graft material. They have a wide spectrum of marker expression crossing between M1 and M2 making them challenging to divest into a singular phenotype.[42] In implanted biomaterials, a significant portion of macrophages involved in the foreign body response have been characterized to simultaneously express both M1+ and M2+ markers. [43]
H&E in combination with PSR staining was utilized to assess tissue and collagen development in explanted TEVGs. Unsurprisingly, IL-10 KO recipient mice had significantly reduced luminal area compared to wild-type recipient mice primarily. The effect of cell seeding in the IL-10 KO mice rescued this phenotype providing similar luminal area and neotissue area to its counterpart in wild-type recipient mice.
For total collagen in the graft, all TEVGs in IL-10 KO recipient mice had significantly reduced deposition compared to their respective counterpart TEVGs in wild-type mice. The collagen that was deposited showed significantly reduced maturity based on a ratio of mature/immature collagen. From a total collagen perspective, mannose receptor positive (CD206+) M2 macrophages have been linked as a driving macrophage phenotype responsible for increased collagen degradation in wound repair[44]. While we did see some increased CD206+ staining and decreased iNOS+ / CD206+ staining ratio in IL-10 KO recipient mice compared to wild-type recipient mice, this observation was not uniform across all seeding groups. In absence of a uniform macrophage phenotype difference explaining the collagen observations, it is possible that it is attributable to an inherent phenotype of IL-10 KO mice. Notably, though, the total collagen differences stand in contrast to reports on IL-10 KO mice having increased collagen deposition in both skin wound beds and in vascular tissue.[45,46] However, IL-10 KO mice are reported to have defects in collagen maturation in agreement with our results.[45] The uniqueness of the TEVG environment would require follow-up investigation to explore the potential mechanism behind the decreased collagen deposition. Additionally, we found TEVGs in IL-10 KO recipient mice had significantly reduced CD31+ neovascularization compared to wild-type recipient mice, indicating impaired angiogenesis in IL-10 KO mice. Indeed, IL-10 KO mice have been shown in a carotid injury model to have delayed re-endothelialization.[47] Taken together, decreased neoangiogenesis and decreased collagen deposition indicate that IL-10 KO mice may have an impairment in new tissue formation during neovessel development. However, the 14-day time course limits our ability to investigate this further. Future work including a serial time course of graft explant until total material degradation would provide better insight into how these properties change over time. A longer time course with ex-vivo graft analysis of biomechanical properties and vessel functionality would also further our understanding of potential long term, functional consequences of TEVG development in IL-10 KO mice and the role of IL-10 in neovessel growth and remodeling.
Recombinant IL-10 treatment decreased the extent of proliferating cells within the graft compared to unseeded grafts. Despite this, IL-10 treated mice had significantly increased macrophage infiltration compared to untreated mice. In context of other work, IL-10 treatment in blood vessels following injury has been shown to decrease the overall inflammatory status as well as decreasing the number of macrophages and proliferating cells.[21,48] For macrophage phenotype polarization, IL-10 treatment significantly increased the number of M2 macrophages in the graft and decreased the ratio of M1 to M2 positive macrophages. Despite these effects of IL-10 treatment, we failed to see any significant improvements in graft development from either reduced occlusion, reduced neotissue formation, or increased vascularization (CD31+ staining). Part of this limited impact of IL-10 treatment could relate to the method of drug delivery itself and dosing. We chose intravenous injections given previous reports demonstrating success in mitigating thrombosis in a murine IVC thrombosis model.[30] Our TEVG surgical model is unique and may require additional fine-tuning to optimize for IL-10 treatment.
There are several additional limitations and points to consider for future investigations utilizing our murine TEVG model. An inability to achieve 100% cell seeding efficiency should be noted when considering the potential difference in effects from wild-type seeded and IL-10 KO seeded BM-MNCs. The static seeding method utilized in this study has been previously shown by our group to have a seeding efficiency of 25%.[49] This represents areas of the graft remaining exposed. In larger diameter TEVGs, such as the ones utilized in our large animal studies, vacuum seeding is able to improve seeding efficiency to approximately 40%.[50] However, this more efficient seeding method is not currently feasible in the smaller murine TEVGs. While the seeding efficiency is not perfect, the cells that are seeded are observed to attach in the luminal side of the porous scaffold.[50] Another consideration is the location of histological analysis. In this present work, we elected to perform all histological analysis and quantification utilizing cross-sections from the middle of the graft. We chose to utilize mid-graft sections to limit analysis to the body of the TEVG only and isolate out the variables introduced by surgical injury, suture presence, and native IVC presence that would all be captured and confounded on histological analysis. Additionally, this section of graft reflects the location of occlusion we recently observed in a large animal study.[12] However, future work seeking to address the influence of IL-10 host status on additional sites adjacent to the TEVG could indeed benefit from a histological analysis of the anastomoses. An alternative approach in this future work could potentially utilize a longitudinal sectioning method allowing quantification of inflammatory and other histological markers along the graft’s entire length.
4. Conclusion
In summary, we found that IL-10 secreted from BM-MNCs seeded onto TEVGs does not play a significant role in graft patency, graft macrophage infiltration, and macrophage phenotype polarization. While recombinant IL-10 treatment did affect the inflammatory status and decrease the ratio of M1 (iNOS+) to M2 (CD206+) macrophages in the graft, this did not translate to changes in graft patency, neotissue deposition, or graft vascularization. Our findings, however, demonstrate that host IL-10 is critical for TEVG patency and neovessel development. BM-MNC cell seeding was able to rescue grafts in the absence of host IL-10 through a potential anti-thrombotic mechanism. These results highlight the importance of understanding the complicated host/TEVG interface when translating TEVGs to patient use.
5. Experimental Section
5.1. Animal Welfare Statement
All surgeries, procedures, and experiments involving the use of animals in this study were done with approval from the Abigail Wexner Research Institute (AWRI) at Nationwide Children’s Hospital Institutional Animal Care and Use Committee (IAUCUC). The AWRI at Nationwide Children’s Hospital is registered as a research facility with the U.S Department of Agriculture and is fully accredited by the AAALAC (#31-R-0166). An animal welfare assurance is on file with OPRR-NIH. Additionally, all surgeries, procedures, and experiments with animals were done in accordance with the National Institutes of Health (NIH) and the American Veterinary Medical Association (AVMA) guidelines.
5.2. Mouse Models
C57BL/6J mice (Stock No: 000664) and IL-10 knock out mice (B6.129P2-Il10tm1Cgn/J, Stock No: 002251) were purchased from The Jackson Laboratory (Bar Harbor, ME).[51]
5.3. TEVG Fabrication
Tissue Engineered Vascular Grafts (TEVGs) were constructed using a dual chamber method as described previously.[52] Briefly, a dual chamber was created by using a non-tapered, non-graduated polypropylene tube as the outer chamber. The inner chamber was made with a 21g hypodermic needle guided through the polypropylene tube. Polyglycolic acid (PGA) mesh (Concordia Fibers, Conventry, RI) was cut into a 3mm × 5mm sheet and wrapped around the inner needle. A poly-L-lactide/- ε-caprolactone sealant (Gunze, Kyoto, Japan) was combined with 1,4-Dioxane (Fisher Chemical) and vortexed for two hours before applying to the mesh in the dual chamber system. Grafts were lyophilized overnight and removed from the dual chamber. Each graft was controlled for quality via inspection under a microscope for uniform diameter, uniform thickness, and any additional signs of gross defects. Grafts were stored in a desiccator until implantation.
5.4. Bone-Marrow Harvest, Scaffold Seeding, and Surgery
Murine bone-marrow was collected as previously described.[53] Harvested cells were resuspended in DMEM (Sigma-Aldrich). Nucleated cells were separated from the cell suspension through density-based centrifugation utilizing Histopaque 1083 (1.083 g/mL, Millipore Sigma, Burlington, MA). Cell concentrations were determined with an ABX Micros 60 hematology analyzer (Horiba, Edison, NJ). Isolated cells were spun down at 300g for 5 minutes and resuspended in DMEM + 1% Pen/Step to a final concentration of 1 million cells / 4μL. Scaffolds were sterilized via exposure to UV light for 15 minutes prior to cell seeding. Grafts were pre-moistened with DMEM + 1% Pen/Strep for 5 minutes. DMEM was removed and 4uL (~1 million cells) was added to the graft. Grafts were incubated at 37°C overnight in a 24-well plate with 1mL of DMEM + 1% Pen/Strep. Scaffolds were implanted into 8–12 week-old female mice as interposition inferior vena cava grafts using standard microsurgical techniques.[52] Mice were recovered from anesthesia without administration of anti-platelet or anti-coagulant drugs. No mortalities during the surgery or due to graft failure occurred. One IL-10 KO mice with an IL-10 KO seeded graft died from surgical site dehiscence.
5.5. Histology and Immunohistochemistry
After two weeks, mice were sacrificed and the abdominal cavity was opened to expose the IVC. TEVGs were flushed with saline, excised, and cut in half through the middle of the graft. Samples were formalin fixed and paraffin embedded and cut cross-sectionally through the middle face of the graft creating 4μm tissue sections onto microscope slides. For histology, H&E and Picrosirius Red/Fast Green (PSR) were performed using standard techinque. For immunohistochemistry, paraffin embedded tissue sections were deparaffinized through xylene and graded alcohol washes. Antigen retrieval was performed in citrate buffer pH 6.1 (Agilent Dako, Target Retrieval Solution, Citrate pH 6.1 (10x)) with constant temperature and pressure from a pressure cooker. Endogenous peroxide blocking was performed by incubating samples in 3% H202 followed by water and PBS-Tween20 washes. Background blocking was performed with a cocktail of Background Sniper (BioCare Medical, Background Sniper) + 3% goat serum (Vector Laboratories, Normal Goat Serum Blocking Solution). Overnight incubation at 4°C was performed for the following primary antibodies in their respective concentrations diluted in Antibody Diluent, Background Reducing (Agilent Dako): CD68 1:2000 (abcam, ab125212), PCNA 1:750 (abcam, ab2426, *Discontinued), CD31 1:500 (abcam, ab28364), iNOS 1:1000 (abcam, ab15323), Mannose Receptor 1:500 (CD206, abcam, ab64693). Chromogenic developed stains were incubated with species appropriate biotinylated secondary antibodies, followed by incubation in avidin/peroxidase (VECTASTAIN Elite ABC HRP Reagent, Vector Laboratories) prior to color development with ImmPACT DAB Peroxidase (HRP) substrate (Vector Laboratories). Optimal color development time was determined individually for each antibody. Samples were counterstained in Gill’s Hematoxylin (Vector Laboratories) and dehydrated followed by permanent mounting (VectaMount Permanent Mounting Medium, Vector Laboratories). For immunofluorescence staining with Mannose Receptor, the same protocol was followed above but modified using a fluorometric anti-rabbit secondary antibody at 1:300 concentration (Thermofisher, Alexa Fluor 647). The immunofluorescence stained slides were then mounted with Slowfade Gold Antifade containing DAPI (Thermofisher, S36938) and cover slipped.
5.6. Ultrasonography
Serial ultrasound was performed with a Vevo 2100 Imaging Platform (Visualsonics, Toronto, Canda). Mice were initially anesthetized with 3.5% isoflurane and then maintained at 2% isoflurane. Mice were placed on a heated platform with limbs positioned on ECG leads in a supine position. The abdomen was treated with Nair to remove any hair and a thin layer of ultrasound gel was applied. Long-axis images were obtained utilizing the B-mode, pulse-wave, and color Doppler functions. A 32MHz frequency transducer was used with pulse-wave readings taken at a sweep speed of 1200Hz.
5.7. IL-10 Drug Treatment
C57BL/6J mice were treated with 1μg doses of recombinant mouse IL-10 (Biolegend, catalog #575804) re-suspended in 100μL of sterile PBS. Four total doses were administered at 2 hours prior to surgery, during surgery, 24 hours post-surgery, and 48 hours post-surgery. Doses were administrated via jugular vein injections for pre- and post-surgery while the intraoperative dose was given immediately via the inferior vena cava following TEVG implantation.
5.8. Quantitative Image Analyses
Histological images were acquired on a Zeiss AxioObserver.Z1 inverted microscope, 89-North PhotoFluor LM-75 light source with appropriate filters and a Zeiss Axiocam 105 coupled with a (color) digital camera. Images were taken using the Zen software program and processed using Fiji, a bundled version of Image J (National Institutes of Health). For quantification of CD31, CD68, iNOS, and CD206, a total of (4) random high-power field (40x) images were taken per sample section, quantified by hand-counting, and averaged. For quantification of PCNA, and PSR, pixels were calculated by specific thresholding based on hue, saturation, and brightness.
5.9. Blood Collection and Analysis
For whole blood collection, mice were initially anesthetized with 3.5% isoflurane and then maintained at 2% isoflurane. Sterile 1.5mL collection tubes were pre-coated with 50uL of 0.5M EDTA as well as 1ml syringes pre-coated with 50uL of 0.5M EDTA. A single incision was made along the abdomen exposing the peritoneum. The peritoneum was separated exposing the intestines, which were coiled and set aside. The inferior vena cava was identified and separated from the aorta. The pre-coated syringe along with a 25g needle was used to puncture the IVC and draw blood until the mouse was exsanguinated. Blood was collected into the pre-coated 1.5mL collection tubes and gently mixed prior to analyses on a Horiba ABX Micros 60 providing both platelet and white blood cell counts. Two measurements were taken per collected sample and averaged.
5.10. Statistical Analysis
Statistical analysis was performed on GraphPad Prism Version 8.4.2(464). A p value of less than .05 was deemed statistically significant. For ultrasound and patency outcomes, a modified Fisher’s Exact test was utilized with Freeman-Halton extension with the following samples sizes: n = 18 / seeding group and IL-10 treatment group in wild-type recipient mice, n = 12 / wild-type seeded and unseeded and 11 / IL-10 KO seeded in IL-10 KO recipient mice. For statistical analysis of histological quantification in wild-type recipient vs IL-10 KO recipient comparisons, a two-way ANOVA was performed separated by (1) seeding group and (2) recipient mouse genotype. If a statistically significant difference in 1 or both effects was observed, a multiple comparison test was performed with either Holm-Sidak’s adjustment for normally distributed data, or with Dunn’s adjustment for non-normally distributed data. For comparison of unseeded TEVGs in mice treated with IL-10, unseeded TEVGs in wild-type recipient mice, and wild-type seeded TEVGs in wild-type recipient mice, Ordinary one-way ANOVA was utilized for normally distributed data or Kruskal-Wallis test for non-normally distributed data. Multiple comparison test was performed and adjusted with Tukey’s adjustment for normally distributed data or Dunn’s adjustment for non-normally distributed data.
Acknowledgements
This work was in part supported by U.S NIH grants R01HL128847 and R01HL098228. J.C.B. was supported by NIH T32AI106704, NIH F32HL144120, and the Ohio State University President’s Postdoctoral Scholar Program. Y.C was supported in part by the PhRMA Foundation Predoctoral Fellowship. C.A.B was supported in part by NIH F31HL145962. J.W.R was supported in part by the American Heart Association under Award Number 18POST33990231. The authors thank the Animal Resource Core at The Abigail Wexner Research Institute at Nationwide Children’s Hospital for their assistance in animal welfare and care. We also thank the Histology Core for its assistance with graft processing and sectioning and Cassidy Duquaine for drawings utilized in this manuscript.
Footnotes
Conflicts of Interest
C.K.B. is an inventor on patent/patent applications [2015252805 (Australia), 2016565483 (Japan), 855,370, 9,446,175, 9,782,522, 10,300,082, 61/987,910, 62/266,309, 62/309,285, 62/209,990, 62/936,225] submitted by Yale University and/or Nationwide Children’s Hospital that cover methods of improving the design, manufacturing, or performance of tissue-engineered vascular grafts. C.K.B. is a founder of Lyst Therapeutics
References
- [1].Hibino N, McGillicuddy E, Matsumura G, Ichihara Y, Naito Y, Breuer C, Shinoka T, The Journal of Thoracic and Cardiovascular Surgery 2010, 139, 431. [DOI] [PubMed] [Google Scholar]
- [2].Shin’oka T, Matsumura G, Hibino N, Naito Y, Watanabe M, Konuma T, Sakamoto T, Nagatsu M, Kurosawa H, The Journal of Thoracic and Cardiovascular Surgery 2005, 129, 1330. [DOI] [PubMed] [Google Scholar]
- [3].Naito Y, Imai Y, Shin’oka T, Kashiwagi J, Aoki M, Watanabe M, Matsumura G, Kosaka Y, Konuma T, Hibino N, et al. , The Journal of Thoracic and Cardiovascular Surgery 2003, 125, 419. [DOI] [PubMed] [Google Scholar]
- [4].Reinhardt JW, Rosado J. de D. R., Barker JC, Lee Y-U, Best CA, Yi T, Zeng Q, Partida-Sanchez S, Shinoka T, Breuer CK, Regenerative Medicine 2019, 14, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Villalona GA, Udelsman B, Duncan DR, McGillicuddy E, Sawh-Martinez RF, Hibino N, Painter C, Mirensky T, Erickson B, Shinoka T, et al. , Tissue Engineering Part B: Reviews 2010, 16, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schulte J, Friedrich A, Hollweck T, König F, Eblenkamp M, Beiras-Fernandez A, Fano C, Hagl C, Akra B, Processes 2014, 2, 526. [Google Scholar]
- [7].Melchiorri AJ, Bracaglia LG, Kimerer LK, Hibino N, Fisher JP, Tissue Engineering Part C: Methods 2016, 22, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Radke D, Jia W, Sharma D, Fena K, Wang G, Goldman J, Zhao F, Adv. Healthcare Mater 2018, 7, 1701461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rotmans JI, Heyligers JM, Stroes ES, Pasterkamp G, Can J Cardiol. 2006, 22, 4. [Google Scholar]
- [10].Roh JD, Sawh-Martinez R, Brennan MP, Jay SM, Devine L, Rao DA, Yi T, Mirensky TL, Nalbandian A, Udelsman B, et al. , Proceedings of the National Academy of Sciences 2010, 107, 4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Breuer C, n.d., NCT01034007. https://clinicaltrials.gov/ct2/show/NCT0103400. Accessed 10-Oct-2020.
- [12].Drews JD, Pepper VK, Best CA, Szafron JM, Cheatham JP, Yates AR, Hor KN, Zbinden JC, Chang Y-C, Mirhaidari GJM, et al. , Sci. Transl. Med 2020, 12, eaax6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Miyachi H, Reinhardt JW, Otsuru S, Tara S, Nakayama H, Yi T, Lee Y-U, Miyamoto S, Shoji T, Sugiura T, et al. , International Journal of Cardiology 2018, 266, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hibino N, Yi T, Duncan DR, Rathore A, Dean E, Naito Y, Dardik A, Kyriakides T, Madri J, Pober JS, et al. , FASEB j. 2011, 25, 4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].de Dios Ruiz‐Rosado J, Lee Y, Mahler N, Yi T, Robledo‐Avila F, Martinez‐Saucedo D, Lee AY, Shoji T, Heuer E, Yates AR, et al. , FASEB j. 2018, 32, 6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hibino N, Mejias D, Pietris N, Dean E, Yi T, Best C, Shinoka T, Breuer C, FASEB j. 2015, 29, 2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Harrington JK, Chahboune H, Criscione JM, Li AY, Hibino N, Yi T, Villalona GA, Kobsa S, Meijas D, Duncan DR, et al. , FASEB j. 2011, 25, 4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Patterson JT, Gilliland T, Maxfield MW, Church S, Naito Y, Shinoka T, Breuer CK, Regenerative Medicine 2012, 7, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Best C, Tara S, Wiet M, Reinhardt J, Pepper V, Ball M, Yi T, Shinoka T, Breuer C, ACS Biomater. Sci. Eng 2017, 3, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R, Science 2017, 356, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Downing LJ, Strieter RM, Kadell AM, Austin JC, Hare BD, Burdick MD, Greenfield J, Wakefield TW, J. Immunol 1998, 161, 1471. [PubMed] [Google Scholar]
- [22].Pajkrt D, van der Poll T, Levi M, Cutler DL, Affrime MB, van den Ende A, Wouter ten Cate J, van Deventer SJH, Blood 1997, 89, 2701. [PubMed] [Google Scholar]
- [23].Kamimura M, Viedt C, Dalpke A, Rosenfeld ME, Mackman N, Cohen DM, Blessing E, Preusch M, Weber CM, Kreuzer J, et al. , Circulation Research 2005, 97, 305. [DOI] [PubMed] [Google Scholar]
- [24].Ramani M, Ollivier V, Khechai F, Vu T, Ternisien C, Bridey F, de Prost D, FEBS Letters 1993, 334, 114. [DOI] [PubMed] [Google Scholar]
- [25].Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW, Vunjak-Novakovic G, Biomaterials 2014, 35, 4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lee WJ, Tateya S, Cheng AM, Rizzo-DeLeon N, Wang NF, Handa P, Wilson CL, Clowes AW, Sweet IR, Bomsztyk K, et al. , Diabetes 2015, 64, 2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sridharan R, Cameron AR, Kelly DJ, Kearney CJ, O’Brien FJ, Materials Today 2015, 18, 313. [Google Scholar]
- [28].Hall PA, Levison DA, Woods AL, Yu CC-W, Kellock DB, Watkins JA, Barnes DM, Gillett CE, Camplejohn R, Dover R, et al. , J. Pathol 1990, 162, 285. [DOI] [PubMed] [Google Scholar]
- [29].Caligiuri G, Rudling M, Ollivier V, Jacob M-P, Michel J-B, Hansson GK, Nicoletti A, Mol Med 2003, 9, 10. [PMC free article] [PubMed] [Google Scholar]
- [30].Myers DD, Hawley AE, Farris DM, Chapman AM, Wrobleski SK, Henke PK, Wakefield TW, Journal of Surgical Research 2003, 112, 168. [DOI] [PubMed] [Google Scholar]
- [31].Grover SP, Mackman N, Arterioscler Thromb Vasc Biol. 2018, 38, 709. [DOI] [PubMed] [Google Scholar]
- [32].Palta S, Saroa R, Palta A, Indian J Anaesth. 2014, 58, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Swystun LL, Liaw PC, Blood 2016, 128, 753. [DOI] [PubMed] [Google Scholar]
- [34].Biro E, Sturk-Maquelin KN, Vogel GMT, Meuleman DG, Smit MJ, Hack CE, Sturk A, Nieuwland R, J Thromb Haemost 2003, 1, 2561. [DOI] [PubMed] [Google Scholar]
- [35].Geelhoed WJ, Moroni L, Rotmans JI, of Cardiovasc J. Trans. Res 2017, 10, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gordon S, Plüddemann A, Martinez Estrada F, Immunol Rev 2014, 262, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].McGarry MP, Stewart CC, J Leukoc Biol 1991, 50, 471. [DOI] [PubMed] [Google Scholar]
- [38].Li W, Kapadia S, Sonmez-Alpan E, Swerdlow S, Mod Pathol 1996, 9, 982. [PubMed] [Google Scholar]
- [39].van Putten SM, Ploeger DTA, Popa ER, Bank RA, Acta Biomaterialia 2013, 9, 6502. [DOI] [PubMed] [Google Scholar]
- [40].Brown BN, Sicari BM, Badylak SF, Front. Immunol 2014, 5, 1. 10.3389/fimmu.2013.00510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Atri C, Guerfali F, Laouini D, IJMS 2018, 19, 1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Noh I, Ed. , Biomimetic Medical Materials: From Nanotechnology to 3D Bioprinting, Springer Singapore, Singapore, 2018. [Google Scholar]
- [43].Sussman EM, Halpin MC, Muster J, Moon RT, Ratner BD, Ann Biomed Eng 2014, 42, 1508. [DOI] [PubMed] [Google Scholar]
- [44].Madsen DH, Leonard D, Masedunskas A, Moyer A, Jürgensen HJ, Peters DE, Amornphimoltham P, Selvaraj A, Yamada SS, Brenner DA, et al. , The Journal of Cell Biology 2013, 202, 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Eming SA, Werner S, Bugnon P, Wickenhauser C, Siewe L, Utermöhlen O, Davidson JM, Krieg T, Roers A, The American Journal of Pathology 2007, 170, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dammanahalli JK, Wang X, Sun Z, Journal of Hypertension 2011, 29, 2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Verma SK, Garikipati VNS, Krishnamurthy P, Khan M, Thorne T, Qin G, Losordo DW, Kishore R, PLoS ONE 2016, 11, e0147615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Feldman LJ, Aguirre L, Ziol M, Bridou J-P, Nevo N, Michel J-B, Steg PG, Circulation 2000, 101, 908. [DOI] [PubMed] [Google Scholar]
- [49].Roh JD, Nelson GN, Udelsman BV, Brennan MP, Lockhart B, Fong PM, Lopez-Soler RI, Saltzman WM, Breuer CK, Tissue Engineering 2007, 13, 2743. [DOI] [PubMed] [Google Scholar]
- [50].Best C, Strouse R, Hor K, Pepper V, Tipton A, Kelly J, Shinoka T, Breuer C, J Tissue Eng 2018, 9, 204173141876470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W, Cell. 1993, 75, 263. [DOI] [PubMed] [Google Scholar]
- [52].Roh JD, Nelson GN, Brennan MP, Mirensky TL, Yi T, Hazlett TF, Tellides G, Sinusas AJ, Pober JS, Saltzman WM, et al. , Biomaterials 2008, 29, 1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Amend SR, Valkenburg KC, Pienta KJ, J. Visualized Exp 2016, 110, e53936. [DOI] [PMC free article] [PubMed] [Google Scholar]