

Abstract
CGG repeat expansions within the premutation range (55–200) of the FMR1 gene can lead to Fragile X-associated tremor/ataxia syndrome and Fragile X-associated neuropsychiatric disorders. These CGG repeats are translated into a toxic polyglycine-containing protein, FMRpolyG. Pathology of Fragile X-associated tremor/ataxia syndrome and Fragile X-associated neuropsychiatric disorders comprises FMRpolyG- and p62-positive intranuclear inclusions. Diagnosing a FMR1-premutation carrier remains challenging, as the clinical features overlap with other neurodegenerative diseases. Here, we describe two male cases with Fragile X-associated neuropsychiatric disorders-related symptoms and mild movement disturbances and novel pathological features that can attribute to the variable phenotype. Macroscopically, both donors did not show characteristic white matter lesions on MRI; however, vascular infarcts in cortical- and sub-cortical regions were identified. Immunohistochemistry analyses revealed a high number of FMRpolyG intranuclear inclusions throughout the brain, which were also positive for p62. Importantly, we identified a novel pathological vascular phenotype with inclusions present in pericytes and endothelial cells. Although these results need to be confirmed in more cases, we propose that these vascular lesions in the brain could contribute to the complex symptomology of FMR1-premutation carriers. Overall, our report suggests that Fragile X-associated tremor/ataxia syndrome and Fragile X-associated neuropsychiatric disorders may present diverse clinical involvements resembling other types of dementia, and in the absence of genetic testing, FMRpolyG can be used post-mortem to identify premutation carriers.
Keywords: FMR1-premutation, FXAND, FXTAS, neuropathology, nuclear inclusions
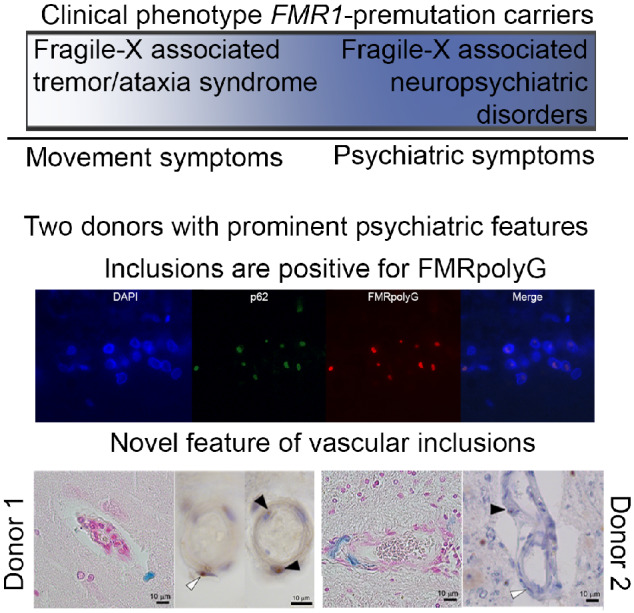
FMR1-premutation carriers can present clinically with a range of symptoms, including neuropsychiatric symptoms. Pathologically, the nuclear inclusions are positive for FMRpolyG and in the donors discussed here, also present in the vasculature. This has not been described before and this finding possibly contributes to the complex phenotype.
Graphical Abstract
Graphical Abstract.
Introduction
The Fragile X mental retardation 1 (FMR1) gene contains a CGG dynamic trinucleotide repeat sequence. Within the general population, this number of CGG repeats is present under 55 repeats (Hagerman and Hagerman, 2015). Expansions of the repeat sequence to more than 200 CGG repeats result in the absence of the FMR1 protein, and in the Fragile X syndrome (Murray et al., 1997). Smaller expansions (55–200 CGG repeats) are in the premutation range (Brouwer et al., 2009) and can lead to Fragile X-associated diseases, including the neurodegenerative diseases Fragile X-associated tremor/ataxia syndrome (FXTAS) (Berry-Kravis et al., 2007a) and the Fragile X-associated neuropsychiatric disorders (FXAND) (Hagerman et al., 2018). CGG-repeats in the premutation range are transcribed and translated into a toxic polyglycine-containing protein, named FMRpolyG protein (Todd et al., 2013). FMRpolyG is considered to play a major role in the pathogenesis of FXTAS (Sellier et al., 2017; Boivin et al., 2018).
The FXTAS phenotype is found in 30% of male carriers and 11–18% of female carriers of the FMR1-premutation and is characterized primarily by intention tremors and cerebellar gait ataxia, and in severe cases accompanied by executive function, memory deficits and Parkinsonism (Jacquemont, 2004; Leehey et al., 2016; Robertson et al., 2016). The onset of the motor symptoms may present after 50 years of age, with longer expansions correlating with an earlier onset and more severe disease (Tassone et al., 2007). However, additional clinical features have been recently described in premutation carriers that include anxiety and depression, which has led to the recent identification of FXAND, encompassing a large group of premutation carriers presented with neuropsychiatric symptoms (Hagerman et al., 2018). Consequently, diagnosing a FMR1-premutation carrier remains challenging, as the clinical features may overlap with other neurodegenerative diseases (Grigsby et al., 2007; Seritan et al., 2008). A genetic screening for CGG expansion can provide the conclusive answer, especially when a grandchild or child is diagnosed with a full mutation, leading to the Fragile X syndrome (Young, 1993).
The pathology of an FMR1-premutation includes p62-positive and mostly solitary, spherical intranuclear inclusions, present in both neurons and astrocytes and found in broad distribution throughout the brain and brainstem (Greco et al., 2006; Tassone et al., 2012). Post-mortem studies in FXTAS donors also revealed cerebellar and cerebral white matter abnormalities with inclusions in astrocytes, particularly evident in sub-cortical cerebral white matter (Greco et al., 2006). This is consistent with MRI studies that reveal global brain atrophy and white matter signal changes with characteristic feature middle cerebral peduncle presents in about 40% of the patients with FXTAS (Famula et al., 2018). Here, we describe novel pathological features of two cases with a FMR1-premutation and a clinical profile of FXAND with mild movement impairments. Our study supports recent studies, showing that testing for FMRpolyG immunoreactivity can confirm the presence of the FMR1-premutation (Todd et al., 2013; Krans et al., 2019).
Materials and methods
Magnetic resonance imaging
Donors were scanned on a 3T whole-body magnetic resonance system (General Electric, Milwaukee, WI). The protocol included a 3D T1-weighted fast spoiled gradient echo sequence for volumetric measurements and for Donor 2, a 3D T2-weighted fluid-attenuated inversion recovery sequence for white matter lesion segmentation.
Post-mortem tissue
Post-mortem tissue was obtained through the Netherlands Brain Bank (NBB) and the department of Pathology, University Medical Center Groningen, The Netherlands. Tissue of a spinocerebellar ataxia donor type 3 (SCA3) was obtained through the NBB and used as a negative control for FMRpolyG. Tissue was fixed in 4% PFA for 4 weeks and dissected in 24 diagnostic regions. Sections of 8 μm of each region were cut and stained using haematoxylin and Eosin (H&E) and Perls staining for iron deposits. Donors were routinely immunostained for amyloid-beta (1:800; mouse, clone IC16 kind gift of Dr Korth, Heinrich Heine University, Düsseldorf, Germany), tau (1:800; mouse, clone AT8, Thermo Fisher Scientific, MA, USA), alpha-synuclein (1:1000; mouse, Zymed; Thermo Fisher Scientific, Bleiswijk, The Netherlands), TDP-43 (pTDP43, 1:8000; mouse, clone 11‐9, Cosmo Bio, Japan) and polyQ (1:800; mouse, clone 5TF1-1C2, Merck Millipore, Burlington, WI, USA).
Immunohistochemical procedures
Sections were deparaffinized and then incubated in 0.3% H2O2 in phosphate buffer saline (pH = 7.4) for 30 min. Sections were then washed with phosphate buffer saline (3 × 5 min) and antigen retrieval was performed in citrate buffer (pH = 6.0) using an autoclave (121°C for 5 min). After washing, sections were incubated in primary antibody (p62, 1:1000; mouse, clone 3/P62 lck ligand, BD Transduction Laboratories, San Jose, CA, USA) FMRpolyG (NTF1), 1:200; Rabbit, antibody developed by Peter Todd; FMRpolyG (1C5), 1:500; mouse developed by Nicolas Charlet-Berguerand; for 1 h at room temperature. After washing, sections were incubated with HRP-labelled Envision (K5007; DAKO, Glostrup, Denmark), washed and visualization of immunostaining was performed with chromogen 3,3′-diaminobenzidine (K5007; DAKO). Finally, sections were counterstained with haematoxylin, dehydrated and cover slipped with Quick-D (Klinipath, Duiven, The Netherlands). Negative controls were included by omitting the primary antibody and showed no immunoreactivity.
Double-labelling immunofluorescence
For double-immunofluorescence, sections were incubated for 1 h with a combination of primary antibodies; p62 (1:1000) and FMRpolyG (NTF1) (1:200). Subsequently, sections were incubated with fluorescent-probed secondary antibodies 1:250 (1 h); Goat Anti-Rabbit-594 (Invitrogen, Carlsbad, CA, USA), and Goat Anti-Mouse 488 (Life Technologies, Carlsbad, CA, USA). Auto-fluorescence was blocked with 0.2% Sudan Black for 5 min at room temperature and mounted with DAPI Fluoromount G (Southern Biotech, AL, USA).
Statistical analysis
No analyses were performed.
Data availability
Data are available upon request.
Results
Clinical profile
Donor 1
At the age of 57 years, the patient complained about pain in his legs and subsequently polyneuropathy was diagnosed. At 59 years, he was seen again at the neurological outpatient clinic due to a minor stroke. Besides the sub-acute stroke-related neurological signs, the patient complained about progressive cognitive problems (memory and slowness). Additionally, behavioural changes (apathic and passive) were mentioned. The patient spent whole days just listening to music and was easily agitated and not co-operative. Neurological examination showed several signs of Parkinsonism. Initially, vascular dementia/vascular Parkinsonism or Parkinson’s disease were mentioned as diagnosis. The MRI showed mild vascular changes, and vascular dementia seemed unlikely. As disease progressed, other symptoms became more apparent such as executive problems, increased cognitive decline but also significant bradyphrenia. Clinically, the patient was demented and atypical Alzheimer’s disease (AD) and frontotemporal dementia were added to the differential diagnosis. Biomarkers did not provide any indication for AD. Additionally, an FDG-PET scan was performed, showing a pattern which partially suited corticobasal degeneration or less likely progressive supranuclear palsy (PSP). However, corticobasal degeneration or PSP were clinically less evident, since dementia with significant frontal characteristics was pronounced. The patient showed increased psychiatric problems, including aggressiveness towards partner, apathy and compulsive behaviour. Due to an acute unsustainable situation at home, the patient was taken into a psychiatric hospital where he committed suicide at the age of 62 years. After autopsy, the donor presented intranuclear inclusion pathology. FMRpolyG immunohistochemistry was positive and subsequent genetic testing revealed a 107 CGG repeat within the FMR1 gene.
Donor 2
At the age of 43 years, this patient came to the memory clinic with memory problems and aphasia, whereas his partner noted that he became aggressive towards her. He showed signs of social anxiety and suffered from chronic depression. He was initially diagnosed with Korsakoff’s syndrome based on earlier excessive alcohol consumption and the early onset of the symptoms. At 51 years, he complained of memory loss and concentration problems and character changes. At 53 years, he suffered from cardiac palpitations and cramps in his fingers, but no cause was found. He had visual hallucinations, in which he saw persons and felt drawn towards the cemetery. A year later, he experienced a change in gait, and dragged his feet. At 55 years, he had a major stroke in the right occipital region, reporting loss of strength and co-ordination in his left arm and leg, followed by a spontaneous recovery. A year later, he suffered from falling and increased bradykinesia. He had a light tremor in both hands, started snoring and developed sleep apnoea. Around this time, his grandchildren were born and diagnosed with Fragile X-syndrome. Genetic testing revealed a 77 CGG repeat in the FMR1 gene, and his initial diagnosis of Korsakoff’s syndrome was retracted. Neuropsychological tests showed cognition disorders concerning short- and long-term memory, feelings of depression, fear, paranoia and anger, agitation, loss of concentration and initiative, and difficulties with divided attention. Over the years, his symptoms worsened, with increasing cognitive problems, ataxia, tremor, hallucinations and dependency on others for his personal care and hygiene. He was well aware of his deterioration and started a euthanasia trajectory and passed away at 65 years. The disease course of both donors is summarized in Fig. 1A.
Figure 1.

Clinical course and imaging results of Donors 1 and 2. Clinical course (A) shows moment in time of MRI. General mild atrophy is observed (B, F). Ventricles are slightly dilated and Donor 1 shows periventricular white matter lesions (C), Donor 2 shows an increased signal due to an infarct (G). Slight cerebellar atrophy can be seen in both donors (D, E, H, I). Enlargement of the fourth ventricle, but no FXTAS-characteristic middle cerebral peduncle sign can be seen. T2-fluid-attenuated inversion recovery of Donor 2 does not show increased insular and middle cerebral peduncle signal intensities (J, K), no abnormalities in vasculature (K) and no thinning of the corpus callosum (M). CVA, cerebrovascular accident; PNP, polyneuropathy; VD, vascular dementia; PD, Parkinson’s disease; AD, Alzheimer’s disease; CBD, corticobasal degeneration; FTD, frontotemporal dementia; PSP, progressive supranuclear palsy.
MRI findings
Both donors showed general athrophy throughout the cortex (Fig. 1B, C, F and G) and dilation of ventricles (Fig. 1C, E and G). White matter intensity changes were observed around the ventricles, and small lesions were observed (Fig. 1B, C, D, E and M Donor 1 (Fig. 1E) and Donor 2, (Fig. 1K). While Donor 2 suffered from a stroke 10 years prior to his passing, a susceptibility weighted imaging for Donor 2 revealed no large vascular damage (Fig. 1L). No obvious thinning of the corpus callosum was seen (Fig. 1M). Overall, both donors revealed mild abnormalities on MRI including periventricular white matter intensities and mild global athrophy fitting with several neurodegenerative diseases.
P62-inclusion pathology
Intranuclear p62-positive inclusions were found in all brain structures, similar as reported before for FXTAS (Greco et al., 2006). Cortical structures such as the frontal cortex showed inclusions in neurons and astrocytes in grey and white matter (Fig. 2A–D). Hippocampus showed many inclusions in CA and dentate gyrus granular cells (Fig. 2E and F). In the cerebellum, inclusions were observed in the white matter and granular cell layer, and in most of the Bergmann glia but not in the Purkinje cells (Fig. 2G and H). The substantia nigra (SN) showed inclusions in astrocytes and dopaminergic neurons (Fig. 2I). Similar to earlier reported findings in FXTAS, inclusions were also found in the ependymal layer (Fig. 2J) and choroid plexus (Fig. 2K). Additional findings include the presence of inclusions in the extra-axial part of nerve 3, but not intra-axial (Fig. 2L).
Figure 2.

p62-Pathology in Donor 2. Abundant p62-positive nuclear inclusions in the frontal cortex grey matter (A, B) and white matter (C, D). The hippocampus shows nuclear inclusions in the CA4 neuron (white arrowhead) and the dentate gyrus (E, F). In the cerebellum, inclusions are seen in the white matter and granular layer (G, H). No inclusions were seen in the Purkinje cells, whereas most Bergmann glia showed inclusions (H). Pathology was also seen in the dopaminergic neurons (white arrowhead) and astrocytes (black arrowhead) in the substantia nigra (I), ependymal (J), extra-axial cells of the third nerve (K) and choroid plexus cells (L). Scale bar A is 50 μm; Scale bar B, D, F, H, I–L is 10 μm; scale bar C, E, G is 100 μm.
Vascular pathology
Both donors showed vascular lesions such as lacunae and strokes throughout the brain. In the H&E staining, brown iron deposits were appreciated (Fig. 3A and E). Perls staining showed that the disrupted vasculature resulted in iron deposits, found throughout the cortex and hippocampus in normal age-related amounts (Fig. 3B and F), and an abnormal increased amount of iron deposits in the ventral region of the SN (Fig. 3C and G). Small p62-positive intranuclear inclusions were observed in the endothelial cells of the blood vessels, as well as in the pericytes (Fig. 3D and H). An overview of p62-inclusions in different cell types and iron deposits visualized by Perls staining is provided per region in Table 1.
Figure 3.

Vascular pathology of Donors 1 and 2. Brown iron-like deposits are seen in both donors (A, E), which stain blue in a Perls staining (B, F). Both donors showed abnormal increased iron deposits in the ventral part of the substantia nigra (C, G). Throughout the brain, inclusions were seen in the endothelial cells (black arrowhead) and the pericytes (white arrowhead) (D, H). Scale bar A, B, D, E, F, H is 10 µm; scale bar C, G is 0.5 cm.
Table 1.
Distribution and burden of inclusion pathology and iron deposits throughout the brain of Donors 1 and 2
| Donor 1 |
Donor 2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| p62 |
p62 |
||||||||
| Area | Region | Neuron | Astrocytes | Vessel | Perls | Neuron | Astrocytes | Vessel | Perls |
| Cortex | Frontal cortex | ••• | ••• | •• | • | ••• | ••• | ••• | •• |
| Temporal cortex | ••• | ••• | •• | • | ••• | ••• | •• | •• | |
| Parietal | •• | •• | • | • | ••• | ••• | •• | • | |
| Occipital cortex | •• | •• | • | • | •• | ••• | •• | • | |
| Insular cortex | •• | ••• | • | • | •• | ••• | • | • | |
| Limbic | Amygdala | •• | •• | • | – | •• | ••• | – | – |
| Hippocampus | ••• | ••• | •• | – | ••• | ••• | ••• | – | |
| Entorhinal cortex | • | •• | • | • | •• | ••• | • | – | |
| Sub-cortical | Putamen | ••• | ••• | ••• | ••• | •• | •• | • | ••• |
| Globus pallidus | ••• | ••• | ••• | ••• | • | •• | • | ••• | |
| Caudatus | ••• | ••• | •• | ••• | •• | ••• | •• | ••• | |
| Thalamus | •• | •• | • | •• | •• | •• | • | • | |
| Cerebellum | Dentate nucleus | • | •• | • | – | • | ••• | • | – |
| Folia | – | ••• | • | – | – | ••• | – | – | |
| Brainstem | Substantia nigra | •• | •• | ••• | •• | ••• | ••• | ••• | ••• |
| Locus ceruleus | ••• | ••• | • | – | ••• | ••• | – | – | |
p62, not present; •, few inclusions present; ••, moderate amount of inclusions present; •••, abundant amount of inclusion present. Perls, not present; •, few iron deposits; ••, moderate amount of iron deposits; •••, abundant amount of iron deposits.
FMRpolyG pathology
FMRpolyG was detected in all regions of the brain. FMRpolyG intranuclear inclusions are shown in the frontal cortex (Fig. 4A), the hippocampus (Fig. 4B), the astrocytes in SN (Fig. 4C) and in the Bergmann glia in the cerebellum (Fig. 4D). FMRpolyG positivity was also observed in the vasculature. Double-immunofluorescence for FMRpolyG and p62 showed that FMRpolyG protein co-localizes with p62 inclusions in the cerebellum (Fig. 4E–H), where an occasional p62-inclusion did not show FMRpolyG positivity. As a negative control, no immunoreactivity of FMRpolyG was observed in a case of spinocerebellar ataxia 3 (SCA3) (Fig. 4I–L), confirming the specificity of our antibodies for the FMR1-premutation-translated product, FMRpolyG.
Figure 4.

Figure 4 FMRpolyG-positivity in Donor 2. FMRpolyG (1C5)-positive inclusions are seen in the frontal cortex (A), hippocampus (B), substantia nigra (C) and cerebellum (D). Inclusions are indicated with white arrowheads. Co-localization of p62 and FMRpolyG (NTF1) immunopositivity in the Bergmann glia of the cerebellum in Donor 2 and spinocerebellar ataxia 3 (SCA3) donor. Bergmann glia shown with DAPI (E, I) showed inclusions with p62 (F, J) and FMRpolyG (G, K). Co-localization of the immunopositivity shows overlap in FXAND (H) and not in SCA3 (L). Scale bar is 10 μm.
Discussion
Here, we report two cases with dementia and psychiatric symptoms who were diagnosed as FMR1-premutation carriers either post-mortem or in advanced disease state. While initial symptoms suggested dementia, both donors showed mild movement disturbances during the progression of the disease. MRI did not reveal the characteristic FXTAS-related imaging profile, which, in combination with the symptoms, posed a challenge for the attending clinicians to diagnose the patients as potential FMR1-premutation carriers. Interestingly, both donors experienced cerebrovascular accidents which could be explained by the post-mortem findings of FMRpolyG inclusions in the endothelial cells and pericytes. We propose that these brain vascular lesions may contribute to the complex symptomology of FMR1-premutation carriers.
Clinical findings
The clinical phenotype for FXTAS has been described in detail, where movement symptoms are the most prominent features of the disease (Hagerman, 2013; Leehey et al., 2016; Robertson et al., 2016). However, the two FMR1-premutation individuals described here exhibited only mild movement symptoms such as rigidity and bradykinesia, and showed predominantly psychiatric problems, matching the recent description of FXAND (Hagerman et al., 2018). Interestingly, Donor 1 experienced polyneuropathy as one of the first symptoms, which can be a prevalent sign in FMR1-premutation carriers with FXTAS presentation (Hagerman et al., 2007; Berry-Kravis et al., 2007b). Of clinical importance, the two donors did not show the typical middle cerebral peduncle imaging feature of FXTAS; however, they did show mild atrophy, similar to other dementias. This study highlights the challenge for clinicians to diagnose FMR1-premutation carriers, and stresses the importance of genetic screening for CGG repeats within FMR1 for suspected dementia with behavioural features.
Inclusion pathology
The p62-inclusion pathology of the two cases described here shows a similar distribution pattern as described in FXTAS (Tassone, 2004; Greco et al., 2006). However, we describe here an additional feature with inclusions present in the endothelial cells and pericytes that can compromise vascular function. As both donors also suffered from multiple vascular incidents in the brain, this suggests that apart from cellular dysfunction, problems in the brain vasculature can contribute to the FMR1-premutation phenotype where infarcts or transient ischaemic attacks can disrupt blood supply. Consistent with our hypothesis, a high incidence of cardiac abnormalities and cerebrovascular disease has been observed in FXTAS donors (Hall et al., 2005; Hamlin et al., 2012), which could be linked to a decline in vascular integrity due to inclusion pathology. In addition, inclusions have been observed in cardiomyocytes (Hunsaker et al., 2011), suggesting that the peripheral vasculature could also be compromised. Furthermore, inclusions were found in the extra-axial part of the nerve sheet, it is possible that the neuropathy can be explained by nerve involvement in the sensory nerves, which has to be studied in future autopsies of carriers.
Conclusion
Our study also confirms that the inclusion pathology in carriers of the FMR1 premutation can be visualized with antibodies directed against FMRpolyG (Krans et al., 2019), with almost all p62-positive inclusions also positive for FMRpolyG. However, we noted that the dopaminergic neurons of the SN showed multiple p62-positive inclusions in one cell that were not positive for FMRpolyG. These inclusions are most likely Marinesco bodies (Beach et al., 2004), which are age-related nuclear inclusions restricted to the SN. FMRpolyG labelling is specific to the protein translated from the FMR1-premutation, and it is not present in the polyQ aggregates found in SCA3. Importantly, we propose that FMRpolyG antibodies can be used as diagnostic tools to identify FMR1-premutation carriers post-mortem, notably in large retrospective brain bank cohorts.
Acknowledgements
The authors thank the Netherlands Brain Bank for providing tissue and Irene van Dijken for her input on the immunofluorescence experiments.
Glossary
- FXAND =
Fragile X-associated neuropsychiatric disorders
- FXPOI =
Fragile X-associated primary ovarian insufficiency
- FXTAS =
Fragile X-associated tremor/ataxia syndrome
- H&E =
haematoxylin and eosin
- NBB =
Netherlands Brain Bank
- RAN =
repeat-associated non-AUG-initiated
- SCA3 =
spinocerebellar ataxia type 3
- SN =
substantia nigra
Funding
This study was funded by Memorabel Zorgonderzoek Nederland, gebied Medische wetenschappen (733050507). Peter Todd was supported by the National Institutions of Health (R01NS086810, R01NS099280) and the Veterans affairs (BLRD BX004842).
Competing interests
The authors report no competing interests.
References
- Beach TG, Walker DG, Sue LI, Newell A, Adler CC, Joyce JN, et al. Substantia nigra Marinesco bodies are associated with decreased striatal expression of dopaminergic markers. J Neuropathol Exp Neurol 2004; 63: 329–37. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Abrams L, Coffey SM, Hall DA, Greco C, Gane LW, et al. Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord 2007a; 22: 2018–30. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Goetz CG, Leehey MA, Hagerman RJ, Zhang L, Li L, et al. Neuropathic features in fragile X premutation carriers. Am J Med Genet 2007b; 143A: 19–26. [DOI] [PubMed] [Google Scholar]
- Boivin M, Willemsen R, Hukema RK, Sellier C.. Potential pathogenic mechanisms underlying Fragile X Tremor Ataxia Syndrome: RAN translation and/or RNA gain-of-function? Eur J Med Genet 2018; 61: 674–9. [DOI] [PubMed] [Google Scholar]
- Brouwer JR, Willemsen R, Oostra BA.. The FMR1 gene and fragile X-associated tremor/ataxia syndrome. Am J Med Genet B Genet 2009; 150B: 782–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famula JL, McKenzie F, McLennan YA, Grigsby J, Tassone F, Hessl D, et al. Presence of middle cerebellar peduncle sign in FMR1 premutation carriers without tremor and ataxia. Front Neurol 2018; 9: 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 2006; 129: 243–55. [DOI] [PubMed] [Google Scholar]
- Grigsby J, Brega AG, Leehey MA, Goodrich GK, Jacquemont S, Loesch DZ, et al. Impairment of executive cognitive functioning in males with fragile X-associated tremor/ataxia syndrome. Mov Disord 2007; 22: 645–50. [DOI] [PubMed] [Google Scholar]
- Hagerman PJ. Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol 2013; 126: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman PJ, Hagerman RJ.. Fragile X-associated tremor/ataxia syndrome. Ann NY Acad Sci 2015; 1338: 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Coffey SM, Maselli R, Soontarapornchai K, Brunberg JA, Leehey MA, et al. Neuropathy as a presenting feature in fragile X-associated tremor/ataxia syndrome. Am J Med Genet A 2007; 143A: 2256–60. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ, Protic D, Rajaratnam A, Salcedo-Arellano MJ, Aydin EY, Schneider A, et al. Fragile X-associated neuropsychiatric disorders (FXAND). Front Psychiatry 2018; 9: 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Berry-Kravis E, Jacquemont S, Rice CD, Cogswell J, Zhang L, et al. Initial diagnoses given to persons with the fragile X associated tremor/ataxia syndrome (FXTAS). Neurology 2005; 65: 299–301. [DOI] [PubMed] [Google Scholar]
- Hamlin AA, Sukharev D, Campos L, Mu Y, Tassone F, Hessl D, et al. Hypertension in FMR1 premutation males with and without fragile X-associated tremor/ataxia syndrome (FXTAS). Am J Med Genet A 2012; 158A: 1304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Greco CM, Spath MA, Smits APT, Navarro CS, Tassone F, et al. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol 2011; 122: 467–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004; 291: 460–9. [DOI] [PubMed] [Google Scholar]
- Krans A, Skariah G, Zhang Y, Bayly B, Todd PK.. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta Neuropathol Commun 2019; 7: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leehey MA, Berry-Kravis E, Goetz CG, Hagerman RJ.. Clinical neurological phenotype of FXTAS. In: Tassone F, Hall DA, editors. FXTAS, FXPOI, and other premutation disorders. Cham: Springer International Publishing; 2016. p. 1–24. [Google Scholar]
- Murray J, Cuckle H, Taylor G, Hewison J.. Screening for fragile X syndrome: information needs for health planners. J Med Screen 1997; 4: 60–94. [DOI] [PubMed] [Google Scholar]
- Robertson EE, Hall DA, McAsey AR, O’Keefe JA.. Fragile X-associated tremor/ataxia syndrome: phenotypic comparisons with other movement disorders. Clin Neuropsychol 2016; 30: 849–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Buijsen RAM, He F, Natla S, Jung L, Tropel P, et al. Translation of expanded CGG repeats into FMRpolyG is pathogenic and may contribute to Fragile X tremor ataxia syndrome. Neuron 2017; 93: 331–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seritan AL, Nguyen DV, Farias ST, Hinton L, Grigsby J, Bourgeois JA, et al. Dementia in fragile X-associated tremor/ataxia syndrome (FXTAS): comparison with Alzheimer’s disease. Am J Med Genet B Genet 2008; 147B: 1138–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet 2004; 41: E43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Adams J, Berry-Kravis EM, Cohen SS, Brusco A, Leehey MA, et al. CGG repeat length correlates with age of onset of motor signs of the fragile X-associated tremor/ataxia syndrome (FXTAS). Am J Med Genet 2007; 144B: 566–9. [DOI] [PubMed] [Google Scholar]
- Tassone F, Greco CM, Hunsaker MR, Seritan AL, Berman RF, Gane LW, et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav 2012; 11: 577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013; 78: 440–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ID. Diagnosing Fragile-X Syndrome. Lancet 1993; 342: 1004–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available upon request.