

Graphical abstract

Abbreviations: ZIKV, Zika virus; SAR, structure-activity relationship; RLM, rat liver microsomal stability; ADME, absorption; distribution, metabolism and excretion; PAMPA, parallel artificial membrane permeability assay; MOI, multiplicity of infection
Keywords: Niclosamide, Flavivirus, Zika virus, Salicylanilide, Small molecule, NS-1 assay
Abstract
Zika virus has emerged as a potential threat to human health globally. A previous drug repurposing screen identified the approved anthelminthic drug niclosamide as a small molecule inhibitor of Zika virus infection. However, as antihelminthic drugs are generally designed to have low absorption when dosed orally, the very limited bioavailability of niclosamide will likely hinder its potential direct repurposing as an antiviral medication. Here, we conducted SAR studies focusing on the anilide and salicylic acid regions of niclosamide to improve physicochemical properties such as microsomal metabolic stability, permeability and solubility. We found that the 5-bromo substitution in the salicylic acid region retains potency while providing better drug-like properties. Other modifications in the anilide region with 2′-OMe and 2′-H substitutions were also advantageous. We found that the 4′-NO2 substituent can be replaced with a 4′-CN or 4′-CF3 substituents. Together, these modifications provide a basis for optimizing the structure of niclosamide to improve systemic exposure for application of niclosamide analogs as drug lead candidates for treating Zika and other viral infections. Indeed, key analogs were also able to rescue cells from the cytopathic effect of SARS-CoV-2 infection, indicating relevance for therapeutic strategies targeting the COVID-19 pandemic.
The Zika virus (ZIKV) was first isolated in the forests of Uganda in 1947. It was circulating predominantly between primates and mosquitoes and had caused limited human infections in Asia and Africa. However, the recent outbreak of ZIKV infection in humans in the Americas and its potential link to fetal microcephaly poses an urgent global concern.1, 2, 3, 4, 5, 6, 7 ZIKV is a vector-borne virus and is transmitted to humans mainly by Aedes aegypti, a mosquito known also to transmit yellow fever, dengue fever and chikungunya virus. ZIKV belongs to the genus Flavivirus in the Flaviviridae family of RNA viruses and consists of a 10.7-kb single-stranded RNA genome that encodes a single polyprotein, which is believed to be cleaved by host-cell proteases and viral NS2B/NS3 protease into three structural (C, prM/M and E) and 7 non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5). ZIKV infection has been linked with multiple adverse effects such as testis damage,8, 9 ocular damage,10, 11 Guillain-Barre syndrome12, 13, 14 and other neural complications in infected adults.15, 16 Several screening efforts had been directed towards employing synthetic small molecules, natural products and FDA approved drugs for progression into clinic, but no candidate has been progressed thus far.17, 18, 19, 20, 21
In a repurposing screen with existing drugs, we had identified the antihelminthic drug niclosamide as a small molecule inhibitor of ZIKV. Niclosamide inhibited ZIKV-induced caspase-3 activation and host cell death at sub micromolar concentrations.2, 22 The antiviral activity of niclosamide was confirmed via a concentration-dependent reduction of ZIKV NS1 protein. It inhibited replication of all three ZIKV strains (MR766, FSS13025 and PRVABC59) as measured by ZIKV-NS1 expression in a concentration-dependent manner.22
Niclosamide was approved by FDA for use in humans to treat tapeworm infections in 198223, 24 and is included on World Health Organization’s list of essential medicines.25 Niclosamide is also categorized as a FDA Category B drug which implies that it did not demonstrate risk to the fetus in animal reproduction studies, although there are no adequate and well-controlled studies in pregnant women. Niclosamide has been reported to have a number of biological activities such as anti-tuberculosis activity,26 anti-bacterial activity,27 drug-resistant Staphylococcus aureus activity,28 anti-cancer activity,29, 30, 31, 32 and has also displayed broad anti-viral activity against coronaviruses (SARS-CoV, MERS-CoV), mosquito-borne flaviviruses (ZIKV, dengue virus (DENV), West Nile virus (WNV), yellow fever virus (YFV), Japanese encephalitis virus (JEV)), hepatitis C virus (HCV), ebola virus (EBOV), human rhinovirus (HRV) and human adenovirus (HAdV) with micromolar to nanomolar potencies.33, 34, 35 Niclosamide was shown to inhibit MERS-CoV replication via SKP2 inhibition and subsequent autophagy induction.36 Similarly, niclosamide demonstrated SARS-CoV-2 inhibition while rescuing autophagy signaling. SARS CoV-2 infection was shown to hamper autophagosome/lysosome fusion efficiency and impede host cell autophagy.37 The broad spectrum anti-viral response of niclosamide appears to be related to its protonophoric activity which disrupts membrane pH gradients and is also proposed to block endosomal acidification, an important step for viral entry.33
At NCATS, niclosamide was also discovered in a screen for orthosteric inhibitors that directly target the interaction between the ZIKV flavivirus proteases NS2B and NS3.38, 39, 40 Viral non-structural NS3 protein is the main driver for the viral protease activity and is conserved among the flavivirus species. As NS2B co-factor binding is essential for NS3 protease function, inhibitors preventing the NS2B-NS3 interaction presented a novel approach. Niclosamide was shown to inhibit NS3-NS2B interaction, inhibit ZIKV growth at early stages and 24 h post infection and reduce ZIKV titer in human placental epithelial cells and iPSC-derived human neural progenitor cells.38
Although the pharmacokinetic properties of niclosamide are appropriate for its use as an anthelmintic agent, its low solubility, low bioavailability, and poor plasma exposure limit its use in studies that require oral dosing and systemic exposure.29, 34, 41, 42, 43, 44 Nevertheless, niclosamide has been found to be potent and effective against a variety of human diseases and at present is being investigated in multiple clinical trials towards ulcerative colitis, diabetic nephropathy, colorectal cancer, metastatic and recurrent prostrate carcinoma and now in COVID-19.45 This emphasizes the need for potent and effective niclosamide analogs with better physicochemical properties for a swift translation to clinic.
In the present study, our goal was to systematically modify the structure of niclosamide and examine the effect of these changes on in vitro anti-ZIKV activity and ADME properties, such as aqueous solubility, rat liver microsome (RLM) stability, and PAMPA permeability. Although niclosamide displays good RLM stability (t1/2 > 30 min), it displays poor kinetic solubility (1.1 µg/mL) and poor permeability (PAMPA < 1X 10−6cm/s) (see Supporting information).46, 47, 48 Thus, it is desirable to improve the aqueous solubility and passive permeability as a structure optimization strategy.
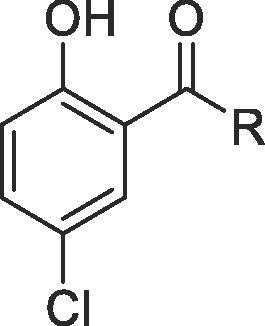
We divided the structure of niclosamide into its salicylic acid and the aniline motifs and systematically examined the effects of electron-donating and electron-withdrawing substitutions in both regions (Fig. 1 ). We also investigated the role of the hydroxyl group on the salicylic acid side chain envisaging an internal hydrogen bonding interaction between this hydroxyl and the amide carbonyl. In addition, we also probed to find suitable replacement of the aniline 4′-NO2 group as this group might be reduced and glucuronidated in vivo.24, 49 A suitable substitution pattern and electronic properties might provide a strategy to discover niclosamide based potent ZIKV inhibitors for improving NS-1 potency and developing compounds with better ADME properties. We used two complimentary assays to measure the expression of viral proteins as a surrogate marker for Zika replication. The non-structural protein NS-1 assay was used to drive SAR and the E-protein (envelope protein) read out was used as a secondary confirmatory assay. While the ZIKV envelope (E) protein, covers the surface of the virus and is responsible for membrane fusion and viral entry, the non-structural NS-1 protein is needed for viral RNA replication and immune evasion. Dose response inhibition of the viral replication was as was measured in these phenotypic assays. The details of these assays are provided in the Supplementary information.
Fig. 1.
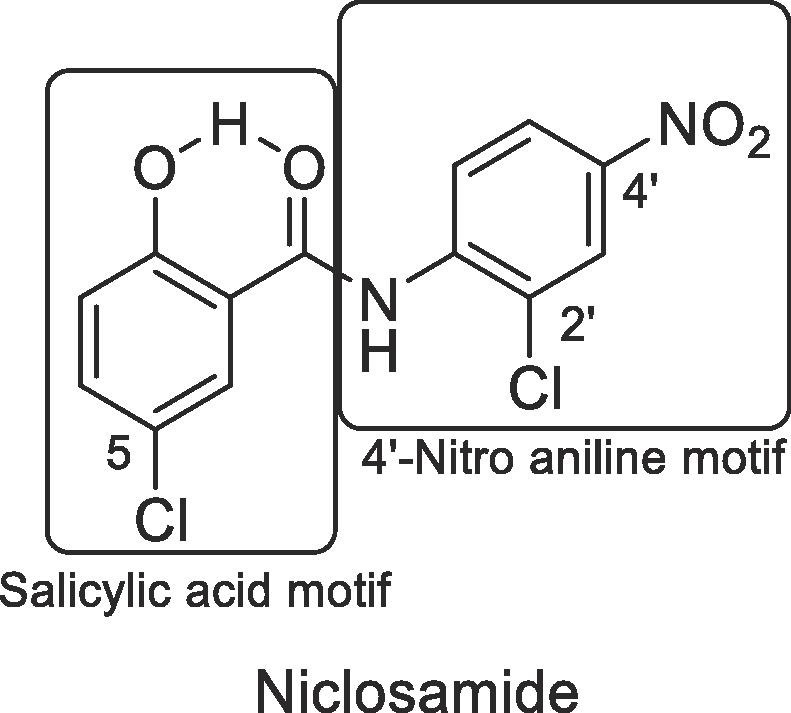
SAR regions of Niclosamide.
To systematically evaluate the SAR associated with niclosamide against Zika virus, we first focused our attention on the salicylic acid region of the molecule (Table 1 ). Compounds were synthesized via PCl3-mediated coupling of the suitably substituted salicylic acids with 2-chloro-4-nitroaniline (Scheme 1 ).50, 51 Experimental procedures and analytical data are provided in the Supplementary Information.
Table 1.
 |
 |
IC50 values are average from three biological replicates, NT = Not tested, ND = Not Determined.
Scheme 1.

Synthesis of anilide derivatives.
As shown in Table 1, replacement of the hydroxyl group of salicylic acid with a 2-methoxy substituent in 2 rendered the compound inactive. Also 3 and 4 (O-ethylamine and O-piperidine substitutions) lost potency in both NS-1 and E-protein assays thus validating that the hydroxyl group is essential for activity. Halide substitutions were then evaluated at the 5-chloro position of 5-chloro-2-hydroxybenzoicacid. It was replaced by 5-F and 5-Br counterparts (5 and 6) and in both cases their activities were similar to niclosamide. Compound 6 also showed improvement in the ADME properties exhibiting better solubility (8 µg/mL), rat liver microsomal stability (t1/2 > 30 min) and PAMPA permeability (67 × 10−6 cm/s). Moving the chloro substituent to the 4- position (4-chloro-2-hydroxy benzamide) in compound 7 also seemed acceptable. Though RLM stability (t1/2 = 21 min) was acceptable, this change could not improve the solubility (<1 µg/mL) and permeability issues associated with niclosamide. Electron-donating –OMe substituents either at the 4/5-position in 8 and 9 proved detrimental as the IC50s increased to 6 µM in both cases. A smaller methyl substituent at the 5-position in 10 led to similar potency (IC50 = 3 µM) compared to niclosamide but did not provide the required improvement in solubility (<1 µg/mL). The 2, 6-dihydroxy benzamide substitution in 11, on the other hand, resulted in good potencies in the NS-1 and E-protein assays and showed better solubility (>7 µg/mL). Introduction of electron-withdrawing substituents though, such as in the nitro substituent para to the hydroxy in 12 and the acetyl substituent in 13 proved detrimental towards activity. These results were confirmed with IC50 values obtained from the E-protein assay.
Fused or extended aromatic groups in the salicylic acid region were evaluated with 14, 15, 16 and 17. 4-hydroxyquinoline 14 was inactive but the other two naphthalene derivatives 15 and 16 showed acceptable potencies (2.6 and 1.6 µM, respectively). The biphenyl derivative 17 showed an IC50 value of 4.1 µM in NS-1 assay.
We introduced an amine in lieu of the phenolic hydroxyl anticipating that it would be capable of forming an internal hydrogen bond. However, this rendered the compound inactive (18 and 19). However, while the acetamide derivative 20 was also inactive, the derivatization of the amine to the corresponding methyl and phenyl sulfonamides 21 and 22 respectively restored activity to the single digit micromolar range. This suggested that the pKa of functional group in C-2 is important for activity and for the analog’s ability to participate in an internal hydrogen bond.
Thus, our initial efforts in the salicylic acid region provided promising leads (5, 6, 7, 11, 16, 22) in terms of potency in the primary NS-1 assay and compounds 6 and 22 also exhibited better in vitro ADME characteristics than niclosamide.
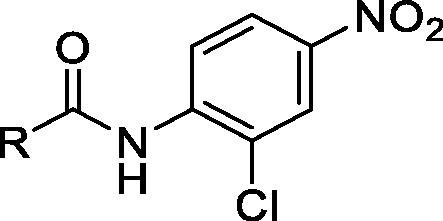
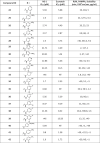
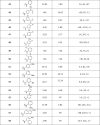
Next, we turned our attention to the anilide region (Table 2 ). Compounds were synthesized either via PCl3-mediated coupling of 5-chloro-2-hydroxybenzoic acid with corresponding amines (Scheme 1) or reacting 5-chloro-2-hydroxy-benzoyl chloride with the corresponding amines using pyridine as the solvent (Scheme 2 ) (see Supplementary Information for experimental procedures).
Table 2.
 |
 |
IC50 values are average from three biological replicates, NT = Not tested, ND = Not Determined.
Scheme 2.

Synthesis of anilide derivatives.
First, we sought to identify bioisosteres of the 4′-NO2 group on the aniline region. As we were aware of potential liabilities of nitroanilines,24, 49 we initiated our efforts with the removal of the NO2 group to assess the physicochemical properties of the molecule in its absence. This resulted in compound 24 which lost some potency (IC50 = 5 µM) as compared to the previous bromide lead compound 6 (IC50 = 0.8 µM) but gained in solubility (5 µg/mL) and PAMPA permeability (118 × 10−6 cm/s). This result confirmed our hypothesis that a suitable replacement of the nitro group might improve the physicochemical properties of niclosamide. Encouraged by this result, we also tried the ‘H’ replacement of 2′-chloro position of the aniline and synthesized 25. This analog exhibited the desired ADME characteristics (RLM t1/2 = 22 min, solubility = 6 µg/mL, PAMPA permeability = 127 × 10−6 cm/s) while maintaining potency in the NS-1 assay (IC50 = 2.5 µM), thus demonstrating that electronic properties can play an important role in providing the best combination of potency and ADME property requirements.
Encouraged by these results, we began investigating the effects of electronic properties of substituents in the anilide ring system. Introduction of an additional mildly electron-withdrawing chloro substituent on niclosamide at the 6′ position in 26 dramatically increased solubility (53 µg/mL) though showed a slight decrease in NS-1 inhibition (IC50 = 6 µM). Shifting the position of the chloro substituent from 2′ to the 3′ position (27) was tolerated. Changing the 2′-chloro substituents to the electron-donating 2′-methoxy in the anilide region 28 provided a promising compound in terms of potency and improved ADME characteristics, with RLM t1/2 > 30 min and solubility at 9 µg/mL and a PAMPA permeability of 334 X 10−6 cm/s. Thus, by addressing the electronic properties in the anilide region we could identify 25 and 28 exhibiting similar IC50s in NS-1 assay but much better physicochemical properties than the parent niclosamide.
We next turned our attention to potential replacements of the nitro group and other heteroaromatic/ aliphatic ring system replacements of the anilide moiety. Introduction of the 5-isopropylthiazol-2-amine in 29 and of 5-nitrothiazol-2-amine in 30 as heterocyclic anilide replacements proved ineffective, as did the 3-chloropyridin-4-amine in 31. A 4′-amino substitution replacing 4′-NO2 on the aniline in 32 and the aliphatic 1-methyl-piperidin-4-amine in 33 also produced inactive compounds.
Replacing the 4′-NO2 substituent with the electron-withdrawing 4′-cyano substituent in 34, however, proved beneficial for potency and showed an IC50 of 1.7 µM in the NS-1 inhibition assay.
The 4′-fluoro replacement was then tried, and 5 analogs were synthesized. Simple replacement of 4′-NO2 with 4′-F in 35 seemed to be tolerated as was the 2′, 4′-difluoro analog 36, but the other fluorinated analogs such as 37 and 38 were less potent in both NS-1 and E-protein assays and compound 39 was inactive. Compound 40 with a 4′-sulfonamide replacement of the nitro moiety was also tried but failed to improve potency compared to niclosamide as measured in the NS-1 assay.
A CF3 replacement of the 4′-NO2 group was then attempted and compounds 41 and 42 were synthesized. This modification proved superior and produced more potent compounds in both NS-1 and E-protein assays (41, NS-1 = 0.8 µM and E-protein = 1.4 µM, and 42, NS-1 = 1.6 µM and E-protein = 1.7 µM) thus showing that the potential liability associated with the nitro group in niclosamide can be removed without compromising potency. The 2′-CF3 analog 43 had modest NS-1 and E-protein inhibition values.
Introduction of a 4′-acetamido electron-withdrawing group to replace the 4′-NO2 in 44 or a 3′-dimethyl carbamoyl group in 45 produced inactive compounds. A 4′-chloro substitution in the anilide was then examined in 46, 47, and 48 but these compounds did not show improvements in potencies over niclosamide. Compounds 49 and 50 with 2′-6′-dichloro and 2′-6′-difluoro substitution were inactive.
With electron-donating 4′-OMe and 3′-OMe substitutions in 51 and 52 the IC50s for inhibition were adversely affected. The same trend continued in 53, 54, 55 and 56 where reduced potencies were observed. Introduction of bicyclic nitro naphthalenes in the anilide region were also investigated with compounds 57 and 58; these both showed desirable inhibition values in the NS-1 assay.
Thus, by modifying the anilide region we found 4′-CN and 4′-CF3 as suitable 4′-NO2 replacements in compounds 34, 41 and 42, all exhibiting similar potencies as niclosamide in the NS-1 inhibition assay. Modifying the electronic properties in the anilide region in 25 and 28 resulted in similar IC50s for inhibition in the NS-1 assay compared to niclosamide but provided improved physicochemical characteristics in terms of RLM stability, solubility, and PAMPA permeability.
To understand further the binding interaction of niclosamide and the structure–activity relationship of the modified derivatives, we generated a binding model of niclosamide with viral protein target NS3 using an ensemble docking approach.52 Our model as depicted in Fig. 2 predicts that niclosamide binds to the NS3-NS2B interface in an inner, well-defined hydrophobic pocket adjacent to a highly polar region. The salicylic acid motif inserts into the pocket by forming extensive hydrophobic and aromatic stacking interactions with the surrounding residues Tyr-23, Phe-46, Leu-58, Leu-65, and Tyr-79. The OH group on C-2 forms a hydrogen bond with the backbone O atom of Arg-24 while the 5-Cl points downward and forms halogen-binding interactions inside the pocket. Both interactions are predicted to be key for the binding affinity and activity of niclosamide. On the other side of the molecule, the anilide motif points out of the pocket to the solvent exposed region with the nitro group forming salt bridge with Arg-28 and Arg-59. These binding interactions compares well to the NS2B-NS3 protein–protein interaction (PPI) site53 with NS2B forming key interactions with residues Tyr-52, Ile-53, and Glu-54 at the PPI interface (Also see Supplementary information Suppl. S1).
Fig. 2.

A) Predicted binding-model for niclosamide to NS3 at the NS3-NS2B PPI site. B) Key interactions of NS2B at NS2B-NS3 PPI site.
These docking studies of the niclosamide derivatives with NS3 provided an understanding of the structural basis for the observed SAR. As shown in Fig. 3 , these analogs were predicted to adopt the same binding conformation as niclosamide at the binding site. As the salicylic acid ring was accommodated in a deep and restricted pocket, it was postulated that substituents with bulky groups would pose steric hindrance and thereby restrict potency. As expected, replacement of the OH group on the salicylic acid ring significantly impaired potency (compounds 2, 3, and 4). Similarly, replacement of the Cl with Me or OMe (9 and 10) resulted in decreased potency, but substituents with 5-F and 5-Br (5 and 6) exhibited comparable potencies as niclosamide. Interestingly, other substitutions on salicylic acid were tolerated (7, 11 and 16). For the anilide motif the binding interaction with Arg-28 was predicted to be key, and in fact replacement of the 4′-NO2 group with 4′-CN and 4′-CF3 (34, 41 and 42) showed comparable potencies as niclosamide, whereas other substitutions disrupting the H-bond interaction dramatically decreased activity (44, 45, 51, 55).
Fig. 3.

Docking-based SAR modification of the salicylic acid motif at the inner hydrophobic pocket and of the anilide motif at the open Arg-site.
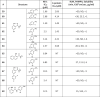
With these insights from our docking studies, advantageous modifications in the salicylamide and anilide region were then combined (Table 3 ). To this end, the 5-bromo-2-hydroxybenzoic acid from 6 that had shown good potency and ADME characteristics was combined with the 4′-CF3 and 4′-CN replacements of 4′-NO2 on anilide (59–64). We were encouraged to see that all of these compounds showed NS-1 IC50s in the low micromolar range. However, these compounds did not show improved solubility and PAMPA permeability over niclosamide. Bicyclic naphthalene in the salicylic acid region and the biphenyl compounds 67 and 68 with extra aromatic appendage showed decreases in potency. An indole replacement in 69 was also not effective in improving potency.
Table 3.
Combining salicylic acid and aniline SAR.
 |
IC50 values are average from three biological replicates. NT = Not tested, ND = Not Determined.
Homologation in the amine motif of niclosamide was also tried and the results are shown in Table 1 in the Supplementary information.
Finally, we wanted to corroborate the SAR we had generated with a Zika virus focus-forming assay to demonstrate actual reductions in viral titer. To test this, we assayed a set of analogs that varied over a wide range of potencies in the NS-1 and E-protein assays. As shown in Fig. 4 and Table 4 , there was a good correlation between relative potencies observed in the molecular assays compared to those observed in the viral titer reduction assay. Viral titer IC50 comparison with cell viability in SNB-19 cells is shown in Table 4 and plotted in Fig 5 .
Fig. 4.

Dose dependent reduction of virus production (as measured in focal forming units) in compound-treated SNB-19 cells.
Table 4.
Correlation of virus NS-1 and virus titer assay.
| Compound ID | NS-1 IC50 (µM) |
E-protein IC50 (µM) | Viral titer reduction IC50 (µM) |
Cell Viability-Nuclei Counta |
|---|---|---|---|---|
| 1 | 0.6 | 0.88 | 0.12 | 57% at 30 µM |
| 6 | 0.84 | 0.99 | 0.11 | 59% at 30 µM |
| 25 | 2.5 | 2.10 | 0.82 | 61% at 30 µM |
| 41 | 0.8 | 1.38 | 0.84 | 59% at 30 µM |
| 34 | 1.7 | 2.34 | 0.81 | 63% at 30 µM |
| 28 | 2.5 | 3.73 | 0.82 | 59% at 30 µM |
| 61 | 1.6 | 1.49 | 0.4 | 55% at 30 µM |
| 38 | 11.4 | 8.32 | 1.8 | 68% at 30 µM |
| 31 | 12.9 | 5.5 | 8.6 | 73% at 30 µM |
Cytotoxicity was performed in SNB-19 cells, activity is normalized to DMSO control.
Fig. 5.

Dose dependent reduction of virus production (as measured in focal forming units) in compound treated SNB-19 host cells for niclosamide and compound 6 (purple), overlaid with cytotoxicity curves (blue) as measured by cell count in host SNB-19 cells.
As we were preparing to report these SAR findings around niclosamide against the Zika virus, our labs were also redirecting drug screening and development resources against the SARS-CoV-2 virus, responsible for the COVID-19 pandemic. In high-throughput drug repurposing screens we found that niclosamide displayed potent in vitro activity against SARS-CoV-2.37, 54 In a recent communication, an independent group also confirmed niclosamide as a potent inhibitor of SARS-CoV-2 (IC50 = 280 nM) with potency > 25X greater than chloroquine and > 40X greater than remdesivir.55 We thus extended our studies to explore the activity of key potent analogs against this strain of the novel beta coronavirus.
Compounds 4, 6, 34, 41, 61, 28, 25 and niclosamide were examined for activity against the SARS-CoV-2 infection. As SARS-CoV-2 can induce cell death after 48 to 72 h of infection, the activity of the compounds to protect against virus-induced cell death was evaluated in a CPE (cytopathic effect) assay.56 Vero-E6 cells were chosen as host cells and infected with SARS-CoV-2 strain (USA_WA1/2020) at MOI of 0.002 which resulted in approximately 5% cell viability 72 h post infection. The detailed assay protocol is described in the Supplementary information, but it should be noted that cell culture conditions, MOI and the incubation time can differ from reported literature protocols, and thus can affect reported potencies. In our hands, niclosamide showed an IC50 of 112 nM with an efficacy of 30% (Fig. 6 ), whereas Jeon et al. have reported 100% efficacy in a similar CPE assay with niclosamide (IC50 = 280 nM and a selectivity index of >176).55 Also, in our hands, the bromo analog of niclosamide, compound 6, appeared slightly improved with an IC50 of 101 nM and 27% efficacy. Compounds 34 and 41 displayed IC50s of 256 and 128 nM with 27% and 21% efficacy. Compounds 61, 28 and 25 showed activity that followed a similar trend to their ability to reduce Zika virus titers with IC50s of 140 nM, 380 nM and 320 nM (with 24%, 44% and 27% corresponding efficacies) respectively. Compound 4, used as a negative control, failed to show significant response in this assay (Fig. 6).
Fig. 6.

Concentration response rescue curves for the key niclosamide analogs against SARS-CoV-2 CPE activity in Vero-E6 cells.
General cytotoxicity of the selected compounds was measured in a counter assay and is reported in the Supplemental information (Supp. S2).
Taken together, our SAR analyses in the NS-1, E-protein, and Zika viral titer assays identified compounds 6, 22, 25 and 28 that showed comparable potencies to niclosamide but significantly improved in vitro drug-like properties such as microsomal stability, solubility, and PAMPA permeability for systemic exposure. Compound 6 had the best combination of activity in all three activity assays and developability assays, positioning it as a promising candidate for in vivo evaluation. We also discovered that the NO2 group, which is generally considered a liability in drug-like compounds, on niclosamide could be replaced with a cyano group (34) or a trifluoromethyl group (41 and 61) without losing potency. We also found that 25 and 28 were equipotent in the viral titer assay as the 4′-CN and 4′-CF3 replacements of the 4′-NO2 substituent (41 and 34). Advantageously, some of these novel leads have improved developability properties as candidates for further in vivo pharmacokinetic profiling studies and advancing promising compounds to animal models of Zika virus infection.
We further tested potent analogs (compounds 6, 25, 28, 34, 41 and 61) along with a negative control (compound 4) against the novel beta corona virus SARS-CoV-2. We observed that the broad anti-viral properties of niclosamide and its analogs were also carried against SARS-CoV-2; the analogs with better drug-like properties will be suitable candidates to be tested in in vivo models of COVID-19.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the Intramural Research Program of the National Center for Advancing Translational Sciences (NCATS) of the NIH and NIH grant U19AI131130 (H.T. and G-I.M.). The authors thank members of NCATS Analytical Chemistry, Drug Metabolism and Pharmacokinetics and Compound Management (CoMa) for their support and collaboration.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2021.127906.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Calvet G., Aguiar R.S., Melo A.S.O., et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis. 2016;16:653–660. doi: 10.1016/S1473-3099(16)00095-5. [DOI] [PubMed] [Google Scholar]
- 2.Tang H., Hammack C., Ogden S.C., et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell. 2016;18:587–590. doi: 10.1016/j.stem.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas D.L., Sharp T.M., Torres J., et al. Local transmission of Zika virus-Puerto Rico, November 23, 2015-January 28, 2016. MMWR Morb Mortal Wkly Rep. 2016;65:154–158. doi: 10.15585/mmwr.mm6506e2. [DOI] [PubMed] [Google Scholar]
- 4.Martines R.B., Bhatnagar J., Keating M.K., et al. Notes from the Field: evidence of Zika virus infection in brain and placental tissues from two congenitally infected newborns and two fetal losses-Brazil, 2015. MMWR Morb Mortal Wkly Rep. 2016;65:159–160. doi: 10.15585/mmwr.mm6506e1. [DOI] [PubMed] [Google Scholar]
- 5.Mlakar J., Korva M., Tul N., et al. Zika virus associated with microcephaly. N Engl J Med. 2016;374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 6.Rasmussen S.A., Jamieson D.J., Honein M.A., Petersen L.R. Zika virus and birth defects-reviewing the evidence for causality. N Engl J Med. 2016;374:1981–1987. doi: 10.1056/NEJMsr1604338. [DOI] [PubMed] [Google Scholar]
- 7.Wahid B., Ali A., Rafique S., Idrees M. Zika: As an emergent epidemic. Asian Pacific J Trop Med. 2016;9:723–729. doi: 10.1016/j.apjtm.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 8.Ma W., Li S., Ma S., et al. Zika virus causes testis damage and leads to male infertility in mice. Cell. 2016;167:1511–1524 e10. doi: 10.1016/j.cell.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 9.Govero J., Esakky P., Scheaffer S.M., et al. Zika virus infection damages the testes in mice. Nature. 2016;540:438–442. doi: 10.1038/nature20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miner J.J., Sene A., Richner J.M., et al. Zika virus infection in mice causes Panuveitis with shedding of virus in tears. Cell reports. 2016;16:3208–3218. doi: 10.1016/j.celrep.2016.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yepez J.B., Murati F.A., Pettito M., et al. Ophthalmic manifestations of congenital zika syndrome in Colombia and Venezuela. JAMA Ophthalmol. 2017;135:440–445. doi: 10.1001/jamaophthalmol.2017.0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao-Lormeau V.M., Blake A., Mons S., et al. Guillain-Barre Syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet. 2016;387:1531–1539. doi: 10.1016/S0140-6736(16)00562-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Araujo L.M., Ferreira M.L., Nascimento O.J. Guillain-Barre syndrome associated with the Zika virus outbreak in Brazil. Arq Neuropsiquiatr. 2016;74:253–255. doi: 10.1590/0004-282X20160035. [DOI] [PubMed] [Google Scholar]
- 14.Fontes C.A., Dos Santos A.A., Marchiori E. Magnetic resonance imaging findings in Guillain-Barre syndrome caused by Zika virus infection. Neuroradiology. 2016;58:837–838. doi: 10.1007/s00234-016-1687-9. [DOI] [PubMed] [Google Scholar]
- 15.Mécharles S., Herrmann C., Poullain P., et al. Acute myelitis due to Zika virus infection. Lancet. 2016;387:1481. doi: 10.1016/S0140-6736(16)00644-9. [DOI] [PubMed] [Google Scholar]
- 16.Carteaux G., Maquart M., Bedet A., et al. Zika virus associated with meningoencephalitis. N Engl J Med. 2016;374:1595–1596. doi: 10.1056/NEJMc1602964. [DOI] [PubMed] [Google Scholar]
- 17.Tong X., Smith J., Bukreyeva N., et al. Merimepodib, an IMPDH inhibitor, suppresses replication of Zika virus and other emerging viral pathogens. Antiviral Res. 2018;149:34–40. doi: 10.1016/j.antiviral.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Delvecchio R., Higa L.M., Pezzuto P., et al. An endocytosis blocking agent, inhibits Zika virus infection in different cell models. Viruses. 2016;8 doi: 10.3390/v8120322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adcock R.S., Chu Y.K., Golden J.E., Chung D.H. Evaluation of anti-Zika virus activities of broad-spectrum antivirals and NIH clinical collection compounds using a cell-based, high-throughput screen assay. Antiviral Res. 2017;138:47–56. doi: 10.1016/j.antiviral.2016.11.018. [DOI] [PubMed] [Google Scholar]
- 20.Abrams R.P.M., Yasgar A., Teramoto T., et al. Therapeutic candidates for the Zika virus identified by a high-throughput screen for Zika protease inhibitors. Proc Natl Acad Sci U S A. 2020;117:31365–31375. doi: 10.1073/pnas.2005463117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y., Cao L., Gao H., et al. Discovery, optimization, and target identification of novel potent broad-spectrum antiviral inhibitors. J Med Chem. 2019;62:4056–4073. doi: 10.1021/acs.jmedchem.9b00091. [DOI] [PubMed] [Google Scholar]
- 22.Xu M., Lee E.M., Wen Z., et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat Med. 2016;22:1101–1107. doi: 10.1038/nm.4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearson R.D., Hewlett E.L. Niclosamide therapy for tapeworm infections. Ann Intern Med. 1985;102:550–551. doi: 10.7326/0003-4819-102-4-550. [DOI] [PubMed] [Google Scholar]
- 24.Andrews P., Thyssen J., Lorke D. The biology and toxicology of molluscicides, Bayluscide. Pharmacol Ther. 1982;19:245–295. doi: 10.1016/0163-7258(82)90064-x. [DOI] [PubMed] [Google Scholar]
- 25.WHO, The selection and use of Essential Medicines. World Health Organisation Geneva, 2007.
- 26.Piccaro G., Giannoni F., Filippini P., Mustazzolu A., Fattorini L. Activities of drug combinations against Mycobacterium tuberculosis grown in aerobic and hypoxic acidic conditions. Antimicrob Agents Chemother. 2013;57:1428–1433. doi: 10.1128/AAC.02154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imperi F., Massai F., Ramachandran Pillai C., et al. New life for an old drug: the anthelmintic drug niclosamide inhibits Pseudomonas aeruginosa quorum sensing. Antimicrob Agents Chemother. 2013;57:996–1005. doi: 10.1128/AAC.01952-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajamuthiah R., Fuchs B.B., Conery A.L., et al. Repurposing salicylanilide anthelmintic drugs to combat drug resistant Staphylococcus aureus. PLoS ONE. 2015;10 doi: 10.1371/journal.pone.0124595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osada T., Chen M., Yang X.Y., et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011;71:4172–4182. doi: 10.1158/0008-5472.CAN-10-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sack U., Walther W., Scudiero D., et al. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. J Natl Cancer Inst. 2011;103:1018–1036. doi: 10.1093/jnci/djr190. [DOI] [PubMed] [Google Scholar]
- 31.Li Y., Li P.K., Roberts M.J., Arend R.C., Samant R.S., Buchsbaum D.J. Multi-targeted therapy of cancer by niclosamide: a new application for an old drug. Cancer Lett. 2014;349:8–14. doi: 10.1016/j.canlet.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mook R.A., Jr., Wang J., Ren X.R., et al. Structure-activity studies of Wnt/β-catenin inhibition in the Niclosamide chemotype: Identification of derivatives with improved drug exposure. Bioorg Med Chem. 2015;23:5829–5838. doi: 10.1016/j.bmc.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jurgeit A., McDowell R., Moese S., Meldrum E., Schwendener R., Greber U.F. Niclosamide is a proton carrier and targets acidic endosomes with broad antiviral effects. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang Y.-W., Yeh T.-K., Lin K.-T., et al. Pharmacokinetics of Anti-SARS-CoV Agent Niclosamide and Its Analogs in Rats. J Food Drug Anal. 2006;14:329–333. [Google Scholar]
- 35.Xu J., Shi P.Y., Li H., Zhou J. Broad spectrum antiviral agent niclosamide and its therapeutic potential. ACS Infect Dis. 2020;6:909–915. doi: 10.1021/acsinfecdis.0c00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gassen N.C., Niemeyer D., Muth D., et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat Commun. 2019;10:5770. doi: 10.1038/s41467-019-13659-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gassen NC, Papies J, Bajaj T, et al., Analysis of SARS-CoV-2-controlled autophagy reveals spermidine, MK-2206, and niclosamide as putative antiviral therapeutics. bioRxiv 2020, 2020.04.15.997254.
- 38.Li Z., Brecher M., Deng Y.Q., et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017;27:1046–1064. doi: 10.1038/cr.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chappell K.J., Stoermer M.J., Fairlie D.P., Young P.R. Mutagenesis of the West Nile virus NS2B cofactor domain reveals two regions essential for protease activity. J General Virol. 2008;89:1010–1014. doi: 10.1099/vir.0.83447-0. [DOI] [PubMed] [Google Scholar]
- 40.Niyomrattanakit P., Winoyanuwattikun P., Chanprapaph S., Angsuthanasombat C., Panyim S., Katzenmeier G. Identification of residues in the dengue virus type 2 NS2B cofactor that are critical for NS3 protease activation. J Virol. 2004;78:13708–13716. doi: 10.1128/JVI.78.24.13708-13716.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kasim N.A., Whitehouse M., Ramachandran C., et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharm. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 42.Ye Y., Zhang X., Zhang T., Wang H., Wu B. Design and evaluation of injectable niclosamide nanocrystals prepared by wet media milling technique. Drug Dev Ind Pharm. 2015;41:1416–1424. doi: 10.3109/03639045.2014.954585. [DOI] [PubMed] [Google Scholar]
- 43.Chen H., Yang Z., Ding C., et al. Discovery of O-alkylamino tethered niclosamide derivatives as potent and orally bioavailable anticancer agents. ACS Med Chem Lett. 2013;4:180–185. doi: 10.1021/ml3003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.World Health Organisation, In WHO Specifications and Evaluations for Public Health Pesticides: WHO. Geneva 2002, http://www.who.int/whopes/quality/en/Niclosamide.pdf.
- 45.Clinical Trials.gov, Other terms: niclosamide https://www.clinicaltrials.gov/ct2/results?recrs=&cond=&term=niclosamide&cntry=&state=&city=&dist=.
- 46.Siramshetty V.B., Shah P., Kerns E., et al. Retrospective assessment of rat liver microsomal stability at NCATS: data and QSAR models. Sci Rep. 2020;10:20713. doi: 10.1038/s41598-020-77327-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun H., Nguyen K., Kerns E., et al. Highly predictive and interpretable models for PAMPA permeability. Bioorg Med Chem. 2017;25:1266–1276. doi: 10.1016/j.bmc.2016.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun H., Shah P., Nguyen K., et al. Predictive models of aqueous solubility of organic compounds built on A large dataset of high integrity. Bioorg Med Chem. 2019;27:3110–3114. doi: 10.1016/j.bmc.2019.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X.Y., Cui J.N., Ren W.M., Zhao G.Q., Li F., Qian X.H. Reductive metabolism of Nitroaromatic compounds by various liver microsomes. Chem Res Chinese Universities. 2010;26:981–985. [Google Scholar]
- 50.Dubey S.K., Singh A.K., Singh H., et al. Synthesis of substituted 1-hydroxy-2-naphthanilides as potential cestodicidal agents. J Med Chem. 1978;21:1178–1181. doi: 10.1021/jm00209a020. [DOI] [PubMed] [Google Scholar]
- 51.Mook R.A., Jr., Chen M., Lu J., Barak L.S., Lyerly H.K., Chen W. Small molecule modulators of Wnt/β-catenin signaling. Bioorg Med Chem Lett. 2013;23:2187–2191. doi: 10.1016/j.bmcl.2013.01.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu X., Legler P.M., Southall N., Maloney D.J., Simeonov A., Jadhav A. Structural insight into exosite binding and discovery of novel exosite inhibitors of botulinum neurotoxin serotype A through in silico screening. J Comput Aided Mol Des. 2014;28:765–778. doi: 10.1007/s10822-014-9758-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lei J., Hansen G., Nitsche C., Klein C.D., Zhang L., Hilgenfeld R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science. 2016;353:503–505. doi: 10.1126/science.aag2419. [DOI] [PubMed] [Google Scholar]
- 54.Mirabelli C, Wotring JW, Zhang CJ, et al., Morphological Cell Profiling of SARS-CoV-2 Infection Identifies Drug Repurposing Candidates for COVID-19. bioRxiv 2020. [DOI] [PMC free article] [PubMed]
- 55.Jeon S, Ko M, Lee J, et al., Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. bioRxiv 2020, 2020.03.20.999730. [DOI] [PMC free article] [PubMed]
- 56.Gorshkov K, Chen CZ, Bostwick R, et al., The SARS-CoV-2 cytopathic effect is blocked with autophagy modulators. bioRxiv 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.