

Abstract
Purpose
The involvement of the intestinally expressed xenobiotic transporters P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP) have been implicated in apixaban disposition based on in vitro studies. Recommendations against co-administration of apixaban with inhibitors of these efflux transporters can be found throughout the literature as well as in the apixaban FDA label. However, the clinical relevance of such findings is questionable due to the high permeability and high solubility characteristics of apixaban.
Methods
Using recently published methodologies to discern metabolic- from transporter- mediated drug-drug interactions, a critical evaluation of all published apixaban drug-drug interaction studies was conducted to investigate the purported clinical significance of efflux transporters in apixaban disposition.
Results
Rational examination of these clinical studies using basic pharmacokinetic theory does not support the clinical significance of intestinal efflux transporters in apixaban disposition. Further, there is little evidence that efflux transporters are clinically significant determinants of systemic clearance.
Conclusions
Inhibition or induction of intestinal CYP3A4 can account for exposure changes of apixaban in all clinically significant drug-drug interactions, and lack of intestinal CYP3A4 inhibition can explain all studies with no exposure changes, regardless of the potential for these perpetrators to inhibit intestinal or systemic efflux transporters.
Keywords: apixaban, bioavailability, clearance, complex drug-drug interactions, mean absorption time
INTRODUCTION
Apixaban (Fig. 1) is an anticoagulant factor Xa inhibitor approved for a number of indications including stroke or blood clot prevention and treatment of deep vein thrombosis or pulmonary embolism [1]. Apixaban is primarily metabolized by cytochrome P450 (CYP) 3A4 (with minor contributions of other isoforms such as CYP 1A2, 2C8, 2C9, 2C19, and 2J2) [1]. The involvement of the intestinally-expressed efflux transporters P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP) has also been suggested throughout the literature [2, 3], as well as in the apixaban Food and Drug Administration (FDA) label [1]. However, in vitro susceptibility to transporters does not always translate to clinically significant outcomes, and this is particularly true for high-solubility drugs that display high membrane permeability characteristics (i.e. Biopharmaceutics Drug Disposition Classification System (BDDCS) Class 1 drugs) [4] for which a significant degree of passive passage across biological membranes is achieved, potentially rendering any transporter-assisted passage clinically insignificant. Thus, the purported clinically significant involvement of efflux transporters in the disposition of apixaban (BDDCS Class 1) is questionable. Understanding major contributors to drug disposition is critical in the clinical setting to allow for appropriate dosing and in particular, how to adjust dose based on disease state, due to pharmacogenomic variance, or in anticipation of a drug-drug interaction (DDI).
Fig. 1.
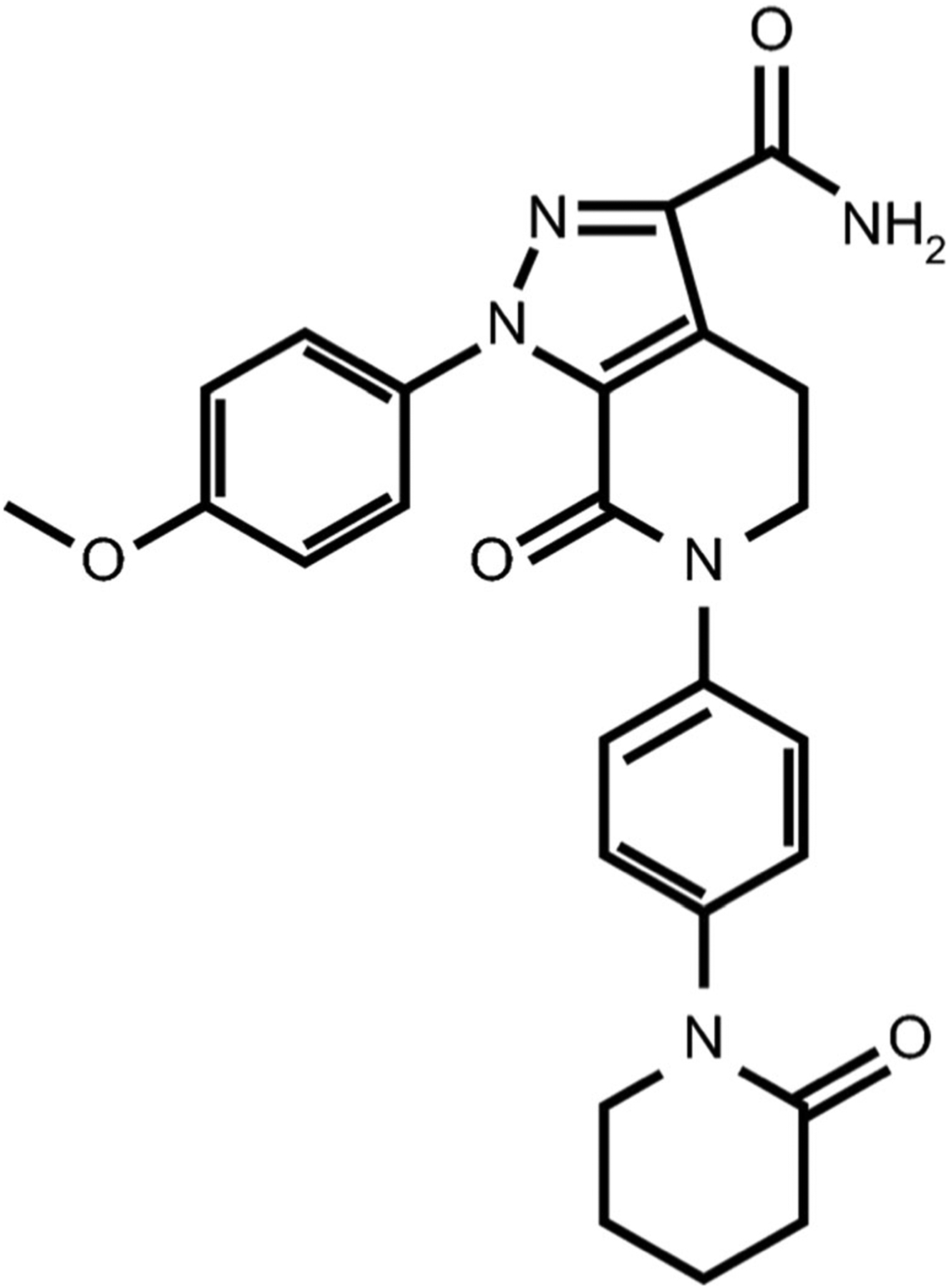
Chemical structure of apixaban.
Clearance (CL) is a critical determinant of drug dosing regimens, as it is inversely related to drug exposure (AUC; area under the concentration-time curve) that ultimately is believed to drive the therapeutic efficacy and potential toxicity of a drug (Eq. 1)
| (1) |
where F denotes fractional bioavailability following an oral dose, and is assumed to be 1 for an intravenous (IV) dose. Characterization of the contributors to clearance pathways, i.e., metabolic enzymes and/or xenobiotic transporters, is crucial in anticipating potential changes in clearance due to DDIs or pharmacogenomic variance of metabolic enzymes or transporters. Our laboratory has thoroughly detailed and documented the expected changes in pharmacokinetic parameters for interactions involving purely metabolic enzymes [5, 6] versus xenobiotic transporters [7, 8]. Inhibition or induction of metabolic enzymes results in changes in CL and AUC that are directionally intuitive and translate to rational changes in mean residence time (MRT) and terminal half-life (t1/2,z), as volume of distribution (Vss) remains unchanged for metabolic interactions [5, 6], as depicted in Eq. 2 [9]
| (2) |
It is considered reasonable to predict strictly metabolic interactions based on in vitro studies [10] due to a strong understanding by the field of the metabolizing enzymes commonly implicated in drug metabolism, which is further bolstered by well-characterized clinical specificities of routinely used metabolic inhibitors and inducers [11].
The FDA has provided guidance on predicting clinically significant transporter interactions [10], however, such predictions are not as straightforward and are even more challenging when both enzymes and transporters are involved in drug disposition, i.e., in so-called “complex DDIs”. We have recently thoroughly discussed how to appropriately predict changes in exposure when transporters are involved using the Extended Clearance Model, which not only requires understanding of how transporter-mediated active influx and efflux intrinsic clearances will potentially change, but also requires estimation of passive diffusion and changes in metabolic and biliary elimination [12]. The methodologies employed to estimate each of these elimination processes are not trivial, and each requires a different set of experimental conditions. Further, the susceptibility of a drug to uptake or efflux transporters in vitro does not always translate to clinically significant in vivo involvement [4]. Additionally, validated clinical transporter probe substrates and inhibitors are lacking [11]; routinely-used inhibitors are often not specific and may have inhibitory potential towards both enzymes and transporters [13], and additional xenobiotic transporters are continuously emerging and suggested to be clinical relevant by the field [14–16]. Furthermore, clinically significant transporter interactions can affect Vss for victim drugs [8] in addition to potential CL changes, resulting in counterintuitive changes in changes in MRT and t1/2,z that are not necessarily opposite in magnitude of CL changes [7], further complicating pharmacokinetic predictions (Eq. 2). Thus, the challenge in predicting exposure changes for complex DDIs is beyond simply accurately estimating the contribution of metabolism versus transporters, is further complicated by the potential for enzyme-transporter interplay, and is currently an area of significant efforts by the field [17, 18].
Oral dosing changes in F (due to altered absorption or first pass extraction) are often underemphasized as an important contributor in DDI-related exposure changes as compared to CL changes (Eq. 1). Discriminating changes in CL from changes in F has been believed not possible without also performing an IV DDI study to estimate changes in CL alone; however, most orally approved drugs have only been studied when orally administered. We have recently discussed that for low extraction ratio drugs, the minimal first pass elimination can indicate that changes in apparent clearance (CL/F) are primarily due to changes in CL alone [5]. Further, for purely metabolic interactions, knowledge that Vss is unchanged can allow for estimation of F changes by examining the change in apparent volume of distribution (Vss/F), which can further be utilized to predict changes in CL alone [19]. For clinically significant intestinal transporter substrates, alteration of transporter activity or expression will result in significant changes in absorption rate and we maintain that such changes should always be used to implicate transporter involvement in vivo [20]. However, changes in absorption rate may not necessarily translate to changes in extent of absorption if there is still sufficient time for absorption to occur, an additional consideration that complicates pharmacokinetic predictions of intestinal transporter substrates.
Utilization of these guiding principles in analyzing clinical data of purported complex DDIs, such as examining changes in absorption rate or Vss, can allow validation of the clinical significance of transporter involvement based on in vitro predictions. We have recently introduced these concepts [19, 20] in evaluating the apixaban-rifampin interaction [29] Here, we critically evaluate all published apixaban clinical DDI studies using the guiding principles mentioned above to investigate the purported clinical significance of P-gp and BCRP in apixaban disposition.
MATERIALS AND METHODS
To determine if intestinal transporter involvement is clinically significant in oral DDIs, changes in mean absorption time (MAT) or time to maximum concentration (tmax) can be compared between the interaction versus control phase of clinical DDI studies [20]. For clinically significant intestinal transporter DDIs, inhibition would result in decreased MAT and tmax, and induction would result in increases in these values. Values of tmax are routinely reported, however, MAT values are less frequently reported and therefore were estimated by digitizing published pharmacokinetic concentration-time profiles using WebPlotDigitizer Version 4.2 (San Francisco, CA) and fitting resulting data to a 2-compartmental model with first-order absorption from the gut using WinNonlin Professional Edition Version 2.1 (Pharsight, Mountain View, CA) to estimate absorption rate (ka; MAT = 1 / ka), as we have previously described [21]. If pharmacokinetic curves were not published, MAT was calculated using published tmax and t1/2,z values using the single-dose relationship between the three parameters, as we recently described in detail [20]. It should be noted that tmax values are observed values and these values depend heavily on the sampling scheme employed by the clinical investigators. However, any such errors have much less impact on drugs with large tmax values such as apixaban (3–4 h) [1]. Recent simulations illustrating the impact of 15 min errors in MAT (which could occur due to minimal absorption phase sampling) for both a rapidly absorbed drug (MAT = 0.5 h; tmax = 1.33 h) and a less-rapidly absorbed drug (MAT = 2 h; tmax = 3.2 h) highlight that such errors have markedly less impact on drugs with larger tmax values [20].
Changes in AUC, CL/F, Vss/F, MRT, t1/2,z are reported as ratios of interaction to control, where ratios of AUC were dose-normalized. Percent AUC extrapolation is also examined as a potential indication of the accuracy of any parameters derived from AUC, with the understanding that high percent extrapolations are only indicative of inaccuracies if terminal half-life is not adequately captured. MRT was calculated using Eq. 3:
| (3) |
where AUMC is area under the moment curve, and both AUC and AUMC are extrapolated to infinity since all clinical investigations were conducted for a single-dose of apixaban. Vss/F is calculated using Eq. 2. Published clinical values are utilized in calculation of ratios in priority, with digitization utilized only to supplement any unreported parameters-of-interest.
Ratios of change in MAT or tmax that indicated greater than 30% change (i.e. ratios outside of the range of 0.77 and 1.30) were considered to be potential evidence of a clinically significant intestinal transporter interaction. If MAT does not significantly change, it can be inferred that either xenobiotic transporters expressed in the intestine are not clinically significant determinants of apixaban disposition or that intestinal transporters are not inhibited or induced in that particular DDI [20].
A comprehensive literature search identified clinical apixaban DDI studies with the perpetrators atenolol [22], cyclosporine [23], diltiazem [24], enoxaparin [25], famotidine [26], ketoconazole [24, 27], naproxen [28], rifampin [29], and tacrolimus [23]. In addition, a study with activated charcoal [30] and two studies investigating the influence of pharmacogenomic variance with respect to CYP3A5, P-gp and/or BCRP [31, 32] were identified and critically discussed to compliment the analysis of clinical DDI studies.
Inhibitory or induction-related specificities of each perpetrator were documented to assess potential alteration of CYP3A, P-gp and/or BCRP activity or expression based on a recent compilation of clinically recommended index inhibitors of drug metabolizing enzymes and drug transporters [11]. In addition, the inhibitory potential of perpetrator drug in the intestine and systemic circulation was investigated by considering the maximum perpetrator concentration in the gut [Igut] or systemic circulation (Cmax) with respect to its half maximal inhibitory concentration (IC50) for CYP3A4, P-gp and BCRP. Values of [Igut] are estimated by considering perpetrator dose divided by the volume of water with which the perpetrator drug was dosed (and if unreported a standard value of 250 mL was utilized in calculations). Reported values of perpetrator Cmax were utilized; however if unreported, these values were referenced from the literature for a similar perpetrator dosing scheme. Fraction unbound in plasma (fu,plasma) values were also tabulated to further contextualize systemic inhibitory potential based on unbound concentrations and were cited from reference [33] unless otherwise noted. Based on the FDA DDI Guidance, values of [Igut]/IC50 > 10 indicate a potentially significant intestinal interaction, and values of Cmax > 0.1 indicate a potentially significant systemic interaction [10].
The rifampin-apixaban DDI study was conducted following both oral and IV administration [29], therefore the recently published clearance versus bioavailability differentiation methodology for metabolic DDIs [19] was utilized to predict changes in CL versus F. This analysis from our previous publications [19, 20] is included for reference. Predicted changes in pharmacokinetic parameters were compared to actual changes based on IV dosing, and provided further insight into the hypothesis that the reported in vitro susceptibility to efflux transporters by apixaban may be clinically insignificant. In addition, predictions of changes in CL versus F were performed for all clinically significant DDIs to characterize the contribution of changes in F versus CL, and the major site of interaction (intestine versus liver), for each interaction.
RESULTS
Implicating intestinal transporter involvement in apixaban disposition proceeded via examination of changes in apixaban absorption rate in clinical DDIs, based on our recently published methodology to identify clinically significant intestinal transporter interactions [20]. Table I details the inhibitory specificities of the nine perpetrators investigated against CYP3A4, P-gp and BCRP, and summarizes the expected intestinal or systemic inhibitory outcomes based on calculations of [Igut] or Cmax divided by IC50. Clinically significant alterations in intestinal efflux capacity (based on values of [Igut]/IC50 > 10) were expected for cyclosporine, diltiazem, ketoconazole, rifampin, and tacrolimus, and not expected or unknown for atenolol, enoxaparin, famotidine, and naproxen. Clinically significant inhibition of systemic efflux transporters based on values of Cmax/IC50 > 0.1 were expected for cyclosporine and diltiazem, however, consideration of unbound plasma systemic concentrations (Cmax,u) of these inhibitors does not support systemic inhibitory potential, as unbound perpetrator concentrations are not sufficiently high. Based on multiple dosing of rifampin, clinically significant induction in both intestinal and systemic P-gp is expected.
Table I.
Inhibitory Potential of Perpetrator Drugs from Apixaban Drug-Drug Interaction Studies for Metabolic Enzymes (CYP3A4) and Xenobiotic Transporters (P-gp and BCRP) Reported to be Clinical Determinants of Apixaban Disposition
| Perpetrator Drug | Perpetrator Dose | BDDCS Class | [lgut]a (μM) | fu plasmab | Cmaxc (μM) | CYP3A4 IC50 (μM) | P-gp IC50 (μM) | BCRP IC50 (μM) | Apixaban-Relevant Inhibitory Potentiald | References |
|---|---|---|---|---|---|---|---|---|---|---|
| Atenolol | 100 mg PO (Single Dose) | 3 | 1500 | 0.94 | 2.25 | > 100 [34] | Unknown | Unknown | Unknown / Not CYP3A4 | [22, 34] |
| Cyclosporine | 100 mg PO (3 Days) | 2 | 333 | 0.068 | 0.29 [35], e | 1.5–20 [36] Clinically Significant[11] |
0.74–6.18 [37] Clinically Significant [11] |
3.2 [38] Clinically Significant[11] |
CYP3A4 (I/S) P-gp (I/S) BCRP (I) |
[11, 23, 35–38] |
| Diltiazem | 360 mg PO (10 Days) | 1 | 3470 | 0.18 | 0.63 [[39], f | 7.8[40],78[41], 120[42] Clinically Significant[45] |
5.0–177 [43] Clinically Significant[45] |
>10[44] | CYP3A4 (I) P-gp (I/S) |
[24, 39–45] |
| Enoxaparin | 40 mg PO (Single Dose) | 1 | 35.6g | Unknown | 0.61 [46], g | Unknown | Unknown | Unknown | Unknown | [25, 46] |
| Famotidine | 40 mg PO (Single Dose) | 3 | 474 | 0.84 | 0.24 [47] | 190–390 [48] | Unknown | Unknown | Unknown / Not CYP3A4 | [26, 47, 48] |
| Ketoconazole | 400 mg PO (6 Days)[24] | 2 | 3010 [24] | 0.017 [49] | 0.021 [50] | 0.08 [41], 0.12[[41, 42] Clinically Significant[11] |
0.65–10.1 [37], 5.6[51] Clinically Significant[11] |
12 [51] | CYP3A4 (I/S) P-gp (I) BCRP (I) |
[11, 24, 27, 37, 41, 42, 49–51] |
| 400 mg PO (1.5 Days)[27] | 7530[27], h | |||||||||
| Naproxen | 500 mg PO (Single Dose) | 2 | 8690 | 0.002 | 0.293 | 9–695 [52] | >8000 [3] | >8000 [3], > 10 [53] | CYP3A4 (I) | [3, 28, 52, 53] |
| Rifampin | Rifampin (600 mg PO; 11 Days) | 2 | 3040 | 0.20 | 10.3 [54] | Clinically Significant Inducer[55] | Clinically Significant Inducer[55] | In vitro induced[56, 57] Clinical relevance has not yet been established[57] |
CYP3A4 (I/S) P-gp (I/S) Inducer |
[29, 54–57] |
| Tacrolimus | 5 mg PO (3 Days) | 2 | 24.9 | 0.01 | 0.033 [58], i | 0.62 [59] | 0.66 [60], 0.84 [61] | 10 [53], 3.6 [62] | CYP3A4 (I) P-gP (I) |
[23, 53, 58–62] |
BCRP, Breast Cancer Resistance Protein; BDDCS, Biopharmaceutics Drug Disposition Classification System; Cmax, maximum perpetrator concentration in systemic circulation; CYP, cytochrome P450; I, Intestine; IC50, half maximal inhibitory concentration; [Igut], maximum perpetrator concentration in the gut; fu,plasma, fraction unbound in plasma; P-gp, P-glycoprotein; PO, oral administration; S, Systemic
[Igut] values are calculated by perpetrator dose divided by volume of water utilized in each clinical study. If unreported, the standard volume of 250 mL was assumed
fu,plasma values are referenced from Lombardo et al. [33] unless otherwise noted
Cmax values are associated with maximum concentration of perpetrator drug reported in original clinical study, however, if unreported the value was referenced from a comparable study with a similar dosing scheme
Apixaban-relevant intestinal (I) and/or systemic (S) inhibitory potential is indicated; [Igut]/IC50>10 indicates intestinal inhibitory potential and Cmax/IC50>0.1 indicates systemic inhibitory potential, with recognition that unbound plasma concentrations may further diminish systemic inhibitory potential
Referenced Cmax is associated with a single dose of 100 mg cyclosporine, and likely underpredicts the true Cmax within this study
Referenced Cmax is also associated with a steady-state 360 mg PO dose, however, this study utilized an extended release formulation of diltiazem and thus may underpredict the true Cmax within this study
Utilized average molecular weight of 4500 g/mol to calculate [Igut] and Cmax; Cmax is associated with a pharmacodynamic measurement from a 40 mg subcutaneous dose
Differing [Igut] values between studies for the same ketoconazole dose are due to the latter study using only 100 mL to dose PO ketoconazole versus the standard value of 250 mL
Referenced Cmax is associated with a single dose of 5 mg tacrolimus, and likely underpredicts the true Cmax within this study
Clinically insignificant DDI changes in pharmacokinetic parameters are presented in Table II (atenolol, cyclosporine, enoxaparin, famotidine, tacrolimus). Clinically significant DDIs are listed in Table III (diltiazem, ketoconazole, naproxen, rifampin). No changes in MAT values were observed for 10 of the 11 interactions studied, with ratios of interaction to control ranging from 0.92–1.12, indicating that intestinal transporters are not clinically significant in these DDIs with a number of potent inhibitors (and one inducer) of P-gp and/or BCRP. A modest prolongation of MAT and tmax was observed only for the diltiazem-apixaban interaction [24], with an MAT ratio of 1.38 and a tmax ratio of 1.33.
Table II.
Clinically Insignificant Changes in Pharmacokinetic Parameters (Expressed as Ratios of Interaction / Control) in Drug-Drug Interaction (DDI) Studies with Apixaban as the Victim Drug
| Victim Drug | Perpetrator Drug | Apixaban-Relevant Inhibitory Potential | Population | Percent ADC Extrapolation (DDI/Con) | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Apixaban (10 mg PO; Single Dose) | Atenolol (100 mg PO; Single Dose) | Unknown / Not CYP3A4 | Healthy Subjects (n = 15)a | 1.07b | 1.33 | 0.87 | 1.15c | 0.91b | 0.77b | 0.84 | 2%/3% | [22] |
| Apixaban (10 mg PO; Single Dose) | Cyclosporine (100 mg PO; 3 Days) | CYP3A4 (I/S) p-gp (I/S) BCRP (I) |
Healthy Males (n = l2)d | 0.98b | 1.00 | 1.19 | 0.79 | 0.54b | 0.64b | 0.56 | 10%/10% | [23] |
| Apixaban (5 mg PO; Single Dose) | Enoxaparin (40 mg PO; Single Dose; Concurrent Dose) | Unknown | Healthy Subjects (n = 20) | 1.01e | 1.00 | 1.07 | 0.93c | –e | –e | 0.98 | 3%/3% | [25] |
| Apixaban (5 mg PO; Single Dose | Enoxaparin (40 mg PO; Single Dose; 6 h after Apixaban) | Unknown | Healthy Subjects (n = 20)f | 0,95e | 1.00 | 1.12 | 0.89c | –e | –e | 1.12 | 3%/3% | [25] |
| Apixaban (10 mg PO; Single Dose) | Famotidine (40 mg PO; Single Dose) | Unknown / Not CYP3A4 | Healthy Subjects (n = 18) | 1.03b | 0.67 | 1.01 | 0.99c | 0.97b | 0.98b | 1.20 | 1%/2% | [26] |
| Apixaban (10 mg PO; Single Dose) | Tacrolimus (5 mg PO; 3 Days) | CYP3A4 (I) P-gp (I) |
Healthy Males (n = 12)d | 0.99b | 1.00 | 0.77 | 1.26 | 0.83b | 0.66b | 0.58 | 9%/10% | [23] |
Pharmacokinetic values reported in the table are based on published average values, unless otherwise noted
AUC, area under the curve; BCRP, Breast Cancer Resistance Protein; Con, control; CL/F, apparent clearance; CYP, cytochrome P450; DDI, drug-drug interaction; I, Intestine; MAT, mean absorption time; MRT, mean residence time; P-gp, P-glycoprotein; PO, oral administration; S, Systemic; tmax, time to maximal concentration; t1/2,z, terminal half-life; Vss/F, apparent volume of distribution at steady state
Interaction phase was n = 14
Ratios are calculated by digitization of published average plasma concentration-time profiles and performing non-compartmental and/or compartmental analysis
CL/F was calculated using known dose, reported AUC and Eq. 1
Subjects were the same for the cyclosporine and tacrolimus DDI studies
Pharmacokinetic curves were not published, thus neither Vss nor MRT could be calculated. However, MAT was calculated by utilization of the single dose relationship between reported tmax and t1/2,z, as previously described [20]
Interaction phase was n = 19
Table III.
Clinically Significant Changes in Pharmacokinetic Parameters (Expressed as Ratios of Interaction / Control) in Drug-Drug Interaction (DDI) Studies with Apixaban as the Victim Drug
| Victim Drug: | Perpetrator Drug: | Relevant Enzyme or Transporter | Population | Percent AUC Extrapolation (DDI/Con) | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Apixaban (10 mg PO; Single Dose) | Diltiazem (360 mg PO; 10 Days) | CYP3A4 (I) P-gP (I/S) |
Healthy Subjects (n = 18) | 1.38a | 1.33 | 1.40b | 0.73c | 0.70a | 1.05a | 0.95b | 5%/6% | [24] |
| Apixaban (10 mg PO; Single Dose) | Ketoconazole (400 mg PO; 6 Days) | CYP3A4 (I/S) P-gP (I) BCRP (I) |
Healthy Males (n = 18) | 1.12a | 1.00 | 1.99 | 0.50c | 0.56a | 1.08a | 1.22 | 3%/2% | [24] |
| Apixaban (25 μg PO; Single Dose) | Ketoconazole (400 mg PO; 1.5 Days) | CYP3A4 (I/S) P-gP (I) BCRP (I) |
Healthy Subjects (n = 18) | 0.96d | 0.88 | 1.90 | 0.53 | 0.69a | 1.24a | 1.14 | 0%/0%a | [27] |
| Apixaban (10 mg PO; Single Dose) | Naproxen (500 mg PO; Single Dose) | CYP3A4 (I) | Healthy Males (n = 21) | 0.96a | 1.00 | 1.54 | 0.65c | 0.62a | 1.14a | 1.00 | 2%/2% | [28] |
| Apixaban (10 mg PO; Single Dose) | Rifampin (600 mg PO; 11 Days) | CYP3A4 (I/S) P-gP (I/S) Inducer |
Healthy Subjects (n = 20)e | 0.92a | 1.00 | 0.48 | 2.14 | 1.42a | 0.65a | 1.03 | 10%/9% | [29] |
Pharmacokinetic values reported in the table are based on published average values, unless otherwise noted
AUC, area under the curve; BCRP, Breast Cancer Resistance Protein; Con, control; CL/F, apparent clearance; CYP, cytochrome P450; DDI, drug-drug interaction; I, Intestine; MAT, mean absorption time; MRT, mean residence time; P-gp, P-glycoprotein; PO, oral administration; S, Systemic; tmax, time to maximal concentration; t1/2,z, terminal half-life; Vss/F, apparent volume of distribution at steady state
Ratios are calculated by digitization of published average plasma concentration-time profiles and performing non-compartmental and/or compartmental analysis
Control phase was n = 17
CL/F was calculated using known dose, reported AUC and Eq. 1
MAT was calculated by utilization of the single dose relationship between reported tmax and t1/2,z, as previously described [20]
Interaction phase was n = 18
Table IV displays the ratios of change in IV and oral apixaban pharmacokinetics following multiple dosing of rifampin [29] that we previously reported [19, 20]. By assuming that this interaction is purely metabolic, and based on the recently published clearance versus bioavailability differentiation methodology [19], the observed 52% reduction in oral apixaban exposure following multiple dosing of rifampin was estimated to be a result of a 1.5-fold increase in CL and a 30% reduction in F. These estimates were compared to actual changes in CL and F based on the IV interaction data, indicating that the observed change in CL was 1.64-fold yielding a 24% reduction in F, supporting the accuracy of our method for predicting the differentiation of changes in clearance from changes in bioavailability for oral metabolic DDIs.
Table IV.
Utilization of the Sodhi and Benet [19] Methodology to Discriminate Clearance from Bioavailability Changes for Orally Dosed Apixaban (Victim) and Rifampin (Perpetrator) from the Study of Vakkalagadda et al. [29]
| Victim | Perpetrator | Reference | ||||||
|---|---|---|---|---|---|---|---|---|
| Apixaban (IV) (Single Dose) | Rifampin (Multiple Dose) | Observed: 0.61 | – | Observed: 0.87 | Observed: 0.76 | – | Observed: 1.64 | [29] |
| Apixaban (Oral) (Single Dose) | Rifampin (Multiple Dose) | Observed: 0.48 | Observed: 1.42a | Assumed:1 | Estimated: 0.70 | Observed: 2.14 | Estimated: 1.50 | [29] |
Pharmacokinetic values reported in the table are based on published average values, unless otherwise noted
AUC, area under the curve; CL, clearance; CL/F, apparent clearance; DDI, drug-drug interaction; F, bioavailability; Vss, volume of distribution at steady state; Vss/F, apparent volume of distribution at steady state
Ratios are calculated by digitization of published average plasma concentration-time profiles and performing non-compartmental and/or compartmental analysis
Although confirming IV data were not available for the remaining four clinically significant DDIs, Table V displays the predicted changes in CL versus F for these interactions with the assumption that all interactions are purely metabolic, based on the recently described CL versus F discrimination methodology [19]. Predicted changes in systemic CL were minimal, ranging from 0.77–1.04, while predicted changes in F ranged from 1.43–1.79. Additionally, estimates of [Igut]/IC50, Cmax/IC50, and Cmax,u/IC50 were calculated, suggesting that all four interactions are predicted to be primarily intestinal, rather than systemic.
Table V.
Estimated Changes in CL versus F (Expressed as Ratios of Interaction / Control) in Clinically Significant Drug-Drug Interactions (DDIs) with Apixaban as the Victim Drug, Utilizing the Methodology of Sodhi and Benet [19]
| Victim Drug: | Perpetrator Drug: | CYP3A4 [Igut]/IC50 |
CYP3A4 Cmax/IC50 |
CYP3A4 Cmax,u/IC50 |
Observed |
Observed |
Estimated |
Observed |
Estimated |
Primary Site of Interaction | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Apixaban (10 mg PO) | Ketoconazole (400 mg PO; 6 Days) | 25,000 – 37,600 | 0.18–0.26 | < 0.004 | 1.99 | 0.56a | 1.79 | 0.50b | 1.00 | Intestine | [24] |
| Apixaban (25 μg PO) | Ketoconazole (400 mg PO; 1.5 Days) | 62,800 – 94,100 | 0.18–0.26 | < 0.004 | 1.90 | 0.69a | 1.45 | 0.53 | 0.77 | Intestine | [27] |
| Apixaban (10 mg PO) | Naproxen (500 mg PO; Single Dose) | 12.5–966 | < 0.3 | < 0.00007 | 1.54 | 0.62a | 1.61 | 0.65b | 1.00 | Intestine | [28] |
| Apixaban (10 mg PO) | Diltiazem (360 mg PO; 10 Days) | 28.9–445 | < 0.08 | < 0.014 | 1.40c | 0.70a | 1.43 | 0.73b | 1.04 | Intestine | [24] |
Pharmacokinetic values reported in the table are based on published average values, unless otherwise noted
AUC, area under the curve; BCRP, Breast Cancer Resistance Protein; Con, control; CL, clearance; CL/F, apparent clearance; CYP, cytochrome P450; DDI, drug-drug interaction; F, bioavailability; I, Intestine; P-gp, P-glycoprotein; PO, oral administration; S, Systemic; Vss, volume of distribution at steady state; Vss/F, apparent volume of distribution at steady state
Ratios are calculated by digitization of published average plasma concentration-time profiles and performing non-compartmental and/or compartmental analysis
CL/F was calculated using known dose, reported AUC and Eq. 1
Control phase was n = 17
In addition, a clinical study with activated charcoal dosed both 2 h and 6 h post apixaban oral dosing was identified [30], where no change in Cmax or tmax was observed, however AUC and t1/2,z decreased respectively to ratios of 0.49 and 0.40 (2 h dose) and to 0.71 and 0.37 (6 h dose).
Two pharmacogenomic studies were identified in which differences in apixaban disposition were investigated with respect to CYP3A5, P-gp and/or BCRP [31, 32]. The first study investigated apixaban disposition in patients with atrial fibrillation and acute stroke with respect to gene polymorphisms in CYP3A5 and ABCB1 (P-gp), concluding that these polymorphisms do not affect the pharmacokinetics of apixaban [31]. The second study investigated dose-normalized apixaban plasma trough concentrations in 70 measurements from 44 patients with atrial fibrillation [32]. The investigators concluded that P-gp pharmacogenomics did not impact plasma trough concentrations, however, patients with either ABCG2 (BCRP) or CYP3A5 gene polymorphisms had higher plasma trough concentrations.
DISCUSSION
Discerning involvement of transporters versus metabolic enzymes is challenging, particularly because the susceptibility of drug to efflux or uptake transporters in vitro does not always translate to clinically significant in vivo involvement [4]. Further, following oral dosing DDIs, separating changes in CL or Vss from F, as well as consideration of the impact of both CL and Vss on MRT and half-life, makes discerning clinically significant transporter involvement a difficult task. Based on the recognition that significant intestinal transporter interactions will result in discernable changes in MAT (and therefore tmax) [20], it is possible to implicate intestinal transporters in oral DDI studies, with no change indicating that intestinal transporters are not relevant. Apixaban tmax occurs approximately 3–4 h after oral dosing [1, 63], a value large enough to sensitively detect changes in absorption rate under standard pharmacokinetic sampling schemes [20].
No change in apixaban absorption rate was observed in 10 of 11 oral DDI studies with MAT ratios ranging from 0.92–1.12 (Tables II and III), which included perpetrator drugs with significant potential to inhibit P-gp and BCRP based on in vitro data (Table I). These results are consistent with the BDDCS class 1 designation of apixaban (high permeability, high solubility), which proposes that such drugs’ high solubility characteristics allows very high concentrations of drug to passively diffuse, greatly overwhelming any transporter-mediated effects at clinically relevant concentrations [4]. It is noteworthy that the ketoconazole-apixaban interaction was conducted at both a clinically relevant dose (10 mg) and a microdose (25 μg), and thus it may be expected that for the lower dose, transporter effects can no longer be overwhelmed due to lower overall concentrations. However, in both studies no changes in MAT or tmax were observed, and the degree of changes in exposure and clearance were almost identical between both studies, indicating at both apixaban concentrations the interaction was primarily due to a process for which soluble concentrations are irrelevant; i.e., CYP3A4 inhibition. Although these results are striking, conclusions would be further strengthened if it were possible to examine patient data in order to calculate changes in MAT and tmax for each individual.
The diltiazem-apixaban DDI resulted in a 1.38-fold change in MAT and a 1.33-fold change in tmax, both values that are very close to our cutoff of 1.30 but suggesting a potentially significant intestinal transporter interaction. If this result was truly reflective of inhibition of P-gp, then it would be expected that other P-gp inhibitors, in particular more potent inhibitors, should also show similar changes in absorption rate. The diltiazem estimate of [Igut]/IC50 for P-gp ranges from 19.6 to 694 and is not markedly different from estimates for cyclosporine (53.9–450), ketoconazole (298–4630 and 746–11,600), and tacrolimus (29.6–37.7). Ketoconazole also significantly inhibits intestinal BCRP, with [Igut]/IC50 estimates of 251 and 628 for both studies. It is possible that non-transporter mediated changes in absorption rate may be responsible for these results, such as changes in pH or gastric emptying by the perpetrator drug diltiazem. However, apixaban does not contain ionizable groups, and thus potential changes in gastric pH by diltiazem should not alter apixaban solubility or absorption, and this hypothesis was nicely confirmed in the famotidine study, where changes in gastric pH had no effect on apixaban pharmacokinetics [26]. Further, changes in gastric emptying by diltiazem are not expected [64], therefore perhaps this outcome is related to limitations associated with utilizing published average pharmacokinetic profiles, as such graphical representations do not necessarily represent any single subject within the study. The study authors indicate diltiazem had no effect on tmax [24], however since we only had access to published median tmax values our calculated tmax ratio was 1.33. Thus, we again highlight that conclusions from utilization of our methodology [20] will be strengthened if absorption rate is calculated for each individual in the study. It should also be recognized that tmax is influenced by both absorption rate and elimination rate parameters, and we have recently published the single dose and steady-state mathematical relationships for reference [20]. Therefore, implicating intestinal transporter involvement based on tmax ratios alone may mislead an investigator, such as in the atenolol or famotidine results where tmax ratios are1.33 and 0.67, respectively, while the respective MAT ratios of 1.07 and 1.03 show no change in absorption rate.
As intestinal efflux transporter involvement is unlikely to contribute to apixaban bioavailability, we further investigate the potential involvement of systemic P-gp/BCRP inhibition to affect apixaban disposition. Examination of the inhibitory potential of perpetrators associated with clinically insignificant DDIs (Table II) reveals that only cyclosporine had the potential to inhibit systemic P-gp with a calculated Cmax/IC50 value of >0.39 and a Cmax,u/IC50 value of >0.027 (based on values presented in Table I), yet no change in apixaban exposure was observed. Of the clinically significant inhibitory DDIs, only diltiazem was expected to achieve systemic concentrations capable of inhibiting P-gp, with similar Cmax/IC50 values of >0.17 and Cmax,u/IC50 of >0.023, highlighting when compared to cyclosporine that the observed diltiazem AUC ratio of 1.4 is likely not due to inhibition of P-gp. Further, significant transporter interactions are expected to result in marked changes in Vss of victim drug [7, 8], however changes in Vss in the IV rifampin-apixaban DDI were minimal (ratio 0.87) (Table IV). Purely metabolic DDIs do not affect the Vss of victim drug [5, 6], thus following oral dosing it is possible to estimate the relative change in CL versus F by attributing the observed change in Vss/F to F alone [19]. Table IV demonstrates that utilization of this methodology for the oral interaction data results in remarkably accurate predictions of CL versus F change, further supporting that for an interaction with a potent inducer of CYP3A4 and P-gp, apixaban is primarily susceptible to alterations in metabolic enzymes rather than transporters.
Examination of the clinically significant DDIs listed in Table III show that in general, changes in CL/F were similar in magnitude to Vss/F, resulting in unchanged MRT and t1/2,z, suggesting that these significant DDIs are primarily due to changes in F. Table V utilizes the CL versus F differentiation methodology [19] to predict the extent of change in CL and F to understand if the observed exposure changes are primarily due to an intestinal or systemic effect. Based on this analysis, predicted changes in systemic CL were minimal (0.77–1.04) whereas predicted changes in F ranged from 1.43–1.79. These results suggest that these significant exposure changes are primarily driven by intestinal interactions, and taken together with the unchanged absorption rates associated with these interactions, we conclude intestinal CYP3A4 is responsible for all significant apixaban DDIs. This conclusion is further rationalized by examining the intestinal versus systemic CYP3A4 inhibitory potential listed in Table V, as all four perpetrators have [Igut]/IC50 values greater than 10, however, Cmax,u/IC50 is only greater than 0.1 for diltiazem.
It is noteworthy that the cyclosporine and tacrolimus DDI studies did not result in clinically significant changes in exposure [23], given their potential to inhibit intestinal CYP3A4. It is possible that since the aim of this DDI study was to examine the impact of clinically relevant systemic cyclosporine and tacrolimus concentrations achieved in transplant patients on apixaban disposition, the oral dosing of these perpetrators was not necessarily at the same time as apixaban dosing. This aspect was not clearly described within the methods, however the study design scheme published within that article [23] does indicate there was some amount of time between dosing of perpetrator and apixaban. Thus, we hypothesize the true intestinal perpetrator concentrations may be much lower than we report in Table I.
The impact of activated charcoal was also investigated, where activated charcoal was dosed during the absorption phase of apixaban (2 h after dosing) and after apixaban absorption was complete (6 h after dosing) [30]. Activated charcoal is often used in situations of drug overdose, as drug is adsorbed on to activated charcoal in the intestine thus reducing extent of absorption. Activated charcoal studies can also be utilized to investigate the potential of a drug to undergo enterohepatic recycling, as reabsorption of drug is prevented after biliary excretion into the intestine. Between the 2 h and 6 h doses of activated charcoal, AUC decreased with ratios of 0.49 and 0.71, respectively, while t1/2,z decreased similarly with ratios of 0.40 and 0.37, respectively. The differential changes in AUC with respect to dosing time support the expected outcome that a larger decrease in F would be observed when activated charcoal was dosed during the apixaban absorption phase. The modest reduction in exposure associated with the 6 h dose of activated charcoal (AUC ratio of 0.71) is not likely due to prevention of enterohepatic recirculation by activated charcoal, as biliary excretion is a minor elimination pathway [65] and none of the pharmacokinetic profiles in any investigated study displayed the characteristic secondary peaks commonly associated with enterohepatic recirculation. Thus, the study authors hypothesize that apixaban undergoes enteroenteric recycling (recycling between systemic circulation and intestinal lumen via passive diffusion) that is prevented when apixaban is adsorbed on to activated charcoal. This may explain the observed similar reduction in t1/2,z for both the 2 h and 6 h doses, as there may be an increase in extent of direct apixaban elimination into the feces via the intestine when activated charcoal is present. We agree that further mechanistic studies are warranted, however, these results underscore the potential bidirectional ability of apixaban to cross intestinal membranes between gut lumen and systemic circulation via passive diffusion, further countering the hypothesis that apixaban is susceptible to the action of transporters.
We identified two pharmacogenomic studies in which CYP3A5, P-gp and/or BCRP pharmacogenomics were investigated. The first study concluded that differences in CYP3A5 and P-gp pharmacogenomics do not affect the pharmacokinetics of apixaban [31]. The second study investigated BCRP pharmacogenomics in addition to CYP3A5 and P-gp. Pharmacokinetic parameters were not assessed, however, investigators associated pharmacogenomics with dose-normalized trough concentration measurements taken 10–14 h post apixaban dosing, for 70 measurements from 40 patients. The investigators concluded that BCRP and CYP3A5 pharmacogenomics, but not P-gp pharmacogenomics, impacted dose-normalized trough concentrations. However, it is unclear if these results accounted for the differences in sampling time between individuals in each group, or even with respect to multiple samples from the same individual. Thus, we reserve any conclusions related to apixaban pharmacogenomics and suggest further research is warranted.
CONCLUSIONS
Throughout the literature [66–69], and even in the apixaban FDA label [1], authors routinely cite the clinically significant DDI studies listed in Table III as evidence that P-gp and/or BCRP is a clinically significant determinant of apixaban disposition, confirming results of in vitro transporter studies [2, 3]. However, rational examination of these clinical studies using basic pharmacokinetic theory simply does not support the clinical significance of efflux transporters in apixaban disposition. These conclusions are not limited to the involvement of intestinal efflux transporters (based on changes in absorption time) for P-gp and BCRP, there is also little evidence that these transporters are clinically significant determinants of systemic clearance. Inhibition or induction of intestinal CYP3A4 can account for exposure changes of apixaban in all clinically significant DDIs, and lack of intestinal CYP3A4 inhibition can explain all studies with no exposure changes, regardless of the potential for these perpetrators to inhibit intestinal or systemic efflux transporters.
Acknowledgements and Disclosures.
This work was supported in part by a Mary Ann Koda-Kimble Seed Award for Innovation. Ms. Sodhi was supported in part by an American Foundation for Pharmaceutical Education Predoctoral Fellowship, NIGMS grant R25 GM56847 and a Louis Zeh Fellowship. Dr. Benet is a member of the UCSF Liver Center supported by NIH grant P30 DK026743. All authors contributed to the writing and analysis of this manuscript. The authors declare no conflict of interest.
ABBREVIATIONS
- AUC
Area under the concentration-time curve
- AUMC
Area under the moment-time curve
- BCRP
Breast Cancer Resistance Protein
- BDDCS
Biopharmaceutics Drug Disposition Classification System
- CL
Clearance
- CL/F
Apparent clearance
- Cmax
Maximum concentration in systemic circulation
- Cmax,u
Maximum unbound concentration in systemic circulation
- CYP
Cytochrome P450
- DDI
Drug-drug interaction
- F
Bioavailability
- FDA
Food and Drug Administration
- fu,plasma
Fraction unbound in plasma
- IC50
Half maximal inhibitory concentration
- Igut
Maximum perpetrator concentration in the gut
- IV
Intravenous
- ka
Absorption rate constant
- MAT
Mean absorption time
- MRT
Mean residence time
- P-gp
P-glycoprotein
- tmax
Time to maximum concentration
- t1/2,z
Terminal half-life
- Vss
Volume of distribution at steady-state
- Vss/F
Apparent volume of distribution at steady-state
REFERENCES
- 1.ELIQUIS. (apixaban) [package insert] Princeton: Bristol-Myers Squibb Company; 2012. [Google Scholar]
- 2.Jacqueroux E, Mercier C, Margelidon-Cozzolino V, Hodin S, Bertoletti L, Delavenne X. In vitro assessment of P-gp and BCRP transporter-mediated drug-drug interactions of riociguat with direct oral anticoagulants. Fundam Clin Pharmacol. 2020;34(1):109–19. [DOI] [PubMed] [Google Scholar]
- 3.Zhang D, He K, Herbst JJ, Kolb J, Shou W, Wang L, et al. Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab Dispos. 2013;41(4): 827–35. [DOI] [PubMed] [Google Scholar]
- 4.Wu C-Y, Benet LZ. Predicting drug disposition via application of BCS: transport / absorption / elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. [DOI] [PubMed] [Google Scholar]
- 5.Benet LZ, Bowman CM, Koleske ML, Rinaldi CL, Sodhi JK. Understanding drug-drug interaction and pharmacogenomic changes in pharmacokinetics for metabolized drugs. J Pharmacokinet Pharmacodyn. 2019;46(2):155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sodhi JK, Huang CH, Benet LZ. Volume of distribution is unaffected by metabolic drug-drug interactions. Clin Pharmacokinet. [E-pub ahead of print, July 28, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benet LZ, Bowman CM, Sodhi JK. How transporters have changed basic pharmacokinetic understanding. AAPS J. 2019;21(6):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grover A, Benet LZ. Effects of drug transporters on volume of distribution. AAPS J. 2009;11(2):250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benet LZ, Galeazzi RL. Noncompartmental determinations of the steady-state volume of distribution. J Pharm Sci. 1979;68(8):1071–4. [DOI] [PubMed] [Google Scholar]
- 10.U.S. Food and Drug Administration, Center for Drug Evaluation and Research. In vitro drug interaction studies – cytochrome P450 enzyme- and transporter- mediated drug interactions guidance for industry. Silver Spring, MD; 2020. [Google Scholar]
- 11.Tornio A, Filppula AM, Niemi M, Backman JT. Clinical studies on drug-drug interactions involving metabolism and transport: methodology, pitfalls and interpretation. Clin Pharmacol Ther. 2019;105(6):1345–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benet LZ, Bowman CM, Liu S, Sodhi JK. The extended clearance concept following oral and intravenous dosing: theory and critical analyses. Pharm Res. 2018;35(12):242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheong J, Halladay JS, Plise E, Sodhi JK, Salphati L. The effects of drug metabolizing enzymes inhibitors on hepatic efflux and uptake transporters. Drug Metab Lett. 2017;11(2):111–8. [DOI] [PubMed] [Google Scholar]
- 14.Kimoto E, Mathialagan S, Tylaska L, Niosi M, Lin J, Carlo AA, et al. Organic anion transporter 2-mediated hepatic uptake contributes to the clearance of high-permeability-low-molecular-weight acid and zwitterion drugs: evaluation using 25 drugs. J Pharmacol Exp Ther. 2018;367(2):322–34. [DOI] [PubMed] [Google Scholar]
- 15.Sato T, Mishima E, Mano N, Abe T, Yamaguchi H. Potential drug interactions mediated by renal organic anion transporter OATP4C1. J Pharmacol Exp Ther. 2017;362(2):271–7. [DOI] [PubMed] [Google Scholar]
- 16.Zamek-Gliszczynski MJ, Taub ME, Chothe PP, Chu X, Giacomini KM, Kim RB, et al. International transporter consortium. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin Pharmacol Ther. 2018;104(5):890–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alluri RV, Li R, Varma MVS. Transporter-enzyme interplay and the hepatic drug clearance: what have we learned so far? Expert Opin Drug Metab Toxicol. 2020;16(5):387–401. [DOI] [PubMed] [Google Scholar]
- 18.Varma MV, El-Kattan AF. Transporter-enzyme interplay: deconvoluting effects of hepatic transporters and enzymes on drug disposition using static and dynamic mechanistic models. J Clin Pharmacol. 2016;56:S99–S109. [DOI] [PubMed] [Google Scholar]
- 19.Sodhi JK, Benet LZ. A simple methodology to differentiate changes in bioavailability from changes in clearance following oral dosing of metabolized drugs. Clin Pharmacol Ther. 2020;108(2):306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sodhi JK, Benet LZ. The necessity of using changes in absorption time to implicate intestinal transporter involvement in oral drug-drug interactions. AAPS J. 2020;22:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau YY, Huang Y, Frassetto L, Benet LZ. Effect of OATP1B1 transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007;81(2):194–204. [DOI] [PubMed] [Google Scholar]
- 22.Frost C, Song Y, Yu Z, Wang J, Lee LS, Schuster A, et al. The effect of apixaban on the pharmacokinetics of digoxin and atenolol in healthy subjects. Clin Pharmacol. 2017;9:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bashir B, Stickle DF, Chervoneva I, Kraft WK. Drug-drug interaction study of apixaban with cyclosporine and tacrolimus in healthy volunteers. Clin Transl Sci. 2018;11(6):590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frost CE, Byon W, Song Y, Wang J, Schuster AE, Boyd RA, et al. Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Br J Clin Pharmacol. 2015;79(5):838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett YC, Wang J, Song Y, Pursley J, Wastall P, Wright R, et al. A randomized assessment of the pharmacokinetic, pharmacodynamic and safety interaction between apixaban and enoxaparin in healthy subjects. Thromb Haemost. 2012;107(5):916–24. [DOI] [PubMed] [Google Scholar]
- 26.Upreti VV, Song Y, Wang J, Byon W, Boyd RA, Pursley JM, et al. Effect of famotidine on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Clin Pharmacol. 2013;5:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mikus G, Foerster KI, Schaumaeker M, Lehmann M-L, Burhenne J, Haefeli WE. Microdosed cocktail of three oral factor Xa inhibitors to evaluate drug-drug interactions with potential perpetrator drugs. Clin Pharmacokinet. 2019;58(9):1155–63. [DOI] [PubMed] [Google Scholar]
- 28.Frost C, Shenker A, Gandhi MD, Pursley J, Barrett YC, Wang J, et al. Evaluation of the effect of naproxen on the pharmacokinetics and pharmacodynamics of apixaban. Br J Clin Pharmacol. 2014;78(4):877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vakkalagadda B, Frost C, Byon W, Boyd RA, Wang J, Zhang D, et al. Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor Xa. Am J Cardiovasc Drugs. 2016;16(2):119–27. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Mondal S, Wang J, Tirucherai G, Zhang D, Boyd RA, et al. Effect of activated charcoal on apixaban pharmacokinetics in healthy subjects. Am J Cardiovasc Drugs. 2014;14(2):147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kryukov AV, Sychev DA, Andreev DA, Ryzhikova KA, Grishina EA, Ryabova AV, et al. Influence of ABCB1 and CYP3A5 gene polymorphisms on pharmacokinetics of apixaban in patients with atrial fibrillation and acute stroke. Pharmgenomics Pers Med. 2018;11:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ueshima S, Hira D, Fujii R, Kimura Y, Tomitsuka C, Yamane T, et al. Impact of ABCB1, ABCG2 and CYP3A5 polymorphisms on plasma trough concentrations of apixaban in Japanese patients with atrial fibrillation. Pharmacogenet Genomics. 2017;27(9):329–36. [DOI] [PubMed] [Google Scholar]
- 33.Lombardo F, Berellini G, Obach RS. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 1352 drug compounds. Drug Metab Dispos. 2018;46(11):1466–77. [DOI] [PubMed] [Google Scholar]
- 34.Maréchal J-D, Yu J, Brown S, Kapelioukh I, Rankin EM, Wolf CR, et al. In silico and in vitro screening for inhibition of cytochrome P450 CYP3A4 by comedications commonly used by patients with cancer. Drug Metab Dispos. 2006;34(4):534–8. [DOI] [PubMed] [Google Scholar]
- 35.Hulskotte E, Gupta S, Xuan F, van Zutven M, O’Mara E, Feng HP, et al. Pharmacokinetic interaction between the hepatitis C virus protease inhibitor boceprevir and cyclosporine and tacrolimus in healthy volunteers. Hepatology. 2012;56(5):1622–30. [DOI] [PubMed] [Google Scholar]
- 36.Donato MT, Jiménez N, Castell JV, Gómez-Lechón MJ. Fluorescence-based assays for screening nine cytochrome P450 (P450) activities in intact cells expressing individual human P450 enzymes. Drug Metab Dispos. 2004;32(7):699–706. [DOI] [PubMed] [Google Scholar]
- 37.Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR, et al. In vitro p-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates. Drug Metab Dispos. 2006;34(5):786–92. [DOI] [PubMed] [Google Scholar]
- 38.Miyata H, Takada T, Toyoda Y, Matsuo H, Ichida K, Suzuki H. Identification of febuxostat as a new strong ABCG2 inhibitor: potential applications and risks in clinical situations. Front Pharmacol. 2016;7:518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel CG, Li L, Girgis S, Kornhauser DM, Frevert EU, Boulton DW. Two-way pharmacokinetic interaction studies between saxagliptin and cytochrome P450 substrates or inhibitors: simvastatin, diltiazem extended-release, and ketoconazole. Clin Pharmacol. 2011;3:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burt HJ, Galetin A, Houston JB. IC50-based approaches as an alternative method for assessment of time-dependent inhibition of CYP3A4. Xenobiotica. 2010;40(5):331–43. [DOI] [PubMed] [Google Scholar]
- 41.Ma B, Preuksaritanont T, Lin JH. Drug interactions with calcium channel blockers: possible involvement of metabolite-intermediate complexation with CYP3A. Drug Metab Dispos. 2000;28(2):125–30. [PubMed] [Google Scholar]
- 42.Wang J-S, Wen X, Backman JT, Taavitsainen P, Neuvonen PJ, Kivistö KT. Midazolam alpha-hydroxylation by human liver microsomes in vitro: inhibition by calcium channel blockers, itraconazole, and ketoconazole. Pharmacol Toxicol. 1999;85(4):157–61. [DOI] [PubMed] [Google Scholar]
- 43.Ellens H, Deng S, Coleman J, Bentz J, Taub ME, Ragueneau-Majlessi I, Chung SP, Herédi-Szabó K, Neuhoff S, Palm J, Balimane P, Zhang L, Jamei M, Hanna I, O’Connor M, Bednarczyk D, Forsgard M, Chu X, Funk C, Guo A, Hillgren KM, Li L, Pak AY, Perloff ES, Rajaraman G, Salphati L, Taur JS, Weitz D, Wortelboer HM, Xia CQ, Xiao G, Yamagata T, Lee CA. Application of receiver operating characteristic analysis to refine the prediction of potential digoxin drug interactions. Drug Metab Dispos. 2013;41(7):1367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Gupta A, Wang H, Zhou L, Vethanayagam RR, Unadkat JD, et al. BCRP transports dipyridamole and is inhibited by calcium channel blockers. Pharm Res. 2005;22(12):2023–34. [DOI] [PubMed] [Google Scholar]
- 45.Isoherranen N, Lutz JD, Chung SP, Hachad H, Levy RH, Ragueneau-Majlessi I. Importance of multi-P450 inhibition in drug-drug interactions: evidence of incidence, inhibition magnitude, and prediction from in vitro data. Chem Res Toxicol. 2012;25(11):2285–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frydman AM, Bara L, Le Roux Y, Woler M, Chauliac F, Samama MM. The antithrombotic activity and pharmacokinetics of enoxaparine, a low molecular weight heparin in humans given single subcutaneous doses of 20 to 80 mg. J Clin Pharmacol. 1988;28(7): 609–18. [DOI] [PubMed] [Google Scholar]
- 47.Lin JH, Chremos AN, Kanovsky SM, Schwartz S, Yeh KC, KannJ. Effects of antacids and food on absorption of famotidine. Br J Clin Pharmacol. 1987;24(4):551–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moody DE, Liu F, Fang WB. In vitro inhibition of methadone and oxycodone cytochrome P450-dependent metabolism: reversible inhibition by H2-receptor agonists and proton-pump inhibitors. J Anal Toxicol. 2013;37(8):476–85. [DOI] [PubMed] [Google Scholar]
- 49.Brown HS, Galetin A, Hallifax D, Houston JB. Prediction of in vivo drug-drug interactions from in vitro data: factors affecting prototypic drug-drug interactions involving CYP2C9, CYP2D6 and CYP3A4. Clin Pharmacokinet. 2006;45(10):1035–50. [DOI] [PubMed] [Google Scholar]
- 50.Badri PS, Dutta S, Wang H, Podsadecki TJ, Polepally AR, Khatri A, et al. Drug interactions with the direct-acting antiviral combination of ombitasvir and paritaprevir-ritonavir. Antimicrob Agents Chemother. 2015;60(1):105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vermeer LMM, Isringhausen CD, Ogilvie BW, Buckley DB. Evaluation of ketoconazole and its alternative clinical CYP3A4/5 inhibitors as inhibitors of drug transporters: the in vitro effects of ketoconazole, ritonavir, clarithromycin, and itraconazole on 13 clinically relevant drug transporters. Drug Metab Dispos. 2016;44(3):453–9. [DOI] [PubMed] [Google Scholar]
- 52.Kajbaf M, Longhi R, Montanari D, Vinco F, Rigo M, Fontana S, et al. A comparative study of the CYP450 inhibition potential of marketed drugs using two fluorescence based assay platforms routinely used in the pharmaceutical industry. Drug Metab Lett. 2011;5(1):30–9. [DOI] [PubMed] [Google Scholar]
- 53.Saito H, Hirano H, Nakagawa H, Fukami T, Oosumi K, Murakami K, et al. A new strategy of high-speed screening and quantitative structure activity relationship analysis to evaluate human ATP-binding cassette transporter ABCG2-drug interactions. J Pharmacol Exp Ther. 2006;317(3):1114–24. [DOI] [PubMed] [Google Scholar]
- 54.Polk RE, Brophy DF, Israel DS, Patron R, Sadler BM, Chittick GE, et al. Pharmacokinetic interaction between amprenavir and rifabutin or rifampin in healthy males. Antimicrob Agents Chemother. 2001;45(2):502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivistö KT. Pharmacokinetic interactions with rifampicin. Clin Pharmacokinet. 2003;42(9):819–50. [DOI] [PubMed] [Google Scholar]
- 56.Lemmen J, Tozakidis IEP, Galla H-J. Pregnane X receptor upregulates ABC-transporter Abcg2 and Abcb1 at the blood-brain barrier. Brain Res. 2013;1491:1–13. [DOI] [PubMed] [Google Scholar]
- 57.Goreczyca L, Aleksunes LM. Transcription factor-mediated regulation of the BCRP/ABCG2 efflux transporter: a review across tissues and species. Expert Opin Drug Metab Toxicol. 2020;16(3): 239–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bekersky I, Dressler D, Colburn W, Mekki Q. Bioequivalaence of 1 and 5 mg tacrolimus capsules using a replicate study design. J Clin Pharmacol. 1999;39:1032–7. [DOI] [PubMed] [Google Scholar]
- 59.Amundsen R, Åsberg A, Ohm IK, Christensen H. Cyclosporine A-and tacrolimus-mediated inhibition of CYP3A4 and CYP3A5 in vitro. Drug Metab Dispos. 2012;40(4):655–61. [DOI] [PubMed] [Google Scholar]
- 60.Kishimoto W, Ishiguro N, Ludwig-Schwellinger E, Ebner T, Schaefer O. In vitro predictability of drug-drug interaction likelihood of p-glycoprotein-mediated efflux of dabigatran etexilate based on [I]2/IC50 threshold. Drug Metab Dispos. 2014;42(2): 257–63. [DOI] [PubMed] [Google Scholar]
- 61.Patil AG, D’Souza R, Dixit N, Damre A. Validation of quinidine as a probe substrate for the in vitro P-gp inhibition assay in Caco-2 cell monolayer. Eur J Drug Metab Pharmacokinet. 2011;36(3):115–9. [DOI] [PubMed] [Google Scholar]
- 62.Gupta A, Dai Y, Vethanayagam RR, Hebert MF, Thummel KE, Unadkat JD, et al. Cyclosporine A, tacrolimus and sirolimus are potent inhibitors of the human breast cancer resistance protein (ABCG2) and reverse resistance to mitoxantrone and topotecan. Cancer Chemother Pharmacol. 2006;58(3):374–83. [DOI] [PubMed] [Google Scholar]
- 63.Frost C, Nepal S, Wang J, Schuster A, Byon W, Boyd RA, et al. Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor Xa inhibitor, in healthy subjects. Br J Clin Pharmacol. 2013;76(5):776–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yavorski RT, Hallgren SE, Blue PW. Effects of verapamil and diltiazem on gastric emptying in normal subjects. Dig Dis Sci. 1991;36(9):1274–6. [DOI] [PubMed] [Google Scholar]
- 65.Raghavan N, Frost CE, Yu Z, He K, Zhang H, Humphreys WG, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos. 2009;37(1):74–81. [DOI] [PubMed] [Google Scholar]
- 66.Byon W, Garonzik S, Boyd RA, Frost CE. Apixaban: a clinical pharmacokinetic and pharmacodynamic review. Clin Pharmacokinet. 2019;58(10):1265–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prom R, Spinler SA. The role of apixaban for venous and arterial thromboembolic disease. Ann Pharmacother. 2011;45(10):1262–83. [DOI] [PubMed] [Google Scholar]
- 68.Budovich A, Zargarova O, Nogid A. Role of apixaban (eliquis) in the treatment and prevention of thromboembolic disease. Pharm Ther. 2013;38(4):206–31. [PMC free article] [PubMed] [Google Scholar]
- 69.Gong IY, Kim RB. Important of pharmacokinetic profile and variability as determinants of dose and response to dabigatran, rivaroxaban, and apixaban. Can J Cardiol. 2013;29:S24–33. [DOI] [PubMed] [Google Scholar]