

Abstract
Proteoforms contribute functional diversity to the proteome and aberrant proteoforms levels have been implicated in biological dysfunction and disease. Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR MS), with its ultrahigh mass-resolving power, mass accuracy, and versatile tandem mass spectrometry capabilities, has empowered top-down, middle-down, and native MS-based approaches for characterizing proteoforms and their complexes in biological systems. Herein, we review the features which make FT-ICR MS uniquely suited for measuring proteoform mass with ultrahigh resolution and mass accuracy; obtaining in-depth proteoform sequence coverage with expansive tandem MS capabilities; and unambiguously identifying and localizing post-translational and non-covalent modifications. We highlight examples from our body of work in which we have quantified and comprehensively characterized proteoforms from cardiac and skeletal muscle to better understand conditions such as chronic heart failure, acute myocardial infarction, and sarcopenia. Structural characterization of monoclonal antibodies (mAbs) and their proteoforms by FT-ICR MS and emerging applications, such as native top-down FT-ICR MS and high-throughput top-down FT-ICR MS-based proteomics at 21 tesla, are also covered. Historically, the information gleaned from FT-ICR MS analyses have helped provide biological insights. We predict FT-ICR MS will continue to enable the study of proteoforms of increasing size from increasingly complex endogenous mixtures and facilitate the benchmarking of sensitive and specific assays for clinical diagnostics.
Keywords: Proteoform, Post-translational modifications (PTMs), Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR MS), Mass spectrometry (MS), Top-down MS, Tandem mass spectrometry (MS/MS and MSn), Electron capture dissociation (ECD), Activated-ion electron capture dissociation (AI-ECD), Electron-transfer dissociation (ETD), Electrospray ionization (ESI), Stored waveform inverse Fourier transform (SWIFT), Linear ion trap quadrupole (LTQ), Multipole storage device (MSD), Monoclonal antibody (mAb), Middle-down MS, Liquid chromatography-mass spectrometry (LC-MS), Collisional induced dissociation (CID), Infrared-multiphoton dissociation (IRMPD), Cardiac troponin T (cTnT), Collisionally activated dissociation (CAD), Signal-to-noise (S/N), Ultraviolet photodissociation (UVPD), Essential light chain (ELC), Regulatory light chain (RLC), In-source dissociation (ISD)
I. Introduction
Diversity of biological function in our proteome is attributed to proteoforms, or the different forms of a protein that arise from events such as genetic mutation, alternative mRNA splicing, and post-translational modifications (PTMs) (Smith et al., 2013). Since its co-invention by Comisarow and Marshall in 1974 (Marshall et al., 1998), Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR MS) has paved the way for the study of proteoforms, even before they were formally named in 2013 (Bogdanov and Smith, 2005; Smith et al., 2013). One of the most disruptive technologies available for characterizing proteoforms is top-down mass spectrometry (MS), for which FT-ICR MS is an indispensable tool with its ultrahigh mass-resolving power and mass accuracy together with versatile fragmentation capabilities (Breuker et al., 2008). Top-down MS measures the intact mass and abundance of proteoforms and subsequently dissociates them using tandem mass spectrometry (MS/MS or MSn) to determine their primary structure and modifications (Kelleher et al., 1999). The semi-quantitative technique also allows for relative quantification of proteoform abundance within and across biological samples and conditions.
Pre-dating and inspiring the development of the widely used Orbitrap mass spectrometer, FT-ICR MS has been used to provide extremely accurate mass measurements for intact proteins and discern closely-related or overlapping masses in MS and MS/MS spectra (Marshall and Hendrickson, 2008). The community quickly saw the capability of these ultrahigh resolution mass analyzers with increased magnet strength (at the time, 9.4 tesla (T)) to provide unit mass resolution and accurate mass measurements for very large protein molecules, such as the 111- and 112-kDa protein chondroitinase enzymes in 1997 (Kelleher et al., 1997; Senko et al., 1996). The development of stored waveform inverse Fourier transform (SWIFT) technology enabled high resolution isolation of precursor ions, providing the opportunity to selectively isolate single protein isotopologues (Guan and Marshall, 1996; O’Connor and McLafferty, 1995), later opening doors for top-down FT-ICR MS of single proteoforms from complex samples. Early days of top-down MS saw improved sequencing of proteins made possible by the transformative, electron-based fragmentation, electron-capture dissociation (ECD) (Zubarev et al., 1998; Zubarev et al., 1999). In-depth sequence characterization by ECD was limited to smaller proteins until McLafferty and co-workers showed improved dissociation for proteins in the 40-kDa range by disrupting secondary and tertiary protein structure in the gas-phase prior to electron capture (activated-ion ECD; AI-ECD) (Ge et al., 2002; Horn et al., 2000). AI-ECD with FT-ICR MS enabled near-complete sequencing of 29-kDa carbonic anhydrase, such that any PTM could be localized within one residue for 235 of the 258 residues (Sze et al., 2002). Top-down MS was further extended to proteins in the 200-kDa range through the use of electrospray additives and prefolding dissociation of protein molecules following electrospray ionization (ESI) (Han et al., 2006). Hybridization of FT-ICR mass spectrometers with front-end linear ion trap quadrupoles (LTQ), multipole storage devices (MSD), and collision cells afforded opportunities for complex and creative MSn sequences and gas-phase chemistry (Patrie et al., 2004; Schaub et al., 2008; Syka et al., 2004b). Clearly, FT-ICR MS-based technologies hold a special place in the history of top-down MS and continue to enable the comprehensive characterization of proteoforms today.
Several reviews discuss the theory and fundamentals of FT-ICR MS (Hendrickson and Emmett, 1999; Marshall et al., 1998; Nikolaev et al., 2016). Relevant to our present discussion, a review by Bogdanov & Smith comprehensively summarizes the advances in FT-ICR MS with respect to the proteomics field from 1998–2002 (Bogdanov and Smith, 2005). Moreover, there are several recent publications which provide perspective on the technical aspects and applications of top-down MS-based proteomics (Cai et al., 2016b; Chen et al., 2018a; Cupp-Sutton and Wu, 2020; Schaffer et al., 2019; Toby et al., 2016). Here, we specifically review FT-ICR MS applied to the characterization of proteoforms, which are regarded as primary effectors in a majority of biological processes and are considered the next proteomics currency (Smith and Kelleher, 2018). We will first discuss the different approaches available for studying proteoforms in biological systems. Next, we will cover the attributes which make FT-ICR MS uniquely suited for comprehensive characterization of proteoforms. Therein, we will highlight examples from our body of work, in which we have used FT-ICR MS extensively as a tool for studying proteoforms in the sarcomere, which is the basic contractile apparatus in myocytes (i.e. muscle cells), and for which careful control of isoform expression, alternative splicing, and PTMs plays a critical role in regulating cardiac and skeletal muscle function (Cai et al., 2016b; Gregorich et al., 2017; Gregorich and Ge, 2014; Gregorich et al., 2016; Jin et al., 2019a; Jin et al., 2016; Jin et al., 2017; Lin et al., 2018; Peng et al., 2014a; Peng et al., 2013a; Peng et al., 2014b; Peng et al., 2013b; Wei et al., 2018; Zhang et al., 2011b). Alterations to proteoform structure (expression of alternatively-spliced variants, site-specific amino acid changes, and PTMs) have been implicated in heart diseases (Cai et al., 2016b) and cancer (Ntai et al., 2018). Hence, we will further examine how FT-ICR MS has enabled unambiguous identification and localization of PTMs and site-specific amino acid variations in chronic heart failure (Zhang et al., 2011b), acute myocardial infarction (Peng et al., 2014b), and sarcopenia (Gregorich et al., 2016; Jin et al., 2019a; Wei et al., 2018).
We will also review the latest applications of FT-ICR MS for characterizing proteoforms and providing unique insights into living systems. For instance, top-down and middle-down FT-ICR MS has been extensively used to characterize the proteoforms and chemical derivatives of intact monoclonal antibodies (mAbs), which have recently emerged as an important class of therapeutics (Chen et al., 2018b; Jin et al., 2019b). Recently, FT-ICR MS-based assays have been demonstrated for the diagnosis of hemoglobinopathies and plasma cell disorders, representing a path forward for ultrahigh resolution top-down proteomics for the clinic (He et al., 2019a; He et al., 2019b; He et al., 2020). Furthermore, native top-down FT-ICR MS strategies are extending mass limits in analysis of proteoform complexes (up to 1.8 MDa) (Li et al., 2018). We have also witnessed incredible improvements to top-down MS analysis of complex proteoform mixtures, specifically for proteoforms >30 kDa, on a liquid chromatography (LC)-FT-ICR MS timescale with the use of higher magnetic field (21 T) and implementation of an MSD for improved spectral signal-to-noise (S/N) (Anderson et al., 2017).
FT-ICR MS has come a long way since its inception nearly 46 years ago. Even with the wide-spread availability and implementation of Orbitrap mass spectrometers, several academic, industrial, and national labs still own and operate commercial and state-of-the-art FT-ICR mass spectrometers. Notably, two custom-built 21 T FT-ICR mass spectrometers equipped with suites of MS/MS and MSn capabilities at Pacific Northwest National Laboratory (PNNL) (Shaw et al., 2016a) and the National High Magnetic Field Laboratory (NHMFL) (Hendrickson et al., 2015; Weisbrod et al., 2017) have made ultrahigh resolving power and mass accuracy available to users nationally, circumventing the need for individual laboratories to purchase the instruments which are known for being costly to maintain and require specialized training to operate. FT-ICR MS is still a preferred technique for unit mass measurement of large proteoforms and sequence characterization, and we predict, will continue to offer a versatile instrumentation platform for in-depth characterization of proteoforms in the years to come.
II. Approaches for Studying Proteoforms in Biological Systems
The human genome project revealed far fewer genes exist than expected, leading to the realization that much of the functional diversity and complexity observed in biology is represented at the level of proteoforms (Smith et al., 2013). Because proteins transcribed from a single gene can exhibit vastly different properties and functions depending on their DNA-, RNA- and PTM-level variation, it has become clear that studying biological systems at the proteoform-level is critically important in the effort to understand biology and disease (Smith and Kelleher, 2018). Therefore, extensive steps have been taken to benchmark the best practices for intact protein analysis (Donnelly et al., 2019), quantify the number of proteoforms that exist in nature (Aebersold et al., 2018), reduce ambiguity in proteoform nomenclature (LeDuc et al., 2018), and classify proteoforms by level of characterization (Smith et al., 2019).
Proteomics generally aims to identify, quantify, and characterize proteins in living organisms, usually with the goal to better understand biological systems. MS-based proteomic approaches have emerged to facilitate these goals, and can be targeted or discovery-based (Aebersold and Mann, 2003; Aebersold and Mann, 2016). Bottom-up (or shot-gun) proteomics meets this goal through the analysis of peptides resulting from digested protein mixtures (Aebersold and Mann, 2016; Zhang et al., 2013). Methodology for the bottom-up proteomics is well-developed and high-throughput, and more than 340,000 proteins from proteomes across the kingdoms of life have been identified and quantified using this method (Müller et al., 2020; Wilhelm et al., 2014). While this approach offers in-depth proteome coverage at great speed and precision, identity and quantity of proteins are determined indirectly from the resulting peptides and their abundance (Nesvizhskii and Aebersold, 2005). Because the mass of intact proteoforms are never directly measured, and peptide fragments originating from closely-related proteoforms are identified and quantified together, proteoform-level information is lost in bottom-up proteomics. Conversely, the top-down MS approach (i.e. top-down proteomics) measures intact proteoform mass and abundance directly (Chait, 2006; Chen et al., 2018a; Kelleher et al., 1999; Toby et al., 2016). Following intact mass measurement, whole proteoforms are subjected to dissociation by MS/MS. Fragment ions can then be directly associated with precursor proteoform masses, allowing for identification of combinatorial amino acid sequence variants and PTMs (Siuti and Kelleher, 2007; Zhang and Ge, 2011). With precise information about proteoform identity and abundance, one can compare proteoform levels in different cell or tissue types, or across different biological and disease states (Cai et al., 2016b; Gregorich and Ge, 2014; Schaffer et al., 2019). Importantly, this presents an opportunity to understand molecular differences between cells and tissues, discover cellular pathways which are perturbed in disease, and identify proteoform-specific biomarkers.
While top-down MS is useful for studying intact proteoforms, the analysis of larger proteoforms and those with multiple co-existing PTMs is technically challenging. Thus, middle-down MS has emerged to ease the challenge of characterizing larger proteoforms and those with extensive modification (Cannon et al., 2010; Chen et al., 2018b; Garcia, 2010a; Jin et al., 2017; Wu et al., 2012). In contrast to the bottom-up approach, middle-down MS employs enzymes such as Asp-N and Glu-C, which produce large peptides (i.e. polypeptides), which have more co-existing PTMs, preserving some proteoform-level information. In contrast to top-down proteomics, the analysis of polypeptides vs. intact proteoforms results in improved MS signal-to-noise ratio, and enhanced coverage of sequence, especially for proteins >100 kDa and mAbs (Fornelli et al., 2014; Jin et al., 2019b; Jin et al., 2017). For this reason, the middle-down approach can be used to analyze incredibly complex proteoforms with multiple PTMs (e.g. histones) (Sidoli and Garcia, 2017; Sidoli et al., 2015) and can serve as a supplement to top-down MS for comprehensive characterization of very large proteoforms.
III. Comprehensive Characterization of Proteoforms with FT-ICR MS
Comprehensively characterizing proteoforms from living systems relies on 1) the measurement of intact proteoform mass with high precision and accuracy and 2) subsequent acquisition of detailed and accurate information about primary structure (e.g. amino acid sequence and PTMs) using MS/MS. Because proteins fragment in a predictable way, amino acid sequences can be determined de novo, and fragments derived from dissociation of these biomolecules can be compared to DNA- or RNA-predicted sequences. Mass deviations in the experimentally-measured precursor and fragment ion masses compared to predicted protein sequence provide evidence for PTMs and amino acid variations. Highly accurate mass measurements at the MS- and MS/MS-level, together with high sequence coverage, enhance our ability to confidently assign proteoform identities and to site-specifically localize PTMs and variations to the predicted amino acid sequence. As we will discuss, relative abundance of proteoforms and PTMs is also important to consider when comparing biological and disease states.
Ultrahigh resolving power and mass accuracy together with versatile MSn capabilities make FT-ICR MS uniquely suited for characterization of proteoforms. Compared to time-of-flight (TOF) mass spectrometers, which are favored for analysis of proteoform complexes due to their effectively unlimited mass range (Mehmood et al., 2015), FT-ICR mass spectrometers can achieve much higher mass resolution (>1M compared to ~100,000) and the use of these instruments for analysis of megadalton proteoform complexes has been recently realized (vide infra) (Li et al., 2018; Shaw et al., 2016a). Additionally, the non-destructive nature of detection in the ICR cell permits measurement and manipulation of ions in the gas-phase for longer periods of time, compared to TOF MS, in which ions are destroyed at the point of detection. Because ions can be monitored for a longer time in the ICR cell, extremely accurate mass measurements can be carried out (Marshall et al., 1998). A major drawback to FT-ICR MS is the high cost to purchase and to maintain the instruments, which contain superconducting magnets and require liquid helium for super-cooling. Orbitraps mass spectrometers now offer high resolution and mass accuracy at a lower cost, and are favored for large-scale top-down proteomics due to their fast scan rate and sensitivity (Cleland et al., 2017; Michalski et al., 2012). Until recently, Orbitrap MS was limited to analysis of proteoforms <30 kDa in molecular weight, and FT-ICR MS is still preferred for targeted characterization of proteoform primary structure and for acquisition of unit mass resolution measurement of intact proteins >100 kDa (Shaw et al., 2016a).
Of note, top-down FT-ICR MS, specifically using ECD, is also well-suited for localizing non-covalent protein modifications, such as metal atoms and ligands. Since ECD preferentially cleaves covalent bonds in the protein backbone, non-covalent interactions are preserved and metal- or ligand-bound fragment ions can be identified, as has been demonstrated for ligands such as spermine, ATP, and platinum anti-cancer drugs (Li et al., 2011; Xie et al., 2006; Yin and Loo, 2010).
a. Measuring Proteoform Mass and Resolving Complex Spectra with Ultrahigh Resolving Power and Accuracy
Marshall et al. describe mass spectral resolution to progress in a series of plateaus, such that as instrumental resolving power increases, we can advance from resolving ions of the same mass but different charge (charge state resolution) to resolving ions of the same nominal mass but different elemental composition (isotopic fine structure) (2002). Ultrahigh mass-resolving power and accuracy are important features which distinguish FT-ICR mass spectrometers from other mass analyzers (Marshall and Hendrickson, 2008). Not only is high mass-resolving power necessary to differentiate closely-related proteoforms from complex mixtures (further discussed in Section IV), but also for obtaining isotopic (i.e. unit mass) resolution of the highly-charged ions generated by ESI of proteins. Large proteoforms often carry more charges. Because the distance between isotopologues is the inverse of the protein ion charge (1/z), increasing resolving power is required with increasing proteoform molecular weight (MW). Mass-resolving power and accuracy increase linearly and quadratically with increasing magnet field strength, respectively, thus analysis of proteoforms can be improved simply by increasing magnetic strength (Karabacak et al., 2010; Marshall and Guan, 1996). Using a 9.4 T hybrid FT-ICR MS with modifications which improved sensitivity (Kaiser et al., 2011), Marshall and co-workers set the record for highest protein unit mass baseline resolved in 2011, achieving baseline unit mass resolution for the 57+ charge state of a 148-kDa mAb (m/Δm50% = 420,000) in magnitude mode (Valeja et al., 2011). Prior to this achievement, the largest proteoform unit mass resolved was the 115-kDa truncated form of cardiac myosin binding protein C at 7 T (Ge et al., 2009). FT-ICR MS has unique advantages for unit mass resolution of mAbs and their associated glycoforms and derivatives (Jin et al., 2019b; Shaw et al., 2016a; Valeja et al., 2011). Impressively, m/Δm50% = 980,000 was achieved for a 150-kDa mAb glycoform in absorption mode with the 21 T FT-ICR mass spectrometer at PNNL (Shaw et al., 2016a). Measurement of proteoform complexes at unit mass resolution with FT-ICR MS has also been demonstrated for complexes up to 158 kDa at 15 T, allowing for precise determination of metal-binding stoichiometry (Li et al., 2014a; Li et al., 2014b).
Considering the number of possible cleavage events increases with increasing proteoform size, the complexity of MS/MS spectra produced by fragmentation of these molecules also increases, requiring high mass-resolving power to differentiate overlapping fragment ions. Furthermore, precursor and fragment ions tend to condense in a small region of the m/z space (500–2000 m/z) for denatured top-down MS of proteins (Ge et al., 2002; Ge et al., 2009; Horn et al., 2000; Meng et al., 2001). Therefore, several techniques which utilize gas-phase reactions have been developed to circumvent overlapping fragment ions, such as ion-ion proton transfer reactions and ion parking (Chrisman et al., 2006; McLuckey et al., 2002; Stephenson and McLuckey, 1996). Ultrahigh mass-resolving power at 21 T, as expected, significantly improved differentiation of closely-clustered electron-transfer dissociation (ETD) fragment ions for the near-complete sequence characterization of carbonic anhydrase at NHMFL (Weisbrod et al., 2017).
b. Obtaining In-depth Proteoform Sequence Coverage
Top-down MS is uniquely capable of providing complete or near-complete proteoform sequence coverage, lending to the localization of amino acid sequence variants, chemical modifications, and PTMs along the protein backbone (Lin et al., 2018; Sze et al., 2002). Higher sequence coverage is achievable in large-part owing to the development of non-ergodic, electron-based dissociation methods, which typically result in more cleavage events than ergodic, energy-based (or vibrational) dissociation (Coon et al., 2005; Syka et al., 2004a; Zubarev et al., 2000; Zubarev et al., 1998). However, vibrational ion activation strategies such as prefolding dissociation (a variation of nozzle-skimmer dissociation), collisionally activated dissociation (CAD), and infrared multiphoton dissociation (IRMPD) have been used to improve electron-based dissociation efficiency by disrupting inter- and intra-molecular interactions in the gas-phase (Bourgoin-Voillard et al., 2014; Ge et al., 2003; Han et al., 2006; Horn et al., 2000; Riley et al., 2015; Zhai et al., 2005). Combining sequence information obtained from MS/MS experiments performed using multiple dissociation strategies has also increased sequence coverage achievable by top-down MS (Gregorich et al., 2017; Lin et al., 2018). While combining information obtained in multiple experiments is useful for achieving high sequence coverage, drawbacks include increased sample consumption and number of MS experiments. Increased number of fragment ions also increases false discovery rate for their identification. For this reason, manual validation of experimental-theoretical fragment ion matches is often a requirement (Cai et al., 2016a).
Arguably one of the most transformative developments for top-down MS characterization of proteoforms was ECD, which was first introduced by McLafferty and co-workers in 1998 and was exclusively available on the FT-ICR mass spectrometer (Zubarev et al., 1998). The advent of ECD provided an electron-based strategy for dissociating highly-charged molecular ions (Zubarev et al., 2000; Zubarev et al., 1998). ECD rapidly and randomly produces fragments by cleavage of N-Cα bonds in the protein backbone after capture of low-energy electrons by multiply-charged protein molecules (Zubarev et al., 2000). This dissociation method can cleave disulfide bonds, while preserving labile PTMs cleaved preferentially by energy-based dissociation methods, allowing site-specific localization of amino acid variations and PTMs (Siuti and Kelleher, 2007). Shortly after ECD was introduced, electron transfer dissociation (ETD) was developed by Hunt and co-workers, which also produces N-Cα bond dissociation and could be used in ion traps and hybridized with FT-ICR MS (Syka et al., 2004b; Weisbrod et al., 2017).
Energy- and electron-based dissociation methods provide complementary information and can be used in tandem or concurrently to improve fragmentation efficiency and comprehensively characterize proteoforms. The first demonstrations of AI-ECD resulted in improved sequence coverage of larger proteins up to 45-kDa (Ge et al., 2002; Horn et al., 2000). Denaturing of the precursor ions in the gas-phase disrupted non-covalent protein interactions, yielding increased number of backbone cleavages with ECD and improved sequence coverage. McLafferty and co-workers achieved 250/258 cleavages of the 29-kDa carbonic anhydrase and allowed characterization of any possible phosphorylation site within one residue, further demonstrating the capability of top-down proteomics to site-specifically discern mass deviations (Sze et al., 2002). Han et al. extended the previous 50-kDa mass limit for top-down FT-ICR MS to 200-kDa through use of ESI additives and prefolding dissociation to break non-covalent inter- and intra-molecular interactions prior to ECD in the gas-phase (Han et al., 2006). Riley et al. recently applied this concept using IRMPD and ETD concurrently for improved sequence coverage of intact proteins in a modified Orbitrap mass spectrometer (Riley et al., 2015). Collectively, these reports show that the collisional or energy-based activation of proteoforms in the gas-phase before, during, or after electron-based dissociation can significantly increase fragmentation efficiency toward improved coverage of proteoform sequence.
Today’s commercial FT-ICR mass spectrometers come equipped with diverse arrays of fragmentation tools which can be combined to perform desired MSn experiments. Costello and co-workers systematically explored the complementarity of fragmentation capabilities available on a Bruker 12 T SolariX Qh/FT-ICR mass spectrometer for top-down MS of glycoproteins (Bourgoin-Voillard et al., 2014). For the glycosylated RNAse B and its non-glycosylated analog, RNAse A, they sought to investigate differences in fragmentation using ECD and ETD with and without pre-collisional induced dissociation (CID) or post- IRMPD activation (Bourgoin-Voillard et al., 2014). Overall, they compared sequence coverage with pre-CID and post-IRMPD to ECD and ETD alone. Any one experiment did not exceed 71% and 69% sequence coverage for RNAse A and RNAse B, respectively; however, combining data for different vibrational and electron-based activation sequences resulted in 90% and 86% sequence coverage for RNAse A and RNAse B, respectively. They also observed >98% retention of the glycan moiety with ECD and ETD, allowing for unambiguous localization of the high-mannose glycosylation site to Asn34. These studies exemplify how versatility in MS/MS experimentation coupled with FT-ICR mass spectrometry can significantly aid in proteoform sequence characterization and confident mapping of PTMs.
Gregorich et al. found use for the complementarity of ECD and CAD in the top-down FT-ICR MS sequencing of the ventricular and atrial isoforms of the heart protein essential light chain (ELC), which contain actin-binding proline-rich N-termini (Gregorich et al., 2017). ECD cannot cleave the N-terminal to proline due to its cyclic structure, whereas this bond cleavage is favored for CID (Schaaff et al., 2000; Zubarev et al., 2000). By combining spectral information from separate dissociation experiments, they achieved 95% and 90% sequence coverage for the atrial and ventricular ELC isoforms (~21 kDa), respectively. Lin et al. further demonstrated this concept for the deep sequencing of the 35-kDa swine cardiac troponin T (cTnT) using a combination of CAD and ECD experiments acquired through direct infusion with nano-ESI interfaced with a 12 T solariX XR FT-ICR mass spectrometer (Lin et al., 2018). In this work, ECD spectra were acquired with both low- and high- electron energy and the sequence information from multiple dissociation experiments combined to obtain complementary sequence coverage. They found that a low electron energy (0.32 V) enabled detection of larger c-ions (10-30 kDa) compared to a higher electron energy (0.80 V), for which c-ions were much less prevalent (Figure 1A). Use of higher electron energy provided more z•-ions and higher coverage of the C-terminus. Complimentarily, CAD experiments at different collisional energies yielded b- and y-ions and covered cleavages at the N-terminus, which were frequently under-represented in the ECD spectra. They achieved 87% sequence coverage for the 35.2-kDa mono-phosphorylated cTnT1 proteoform and localized the phosphorylation site to Ser-1 after combination of data from 21 offline CAD experiments, 20 offline ECD experiments (up to 2000 summed transients, ~2.2 s transient length), and online LC-quadrupole-time-of-flight (Q-TOF) MS/MS(Figure 1B). Use of direct infusion FT-ICR MS/MS allowed for more transients to be summed which improves S/N of fragment ions and increases sequence coverage and confidence of fragment ion assignments.
Figure 1|. Deep top-down FT-ICR MS sequencing of 35-kDa swine cardiac troponin T using combined data from complementary dissociation strategies.
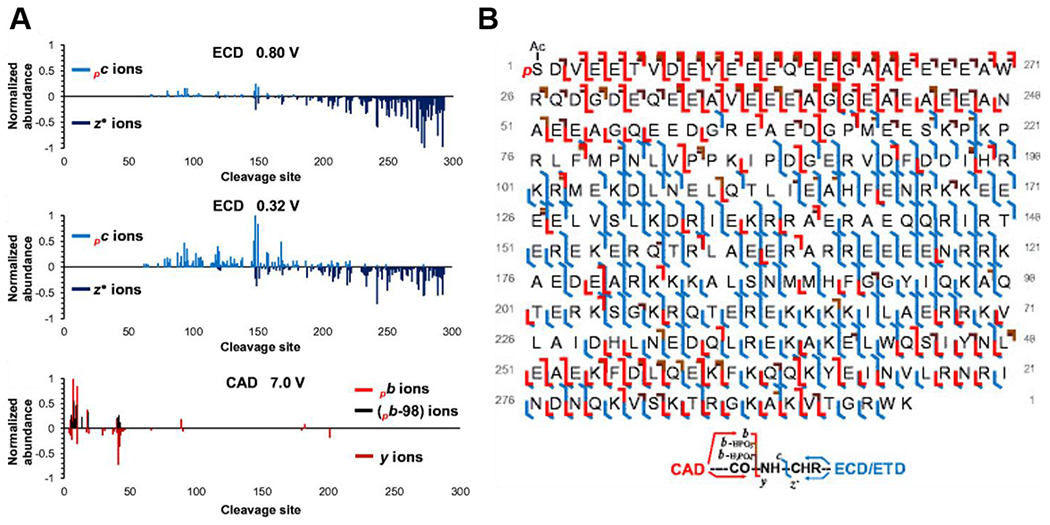
A) cTnT1 cleavage site vs. normalized fragment ion abundance for ECD with 0.8 V and 0.32 V electron energies (2000 transients summed) and representative CAD with 7.0 V collisional energy (300 transients summed). B) Sequence table for mono-phosphorylated swine cTnT1 showing b-, y-, c- and z•- fragment ions from combined offline CAD, offline ECD, and online LC-Q-TOF MS/MS data, with 87% sequence coverage. Reprinted and adapted with permission from Lin, Z., Guo, F., Gregorich, Z.R., Sun, R., Zhang, H., Hu, Y., Shanmuganayagam, D., Ge, Y. J Am Soc Mass Spectrom. 2018. 29, 1284-1294 (Lin et al., 2018). Copyright © 2018 American Chemical Society.
Typically, obtaining comprehensive sequence coverage for proteoforms requires compiling data from multiple MS/MS experiments performed using complementary dissociation strategies. However, Weisbrod et al. obtained 87% sequence coverage for carbonic anhydrase using 21 T FT-ICR MS with front-end ETD (94% if proline cleavages are disregarded) in a single ETD spectrum with 1500 transients acquired (Weisbrod et al., 2017). They achieved even coverage for both the N- and C-termini of the protein by using multiple fragment ion fills in the MSD to improve signal-to-noise (S/N) and short ETD reaction periods to increase the number of large fragment ions detected. Additionally, the high dynamic range and resolving power afforded at 21 T aided in this feat.
More opportunities will arise for in-depth proteoform characterization as new dissociation strategies become available. Recently, ultraviolet photo dissociation (UVPD), which implements ultraviolet light (e.g. 193 nm) to extensively fragment and sequence peptides and proteins, has been implemented on the 15 T FT-ICR mass spectrometer at PNNL (Shaw et al., 2016b) and the 21 T FT-ICR mass spectrometer at NHMFL (Smith et al., 2020), and is promising for increasing our ability to characterize proteoforms with top-down FT-ICR MS. The UVPD technique coupled to with FT-ICR MS for in-depth sequencing of peptides dates back to 1987 (Hunt et al., 1987). This pairing was briefly explored for sequencing of ubiquitin (8.6 kDa) in the early days of top-down FT-ICR MS (Guan et al., 1996), and led to the serendipitous discovery of ECD (Zubarev et al., 1998). Top-down FT-ICR MS of intact proteoforms using UVPD was revisited after the complete sequencing of intact proteins up to 29 kDa was demonstrated using UVPD on an Orbitrap mass spectrometer (Shaw et al., 2013; Shaw et al., 2016b).
Middle-down MS, which employs limited digestion, has been utilized for comprehensive sequencing of larger proteoforms, especially those with MW >200 kDa. The middle-down approach allows for interrogation of polypeptides (~3–20 kDa) of proteoforms without losing the information that is lost by full digestion of the proteoforms in the bottom-up approach (Garcia, 2010b). The middle-down approach can be complementary to the top-down approach, lending an increased ability to sequence the middle regions of larger proteoforms and to site-localize PTMs. Because there are fewer cleavage sites for polypeptides compared to the intact proteins from which they are derived, fewer fragment ions are produced. Fragment ions generated from smaller precursor ions also exhibit higher S/N and, therefore, more sequence information can be gleaned from the MS/MS spectra. Jin et al. employed limited digestion and middle-down FT-ICR MS to characterize the 223-kDa human heart protein, cardiac myosin heavy chain, an important component of the molecular motor which fuels muscle contraction (Jin et al., 2017). Using Glu-C, Asp-N, and trypsin limited digestion, they completely characterized the sequence of β-MHC with high mass accuracy (Figure 2A) and localized N-terminal Gly1 acetylation, Lys34 methylation, and Lys129 trimethylation using ECD (Figure 2B). Middle-down FT-ICR MS also proved to be incredibly useful for characterizing the branching of ubiquitylated proteoforms (Valkevich et al., 2014).
Figure 2|. Middle-down FT-ICR MS characterization of 223-kDa human cardiac myosin heavy chain using combined limited digestion strategies.
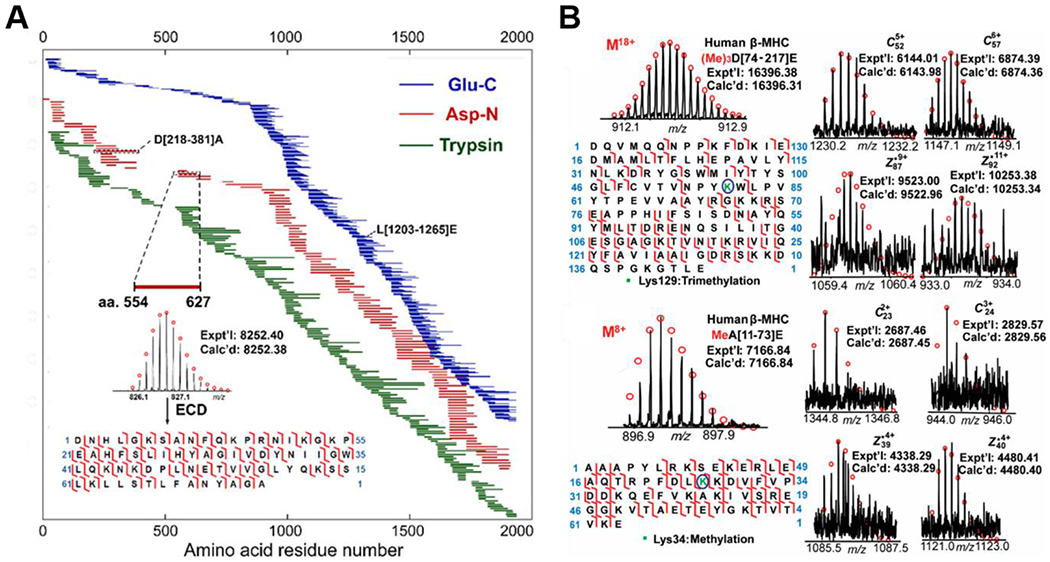
A) Schematic showing complete sequence coverage for human β-MHC achieved by middle-down MS with limited digestion with Glu-C (blue), Asp-N (red), and trypsin (green). Peptide mapping is based on accurate mass measurements and MS/MS experiments B) Localization of trimethylation (Lys129) on polypeptide D[74-217] and methylation (Lys34) on polypeptide A[11-73] from Glu-C digestion of human β-MHC. Reprinted and adapted with permission from Jin, Y., Wei, L., Cai, W., Lin, Z., Wu, Z., Peng, Y., Kohmoto, T., Moss, R., and Ge, Y. Anal. Chem. 2017. 89, 4922-4930 (Jin et al., 2017). Copyright © 2017 American Chemical Society.
c. Identifying, Localizing, and Quantifying Post-translational Modifications
Covalent protein modifications, such as methylation, acetylation, phosphorylation, glycosylation, and ubiquitylation are critical regulators of protein activity and function, playing a crucial role in cellular processes such as gene expression, signaling, and protein degradation (Biggar and Li, 2015; Deribe et al., 2010; Grimes et al., 2018; Jenuwein and Allis, 2001; Karve and Cheema, 2011; Mann and Jensen, 2003; Moremen et al., 2012; Olsen et al., 2006; Webb and Bennett, 2013; Xu and Jaffrey, 2011). Furthermore, unusual modifications such as phosphoglycerolation, S-thiolation, O-mycoloylation have been implicated as key features regulating bacterial infection (Ansong et al., 2013; Carel et al., 2017; Chamot-Rooke et al., 2011). Moreover, altered PTM levels have been associated with cancer and heart diseases, presenting opportunities to discover proteoform biomarkers for diagnosis (An et al., 2009; Chakouri et al., 2018; Krueger and Srivastava, 2006; Mazur et al., 2010; Ntai et al., 2018; Ntai et al., 2016; Peng et al., 2014b; Zhang et al., 2011b). For these reasons, it is not only important to correctly identify and localize PTMs, but also to quantify the proteoform abundance, a unique facet of top-down MS (Siuti and Kelleher, 2007; Zhang and Ge, 2011).
Ultrahigh resolution FT-ICR MS is useful for precisely identifying and locating low-mass modifications such as disulfide bonds (−2.016 Da) or deamidation (+0.984 Da). Peng et al. used top-down FT-ICR MS to characterize the 56-kDa human salivary α-amylase, precisely mapping 5 disulfide bonds (Peng et al., 2012). Initially, they used ECD to identify a mass deviation from the DNA-predicted sequence as a 15-amino acid truncation of the N-terminus and formation of N-terminal pyroglutamic acid. Importantly, they used complete and partial reduction of salivary α-amylase combined with high-resolution 7 T LTQ/FT-ICR MS to determine the participating cysteine residues for 5 disulfide bonds (contributing an overall mass deviation of -10.08 Da). Disulfide bonds are an important PTM which contribute to native proteoform folding and structure. However, it is widely recognized that disulfide bonds are difficult to analyze due to the poor fragmentation efficiency by top-down MS. Loo and co-workers employed the super-charging reagent sulfolane to improve the fragmentation of proteins with multiple intra-molecular disulfide bonds with FT-ICR MS and CAD and ECD (Zhang et al., 2015). By increasing the charge states of the disulfide-bonded proteins, they improved top-down MS characterization of this important PTM.
FT-ICR MS is also uniquely suited for differentiating PTMs which are close in mass, such as phosphorylation (79.966 Da) vs. sulfation (79.957 Da) and acetylation (42.011 Da) vs. trimethylation (42.047 Da). Gregorich et al. uncovered regional proteoform differences for essential light chain (ELC) and regulatory light chain (RLC) in the ventricle and atrium of the human heart using top-down FT-ICR MS with ECD (Gregorich et al., 2017). Importantly, ultrahigh resolving power and mass accuracy allowed for differentiation of Nα-trimethylation and Nα-acetylation for the RLC isoforms. Specifically, swine ventricular RLC isoform (gene MYL2) is known to be N-terminally trimethylated. However, they found that the swine atrial RLC isoform (gene MYL7) is N-terminally acetylated, resolving the 0.036 Da difference between the two PTM identities (Figure 3). In addition, they unambiguously localized the phosphorylation sites for swine atrial RLC and ventricular RLC to Ser22 and Ser14, respectively.
Figure 3|. Top-down FT-ICR MS distinguishes PTMs closely related in mass.
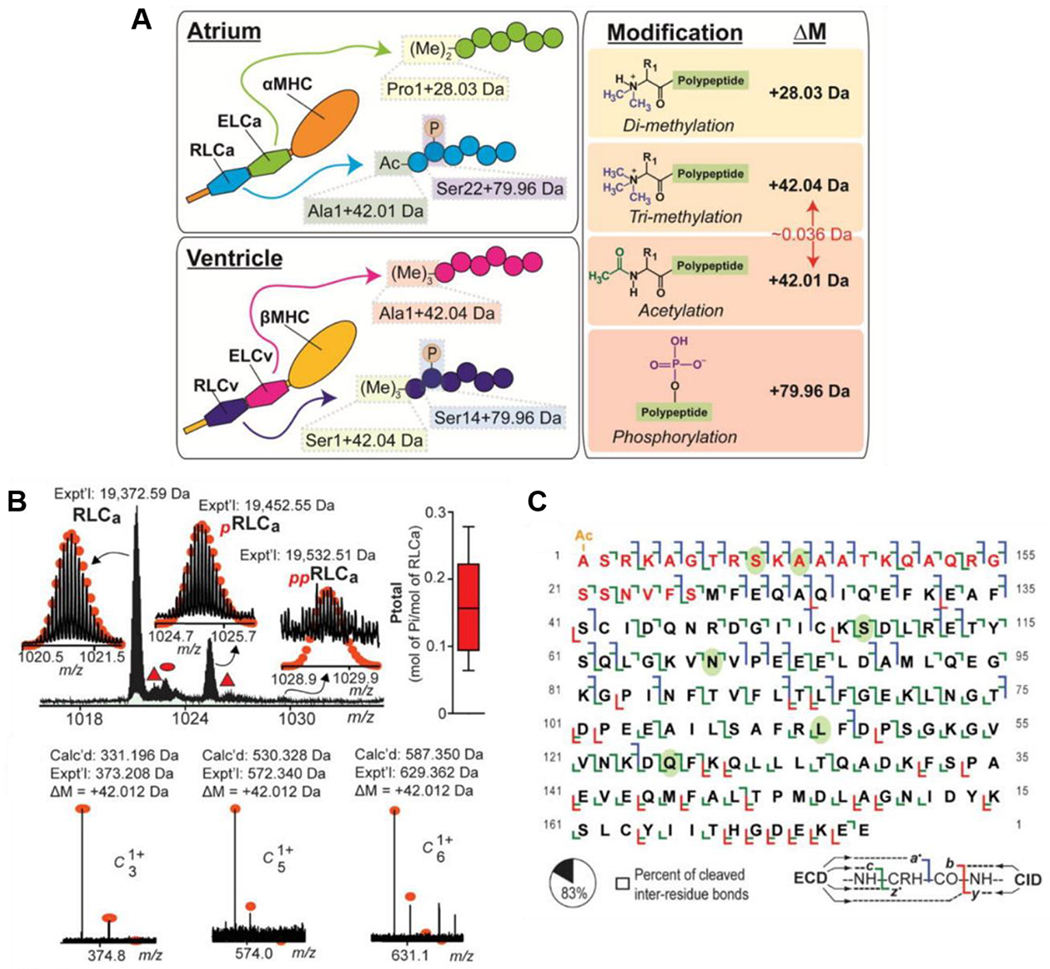
A) Schematic representation of PTM differences found in atrial and ventricular ELC and RLC isoforms B) (Top left) Intact mass spectra of swine atrial RLC (RLCa) proteoforms with experimental mass. Mono-phosphorylation and bis-phosphorylation are denoted by red “p” and “pp” respectively. (Top right) MS-based quantification of total RLCa phosphorylation, represented as mol of inorganic phosphate (Pi)/mol of RLCa with n=5 biological replicates. (Bottom) Representative c-ions showing the +42.010 Da mass deviation of the experimental (Expt’l) compared to calculated (Calc’d) fragment ion masses. C) Sequence table with bond cleavages for RLCa, with 83% of inter-residue bonds cleaved. Red residues deviated from the sequence available in the UniProtKB/Swiss-Prot database and were correctly determined at the time of study. Green ovals indicate residues that differ between human and swine RLCa. Reprinted from J Mol Cell Cardiol, 107, Zachery R. Gregorich, Wenxuan Cai, Ziqing Lin, Albert J. Chen, Ying Peng, Takushi Kohmoto, and Ying Ge, Distinct Sequences and Post-translational Modifications in Cardiac Atrial and Ventricular Myosin Light Chains Revealed by Top-down Mass Spectrometry, 13-21, Copyright © 2017, with permission from Elsevier.
Changes to the PTM status of proteoforms can be important biomarkers of disease, as was shown by Zhang et al. with regard to cardiac troponin I (cTnI) phosphorylation in chronic heart failure (Zhang et al., 2011b). The study, which was the first of its kind, used quantitative top-down proteomics to analyze clinical myocardium tissue samples obtained from 36 human hearts at varying stages of disease, ranging from normal cardiac function to chronic heart failure. Total phosphorylation (%Ptot) of cTnI was calculated by taking the summed abundance of all the phosphorylated cTnI proteoforms relative to the summed abundance of all cTnI proteoforms. Post-mortem hearts with normal cardiac function had a %Ptot of 56.4 +/− 3.5% (n=7) whereas hearts with end-stage chronic heart failure had a %Ptot of 1.0 +/− 0.6% (n=6). Ultimately, they concluded that total phosphorylation of cTnI decreases in the myocardium with various severity of heart diseases compared to hearts with normal cardiac function. The changing phosphorylation sites were unambiguously localized to Ser 22/23 using FT-ICR ECD MS/MS, and the sites were determined to be sequentially phosphorylated/dephosphorylated. Furthermore, since Ser 22/23 are well known PKA-mediated sites, this supports dysregulation of phosphorylation of cTnI by PKA in the disease phenotypes.
In heart diseases, it is well established that sarcomeric protein PTMs regulate cardiac contraction and relaxation (Chakouri et al., 2018; Solaro, 2008; Solaro and Kobayashi, 2011). Furthermore, different abundance of isoforms or alternative-splicing variants play a role in contractile function. Peng et al. used online LC/FT-ICR MS to quantify and characterize the proteoforms constituting the cardiac sarcomere (i.e. the basic contractile apparatus in cardiomyocytes) in a swine model for acute myocardial infarction (AMI), or heart attack (Peng et al., 2014b). Phosphorylation levels of the sarcomeric proteins were quantified by calculating the abundance of phosphorylated proteoforms relative to the summed abundance of all proteoforms (Figure 4A). Tissues analyzed from the infarcted swine hearts showed a concerted decrease in phosphorylation compared to non-infarcted control hearts. Specifically, this trend was observed for cTnI and an unknown 25-kDa protein, which was later identified by FT-ICR ECD MS/MS as enigma homolog isoform 2 (ENH2), which was not previously reported to be a phosphoprotein (Figure 4B–C). Using offline ECD, phosphorylation sites were mapped to Ser 22/23 and Ser118 for cTnI and ENH2, respectively, with an amino acid polymorphism Ser72Cys localized for the ENH2 (Figure 4D–E).
Figure 4|. Relative quantitation of swine cardiac troponin I and enigma homolog phosphorylation in myocardial infarction and subsequent characterization by top-down FT-ICR MS.

A) Relative abundance of sarcomeric proteoforms in AMI and Con. B-C) Representative high resolution mass spectra for cTnI (B) and ENH2 (C) with insets showing a zoom of the most abundant proteoforms and corresponding experimental (Exp) and calculated (Calc) masses for control (Con) and acute myocardial infarction (AMI) swine myocardium. D-E) Sequence table with ECD bond cleavages for cTnI (D) and ENH2 (E) Acetylation sites are denoted by red “Ac”, phosphorylation sites are denoted with yellow circles and red “p”, and sequence variants are denoted by green stars with red font. This research was originally published in Molecular and Cellular Proteomics. Ying Peng, Zachery R. Gregorich, Santosh G. Valeja, Han Zhang, Wenxuan Cai, Yi-Chen Chen, Huseyin Guner, Albert J. Chen, Denise J. Schwahn, Timothy A. Hacker, Xiaowen Liu and Ying Ge. Top-down Proteomics Reveals Concerted Reductions in Myofilament and Z-disc Protein Phosphorylation after Acute Myocardial Infarction. Mol Cell Proteomics. 2014. 13, 2752-2764 (Peng et al., 2014b). Copyright © the American Society for Biochemistry and Molecular Biology.
Sarcopenia, a disease associated with age-related loss of muscle function and strength, plagues the aging population (Cohen et al., 2015; Rolland et al., 2008; Ryall et al., 2008). Quantitative top-down proteomics has been used as a tool to understand this disease at the proteoform-level (Gregorich et al., 2016; Jin et al., 2019a; Wei et al., 2018). Gregorich et al. uncovered significant decrease in phosphorylation of the fast-twitch skeletal isoform of myosin regulatory light chain (RLC) in the skeletal muscle of aging rats (Gregorich et al., 2016). They localized the phosphorylation sites to Ser14/15 using ECD MS/MS, associating this change to age-related contractile impairment using accompanying single-fiber functional measurements. Furthermore, significant age-related proteoform changes in the fast- and slow-twitch skeletal muscles for aging rats were reported by Wei et al., including novel age-related phosphorylation changes for the important Z-disc protein, cypher (Wei et al., 2018). Specifically, they determined that phosphorylation for cypher 4s and cypher 2s significantly decreased in the fast-twitch muscles of aging rats, but observed no change for the phosphorylation of the proteins in the slow-twitch muscles. Protein characterization and PTM localization for the newly identified proteoforms from fast- and slow-twitch skeletal muscle in rats from different age groups was performed using a 7 T LTQ/FT-ICR mass spectrometer. This study included identification and characterization of a novel isoform of the sarcomeric Z-disc protein, enigma. Again, the ultrahigh resolution afforded by FT-ICR was used to differentiate between acetylation and trimethylation, which are common N-terminal protein modifications for sarcomeric proteins. Sarcopenia-related proteoform changes were further investigated in the skeletal muscles of non-human primates by Jin et al., also utilizing the ultrahigh resolution of top-down FT-ICR MS (Jin et al., 2019a).
Quantitative top-down FT-ICR MS has also been used to assess proteoform heterogeneity in cardiac tissues. Gregorich et al. used online LC/FT-ICR MS and offline FT-ICR MS/MS to investigate basal regional heterogeneity in swine hearts (Gregorich et al., 2015). Interestingly, they found that levels of phosphorylation for cTnI were significantly higher in ventricular regions of the heart compared to the atrial regions, whereas phosphorylation of α-tropomyosin (α-Tpm) was found to be higher in the atrial regions compared to the ventricular regions.
IV. Analysis of Complex Proteoform Mixtures
In many cases, the proteoforms we aim to measure come from endogenous protein mixtures such as cell- or tissue-lysates (Aebersold et al., 2018; Brown et al., 2019; Cai et al., 2016b; Smith and Kelleher, 2018; Smith et al., 2013). Proteoforms which are introduced into the mass spectrometer together must be resolved from each other to be identified and quantified as separate features. Several groups have utilized FT-ICR MS resolving power to separate and measure mixtures of proteoforms. Ge et al. showed that FT-ICR MS was an effective complement to liquid chromatography through top-down proteomic analysis of a complex mixture of E. coli proteins (2-30 kDa) (Ge et al., 2002). They also used this strategy to analyze a complex mixture of secreted proteins from Mycobacterium tuberculosis, measuring 689 distinct molecular ions from three mixtures (Ge et al., 2003). Zabrouskov et al. utilized a technique which involved front-end SEC fractionation followed by direct ESI-top-down FT-ICR MS for plant proteomics (Zabrouskov et al., 2003). Meng et al. utilized extensive proteome fractionation of S. cerevisiae using continuous elution electrophoresis and RPLC followed by direct infusion analysis by FT-ICR MS (Meng et al., 2002). Extending this idea to proteoforms in the range of 30-80 kDa, Tucholski et al. followed up with an MS-compatible serial size exclusion strategy, previously shown to dramatically improve detection of proteoforms >60 kDa when interfaced RPLC with Q-TOF mass spectrometry (Cai et al., 2017), to fractionate and directly analyze a complex mixture of cytosolic proteins extracted from heart tissue. They isotopically resolved 31 distinct proteoforms between 30-50 kDa in just a 100 m/z window and identified metabolic enzymes by accurate intact mass (Tucholski et al., 2019). Notably, 6 isotopic peak clusters in this mass range were resolved in just a 5 m/z window (Figure 5).
Figure 5|. sSEC fractionation and FT-ICR MS of metabolic enzymes from complex mixture of cytosolic proteins.
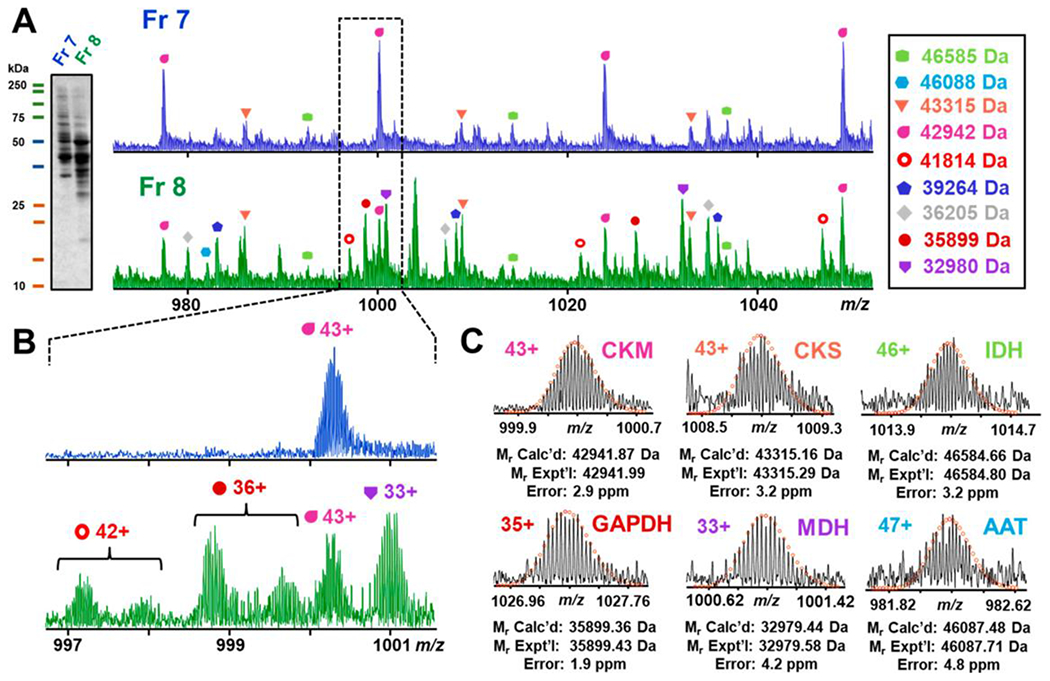
A) SDS-PAGE visualization of fractions 7 and 8 with corresponding FT-ICR mass spectra. B) Enlarged mass spectra of 997-1001 m/z for fractions 7 and 8 showing the difference between the mass spectra of the two fractions. C) Proteins detected by intact mass from top left to bottom right: creatine kinase, M-type (CKM); creatine kinase, S-type (CKS); isocitrate dehydrogenase (IDH); glyceraldehyde-3-phosphate dehydrogenase (GAPDH); malate dehydrogenase, mitochondrial (MDH); aspartate aminotransferase (AAT). Reprinted and adapted with permission from Tucholski, T., Knott, S., Chen, B., Pistono, P., Lin, Z., Ge, Y. Anal. Chem., 2019. 91, 3835-3844 (Tucholski et al., 2019). Copyright © 2019 American Chemical Society.
Nevertheless, given the vast number of proteoforms that exist in biological systems and the complexity of most samples obtained from cell or tissue lysates, LC-based separation prior to top-down or middle-down FT-ICR MS is often required to achieve high proteome coverage. Several large-scale top-down proteomics experiments covering human and yeast proteomes using nanoLC LTQ-FT-ICR MS (Anderson et al., 2017; Kellie et al., 2012; Lee et al., 2009; Lee et al., 2002; Parks et al., 2007; Roth et al., 2008; Tipton et al., 2012; Tran et al., 2011; Wu et al., 2009) and even capillary zone electrophoresis (CZE) LTQ-FT-ICR MS (Li et al., 2014c) have been reported recently, compared to relatively few articles of this type published before the time of the 2005 Smith & Bogdanov review (Bogdanov and Smith, 2005). Notably, Kelleher and co-workers employed solution isoelectric focusing and gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) (Tran and Doucette, 2008) fractionation followed by nanocapillary reversed phase chromatography (RPLC) interfaced with a 12 T LTQ-FT-ICR MS and identified over 3,000 human proteoforms (Tran et al., 2011).
For proteoforms >25 kDa, top-down analysis is inherently challenging as signal is spread across an increased number of isotologues and charge states with increasing molecular weight (Compton et al., 2011). For this reason, most proteomics experiments which aim to observe proteoforms >50 kDa implement size-based fractionation to reduce further signal suppression of high-MW proteoforms by co-eluting low-MW proteoforms (Cai et al., 2017; Tran and Doucette, 2008; Tran et al., 2011; Tucholski et al., 2019; Vellaichamy et al., 2010). Tran et al. identified proteoforms up to 105 kDa in the study which implemented 4-dimensions of proteome separation. Intact proteoform masses were acquired at low resolution and fragmented using in-source dissociation (ISD) (Tran et al., 2011). The technique led to successful identification of high-MW proteoforms, but failed to provide accurate precursor mass measurements (Tran et al., 2011). In an attempt to extend the mass range of large-scale, resolved top-down proteomics, Marshall and co-workers achieved routine and reliable analysis of proteins up to 50 kDa and baseline unit mass resolution of proteoforms up to 72 kDa on an LC-time scale using nanoLC ESI FT-ICR MS at 14.5 T (Tipton et al., 2012). With accurate mass measurements (less than 10 ppm), they improved the throughput of top-down proteomic identifications, and furthered the study of proteoforms by allowing for measurement and characterization of PTMs and closely eluting protein isoforms.
However, on an LC-timescale, obtaining isotopic resolution for high-molecular weight proteoforms still proves challenging. Since mass-resolving power and spectral acquisition rate increase linearly with magnetic field, it is conceivable that using higher-strength magnets would dramatically improve top-down proteomic experiments, especially on an LC-FT-ICR MS timescale (Marshall and Guan, 1996). Taking advantage of these concepts, Anderson et al. demonstrated high-throughput top-down proteomics of human colorectal cell line extract using LC-MS/MS on the 21 T FT-ICR mass spectrometer at the NHMFL (Anderson et al., 2017). The instrument is equipped with an MSD to make significant improvements to the top-down analysis of complex proteoform mixtures on an LC-timescale (Anderson et al., 2017). Impressively, the use of multiple accumulations of precursor or fragment ions in the MSD together with an inherently higher spectral acquisition rate achieved at 21 T allowed for collection of high-quality MS/MS spectra on an LC-timescale, even for ETD spectra. They identified 3,238 unique proteoforms at 1% false discovery rate, including 372 proteoforms >30 kDa detected at isotopic resolution and achieved an impressive 51% sequence coverage of the 20-kDa peptidyl-prolyl cis isomerase for a single ETD MS2 acquisition. Despite more fills needed to achieve good S/N for ETD acquisitions, and thus a longer duty cycle, the proteoforms identified by this strategy had higher characterization scores due to increased sequence coverage, important for site-specific localization of PTMs. Notably, they isotopically resolved proteins >30 kDa on an LC-timescale, which shows that analysis of larger proteoforms by LC-FT-ICR MS is possible at 21 T (Figure 6).
Figure 6|. Top-down proteomics of high-MW proteoforms from colorectal cell line using 21 T LC-FT-ICR MS.
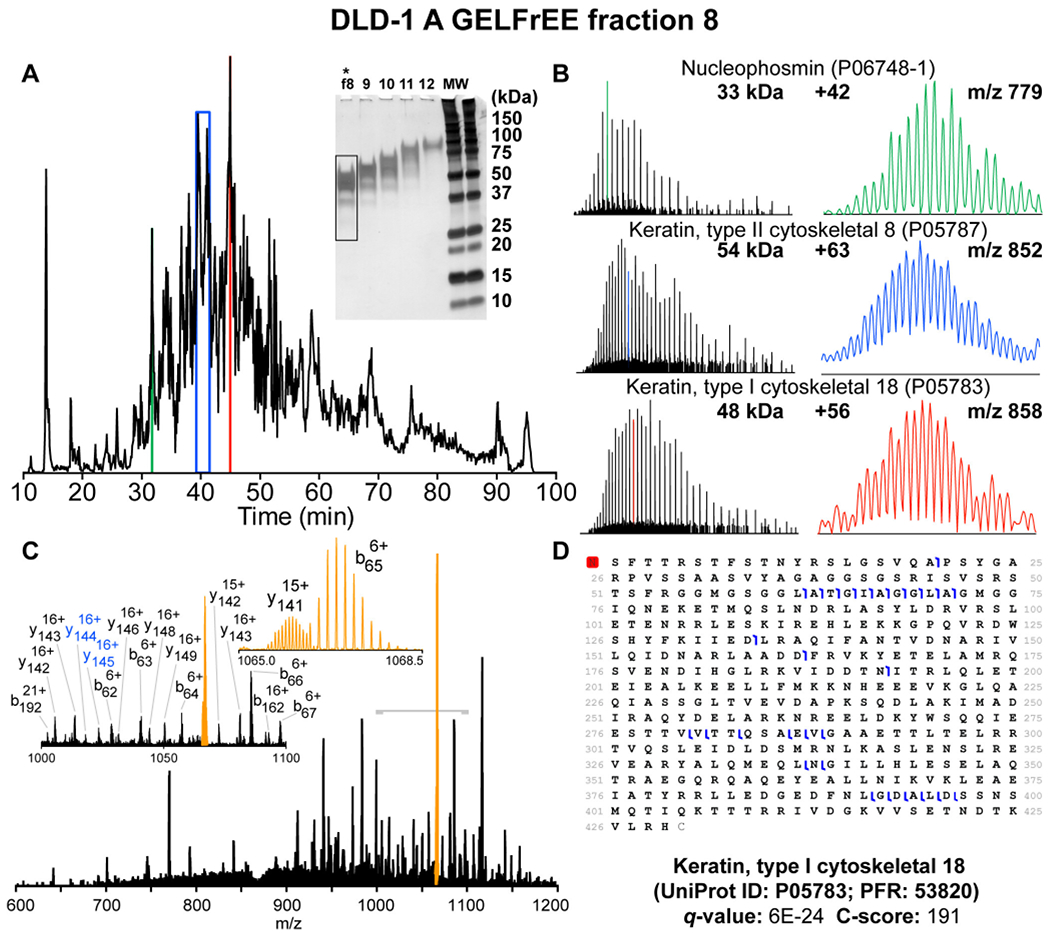
A) Total ion chromatogram obtained by LC-MS/MS for GELFrEE fraction 8 (highlighted) B) Single-scan mass spectra for proteins >30 kDa obtained by 21 T FT-ICR MS on an LC-timescale with zoom of selected isotopically-resolved charge states C) Online CID MS/MS spectrum for 54 kDa keratin type 1 D) Sequence table showing cleavages for keratin type 1. Reprinted and adapted with permission from Anderson, L. C., DeHart, C. J., Kaiser, N. K., Fellers, R. T., Smith, D. F., Greer, J. B., LeDuc, R. D., Blakney, G. T., Thomas, P. M., Kelleher, N. L., Hendrickson, C. L. Identification and Characterization of Human Proteoforms by Top-Down LC-21 Tesla FT-ICR Mass Spectrometry. J Proteome Res 2017 16, 1087-1096 (Anderson et al., 2017). Copyright © 2017 American Chemical Society.
FTICR mass spectrometers provide the highest resolution ion isolation capability, making them inherently suited for gas-phase isolation of proteoforms from complex mixtures. The use of SWIFT with high mass selectivity for isolation of individual proteoforms from complex mixtures prior to fragmentation has allowed higher-confidence identification of proteoforms (Guan and Burlingame, 2010). Most recently, Hendrickson and co-workers demonstrated ultrahigh resolving power isolation of histone proteoforms prior to UVPD using the 21 T FT-ICR mass spectrometer at NHMFL(Smith et al., 2020). They demonstrated the rapid (52.5 ms) isolation of the H2B histone proteoform from a mixture of eight other histone proteoforms which were initially co-isolated in the ion trap with an isolation window of 10 m/z. Using SWIFT, the H2B proteoform was isolated in a 0.6 m/z window with near 100% efficiency for in-cell fragmentation by UVPD, yielding 53% proteoform sequence coverage with 20 transients summed over the proteoform elution time (1.3 min) (Smith et al., 2020). The fast, high resolution isolation of individual proteoforms from mixtures of co-isolated precursors presents opportunities for selective sequence characterization of proteoforms on an LC-MS timescale.
Improving the performance of FT-ICR MS for analysis of complex proteoform mixtures depends on improving instrument duty cycle, since slower acquisition rates are required to achieve high-quality MS/MS spectra at high resolution. Realized that the sequence-coverage gains are obtainable by fragmenting multiple protein charge states at once, Loo and co-workers adapted a data-independent nanoLC-MS/MS strategy on their 15 T FT-ICR mass spectrometer to improve duty cycle for analysis of the 20S and 19S proteasome subunits (Lakshmanan et al., 2014). They utilized continuous accumulation of ions (CASI)-CAD by isolating 6-7 protein charge states together in an external quadrupole and subsequently dissociating them in the collision cell prior to detection in the ICR cell. Using this strategy, they eliminated precursor-ion selection time and thereby improved duty cycle. Fragmenting multiple charge states together proves promising for improving fragment ion S/N for top-down analysis of larger proteoforms for online FT-ICR MS, as has been demonstrated for larger proteoforms with online Q-TOF MS (Cai et al., 2017).
V. Biological Insights Provided by FT-ICR MS
The use of FT-ICR MS to comprehensively characterize proteoform sequence and map PTMs or sequence variations has provided important biological insights (Cai et al., 2016b; Gregorich and Ge, 2014; Peng et al., 2014a). For instance, cardiac myosin binding protein C (cMyBP-C), which plays an important role in regulating muscle contraction; its phosphorylation status is essential for normal muscle contraction (Kensler et al., 2017; Sadayappan et al., 2005). Ge et al. integrated top-down and middle-down FT-ICR MS with ECD, to comprehensively characterize and unambiguously localize phosphorylation sites in full-length mouse cMyBP-C (142 kDa) and truncated proteoforms (28-115 kDa) (Ge et al., 2009). Importantly, the sites for the full-length cMyBP-C were Ser-283, Ser-292, and Ser-312, which are all sites mediated by protein kinase A (PKA), and data suggests sequential phosphorylation of these sites, with Ser-292 being phosphorylated first and inducing conformational change which makes the other phosphorylation sites accessible to PKA (Ge et al., 2009; Kensler et al., 2017; Sadayappan et al., 2005). A comparison of the mass spectra for the full-length and truncated forms revealed that overall phosphorylation state of cMyBP-C was altered in the truncated forms compared to the full-length form. This is important to know because biochemistry and x-ray crystallography experiments are often performed with truncated proteoforms rather than full-length proteoforms.
Owing to its ability to characterize proteoform structure without a priori knowledge of sequence or modification sites, top-down FT-ICR MS coupled with in vitro enzyme assays is a useful tool for characterizing proteoform structure relative to enzyme activity and function (Wu et al., 2019; Yu et al., 2016). Kinases are a class of enzymes which reversibly phosphorylate proteins to control cell growth and signal transduction, and represent a large class of drug targets (Ferguson and Gray, 2018; Hunter, 1995; Manning et al., 2002; Zhang et al., 2009a). Characterization of kinase proteoforms by top-down MS following in vitro reactions allows for sequencing of kinase active site, localization of phosphorylated residues, and elucidation of truncated or otherwise-modified proteoforms that might result in altered enzyme activity. Yu et al. comprehensively characterized the catalytic domain of bacterially-expressed AMP-activated protein kinase (AMPK), a kinase which is important for energy metabolism in all eukaryotic cells (Yu et al., 2016). Comparing AMPK basal phosphorylation state with its state after in vitro phosphorylation by its upstream kinase, liver kinase B1 (LKB), revealed that AMPK was capable of auto-phosphorylation and substrate phosphorylation even before activation with LKB. More recently, Wu et al. characterized the recombinant catalytic sub-unit of cAMP-dependent protein kinase (PKA C-subunit) (Wu et al., 2019). They characterized seven phosphorylation sites on the most abundant PKA C-subunit proteoform using complementary information obtained from ECD and CAD experiments. Both studies are examples of the utility of high-resolution MS to assess in vitro enzyme assays and study kinase/phosphatase reactions (Wu et al., 2019; Yu et al., 2016).
Top-down FT-ICR MS is a useful tool for characterizing isoforms with high sequence homology and their associated proteoforms, such as tropomyosin (Tpm) which is encoded by many genes in humans (TPM1, TPM2, TPM3, TPM4) and has many proteoforms which arise form alternative splicing and PTMs (Jin et al., 2016; Peng et al., 2013a; Peng et al., 2013b). Importantly, differences in the composition of Tpm proteoforms in both cardiac and skeletal muscle are known to affect contractile properties and function. Peng et al. identified and characterized swine Tpm isoforms, including localizing the phosphorylation site of swine α-Tpm (gene TPM1) to Ser-283 and site-specifically localizing two amino acid polymorphisms that deviate from the TPM1-predicted sequence (Peng et al., 2013a). In this study, they also identified the previously un-characterized swine β-Tpm, which was found to have 100% sequence homology with mouse β-Tpm. A later study characterized Tpm proteoforms derived from human hearts, and identified α-Tpm (TPM1), β-Tpm (TPM2), and κ-Tpm (an alternatively spliced form of TPM1) in both atrial and ventricular regions of the heart. Importantly, they found significant differences in the levels of κ-Tpm and the mono-phosphorylated form of α-Tpm between ventricular and atrial regions of non-diseased donor hearts, demonstrating regional proteoform differences (Peng et al., 2013b). Skeletal muscle Tpm proteoforms have also been comprehensively investigated in rat, swine, and human skeletal muscle tissues using top-down FT-ICR MS. These findings support muscle-type specific proteoform differences in the rat and human tissues (Jin et al., 2016). Given the size of Tpm (32 kDa) and the close mass proximity of its genetic isoforms and associated proteoforms, FT-ICR MS is an ideal tool for confident identification and sequence characterization. Together, these studies demonstrate the deep-sequencing ability of top-down FT-ICR and also the advantage of the “bird’s-eye view” for relative quantitation of proteoforms in different tissues (Jin et al., 2016; Peng et al., 2013a; Peng et al., 2013b).
VI. New FT-ICR MS Applications for Studying Proteoforms
Classically, FT-ICR MS has been favored for determination of proteoform primary structure. In recent years, top-down and middle-down FT-ICR MS have been extended to the detailed structural analysis of mAbs and their proteoforms. FT-ICR MS-based assays have also emerged to provide fast and accurate clinical diagnosis for diseases such as hemoglobinopathy. Native FT-ICR MS platforms have proven useful for determining higher order structure and sub-unit composition of proteoform complexes, and have recently been integrated with top-down FT-ICR MS.
a. Structural Characterization of Monoclonal Antibodies
In-depth structural characterization of mAb-based therapeutics is essential given that molecular heterogeneity can affect quality and pharmacological properties. MS-based techniques are favored for characterization of antibody primary structure, but bottom-up proteomics approaches often introduce artifactual modifications and the opportunity for direct quantification of mAb proteoforms is lost with extensive digestion (Srebalus Barnes and Lim, 2007; Zhang et al., 2009b). Top-down and middle-down FT-ICR MS are well-suited for structural characterization and quantification of these high-molecular weight proteoforms. Additionally, ultrahigh resolving power lends the ability to obtain high mass accuracy measurements and confident identifications for mAb proteoforms that exist due to C-terminal processing of the heavy-chain, and amino acid sequence variation, and post-translational/artifactual modification (e.g. disulfide bonds, glycosylation, oxidation, and deamidation) (Chen et al., 2018b; Jin et al., 2019b; Mao et al., 2013; Shaw et al., 2016a; Valeja et al., 2011; Zhang et al., 2009b). Marshall and co-workers performed in-depth structural analysis of an intact monoclonal antibody using a custom-built 9.4 T FT-ICR MS and ECD (Mao et al., 2013). They applied ECD to all charge states 42+ through 58+ simultaneously, and found that this strategy gave more extensive backbone cleavages than dissociation of any one charge state alone. This study benchmarked top-down FT-ICR MS and ECD against Orbitrap and ETD for mAb characterization, and found comparable performance (Mao et al., 2013). Shaw et al. showcased the ultrahigh mass resolving power of the 21 T FT-ICR mass spectrometer at PNNL by measuring mAb glycoforms (55+ charge state of ~148 kDa) at unit mass resolution with mass errors between 0.74 and 1.15 ppm for all glycoforms (Shaw et al., 2016a). Using a 6 s transient (200 averaged transients) and absorption mode processing, they achieved 980,000 resolving power at 2699 m/z for the most abundant glycoform.
Jin et al. used top-down and middle-down mass spectrometry on a 12 T FT-ICR MS to comprehensively characterize monoclonal antibody proteoforms (Jin et al., 2019b). They obtained unit mass resolution for an intact mAb (IgG1, 147 kDa), as well as detailed sequence information following MS/MS of the intact mAb. Glycoforms and micro-variants of the mAb were subsequently characterized in-depth using IdeS digestion (to produce 25 kDa subunits) and middle-down FT-ICR MS. With combined analyses, 76% sequence coverage was achieved. Chen et al. applied hydrophobic interaction chromatography to separate mAb proteoforms coupled to online top-down FT-ICR MS with ECD fragmentation (Chen et al., 2018b). They utilized broadband ECD of all mAb charge-states, and achieved 80 MS/MS scans during the period of mAb elution. Because hydrophobic interaction chromatography (HIC) is operated in a semi-native mobile phase, the charge states for the mAb precursor ions and charge-reduced species (5000-10000 m/z) were well-separated from the fragment ions (1000-3000 m/z) (Figure 7A). They partially characterized the sequence and structure of the complementarity-determining regions using this strategy (Figure 7B–C). Marshall and co-workers have employed a nano-LC FT-ICR MS-based platform for characterization of mAbs from the human serum which will be discussed in Section IVb (vide infra).
Figure 7|. Online HIC-MS/MS with broadband ECD of deglycosylated mAb2 on a 12 T solariX XR FT-ICR mass spectrometer.
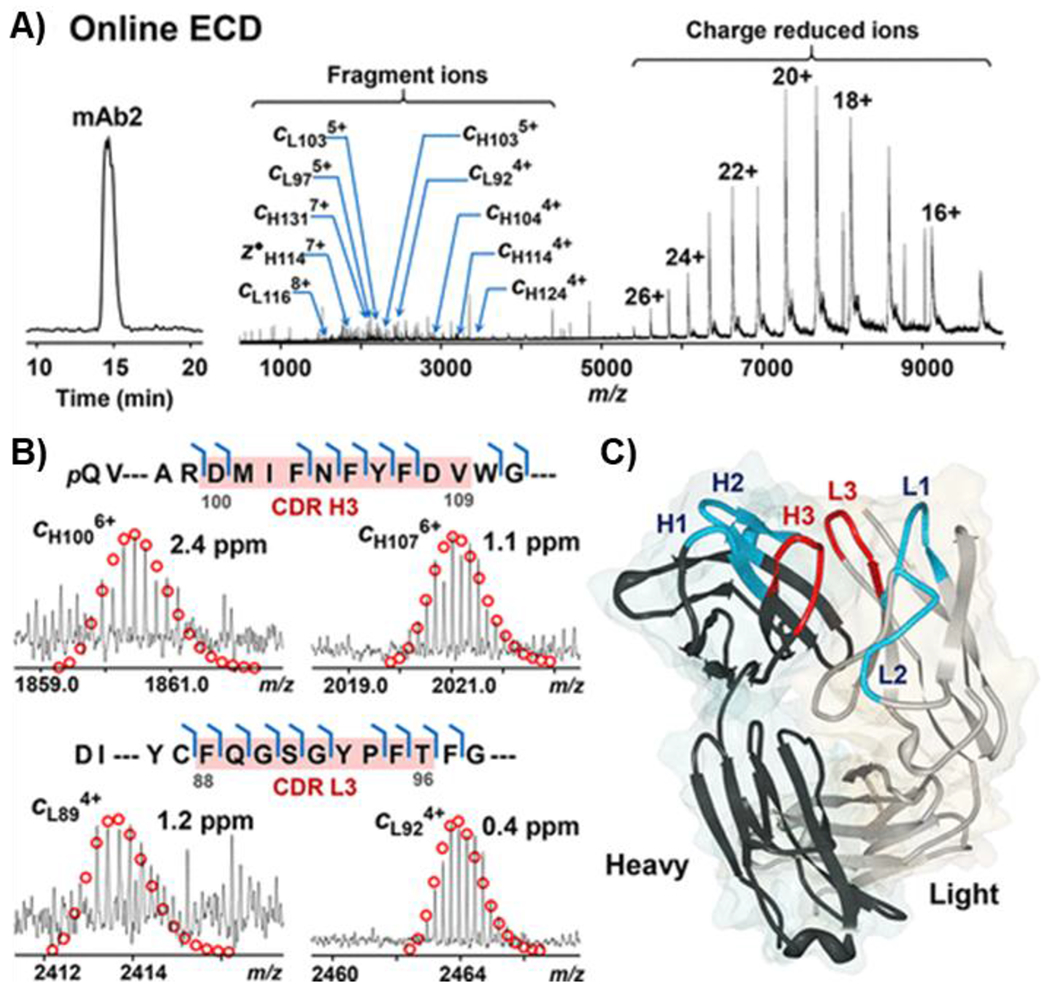
A) Chromatogram and ECD spectrum of mAb2 showing charge reduced species and fragment ions. B) Representative fragment ions from the CDRs H3 and L3 (highlighted in red) of heavy chain (top) and light chain (bottom). C) Crystal structure of the Fab fragment of mAb2 (PDB 5K8A). The CDRs fragmented by ECD (H3 and L3) are highlighted in red, and the other CDRs are highlighted in blue (H1, H2, L1, and L2). Reprinted and adapted with permission from Chen, B., Lin, Z., Alpert, A. J., Fu, C., Zhang, Q., Pritts, W. A., Ge, Y. Anal. Chem., 2018. 90, 7135-7138 (Chen et al., 2018b). Copyright 2018 American Chemical Society.
b. Clinical Diagnostics
MS-based assays afford sensitive and specific tools for clinical diagnoses and precision medicine. Marshall and co-workers employed a sensitive nano-LC 21 T FT-ICR MS/MS platform for high accuracy mass measurements and extensive residue cleavage for mAbs spiked into human serum as a proof-of-concept for biomarker detection in monoclonal gammopathy (He et al., 2017). Using the middle-down approach, they correctly identified five therapeutic mAbs in human serum with polyclonal background at clinically relevant concentrations with up to 50% sequence coverage. Next, they demonstrated the utility of this platform for clinical diagnosis plasma cell disorders through detection and sequence analysis of immunoglobulin light chains from human serum (He et al., 2019a). Applying the middle-down nano-LC FT-ICR MS/MS platform for blind analysis of samples from patients with AL amyloidosis, they correctly classified the light chains with 100% match to gene sequencing results (He et al., 2019a). He et al. demonstrated the benefit of using proteomics (requiring blood draw) over the invasive genomics (requiring bone marrow aspiration) for the diagnosis of AL amyloidosis. Recently, He et al. also developed a fast and accurate top-down MS assay using the 21 T FT-ICR mass spectrometer at NHMFL for diagnosis of hemoglobinopathies (e.g. sickle cell disease) and β-thalassemia, which are prevalent genetic disorders (He et al., 2019b). The top-down MS-based assay features ultrahigh mass accuracy, and extensive residue cleavage of Hb variants for de novo sequencing and determination of mutations. Current diagnostic technology involves cation-exchange high pressure liquid chromatography (HPLC) and electrophoresis for the determination of hemoglobin (Hb) variants, which are limited in resolution compared to a MS-based assay and often require further genetic testing to confirm the results of HPLC or electrophoretic methods. The FT-ICR MS-based assay allowed the resolution of and subsequent extensive de novo sequencing to locate sequence variants, based on measurement of precursor Hb mass at ultrahigh resolving power and mass accuracy. They correctly identified 18 Hb variants in a blind analysis, and reported the identification of the homozygous Hb Himeji variant. They also use relative quantification to diagnose β-thalassemia by calculating the intensity ratio of δ/β Hb subunits, with an increase in this ratio being a sensitive biomarker for β-thalassemia. Collectively, this work by He et al. demonstrated that top-down proteomics powered by FT-ICR MS with its ultrahigh resolving power and mass accuracy can provide rich clinically relevant information and may be used as a sensitive and specific tool for precision diagnostics (He et al., 2019b; He et al., 2020).
c. Characterizing Proteoforms in Complexes
Proteoforms often interact and assemble to form non-covalent complexes in cells that execute a diverse array of biological processes (Alberts, 1998). Native MS and native top-down MS have emerged as sensitive tools for studying proteoforms in complexes, and are complementary to structural biology approaches such as X-ray crystallography, NMR, and cryoEM (Heck, 2008; Kumar et al., 2020; Loo, 1997). FT-ICR MS, with its high mass-resolving power and versatile options for ion isolation and MSn, is an attractive tool for determining intact proteoform complex mass, stoichiometry, and most recently, sequence information and location of covalent and non-covalent structural features (Li et al., 2018; Li et al., 2014a; Li et al., 2014b; Zhang et al., 2011a).
Gross and co-workers launched an approach which used ECD for top-down MS sequencing of complexes introduced to a hybrid 12 T FT-ICR mass spectrometer by native ESI (Zhang et al., 2011a). While previous native MS studies were limited to measurement of the intact protein complex, and relied on bottom-up MS for sequence characterization, this technique allowed for native top-down MS of protein complexes. They showed that fragmentation of native protein complexes with ECD and CAD could provide important structural information, such as the location of metal binding sites of the concanavalin A complex (103 kDa). Loo and co-workers similarly showed that FT-ICR MS could be uniquely used to obtain native complex mass measurements and subunit sequence information in a single experiment (Li et al., 2014b). They obtained mass measurements at isotopic resolution for the 147-kDa yeast alcohol dehydrogenase (ADH), revealing binding of precisely two zinc atoms per subunit. They identified non-covalent NAD+/NADH ligand binding for horse liver ADH using in-source dissociation (ISD) and localized zinc binding and site-mutations for yeast ADH using ECD. These studies together demonstrate that FT-ICR MS is an all-in-one tool uniquely suited to provide multiple layers of information for proteoforms in a complex.
Loo and co-workers very recently established an FT-ICR MS-based platform to integrate proteomics and structural biology (Li et al., 2018). Notably, they addressed previous concerns about the transmission and detection of high m/z ions by FT-ICR MS and demonstrated detection of the 1.8-MDa aggregated β-galactosidase complex (at 18,000 m/z), where previously the largest noncovalent native complexes studied by FT-ICR MS were smaller than 160 kDa (Li et al., 2014a). With trapping and transmission of large ions with high m/z in question, Li et al. first calculated the trapping limit (Marshall and Guan, 1996) of their Bruker SolariX 15 T FT-ICR mass spectrometer and determined that it theoretically has the ability to trap molecules greater than 1 MDa. Next, they experimentally determined that cesium iodide cluster ions up to 17,000 m/z (first observed by (Castro et al., 1986)) could be effectively transmitted after adjustments of instrumental parameters such as radio frequencies (RF) and RF amplitudes. They went on to test the capabilities of this instrument, which is equipped with ISD, CAD, IRMPD, and ECD, for both native MS and top-down MS of various macromolecular complexes. They obtained information about complex stoichiometry together with reasonably high proteoform-subunit sequence coverage across the complexes analyzed (up to 87%), enabling mapping of PTMs and determination of interfacial and surface residues. In their analysis of glutamate dehydrogenase, they showed good correlation between the residues cleaved by native top-down MS and solvent-exposed regions according to the crystal structure, indicating the MS-based technique can provide insight about higher-order structure. This study further demonstrates the unique versatility of hybrid FT-ICR mass spectrometers to provide stoichiometry, higher-order structural information, and amino-acid sequences of proteoform complexes.
VII. Conclusions and Future Outlooks
With unmatched mass resolution and mass accuracy, FT-ICR MS has been a valuable tool for “top-down” characterization of proteoforms as demonstrated in numerous publications in the past decades, which have been highlighted in this review. The versatile fragmentation capabilities available in the FT-ICR mass spectrometer (and hybrid instruments based on FT-ICR MS) allowed comprehensive sequence characterization and mapping PTMs for individual proteoforms. The incorporation of ECD/ETD greatly enhances the top-down MS capabilities, especially for mapping labile PTMs such as phosphorylation and glycosylation. In the past few years, significant advances have been made in the use of FT-ICR MS for analyzing complex proteoform mixtures on a chromatographic time-scale, for characterizing very large proteoforms and native proteoforms in their high-order structure.
We foresee that FT-ICR MS will continue to be used as a tool to provide in-depth characterization of proteoform primary structure, toward localizing sequence variants, PTMs, and ligand binding sites. It will continue to be employed as a sensitive tool for characterization of antibodies and higher-order structure of proteoforms incorporated into protein complexes to establish structure-function relationships. Theoretical prediction (Marshall and Guan, 1996) of FT-ICR MS performance increase with increasing magnetic field strength has been proven by experimental results. Therefore, one could foresee an increased prevalence of high field instruments for use to answer increasingly complex biological questions.
In terms of discovery-based study of proteoforms, improvements in the last 10 years for LC-FT-ICR MS are promising, and will hopefully lead to large-scale proteomics projects which are capable of routinely identifying proteoforms >50 kDa. We are also optimistic that native top-down FT-ICR MS platforms will be extended to the large-scale study of endogenous proteoform complexes, as recently demonstrated using a quadrupole-Orbitrap mass spectrometer (Skinner et al., 2018). Conceivably, future developments in FT-ICR MS will help to accelerate high-throughput, comprehensive characterization of large numbers of proteoforms in their endogenous state to improve our understanding of proteoform biology. Additionally, recent advances in top-down and middle-down 21 T FT-ICR MS for biomarker detection will undoubtedly lead to the development of faster, more specific, and sensitive assays for clinical diagnostics, enabling precision medicine.
VIII. Acknowledgements
Y.G. and T.T. would like to acknowledge funding from NIH R01 grants, GM117058, GM125085, HL109810, and HL096971 and the high-end instrument grant S10 OD018475 (to Y.G.). T.T. would like to acknowledge support of the NIH Chemistry and Biology Interface Training Program (T32 GM008505).
Footnotes
This paper is dedicated to Alan Marshall in recognition of his contributions to the field of mass spectrometry
IX. References
- Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, Costello CE, Cravatt BF, Fenselau C, Garcia BA et al. (2018). How many human proteoforms are there? Nat Chem Biol 14, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R and Mann M (2003). Mass spectrometry-based proteomics. Nature 422, 198–207. [DOI] [PubMed] [Google Scholar]
- Aebersold R and Mann M (2016). Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355. [DOI] [PubMed] [Google Scholar]
- Alberts B (1998). The Cell as a Collection of Protein Machines: Preparing the Next Generation of Molecular Biologists. Cell 92, 291–294. [DOI] [PubMed] [Google Scholar]
- An HJ, Kronewitter SR, de Leoz ML and Lebrilla CB (2009). Glycomics and disease markers. Curr Opin Chem Biol 13, 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LC, DeHart CJ, Kaiser NK, Fellers RT, Smith DF, Greer JB, LeDuc RD, Blakney GT, Thomas PM, Kelleher NL et al. (2017). Identification and Characterization of Human Proteoforms by Top-Down LC-21 Tesla FT-ICR Mass Spectrometry. J Proteome Res 16, 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansong C, Wu S, Meng D, Liu X, Brewer HM, Deatherage Kaiser BL, Nakayasu ES, Cort JR, Pevzner P, Smith RD et al. (2013). Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc Natl Acad Sci U S A 110, 10153–10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggar KK and Li SSC (2015). Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol 16, 5–17. [DOI] [PubMed] [Google Scholar]
- Bogdanov B and Smith RD (2005). Proteomics by FTICR mass spectrometry: top down and bottom up. Mass Spectrom Rev 24, 168–200. [DOI] [PubMed] [Google Scholar]
- Bourgoin-Voillard S, Leymarie N and Costello CE (2014). Top-down tandem mass spectrometry on RNase A and B using a Qh/FT-ICR hybrid mass spectrometer. Proteomics 14, 1174–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuker K, Jin M, Han X, Jiang H and McLafferty FW (2008). Top-down identification and characterization of biomolecules by mass spectrometry. J Am Soc Mass Spectrom 19, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KA, Chen B, Guardado-Alvarez TM, Lin Z, Hwang L, Ayaz-Guner S, Jin S and Ge Y (2019). A photocleavable surfactant for top-down proteomics. Nat Methods 16, 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Guner H, Gregorich ZR, Chen AJ, Ayaz-Guner S, Peng Y, Valeja SG, Liu X and Ge Y (2016a). MASH Suite Pro: A Comprehensive Software Tool for Top-Down Proteomics. Mol Cell Proteomics 15, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Tucholski T, Chen B, Alpert AJ, McIlwain S, Kohmoto T, Jin S and Ge Y (2017). Top-Down Proteomics of Large Proteins up to 223 kDa Enabled by Serial Size Exclusion Chromatography Strategy. Anal Chem 89, 5467–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Tucholski TM, Gregorich ZR and Ge Y (2016b). Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert Rev Proteomics 13, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon J, Lohnes K, Wynne C, Wang Y, Edwards N and Fenselau C (2010). High-Throughput Middle-Down Analysis Using an Orbitrap. J Proteome Res 9, 3886–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carel C, Marcoux J, Reat V, Parra J, Latge G, Laval F, Demange P, Burlet-Schiltz O, Milon A, Daffe M et al. (2017). Identification of specific posttranslational O-mycoloylations mediating protein targeting to the mycomembrane. Proc Natl Acad Sci U S A 114, 4231–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro ME, Russell DH, Amster IJ and McLafferty FW (1986). Detection of mass 16241 ions by Fourier-transform mass spectrometry. Analytical Chemistry 58, 483–485. [DOI] [PubMed] [Google Scholar]
- Chait BT (2006). Chemistry. Mass spectrometry: bottom-up or top-down? Science 314, 65–66. [DOI] [PubMed] [Google Scholar]
- Chakouri N, Reboul C, Boulghobra D, Kleindienst A, Nottin S, Gayrard S, Roubille F, Matecki S, Lacampagne A and Cazorla O (2018). Stress-induced protein S-glutathionylation and phosphorylation crosstalk in cardiac sarcomeric proteins - Impact on heart function. Int J Cardiol 258, 207–216. [DOI] [PubMed] [Google Scholar]
- Chamot-Rooke J, Mikaty G, Malosse C, Soyer M, Dumont A, Gault J, Imhaus AF, Martin P, Trellet M, Clary G et al. (2011). Posttranslational modification of pili upon cell contact triggers N. meningitidis dissemination. Science 331, 778–82. [DOI] [PubMed] [Google Scholar]
- Chen B, Brown KA, Lin Z and Ge Y (2018a). Top-Down Proteomics: Ready for Prime Time? Anal Chem 90, 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Lin Z, Alpert AJ, Fu C, Zhang Q, Pritts WA and Ge Y (2018b). Online Hydrophobic Interaction Chromatography–Mass Spectrometry for the Analysis of Intact Monoclonal Antibodies. Anal Chem 90, 7135–7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrisman PA, Pitteri SJ and McLuckey SA (2006). Parallel Ion Parking of Protein Mixtures. Anal Chem 78, 310–316. [DOI] [PubMed] [Google Scholar]
- Cleland TP, DeHart CJ, Fellers RT, VanNispen AJ, Greer JB, LeDuc RD, Parker WR, Thomas PM, Kelleher NL and Brodbelt JS (2017). High-Throughput Analysis of Intact Human Proteins Using UVPD and HCD on an Orbitrap Mass Spectrometer. J Proteome Res 16, 2072–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Nathan JA and Goldberg AL (2015). Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 14, 58–74. [DOI] [PubMed] [Google Scholar]
- Compton PD, Zamdborg L, Thomas PM and Kelleher NL (2011). On the scalability and requirements of whole protein mass spectrometry. Anal Chem 83, 6868–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J and Hunt DF (2005). Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc Natl Acad Sci U S A 102, 9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupp-Sutton KA and Wu S (2020). High-throughput quantitative top-down proteomics. Mol Omics 16, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deribe YL, Pawson T and Dikic I (2010). Post-translational modifications in signal integration. Nat Struct Mol Biol 17, 666–72. [DOI] [PubMed] [Google Scholar]
- Donnelly DP, Rawlins CM, DeHart CJ, Fornelli L, Schachner LF, Lin Z, Lippens JL, Aluri KC, Sarin R, Chen B et al. (2019). Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat Methods 16, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson FM and Gray NS (2018). Kinase inhibitors: the road ahead. Nat Rev Drug Discov 17, 353–377. [DOI] [PubMed] [Google Scholar]
- Fornelli L, Ayoub D, Aizikov K, Beck A and Tsybin YO (2014). Middle-down analysis of monoclonal antibodies with electron transfer dissociation orbitrap fourier transform mass spectrometry. Anal Chem 86, 3005–12. [DOI] [PubMed] [Google Scholar]
- Garcia BA (2010a). What Does the Future Hold for Top Down Mass Spectrometry? J Am Soc Mass Spectrom 21, 193–202. [DOI] [PubMed] [Google Scholar]
- Garcia BA (2010b). What does the future hold for Top Down mass spectrometry? J Am Soc Mass Spectrom 21, 193–202. [DOI] [PubMed] [Google Scholar]
- Ge Y, El-Naggar M, Sze SK, Oh HB, Begley TP, McLafferty FW, Boshoff H and Barry CE 3rd. (2003). Top down characterization of secreted proteins from Mycobacterium tuberculosis by electron capture dissociation mass spectrometry. J Am Soc Mass Spectrom 14, 253–61. [DOI] [PubMed] [Google Scholar]
- Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park JH, Begley TP and McLafferty FW (2002). Top down characterization of larger proteins (45 kDa) by electron capture dissociation mass spectrometry. J Am Chem Soc 124, 672–8. [DOI] [PubMed] [Google Scholar]
- Ge Y, Rybakova IN, Xu Q and Moss RL (2009). Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proc Natl Acad Sci U S A 106, 12658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorich ZR, Cai W, Lin Z, Chen AJ, Peng Y, Kohmoto T and Ge Y (2017). Distinct sequences and post-translational modifications in cardiac atrial and ventricular myosin light chains revealed by top-down mass spectrometry. J Mol Cell Cardiol 107, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorich ZR and Ge Y (2014). Top-down proteomics in health and disease: challenges and opportunities. Proteomics 14, 1195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorich ZR, Peng Y, Cai W, Jin Y, Wei L, Chen AJ, McKiernan SH, Aiken JM, Moss RL, Diffee GM et al. (2016). Top-Down Targeted Proteomics Reveals Decrease in Myosin Regulatory Light-Chain Phosphorylation That Contributes to Sarcopenic Muscle Dysfunction. J Proteome Res 15, 2706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorich ZR, Peng Y, Lane NM, Wolff JJ, Wang S, Guo W, Guner H, Doop J, Hacker TA and Ge Y (2015). Comprehensive assessment of chamber-specific and transmural heterogeneity in myofilament protein phosphorylation by top-down mass spectrometry. J Mol Cell Cardiol 87, 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes M, Hall B, Foltz L, Levy T, Rikova K, Gaiser J, Cook W, Smirnova E, Wheeler T, Clark NR et al. (2018). Integration of protein phosphorylation, acetylation, and methylation data sets to outline lung cancer signaling networks. Sci Signal 11, doi: 10.1126/scisignal.aaq1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan S and Burlingame AL (2010). High mass selectivity for top-down proteomics by application of SWIFT technology. J Am Soc Mass Spectrom 21, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan S and Marshall AG (1996). Stored waveform inverse Fourier transform (SWIFT) ion excitation in trapped-ion mass spectometry: Theory and applications. Int J Mass Spectrom 157–158, 5–37. [Google Scholar]
- Guan Z, Kelleher NL, O'Connor PB, Aaserud DJ, Little DP and McLafferty FW (1996). 193 nm photodissociation of larger multiply-charged biomolecules. Int J Mass Spectrom 157–158, 357–364. [Google Scholar]
- Han X, Jin M, Breuker K and McLafferty FW (2006). Extending top-down mass spectrometry to proteins with masses greater than 200 kilodaltons. Science 314, 109–112. [DOI] [PubMed] [Google Scholar]
- He L, Anderson LC, Barnidge DR, Murray DL, Dasari S, Dispenzieri A, Hendrickson CL and Marshall AG (2019a). Classification of Plasma Cell Disorders by 21 Tesla Fourier Transform Ion Cyclotron Resonance Top-Down and Middle-Down MS/MS Analysis of Monoclonal Immunoglobulin Light Chains in Human Serum. Anal Chem 91, 3263–3269. [DOI] [PubMed] [Google Scholar]
- He L, Anderson LC, Barnidge DR, Murray DL, Hendrickson CL and Marshall AG (2017). Analysis of Monoclonal Antibodies in Human Serum as a Model for Clinical Monoclonal Gammopathy by Use of 21 Tesla FT-ICR Top-Down and Middle-Down MS/MS. J Am Soc Mass Spectrom 28, 827–838. [DOI] [PubMed] [Google Scholar]
- He L, Rockwood AL, Agarwal AM, Anderson LC, Weisbrod CR, Hendrickson CL and Marshall AG (2019b). Diagnosis of Hemoglobinopathy and β-Thalassemia by 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometry and Tandem Mass Spectrometry of Hemoglobin from Blood. Clin Chem 65, 986–994. [DOI] [PubMed] [Google Scholar]
- He L, Rockwood AL, Agarwal AM, Anderson LC, Weisbrod CR, Hendrickson CL and Marshall AG (2020). Top-down proteomics—a near-future technique for clinical diagnosis? Ann Transl Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck AJ (2008). Native mass spectrometry: a bridge between interactomics and structural biology. Nat Methods 5, 927–33. [DOI] [PubMed] [Google Scholar]
- Hendrickson CL and Emmett MR (1999). Electrospray Ionization Fourier-transform Ion Cyclotron Resonance Mass Spectrometry. Annu Rev Phys Chem 50, 517–536. [DOI] [PubMed] [Google Scholar]
- Hendrickson CL, Quinn JP, Kaiser NK, Smith DF, Blakney GT, Chen T, Marshall AG, Weisbrod CR and Beu SC (2015). 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometer: A National Resource for Ultrahigh Resolution Mass Analysis. J Am Soc Mass Spectrom 26, 1626–1632. [DOI] [PubMed] [Google Scholar]
- Horn DM, Ge Y and McLafferty FW (2000). Activated ion electron capture dissociation for mass spectral sequencing of larger (42 kDa) proteins. Anal Chem 72, 4778–4784. [DOI] [PubMed] [Google Scholar]
- Hunt DF, Shabanowitz J and Yates JR (1987). Peptide sequence analysis by laser photodissociation Fourier transform mass spectrometry. Chem Comm 8, 548–550. [Google Scholar]
- Hunter T (1995). Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80, 225–236. [DOI] [PubMed] [Google Scholar]
- Jenuwein T and Allis CD (2001). Translating the Histone Code. Science 293, 1074. [DOI] [PubMed] [Google Scholar]
- Jin Y, Diffee GM, Colman RJ, Anderson RM and Ge Y (2019a). Top-down Mass Spectrometry of Sarcomeric Protein Post-translational Modifications from Non-human Primate Skeletal Muscle. J Am Soc Mass Spectrom 30, 2460–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Lin Z, Xu Q, Fu C, Zhang Z, Zhang Q, Pritts WA and Ge Y (2019b). Comprehensive characterization of monoclonal antibody by Fourier transform ion cyclotron resonance mass spectrometry. MAbs 11, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Peng Y, Lin Z, Chen YC, Wei L, Hacker TA, Larsson L and Ge Y (2016). Comprehensive analysis of tropomyosin isoforms in skeletal muscles by top-down proteomics. J Muscle Res Cell Motil 37, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Wei L, Cai W, Lin Z, Wu Z, Peng Y, Kohmoto T, Moss RL and Ge Y (2017). Complete Characterization of Cardiac Myosin Heavy Chain (223 kDa) Enabled by Size-Exclusion Chromatography and Middle-Down Mass Spectrometry. Anal Chem 89, 4922–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser NK, Quinn JP, Blakney GT, Hendrickson CL and Marshall AG (2011). A Novel 9.4 Tesla FTICR Mass Spectrometer with Improved Sensitivity, Mass Resolution, and Mass Range. J Am Soc Mass Spectrom 22, 1343–1351. [DOI] [PubMed] [Google Scholar]
- Karabacak NM, Easterling ML, Agar NYR and Agar JN (2010). Transformative effects of higher magnetic field in Fourier transform ion cyclotron resonance mass spectrometry. J Am Soc Mass Spectrom 21, 1218–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karve TM and Cheema AK (2011). Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J Amino Acids 2011, 207691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK and McLafferty FW (1999). Top Down versus Bottom Up Protein Characterization by Tandem High-Resolution Mass Spectrometry. J Am Chem Soc 121, 806–812. [Google Scholar]
- Kelleher NL, Senko MW, Siegel MM and McLafferty FW (1997). Unit resolution mass spectra of 112 kDa molecules with 3 Da accuracy. J Am Soc Mass Spectrom 8, 380–383. [Google Scholar]
- Kellie JF, Catherman AD, Durbin KR, Tran JC, Tipton JD, Norris JL, Witkowski CE 2nd, Thomas PM and Kelleher NL (2012). Robust analysis of the yeast proteome under 50 kDa by molecular-mass-based fractionation and top-down mass spectrometry. Anal Chem 84, 209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler RW, Craig R and Moss RL (2017). Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments. Proc Natl Acad Sci U S A 114, E1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger KE and Srivastava S (2006). Posttranslational protein modifications: current implications for cancer detection, prevention, and therapeutics. Mol Cell Proteomics 5, 1799–810. [DOI] [PubMed] [Google Scholar]
- Kumar P, Wang Y, Zhang Z, Zhao Z, Cymes GD, Tajkhorshid E and Grosman C (2020). Cryo-EM structures of a lipid-sensitive pentameric ligand-gated ion channel embedded in a phosphatidylcholine-only bilayer. Proc Natl Acad Sci U S A 117, 1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan R, Wolff JJ, Alvarado R and Loo JA (2014). Top-down protein identification of proteasome proteins with nanoLC-FT-ICR-MS employing data-independent fragmentation methods. Proteomics 14, 1271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDuc RD, Schwämmle V, Shortreed MR, Cesnik AJ, Solntsev SK, Shaw JB, Martin MJ, Vizcaino JA, Alpi E, Danis P et al. (2018). ProForma: A Standard Proteoform Notation. J Proteome Res 17, 1321–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Kellie JF, Tran JC, Tipton JD, Catherman AD, Thomas HM, Ahlf DR, Durbin KR, Vellaichamy A, Ntai I et al. (2009). A robust two-dimensional separation for top-down tandem mass spectrometry of the low-mass proteome. J Am Soc Mass Spectrom 20, 2183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Berger SJ, Martinovic S, Pasa-Tolic L, Anderson GA, Shen Y, Zhao R and Smith RD (2002). Direct mass spectrometric analysis of intact proteins of the yeast large ribosomal subunit using capillary LC/FTICR. Proc Natl Acad Sci U S A 99, 5942–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lin T-Y, Van Orden SL, Zhao Y, Barrow MP, Pizarro AM, Qi Y, Sadler PJ and O’Connor PB (2011). Use of Top-Down and Bottom-Up Fourier Transform Ion Cyclotron Resonance Mass Spectrometry for Mapping Calmodulin Sites Modified by Platinum Anticancer Drugs. Anal Chem 83, 9507–9515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Nguyen HH, Ogorzalek Loo RR, Campuzano IDG and Loo JA (2018). An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat Chem 10, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wolff JJ, Van Orden SL and Loo JA (2014a). Native Top-Down Electrospray Ionization-Mass Spectrometry of 158 kDa Protein Complex by High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal Chem 86, 317–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wongkongkathep P, Van Orden SL, Ogorzalek Loo RR and Loo JA (2014b). Revealing Ligand Binding Sites and Quantifying Subunit Variants of Noncovalent Protein Complexes in a Single Native Top-Down FTICR MS Experiment. J Am Soc Mass Spectrom 25, 2060–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Compton PD, Tran JC, Ntai I and Kelleher NL (2014c). Optimizing capillary electrophoresis for top-down proteomics of 30–80 kDa proteins. Proteomics 14, 1158–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Guo F, Gregorich ZR, Sun R, Zhang H, Hu Y, Shanmuganayagam D and Ge Y (2018). Comprehensive Characterization of Swine Cardiac Troponin T Proteoforms by Top-Down Mass Spectrometry. J Am Soc Mass Spectrom 29, 1284–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo JA (1997). Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom Rev 16, 1–23. [DOI] [PubMed] [Google Scholar]
- Mann M and Jensen ON (2003). Proteomic analysis of post-translational modifications. Nat Biotechnol 21, 255–61. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T and Sudarsanam S (2002). The protein kinase complement of the human genome. Science 298, 1912–34. [DOI] [PubMed] [Google Scholar]
- Mao Y, Valeja SG, Rouse JC, Hendrickson CL and Marshall AG (2013). Top-down structural analysis of an intact monoclonal antibody by electron capture dissociation-Fourier transform ion cyclotron resonance-mass spectrometry. Anal Chem 85, 4239–46. [DOI] [PubMed] [Google Scholar]
- Marshall AG and Guan S (1996). Advantages of High Magnetic Field for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Rapid Commun Mass Spectrom 10, 1819–1823. [DOI] [PubMed] [Google Scholar]
- Marshall AG, Hendrickson CL, and Shi SD-H (2002). Peer Reviewed: Scaling MS Plateaus with High-Resolution FT-ICRMS. Anal Chem 74, 252–9. [DOI] [PubMed] [Google Scholar]
- Marshall AG and Hendrickson CL (2008). High-Resolution Mass Spectrometers. Annu Rev Anal Chem 1, 579–599. [DOI] [PubMed] [Google Scholar]
- Marshall AG, Hendrickson CL and Jackson GS (1998). Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrom Rev 17, 1–35. [DOI] [PubMed] [Google Scholar]
- Mazur MT, Cardasis HL, Spellman DS, Liaw A, Yates NA and Hendrickson RC (2010). Quantitative analysis of intact apolipoproteins in human HDL by top-down differential mass spectrometry. Proc Natl Acad Sci U S A 107, 7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckey SA, Reid GE and Wells JM (2002). Ion parking during ion/ion reactions in electrodynamic ion traps. Anal Chem 74, 336–46. [DOI] [PubMed] [Google Scholar]
- Mehmood S, Allison TM and Robinson CV (2015). Mass Spectrometry of Protein Complexes: From Origins to Applications. Annu Rev Phys Chem 66, 453–474. [DOI] [PubMed] [Google Scholar]
- Meng F, Cargile BJ, Miller LM, Forbes AJ, Johnson JR and Kelleher NL (2001). Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat Biotechnol 19, 952–957. [DOI] [PubMed] [Google Scholar]
- Meng F, Cargile BJ, Patrie SM, Johnson JR, McLoughlin SM and Kelleher NL (2002). Processing complex mixtures of intact proteins for direct analysis by mass spectrometry. Anal Chem 74, 2923–9. [DOI] [PubMed] [Google Scholar]
- Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Müller M, Viner R, Schwartz J, Remes P, Belford M et al. (2012). Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics 11, O111.013698–O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen KW, Tiemeyer M and Nairn AV (2012). Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller JB, Geyer PE, Colaço AR, Treit PV, Strauss MT, Oroshi M, Doll S, Virreira Winter S, Bader JM, Köhler N et al. (2020). The proteome landscape of the kingdoms of life. Nature. doi.org/10.1038/s41586-020-2402-x [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI and Aebersold R (2005). Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics 4, 1419–40. [DOI] [PubMed] [Google Scholar]
- Nikolaev EN, Kostyukevich YI and Vladimirov GN (2016). Fourier transform ion cyclotron resonance (FT ICR) mass spectrometry: Theory and simulations. Mass Spectrom Rev 35, 219–58. [DOI] [PubMed] [Google Scholar]
- Ntai I, Fornelli L, DeHart CJ, Hutton JE, Doubleday PF, LeDuc RD, van Nispen AJ, Fellers RT, Whiteley G, Boja ES et al. (2018). Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc Natl Acad Sci U S A 115, 4140–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntai I, LeDuc RD, Fellers RT, Erdmann-Gilmore P, Davies SR, Rumsey J, Early BP, Thomas PM, Li S, Compton PD et al. (2016). Integrated Bottom-Up and Top-Down Proteomics of Patient-Derived Breast Tumor Xenografts. Mol Cell Proteomics 15, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor PB and McLafferty FW (1995). High-resolution ion isolation with the ion cyclotron resonance capacitively coupled open cell. J Am Soc Mass Spectrom 6, 533–535. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P and Mann M (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–48. [DOI] [PubMed] [Google Scholar]
- Parks BA, Jiang L, Thomas PM, Wenger CD, Roth MJ, Boyne MT 2nd, Burke PV, Kwast KE and Kelleher NL (2007). Top-down proteomics on a chromatographic time scale using linear ion trap fourier transform hybrid mass spectrometers. Anal Chem 79, 7984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrie SM, Charlebois JP, Whipple D, Kelleher NL, Hendrickson CL, Quinn JP, Marshall AG and Mukhopadhyay B (2004). Construction of a hybrid quadrupole/Fourier transform ion cyclotron resonance mass spectrometer for versatile MS/MS above 10 kDa. J Am Soc Mass Spectrom 15, 1099–108. [DOI] [PubMed] [Google Scholar]
- Peng Y, Ayaz-Guner S, Yu D and Ge Y (2014a). Top-down mass spectrometry of cardiac myofilament proteins in health and disease. Proteom Clin Appl 8, 554–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Chen X, Sato T, Rankin SA, Tsuji RF and Ge Y (2012). Purification and high-resolution top-down mass spectrometric characterization of human salivary alpha-amylase. Anal Chem 84, 3339–46. [DOI] [PubMed] [Google Scholar]
- Peng Y, Chen X, Zhang H, Xu Q, Hacker TA and Ge Y (2013a). Top-down targeted proteomics for deep sequencing of tropomyosin isoforms. J Proteome Res 12, 187–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Gregorich ZR, Valeja SG, Zhang H, Cai W, Chen Y-C, Guner H, Chen AJ, Schwahn DJ, Hacker TA et al. (2014b). Top-down proteomics reveals concerted reductions in myofilament and Z-disc protein phosphorylation after acute myocardial infarction.Mol Cell Proteomics 13, 2752–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Yu D, Gregorich Z, Chen X, Beyer AM, Gutteran DD and Ge Y (2013b). In-depth proteomic analysis of human tropomyosin by top-down mass spectrometry. J Muscle Res Cell Motil 34, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley NM, Westphall MS and Coon JJ (2015). Activated Ion Electron Transfer Dissociation for Improved Fragmentation of Intact Proteins. Anal Chem 87, 7109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland Y, Czerwinski S, Abellan Van Kan G, Morley JE, Cesari M, Onder G, Woo J, Baumgartner R, Pillard F, Boirie Y et al. (2008). Sarcopenia: its assessment, etiology, pathogenesis, consequences and future perspectives. J Nutr Health Aging 12, 433–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MJ, Parks BA, Ferguson JT, Boyne MT 2nd and Kelleher NL (2008). "Proteotyping": population proteomics of human leukocytes using top down mass spectrometry. Anal Chem 80, 2857–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall JG, Schertzer JD and Lynch GS (2008). Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology 9, 213–28. [DOI] [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW 2nd, Klevitsky R, Seidman CE, Seidman JG and Robbins J (2005). Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res 97, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaff TG, Cargile BJ, Stephenson JL and McLuckey SA (2000). Ion Trap Collisional Activation of the (M + 2H)2+ – (M + 17H)17+ Ions of Human Hemoglobin β-Chain. Anal Chem 72, 899–907. [DOI] [PubMed] [Google Scholar]
- Schaffer LV, Millikin RJ, Miller RM, Anderson LC, Fellers RT, Ge Y, Kelleher NL, LeDuc RD, Liu X, Payne SH et al. (2019). Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 19, e1800361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub TM, Hendrickson CL, Horning S, Quinn JP, Senko MW and Marshall AG (2008). High-Performance Mass Spectrometry: Fourier Transform Ion Cyclotron Resonance at 14.5 Tesla. Anal Chem 80, 3985–3990. [DOI] [PubMed] [Google Scholar]
- Senko MW, Hendrickson CL, Pasa-Tolic L, Marto JA, White FM, Guan S and Marshall AG (1996). Electrospray ionization Fourier transform ion cyclotron resonance at 9.4 T. Rapid Commun Mass Spectrom 10, 1824–8. [DOI] [PubMed] [Google Scholar]
- Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL and Brodbelt JS (2013). Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J Am Chem Soc 135, 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JB, Lin TY, Leach FE 3rd, Tolmachev AV, Tolic N, Robinson EW, Koppenaal DW and Pasa-Tolic L (2016a). 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometer Greatly Expands Mass Spectrometry Toolbox. J Am Soc Mass Spectrom 27, 1929–1936. [DOI] [PubMed] [Google Scholar]
- Shaw JB, Robinson EW and Pasa-Tolic L (2016b). Vacuum Ultraviolet Photodissociation and Fourier Transform-Ion Cyclotron Resonance (FT-ICR) Mass Spectrometry: Revisited. Anal Chem 88, 3019–23. [DOI] [PubMed] [Google Scholar]
- Sidoli S and Garcia BA (2017). Middle-down proteomics: a still unexploited resource for chromatin biology. Expert rev proteomic 14, 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Lin S, Karch KR and Garcia BA (2015). Bottom-Up and Middle-Down Proteomics Have Comparable Accuracies in Defining Histone Post-Translational Modification Relative Abundance and Stoichiometry. Anal Chem 87, 3129–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuti N and Kelleher NL (2007). Decoding protein modifications using top-down mass spectrometry. Nat Methods 4, 817–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner OS, Haverland NA, Fornelli L, Melani RD, Do Vale LHF, Seckler HS, Doubleday PF, Schachner LF, Srzentic K, Kelleher NL et al. (2018). Top-down characterization of endogenous protein complexes with native proteomics. Nat Chem Biol 14, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DF, Blakney GT, Beu SC, Anderson LC, Weisbrod CR and Hendrickson CL (2020). Ultrahigh Resolution Ion Isolation by Stored Waveform Inverse Fourier Transform 21 T Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal Chem 92, 3213–3219. [DOI] [PubMed] [Google Scholar]
- Smith LM and Kelleher NL (2018). Proteoforms as the next proteomics currency. Science 359, 1106–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Kelleher NL and Consortium for Top Down, P. (2013). Proteoform: a single term describing protein complexity. In Nat Methods, vol. 10, pp. 186–7. United States. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Thomas PM, Shortreed MR, Schaffer LV, Fellers RT, LeDuc RD, Tucholski T, Ge Y, Agar JN, Anderson LC et al. (2019). A five-level classification system for proteoform identifications. Nat Methods 16, 939–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ (2008). Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. J Biol Chem 283, 26829–26833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ and Kobayashi T (2011). Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem 286, 9935–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srebalus Barnes CA and Lim A (2007). Applications of mass spectrometry for the structural characterization of recombinant protein pharmaceuticals. Mass Spectrom Rev 26, 370–388. [DOI] [PubMed] [Google Scholar]
- Stephenson JL and McLuckey SA (1996). Ion/Ion Proton Transfer Reactions for Protein Mixture Analysis. Anal Chem 68, 4026–4032. [DOI] [PubMed] [Google Scholar]
- Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J and Hunt DF (2004a). Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syka JEP, Marto JA, Bai DL, Horning S, Senko MW, Schwartz JC, Ueberheide B, Garcia B, Busby S, Muratore T et al. (2004b). Novel Linear Quadrupole Ion Trap/FT Mass Spectrometer: Performance Characterization and Use in the Comparative Analysis of Histone H3 Post-translational Modifications. J Proteome Res 3, 621–626. [DOI] [PubMed] [Google Scholar]
- Sze SK, Ge Y, Oh H and McLafferty FW (2002). Top-down mass spectrometry of a 29-kDa protein for characterization of any posttranslational modification to within one residue. Proc Natl Acad Sci U S A 99, 1774–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipton JD, Tran JC, Catherman AD, Ahlf DR, Durbin KR, Lee JE, Kellie JF, Kelleher NL, Hendrickson CL and Marshall AG (2012). Nano-LC FTICR tandem mass spectrometry for top-down proteomics: routine baseline unit mass resolution of whole cell lysate proteins up to 72 kDa. Anal Chem 84, 2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toby TK, Fornelli L and Kelleher NL (2016). Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu Rev Anal Chem 9, 499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran JC and Doucette AA (2008). Gel-eluted liquid fraction entrapment electrophoresis: an electrophoretic method for broad molecular weight range proteome separation. Anal Chem 80, 1568–1573. [DOI] [PubMed] [Google Scholar]
- Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M et al. (2011). Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 480, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucholski T, Knott SJ, Chen B, Pistono P, Lin Z and Ge Y (2019). A Top-Down Proteomics Platform Coupling Serial Size Exclusion Chromatography and Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal Chem 91, 3835–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeja SG, Kaiser NK, Xian F, Hendrickson CL, Rouse JC and Marshall AG (2011). Unit mass baseline resolution for an intact 148 kDa therapeutic monoclonal antibody by Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem 83, 8391–8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkevich EM, Sanchez NA, Ge Y and Strieter ER (2014). Middle-Down Mass Spectrometry Enables Characterization of Branched Ubiquitin Chains. Biochemistry 53, 4979–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellaichamy A, Tran JC, Catherman AD, Lee JE, Kellie JF, Sweet SM, Zamdborg L, Thomas PM, Ahlf DR, Durbin KR et al. (2010). Size-sorting combined with improved nanocapillary liquid chromatography-mass spectrometry for identification of intact proteins up to 80 kDa. Anal Chem 82, 1234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb K and Bennett EJ (2013). Eavesdropping on PTM cross-talk through serial enrichment. Nat Methods 10, 620–621. [DOI] [PubMed] [Google Scholar]
- Wei L, Gregorich ZR, Lin Z, Cai W, Jin Y, McKiernan SH, McIlwain S, Aiken JM, Moss RL, Diffee GM et al. (2018). Novel Sarcopenia-related Alterations in Sarcomeric Protein Post-translational Modifications (PTMs) in Skeletal Muscles Identified by Top-down Proteomics. Mol Cell Proteomics 17, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod CR, Kaiser NK, Syka JEP, Early L, Mullen C, Dunyach JJ, English AM, Anderson LC, Blakney GT, Shabanowitz J et al. (2017). Front-End Electron Transfer Dissociation Coupled to a 21 Tesla FT-ICR Mass Spectrometer for Intact Protein Sequence Analysis. J Am Soc Mass Spectrom 28, 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H et al. (2014). Mass-spectrometry-based draft of the human proteome. Nature 509, 582–7. [DOI] [PubMed] [Google Scholar]
- Wu C, Tran JC, Zamdborg L, Durbin KR, Li M, Ahlf DR, Early BP, Thomas PM, Sweedler JV and Kelleher NL (2012). A protease for 'middle-down' proteomics. Nat Methods 9, 822–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Yang F, Zhao R, Tolic N, Robinson EW, Camp DG 2nd, Smith RD and Pasa-Tolic L (2009). Integrated workflow for characterizing intact phosphoproteins from complex mixtures. Anal Chem 81, 4210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Jin Y, Chen B, Gugger MK, Wilkinson-Johnson CL, Tiambeng TN, Jin S and Ge Y (2019). Comprehensive Characterization of the Recombinant Catalytic Subunit of cAMP-Dependent Protein Kinase by Top-Down Mass Spectrometry. J Am Soc Mass Spectrom 30, 2561–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Zhang J, Yin S and Loo JA (2006). Top-Down ESI-ECD-FT-ICR Mass Spectrometry Localizes Noncovalent Protein-Ligand Binding Sites. J Am Chem Soc 128, 14432–14433. [DOI] [PubMed] [Google Scholar]
- Xu G and Jaffrey SR (2011). The new landscape of protein ubiquitination. Nat Biotechnol 29, 1098–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S and Loo JA (2010). Elucidating the site of protein-ATP binding by top-down mass spectrometry. J Am Soc Mass Spectrom 21, 899–907. [DOI] [PubMed] [Google Scholar]
- Yu D, Peng Y, Ayaz-Guner S, Gregorich ZR and Ge Y (2016). Comprehensive Characterization of AMP-Activated Protein Kinase Catalytic Domain by Top-Down Mass Spectrometry. J Am Soc Mass Spectrom 27, 220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabrouskov V, Giacomelli L, van Wijk KJ and McLafferty FW (2003). A new approach for plant proteomics: characterization of chloroplast proteins of Arabidopsis thaliana by top-down mass spectrometry. Mol Cell Proteomics 2, 1253–60. [DOI] [PubMed] [Google Scholar]
- Zhai H, Han X, Breuker K and McLafferty FW (2005). Consecutive Ion Activation for Top Down Mass Spectrometry: Improved Protein Sequencing by Nozzle–Skimmer Dissociation. Anal Chem 77, 5777–5784. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cui W, Wen J, Blankenship RE and Gross ML (2011a). Native Electrospray and Electron-Capture Dissociation FTICR Mass Spectrometry for Top-Down Studies of Protein Assemblies. Anal Chem 83, 5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H and Ge Y (2011). Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ Cardiovasc Genet 4, 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Guy MJ, Norman HS, Chen YC, Xu Q, Dong X, Guner H, Wang S, Kohmoto T, Young KH et al. (2011b). Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res 10, 4054–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ogorzalek Loo RR and Loo JA (2015). Increasing Fragmentation of Disulfide-Bonded Proteins for Top-Down Mass Spectrometry by Supercharging. Int J Mass Spectrom 377, 546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL and Gray NS (2009a). Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9, 28–39. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Fonslow BR, Shan B, Baek M-C and Yates JR (2013). Protein Analysis by Shotgun/Bottom-up Proteomics. Chem Rev 113, 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Pan H and Chen X (2009b). Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev 28, 147–76. [DOI] [PubMed] [Google Scholar]
- Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK and McLafferty FW (2000). Electron capture dissociation for structural characterization of multiply charged protein cations. Anal Chem 72, 563–73. [DOI] [PubMed] [Google Scholar]
- Zubarev RA, Kelleher NL and McLafferty FW (1998). Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J Am Chem Soc 120, 3265–3266. [DOI] [PubMed] [Google Scholar]
- Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK and McLafferty FW (1999). Electron Capture Dissociation of Gaseous Multiply-Charged Proteins Is Favored at Disulfide Bonds and Other Sites of High Hydrogen Atom Affinity. J Am Chem Soc 121, 2857–2862. [Google Scholar]