

Abstract
Sepsis is a leading cause of mortality in intensive care unit worldwide, it's accompanied by immune cell dysfunction induced by multiple factors. However, little is known about the specific alterations in immune cells in the dynamic pathogenesis of sepsis secondary to bacterial pneumonia. Here, we used single cell RNA sequencing (scRNA-seq) to profile peripheral blood mononuclear cells (PBMCs) in a healthy control and two patients with sepsis secondary to bacterial pneumonia, including acute, stable and recovery stage. We analyzed the quantity and function of immune cells. During disease course, interferon gamma response was upregulated; T/NK cell subtypes presented activation and exhaustion properties, which might be driven by monocytes through IL-1β signaling pathways; The proportion of plasma cells was increased, which might be driven by NK cells through IFN signaling pathways; Additionally, interferon gamma response was upregulated to a greater degree in sepsis secondary to pneumonia induced by SARS-COV-2 compared with that induced by influenza virus and bacteria.
Keywords: Single cell transcriptomics, Sepsis, Immune cells
1. Introduction
Sepsis is a complex and heterogeneous syndrome with highly variable clinical manifestations [1]. Pneumonia is one of the most common primary cause for sepsis in humans [2,3]. During sepsis, dysregulated host immune response to infecting pathogens lead to lethal organ dysfunction [4]. Among the known infecting pathogens, gram-negative bacteria were the predominant contributor, accounting for 62% positive cultures in patients with severe sepsis [3]. Therefore, the investigation of sepsis secondary to pneumonia infected by gram-negative bacteria is of great significance to understand and treatment of sepsis.
During sepsis, both innate and adaptive immune cells showed long-term dysfunctional signatures even after clinical “recovery” due to immunosuppression [5]. PBMC was the most commonly studied sample for the assessment of systematic immune status in sepsis. The immunosuppression condition was reflected by the reduced quantity and functional defects of immune cells, especially for lymphocytes, including T cells, B cells and NK cells [6]. Among these, T cells with diverse subpopulations including CD4+ T cells and CD8+ T cells present the most notably immunosuppression features [7,8]. Myeloid cells were increased in number but defected in function during sepsis [9]. Among them, monocytes negatively induced T cell proliferation upon endotoxin stimulation [10]. However, these studies were carried out using flow cytometry based on known surface marker genes or in vitro experiments, and could only partially reveal the molecular alterations of immune cells in sepsis.
With the development of transcriptome, the expression alterations of immune cells in sepsis were disclosed comprehensively [[11], [12], [13]]. However, the whole picture of the immune cell composition and the cellular interactions of immune cell subpopulations are still unknown. The emerging of scRNA-seq technology contributes to more comprehensive knowledge about the global landscape of immune cell alterations in sepsis. Although recent studies have employed scRNA-seq to investigate the immune cell signatures in sepsis secondary to urinary-tract infection [14], and sepsis with acute respiratory distress syndrome [15], the specific pathogen and the dynamic immune response associated with disease course have not been fully investigated. In this study, we explored the dynamic alterations in circulating immune cells, from the aspect of composition and function, and dissected the immune response during the pathogenesis of sepsis secondary to pneumonia infected by gram-negative bacteria. Furthermore, we also investigated the mechanisms implicated in the different immunological response of different causes of sepsis including bacteria and viruses.
2. Results
2.1. Global landscape of the immune cell composition during the pathogenesis of sepsis secondary to bacterial pneumonia
To explore the dynamic alterations of the immune cell landscape driven by sepsis secondary to bacterial pneumonia, PBMC was isolated from two patients with sepsis secondary to pneumonia infected by gram-negative bacteria, followed by scRNA-seq with 10× Genomics platform. The clinical features of the two patients were listed in Table S1. Patient samples were collected with three disease stages including acute stage, stable stage and recovery stage. However, sample from the first patient in the acute stage was aborted due to low cell quality. For comparison, we conducted scRNA-seq of PBMC from a healthy control (ID: HC-1), in combination of another four scRNA-seq datasets from healthy controls (ID: HC-2, HC-3, HC-4, HC-5) downloaded from 10× Genomics (https://www.10xgenomics.com/cn/). After stringent quality control, cells from all samples were integrated after batch effect corrected (Fig. 1A, B). Subsequently, a total of 52,412 cells were subjected to further analysis (Fig S1A). Unsupervised clustering identified three clusters, which were dimensionality reduction by uniform manifold approximation and projection (UMAP) (Fig. 1B). All of the three clusters were composed of cells from every sample, indicating well-corrected batch effect (Fig. 1C). These clusters were further annotated as T/NK cells (CD3D + IL32 + NKG7 +), myeloid cells (CD14 + FCGR3A + S100A9 +) and B cells (CD79B + CD79A + IGHM +) based on their canonically cell marker genes (Fig. 1D, E). T/NK cells were involved in lymphocyte activation and adaptive immune response (Fig. 1D). Myeloid cells functioned in myeloid leukocyte activation, cytokine-mediated signaling pathway and response to bacterium (Fig. 1D). B cells were implicated in B cell activation and B cell proliferation (Fig. 1D). Next, we quantified the composition of immune cells in each disease stages and healthy controls to reveal substantial changes during disease progression. T/NK cells and B cells were reduced, whereas myeloid cells were expanded in sepsis secondary to bacterial pneumonia compared with healthy controls (Fig. 1F). Interestingly, unexpected changes were occurred during the disease process. The proportion of T/NK cells was sustained declined, whereas that of myeloid cells was increased continuously across disease stages especially in the recovery stage (Fig. 1F, Fig S1B), which suggested that the disorder of immune cells still existed in the recovery stage.
Fig. 1.

Study design and single-cell transcriptomic landscape of PBMCs from patients with sepsis secondary to bacterial pneumonia and healthy controls.
A. An overview of the study design and workflow. B. Overall cell type composition of 52,412 cells from sepsis patient with acute stage (Acu, n = 1), stable stage (Sta, n = 2) and recovery stage (Rec, n = 2), as well as healthy controls (HCs, n = 5) were visualized with UMAP projection. C. The same UMAP plot related to Fig. 1.B and cells were colored based on disease stage and individual. D. Heatmap showed the differentially expressed genes in three major types of PBMCs. The colour key from purple to yellow indicated low to high expression levels. Enriched GO terms for each cell type (biological processes) were showed at the right. E. Feature plots showed the expression of canonically cell marker genes used to define each cluster. F. Pie plots showed the ratio of each cell type constitution in HCs and sepsis patients at three disease stages. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. S1.

Pathway enrichment analysis in all PBMCs and cell subpopulations
A. The number of cells per sample (upper panel) and mean genes per cells (lower panel) that passed quality control. The error bar indicated mean with standard error. B. Histogram showed the proportion of the major cell types in each healthy controls (HCs) and patient sample. C. Pathway enrichment analysis of significant hallmark gene sets comparing PBMCs from recovery stage with those from stable stage. NES, normalized enrichment score. D. GSEA enrichment plots of interferon gamma response of T/NK cells (stable stage v.s acute stage; recovery stage v.s stable stage). E. GSEA enrichment plots of TNFA signaling via NFκB of myeloid cells (stable stage v.s acute stage; recovery stage v.s stable stage). F. GSEA enrichment plots of MTORC1 signaling of B cells in each disease stage versus HCs, recovery stage versus stable stage.
2.2. Interferon response and TNFA signaling were upregulated during the pathogenesis of sepsis secondary to pneumonia
To further dissect the molecular mechanisms of the dynamic changes underlying the pathogenesis of sepsis secondary to pneumonia, we performed pathway enrichment analysis with all differentially expressed genes between each disease stages and healthy controls, to find biological pathways altered in disease condition at a global insight (Fig. 2A, B, C). Patients in all of the three stages showed upregulated interferon gamma response compared to healthy controls. Interestingly, patients in the recovery stage also showed upregulated interferon gamma response and interferon alpha response compared with stable stage (Fig S1C). Moreover, IFN response genes including IFNGR1, IFITM2, IFITM3, IFI6, ISG15 and ISG20 were increased in disease progression (Fig. 2D), suggesting sustained effects of interferon response in the disease course. In addition, TNFA signaling via NFκB was also upregulated in the acute and recovery stages compared to healthy controls (Fig. 2A, C). Patients in recovery stage also showed upregulated TNFA signaling via NFκB compared with stable stage. Genes involved in this pathway such as NFκBIA, JUNB, TNFAIP3, CCL4 and SOCS3 were highly expressed in disease stages compared with healthy controls (Fig. 2D). To explore whether these pathway alterations among disease course and healthy controls were driven by certain cell subtypes, we further expanded pathway enrichment analysis in all immune cell subpopulations. Surprisingly, interferon gamma response was upregulated in all disease stages in T/NK cells (Fig. 2E, Fig S1D), indicating T/NK cells may contribute to the upregulation of interferon gamma response in disease progression. In contrast, TNFA signaling via NFκB was upregulated in the acute and recovery stages of myeloid cells compared with healthy controls (Fig. 2F), suggesting that myeloid cells may be associated with the upregulation of TNFA signaling via NFκB in disease stages. Interestingly, TNFA signaling via NFκB was downregulated in stable stage compared with the acute stage, whereas was upregulated in the recovery stage compared with stable stage, suggesting the resolution of inflammation in stable stage but unexpected aggregation of inflammatory features in the recovery stage of myeloid cells (Fig S1E). However, in B cells, the MTORC1 signaling was upregulated in all disease stages, and was downregulated in the recovery stage compared to the stable stage (Fig S1F). This signaling was only upregulated in the acute stages compared with healthy controls for all PBMCs (Fig. 2A). All of these data indicated the global alterations of signaling pathways in sepsis secondary to pneumonia was possibly associated with T/NK cells and myeloid cells.
Fig. 2.

Interferon response and TNFA signaling were upregulated during the pathogenesis of sepsis secondary to bacterial pneumonia.
Pathway enrichment analysis of significant hallmark gene sets comparing PBMCs from A. acute stage, B. stable stage and C. recovery stage with those from healthy controls (HCs). NES, normalized enrichment score. D. Dot plots showed the scaled expression level and percentage of key genes involved in interferon gamma response and TNFA signaling via NFκB and of PBMCs in HCs versus disease courses. The colour key from gray to purple indicated low to high expression levels. The dot size indicated the percentage of cells that expressed genes. E. GSEA enrichment plot of interferon gamma response of T/NK cells in disease courses versus HCs. F. GSEA enrichment plot of TNFA signaling via NFκB of myeloid cells in disease courses versus HCs. G. Genes were clustered according to their expression patterns during disease progression. Enriched GO terms for each cell type (biological processes) were showed at the right. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Next, we explored the dynamically transcriptional alterations in gene expression during disease progression. Eight clusters with different time-dependent expression patterns were found, the biological functions of genes in these clusters were also assessed (Fig. 2G). Cluster1 comprised 2989 genes with declined expression along disease course. The function of genes in cluster1 were enriched in autophagy (Fig. 2G). Previous studies demonstrated that the repair of autophagy lead to the dysfunction of proximal tubular in sepsis [16], indicating the dynamic gene expression in cluster1 may be associated with kidney injury in sepsis. Cluster 6 contained 2283 genes with sustained increased expression during disease stages. These genes were enriched in regulation of type I interferon production (Fig. 2G), which was consistent with the sustained upregulation of the interferon response genes in disease stages (Fig. 2D).
2.3. T and NK cells presented sustained exhaustion and apoptosis features in sepsis secondary to bacterial pneumonia
Given that T and NK cells were the major populations in healthy controls, and showed a sustained decrease in the process of sepsis secondary to bacterial pneumonia, the compositional and molecular changes of them across the disease stages may reflect the severity of disease progression. Thus, we next re-clustered the T/NK cells to dissect the dynamic changes of them with a finer resolution. Ten clusters were generated and were visualized via UMAP plot (Fig. 3A). These clusters were classified with canonically cell marker genes into four CD4+ T cell subtypes (CD3D + CD4 +), three CD8+ T cell subtypes (CD3D + CD8A + CD8B +), NKT cells (CD3D + NCR3 +), NK cells (FGFBP2 + NKG7 +) and proliferating T cells (Pro-T, MKI67 + STMN1 +). Among these, CD4+ T cell subtypes included CD4+ Tn cells (CD4 + CCR7 +), CD4+ Tpm cells (LTB + IL7R +), Treg cells (IL2RA + FOXP3 +) and CD4+ CD28+ T cells (CD4 + CD28 +). Additionally, CD8+ T cells were divided into CD8+ Tn cells (CD8A + CCR7 +), CD8+ Te cells (CCL5 + GZMB +) and CD8+ Tem cells (GZMA + GZMK +) (Fig. 3B). To further understand the function of each subtype, we performed Gene Ontology (GO) analysis. CD8+ Tem cells, Treg cells, pro-T cells, CD8+ Te cells and NK cells were showed to be involved in response to interferon-gamma (Fig. 3C). Additionally, the expression patterns of interferon-gamma response genes including interferon receptor (IFNGR1), ISG15, IFI44L, IFI6 and IFITM3 and were different, with an elevated expression levels in almost all T and NK cell subtypes during disease course compared with healthy controls (Fig S.2A). All subtypes were enriched in lymphocyte activation (Fig. 3C). Meanwhile, CD69, a marker of T cell activation, was also highly expressed in disease conditions compared with healthy controls, suggesting the activated immunological response in T and NK cells (Fig. 3D). However, CD4+ Tpm cells, NKT cells, CD4+ CD28+ T cells, CD8+ Te cells and NK cells were involved in positive regulation of apoptotic process, suggesting immunosuppression states of T cells (Fig. 3C). Immune suppression in sepsis was mainly characterized by the apoptosis and exhaustion of lymphocytes, especially T cells [17]. Further examination of the expression of pro-apoptotic associated genes, such as TNFSF10, TNFRSF1B, BCL2L11 and CASP3 and T cell exhaustion associated markers including PRDM1 and LAG3 were upregulated during disease progression in all T and NK cell subtype expect for CD4+ CD28+ T cells (Fig. 3D).
Fig. 3.

T and NK cells presented sustained exhaustion and apoptosis features in sepsis secondary to bacterial pneumonia.
A. Sub-clustering of T and NK cells in healthy controls (HCs) and sepsis patients. B. Violin plots showed the scaled expression levels of canonical markers used to identify each cell types. C. Heatmap of enriched GO terms for each cell type (biological processes). D. Dot plots showed the expression of genes that involved in T cell exhaustion, apoptosis and activation in HCs and disease stages. The colour key from gray to purple indicated low to high expression levels. The dot size indicated the percentage of cells that expressed genes. E. Histogram showed the proportion of each cell type in HCs and sepsis patients at three disease stages. F. Sub-clustering of NK cells in HC and sepsis patients. G. Heatmap of enriched GO terms for each NK cell type (biological processes). H. Histogram showed the proportion of each NK cell type in HCs and sepsis patients at three disease stages. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We next investigated the immunological changes implicated in T and NK cell subtypes during the progression of sepsis secondary to bacterial pneumonia. For the perspective of cell composition, T and NK cells displayed a divergent landscape between disease conditions and healthy controls. CD4+ Tn cells and CD8+ Tn cells were attenuated in sepsis patients, indicating the immunosuppression status of these cell subpopulations. Both of these two cell types showed a similar decreasing trend along disease progress, with initially declined in the acute stage, slightly increased in the stable stage and further decreased in the recovery stage (Fig. 3E). By contrast, the proportion of CD8+ Te cells and CD8+ Tem cells were risen in disease course, which agreed with their function in lymphocyte activation, indicating enhanced immune effector effects of them. Similar ascension trend was observed in CD8+ Te cells and CD8+ Tem cells along disease stages, which increased in the acute stage, slightly dropped in the stable stage, and increased continually in the recovery stage (Fig. 3E). Additionally, the proportion of NK cells was increased in disease process compared to healthy controls. While the proportion of NK cells were comparable between healthy controls and the acute stage, it was gradually increased in the following two disease stages (Fig. 3E).
To further explore the cellular and functional alterations in NK cells. We performed re-cluster again and divided NK cells into two clusters: NK1 and NK2 (Fig. 3F). NK1 highly expressed FGFBP2, GZMB and FCGR3A, involved in response to bacterium, phagocytosis and I-kappaB kinase/NF-kappaB signaling (Fig S2B, Fig. 3G). NK2 highly expressed GZMK, XCL1 and KLRC1, involved in lymphocyte activation, cytokine-mediated signaling pathway and cell killing (Fig S2B, Fig. 3G). Cellular composition analysis revealed the dynamic alterations of NK cell subtypes. NK1 was the predominant subtype of NK cells. Surprisingly, NK1 was increased in the acute and stable stage, then dropped to similar level in healthy controls, whereas NK2 showed the reverses trend, indicating NK cells recovered in the aspect of cell proportion in convalescent patients (Fig. 3H). However, NK cells expressed high levels of exhaustion and apoptosis marker genes in disease stages even though in the recovery stage, indicating the dysfunction of NK cells still remained in convalescent patients despite the recovered cell composition (Fig. 3D). With regard to the functional features of T/NK cells that induced their phenotypic shifting during the disease progression, regulation of cell activation was significantly upregulated in the acute stage and recovery stage (Fig S2C, E). Granzyme-mediated apoptotic signaling pathway was markedly unregulated in the stable and recovery stage (Fig S2D, E). Particularly, positive regulation of apoptotic process showed upregulated in the stable stage, reinforcing the immunosuppression status of T/NK cells in the stable stage (Fig S2D). Moreover, positive regulation of cytokine production, antigen processing and presenting of exogenous peptide antigen via MHC class II were upregulated in the recovery stage (Fig S2E).
Fig. S2.

Functional features of T/NK cells that upregulated in patients with sepsis secondary to bacterial pneumonia
A. Dot plots showed the expression level and percentage of interferon gamma response genes of T/NK cells in healthy controls (HCs) and disease stages. The colour key from gray to purple indicates low to high expression levels. The dot size indicated the percentage of cells that expressed genes. B. Violin plots showed the scaled expression levels of canonically cell marker genes used to identify each NK cell types. Upregulated GO terms in disease stages (C. acute stage, D. stable stage, E. recovery stage) versus healthy controls of T/NK cells. GO terms were labeled with name and ID, and sorted by -log10(P) value.
2.4. Myeloid cells displayed dysregulation mainly in the recovery stage
In our data, myeloid cells were the predominantly altered cell population at different disease stages, indicating a strongly immunological response to sepsis. We then re-clustered the total myeloid cells to further explore the dynamically transcriptional alterations induced by sepsis secondary to bacterial pneumonia. Subsequently, eight clusters were obtained and were visualized using UMAP plot (Fig. 4A). These clusters were defined as six monocyte subtypes (CD14 + /FCGR3A +), megakaryocytes (MK, PF4 + PPBP +), and dendritic cells (DC, JCHAIN + SEC11C +) according to their canonically cell marker genes. Furthermore, the monocytes population consisted of CD14+ Monocyte1 (CD14+ Mono1: CD14 + CPVL +), CD14+ Monocyte2 (CD14+ Mono2: S100A8 + VCAN +), CD16+ Monocyte (CD16+ Mono: FCGR3A + MS4A7 +), HLA-DR+ Monocyte (HLA-DR+ Mono: HLA-DR + CD74 +), CD24+ Monocyte (CD24+ Mono: CD24 + CAMP +) and LDHB+ Monocyte (LDHB+ Mono: LDHB + STMN1 +) (Fig. 4B). The functional features of these cell subpopulations were diverged and were revealed by GO analysis. Almost all cell subtypes were involved in regulation of cytokine production and response to bacterium, indicating the activated immunological status of monocytes (Fig. 4C). CD14+ Mono2, CD16+ Mono, CD24+ Mono, HLA-DR+ Mono and LDHB+ Mono were involved in lymphocyte activation (Fig. 4C). However, CD14+ Mono2 and CD16+ Mono were enriched in positive regulation of cell death, apoptotic signaling pathway, negative regulation of cell proliferation and negative regulation of immune system process (Fig. 4C), indicating that the myeloid cells particularly monocytes may be involved in the regulation of both activation and apoptosis of lymphocyte.
Fig. 4.

Myeloid cells displayed functional dysregulation mainly in the recovery stage.
A. Sub-clustering of myeloid cells in healthy controls (HCs) and sepsis patients. B. Violin plots showed the scaled expression levels of canonically cell marker genes used to identify each cell types. C. Heatmap of enriched GO terms for each cell type (biological processes). D. Histogram showed the proportion of each myeloid cell type in HCs group and different stages of patient groups. E. Enriched GO terms for genes that upregulated in the recovery stage compared with HCs.
As myeloid cells showed a continuously increasing trend across three disease stages compared with healthy controls, we next investigated whether this phenotypic remodeling was existed with more meticulous scenario. Thus, we gained insights into the compositional alterations during disease progression. Unexpectedly, the proportion of myeloid cell subtypes was comparable in the acute and stable stages compared with healthy controls. A newly discovered cell type of monocytes named CD24+ Mono, was specifically emerged in the recovery stage, which functioned in regulated exocytosis, response to bacterium and defense response to other organisms (Fig. 4C, D). However, this new cell type was mainly derived from the first patient, indicating heterogeneous properties among individual patient with sepsis (Fig S3). Consistently, functional alterations of myeloid cells, with differentially expressed genes between disease stages and healthy controls, were concentrated on the recovery stage, specifically, the function of myeloid cells in the recovery stage were myeloid leukocyte activation, positive regulation of cytokine production, macrophage and response to molecule of bacterial origin (Fig. 4E), suggesting activated immunological response implicated in myeloid cells still remained in the convalescent patient.
Fig. S3.
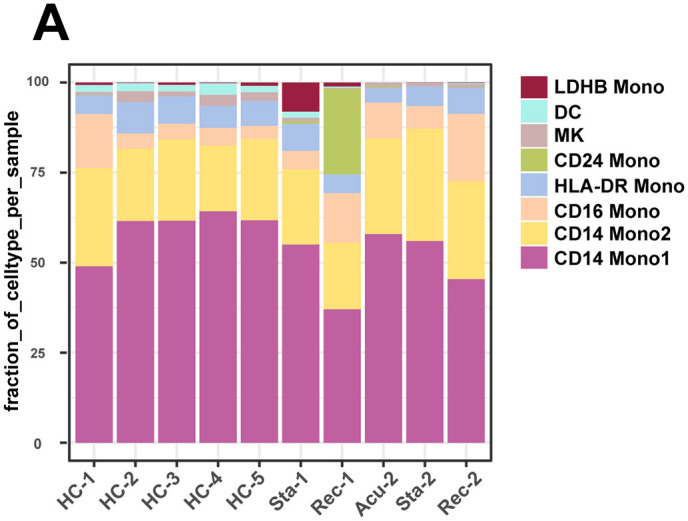
Myeloid cells composition in each healthy controls and patients
2.5. Expansion of plasma cells in the process of sepsis secondary to pneumonia
B cells were depleted in sepsis secondary to bacterial pneumonia. However, there was no clear alterations in the proportion of them among disease stages, which prompted us to explore the cellular and molecular changes in a finer fashion. Thus, B cells were re-clustered into three clusters and dimensionality reduction with UMAP (Fig. 5A). Cluster 0 expressed high levels of TCL1A, IGHD and IL4R, involved in regulation of immune effector process, B cell proliferation and lymphocyte, was defined as B naïve cells (Bn) (Fig. 5B, C). Cluster 1 highly expressed AIM2, TNFRSF13B and CD27, associated with B cell differentiation, was identified as B memory cells (Bm) (Fig. 5B, C). Cluster 2 was annotated as plasma cells due to high expression level of MZB1, IGHG3 and JCHAIN (Fig. 5B). Moreover, plasma cells were enriched with varies functions including antigen processing and presentation, signal peptide processing, antigen processing and presentation of peptide antigen via MHC class I and leukocyte activation involved in immune response (Fig. 5C). In accordance with the multiple functions implicated in plasma cells, the proportion of them was observed to be increased in different disease stages compared with healthy controls, and reached a peak in the stable stage then relatively declined in the recovery stage (Fig. 5D). In line with this, with regard to the activation of plasma cells in disease course versus healthy controls, leukocyte activation involved in immune response was conserved (Fig S4A-C), reinforcing the essential roles of plasma cells proliferation in the disease process.
Fig. 5.

Expansion of plasma cells in the process of sepsis secondary to pneumonia.
A. Sub-clustering of B cells in healthy controls (HCs) and sepsis patients. B. Violin plots showed the scaled expression levels of canonically cell marker genes used for annotating each cell types. C. Heatmap of enriched GO terms for each cell type (biological processes). D. Histogram showed the proportion of each B cell type in HCs groups and different courses of disease. E. Violin plots showed the log2(TPM/10 + 1) expression levels of HLA-DRA in disease stages versus HCs. F. Network of the potential signaling paths between the ligand IFNG and its predicted target gene, and the signaling/transcriptional regulators in these paths were visualized (blue: ligand; red: target genes; yellow: signaling/transcriptional regulators). The thickness of edge lines represented the weight of the interactions in the weighted integrated networks. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. S4.

Functional alterations of plasma cells in the process of sepsis secondary to bacterial pneumonia
Upregulated GO terms in disease stages (A. acute stage, B. stable stage, C. recovery stage) versus HCs of T/NK cells. GO terms were labeled with name and ID, and sorted by -log10(P) value. Heatmap showed the predicted ligand activity and the regulatory potential of the ligand and their target genes. Ligands of NK1 cells and NK2 cells and their target genes in plasma cells in the D. acute stage and E. stable stages compared with healthy controls (orange: ligand; purple: target genes).
Next, we sought to investigate the potential mechanisms underlying the proliferation of plasma cells in the acute and stable stage. Intriguingly, NK cells were observed to be enriched in B cell proliferation and leukocyte activation involved in immune response, as well as positive regulation of apoptotic process (Fig. 3C). Therefore, we tried to explore the potential regulation network between NK cells and plasma cells to dissect the effects of NK cells on plasma cells proliferation. In the acute and stable stages, IFNG and HLA-DRA showed enriched regulatory potential of ligand and target genes (Fig S4D, E). Interestingly, the change patterns of HLA-DRA expression level in three disease stages compared with healthy controls were similar with the alterations of plasma cells proportion (Fig. 5E), suggesting that the alterations of HLA-DRA expression were parallel with the proliferation of plasma cells. To explore the regulatory mechanisms of IFNG on HLA-DRA, the potential signaling pathways were inferred between IFNG and HLA-DRA, IFNG may regulate HLA-DRA through EP300, STAT1, SPI1 and RFX5 (Fig. 5F), indicating the potential cell-cell interactions of NK cells on plasma cells exert crucial roles in the proliferation of plasma cells through IFNG signaling pathways.
2.6. Monocytes might facilitate the proliferation and exhaustion of T cells through IL1B signaling pathways
T cells presented both immunological activation and suppression status, varies subpopulations of monocytes were observed to regulate lymphocyte activation and involved in apoptosis pathways. Additionally, it was shown that T cell function was suppressed in a monocyte dependent fashion in sepsis patients [18]. We next aimed to explore whether intracellular interaction exists between monocytes and T cells. Thus, we constructed a putative cellular interaction network between monocytes and T cells in different disease stages compared with healthy controls to investigate whether monocytes impact the activation and suppression of T cells.
For the purpose of exploring the roles of monocytes on T cell activation, we selected subsets of monocytes including CD14+ Mono2, CD16+ Mono, CD24+ Mono, HLA-DR+ Mono and LDHB+ Mono as sender cells because they were highly associated with lymphocyte activation (Fig. 4C). CD8+ Te cells and CD8+ Tem cells were chosen as receiver cells due to their sustained increasing proportion across disease stages versus healthy controls. NicheNet [19] was employed to predict interactions between selected monocytes and T cells based on differential expressed genes in CD8+ Te cells and CD8+ Tem cells upon disease induction. For CD8+ Te cells, ligand -target interactions were centered on the recovery stage, whereas were sparse in the acute stages (Fig. 6A, Fig S5A). Concurrently, the ligand-target interactions between selected monocytes and CD8+ Tem cells were also enriched in the recovery stage (Fig. 6B). Thus, we focused on the ligand-mediated intracellular interactions mainly in the recovery stage. Interestingly, among the top predicted ligands, we found IL1B was expressed by selected monocytes in regulation of both CD8+ Te cells and CD8+ Tem cells. Moreover, S100A9, JUN and S100A8 were the common target genes in CD8+ Te cells and CD8+ Tem cells potentially regulated by IL1B (Fig. 6A, B). Further analysis of inferring the potential signaling pathways between IL1B and its target genes discovered some transcriptional regulators including SPl1, RELA, FOS, EP300, STAT3, IRAK1 and MYC (Fig. 6C).
Fig. 6.

Monocytes might facilitate the proliferation and exhaustion of T cells through IL1B signaling pathways.
Heatmap showed the predicted ligand activity and the regulatory potential of the ligand and their target genes. Ligands of CD14+ Mono2, CD16+ Mono, CD24+ Mono, HLA-DR+ Mono and LDHB+ Mono and their target genes in A. CD8+ Te cells, B. CD8+ Tem cells in the recovery stage compared with healthy controls (HCs) (orange: ligand; purple: target genes). C. Network of the potential signaling pathways between the ligand IL1B and it predicted target genes, and the signaling/transcriptional regulators in these paths were visualized in recovery stage. (red: ligand; yellow: target genes; green: signaling/transcriptional regulators; blue: receptor). The thickness of edge line represented the weight of the interactions in the weighted integrated networks. Ligands of CD14+ Mono2, CD16+ Mono and their target genes in D. CD4+ Tn cells, E. CD8+ Tn cells in the acute stage compared with HCs (orange: ligand; purple: target genes). F. Network of the potential signaling pathways between the ligand IL1B and it predicted target genes, and the signaling/transcriptional regulators in these paths were visualized in acute stage. (red: ligand; yellow: target genes; green: signaling/transcriptional regulators; blue: receptor). The thickness of edge line represented the weight of the interactions in the weighted integrated networks. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. S5.

Monocytes may regulate the proliferation and exhaustion of T cells during disease course
Heatmap showed the predicted ligand activity and the regulatory potential of the ligand and their target genes. A. Ligands of CD14+ Mono2, CD16+ Mono, CD24+ Mono, HLA-DR+ Mono and LDHB+ Mono and their target genes in CD8+ Te cells in the acute stage compared with healthy controls (HCs).
B. Ligands of CD14+ Mono2, CD16+ Mono and their target genes in CD4+ Tn cells in the recovery stage compared with HCs. C. Ligands of CD14+ Mono2, CD16+ Mono and their target genes in CD8+ Tn cells in the stable stage versus healthy controls (orange: ligand; purple: target genes). D. Ligands of CD14+ Mono2, CD16+ Mono and their target genes in CD8+ Tn cells in the recovery stage compared with HCs.
To investigate the regulatory relationship and potential mechanisms in monocytes on T cells suppression, we selected CD14+ Mono2 and CD16+ Mono as sender cells, due to their function in positive regulation of cell death, apoptotic signaling pathway, negative regulation of cell proliferation and negative regulation of immune system process (Fig. 4C). Additionally, CD4+ Tn cells and CD8+ Tn cells were served as the receiver cells due to their continuously declined proportion across disease course (Fig. 3E).

Fig. 7. Different immunological response presented between sepsis secondary to pneumonia induced by bacteria, influenza virus and SARS-COV-2. A. Violin plots showed the log2(TPM/10 + 1) expression of genes enriched in interferon gamma response of sepsis secondary to pneumonia induced by bacteria, influenza virus and SARS-COV-2 for each patient. B. Pie plots showed the ratio of major cell type composition in sepsis secondary to pneumonia induced by bacteria, SARS-COV-2 and influenza virus. C. Histogram showed the proportion of each cell type in sepsis secondary to pneumonia induced by bacteria, SARS-COV-2 and influenza virus. D. GSEA enrichment plot of interferon gamma response of CD14+ Mono1 cells, NK1 cells, CD4+ Tpm cells, CD8+ Tem cells, CD8+ Te cells, Bm cells and Bn cells in sepsis secondary to pneumonia induced by bacteria versus SARS-COV-2. E. Dot plots showed the scaled expression level and percentage of exhaustion and apoptotic genes of T/NK cells in sepsis secondary to pneumonia induced by influenza virus, SARS-COV-2 and bacteria. The colour key from gray to blue indicated low to high expression levels. The dot size indicated the percentage of cells that expressed genes. Bac, bacteria; SARS, SARS-COV-2; Influ, influenza virus. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
For CD4+ Tn cells, ligand-target interactions were concentrated in the acute and stable stages, but were not predicted in the recovery stage for no differential expression genes were found between recovery stage and healthy controls (Fig. 6D, Fig S5B). Further analysis on CD8+ Tn cells showed that ligand-target interactions were enriched in all disease stages, suggesting the selected monocytes may exert their regulatory functions on CD8+ Tn cells during disease progression (Fig. 6E, Fig S5C, D). To dissect the common mechanisms of regulatory relationships underlying the suppression of CD4+ Tn cells and CD8+ Tn cells induced by monocytes, we looked for the shared ligands and their target genes. Surprisingly, IL1B was also expressed by selected monocytes in regulating both CD4+ Tn cells and CD8+ Tn cells (Fig. 6D, E). Concurrently, PRDM1 and ITGB1 were among the commonly predicted target genes in CD4+ Tn cells and CD8+ Tn cells driven by IL1B (Fig. 6D, E). Of particular interest, PRDM1 was found to be upregulated in T cell subtypes across disease stages, supporting that monocytes may promote the exhaustion of both CD4+ Tn cells and CD8+ Tn cells with the target of PRDM1. Furthermore, some transcription regulators such as SPl1, IRAK1, STAT3, RELA, SMAD3 and PRTN3 were observed to be implicated in the IL1B-PRDM1 signaling pathways (Fig. 6F). Altogether, these data indicated that monocytes may stimulate both the activation and exhaustion of T cell through IL1B signaling pathway with different target genes.
2.7. Different immunological response presented between sepsis secondary to pneumonia induced by bacteria and viruses
A recent study found that sepsis patients induced by bacteria and severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) were different in organ dysfunction, outcome, and counts of T lymphocytes and their subtypes [20]. However, their difference in immunological response was unclear. Thus, it is vital for comparing the immunological difference of different cause of sepsis in the acute disease stage. First, we downloaded the scRNA-seq data of two severe coronavirus distress syndrome (COVID-19) patients from a previously published study [21]. One severe COVID-19 patient was diagnosed with septic shock, while another one presented multi-organ failure. Second, to gain a more comprehensive knowledge about the difference in immunological response in sepsis of different causes, we also downloaded a public scRNA-seq dataset of sepsis secondary to pneumonia induced by influenza virus [15]. Thus, these three scRNA-seq datasets were further integrated with two scRNA-seq datasets of the acute stage from sepsis secondary to bacterial pneumonia, of which, one dataset was from our data, another one was downloaded from a recently published study [15].
Pathway enrichment analysis showed that interferon gamma response was upregulated in SARS-COV-2 induced sepsis compared with bacterial sepsis, and in SARS-COV-2 induced sepsis compared with influenza virus induced sepsis, as well as in influenza virus induced sepsis compared with bacterial sepsis (Fig S6A-C). Additionally, the interferon response related genes such as IFITM2, IFITM3, ISG20 and ISG15 were highly expressed in all of the three causes of sepsis, among which, the altered expression of IFITM2 and IFITM3 among three causes of sepsis were in line with the alterations of interferon gamma response among them (Fig. 7A). These data suggested that the interferon gamma response may be aberrant with highest degree in SARS-COV-2 induced sepsis, secondly in influenza virus induced sepsis and last in bacterial sepsis.
Fig. S6.

Pathway enrichment analysis of significant hallmark gene sets with pairwise comparison in sepsis secondary to pneumonia induced by bacteria, influenza virus and SARS-COV-2.
Pathway enrichment analysis of significant hallmark gene sets with pairwise comparison in A. sepsis induced by SARS-COV-2 versus sepsis induced by bacteria, B. sepsis induced by SARS-COV-2 versus sepsis induced by influenza virus, C. sepsis induced by influenza virus versus sepsis induced by bacteria.
Next, we attempted to explore the cellular immunological difference behind viral sepsis and bacterial sepsis. For the purpose of an unbiased comparison, the cell subtypes of these downloaded scRNA-seq datasets were annotated according to the acute stage of sepsis secondary to bacterial pneumonia in our study with SciBet [22]. As a result, all of the cell subpopulations were annotated in these downloaded scRNA-seq datasets (Fig. 7B, C). The proportion of T/NK cells was lower in both SARS-COV-2 induced sepsis and influenza virus induced sepsis compared with bacterial sepsis (Fig. 7B). GO analysis showed that defense response to other organism; response to bacterium and apoptotic signaling pathway were enriched when differentially expressed genes were compared with SARS-COV-2 induced sepsis and bacterial sepsis (Fig S7A). Apoptotic signaling pathway and regulation of immune effector process were involved when differentially expressed genes were compared with SARS-COV-2 induced sepsis and influenza virus induced sepsis (Table S4). Moreover, cellular response to type I interferon and type I interferon signaling pathway were upregulated when differentially expressed genes were compared with influenza virus induced sepsis and bacterial sepsis (Table S4). However, the proportion of myeloid cells and B cells were higher in both SARS-COV-2 induced sepsis and influenza virus induced sepsis compared with bacterial sepsis (Fig. 7C). GO analysis showed that antimicrobial humoral immune response and response to virus were enriched in myeloid cells when compared with SARS-COV-2 induced sepsis and bacterial sepsis (Fig S7B). Apoptotic signaling pathway and process utilizing autophagic mechanism were involved when differentially expressed genes were compared with SARS-COV-2 induced sepsis and influenza virus induced sepsis (Table S4). In addition, activation of immune response and regulation of acute inflammatory response were upregulated when differentially expressed genes were compared with influenza virus induced sepsis and bacterial sepsis (Table S4). In B cells, complement activation, classical pathway; B cell receptor signaling pathway and B cell proliferation were enriched when compared with SARS-COV-2 induced sepsis and bacterial sepsis (Fig S7C). Regulation of lymphocyte activation and immune response-activating signal transduction were involved when differentially expressed genes were compared with SARS-COV-2 induced sepsis and influenza virus induced sepsis (Table S4). Furthermore, interferon-gamma-mediated signaling pathway and type I interferon signaling pathway were upregulated when differentially expressed genes were compared with influenza virus induced sepsis and bacterial sepsis (Table S4). We then focused on the immunological difference underlying cell subpopulations with a finer resolution. For innate immune response, the percentage of CD14+ Mono1 and NK cells especially NK1 cells was highest in SARS-COV-2 induced sepsis, secondly in bacterial sepsis, and last in influenza virus induced sepsis (Fig. 7C). Moreover, the proportion of CD16+ Mono was higher in influenza virus induced sepsis compared with SARS-COV-2 induced sepsis and bacterial sepsis (Fig. 7C). However, with regard to adaptive immune response, the proportion of CD4+ Tn cells was highest in bacterial sepsis, secondly in SARS-COV-2 induced sepsis and last in influenza virus induced sepsis, whereas the percentage of Bm cells was highest in influenza virus induced sepsis, secondly in SARS-COV-2 induced sepsis and last bacterial sepsis (Fig. 7C). Additionally, the proportion of CD4+ Tpm cells, CD8+ Tn cells, and CD8+ Tem cells were highest in bacterial sepsis, secondly in influenza virus induced sepsis and last in SARS-COV-2 induced sepsis (Fig. 7C). Conversely, the percentage of Bn cells were highest in SARS-COV-2 induced sepsis, secondly in influenza virus induced sepsis and last in bacterial sepsis (Fig. 7C). Furthermore, the proportion of CD8+ Te cells was lower in SARS-COV-2 induced sepsis compared with bacterial sepsis and influenza virus induced sepsis (Fig. 7C).
Fig. S7.

Functional difference of immune cells between sepsis secondary to pneumonia induced by bacteria and SARS-COV-2.
GO terms of differentially expressed genes of sepsis secondary to pneumonia induced by bacteria and SARS-COV-2 in A.T/NK cells, B. myeloid cells and C. B cells.
Interestingly, pathway enrichment analysis revealed upregulated interferon gamma response in the above-mentioned cell subtypes when comparing three causes of sepsis. Interferon gamma response was upregulated in CD14+ Mono1, NK1 cells, CD4+ Tpm cells, CD8+ Tem cells, CD8+ Te cells, Bm cells and Bn cells in SARS-COV-2 induced sepsis compared with bacterial sepsis (Fig. 7D), whereas was upregulated in CD4+ Tn cells, CD4+ Tpm cells, CD16+ Mono, and Bn cells in SARS-COV-2 induced sepsis compared with influenza virus induced sepsis (Fig S8A). In addition, interferon gamma response was also upregulated in CD8+ Tem cells, NK1 cells, CD4+ Tn cells, Bn cells, CD4+ Tpm cells and CD8+ Tn cells in bacterial sepsis compared with influenza virus induced sepsis (Fig S8B), indicating that interferon gamma response may contribute to the immunological difference underlying sepsis secondary to pneumonia induced by gram-negative bacteria, SARS-COV-2 and influenza virus. Previous studies have suggested that T cells of SARS-COV-2 patients showed immune exhaustion and apoptotic features [23,24]. Thus, we investigated whether exhaustion and apoptotic features presented difference in T cells of bacterial sepsis, SARS-COV-2 induced sepsis and influenza virus induced sepsis. Surprisingly, the exhaustion and apoptotic levels were highest in SARS-COV-2 induced sepsis, secondly in influenza virus induced sepsis and last in bacterial sepsis as reflected by the expression of PRDM1, CASP1, TNFSF10 and TNFRSF1B (Fig. 7E).
Fig. S8.

Pathway enrichment analysis of cell subpopulations in sepsis secondary to pneumonia induced by bacteria, influenza virus and SARS-COV-2.
A. GSEA enrichment plot of interferon gamma response of CD4+ Tn cells, CD4+ Tpm cells, CD16+ Mono cells and Bn cells in sepsis secondary to pneumonia induced by SARS-COV-2 versus influenza virus. B. GSEA enrichment plot of interferon gamma response of CD8+ Tem cells, NK1 cells, CD4+ Tn cells, Bn cells, CD4+ Tpm cells and CD8+ Tn cells in sepsis secondary to pneumonia induced by bacteria versus influenza virus.
3. Discussion
It is established that sepsis altered both the innate and adaptive immune response for a long period after clinical “recovery” [5], which prompted us to explore the detailed alterations of immune response and discover potential therapeutic treatment. However, global picture of immune cell dysfunction cannot be obtained with regard to all subpopulations using conventional bulk RNA sequencing (bulk RNA-seq). The emerging of scRNA-seq technology helps us to understand the cellular and molecular features with higher resolution and accuracy in sepsis-induced immune dysregulation [14,15,25]. However, considering the highly heterogeneous properties of sepsis regarding primary cause and infecting pathogens, their immune response may be divergent. A study has shown that intra-abdominal sepsis and pneumonia-derived sepsis presented different immune response, manifested by different number of immune cell subpopulations [7]. Thus, it is difficult to popularize findings of certain kind of cause to sepsis to the other divergent ones related to therapeutic target or mechanisms. Furthermore, the timeline of immunological alterations during sepsis progression was not known in single cell resolution, which is essential for the discovery of effective therapeutic target and diagnostic biomarkers. To address these limitations, we performed scRNA-seq to explore the global immunological changes in sepsis secondary to pneumonia infected by gram-negative bacteria, elucidating the dynamic cellular and molecular signatures along disease course, as well as the cellular interactions among immune cell subpopulations.
The novelty of our work was that we illustrated the dynamic immune signatures in patients with sepsis secondary to pneumonia infected by gram-negative bacteria. First, the major immune cell subpopulations were changed during disease progression, with myeloid cell escalated, T/NK cells and B cells decreased, suggesting shifted phenotypic alterations of immune cells induced by sepsis. Second, patients with sepsis showed strong and sustained upregulation of IFN gamma response and TNFA signaling via NFκB in the whole immune cells and major cell subtypes. Third, immune activation was observed in many cell subtypes including CD8+ Te cells, CD8+ Tem cells and plasma cells, manifested by elevated proportions of these cell types. Interestingly, NK cells were also expanded in disease course, although slightly decreased in the acute stage, which was contradicted with a previous study suggesting the apoptosis of NK cells in sepsis [26]. However, this might because only the acute stage of sepsis was focused on in these studies, prompting further studies on why NK cells were expanded in the disease progression especially at later recovery stage and their potential effects on sepsis. Here, we found NK cells might promote the proliferation of plasma cells, the potential mechanisms may depend on IFNG signaling pathway. Meanwhile, extensive immune exhaustion was observed in almost all T/NK cells subpopulations particularly in CD4+ Tn cells and CD8+ Tn cells across disease stages, further coupled with declined proportions of T/NK cells, indicating both quantitative and qualitative defects of T/NK cells in disease progression. Fourth, intracellular interaction analysis showed that monocytes subsets may facilitate the activation and exhaustion of T cells through IL1B signaling pathways with different target genes during recovery stage and acute stage, respectively. Fifth, the newly discovered CD24+ monocytes were almost exclusively existed in the recovery stage of the first patient, further cohort including sepsis patients of three stages with flow cytometry was also unable to validate the high proportions of this subset in the recovery stage (data not shown), indicating the heterogeneous nature of sepsis and emphasizing the importance of personalized and precision medicine. Finally, the immunological response in sepsis induced by gram-negative bacteria, SARS-COV-2 and influenza virus was different in the aspect of cellular composition and gene features, which indicated that further investigations on sepsis induced by different pathogens are warranted for the effective treatment of sepsis.
The immunological features were different in sepsis secondary to pneumonia infected by gram-negative bacteria, SARS-COV-2 and influenza virus in the aspect of cellular composition and enriched pathways. Our data indicated that beside bacterial sepsis, interferon gamma response was also upregulated in sepsis induced bySARS-COV-2 and influenza virus, which was in agreement with previous findings in SARS-COV-2 and influenza virus. For instance, interferon related signaling pathways were also upregulated in severe COVID19 patients [27]. In addition, interferon gamma signaling was enriched in influenza virus-infected human nasal epithelial cells [28]. These findings indicated that interferon gamma response might be conserved in different cause induced sepsis with varying degrees, which might be serves as a promising therapeutic target in treating sepsis induced by different causes. Moreover, features of immune exhaustion and apoptosis were highest in T and NK cells of SARS-COV-2 induced sepsis, secondly in influenza virus induced sepsis and last in bacterial sepsis, suggesting different degrees of immunosuppression may exist in different causes induced sepsis.
Although the alterations of cell composition in major cell subtypes including T/NK cells, myeloid cells and B cells during disease progression were consistent in the two sepsis patients (Fig S1B), the heterogeneity still existed in subpopulations of T/NK cells, B cells and NK cells at a higher resolution (Fig S9A, B, C), emphasizing the significance of studying cellular immune response at a higher resolution. However, different from T/NK cells, B cells and NK cells, the alterations of cell composition in myeloid cells were in line with that in major cell subtypes (Fig S3), indicating T/NK cells, B cells and NK cells may endow with more diversity than myeloid cells.
Fig. S9.

Cell composition in sepsis secondary to pneumonia induced by bacteria
Histogram showed the cell composition of A.T/NK cells, B. NK cells and C. B cells in each patient sample and healthy control in scRNA-seq data. D. boxplot showed the cell composition of three major cell types in each patient sample in bulk RNA-seq data.
There were several limitations existed in our study. First, the sample size was relatively small and, regretfully, the sample of the acute stage for the first patient was not included for analysis due to bad cell quality. To address this limit, we increased the number of patient samples in three disease stages with bulk RNA-seq to infer the cell composition of major cell subtypes due to their consistency between two patients for scRNA-seq during disease course. However, the results from bulk RNA-seq was not coincide with that in scRNA-seq (Fig S9.D), which might be attributed to two reasons. First, inferring the cell composition with bulk RNA-seq may be less precise than that with scRNA-seq. Second, the patient cohort for bulk RNA-seq in three disease stage was collected from different patients, which was different from that for scRNA-seq. Thus, future studies with larger sample size along disease course would be beneficial for demonstrating the links of host immune response and specific pathogen, accordingly improving organ injury and prognosing disease outcome. In addition, the regulatory mechanisms of intracellular network revealed by cell-cell interaction analysis were not validated experimentally; because it is difficult to isolate so many immune cell subpopulations and co-cultured them to explore their interactions; on the other hand, due to methodology limitations, the analysis for cellular interactions only predicts ligand and its target genes, therefore, it's hard to find a specific receptor for intervention. Further studies are warranted to explore the pathogenesis of sepsis secondary to bacterial pneumonia with more complete bioinformatics and experimental technologies.
In conclusion, our study explored the dynamic alterations of the circulating immune cells in sepsis secondary to pneumonia induced by gram-negative bacteria in single cell resolution, which provided the preliminary findings about the potential mechanisms underlying immunological response during disease progression.
4. Methods and materials
4.1. Ethics statement
The ethical consent of this study was approved by the Ethics Committee of Zhujiang Hospital of Southern Medical University (Guangzhou, China).
4.2. Patients information
Whole blood samples were collected from patients who were diagnosed as sepsis secondary to pneumonia induced by gram-negative bacterial infection with informed consent at Zhujiang Hospital, Southern Medical University. The criterions of enrolled patients were as following: infection-induced organ dysfunction with SOFA sore ≥2 according to the international guidelines for management of sepsis and septic shock of 2016, and in combination of procalcitonin (PCT) ≥ 2 ng/mL. For scRNA-seq, the patients came from two females. The first one was 48 years old while the second one was 68 years old. For bulk RNA-seq, the patients came from 6 females and 13 males. In Table S1, the definitions of different disease stages were as following: 1) acute stage: the same as the diagnostic criteria of SIRS, meeting over two criterions as followings: ① Body temperature: >38 °C or <36 °C; ② Heart rate: >90 beats/min; ③Respiratory function: >20 times/min or PaCO2 < 32 mmHg made by high ventilation; ④ Peripheral blood picture: immature WBC > 12 × 10 [9]/L or < 4 × 10 [9]/L; 2) stable stage: lymphocyte count <1.1 × 10 [9]/L; 3) recovery stage: resolution of infection and respiratory function, ascension of lymphocyte count. Detailed information of each sepsis patient and healthy control for scRNA-seq and bulk RNA-seq were listed in Table S1.
4.3. PBMC isolation
2 mL blood was collected from each patient and healthy control; PBMC was prepared with density-gradient centrifugation. Briefly, whole blood cells were diluted with 1× DPBS (1:1), layered on top of Ficoll-Paque Plus and then centrifuged at 500g for 20 min at room temperature. The white layer containing PBMC was isolated and washed twice with DPBS, followed by removing erythrocyte with red blood cell lysate on ice. After centrifuged at 1000 rpm for 5 min, cells were resuspended and filtered with a 40 μm cell strainer. Cell viability was verified with trypan blue and was approached to 90% per sample. Cells with high cell viability were subjected to library preparation.
4.4. ScRNA-seq library preparation and sequencing
The prepared cell suspensions were subjected to 10× Chromium Single Cell 3′ library with Chromium Single Cell 30 v3 reagent (10× Genomics) according to the manufacturer's instructions. About 8000 cells were loaded per sample. The followed sequencing was carried out with Illumina (Nova 6000) according to the manufacturer's instructions.
4.5. ScRNA-seq data preprocessing and quality control
The single cell transcriptome data was processed with Cell Ranger Software Suite (Version 3.1.0) [29] to perform alignment, filtering, barcode separating, and UMI counting with default parameters. Raw reads were aligned to the human reference genome GRCh38 using Cell Ranger. Feature-barcode matrices per sample were generated for secondary analysis. For quality controls, each sample was initially subjected to remove ambient RNA and doublets with SoupX R package [30]and scDblFinder R package(https://github.com/plger/scDblFinder), respectively. Then we filtered cells with the following criterions: 1) 500 < nFeature_RNA < 5000; 2) 1000 ≤ nCount_RNA ≤ 20,000; 3) percent of mitochondrial genes <30%; 4) percent of hemoglobin genes <0.01%. The mitochondrial genes and ribosome genes were removed. After filtering, a total of 52,412 cells with high quality from 5 patient samples, a healthy control combined with other 4 healthy controls downloaded from 10× Genomic (https://www.10xgenomics.com/cn/) were obtained for further integrated analysis using integrated pipeline of Seurat R package with default settings [31] .
4.6. Bulk RNA-seq library construction and sequencing
3 μg RNA per sample was employed for library preparations with NEBNext® UltraTMRNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer's instructions. Briefly, mRNA was extracted from total RNA with poly-T oligo-coupled magnetic beads. Then, fragmentation was performed under heating to obtain 180 nt to 250 nt RNAs. After the first strand cDNA and the second strand cDNA were synthesized, the DNAs fragments were subjected to adenylation of 3′ ends, ligation of adaptor. Then, the DNAs library were purified with AMPure XP system (Beckman Coulter, Beverly, USA), followed by PCR amplification. Lastly, the Agilent Bioanalyzer 2100 system were applied to purify PCR products and evaluate library quality. The followed sequencing was performed with Illumina (Nova 6000) according to the manufacturer's recommendations.
4.7. Bulk RNA-seq data processing and quality control
The raw data was filtered to obtain clean data with FASTQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The clean data was then mapped to the human reference genome GRCh38 with STAR (version = 2.7.6a) [32]. The read counts per gene were counted with “--quantMode” parameter implicated in STAR.
4.8. Infer cell composition in bulk RNA-seq
The Bisque r packages [33]were used to infer the cell composition in bulk RNA-seq of sepsis patients with the data of scRNA-seq in our study as reference, the default parameters were employed during analysis.
4.9. Dimensionality reduction and clustering
2000 highly variable features were obtained after integrated analysis. The integrated data was subjected to scale and calculate PCA. The first 30 PCs were selected for the unsupervised cluster with “RunUMAP” function, the “FindClusters” function (resolution = 0.015) in the Seurat R package [24] was used to generate 3 clusters. To re-cluster the T/NK cells, “FindClusters” function (resolution = 0.4) was applied again and obtain 10 clusters. To re-cluster the NK cells, “FindClusters” function (resolution = 0.15) was used again to obtain 2 clusters. To re-cluster the myeloid cells, “FindClusters” function (resolution = 0.1) was applied again to obtain 8 clusters. To re-cluster the B cells, “FindClusters” function (resolution = 0.2) was applied again to obtain 3 clusters. UMAP was used to visualize the result of clustering.
4.10. Differential expression analysis
Differential expression analysis was performed with “FindMarkers” function in Seurat packages with default parameters. Adjusted p < 0.05 and |log2FC| > 0.25 was used to defined significant DEGs.
4.11. GO analysis
DEGs of different groups in this study were subjected to Metascape webtools (https://metascape.org/gp/index.html#/main/step1) to perform GO analysis with the “biological processes” annotations [34] . P < 0.01 was considered as significantly enriched.
4.12. Pathway enrichment analysis
Pathway enrichment analysis was performed with fgsea R packages for all DEGs between groups. The hallmark gene sets were downloaded from Molecular Signature Database (MsigDB; https://www.gsea-msigdb.org/gsea/msigdb/collections.jsp#C2) [35] . Then, ‘fgsea’ function (nperm = 1000, minSize = 10, maxSize = 500) was performed to enrich biological pathways with P < 0.05, the results were explained by normalized enrichment score (NES) and adjusted p-value [36]. All of the gene signature list for GSEA Pathway enrichment analysis was provided in Table S3.
4.13. Time-dependent expression pattern analysis
TCseq [37] packages were exploited to identify time-dependent transcriptional alterations in disease progression. First, the average expression level log2(TPM/10 + 1) of each gene in each stage among single cells was calculated. Then, the ‘timeclust’ function was used to cluster different expression patterns. All of the gene signature list for 8 clusters by TCseq was provided in Table S3.
4.14. Cell-cell interaction
The NicheNetr package was used to explore the intracellular communication potentially lead to the differential gene expression between different disease stages and healthy controls [19]. Nichenet could specifically predict prioritization from sender cells and their target genes in receiver cells that altered in expression level upon disease induction. Differential expression genes in receiver cells between disease condition and healthy controls were served as target genes, which were obtained with “Findmarkers” function. Genes with adjusted P < 0.05, average log2fold change >0.25, and expressed in more than 10% receiver cells were considered as differential expression genes. The ligand activity analysis was performed based on their target genes with default parameters. The ligand activity was ranked with their Pearson correlation coefficient, the top20 ligand was selected for further analysis of ligand-target network. Only ligands from the top20 with enough regulatory potential scores will be predicted to have target genes (cut.off = 0.33). Ligands and target genes of interest were subjected to infer the potential signaling paths between them. The predicted signaling paths were visualized with Cytoscape (version 3.8.0) [38].
4.15. Supervised cell type annotation
The SciBet package [22] was used to perform the supervised cell type annotation of SARS-COV-2 scRNA-seq data according to data of the acute stage of bacterial sepsis in this study. The ‘SelectGene’ function was used to supervised gene selection with k = 50. The ‘SciBet’ function with default parameters was used to single cell identification.
4.16. Statistical analysis
The statistical method and associated threshold for each analysis were described in the above method sections.
The following are the supplementary data related to this article.
Patient information.
Gene signature list for 8 clusters of TCseq.
GSEA gene signature of PBMC and cell subtypes.
GO terms for diff sepsis.
Data availability
The raw sequence data reported in this paper has been deposited in the Genome Sequence Archive of the Beijing Institute of Genomics (BIG) Data center, BIG, Chinese Academy of Sciences, under accession number HRA000287 at http://bigd.big.ac.cn/gsa-human.The scRNA-seq data for COVID patients were downloading from http://bigd.big.ac.cn/gsa-human with the accession number: HRA000150. The scRNA-seq data for sepsis patients secondary to pneumonia were downloading from the GEO database with the accession number: GSE151263.
Author state funding sources
Teng Wang: Investigation, Formal analysis, Writing - Original Draft. Xianglong Zhang: Formal analysis. Zhanguo Liu: Investigation. Tong Yao: Investigation. Dongying Zheng: Investigation. Jianwei Gan: Resources. Shuang Yu: Resources. Lin Li: Conceptualization, Supervision, Writing- Reviewing and Editing. Peng Chen: Conceptualization, Supervision, Writing- Reviewing and Editing. Jian Sun: Conceptualization, Supervision.
Funding sources
The current research was supported by National Science and Technology Major Project of China (2017ZX10202202), Guangzhou Science and Technology Plan Project (201804020001), and Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01S131) to JS, the National Natural Science Foundation of China (81873926) to PC, and the National Natural Science Foundation of China (32070833) and Key-Area Research and Development Program of Guangdong Province, Modernization of Chinese medicine in Lingnan (2020B1111100011)to LL.
Declaration of Competing Interest
The authors declare that they have no conflict of interest.
References
- 1.Angus D.C., et al. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- 2.Ranieri V.M., et al. Drotrecogin alfa (activated) in adults with septic shock. New England Journal of Medicine. 2012;366(22):2055–2064. doi: 10.1056/NEJMoa1202290. [DOI] [PubMed] [Google Scholar]
- 3.Vincent J.L., et al. International study of the prevalence and outcomes of infection in intensive care units. Jama. 2009;302(21):2323–2329. doi: 10.1001/jama.2009.1754. [DOI] [PubMed] [Google Scholar]
- 4.Synger M., et al. The Third International Consensus Definitions for Sepsis and Septic Shock. Jama. 2016;315(8):801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delano M.J., et al. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? The Journal of clinical investigation. 2016;126(1):23–31. doi: 10.1172/JCI82224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hohlstein P., et al. Prognostic relevance of altered lymphocyte subpopulations in critical illness and sepsis. Journal of clinical medicine. 2019;8(3):353. doi: 10.3390/jcm8030353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoser G.A., et al. Absolute counts of peripheral blood leukocyte subpopulations in intraabdominal sepsis and pneumonia-derived sepsis: a pilot study. Folia histochemica et cytobiologica. 2012;50(3):420–426. doi: 10.5603/19751. [DOI] [PubMed] [Google Scholar]
- 8.Danahy D.B., et al. Clinical and experimental sepsis impairs CD8 T-cell-mediated immunity. Critical Reviews™ in Immunology. 2016;36(1) doi: 10.1615/CritRevImmunol.2016017098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venet F., et al. Myeloid cells in sepsis-acquired immunodeficiency. Annals of the New York Academy of Sciences. 2020 doi: 10.1111/nyas.14333. [DOI] [PubMed] [Google Scholar]
- 10.Wolk K., et al. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood, The Journal of the American Society of Hematology. 2000;96(1):218–223. [PubMed] [Google Scholar]
- 11.Wu T., et al. Comprehensive Transcriptome Profiling of Peripheral Blood Mononuclear Cells from Patients with Sepsis. International journal of medical sciences, 2020;17(14):2077. doi: 10.7150/ijms.46910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Washburn M.L., et al. T cell–and monocyte-specific RNA-sequencing analysis in septic and nonseptic critically ill patients and in patients with cancer. The Journal of Immunology. 2019;203(7):1897–1908. doi: 10.4049/jimmunol.1900560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liepelt A., et al. Differential Gene Expression in Circulating CD14+ Monocytes Indicates the Prognosis of Critically Ill Patients with Sepsis. Journal of clinical medicine. 2020;9(1):127. doi: 10.3390/jcm9010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reyes M., et al. An immune-cell signature of bacterial sepsis. Nature medicine. 2020;26(3):333–340. doi: 10.1038/s41591-020-0752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang Y., et al. Single cell RNA sequencing identifies an early monocyte gene signature in acute respiratory distress syndrome. JCI insight. 2020;5(13) doi: 10.1172/jci.insight.135678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsiao H.W., et al. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 2012;37(3):289–296. doi: 10.1097/SHK.0b013e318240b52a. [DOI] [PubMed] [Google Scholar]
- 17.van der Poll T., et al. The immunopathology of sepsis and potential therapeutic targets. Nature Reviews Immunology. 2017;17(7):407. doi: 10.1038/nri.2017.36. [DOI] [PubMed] [Google Scholar]
- 18.Albertsmeier M., et al. Monocyte-dependent suppression of T-cell function in postoperative patients and abdominal sepsis. Shock. 2017;48(6):651–656. doi: 10.1097/SHK.0000000000000924. [DOI] [PubMed] [Google Scholar]
- 19.Browaeys R. NicheNet: modeling intercellular communication by linking ligands to target genes. Nature methods. 2020;17(2):159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 20.Ren C., et al. Comparison of clinical laboratory tests between bacterial sepsis and SARS-CoV-2-associated viral sepsis. Military Medical Research. 2020;7(1):1–3. doi: 10.1186/s40779-020-00267-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J.Y., et al. Single-cell landscape of immunological responses in patients with COVID-19. Nature immunology. 2020;21(9):1107–1118. doi: 10.1038/s41590-020-0762-x. [DOI] [PubMed] [Google Scholar]
- 22.Li C., et al. SciBet as a portable and fast single cell type identifier. Nature communications. 2020;11(1):1–8. doi: 10.1038/s41467-020-15523-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu L., et al. Single-cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID-19 and influenza patients. Immunity. 2020;53(3):685–696. doi: 10.1016/j.immuni.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeannet R., et al. Severe COVID-19 is associated with deep and sustained multifaceted cellular immunosuppression. Intensive Care Medicine. 2020;46:1769–1771. doi: 10.1007/s00134-020-06127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen M., et al. Single-cell transcriptomics reveals the alteration of peripheral blood mononuclear cells driven by sepsis. Annals of translational medicine. 2020;8(4) doi: 10.21037/atm.2020.02.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venet F., et al. Early assessment of leukocyte alterations at diagnosis of septic shock. Shock. 2010;34(4):358–363. doi: 10.1097/SHK.0b013e3181dc0977. [DOI] [PubMed] [Google Scholar]
- 27.Lee J.S., et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Science immunology. 2020;5(49) doi: 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan K.S., et al. RNA sequencing of H3N2 influenza virus-infected human nasal epithelial cells from multiple subjects reveals molecular pathways associated with tissue injury and complications. Cells. 2019;8(9):986. doi: 10.3390/cells8090986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng G.X., et al. Massively parallel digital transcriptional profiling of single cells. Nature communications. 2017;8(1):1–12. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young M.D., et al. SoupX removes ambient RNA contamination from droplet based single cell RNA sequencing data. GigaScience. 2020;9(12) doi: 10.1093/gigascience/giaa151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butler A., et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology. 2018;36(5):411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobin A., et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jew B., et al. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nature communications. 2020;11(1):1–11. doi: 10.1038/s41467-020-15816-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y., et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nature communications. 2019;10(1):1–10. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liberzon A., et al. The molecular signatures database hallmark gene set collection. Cell systems. 2015;1(6):417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korotkevich G., et al. Fast gene set enrichment analysis. BioRxiv. 2019:060012. [Google Scholar]
- 37.Mengjun L.G. 2019. TCseq: Time Course Sequencing Data Analysis. [Google Scholar]
- 38.Shannon P., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Patient information.
Gene signature list for 8 clusters of TCseq.
GSEA gene signature of PBMC and cell subtypes.
GO terms for diff sepsis.
Data Availability Statement
The raw sequence data reported in this paper has been deposited in the Genome Sequence Archive of the Beijing Institute of Genomics (BIG) Data center, BIG, Chinese Academy of Sciences, under accession number HRA000287 at http://bigd.big.ac.cn/gsa-human.The scRNA-seq data for COVID patients were downloading from http://bigd.big.ac.cn/gsa-human with the accession number: HRA000150. The scRNA-seq data for sepsis patients secondary to pneumonia were downloading from the GEO database with the accession number: GSE151263.