

Abstract
From a genomics perspective, bivalves (Mollusca: Bivalvia) have been poorly explored with the exception for those of high economic value. The bivalve order Unionida, or freshwater mussels, has been of interest in recent genomic studies due to their unique mitochondrial biology and peculiar life cycle. However, genomic studies have been hindered by the lack of a high-quality reference genome. Here, I present a genome assembly of Potamilus streckersoni using Pacific Bioscience single-molecule real-time long reads and 10X Genomics-linked read sequencing. Further, I use RNA sequencing from multiple tissue types and life stages to annotate the reference genome. The final assembly was far superior to any previously published freshwater mussel genome and was represented by 2,368 scaffolds (2,472 contigs) and 1,776,755,624 bp, with a scaffold N50 of 2,051,244 bp. A high proportion of the assembly was comprised of repetitive elements (51.03%), aligning with genomic characteristics of other bivalves. The functional annotation returned 52,407 gene models (41,065 protein, 11,342 tRNAs), which was concordant with the estimated number of genes in other freshwater mussel species. This genetic resource, along with future studies developing high-quality genome assemblies and annotations, will be integral toward unraveling the genomic bases of ecologically and evolutionarily important traits in this hyper-diverse group.
Keywords: Pacific Biosciences, 10XGenomics, freshwater mussel, phylogenomics, Potamilus
Significance
The global decline of freshwater mussels has emphasized the need to better understand the biology, ecology, and evolution of the group. The basic genetics of freshwater mussels remain poorly understood despite a recent push to understand factors that contribute to their demise. Recent investigations in freshwater mussels to determine the genomic mechanisms involved with ecologically important traits such their life cycle and sex determination have been hindered by the lack of a high-quality reference genome. Here, I develop a high-quality reference genome for the freshwater mussel species Potamilus streckersoni or Brazos Heelsplitter. This genome assembly will facilitate future genome wide association studies to better understand these highly imperiled organisms.
Introduction
From a genomics perspective, bivalves (Mollusca: Bivalvia) have been poorly explored with the exception for those of high economic value (Liu et al. 2021). One bivalve order of considerable interest is Unionida or freshwater mussels, which consists of over 800 species (Graf and Cummings 2007). Freshwater mussels have a unique mitochondrial (mt) biology: they deviate from strictly maternal inheritance of mt and have a unique mode of mt inheritance called doubly uniparental inheritance (DUI) (e.g., Breton et al. 2007, 2018; Zouros 2013). They also display a peculiar life history that involves a parasitic larval stage (glochidia) that must attach to vertebrate hosts (primarily fish) to complete metamorphosis (Barnhart et al. 2008). While numerous genomic studies have sought to identify genes associated with inherent biological characteristics of mussels (e.g., Luo et al. 2014; Shi et al. 2015; Patnaik et al. 2016; Bertucci et al. 2017; Renaut et al. 2018; Capt et al. 2019), these studies have been hindered by the lack of a high-quality reference genome. Here, I present a genome assembly for the freshwater mussel species Potamilus streckersoni, or Brazos Heelsplitter, using Pacific Bioscience single-molecule real-time (PacBio; Menlo Park, CA) long reads and 10X Genomics-linked read sequencing (10X; San Francisco, CA). Further, I use RNA sequencing (RNA-seq) from multiple tissue types and life stages to provide a functional annotation for the assembly. This study will be integral toward unraveling the basic biology of freshwater mussels and the genomic bases of evolutionarily important traits contributing to their diversification.
Materials and Methods
Data Generation
One gravid female P. streckersoni was collected from the type locality and deposited at Florida Museum (UF439535). Whole genomic DNA was extracted from fresh mantle tissue using the Qiagen PureGene Kit (Hilden, Germany) with standard protocols. High-molecular weight DNA was ensured by visualizing the isolation on a 1% agarose gel stained with GelRed nucleic acid stain (Biotium, Hayward, CA). Isolation quantity and quality was assessed using a QubitTM fluorometer and a NanoDropTM One (ThermoFisher Scientific; Waltham, MA), respectively. Before PacBio and 10X sequencing, the initial identification based on external shell morphology was confirmed by amplifying and sequencing the mt gene NADH dehydrogenase subunit 1 as described in Serb et al. (2003). A PacBio library was size selected with 10 kb cut-off following the SMRT bell construction protocol. The library was sequenced on a single-molecule real-time cell of a PacBio Sequel II using the v 2.0 chemistry. For 10X sequencing, a Chromium 10X library was constructed from high-molecular-weight DNA according to manufacturers recommended protocols. The resulting library was quantitated by qPCR and sequenced on an Illumina NovaSeq 6000 (San Diego, CA).
Total RNA was extracted from five different tissue types from the adult female: foot, gill, gonad, mantle, and stomach. In addition to total RNA extraction of adult somatic and gonadal tissue, RNA was isolated from a pool of fully developed glochidia to pick up additional transcripts expressed at the larval stage. All RNA was extracted using QIAshredder and RNeasy kits as described by the manufacturer (Qiagen). RNA quality and integrity were determined using a NanoDropTM and an Agilent Bioanalyzer (Santa Clara, CA), respectively. Messenger RNA (mRNA) was purified from approximately 200 ng of total RNA and a mRNA library was prepared using KAPA mRNA HyperPrep Kits (Roche; Basel, Switzerland). Indexed libraries that met appropriate cut-offs were quantified by qRT-PCR and sequenced using 100 bp paired-end sequencing on an Illumina NovaSeq 6000.
Genome Assembly
A female mitogenome for P. streckersoni was de novo assembled with 10X reads using MitoZ v 2.4 (Meng et al. 2019). After assembly, the mitogenome was annotated using MITOS (Bernt et al. 2013) and final limits of transfer RNAs (tRNAs) were assessed using ARWEN (Laslett and Canback 2008).
Before de novo assembly of the nuclear genome, mt reads were removed from 10X and PacBio data using BWA-MEM (Li 2013), minimap2 v 2.17-r941 (Li 2018), and SAMtools v 1.9 (Li et al. 2009). Overall characteristics of the genome (e.g., genome size, repetitive elements) were estimated using 10X data by k-mer spectrum distribution analysis for k = 21 in JELLYFISH v 2.2.10 (Marçais and Kingsford 2011) and GENOMESCOPE v 2.0 (Ranallo-Benavidez et al. 2020).
A draft genome assembly was derived from 10X reads using Supernova v 2.1.1 with default parameters (Weisenfeld et al. 2017). For PacBio reads, wtdbg2 v 2.5 (Ruan and Li 2019) was used to assemble a draft genome using the commands “-x rsII -AS 4 -p 19 –tidy-reads 5000 –edge-min 4 –rescue-low-cov-edges” to account for high error rate and coverage. The PacBio assembly had superior contiguity and rather than use the 10X assembly, a pipeline was developed using linked reads to correct and scaffold the PacBio assembly similar to previous studies (e.g., Li et al. 2019; Wallberg et al. 2019).
The wtdbg2 assembly was polished using two iterations of arrow in GCpp v 1.9.0 (Pacific Biosciences 2019) and NextPolish v 1.2.3 (Hu et al. 2020), per developers recommendation for accuracy. The Tigmint+ARKS pipeline in ARCS v 1.1.0 (Coombe et al. 2018; Jackman et al. 2018; Yeo et al. 2018) was used to correct assembly errors and scaffold contigs using 10X data. Default parameters were used except for the barcode read frequency range (-m 20–20,000). LINKS v1.8.7 (Warren et al. 2015) was used to process ARKS results and construct scaffolds using the default parameters except for the ratio of barcode links between two most supported graph edges (-a 0.9). Gaps were introduced within scaffolds due to joins made by ARKS, and TGS-GapCloser v 1.0.1 (Xu et al. 2020) was used to fill gaps with PacBio reads.
Purge Haplotigs (Roach et al. 2018) was used to remove highly heterozygous haplotypes and trim contigs with overlapping ends. Coverage thresholds of 20, 190, and 195 were used to allow Purge Haplotigs to examine all contigs for suspected haplotigs. The curated assembly was subjected to two iterations of polishing in NextPolish. MegaBLAST v 2.10.0 was used to screen for possible contaminated scaffolds by identifying hits with >98% homology to available prokaryotic genomes from NCBI (https://www.ncbi.nlm.nih.gov). I used QUAST v 5.0.2 (Mikheenko et al. 2018) to generate assembly statistics and BUSCO v 4.0.6 (Seppey et al. 2019) to evaluate the completeness of the assembly using the 954 conserved genes in the Metazoan lineage after each step of the pipeline.
Genome Annotation
RNA-Seq reads were trimmed using TRIM GALORE! v0.6.4 (www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with default parameters except for minimum read length (35 bp). Data quality was ensured in FastQC v 0.11.9 (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Prior to read mapping, repeats in the P. streckersoni curated genome assembly were identified and masked using RepeatModeler v 2.0.1 (Flynn et al. 2020) and RepeatMasker v 4.0.9 (Smit et al. 2015), respectfully. RNA-seq reads were mapped to the masked assembly using HISAT2 v 2.1.0 (Kim et al. 2015), and aligned reads were used to train AUGUSTUS v 3.3.3 (Stanke et al. 2008). To incorporate cDNA evidence, rnaSPAdes (Bushmanova et al. 2019) was used to de novo assemble transcriptomes from RNA samples of adult tissues and pooled glochidia, respectively. Transcripts less than 200 bp were removed and remaining transcripts were aligned to the genome assembly using minimap2. Metazoan reference protein sequences were compiled from the OrthoDB protein database v 10 (Kriventseva et al. 2019), and ProtHint v 2.4.0 (Brůna et al. 2020) was used to generate potential borders between coding and noncoding regions.
Structural and function annotation was performed using the Funannotate pipeline v 1.8.0 (Palmer and Stajich 2017). Consensus gene models were produced by EVidenceModeler v 1.1.1 (EVM; Haas et al. 2008) using protein evidence, transcript evidence, and ab initio predictions from AUGUSTUS, GeneMark-ES v 4.61 (Lomsadze 2005), GlimmerHMM (Majoros et al. 2004), and SNAP (Korf 2004). tRNAscan-SE v 2.0.5 (Chan and Lowe 2019) was used to generate tRNA models. Protein annotations were assessed using BUSCO, CAZy (Cantarel et al. 2009), eggNOG (Huerta-Cepas et al. 2019), MEROPS (Rawlings et al. 2018), Pfam (Finn et al. 2014), and UniProt (The UniProt Consortium 2017) domains by eggNOG-mapper v 2 (Huerta-Cepas et al. 2017), InterProScan v 5.47-82.0 (Jones et al. 2014), and BLASTP v 2.10.0.
Phylogenomic Analysis
RNA-seq reads were compiled for 19 additional taxa distributed across the subclass Paleoheterodonta from the SRA database (supplementary table S2, Supplementary Material online). The larval transcriptome for P. streckersoni depicted a higher duplication rate when compared to the adult transcriptome (supplementary table S1, Supplementary Material online); therefore, the adult transcriptome was used for phylogenomic analyses to minimize impacts of heterozygosity. Dataset generation followed similar methods as Kocot et al. (2011, 2019). Reads were quality trimmed using TRIM GALORE! and transcriptomes were de novo assembled in rnaSPAdes. Nucleotide sequences were translated with TRANSDECODER v 5.5.0 (http://transdecoder.sourceforge.net/) and HAMSTR v 13.2.6 (Ebersberger et al. 2009) was used to generate orthologs. Genes were aligned with MAFFT v 7.471 (Katoh and Standley 2013) and were trimmed using ALISCORE (Misof and Misof 2009) and ALICUT v 2.31 (Kück 2009) to remove ambiguously aligned regions. Gene trees were generated in FASTTREE v 2.1.10 (Price et al. 2010) and PHYLOTREEPRUNER (Kocot et al. 2013) was used to select the best sequence for each taxon. Genes represented by 10 or more taxa were retained and concatenated using FASconCAT-G v 1.04 (Kück and Longo 2014). Phylogenetic reconstruction was performed in IQ-TREE v 2.1.2 (Chernomor et al. 2016; Minh et al. 2020) and ModelFinder (Kalyaanamoorthy et al. 2017) was used to select the partitioning scheme and best amino acid substitution models for the analysis. IQ-TREE analyses conducted 10 repetitions of an initial tree search and 1,000 ultrafast bootstrap replicates (ufBS) for nodal support (Hoang et al. 2018), per developers recommendation for accuracy.
Results and Discussion
PacBio sequencing generated ∼225 Gb with a subread N50 of 29 kb, representing >100× coverage of the genome assembly. 10X sequencing generated 672.03 million 150-bp paired end reads, roughly ∼48× coverage of the assembled genome. The female-type mitogenome for P. streckersoni was recovered as a single contig represented by 16,293 bp (supplementary fig. S1, Supplementary Material online) and consisted of 37 genes similar to mitogenomes derived from other Potamilus spp. (Feng et al. 2016; Wen et al. 2017): 13 protein-coding genes, 2 ribosomal RNAs, and 22 tRNAs.
The final curated genome assembly was represented by 2,368 scaffolds (2,472 contigs), 1,776,755,624 bp, and a scaffold N50 of 2,051,244. Additional information about genome statistics is presented in Table 1. The assembled genome size was concordant with the estimated genome size generated from 10X data (∼1.81 Gb; supplementary fig. S2, Supplementary Material online), and was also similar to the estimated genome size of Venustaconcha ellipsiformis (∼1.80 Gb), the most closely related freshwater mussel species with genomic resources (Renaut et al. 2018) (fig. 1; table 2). Overall, 94.6% of the 954 expected metazoan genes were identified in the nuclear genome assembly by BUSCO, which supported the assembly as largely complete. RepeatModeler marked 51.03% of the assembly as repetitive elements (∼0.96 Gb), which was larger than the repetitive element content estimated by JELLYFISH and GENOMESCOPE (32.5%); however, aligns with the percentage of repetitive elements in other bivalve genomes (e.g., Wang et al. 2017; Bai et al. 2019; Gomes-dos-Santos et al. 2020) (table 2). The majority of repetitive elements in the P. streckersoni genome assembly were uncharacterized (25.5%; ∼453 Mb) similar to other studies in freshwater mussels (Gomes-dos-Santos et al. 2020), but notable categorizable transposable element compositions were as follows: DNA transposons—13.71% (∼243 Mb), long interspersed nuclear elements—5.01% (∼89 Mb), long terminal repeats—3.09% (∼55 Mb), Penelope-like elements—1.14% (∼20 Mb), and short interspersed nuclear elements—0.01% (∼134 kb).
Table 1.
QUAST Scaffold Statistics and BUSCO Profiles Based on the Metazoan and mollusca_odb10 Lineage for Each Assembly
| Value | PacBio | PacBio + 10X | Final Assembly |
|---|---|---|---|
| Scaffolds | 6,770 | 6,591 | 2,368 |
| Contigs | 6,770 | 6,877 | 2,472 |
| Total length | 1,847,057,843 | 1,860,819,131 | 1,776,755,624 |
| Scaffold N50 | 1,241,163 | 1,977,022 | 2,051,244 |
| Scaffold N75 | 570,763 | 915,592 | 1,056,644 |
| Scaffold L50 | 436 | 265 | 245 |
| Scaffold L75 | 981 | 608 | 546 |
| N’s per 100 kbp | 0 | 0.55 | 0.53 |
| Complete single copy | 892 (93.5%) | 891 (93.4%) | 894 (93.7%) |
| Complete duplicated | 11 (1.2%) | 12 (1.3%) | 9 (0.9%) |
| Fragmented | 10 (1.0%) | 10 (1.0%) | 11 (1.2%) |
| Missing | 41 (4.3%) | 41 (4.3%) | 40 (4.2%) |
Note.—The three steps columns represent the polished assembly derived from wtdbg2 (PacBio), the polished assembly generated from the Tigmint+ARKS pipeline (PacBio + 10X), and the final assembly after removing highly heterozygous haplotypes (final assembly).
Fig. 1.
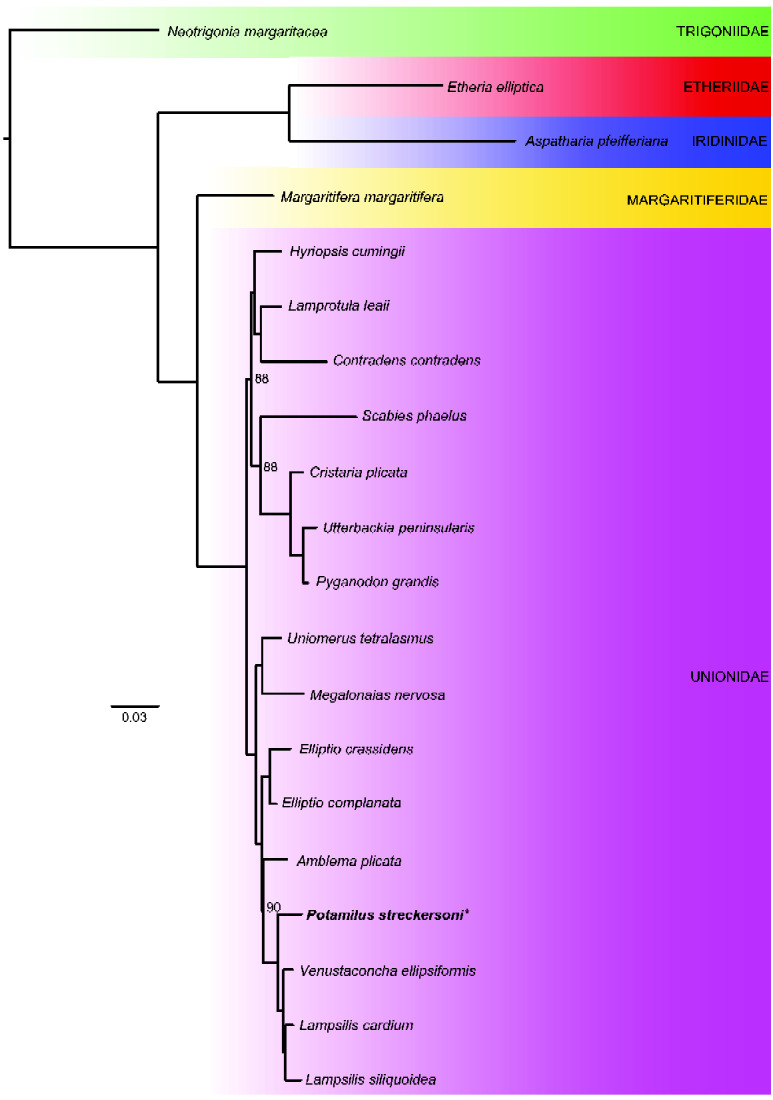
Phylogenetic reconstruction generated from select transcriptomes from subclass Paleoheterodonta and family designations for selected taxa. All but three nodes were represented by full ultrafast bootstrap support.
Table 2.
Summary Statistics of Annotated Genome Assemblies Available for Bivalvia
| Order | Taxa | Genome Size (Mb) | Scaffold N50 (Kb) | Repeat Content (%) | Number of Genes | Source |
|---|---|---|---|---|---|---|
| Adapedonta | Sinonovacula constricta | 1,332 | 57,990 | 36.65 | 26,273 | Dong et al. (2020) |
| Arcida | Scapharca broughtonii | 885 | 4,500 | 46.41 | 24,045 | Bai et al. (2019) |
| Mytilida | Bathymodiolus platifrons | 1,660 | 343 | 47.25 | 33,584 | Sun et al. (2017) |
| Limnoperna fortune | 1,670 | 312 | 33.40 | 60,717 | Uliano-Silva et al. (2018) | |
| Modiolus philippinarum | 2,630 | 100 | 59.66 | 36,549 | Sun et al. (2017) | |
| Ostreida | Crassostrea gigas | 559 | 401 | 34.71 | 28,072 | Zhang et al. (2012) |
| Crassostrea virginica | 685 | 75,944 | 39.69 | 34,596 | Gómez-Chiarri et al. (2015) | |
| Saccostrea glomerate | 788 | 804 | 45.39 | 29,738 | Powell et al. (2018) | |
| Pectinida | Argopecten purpuratus | 725 | 1,020 | 32.04 | 26,256 | Li et al. (2018) |
| Chlamys farreri | 780 | 602 | 27.73 | 28,602 | Li et al. (2017) | |
| Patinopecten yessoensis | 988 | 804 | 27.85 | 24,738 | Wang et al. (2017) | |
| Pteriida | Pinctada fucata | 1,024 | 167 | 43.35 | 31,447 | Takeuchi et al. (2012, 2016) |
| Pinctada fucata martensii | 991 | 324 | 48.01 | 30,815 | Du et al. (2017) | |
| Unionida | Margaritifera margaritifera | 2472 | 289 | 59.07 | 35,119 | Gomes-dos-Santos et al. (2020) |
| Megalonaias nervosa | 2,360 | 53 | ∼25 | 49,149 | Rogers et al. (2020) | |
| Potamilus streckersoni | 1,776 | 2,051 | 50.13 | 41,065 | This study | |
| Venustaconcha ellipsiformis | ∼1,800 | 7 | 36.29 | 201,068 | Renaut et al. (2018) | |
| Venerida | Archivesica marissinica | 1,520 | 73,300 | 42.20 | 28,949 | Ip et al. (2021) |
| Cyclina sinensis | 903 | 46.5 | 43.14 | 27,564 | Wei et al. (2020) | |
| Ruditapes philippinarum | 1,129 | 345 | 38.30 | 27,652 | Yan et al. (2019) |
RNA-seq generated ∼13 Gb and transcriptomes based on adult tissue and larvae (96,843 and 104,614 transcripts, respectively) were largely complete based on BUSCO analyses, with each transcriptome having more than 96% of metazoan genes (supplementary table S1, Supplementary Material online). A phylotranscriptomic approach nested P. streckersoni within the family Unionidae and a close relative to Lampsilis and Venustaconcha ellipsiformis (fig. 1), aligning with previous phylogenetic studies (Smith et al. 2019, 2020). The functional annotation returned 52,407 gene models (41,065 protein-coding genes, 11,342 tRNAs). The estimated number of protein-coding genes was slightly more than other mollusk genomes of similar size (e.g., Nam et al. 2017; Wang et al. 2017), but was similar to the recent estimates in freshwater mussels (Gomes-dos-Santos et al. 2020; Rogers et al. 2020) (table 2). Coding sequences from the 41,065 predicted genes had a mean length of 375.84 amino acids and 22,568 were annotated by available protein databases. The number of annotated genes aligns with other bivalve genomes (e.g., Bai et al. 2019; Dong et al. 2020), but suggests some hypothetical proteins may be lineage specific.
In this study, I present the first high quality genome assembly for a freshwater mussel, which will be an invaluable resource for understating their biology, ecology, and evolution. Future efforts to develop a chromosome level assembly (e.g., HiC sequencing) will be useful to determine the karyotype of P. streckersoni and further investigate gene superfamilies. Nonetheless, this resource will facilitate genome wide association studies to screen for the genomic bases of ecologically and evolutionarily important traits such as sex determining pathways and mechanisms involved with immune evasion. Freshwater mussels also represent one of the most imperiled groups of organisms globally (Lopes-Lima et al. 2018), and this resource, along with future studies developing high-quality genome assemblies and annotations, may shed light on possible genomic characteristics that contribute to imperilment and terminal extinction.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
I wish to thank Allison Banse, Christopher Castaldi, Eric Johnson, Lauren Machado, Emma Sikes, Guilin Wang, SNPsaurus, and Yale Center for Genome Analysis for assistance in the laboratory, Melissa Mullins for assistance with collections, Robert Doyle for contributing funding, along with Kevin Kocot, Mark Kirkpatrick, Chad Mansfield, Cole Matson, Brendan Pinto, Chi‐Yen Tseng, and Nathan Whelan for discussions regarding this research. This work was supported by the Baylor University Glasscock Endowment Fund (Grant No. 032MBCU-63); and the University of Texas at Austin Stengl-Wyer Endowment.
Data Availability
The final genome assembly and annotation, as well as all 10X, PacBio, RNA-seq reads, and pooled adult and larval transcriptomes are accessible under NCBI BioProject PRJNA681676 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA681676). The de novo assembled female mitogenome is available on GenBank (MW413895).
Literature Cited
- Bai C-M, et al. 2019. Chromosomal-level assembly of the blood clam, Scapharca (Anadara) broughtonii, using long sequence reads and Hi-C. GigaScience 8(7):giz067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhart MC, Haag WR, Roston WN.. 2008. Adaptations to host infection and larval parasitism in Unionoida. J. N. Am. Benthol. Soc. 27(2):370–394. [Google Scholar]
- Bernt M, et al. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69(2):313–319. [DOI] [PubMed] [Google Scholar]
- Bertucci A, et al. 2017. Transcriptomic responses of the endangered freshwater mussel Margaritifera margaritifera to trace metal contamination in the Dronne River. Environ. Sci. Pollut. Res. 24(35):27145–27159. [DOI] [PubMed] [Google Scholar]
- Breton S, Beaupré HD, Stewart DT, Hoeh WR, Blier PU.. 2007. The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends Genet. 23(9):465–474. [DOI] [PubMed] [Google Scholar]
- Breton S, Capt C, Guerra D, Stewart DT.. 2018. Sex-determining mechanisms in bivalves. In: Leonard JL, editor. Transitions between sexual systems. New York: Springer International Publishing. p. 165–192. [Google Scholar]
- Brůna T, Lomsadze A, Borodovsky M.. 2020. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom. Bioinformatics 2(2):lqaa026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushmanova E, Antipov D, Lapidus A, Prjibelski AD.. 2019. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8(9):giz100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, et al. 2009. The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37(Database):D233–D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capt C, Renaut S, Stewart DT, Johnson NA, Breton S.. 2019. Putative mitochondrial sex determination in the Bivalvia: insights from a hybrid transcriptome assembly in freshwater mussels. Front. Genet. 10:840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, Lowe TM.. 2019. tRNAscan-SE: searching for tRNA genes in genomic sequences. In: Kollmar M, editor. Gene prediction. Vol. 1962. New York: Springer. p. 1–14. Available from: 10.1007/978-1-4939-9173-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomor O, von Haeseler A, Minh BQ.. 2016. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 65(6):997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombe L, et al. 2018. ARKS: chromosome-scale scaffolding of human genome drafts with linked read kmers. BMC Bioinformatics 19(1):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, et al. 2020. The chromosome-level genome assembly and comprehensive transcriptomes of the razor clam (Sinonovacula constricta). Front. Genet. 11:664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, et al. 2017. The pearl oyster Pinctada fucata martensii genome and multi-omic analyses provide insights into biomineralization. GigaScience 6(8):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersberger I, Strauss S, von Haeseler A.. 2009. HaMStR: profile hidden markov model based search for orthologs in ESTs. BMC Evol. Biol. 9(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Zhang X, Zhao G-F.. 2016. The complete mitochondrial genome of the scaleshell Leptodea leptodon (Bivalvia: Unionidae). Conserv. Genet. Resourc. 8(4):443–445. [Google Scholar]
- Finn RD, et al. 2014. Pfam: the protein families database. Nucleic Acids Res. 42(D1):D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, et al. 2020. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U.S.A. 117(17):9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-dos-Santos A, et al. 2020. The Crown Pearl: a draft genome assembly of the European freshwater pearl mussel Margaritifera margaritifera (Linnaeus, 1758). BioRxiv. 2020. Available from: 10.1101/2020.12.06.413450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Chiarri M, Warren WC, Guo X, Proestou D.. 2015. Developing tools for the study of molluscan immunity: the sequencing of the genome of the eastern oyster, Crassostrea virginica . Fish Shellfish Immunol. 46(1):2–4. [DOI] [PubMed] [Google Scholar]
- Graf DL, Cummings KS.. 2007. Review of the systematics and global diversity of freshwater mussel species (Bivalvia: Unionoida). J. Mollusc. Stud. 73(4):291–314. [Google Scholar]
- Haas BJ, et al. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang DT, Chernomor O, von Haeseler A, Quang Minh B, Sy Vinh L.. 2018. Ufboot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35(2):518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Fan J, Sun Z, Liu S.. 2020. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36(7):2253–2255. [DOI] [PubMed] [Google Scholar]
- Huerta-Cepas J, et al. 2017. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34(8):2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, et al. 2019. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47(D1):D309–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip JC-H, et al. 2021. Host–endosymbiont genome integration in a deep-sea chemosymbiotic clam. Mol. Biol. Evol. 38(2):502–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SD, et al. 2018. Tigmint: correcting assembly errors using linked reads from large molecules. BMC Bioinformatics 19(1):393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9):1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS.. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14(6):587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL.. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12(4):357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocot KM, et al. 2011. Phylogenomics reveals deep molluscan relationships. Nature 477(7365):452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocot KM, Citarella MR, Moroz LL, Halanych KM.. 2013. PhyloTreePruner: a phylogenetic tree-based approach for selection of orthologous sequences for phylogenomics. Evol. Bioinform. 9:EBO.S12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocot KM, Todt C, Mikkelsen NT, Halanych KM.. 2019. Phylogenomics of Aplacophora (Mollusca, Aculifera) and a solenogaster without a foot. Proc. R. Soc. B 286(1902):20190115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva EV, et al. 2019. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47(D1):D807–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kück P. 2009. ALICUT: a PerlScript which cuts ALISCORE identified RSS (Version 2) [Computer software]. Department of Bioinformatics, Zoologisches Forschungsmuseum A Koenig (ZFMK).
- Kück P, Longo GC.. 2014. FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front. Zool. 11(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D, Canback B.. 2008. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24(2):172–175. [DOI] [PubMed] [Google Scholar]
- Li C, et al. 2018. Draft genome of the Peruvian scallop Argopecten purpuratus. GigaScience 7(4):giy031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv:1303.3997 [q-Bio]. Available from: http://arxiv.org/abs/1303.3997.
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18):3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, et al. 2019. A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). GigaScience 8(6):giz072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. 2017. Scallop genome reveals molecular adaptations to semi-sessile life and neurotoxins. Nat Commun. 8(1):1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, et al. 2021. MolluscDB: an integrated functional and evolutionary genomics database for the hyper-diverse animal phylum Mollusca. Nucleic Acids Res. 49(D1):D988–D997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomsadze A. 2005. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33(20):6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Lima M, et al. 2018. Conservation of freshwater bivalves at the global scale: diversity, threats and research needs. Hydrobiologia 810(1):1–14. [Google Scholar]
- Luo Y, et al. 2014. Transcriptomic profiling of differential responses to drought in two freshwater mussel species, the Giant Floater Pyganodon grandis and the Pondhorn Uniomerus tetralasmus. PLoS One 9(2):e89481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Salzberg SL.. 2004. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20(16):2878–2879. [DOI] [PubMed] [Google Scholar]
- Marçais G, Kingsford C.. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27(6):764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G, Li Y, Yang C, Liu S.. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A.. 2018. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34(13):i142–i150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, et al. 2020. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37(5):1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misof B, Misof K.. 2009. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst. Biol. 58(1):21–34. [DOI] [PubMed] [Google Scholar]
- Nam B-H, et al. 2017. Genome sequence of pacific abalone (Haliotis discus hannai): the first draft genome in family Haliotidae. GigaScience 6(5):gix014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacific Biosciences. 2019. GCpp (1.0.0) [Computer software]. Available from: https://github.com/PacificBiosciences/gcpp.
- Palmer J, Stajich J.. 2017. Funannotate: Eukaryotic genome annotation pipeline. Available from: 10.5281/zenodo.1134477. [DOI]
- Patnaik BB, et al. 2016. Sequencing, de novo Assembly, and annotation of the transcriptome of the endangered freshwater pearl bivalve, Cristaria plicata, provides novel insights into functional genes and marker discovery. PLoS One. 11(2):e0148622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell D, et al. 2018. The genome of the oyster Saccostrea offers insight into the environmental resilience of bivalves. DNA Res. 25(6):655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP.. 2010. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5(3):e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranallo-Benavidez TR, Jaron KS, Schatz MC.. 2020. GenomeScope 2.0 and Smudgeplots: reference-free profiling of polyploid genomes. Nat. Commun. 11(1):1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, et al. 2018. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46(D1):D624–D632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaut S, et al. 2018. Genome survey of the freshwater mussel Venustaconcha ellipsiformis (Bivalvia: Unionida) using a hybrid de novo assembly approach. Genome Biol. Evol. 10(7):1637–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach MJ, Schmidt SA, Borneman AR.. 2018. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19(1):460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, et al. 2020. Gene family amplification facilitates adaptation in freshwater unionid bivalve Megalonaias nervosa. Mol. Ecol. Available from: 10.1111/mec.15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J, Li H.. 2020. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17(2):155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppey M, Manni M, Zdobnov EM.. 2019. BUSCO: assessing genome assembly and annotation completeness. In: Kollmar M, editor. Gene prediction: methods and protocols. New York: Springer. p. 227–245. Available from: 10.1007/978-1-4939-9173-0_14. [DOI] [PubMed] [Google Scholar]
- Serb JM, Buhay JE, Lydeard C.. 2003. Molecular systematics of the North American freshwater bivalve genus Quadrula (Unionidae: Ambleminae) based on mitochondrial ND1 sequences. Mol. Phylogenet. Evol. 28(1):1–11. [DOI] [PubMed] [Google Scholar]
- Shi J, Hong Y, Sheng J, Peng K, Wang J.. 2015. De novo transcriptome sequencing to identify the sex-determination genes in Hyriopsis schlegelii. Biosci. Biotechnol. Biochem. 79(8):1257–1265. [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P.. 2015. RepeatMasker Open-4.0 (4.0.9) [Computer software]. Available from: http://www.repeatmasker.org.
- Smith CH, Johnson NA, Inoue K, Doyle RD, Randklev CR.. 2019. Integrative taxonomy reveals a new species of freshwater mussel, Potamilus streckersoni sp. nov. (Bivalvia: Unionidae): implications for conservation and management. Syst. Biodiv. 17(4):331–348. [Google Scholar]
- Smith CH, Pfeiffer JM, Johnson NA.. 2020. Comparative phylogenomics reveal complex evolution of life history strategies in a clade of bivalves with parasitic larvae (Bivalvia: Unionoida: Ambleminae). Cladistics 36(5):505–520. [DOI] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D.. 2008. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24(5):637–644. [DOI] [PubMed] [Google Scholar]
- Sun J, et al. 2017. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 1(5):0121. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, et al. 2012. Draft genome of the pearl oyster Pinctada fucata: a platform for understanding bivalve biology. DNA Res. 19(2):117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, et al. 2016. Bivalve-specific gene expansion in the pearl oyster genome: implications of adaptation to a sessile lifestyle. Zool. Lett. 2(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uliano-Silva M, et al. 2018. A hybrid-hierarchical genome assembly strategy to sequence the invasive golden mussel, Limnoperna fortunei. GigaScience 7(2):gix128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. 2017. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45(D1):D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallberg A, et al. 2019. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genomics 20(1):275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, et al. 2017. Scallop genome provides insights into evolution of bilaterian karyotype and development. Nat. Ecol. Evol. 1(5):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RL, et al. 2015. LINKS: scalable, alignment-free scaffolding of draft genomes with long reads. GigaScience 4(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, et al. 2020. Chromosome-level clam genome helps elucidate the molecular basis of adaptation to a buried lifestyle. IScience 23(6):101148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB.. 2017. Direct determination of diploid genome sequences. Genome Res. 27(5):757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen HB, et al. 2017. The complete maternally and paternally inherited mitochondrial genomes of a freshwater mussel Potamilus alatus (Bivalvia: Unionidae). PLoS One 12(1):e0169749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, et al. 2020. TGS-GapCloser: fast and accurately passing through the Bermuda in large genome using error-prone third-generation long reads. GigaScience 9(9):giaa094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, et al. 2019. Clam genome sequence clarifies the molecular basis of its benthic adaptation and extraordinary shell color diversity. IScience 19:1225–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo S, Coombe L, Warren RL, Chu J, Birol I.. 2018. ARCS: scaffolding genome drafts with linked reads. Bioinformatics 34(5):725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, et al. 2012. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490(7418):49–54. [DOI] [PubMed] [Google Scholar]
- Zouros E. 2013. Biparental inheritance through uniparental transmission: the doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol. Biol. 40(1):1–31. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The final genome assembly and annotation, as well as all 10X, PacBio, RNA-seq reads, and pooled adult and larval transcriptomes are accessible under NCBI BioProject PRJNA681676 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA681676). The de novo assembled female mitogenome is available on GenBank (MW413895).