

Abstract
Most cellular stress responses converge on the mitochondria. Consequently, the mitochondria must rapidly respond to maintain cellular homeostasis and physiological demands by fine-tuning a plethora of mitochondria-associated processes. The outer mitochondrial membrane (OMM) proteins are central to mediating mitochondrial dynamics, coupled with continuous fission and fusion. These OMM proteins also have vital roles in controlling mitochondrial quality and serving as mitophagic receptors for autophagosome enclosure during mitophagy. Mitochondrial fission segregates impaired mitochondria in smaller sizes from the mother mitochondria and may favor mitophagy for eliminating damaged mitochondria. Conversely, mitochondrial fusion mixes dysfunctional mitochondria with healthy ones to repair the damage by diluting the impaired components and consequently prevents mitochondrial clearance via mitophagy. Despite extensive research efforts into deciphering the interplay between fission–fusion and mitophagy, it is still not clear whether mitochondrial fission essentially precedes mitophagy. In this review, we summarize recent breakthroughs concerning OMM research, and dissect the functions of these proteins in mitophagy from their traditional roles in fission–fusion dynamics, in response to distinct context, at the intersection of the OMM platform. These insights into the OMM proteins in mechanistic researches would lead to new aspects of mitochondrial quality control and better understanding of mitochondrial homeostasis intimately tied to pathological impacts.
Subject terms: Macroautophagy, Protein quality control
Facts
Mitochondria undergo continuous fission and fusion events, referred as mitochondrial dynamics.
Mitochondrial quality control is precisely governed by autophagic degradation, termed as “mitophagy”.
Whether the interplay of mitochondrial dynamics and mitochondrial quality control simply bases on mitochondrial morphology or size remains to be answered.
Fused mitochondria might be permissible to mitochondrial elimination, mostly relying on the orchestration of OMM proteins.
The crosstalk between mitochondrial dynamics and mitophagy should be an emerging area to be addressed.
Open questions
How does specific OMM proteins engage in regulating mitochondrial homeostasis by coordinating mitochondrial dynamics and mitochondrial quality control?
How are tubular mitochondria segregated and directly pinched off to be engulfed by the autophagy isolation membrane?
Given the interplay of distinct molecular pathways of mitophagy, what signal in physiological contexts would stimulate specific route of mitochondrial elimination?
Is mitochondrial fission a prerequisite for mitochondrial clearance?
In tight coordination with damaged mitochondrial degradation, how would mitochondrial biogenesis be involved to maintain proper organelle homeostasis, coupled with mitochondrial dynamics?
In light of the actions of mitophagic modulators, what therapeutic strategy would be administrated to target mitochondria-related diseases?
Introduction
Mitochondria are double-membrane-bound subcellular organelles found within the cells of all multicellular eukaryotes. This organelle is considered a “powerhouse” at the heart of cell metabolism, catalyzing the production of adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS) [1, 2]. The outer and inner membranes of the mitochondria are composed of a mosaic of proteins and phospholipids, with a distinct intermembrane space between them. The inner membrane invaginates and curves inward into the mitochondrial matrix to form the cristae [3, 4]. A number of assembled complexes involved in OXPHOS and ATP synthesis are located on the cristae [5, 6].
Mitochondria undergo continuous cycles of fusion, during which segregated mitochondria join; and fission, during which the mitochondria divide. Collectively, this process is referred to as “mitochondrial dynamics” [7–13]. The mitochondrial fission and fusion adapt well to maintain the mitochondrial size, morphology, and position [14, 15]. Adapting to distal metabolic demand, mitochondrial fission facilitates mitochondrial traffic and is crucial to maintain organelle distribution [16, 17]. In contrast, mitochondrial fusion serves in the communication of mitochondrial content, including mitochondrial DNA (mtDNA) and mitochondrial proteins involved in OXPHOS [18, 19], as well as buffering acute damages within mitochondria [20, 21]. Unrestricted fission in response to cellular stress leads to small and fragmented mitochondria, during which calcium signaling and cell death pathways are activated [22]. Conversely, excessive fusion not only leads to a hyper-fused mitochondrial network, that is proposed to counteract environmental insults and maintain cellular integrity, but also confers an abnormal copy number of mtDNA [18, 21].
The mitochondrion has acquired a myriad of functions over its evolution, such as regulating calcium homeostasis, redox signaling, and heme synthesis [23–25]. In addition, mitochondria are essential hubs for several inflammatory processes, including NLRP3 inflammasome activation [26–28] and cGAS-STING pathways [29]. Moreover, increasing evidence supports that mitochondria can sense cellular stress challenges and collaborate with various cellular processes, including apoptosis, autophagy and mitophagy, to maintain cellular homeostasis [1, 30–34]. Notably, mitochondrial turnover, achieved by selective autophagy (mitophagy), is one such terminal response to extreme stress. Mitophagy is a highly hierarchical process that is composed of isolation membrane enclosure of mitochondria, formation of mitophagosome and digestion of mitochondria within lysosome [35–39].
Up till now, most studies have considered mitophagy to go in-hand with mitochondrial dynamics: short mitochondria generated by mitochondrial fragmentation are easily targeted by mitophagy, as these small and isolated mitochondria are readily engulfed by the autophagy machinery and permissible for elimination [40–43]. Yet, our understanding on their interplay remains limited. Of note, many proteins accounting for mitochondrial dynamics are also involved in mitophagic progression [42, 44, 45]. Specifically, the outer mitochondrial membrane (OMM) proteins have been intensively studied for their roles in mitochondrial morphology and mitochondrial elimination [42, 43, 46]. However, whether OMM proteins mediate mitochondrial division, while simultaneously coordinating mitophagy as a coherent mechanism remains unclear. In this review, we aim to summarize the recent breakthroughs on the functions of OMM proteins in mediating the crosstalk of mitochondrial dynamics and mitochondrial quality control. We then convey our insights into how mitochondrial dynamics and mitophagy are coordinated and provide our perspectives on the future research to advance this field.
Mitochondrial dynamics
Mitochondria are dynamic and mobile organelles [22], which are transported by various motor proteins along the cytoskeleton [47]. Fundamentally, the process of mitochondrial fusion allows for the mixing of numerous mitochondrial contents between neighboring mitochondria. The complementation process of fusion between damaged mitochondrion and healthy mitochondrion helps buffer transient stresses or defects within a mitochondrion by diluting toxins [18, 48, 49]. Moreover, any imbalance to fission or fusion during cell division can impair mtDNA segregation. Consequently, the dysregulation of the mitochondrial fission–fusion leads to defects in a recalibration of cellular responses and transmission of diffusible signals within mitochondria, which subsequently cause mitochondria-associated diseases at the organismal level [50–54].
Mitochondrial fission machinery
Mitochondrial division is primarily mediated by dynamin-related guanosine triphosphatases (GTPase) protein, dynamin 1 (Dnm1) in yeast [15] and dynamin-related protein 1 (Drp1) in mammals [55]. Drp1 predominantly localizes to the cytoplasm and is recruited onto the OMM by accessory receptors [51, 56]. This oligomeric ring-like Drp1 structure forces the OMM to furrow; as a result, its GTPase activity at the scission sites causes the mitochondria to divide [57]. Despite the importance of Drp1 for the OMM constriction, Drp1 lacks a transmembrane domain to target the OMM directly. Several other OMM proteins must help recruit Dnm1/Drp1: in yeast, the mitochondrial outer membrane-anchored mitochondrial fission 1 protein (Fis1) [7], mitochondrial division protein 1 (Mdv1) [58] and CCR4-associated factor 4 (Caf4) [59] are responsible for Dnm1 accumulation on the mitochondria. However, to date, no Mdv1 and Caf4 orthologs have been identified in mammals. Rather, it seems that mammals employ different OMM proteins to achieve Drp1-mediated fission (Fig. 1A), including the mitochondrial fission factor (Mff) [10, 60], mitochondrial dynamics proteins 49 and 51 (MiD49 and MiD51) [11, 12, 61]. Although these proteins are all crucial for mitochondrial fission, they differentially modulate the activity of Drp1.
Fig. 1. Molecular mechanisms of mammalian mitochondrial dynamics.
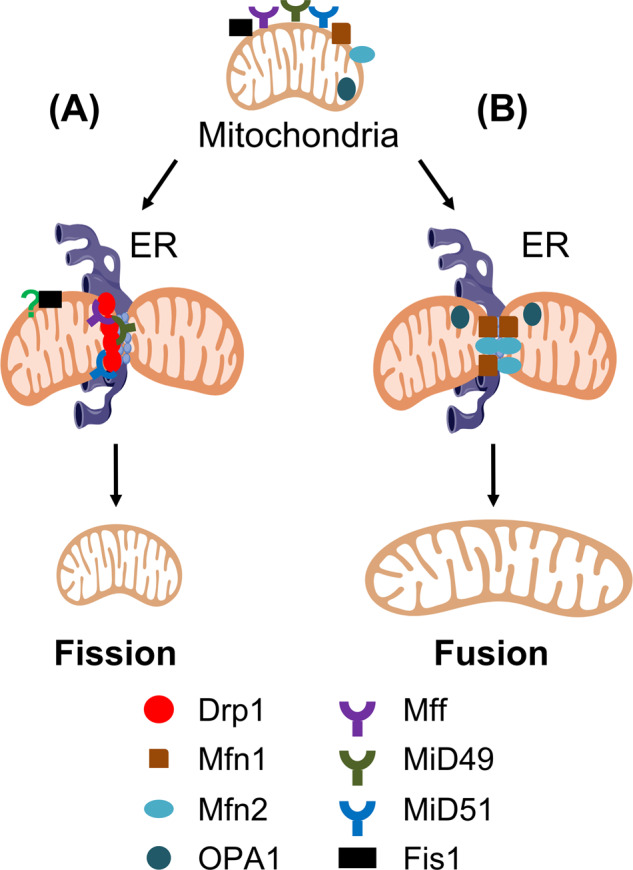
A During mitochondrial division, the endoplasmic reticulum (ER) converges with mitochondria. At the constriction sites spatially marked by mitochondria-associated membranes (MAM), Drp1 is recruited by Mff, MiD49 or MiD51 onto the cytosolic surface of mitochondria and acts as a GTPase to complete the scission of outer mitochondrial membrane (OMM). Although Fis1 was originally identified as an essential Drp1 adapter for fission in yeast, it is dispensable for mitochondrial dynamics in mammals. B In contrast to fission, OMM proteins Mfn1 and Mfn2 form mitofusin complexes by homo-dimerization or hetero-dimerization to tether the adjacent outer membranes. Driven by mitofusin proteins GTPase activity, OMM first fuses, followed by the subsequent inner mitochondrial membrane (IMM) fusion, due to the GTPase activity of OPA1.
For example, overexpression of high Mff levels leads to mitochondrial fragmentation, whereas cells depleted of Mff by siRNA or Mff-null cells, have highly-fused mitochondria and dysregulated Drp1 assembly [10, 11]. While Mff exists in all metazoans, MiD49 and MiD51 are chordate-specific to orchestrate mitochondrial dynamics [12]. Cellular overexpression of MiD49 or MiD51 causes excessive inactive Drp1 by inhibiting its GTPase activity, causing them to be sequestered from the OMM [11, 12, 61, 62]. Conversely, cells loosing MiD49 or MiD51, or MiDs-depleted cells show abolished oligomerization of Drp1 on the OMM, resulting in mitochondrial elongation or collapse [11].
The nucleotidyl transferase domain of MiD51 has a high affinity for adenosine diphosphate (ADP): in the presence of ADP as an MiD51 cofactor, Drp1 assembles to form spirals with a high GTPase activity that warps around the mitochondrial tubules to sever the OMM [63, 64]. Thus, ADP binding is structurally indispensable for Drp1-mediated mitochondrial fission. However, MiD49 shows only partial conservation as with the MiD51 ADP-binding sequence and ADP is not considered as a MiD49 cofactor for stimulating mitochondrial fission [64]. In addition to the distinct structures of MiD49 and MiD51, little is known about whether MiD49 and MiD51 differentially regulate mitochondrial homeostasis. Until very recently, we revealed that MiD51, but not MiD49, possesses a specific regulation on cell death and mitophagy, independent of its fission–fusion function [65].
In another mitochondrial fission regulatory protein, the functions of the 16-kDa OMM protein Fis1 on mitochondria dynamics has been of great interest. Fis1 contains a single transmembrane domain that integrates with the OMM [8] and was originally identified in budding yeast Saccharomyces cerevisiae. Fis1 physically interacts with Dnm1 and mediates Dnm1 assembly on the OMM [7]. Although the loss of Fis1 in yeast induces defects in mitochondrial Dnm1 recruitment and contributes to failed fission [66], its role in mammalian cells is debated in recent years [10, 60, 67]. For example, while high levels of Fis1 in HeLa and COS7 cells induce fission [67], conditional knockout of Fis1 in colon carcinoma cells causes Drp1 assembly and mitochondrial fragmentation [10]. Overall, it seems that Mff, MiD49, and MiD51 have a more predominant role than Fis1 in regulating Drp1 recruitment onto mitochondria-associated membranes (MAM) in mammals [10–12, 68].
Mitochondrial fusion machinery
OMM fusion is mediated by the GTPase proteins mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) (Fzo1 in yeast) [69]. Subsequent fusion of the inner membrane is achieved by OPA1 (Mgm1 in yeast) [70] (Fig. 1B). Prior to fusion, Mfn1 and Mfn2 assemble as homotypic and heterotypic dimers to tether to the adjacent OMM. They then undergo a conformational change driven by the GTP hydrolysis that facilitates the fusion process [71]. Interestingly, loss of Mfn1 alone displays short mitochondrial tubules or spheres in uniform sizes; whereas, Mfn2-depleted cells exhibit mitochondrial spheres of widely varying sizes [50]. However, depletion of both Mfn1 and Mfn2 together induces a greater impact of mitochondrial fragmentation with almost uniformed characteristic mitochondrial fragmentation, either with very short mitochondrial tubules or very small spheres. Pathologically, missense mutations of Mfn2 in human can lead to Charcot–Marie–Tooth neuropathy type 2A (CMT2A), an axonal peripheral sensorimotor neuropathy, and autosomal dominant peripheral neuropathy [72]. Additionally, mice lacking mitofusin proteins die during mid-gestation [50], implying that mitochondrial fusion is critical to embryonic development.
OPA1 is the best-studied inner mitochondrial membrane (IMM) fusion protein: it localizes to the inner membrane, where it binds negative charged phospholipids and controls cristae structure [51]. Although intrinsic OPA1 GTPase activity is low, self-oligomerization induced by association with phospholipids, such as cardiolipin, promotes GTP hydrolysis and results in deformation and tubulation of the inner membrane [52]. There are eight OPA1 isoforms that are produced by RNA splicing. These isoforms include the inner membrane-anchored long isoform L-OPA1 and the intermembrane space-localized short isoform S-OPA1 [73]. The IMM peptidase OMA1 mediates OPA1 cleavage, responding to mitochondrial membrane potential dissipation, as a balance between L-OPA1 and S-OPA1 is essential in maintaining mitochondrial morphology [73, 74].
Mitochondrial fusion is presumably to compensate defects by diluting stress inside of mitochondria. However, when the damage reaches to a certain threshold that fusion is uncapable to fix, a terminal response as destined mitochondrial removal may serve to maintain cellular homeostasis. Clearly, how interconnected mitochondria undergo clearance would usher more in-depth understanding of crosstalk among mitochondrial dynamics.
Mitochondria-associated diseases linked to mitochondrial dynamics
Inherited defects in genes serving for the machinery of mitochondrial dynamics contribute to mitochondrial disorders. Charcot–Marie–Tooth neuropathy type 2A (CMT2A) is a well-known genetically inherited neuronal disease, implicated in peripheral nervous system, caused by Mfn2 gene mutations [75–77]. Most of these mutations locate within or close to its GTPase domain and mitochondrial targeting region, which are responsible for mitochondrial fusion competency [76, 78]. OPA1 dysregulation confers susceptibility to dominant optic atrophy, a hereditary optic neuropathy, coupled with unopposed fragmented mitochondria [79, 80]. Imbalance of mitochondrial fission–fusion also causes neurodegenerative diseases. In the case of Parkinsonism, caused by autosomal recessive mutations of PINK1 or Parkin gene, possesses small mitochondria and abnormal accumulation of Drp1 [21, 81, 82]. In fibroblasts derived from patients with Huntington’s disease, dysfunctional huntingtin interacts with Drp1 and facilitates its GTPase activity, resulting in unrestricted mitochondrial division [83]. In addition to cardiomyopathies arising from hyper-fission, the skeletal muscle from obese and type 2 diabetic patients displays small and rounded mitochondria [78, 84–86]. Although significant progresses have been achieved, readout of fission–fusion dysregulation may not be sufficiently precise to understand a variety of pathologic symptoms in mitochondria-associated diseases. Clearly, continued efforts to understand mitochondrial behaviors, linking mitochondrial dynamics together with other actions, such as its quality control pathway, are required. This would essentially contribute to a more comprehensive perspective in therapeutic interventions about mitochondrial diseases.
Mitochondrial quality control
Mitochondrial unfolded protein response
Mitochondria employ different signaling pathways to maintain organelle quality in response to cellular stress. One such pathway is the mitochondrial unfolded protein response (UPRmt), which ensures mitochondrial proteostasis. This response is activated upon the aggregation of unfolded proteins within the mitochondria, or an imbalance of nuclear-encoded and mitochondria-encoded proteins, which confers proteotoxic stress [87–90]. UPRmt is well-characterized in C. elegans, which requires the matrix peptide exporter HAF1 and the bZIP transcription factor ATFS-1 [91]. In cells with healthy mitochondria, ATFS-1 localizes to the mitochondrial matrix, whereby it is constitutively degraded by AAA+-protease LON [92]. However, mitochondrial depolarization impairs ATFS-1 import into the mitochondria, mediated by the transporter protein HAF1 and triggers UPRmt. A large body of evidence suggests that UPRmt activation recovers mitochondria from damage and prolongs life span of organisms and is physiologically implicated in longevity and aging [90, 93, 94]. It remains unknow whether the regulation of UPRmt in C. elegans is developmentally conserved among different species. Therefore, how this process is regulated at the molecular level in mammals remains to be further studied. In addition, given the beneficial impacts of UPRmt as a defense mechanism induced by proteolysis dysfunction, it is important to investigate pharmaceutical approaches to target UPRmt, in a context that life span in higher organisms would be improved. It would shed new insights into promising therapeutic success to treat age-related diseases.
Mitophagy
Mitochondrial homeostasis is also achieved by selective autophagic elimination of mitochondria (mitophagy), which is active during cellular programming and differentiation. One example is the clearance of redundant mitochondria during erythrocyte differentiation [95]. Another instance is evident during embryogenesis, where the offspring mitochondria are exclusively inherited maternally and the paternal mitochondria must be removed by mitophagy [96].
In general, nonselective bulk autophagy is achieved via five characteristic stages: (i) isolation membrane initiation, (ii) phagophore expansion, (iii) autophagosome maturation, (iv) auto-lysosome fusion and (v) lysosomal degradation [32, 97–100]. During mitophagy (selective autophagy), the mitochondria are presumably targeted as autophagic cargoes, in line with these hierarchical steps for mitophagosome formation [36, 38, 101–104]. Intriguingly, the organelle-localized signals that trigger mitophagy or for bulk autophagy may differ. The question as to how the selective autophagic signaling assemble mitophagy, within the steady-state conditions of the whole cell, remains unclear. In addition, whether mitochondrial fission always serves to promote mitochondrial elimination is not yet completely understood [42].
PINK1/Parkin-dependent mitophagy
Mitophagy is differentially activated depending on disparate stimuli and manifested via specific routes. PINK1/Parkin-mediated mitophagy is the most prevalent mitophagy pathway (Fig. 2A). Under basal conditions, PTEN-induced putative kinase 1 (PINK1) is constitutively imported from the cytosol into the mitochondrial intermembrane space, where it is rapidly degraded by mitochondrial proteases and proteasome [105]. However, under stress conditions, mitochondrial depolarization prevents PINK1 import, allowing PINK1 to instead be stabilized on the OMM [34]. As a mitochondria serine/threonine kinase, PINK1 subsequently phosphorylates mitochondrial ubiquitin and the E3 ligase Parkin [106, 107]. In a feed-forward mechanism, phosphorylated Parkin further stimulates Parkin recruitment and activation [34, 108], allowing Parkin to target and ubiquitinate OMM proteins, including Mfn1 [109], Mfn2 [40, 109], translocase of outer mitochondrial membrane 20 (Tom20), and voltage-dependent anion channel [110] for degradation via the ubiquitin–proteasome system [111, 112]. These ubiquitinated OMM proteins yield more substrates for PINK1 phosphorylation, which in turn further activates Parkin in a positive amplification loop. Finally, autophagy receptors sequestosome-1 (p62/SQSTM1) [113], optineurin (OPTN) [114] and calcium-binding and coiled-coil domain-containing protein 2 (CALCOCO2/NDP52) recognize ubiquitin-tagged OMM proteins, and are recruited to sequester the mitochondria as autophagic cargo [37]. These autophagy adapters contain an LC3 interacting region (LIR), which directly interacts with autophagosomal LC3, and instigates mitochondrial elimination via mitophagosome trafficking and delivery into lysosome [115].
Fig. 2. Mammalian mitophagy machinery.

A During PINK1/Parkin-dependent mitophagy, ectopic toxins (such as CCCP) cause mitochondrial damage, in which PINK1 cleavage and import fail. PINK1 thus stabilizes on the outer mitochondrial membrane (OMM) and phosphorylates ubiquitin and Parkin. The attached OMM proteins are subsequently ubiquitinated by the E3 ligase, Parkin. These ubiquitinated OMM proteins serve as binding partners for P62. P62 further recruits LC3 and assembles the isolation membrane for selective autophagic mitochondrial removal. B During receptor-mediated mitophagy, in which cells are under hypoxic or mitochondrial membrane potential dissipation conditions, FUNDC1, Nix or Bcl-L-13 on OMM directly attracts LC3 via an LIR motif for mitophagosome formation. C STX17-induced mitophagy upon Fis1 loss participates in autonomous mitochondrial elimination through a hierarchical autophagic route. Under basal conditions, Fis1 interacts with STX17, preventing the over-translocation of STX17 onto mitochondria, governing the initiation of mitophagy. Fis1 loss primes the dynamic shuffling of STX17 onto mitochondria-associated membranes (MAM) and mitochondrial pools. Mitochondrial STX17 then self-oligomerizes and recruits ATG14. Subsequently, downstream autophagy modulators assemble on the mitochondria to ensure commitment to mitochondrial clearance.
Despite plenty of evidence demonstrating the necessity of PINK1 and Parkin in maintaining mitochondrial fidelity, most studies have used in vitro models of ectopic Parkin overexpression and induced mitochondrial damage with the mitochondrial uncoupler, carbonyl cyanide m-chlorophenyl hydrazine (CCCP) at high doses of 10–20 µM [34, 109, 116, 117]. Less is known about the mitochondrial “vulnerability” threshold to exogenous toxins, consequently undergoing autophagic clearance. Of note, a low dose of CCCP at 5 µM, the time-course of Parkin translocation at 1.5 h, degradation of the OMM proteins and mitochondrial removal at 16 h, can be readily visualized and tracked [65]. Somewhat surprisingly, the OMM proteins possess disparate modulation in the early versus the late stage of mitophagy. For example, Mfn2 was found to brake mitophagy at an early phase [118] but positively serves for mitophagy at a later stage of PINK1/Parkin-dependent mitophagy [65]. Remarkably, our recent study showed that MiD51 depletion, which induces mitochondrial fusion, confers susceptibility to Parkin recruitment and is associated with rapid clearance of the mitochondria [65]. This study was the first to illustrate a novel action of MiD51, and thus functionally segregate MiD51 from MiD49. More importantly, this finding highlighted that mitochondrial fission is not always indispensable for mitochondrial turnover [65].
PINK1/Parkin-independent mitophagy
Mitophagy receptor-mediated mitophagy
Recent intensive studies on alternative PINK1/Parkin-independent mitophagy have made great progress in understanding mitochondrial quality control in cells devoid of Parkin activity [45, 95, 119–122]. Studies have shown that mitophagy receptors directly interact with and recruit Microtubule-associated protein 1A/1B-light chain 3 (LC3) [119, 120] or GABA Type A Receptor-Associated Protein (GABARAP) [122], which are key components of autophagosome membrane, via the LIR motif to help sequester mitochondria for autophagic degradation. These receptors thus serve as a degradation “eat-me” signal for damaged mitochondria (Fig. 2B). Several OMM proteins have been identified as mitophagy receptors, including Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) [123], NIX (also known as BNIP3L) [95], Fun14 Domain-containing 1 (FUNDC1) [119], FK506-binding protein 8 (FKBP8) [120], and Bcl2-like protein 13 (Bcl2-L-13) [45, 46].
In response to hypoxia, BNIP3 and NIX are transcriptionally upregulated, and mediated by Hypoxia-inducible factor 1-alpha (HIF1α) [124, 125] and Forkhead box protein O3 (FOXO3) [125]. They play crucial complementary roles to govern mitochondrial quality and avoid abnormal reactive oxygen species (ROS) accumulation [122, 126]. Moreover, NIX regulates red blood cells development by removing redundant mitochondria [95]. The phosphorylation of BNIP3 and NIX drives mitophagy. The phosphorylation of Ser17 and Ser24 flanking the BNIP3 LIR motif promotes its binding to ATG8 members [127]. The phosphorylated Ser34/35 juxtaposed to the NIX LIR enhances its affinity to LC3A/B [128], and the phosphorylation of NIX at Ser212 accounts for its dimerization and robust recruitment of autophagosome onto mitochondria [129]. Moreover, upon hypoxia, FUNDC1 is phosphorylated and associated with LC3 [119], whereas FKBP8 strongly associates with lipidated LC3A upon mitochondrial depolarization [120]. Consequently, mitochondria are engulfed by autophagosome for autophagic elimination. Mammalian Bcl2-L-13, a homolog of yeast protein ATG32, induces mitochondrial fission and mitophagy by interacting LC3 in HEK293 cells after CCCP treatment [45]. In addition to the mechanisms mentioned above, a novel Parkin- or LC3B-independent but p62/LC3C-dependent piecemeal-type basal mitophagy has also been revealed recently [102]. In this pathway, it is suggested that lysosomal targeting of MTX1 by LC3C is required for the maintenance of mitochondrial network when cells undergo oxidative phosphorylation; however, how exactly MTX1 is recognized by LC3C has not yet been characterized [102].
Interestingly, the phospholipid cardiolipin, synthesized on the IMM, also contains a unique LIR motif and can initiate mitochondrial engulfment by LC3 via its externalization from the IMM to the OMM upon mitochondrial depolarization [130]. In primary cortical neurons and SH-SY5Y cells, rotenone induces the redistribution of cardiolipin onto OMM, and serves as an “eat-me” signal to recruit autophagosome onto mitochondria [130]. Given by the different extent of mitochondrial membrane potential loss caused by rotenone and CCCP treatment, cardiolipin was proposed to have a complementary role of Parkin for mitophagy [131]. The cardiolipin-mediated mitophagy is induced by mild stress caused by rotenone treatment, but CCCP incubation primes cells to PINK1/Parkin-dependent mitophagy. To this end, it would be very interesting to examine whether cardiolipin could function in the Parkin-depleted cells.
Collectively, mitophagy receptors are inactive under resting conditions, and their activity is elicited upon signals triggering mitochondrial damage. These receptors then exhibit a preferential association with LC3 family members, to recruit autophagosome that encapsulates mitochondria. As a consequence, mitochondria are removed via the autophagic route. Over the past several years, this field has gained fruitful achievements in understanding the various mitophagy receptors at the molecular level, particularly focusing on the autophagosome recruitment step. However, questions such as what signal exactly triggers the phagophore to engulf mitochondria would need to be further explored.
Mitophagy receptor-independent mitophagy
Most of the mitophagic pathways described above have been shown using acute and extreme stress signals, such as inducing mitochondrial depolarization with CCCP [34, 45], or mitochondrial respiration damage with oligomycin plus antimycin A [37], or hypoxia [119]. We, however, recently identified an additional mitophagy mechanism without using ectopic inductions [39]. Specifically, we showed that the SNARE protein, Syntaxin 17 (STX17), initiates mitophagy via a macroautophagic pathway that is triggered by the loss of the OMM protein, Fis1 [39]. Under basal conditions, Fis1 acts as a “gatekeeper” to prevent the dynamic trafficking of STX17 from the endoplasmic reticulum to the mitochondria and restraining STX17 self-oligomerization. As a result, basal mitophagy in the resting conditions is minimal. Conversely, loss of Fis1 through genetic approaches disables this protective gatekeeper from the mitochondria, resulting in priming of STX17 to actively translocate onto mitochondria-associated membranes (MAM) and the mitochondria. STX17 further recruits ATG14 onto the mitophagosome formation site and assembles the isolation membrane involving a group of proteins including DFCP1, WIPI-1, ATG5, and ATG16 on mitochondria (Fig. 2C). Subsequently, Rab7 is recruited onto the mitophagosome. As a small GTPase protein, Rab7 cycles between two nucleotide-bound states, a GDP-bound inactive state and a GTP-bound active state [132]. In the presence of an active GTP-bound form, Rab7 drives mitophagosome-lysosome fusion to ensure mitochondrial elimination [39, 133]. In demand of mitophagy activation upon Fis1 deficiency, STX17 also initiates the nuclear translocation of transcription factor EB (TFEB), a master regulator of autophagy and lysosome biogenesis [134]. Consequently, the nuclear localization and transcriptional activity of TFEB serve for the need of mitochondria turnover via autophagy [135].
More surprisingly, we also found that Drp1-modulated mitochondrial fission makes little contribution to the mechanistic regulation of STX17-induced mitophagy. The hyper-fused mitochondria from the loss of Drp1 can still undergo mitophagy [39]. Strikingly, this notion that fused mitochondria are permissible to autophagic clearance casts doubt to the mainstream concept that mitochondrial fission is preliminary for mitophagy [21, 22]. We seek to address this observation by illustrating the potential interplay between mitochondria dynamics and mitophagy; adapting to stress, mitochondrial dynamics and mitophagy are intimately integrated and coordinated to maintain mitochondrial homeostasis. Of note, dominant-negative mutant of Drp1, which facilitates mitochondrial fusion, abrogates mitophagy [41]. A predominant notion in this field is that mitochondrial fission represents an effort to favor mitophagy, given by that smaller mitochondria are easier to be encapsulated by autophagosome to proceed mitophagy [21, 22] (Fig. 3B). However, mitochondrial functions involve beyond the morphological side. In face of stress, mitochondrial fusion may be an initial response to aid in buffering and diluting mitochondrial defects (Fig. 3A). But when the damage reaches to a certain threshold that fusion is incompetent to repair, mitochondria are destined to be cleared wholesale through the autophagic route. Under intense damage such as calcium overload, oxidative stress or toxin accumulation, acute shift to mitochondrial fission may be retarded (Fig. 3B). Consequently, small sizes of mitochondria fail to be achieved. Instead, fused mitochondria acutely undergo mitophagy to terminate and remove the damage. In this case, mitochondrial fusion is permissible to cope with damage removal via mitophagic pathway (Fig. 3C).
Fig. 3. A schematic illustration about interplay between mitochondrial dynamics and mitophagy.

A As a first line of defense to toxins, mitochondrial fusion works as a “transient response”, by mixing and sharing mitochondrial contents, to dilute and buffer mitochondrial damage (black dots), as well as to recover bad mitochondria back to healthy mitochondria. B When deleterious damage accumulates in the mitochondria, fission results in the segregation of impaired mitochondria from healthy ones. Damaged mitochondria in small sizes are destined for autophagic removal. C In response to acute stress, mitochondria fuse in self-repair mechanism as a first-aid. Simultaneously, stress cumulates on the mitochondria. When the damage reaches a threshold at which “transient response” (fusion) is incompetent to repair, in the case that mitochondrial fission is retarded by mitochondria dysfunction or restrained by genetic ablation of fission machinery, mitochondria undergo autophagic elimination at a hyper-fused state. Fused mitochondria are permissible to mitophagy.
By tethering an autophagy molecule (STX17) and an OMM protein (Fis1), our work is the first to bridge mitochondria and hierarchical autophagic route in a perfect coalescence, in which autonomous mitophagy regulation independent of ectopic mitochondrial damages is achieved. The ER-resident protein STX17 has controversial roles in PINK1/Parkin-dependent mitophagy; while it modulates the localization of PGAM5 and the association between PGAM5 and FUNDC1 to facilitate mitophagy [136], STX17 is also reported to be dispensable for depolarized mitochondria removal mediated by PINK1/Parkin [137]. Given a novel role of STX17 in PINK1/Parkin-independent mitophagy [39], future investigations concerning the role of ER for mitophagy are needed. It is also unclear how the spatial position of MAM allows for mitophagosome formation. Another important piece of puzzle at the mechanistic level is what mitochondrial dysfunction intrinsically initiates this type of autonomous mitophagy. Furthermore, subsequent advances in pathological impacts of STX17-induced mitophagy will promise more in-depth clues of mitochondria-related diseases.
Bulk autophagy versus mitophagy
Bulk autophagy is an evolutionarily conserved “self-eating” process, regulated by genetic programming. It is a highly hierarchical pathway that involves the de novo formation of vesicles characterized as “autophagosomes”, which engulf regions of the cytoplasm, damaged or unwanted organelles, protein aggregates, and invading pathogens [32, 33, 100]. In particular, mitophagy is a selective form of autophagy: numerous studies have illustrated that mitophagy employs general autophagic mediators, such as ULK1 [37, 46, 138], FIP200, and ATG13 [46, 104, 139], which are recruited onto damaged mitochondria. It has been reported that Bcl2-L-13, the mammalian homolog of Atg32, is an essential mitophagy receptor in yeast and relies on the ULK1 complex to regulate mitochondrial elimination [46]. However, little is known regarding whether mitophagic formation is distinguishable from the hierarchy of bulk autophagy. Indeed, based on our study of STX17-initiated mitophagy upon Fis1 loss, the mitophagic process directly recruits ATG14 and PI3P (phosphatidylinositol-3-phosphate) binding proteins, but independent of upstream proteins ULK1 and ATG9A [39]. Therefore, we suggest that mitophagy is activated by a specific “organelle-localized” signaling on mitochondria, that differs from the “global signaling-triggered” bulk autophagy. ULK1 is activated at the early stage of bulk autophagy. However, the “organelle-localized” mitophagy does not require ULK1, which is indispensable for “global signaling-triggered” bulk autophagy, though a well-established bulk autophagy modulator ATG13 displays an oscillatory dynamic translocation onto mitochondria and accounts for mitochondrial clearance [104]. Greater insights into the specific mitophagic machinery, that is unique from bulk autophagic machinery may allow for a better understanding of selective autophagy.
Physiological roles of mitophagy
Dysregulated mitophagy is highly implicated in various pathologies, including those affecting neurons [101, 140–142] and muscle [143], as well as aging [144, 145] and cancer [146, 147]. PINK1/Parkin-dependent mitophagy is highly emphasized in neurodegenerative diseases, such as Parkinson’s disease (PD) [101, 140, 148], Alzheimer’s disease (AD) [142], and Huntington’s disease (HD) [149]. Mutations of PINK1 and Parkin have been linked to autosomal recessive PD [101, 140, 141]. Gene mutations cause the inactivity of PINK1 [148] or Parkin [141, 150], which consequently decelerates mitophagy in neuron cells. Lack of mitophagy causes the accumulation of dysfunctional mitochondria, which result in loss of dopaminergic neurons at the early onset of PD [141]. In addition, key research on PINK1/Parkin-mediated mitophagy has led to an explosion of knowledge regarding its significance in AD [142] and HD [149]. While the overexpression of Parkin in AD mice restored activity-dependent synaptic plasticity and rescued behavior abnormalities, including decreasing β-amyloid load [142], mitophagy was found to be defective in the HD mouse model [149]. Clearly, the specific mechanism of mitophagy among different models of neurodegenerative diseases requires to be further explored. How exactly mitophagy dysregulation contributes to diseases is still enigmatic. Furthermore, the effects of mitophagy are prominent in skeletal muscle development. During myogenesis process from immature myoblasts to mature myotubes, mitophagy is upregulated to ensure the metabolic shift from glycolysis to OXPHOS, in support of the increased energetic demand of contractile muscle [143]. Also, dysfunction of mitophagy by depletion of ATG5 or P62 in C2C12 cells retards myotube development [143]. Moreover, the critical role of mitophagy in aging has been manifested by life span analyses conducted in Drosophila [144, 145] and C. elegans [151] models, in which mitophagy prolonged longevity but declined along with aging, most likely because that mitophagy removes dysfunctional mitochondria coupled with mtDNA mutations [152].
In the other spectrum, whether mitophagy positively regulates tumorigenesis remains debatable [124, 146, 153, 154]. Parkin loss is closely associated with cancer progression in various tissues [153], as Parkin knockout mice show spontaneous tumor growth, suggesting that the accumulated dysfunctional mitochondria, as a result of mitophagy defect, generates high ROS levels. Increased ROS production leads to increased transcriptional activation of genes involved in glycolysis, which facilitates the Warburg effect and is optimal for cancer cell survival as well as development [146]. Moreover, the mitophagy receptor BNIP3 is responsible for tumor suppression, supported by that Bnip3 depletion in mice model enables breast cancer metastasis in the lung, liver, and bone [147]. Nevertheless, mitophagy can also confer a supportive role in oncology because hypoxia, a pro-survival microenvironment for cancer cells, induces mitophagy [124]. Up to now, we are still in the early stages of understanding the mechanistic actions of specific molecules in the pathological level, mostly focusing on Parkin. Thus, the precise relevance of mitophagy in potential therapeutic treatments requires more exploration.
Crosstalk between mitochondrial dynamics and mitophagy
Mitochondrial dynamics closely links to mitochondrial quality control, especially mitophagy. A large body of studies have proposed that mitochondrial fission is necessary for the initiation of mitophagy, but fusion serves as a “rescue” mechanism preventing mitophagy [20, 21, 41]. A simple rationale is that mitochondria fission segregates dysfunctional mitochondria into small sizes to be easily cleared (Fig. 3B). Conversely, fusion acts as a “transient response” adapting to mitochondrial stress and dilutes impaired mitochondrial components (Fig. 3A). Depolarization of mitochondria is a well-established driving force for mitophagy, involved in PINK1/Parkin [34], NIX [126], and Bcl2-L-13 [45]-mediated mitochondrial elimination. Clearly, mitochondrial fragmentation is coupled with mitochondrial membrane potential loss and precedes mitophagosome formation [34, 45, 126]. Upon hypoxic stress, FUNDC1-dependent mitophagy also associates with mitochondrial fission [43]. In support of this positive coherence between fission and mitophagy, genetic manipulation of the Drp1 through overexpression of dominant-negative form [41] or Drp1 deficiency [42, 109] both retarded mitophagy. Nevertheless, it still remains debatable whether mitochondrial fission is indeed a precursor for mitochondrial elimination. Recently, it was proposed that mitochondrial division is dispensable for mitophagy [155]. Consistently, our recent studies also found that certain OMM proteins that mediate mitochondrial dynamics do not necessarily involve in mitophagy formation via fission–fusion [39, 65]. As discussed, MiD51 depletion even primes cells for PINK1/Parkin-mediated mitochondrial removal, despite that MiD51 loss triggers mitochondrial fusion. We elucidated that the modulation of mitochondrial quality control uncouples from mitochondrial dynamics [65]. In line with this, we also reveal that over-fusion of mitochondria, resulted from Drp1 or Mff silencing using RNA interference, has no effect on STX17-initiated mitophagy upon Fis1 loss, further substantiating the idea that mitochondrial fission–fusion dynamics is not a prerequisite for and may be split from mitophagy [39]. Undoubtedly, more studies are needed to define the crosstalk between mitochondrial dynamics and mitophagy. The key question as to how tubular mitochondria are directly pinched off from main mitochondrial bodies engulfed by autophagy isolation membrane remains to be answered. Furthermore, autonomous mitochondrial damage at the genetic level, not induced by ectopic toxins, may be a driving force for mitophagy, and closer investigations of how specific OMM proteins engage in regulating mitochondrial homeostasis in this specific context is also necessary. We consider that future work which focuses on developing pathological models, based on the physiological contributions by these mechanisms, will yield a better understanding of translational significances related to mitochondrial functions.
Of note, MAM, the interface between ER and mitochondria, has been highly emphasized in mitochondrial dynamics and mitophagy [156–160], and is responsible for mitochondrial constriction [161]. Mitochondrial Drp1 receptors and Drp1 oligomers assemble on MAM, whereby ER wraps around mitochondria, to form a fission site [68, 162]. In addition, Mfn2 enriched on MAM tightens the juxtaposition of mitochondria and ER, and positively regulates mitochondrial calcium influx [163]. It was reported that Mfn2 ablation in cells disrupted ER morphology and loosened ER and mitochondria interactions. The increased distance between ER and mitochondria couples with mitochondrial fragmentation [163]. In addition, the GTPase protein S-OPA1 also accumulates and localizes on MAM to accelerate fission [164]. All these studies support that MAM is critical to mitochondrial dynamics. On the other hand, the ER-resident protein STX17, shuffles among ER, MAM, and mitochondria in response to nutrient levels, conferring important roles in switching between mitochondrial dynamics and autophagosome formation [158, 165]. Closer investigations of STX17 on MAM have demonstrated that MAM is involved in mitophagy [39, 136, 137], implying a broad role of MAM in mitochondrial homeostasis. Currently, we still lack a comprehensive picture of the coordination between fission and mitophagy initiation on MAM. It will be intriguing to answer whether the Drp1-mediated fission on MAM is a “trigger” or “brake” for mitophagy initiation. Is the site that Drp1 severs mitochondria also accounting for mitophagosome formation? An important question is how exactly fission and mitophagy are orchestrated remains to be elucidated.
Conclusion
In summary, the advent of studies about the multi-facets of mitochondrial behaviors usher in the exciting areas concerning the crosstalk among these actions. OMM proteins distribute on the interface between mitochondria and cytosol, or ER, in a geographic advantage that they crucially play for the interplay of various mitochondrial signaling pathways. Up to now, exciting breakthroughs into deciphering OMM proteins in mitochondrial dynamics and mitophagy have informatively shed light into mitochondrial homeostasis (Table 1). But greater insights into the significance of mitochondrial functions and mitochondria-associated diseases are still needed for further investigation. Given the promising advances of more metabolic and genomic approaches, more in-depth understanding into animal disease models and pathological causes will allow for a better prospect in rational therapeutic application.
Table 1.
Lessons from OMM proteins: mitochondrial fission does not essentially favor mitophagy.
| Protein | Action on mitochondrial dynamics | Role in mitophagy |
|---|---|---|
| Drp1 | A GTPase protein that accounts for mitochondrial fission. |
1. Facilitates PINK1/Parkin-dependent mitophagy [42, 109] and FUNDC1-mediated mitophagy [43]. 2. No effect on STX17-initiated mitophagy [39]. |
| Mff |
An OMM protein; A Drp1 receptor to support mitochondrial division. |
1. Facilitates PINK1/Parkin-dependent mitophagy [166]. 2. No effect on STX17-initiated mitophagy [39]. |
| Fis1 |
An OMM protein; A controversial role in fission–fusion. |
1. Promotes mitophagosome biogenesis for PINK1/Parkin-mediated mitophagy [133]. 2. Retards mitophagy by governing the over-assembly of STX17 on mitochondria. Loss of Fis1 primes cells for STX17-induced mitophagy [39]. |
| MiD49 |
An OMM protein; A Drp1 receptor to support mitochondrial division. |
Positively regulates PINK1/Parkin-dependent mitophagy. Loss of MiD49 decelerates PINK1/Parkin-dependent mitophagy [65]. |
| MiD51 |
An OMM protein; A Drp1 receptor to support mitochondrial division. |
Restrains PINK1/Parkin-dependent mitophagy. MiD51 loss accelerates Parkin translocation onto depolarized mitochondria [65] and mitophagy. |
| Mfn1 |
An OMM protein; GTPase activity required for mitochondrial fusion. |
1. Substrate of UPS induced by Parkin, which is preliminary for PINK1/Parkin-dependent mitophagy [109]; 2. Required for Gp78-dependent mitophagy upon mitochondrial depolarization [167]. |
| Mfn2 |
An OMM protein; GTPase activity required for mitochondrial fusion. |
1. Substrate of UPS induced by Parkin, which is preliminary for PINK1/Parkin-dependent mitophagy [109]; 2. At the early stage of mitochondrial depolarization, Mfn2 functions as an ER-mitochondria tether to prevent PINK1/Parkin-mediated mitophagy [118]; 3. At the late stage of PINK1/Parkin-mediated mitophagy, the UPS activation of Mfn2 is indispensable for mitophagy [65]. |
| OPA1 |
An IMM protein; GTPase activity required for mitochondrial fission or fusion (depending on disparate OPA1 isoforms). |
1. Negatively regulates basal mitophagy [168]; 2. A negative role to gate FUNDC1-mediated mitophagy [43]. |
OMM outer mitochondrial membrane, IMM inner mitochondrial membrane, UPS ubiquitin–proteasome system.
Acknowledgements
We apologize to the authors of any key studies that were omitted due to space limitations. We thank all members of YCL laboratory for valuable discussion. We are grateful to Dr. Yew Mun Lee and Dr. Yuan Liu for their helpful discussion and suggestions. We truly appreciate the two anonymous referees for their valuable comments and suggestions on our paper. This work is financially supported by Tier 2 (MOE-T2-1-131) and Tier1 (R-154-000-B51-114; NUSMed-FoS Joint Research Program) grants from the Ministry of Education (MOE), Singapore, awarded to YCL.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by: G. Del Sal
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–60. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 2.Lane N, Martin W. The energetics of genome complexity. Nature. 2010;467:929–34. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- 3.Palade GE. The fine structure of mitochondria. Anat Rec. 1952;114:427–51. doi: 10.1002/ar.1091140304. [DOI] [PubMed] [Google Scholar]
- 4.Sjostrand FS. Electron microscopy of mitochondria and cytoplasmic double membranes. Nature. 1953;171:30–2.. doi: 10.1038/171030a0. [DOI] [PubMed] [Google Scholar]
- 5.Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci USA. 2010;107:16823–7. doi: 10.1073/pnas.1011099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, et al. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272:1136–44. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 7.Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–80. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–9. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 9.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–20. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–58. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–67. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–73. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 14.Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW, Walter P. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol Biol Cell. 1997;8:1233–42. doi: 10.1091/mbc.8.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michalska BM, Kwapiszewska K, Szczepanowska J, Kalwarczyk T, Patalas-Krawczyk P, Szczepański K, et al. Insight into the fission mechanism by quantitative characterization of Drp1 protein distribution in the living cell. Sci Rep. 2018;8:8122. doi: 10.1038/s41598-018-26578-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pagliuso A, Cossart P, Stavru F. The ever-growing complexity of the mitochondrial fission machinery. Cell Mol life Sci. 2018;75:355–74. doi: 10.1007/s00018-017-2603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–9. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tam ZY, Gruber J, Halliwell B, Gunawan R. Context-dependent role of mitochondrial fusion-fission in clonal expansion of mtDNA mutations. PLoS Comput Biol. 2015;11:e1004183. doi: 10.1371/journal.pcbi.1004183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. 2011;14:1939–51. doi: 10.1089/ars.2010.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505:335–43. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh-hama T. Evolutionary consideration on 5-aminolevulinate synthase in nature. Orig Life Evol Biosph. 1997;27:405–12. doi: 10.1023/a:1006583601341. [DOI] [PubMed] [Google Scholar]
- 24.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, et al. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006;40:553–60. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. 2013;6:19. doi: 10.1186/1756-8722-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42:406–17. doi: 10.1016/j.immuni.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–62. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–42. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 31.Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–94. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- 32.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 33.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–62. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 34.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–74. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 36.Wei H, Liu L, Chen Q. Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim Biophys Acta. 2015;1853:2784–90. doi: 10.1016/j.bbamcr.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 37.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–14. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Qi W, Chen G, Feng D, Liu J, Ma B, et al. Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy. 2015;11:1216–29. doi: 10.1080/15548627.2015.1017180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xian H, Yang Q, Xiao L, Shen H-M, Liou Y-C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat Commun. 2019;10:2059. doi: 10.1038/s41467-019-10096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–5. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol. 2017;216:3231–47. doi: 10.1083/jcb.201612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12:689–702. doi: 10.1080/15548627.2016.1151580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017;18:495–509. doi: 10.15252/embr.201643309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527. doi: 10.1038/ncomms8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murakawa T, Okamoto K, Omiya S, Taneike M, Yamaguchi O, Otsu K. A mammalian mitophagy receptor, Bcl2-L-13, recruits the ULK1 complex to induce mitophagy. Cell Rep. 2019;26:338–45.e6. doi: 10.1016/j.celrep.2018.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–78. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 48.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–62. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 49.Meyer JN, Bess AS. Involvement of autophagy and mitochondrial dynamics in determining the fate and effects of irreparable mitochondrial DNA damage. Autophagy. 2012;8:1822–3. doi: 10.4161/auto.21741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–10. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 52.Ban T, Heymann JA, Song Z, Hinshaw JE, Chan DC. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet. 2010;19:2113–22. doi: 10.1093/hmg/ddq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zuchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol. 2006;59:276–81. doi: 10.1002/ana.20797. [DOI] [PubMed] [Google Scholar]
- 54.Suomalainen A, Battersby BJ. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol. 2018;19:77–92. doi: 10.1038/nrm.2017.66. [DOI] [PubMed] [Google Scholar]
- 55.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–25. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 56.Xiao L, Xian H, Lee KY, Xiao B, Wang H, Yu F, et al. Death-associated protein 3 regulates mitochondrial-encoded protein synthesis and mitochondrial dynamics. J Biol Chem. 2015;290:24961–74. doi: 10.1074/jbc.M115.673343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–7. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tieu Q, Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol. 2000;151:353–66. doi: 10.1083/jcb.151.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Griffin EE, Graumann J, Chan DC. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol. 2005;170:237–48. doi: 10.1083/jcb.200503148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–12. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, et al. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–78. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem. 2013;288:27584–93. doi: 10.1074/jbc.M113.479873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Richter V, Palmer CS, Osellame LD, Singh AP, Elgass K, Stroud DA, et al. Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J Cell Biol. 2014;204:477–86. doi: 10.1083/jcb.201311014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loson OC, Liu R, Rome ME, Meng S, Kaiser JT, Shan SO, et al. The mitochondrial fission receptor MiD51 requires ADP as a cofactor. Structure. 2014;22:367–77. doi: 10.1016/j.str.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xian H, Liou YC. Loss of MIEF1/MiD51 confers susceptibility to BAX-mediated cell death and PINK1-PRKN-dependent mitophagy. Autophagy. 2019;15:2107–25. doi: 10.1080/15548627.2019.1596494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shaw JM, Nunnari J. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 2002;12:178–84. doi: 10.1016/s0962-8924(01)02246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci. 2004;117:1201–10. doi: 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- 68.Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, et al. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci. 2016;129:2170–81. doi: 10.1242/jcs.185165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meeusen S, McCaffery JM, Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305:1747–52. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- 70.Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. 2009;20:3525–32. doi: 10.1091/mbc.E09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–62. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 72.Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–51. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 73.Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–77. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anand R, Langer T, Baker MJ. Proteolytic control of mitochondrial function and morphogenesis. Biochim Biophys Acta. 2013;1833:195–204. doi: 10.1016/j.bbamcr.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 75.Detmer SA, Chan DC. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 2007;176:405–14. doi: 10.1083/jcb.200611080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cho HJ, Sung DH, Kim BJ, Ki CS. Mitochondrial GTPase mitofusin 2 mutations in Korean patients with Charcot-Marie-Tooth neuropathy type 2. Clin Genet. 2007;71:267–72. doi: 10.1111/j.1399-0004.2007.00763.x. [DOI] [PubMed] [Google Scholar]
- 77.Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T, et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. 2005;116:23–7. doi: 10.1007/s00439-004-1199-2. [DOI] [PubMed] [Google Scholar]
- 78.Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, et al. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405–15. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 79.Cohn AC, Toomes C, Potter C, Towns KV, Hewitt AW, Inglehearn CF, et al. Autosomal dominant optic atrophy: penetrance and expressivity in patients with OPA1 mutations. Am J Ophthalmol. 2007;143:656–62. doi: 10.1016/j.ajo.2006.12.038. [DOI] [PubMed] [Google Scholar]
- 80.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–6. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 81.Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lammermann K, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem. 2009;284:22938–51. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ishihara N, Otera H, Oka T, Mihara K. Regulation and physiologic functions of GTPases in mitochondrial fusion and fission in mammals. Antioxid Redox Signal. 2013;19:389–99. doi: 10.1089/ars.2012.4830. [DOI] [PubMed] [Google Scholar]
- 83.Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. 2011;17:377–82. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–76. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–28. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–7. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 87.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–9. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–7. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Hoj PB, et al. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem. 1996;240:98–103. doi: 10.1111/j.1432-1033.1996.0098h.x. [DOI] [PubMed] [Google Scholar]
- 90.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–80. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 91.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–40. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–90. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pulliam DA, Bhattacharya A, Van Remmen H. Mitochondrial dysfunction in aging and longevity: a causal or protective role? Antioxid Redox Signal. 2013;19:1373–87. doi: 10.1089/ars.2012.4950. [DOI] [PubMed] [Google Scholar]
- 94.Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–8. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 95.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–5. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–4. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- 97.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 99.Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr. 2007;27:19–40. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- 100.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 101.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–73. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Le Guerroue F, Eck F, Jung J, Starzetz T, Mittelbronn M, Kaulich M, et al. Autophagosomal content profiling reveals an LC3C-dependent piecemeal mitophagy pathway. Mol Cell. 2017;68:786–96.e6. doi: 10.1016/j.molcel.2017.10.029. [DOI] [PubMed] [Google Scholar]
- 103.Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20:31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dalle Pezze P, Karanasios E, Kandia V, Manifava M, Walker SA, Gambardella Le Novere N, et al. ATG13 dynamics in nonselective autophagy and mitophagy: insights from live imaging studies and mathematical modeling. Autophagy. 2020;1–11. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 105.Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–85. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, et al. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol. 2015;209:111–28. doi: 10.1083/jcb.201410050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–53. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiao B, Goh JY, Xiao L, Xian H, Lim KL, Liou YC. Reactive oxygen species trigger Parkin/PINK1 pathway-dependent mitophagy by inducing mitochondrial recruitment of Parkin. J Biol Chem. 2017;292:16697–708. doi: 10.1074/jbc.M117.787739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–80. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ham SJ, Lee D, Yoo H, Jun K, Shin H, Chung J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc Natl Acad Sci USA. 2020;117:4281–91. doi: 10.1073/pnas.1909814117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–40. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ordureau A, Paulo JA, Zhang J, An H, Swatek KN, Cannon JR, et al. Global landscape and dynamics of Parkin and USP30-dependent ubiquitylomes in ineurons during mitophagic signaling. Mol Cell. 2020;77:1124–42. doi: 10.1016/j.molcel.2019.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Evans CS, Holzbaur ELF. Lysosomal degradation of depolarized mitochondria is rate-limiting in OPTN-dependent neuronal mitophagy. Autophagy. 2020;16:962–4. doi: 10.1080/15548627.2020.1734330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ryan TA, Phillips EO, Collier CL, Jb Robinson A, Routledge D, Wood RE, et al. Tollip coordinates Parkin-dependent trafficking of mitochondrial-derived vesicles. EMBO J. 2020;39:e102539. doi: 10.15252/embj.2019102539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107:378–83. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McLelland GL, Goiran T, Yi W, Dorval G, Chen CX, Lauinger ND, et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife. 2018;7:e32866. doi: 10.7554/eLife.32866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–85. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 120.Bhujabal Z, Birgisdottir AB, Sjottem E, Brenne HB, Overvatn A, Habisov S, et al. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18:947–61. doi: 10.15252/embr.201643147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wei Y, Chiang WC, Sumpter R, Jr., Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–38.e10. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielmann Y, Stangler T, et al. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5:690–8. doi: 10.4161/auto.5.5.8494. [DOI] [PubMed] [Google Scholar]
- 123.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–73. [PubMed] [Google Scholar]
- 125.Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene. 2008;27:S114–27. doi: 10.1038/onc.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–90. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288:1099–113. doi: 10.1074/jbc.M112.399345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, Kawasaki M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep. 2017;7:1131. doi: 10.1038/s41598-017-01258-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Marinkovic M, Sprung M, Novak I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy. 2020;1–13. [DOI] [PMC free article] [PubMed]
- 130.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Williams JA, Ding WX. Mechanisms, pathophysiological roles and methods for analyzing mitophagy - recent insights. Biol Chem. 2018;399:147–78. doi: 10.1515/hsz-2017-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pfeffer SR. Rab GTPase regulation of membrane identity. Curr Opin Cell Biol. 2013;25:414–9. doi: 10.1016/j.ceb.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife. 2014;3:e01612. doi: 10.7554/eLife.01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–7. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 135.Nezich CL, Wang C, Fogel AI, Youle RJ. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol. 2015;210:435–50. doi: 10.1083/jcb.201501002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sugo M, Kimura H, Arasaki H, Amemiya T, Hirota N, Dohmae n, et al. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. EMBO J. 2018;37:e98899. doi: 10.15252/embj.201798899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McLelland GL, Lee SA, McBride HM, Fon EA. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016;214:275–91. doi: 10.1083/jcb.201603105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15:566–75. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci. 2012;125:1488–99. doi: 10.1242/jcs.094110. [DOI] [PubMed] [Google Scholar]
- 140.Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, et al. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6:871–8. doi: 10.4161/auto.6.7.13286. [DOI] [PubMed] [Google Scholar]
- 141.Houlden H, Singleton AB. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012;124:325–38. doi: 10.1007/s00401-012-1013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hong X, Liu J, Zhu G, Zhuang Y, Suo H, Wang P, et al. Parkin overexpression ameliorates hippocampal long-term potentiation and beta-amyloid load in an Alzheimer’s disease mouse model. Hum Mol Genet. 2014;23:1056–72. doi: 10.1093/hmg/ddt501. [DOI] [PubMed] [Google Scholar]
- 143.Sin J, Andres AM, Taylor DJ, Weston T, Hiraumi Y, Stotland A, et al. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy. 2016;12:369–80. doi: 10.1080/15548627.2015.1115172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rana A, Rera M, Walker DW. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci USA. 2013;110:8638–43. doi: 10.1073/pnas.1216197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–83. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015;16:1145–63. doi: 10.15252/embr.201540759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Manka D, Spicer Z, Millhorn DE. Bcl-2/adenovirus E1B 19 kDa interacting protein-3 knockdown enables growth of breast cancer metastases in the lung, liver, and bone. Cancer Res. 2005;65:11689–93. doi: 10.1158/0008-5472.CAN-05-3091. [DOI] [PubMed] [Google Scholar]
- 148.Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. 2001;68:895–900. doi: 10.1086/319522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Khalil B, El Fissi N, Aouane A, Cabirol-Pol MJ, Rival T, Lievens JC. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015;6:e1617. doi: 10.1038/cddis.2014.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wauer T, Komander D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013;32:2099–112. doi: 10.1038/emboj.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Palikaras K, Lionaki E, Tavernarakis N. Coupling mitogenesis and mitophagy for longevity. Autophagy. 2015;11:1428–30. doi: 10.1080/15548627.2015.1061172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, et al. Measuring in vivo mitophagy. Mol Cell. 2015;60:685–96. doi: 10.1016/j.molcel.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hu HH, Kannengiesser C, Lesage S, Andre J, Mourah S, Michel L, et al. PARKIN inactivation links Parkinson’s disease to melanoma. J Natl Cancer Inst. 2016;108:djv340. doi: 10.1093/jnci/djv340. [DOI] [PubMed] [Google Scholar]
- 154.Yeo CW, Ng FS, Chai C, Tan JM, Koh GR, Chong YK, et al. Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res. 2012;72:2543–53. doi: 10.1158/0008-5472.CAN-11-3060. [DOI] [PubMed] [Google Scholar]
- 155.Yamashita SI, Jin X, Furukawa K, Hamasaki M, Nezu A, Otera H, et al. Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol. 2016;215:649–65. doi: 10.1083/jcb.201605093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–21. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607–25. doi: 10.1038/nrm3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell. 2015;32:304–17. doi: 10.1016/j.devcel.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 159.Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016;35:1368–84. doi: 10.15252/embj.201593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wu W, Li W, Chen H, Jiang L, Zhu R, Feng D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy. 2016;12:1675–6. doi: 10.1080/15548627.2016.1193656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–62. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ji WK, Chakrabarti R, Fan X, Schoenfeld L, Strack S, Higgs HN. Receptor-mediated Drp1 oligomerization on endoplasmic reticulum. J Cell Biol. 2017;216:4123–39. doi: 10.1083/jcb.201610057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–10. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 164.Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204:919–29. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–93. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 166.Gao J, Qin S, Jiang C. Parkin-induced ubiquitination of Mff promotes its association with p62/SQSTM1 during mitochondrial depolarization. Acta Biochim Biophys Sin (Shanghai) 2015;47:522–9. doi: 10.1093/abbs/gmv044. [DOI] [PubMed] [Google Scholar]
- 167.Fu M, St-Pierre P, Shankar J, Wang PT, Joshi B, Nabi IL, et al. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell. 2013;24:1153–62. doi: 10.1091/mbc.E12-08-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liao C, Ashley N, Diot A, Morten K, Phadwal K, Williams A, et al. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology. 2017;88:131–42. doi: 10.1212/WNL.0000000000003491. [DOI] [PMC free article] [PubMed] [Google Scholar]