

Abstract
The cecal microbiota plays important roles in host food digestion and nutrient absorption, which may in part affect feed efficiency (FE). To investigate the composition and functional differences of cecal microbiota between high (n = 30) and low (n = 29) feed conversion ratio (FCR; metric for FE) groups, we performed 16S rRNA gene sequencing and predicted the metagenome function using Phylogenetic Investigation of Communities by Reconstruction of Unobserved Species in yellow broilers. The results showed that the 2 groups had the same prominent microbes but with differing abundance. Firmicutes, Bacteroidetes, and Actinobacteria were 3 prominent bacterial phyla in the cecal microbial community. Although there were no differences in microbial diversity, compositional differences related to FCR were found via linear discriminant analysis (LDA) effect size; the genus Bacteroides had a significantly higher abundance (LDA >2) in the high FE (HFE) group than in the low FE group. Furthermore, genus Bacteroides had a negative FCR-associated correlation (P < 0.05). Oscillospira was positively correlated with Bacteroides in both groups, whereas Dorea was negatively correlated with Bacteroides in the HFE group. Predictive functional analysis revealed that metabolic pathways such as “starch and sucrose metabolism,” “phenylalanine, tyrosine and tryptophan biosynthesis,” and “carbohydrate metabolism” were significantly enriched in the HFE group. The relatively subtle differences in FE-associated cecal microbiota composition suggest a possible link between cecal microbiota and FE. Moreover, Bacteroides may potentially be used as biomarkers for FE to improve growth performance in yellow broilers.
Key words: yellow broiler, feed conversion ratio, cecal microbiota, microbial community, 16S rRNA gene
Introduction
Domestic chickens are a commonly used animal model in biological research and a major source of food and protein worldwide (Oakley et al., 2015). Body weight gain (BWG) and broiler performance are the main concerns for producers of chickens for meat. Feed accounts for more than 70% of production costs (Aggrey et al., 2010), which are closely linked to poultry industry profit. Improving feed efficiency (FE) can increase nutrient utilization in feed, while reducing waste, greenhouse gases emission, and excrement effluent (Hume et al., 2011; Liu et al., 2017). The performance of a chicken flock can be evaluated by using the feed conversion ratio (FCR) or residual feed intake (FI), metrics of FE (Aggrey et al., 2010; Willems et al., 2013). Feed conversion ratio is widely used for meat producing poultry and is calculated as FI divided by BWG. Thus, flocks with a low FCR are regarded as having a high FE (HFE). Genetics, health, diet, and rearing environment all influence FCR (Pedroso et al., 2006; Al-Fataftah and Abu-Dieyeh, 2007; Awad et al., 2009; Aggrey et al., 2010). In addition, variation in FCR is closely related to gut microbiota (Singh et al., 2014; Stanley et al., 2016; Yan et al., 2017).
The chicken gastrointestinal tract (GIT) is a place for digestion and nutrition absorption; the complex and diverse microbial communities of the GIT aid in the breakdown and digestion of food (Stanley et al., 2014). The relationship between the microorganisms of each intestinal segment of the GIT and FE has been reported (Stanley et al., 2012, 2016; Yan et al., 2017). High-throughput 16S rRNA-based pyrosequencing analysis of poultry fecal microbiota showed that Cloacibacillus, Helicobacter, and Oscillibacter are more abundant in birds with low FCR (Singh et al., 2012). Poultry fecal metagenomes further revealed that 33 genera are significantly different in high and low FCR birds (Singh et al., 2014).
The chicken cecum is considered to be the most important part in the distal intestine, with the greatest concentration of intestinal microorganisms in mature chickens, affecting health and performance (Johansson et al., 1948; Degolier et al., 1999; Stanley et al., 2014). Digestion in the cecum is associated with cecal microbes (Clench and Mathias, 1995). Digestibility and the ability to metabolize crude fiber or other nutrients are lower in birds with a cecectomy than in normal birds (Chaplin, 1989). Thus, considerable attention has been paid to cecum microbiota (Corrigan et al., 2011; Sergeant et al., 2014), but relatively few studies are available on its relationship with FE. Research on the cecal microbiota found 24 unclassified bacterial species to be differentially abundant between high and low FCR chickens (Stanley et al., 2012). With advancements in sequencing technology, a new method for metagenomic biomarker discovery and a key tool of predictive functional profiling of microbial communities have been widely used (Segata et al., 2011; Langille et al., 2013). Increased information on the community and functional capacity of the cecal microbiota associated with FE enables a more comprehensive HFE characterization. Most researchers agree that the ceca are the primary site for microbial fermentation, where undigested carbohydrates are transformed into short-chain fatty acids (SCFA), lactate, and gases (Marounek et al., 1999; Jamroz et al., 2002).
Here, we sequenced the V4 region of the 16S rRNA gene to describe the cecal microbiota diversity, components, and predicted functionality to further investigate the differences in the microbial community structure and functional capacity between the HFE and low FE (LFE) chickens. By comparing the abundances of microbial populations between these 2 groups, we determined whether the presence of certain bacteria was correlated with broiler production performance. In addition, we performed Spearman's correlation analysis to determine whether there was any correlation between cecal microbiota and FE, and Pearson's correlation analysis to reveal the relationship between bacteria in HFE and LFE groups. This study may increase our understanding of the correlation between cecal microbiota and FE, in addition to providing certain novel insights on improving growth performance in yellow broilers.
Materials and methods
Ethics Statement
All of the experimental procedures were conducted in accordance with the Guidelines for Experimental Animals established by the Animal Care and Use Committee of China Agricultural University. This experiment was approved by the Experimental Animal Welfare Committee of China Agricultural University.
Animal Experiment and Sample Selection
This study used 270 yellow broiler males, raised in the breeding farm of Jiangsu Xingmu Agricultural Science and Technology Co., Ltd. Each broiler was assigned to a cage and raised in the same environment from birth to 63 d. All chickens were fed during the experiment in 3 phases: a starter diet from days 1 to 20, grower diet from days 21 to 40, and finisher diet from days 41 to 63 (Table 1). Diets were formulated to meet the NRC (1994) nutrient requirements. Subjects had individual food containers to ensure free and independent feeding and drinking water. The FI and BW were measured every 5 d. By the age of 63 d, only 213 chickens had complete phenotypic records. Feed conversion ratio was calculated as the ratio of FI to BWG during the feeding period from 5 to 63 d. Broiler FE was ranked by the FCR, after which 30 chickens with the highest FE and 30 with the lowest FE were selected for sampling (Supplementary Figure 1). Significant differences between HFE and LFE were determined using the Wilcoxon rank-sum test.
Table 1.
Ingredients and nutrient composition of diets (as-fed basis, %, unless otherwise indicated).
| Item | Starter diet 1–20 d |
Grower diet 21–40 d |
Finisher diet 41–63 d |
|---|---|---|---|
| Ingredient | |||
| Corn | 52.2 | 56.4 | 64.7 |
| Soybean meal | 29.0 | 22.0 | 11.0 |
| Barley | 10.0 | 10.0 | 10.0 |
| Peanut meal | 2.0 | 3.0 | 3.0 |
| Corn protein flour | 1.0 | 2.0 | 4.0 |
| Soya oil | 0.8 | 2.0 | 3.0 |
| Limestone flour | 1.8 | 1.6 | 1.5 |
| Dicalcium phosphate | 1.2 | 1.0 | 0.8 |
| Premix1 | 2.0 | 2.0 | 2.0 |
| Nutrition composition | |||
| Energy (ME kcal/kg) | 2880 | 3,000 | 3,150 |
| Crude protein | 21.0 | 18.5 | 16.0 |
| Crude fat | 3.0 | 4.3 | 5.5 |
| Crude fiber | 2.5 | 2.2 | 2.0 |
| Calcium | 1.0 | 0.88 | 0.77 |
| Total phosphorus | 0.65 | 0.57 | 0.5 |
| Available phosphorus | 0.41 | 0.36 | 0.3 |
| Lysine | 1.15 | 0.95 | 0.75 |
| Methionine | 0.55 | 0.5 | 0.47 |
| Methionine + cysteine | 0.82 | 0.75 | 0.7 |
| Threonine | 0.71 | 0.65 | 0.5 |
Premix provided the following nutrients per kilogram of diet: vitamin A, 300,000 IU; vitamin D, 150,000 IU; vitamin K, 750 IU; vitamin K3, 75 mg; vitamin B1, 135 mg; vitamin B2, 450 mg; vitamin B6, 90 mg; vitamin B12, 0.6 mg; nicotinic acid, 1.5 g; pantothenic acid, 450 mg; folic acid, 30 mg; biotin, 3 mg; Fe, 1.95 g; Cu, 375 mg; Zn, 3 g; Mn, 3.525 g; I, 30 mg; Se, 6.75 mg.
Sample Collection and DNA Extraction
Sampled chickens were euthanized on the morning of day 64, and cecum contents were aseptically collected after slaughter. Samples were immediately placed in dry ice and stored at −80°C for subsequent analysis. The study ultimately used 59 samples because one sample from the LFE group was contaminated. Microbial genome DNA was extracted and purified from selected samples using the Mag-Bind Stool DNA Kit (Omega Biotek, Norcross, GA) following the manufacturer's instructions. The concentration of the DNA extract was measured using a NanoDrop instrument (Thermo Fisher Scientific, Waltham, MA).
16S rRNA Gene Amplicon Sequencing
The V4 hypervariable region of the 16S rRNA gene was amplified using forward primer 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and reverse primer 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Caporaso et al., 2012). All PCR reactions were performed in 50 μL reaction mixtures using Phusion Master Mixes which contained 2X Phusion Master Mix, 2.5 μL of each primer, and 30 ng DNA template. Thermocycling conditions included an initial denaturation step at 95°C for 3 min; followed by 30 cycles of 95°C for 45 s, 56°C for 45 s, and 72°C for 45 s; and a final extension step at 72°C for 10 min. Amplicons were purified using Agencourt AMPure XP beads and eluted in the elution buffer. Library quality was assessed using an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA). The library was sequenced on an Illumina HiSeq 2500 platform (Illumina Inc., San Diego, CA) and 250 bp paired-end reads were generated.
Statistical Analysis
Raw data were processed and filtered (Fadrosh et al., 2014) to yield clean reads that were then assembled with Fast Length Adjustment of Short Reads, v1.2.11 (FLASH; v1.2.11; http://ccb.jhu.edu/software/FLASH/; Magoc and Salzberg, 2011). These clean tags were clustered at 97% similarity in USEARCH software (v7.0.1090; http://drive5.com/uparse/) (Edgar, 2013), yielding representative sequences of the operational taxonomic unit (OTU). Subsequently, the Quantitative Insights Into Microbial Ecology platform was used (QIIME; v1.9.1; http://qiime.org/; developer, Knight and Caporaso labs; USA; Caporaso et al., 2010). Representative OTU sequences were compared to the Greengenes V13_5 database (Desantis et al., 2006) using the RDP classifier software (v2.2; http://rdp.cme.msu.edu/classifier/classifier.jsp; Michigan 4882, USA; Wang et al., 2007) for OTU species annotation and relative abundance analysis of microorganisms at different classification levels. A Venn diagram was used to represent the relative abundance of OTUs. Alpha diversity values (observed species, Chao, abundance-based coverage estimator [ACE], Shannon, and Simpson indices) of the sample were calculated in Mothur (v1.31.2; http://www.mothur.org/wiki/Classify.seqs) (Schloss et al., 2009). To obtain beta diversity, Bray-Curtis distances were calculated in Quantitative Insights Into Microbial Ecology and subjected to principal coordinate analysis with the ape package in R (Paradis et al., 2004). Based on the nonparametric Kruskal-Wallis sum-rank test, linear discriminant analysis effect size (LEfSe) analysis was performed to determine the community that significantly affected sample division (Segata et al., 2011). A linear discriminant analysis score threshold of >2.0 was selected as significantly different for HFE and LFE. Correlations between FCR and taxonomic relative abundance at the phylum and genus levels were determined using Spearman correlation coefficients. Spearman's rank correlations and P-values were calculated with the psych package (v1.7.2; http://cran.r-project.org/web/packages/psych; author, W. Revelle). We quantified the degree of correlation between predominant microbial genera using Pearson's correlation in R software and visualized the correlation using the package ggcorrplot version 0.1.3 (http://www.sthda.com/english/wiki/ggcorrplot). Functional profiles of microbial communities were determined using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved Species (PICRUSt) (Langille et al., 2013). Taxonomy and OTU assignments were obtained by comparing the 16S rRNA gene to the 13_5 version of the Greengenes database. Taxonomic assignment of OTUs was categorized using functions based on the Kyoto Encyclopedia of Genes and Genomes annotations for level 3 pathways in PICRUSt. Pathway significance was analyzed using nonparametric tests.
Results
Sequencing and Diversity of the Cecal Microbiota
High and low groups had significantly different FCR values (Figure 1). Sequencing of 16S rRNA produced 3,128,989 raw reads from 59 samples. After assembly and filtration, the HFE and LFE samples had an average of 42,019 and 40,166 clean tags, respectively, at a mean length of 253 bp. The remaining reads were classified into 841 OTUs. The Venn diagram (Supplementary Figure 2) shows that 86.82% of all OTUs (737 OTUs) were shared, whereas 5.30 and 7.88% of the OTUs were different in the HFE and LFE, respectively. The rarefaction curves (Supplementary Figure 3) generated from the observed species index, ACE, and Chao indices reflect that the sample sequencing amount was sufficient, and the sequencing depth covered all of the species in the sample. All sample data used were enough for subsequent analyses. We employed 5 indices (observed species, ACE, Chao, Shannon, and Simpson) to estimate the alpha diversity of the HFE and LFE cecum samples (Figure 2), which did not differ significantly. Beta diversity analysis using Bray-Curtis distances did not show specific clustering based on the different FEs (Figure 3).
Figure 1.
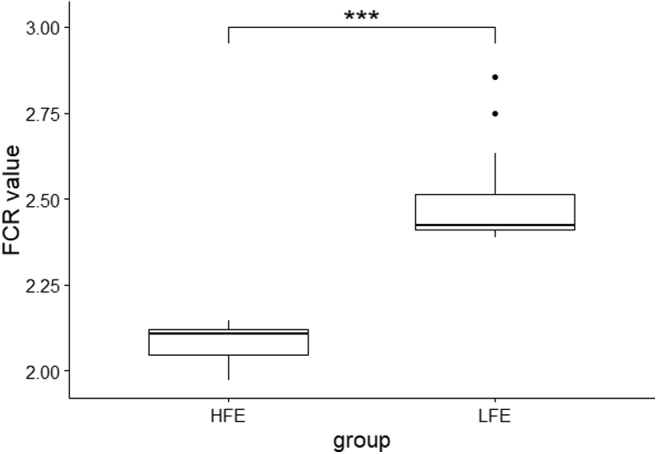
Box plot of FCR values for HFE and LFE groups (∗∗∗P < 0.001). Abbreviations: FCR, feed conversion ratio; HFE, high feed efficiency; LFE, low feed efficiency.
Figure 2.

Alpha diversity index. Five indicator box plots: the observed species index shows the number of OTUs actually observed; ACE and Chao indices were used to estimate the number of OTUs and microbial richness; and Shannon and Simpson indices were used to assess biodiversity. Abbreviations: ACE, abundance-based coverage estimator; HFE, high feed efficiency; LFE, low feed efficiency; OTUs, operational taxonomic units.
Figure 3.

Composition comparison of cecal microbiota between the HFE and LFE groups. PCoA plots (based on OTUs) of beta diversity. For HFE: n = 30 broilers; for LFE: n = 29 broilers. Plot is based on the Bray-Curtis distances. The amount of variance is depicted by the percentages in parentheses on each axis. Abbreviations: HFE, high feed efficiency; LFE, low feed efficiency; OTUs, operational taxonomic units; PCoA, principal coordinate analysis.
Taxonomic Composition of the HFE and LFE Groups
We analyzed phylum- and genus-level relative abundance of the microorganisms annotated with OTUs and then plotted stacked histograms (Figure 4). At the phylum level, Firmicutes was the most prominent microbe, accounting for 83.5% in the HFE group and 85.7% in the LFE group (Figure 4A). Bacteroidetes (HFE: 5.2%, LFE: 6.9%) and Actinobacteria (HFE: 5.9%, LFE: 2.0%) were, respectively, the second and third most abundant phyla based on 16S rRNA sequencing. These 3 bacteria accounted for more than 90% of the microbial flora. However, there were no significant differences between the HFE and LFE groups.
Figure 4.

Average relative abundances of predominant bacteria at the (A) phylum and the (B) genus level in the cecal digesta in high and low FCR groups. Abbreviations: FCR, feed conversion ratio; HFE, high feed efficiency; LFE, low feed efficiency.
Faecalibacterium, Ruminococcus, Oscillospira, Blautia, Bifidobacterium, and Lactobacillus were the top 6 prominent microflora in the 2 groups (Figure 4B). There was no significant difference in the relative abundance of these genera between the HFE and LFE groups. Notably, the relative abundances of Faecalibacterium, Bifidobacterium, and Lactobacillus differed by about 4% between the groups, which was higher than the between-group differences of other dominant genera.
Characterization of Cecal Microbiota in the HFE and LFE Groups
We performed LEfSe analysis to compare unique biomarkers of cecal microbes in the HFE and LFE groups (Figure 5). The results showed that Ruminococcaceae, Rikenellaceae, Bacteroidaceae, and Bacteroides were different between the HFE and LFE groups. The genus Bacteroides could be considered as a potential biomarker for the HFE group.
Figure 5.
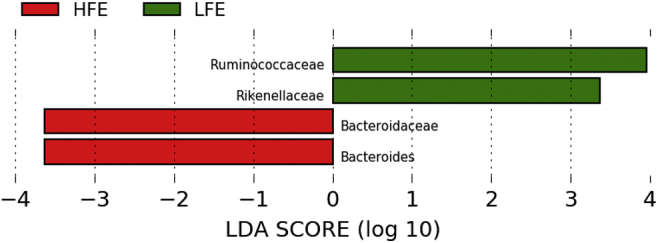
LEfSe results for cecal microbiota of HFE and LFE groups (only LDA scores above 2 are shown). Abbreviations: HFE, high feed efficiency; LDA, linear discriminant analysis; LEfSe, LDA effect size; LFE, low feed efficiency.
Correlation of the Cecal Microbiota With FE
Spearman correlations were used to identify the FE-associated cecal microbiota. Table 2 presents the correlations between FCR and microbial relative abundance. Although no significant FCR-related correlations were found at the phylum level, the genus Bacteroides exhibited a significant negative correlation with FCR (P < 0.05).
Table 2.
Spearman correlation coefficients between main bacterial taxa and feed efficiency.
| Taxa | FCR1 |
|---|---|
| Phylum level | |
| Actinobacteria | 0.077 |
| Bacteroidetes | −0.038 |
| Firmicutes | 0.171 |
| Proteobacteria | 0.224 |
| Tenericutes | −0.102 |
| Genus level (phylum; class; order; family; genus) | |
| Firmicutes; Clostridia; Clostridiales; Ruminococcaceae; Faecalibacterium | 0.253 |
| Firmicutes; Clostridia; Clostridiales; Ruminococcaceae; Ruminococcus | 0.029 |
| Firmicutes; Clostridia; Clostridiales; Ruminococcaceae; Oscillospira | −0.029 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; Blautia | 0.023 |
| Actinobacteria; Actinobacteria; Bifidobacteriales; Bifidobacteriaceae; Bifidobacterium | 0.032 |
| Firmicutes; Bacilli; Lactobacillales; Lactobacillaceae; Lactobacillus | 0.074 |
| Proteobacteria; Gammaproteobacteria; Enterobacteriales; Enterobacteriaceae; Escherichia | 0.162 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; Coprococcus | 0.05 |
| Bacteroidetes; Bacteroidia; Bacteroidales; Bacteroidaceae; Bacteroides | −0.3272 |
| Firmicutes; Erysipelotrichi; Erysipelotrichales; Erysipelotrichaceae; cc_115 | −0.005 |
| Firmicutes; Clostridia; Clostridiales; Lachnospiraceae; Dorea | 0.108 |
| Firmicutes; Clostridia; Clostridiales; Ruminococcaceae; Butyricicoccus | 0.068 |
FCR = feed conversion ratio, metric for feed efficiency trait. Correlations were examined between bacterial taxa (at both the phylum and genus levels) and FCR values were found to be significantly different between low and high feed efficiency chickens (high feed efficiency: n = 30; low feed efficiency: n = 29).
P < 0.05.
Correlation Between Predominant Microbial Genera in HFE and LFE Groups
Pearson's correlation analysis was performed to quantify between-genus relationships based on different FEs. Genera correlations and significance among microbes in the HFE and LFE groups are shown in Figure 6 and Table 3, respectively. The potential biomarker, Bacteroides, was negatively correlated with most of the genera detected in the HFE (Figure 6A) and LFE (Figure 6B) groups, especially with Dorea in the HFE group (P < 0.05); however, it was significantly positively correlated with Oscillospira (HFE: P < 0.01; LFE: P < 0.05) in both groups. In the HFE group, Lactobacillus was significantly negatively correlated with Faecalibacterium, Ruminococcus, and Oscillospira, whereas Lactobacillus was significantly positively correlated with Blautia. Blautia was significantly positively correlated with Dorea and cc_115; similarly, a correlation trend was also observed in Bifidobacterium and Butyricicoccus (Figure 6A). In the LFE group, Ruminococcus was positively correlated with Blautia, Lactobacillus, Coprococcus, and cc_115. Lactobacillus was negatively correlated with Faecalibacterium and Butyricicoccus, but it was significantly positively correlated with Blautia (Figure 6B).
Figure 6.

Pearson's correlations between predominant bacterial genera in the cecum of high (A) and low (B) chicken feed efficiency. Red and blue denote positive and negative association, respectively. The intensity of the colors represents the degree of association between the bacterial genera.
Table 3.
Correlation coefficient and significance (P-value) between bacterial genera in the cecum of high and low feed efficiency groups.
| Genus | Group | Bacteroides | Faecalibacterium | Ruminococcus | Oscillospira | Blautia | Bifidobacterium | Lactobacillus | Escherichia | Coprococcus | cc_115 | Dorea | Butyricicoccus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bacteroides | H | −0.3021 | 0.117 | 0.509 | −0.119 | −0.161 | −0.161 | −0.108 | −0.358 | −0.284 | −0.405 | −0.345 | |
| L | −0.346 | −0.263 | 0.412 | −0.084 | −0.197 | −0.13 | −0.223 | −0.16 | −0.087 | −0.254 | −0.172 | ||
| Faecalibacterium | H | 0.1042 | 0.311 | 0.076 | −0.303 | 0.017 | −0.441 | −0.111 | 0.359 | −0.121 | 0.008 | 0.203 | |
| L | 0.066 | −0.207 | 0.086 | −0.414 | −0.052 | −0.37 | −0.045 | −0.099 | −0.053 | −0.084 | 0.359 | ||
| Ruminococcus | H | 0.538 | 0.095 | 0.283 | −0.464 | −0.027 | −0.519 | 0.256 | 0.018 | −0.211 | −0.2 | 0.17 | |
| L | 0.167 | 0.28 | −0.276 | 0.583 | −0.05 | 0.621 | −0.116 | 0.377 | 0.544 | 0.319 | −0.028 | ||
| Oscillospira | H | 0.004 | 0.69 | 0.13 | −0.446 | −0.318 | −0.422 | −0.026 | −0.106 | −0.5 | −0.548 | −0.369 | |
| L | 0.026 | 0.656 | 0.148 | −0.513 | −0.4 | −0.346 | −0.218 | −0.028 | −0.204 | −0.403 | 0.36 | ||
| Blautia | H | 0.531 | 0.103 | 0.01 | 0.014 | −0.204 | 0.479 | −0.166 | 0.135 | 0.703 | 0.705 | −0.317 | |
| L | 0.666 | 0.026 | 0.001 | 0.004 | −0.111 | 0.792 | −0.143 | 0.066 | 0.322 | 0.343 | −0.389 | ||
| Bifidobacterium | H | 0.395 | 0.928 | 0.888 | 0.086 | 0.278 | −0.198 | −0.089 | −0.172 | 0.124 | −0.031 | 0.702 | |
| L | 0.305 | 0.79 | 0.797 | 0.031 | 0.567 | −0.208 | −0.025 | 0.005 | −0.033 | 0.494 | −0.19 | ||
| Lactobacillus | H | 0.394 | 0.015 | 0.003 | 0.02 | 0.007 | 0.295 | 0.048 | 0.117 | 0.115 | 0.296 | −0.25 | |
| L | 0.501 | 0.048 | 3.26E-04 | 0.066 | 3.07E-07 | 0.278 | −0.166 | −0.025 | 0.246 | 0.24 | −0.448 | ||
| Escherichia | H | 0.571 | 0.559 | 0.172 | 0.893 | 0.381 | 0.641 | 0.799 | 0.112 | 0.106 | −0.158 | 0.093 | |
| L | 0.245 | 0.818 | 0.55 | 0.255 | 0.458 | 0.899 | 0.391 | −0.172 | −0.106 | −0.322 | 0.209 | ||
| Coprococcus | H | 0.052 | 0.052 | 0.924 | 0.578 | 0.478 | 0.364 | 0.538 | 0.557 | 0.194 | 0.433 | −0.017 | |
| L | 0.407 | 0.609 | 0.044 | 0.885 | 0.735 | 0.978 | 0.897 | 0.373 | 0.222 | 0.367 | 0.064 | ||
| cc_115 | H | 0.128 | 0.523 | 0.263 | 0.005 | 1.45E-05 | 0.513 | 0.547 | 0.576 | 0.305 | 0.529 | 0.229 | |
| L | 0.654 | 0.786 | 0.002 | 0.29 | 0.089 | 0.865 | 0.199 | 0.585 | 0.246 | 0.307 | 0.043 | ||
| Dorea | H | 0.026 | 0.967 | 0.29 | 0.002 | 1.35E-05 | 0.87 | 0.112 | 0.404 | 0.017 | 0.003 | 0.009 | |
| L | 0.184 | 0.664 | 0.092 | 0.03 | 0.068 | 0.006 | 0.21 | 0.088 | 0.05 | 0.106 | −0.261 | ||
| Butyricicoccus | H | 0.062 | 0.283 | 0.368 | 0.045 | 0.088 | 1.53E-05 | 0.183 | 0.624 | 0.93 | 0.224 | 0.963 | |
| L | 0.374 | 0.056 | 0.887 | 0.055 | 0.037 | 0.324 | 0.015 | 0.276 | 0.743 | 0.823 | 0.171 |
Abbreviations: H, high feed efficiency group; L, low feed efficiency group.
The upper triangle is Pearson's correlation coefficient.
The lower triangle is the P-value corresponding to significance. P < 0.05 indicated a significant difference, and P < 0.01 showed an extremely significant difference.
Functional Prediction of Cecal Microbiota Between HFE and LFE
To predict how bacteria potentially contribute to differences in host FE, we performed PICRUSt using the Kyoto Encyclopedia of Genes and Genomes database. The results showed that 14 predicted microbial pathways differed significantly in abundance between the HFE and LFE groups (Table 4). The differential abundance prediction pathway of the highest relative abundance was related to metabolic function. In the HFE group, bacterial genes significantly enriched pathways, which were involved in amino acids biosynthesis (phenylalanine, tyrosine, and tryptophan), the metabolism of starch and sucrose, C5-branched dibasic acid, and carbohydrates, and nucleotide excision repair. It is worth noting that the 2 pathways of “starch and sucrose metabolism” and “phenylalanine, tyrosine, and tryptophan biosynthesis” had higher relative abundance than the other pathways.
Table 4.
Significant differences pathways between the HFE and LFE groups.
| Pathways ID | HFE1 | LFE2 | P-value | KEGG pathways annotation |
|---|---|---|---|---|
| Primary immune deficiency | 0.000514 | 0.000457 | 0.004 | Human Diseases; Immune System Diseases; Primary immunodeficiency |
| Glycan biosynthesis and metabolism | 0.000197 | 0.000242 | 0.011 | Unclassified; Metabolism; Glycan Biosynthesis and Metabolism |
| Dioxin degradation | 0.000692 | 0.000642 | 0.015 | Metabolism; Xenobiotics Biodegradation and Metabolism; Dioxin Degradation |
| D-Arginine and D-ornithine metabolism | 2.3E-05 | 1.85E-05 | 0.023 | Metabolism; Metabolism of Other Amino Acids; D-Arginine and D-Ornithine Metabolism |
| Chloroalkane and chloroalkene degradation | 0.002457 | 0.002323 | 0.026 | Metabolism; Xenobiotics Biodegradation and Metabolism; Chloroalkane and Chloroalkene Degradation |
| Xylene degradation | 0.000684 | 0.000636 | 0.029 | Metabolism; Xenobiotics Biodegradation and Metabolism; Xylene Degradation |
| Shigellosis | 8.28E-09 | 6.9E-08 | 0.034 | Human Diseases; Infectious Diseases; Shigellosis |
| Melanogenesis | 0 | 4.08E-08 | 0.037 | Organismal Systems; Endocrine System; Melanogenesis |
| C5-branched dibasic acid metabolism | 0.003539 | 0.003523 | 0.039 | Metabolism; Carbohydrate Metabolism; C5-Branched Dibasic Acid Metabolism |
| Nucleotide excision repair | 0.004145 | 0.004006 | 0.044 | Genetic Information Processing; Replication and Repair; Nucleotide Excision Repair |
| Carbohydrate metabolism | 0.001776 | 0.001744 | 0.044 | Unclassified; Metabolism; Carbohydrate Metabolism |
| Proteasome | 0.000475 | 0.000463 | 0.046 | Genetic Information Processing; Folding, Sorting and Degradation; Proteasome |
| Starch and sucrose metabolism | 0.011162 | 0.011075 | 0.049 | Metabolism; Carbohydrate Metabolism; Starch and Sucrose Metabolism |
| Phenylalanine, tyrosine, and tryptophan biosynthesis | 0.008417 | 0.008315 | 0.049 | Metabolism; Amino Acid Metabolism; Phenylalanine, Tyrosine, and Tryptophan Biosynthesis |
The significance of the gene distribution between the groups was analyzed using nonparametric test with a P-value < 0.05.
Abbreviations: HFE, high feed efficiency; KEGG, Kyoto Encyclopedia of Genes and Genomes; LFE, low feed efficiency.
Relative abundance of functional prediction pathways in the HFE group.
Relative abundance of functional prediction pathways in the LFE group.
Discussion
Feed efficiency is critical for modern commercial broiler production. Although the modern commercial broiler poultry industry embodies a standardized diet strategy, reasonable management measures, and a suitable breeding environment, it still shows considerable difference in the FE of flock chickens from the same breed (Eerden et al., 2004). As a lower FCR represents HFE, we separated experimental chicken flocks into HFE and LFE groups to study the variation in composition of their cecal microbial communities. Identifying consistent differences in these bacterial communities may provide insights on improving commercial poultry FE through the manipulation of microorganisms in the future. Previous research has shown that although the commercial broiler growth rate has increased by over 400%, the FE decreased by 50%, based on the genotypes produced from 1950 to 2005 (Zuidhof et al., 2014). Changes in broiler performance are mainly owing to genetic advancements (Havenstein and Ferket, 2003; Zuidhof et al., 2014). Broilers in this experiment were from the same breed, and the influence of genotype differences on FE may be relatively small or even negligible. Although we cannot fully exclude the influence of factors such as genes, diet, and environment on FE, we can plausibly attribute the observed changes in FE to microbial differences because we used the same breed, the same rearing environment, and consistent nutrition strategies during each phase.
The microbial community in young chicks changes with age, increasing its complexity, as mature birds develop more stable bacterial communities (van der Wielen et al., 2002; Lu et al., 2003). In our study, we inferred that the microbial composition was relatively stable owing to the maturity of the subject broilers. The relationship between community diversity and FE is worthy of attention. Our alpha diversity results were similar to those of a previous study on fecal microflora, which found no significant differences in alpha diversity between high- and low-grade (based on FCR and weight gain) chickens (Diaz-Sanchez et al., 2019). These outcomes were also similar to findings in pigs, showing that intestinal bacterial diversity does not differ among animals with varying FE, and specific bacterial groups could potentially be relevant to porcine FE (Mccormack et al., 2017). Previous studies of gut microbiota in chickens have shown that on farms with good health and FCR history, the variability of microbial communities between chickens is small, whereas on farms with problems, differences in chicken flocks are less uniform (Rinttilä and Apajalahti, 2013). Unlike the link between microbiota diversity and obesity in humans (Turnbaugh et al., 2009), the microbiota composition may not be the main factor affecting FE, and specific microbes may play an important role in our study.
In chickens, Firmicutes dominates the cecal microflora (Mohd Shaufi et al., 2015; Sohail et al., 2015). Our study confirmed that the dominant phylum was Firmicutes, but this result differs from a report on the layer cecum, which found that the main microflora was Bacteroidetes (>50%), whereas Firmicutes accounted for only about 20% (Yan et al., 2017). The different taxonomic composition of the cecal microbiome is linked to the chicken breed used, geographical location, bird age, and common dietary changes (Lu et al., 2003; Singh et al., 2014; Siegerstetter et al., 2017). Firmicutes decomposes polysaccharides that cannot be digested by the host in the intestinal tract, promoting the digestion and absorption of nutrients by the body (Medinger et al., 2010; Lozupone et al., 2012; Johnson et al., 2015). Here, we observed that Bacteroidetes and Actinobacteria were the second and third most abundant phyla, respectively. Bacteroidetes, Firmicutes, and Actinobacteria are the 3 major phyla that inhabit the human large intestine, and these bacteria possess a fascinating array of enzymes that can degrade complex dietary substrates (Scott et al., 2013). In humans, the ratio of Firmicutes to Bacteroidetes (F/B) is known to be correlated with obesity. Obese children reportedly have a higher F/B ratio (Bervoets et al., 2013). Similarly, the HFE group in this study had a higher cecal F/B ratio than the LFE group. Thus, we proposed that the changes in the relative abundance of Firmicutes and Bacteroidetes may be linked to FE.
Faecalibacterium is predominant in the chicken cecum and plays an important role in the generation of volatile fatty acids (Lund et al., 2010). Feed with probiotics containing Lactobacillus cultures can enhance chicken weight and ensure efficient feed absorption (Oakley et al., 2014). Both Lactobacilli and Bifidobacteria have been associated with beneficial effects on the host, such as the promotion of gut maturation, gut integrity, antagonism against pathogens, and immune modulation (Lan et al., 2005). The lack of significant differences in the abundance of these dominant genera indicated that they likely play an important role in maintaining intestinal homeostasis. However, they may not be a crucial contributing factor in differentiating FE.
Our LEfSe results suggested that Bacteroides was a potential biomarker in the HFE groups; similar observations have also been reported for the colon (Tan et al., 2018). Previous research has revealed that fecal bacterial genera, such as Bacteroides and Lactobacillus, were more abundant in high FCR chickens (Singh et al., 2014), in contrast to our results. We speculated that Bacteroides may have different effects on FE in different intestinal segments. Bacteroides are anaerobic, gram-negative rods (Gibson and Roberfroid, 2004) that consume polysaccharides in the colon, characterized by bile resistance and hydrolysis of bile salt (Macy and Probst, 1979; Wexler, 2007). Bacteroides were found to be the main bacteria involved in producing SCFA (Kaakoush et al., 2014). Therefore, we inferred that differences in the amount of cecal SCFA may be causing FE variation. Previous studies in germ-free mice revealed that during the development of the posterior intestine, Bacteroides thetaiotaomicron stimulated angiogenesis, which is related to the formation of the capillary network for efficient distribution of absorbed nutrients (Stappenbeck et al., 2002). Although both groups had low Bacteroides abundance in our study, the different abundances of Bacteroides may impact host nutrient absorption of nutrients, resulting in differences in HFE and LFE.
We did not identify significant correlations between cecal microbiota composition and FE at the phylum level, similar to previous findings in pigs (Mccormack et al., 2017). Since lower FCR reflects satisfactory performance, bacteria negatively correlated with FCR are considered to improve performance. At weaning, the genus Bacteroides was negatively correlated with FCR, suggesting that the genus could improve FE. This negative correlation may be due to the fact that Bacteroides are generally related to polysaccharide degradation, especially of starch and glucans (Degnan et al., 1997; Beckmann et al., 2006). Bacteroides are also linked to SCFA formation and positively correlated with many lipid metabolites (Saxena et al., 2016; Chen et al., 2020). These characteristics may favor the improvement of host FE. In the case of malabsorption of nutrients in the small intestine, the correlation between cecal microbiota and FCR is obvious, but the beneficial bacteria do not directly affect FCR (Rinttilä and Apajalahti, 2013). Further research will be required to determine the exact contributions of Bacteroides to FE.
Bacteroides is a potential biomarker of FE-associated characteristics and was significantly correlated with FE. Pearson's correlation analysis further quantified the degree of correlation between cecal genera in the HFE and LFE groups; notably, the Bacteroides and Oscillospira showed a stronger positive correlation in the HFE group than in the LFE group. Oscillospira has been observed in several studies to be related to leanness or lower body mass index (Tims et al., 2013; Verdam et al., 2013; Goodrich et al., 2014). In addition, researchers believe that Oscillospira relies on fermentation products as a source of growth substrates secreted by other species, such as members of Bacteroides (Konikoff and Gophna, 2016), which may explain the positive correlation between the 2 bacteria in our study. Although Oscillospira was not the FE-related biomarker in this study, it likely had a synergistic effect with Bacteroides to improve host FE. In our study, there was a negative correlation between Bacteroides and Dorea in the HFE group, which could suggest that these 2 genera have a competitive relationship or antagonistic effect. Studies have shown that 3 types of bariatric surgery could cause a significant reduction in the abundance of Dorea, namely Roux-en-Y gastric bypass, sleeve gastroplasty, and bilio-intestinal bypass (Kong et al., 2013; Damms Machado et al., 2015; Patrone et al., 2016). Furthermore, after gastric bypass, most corpulence parameters in patients with obesity disease are positively correlated with Dorea and negatively with Bacteroides (Kong et al., 2013). Similar to our results in the HFE group, a negative and significant correlation between the abundance of Lactobacillus and Ruminococcus has been reported in intermittent hypoxia mouse models (Moreno-Indias et al., 2016); however, in the LFE group of our study, there was a positive correlation between these genera. Therefore, we infer that there may be different microbial relationships within the 2 groups that interact to affect host productivity.
Microbiota in the human large intestine ferments carbohydrates to produce SCFA, which are mostly absorbed (Flint et al., 2012). Microbial genes of Firmicutes and Bacteroidetes mainly encode carbohydrate active enzyme, whose main function is to decompose carbohydrates (Kaoutari et al., 2013). Although small, the relative abundances of “starch and sucrose metabolism,” “phenylalanine, tyrosine, and tryptophan biosynthesis,” and “carbohydrate metabolism” pathways were significantly more enriched in the HFE groups than in the LFE groups. Our results were similar to previous studies in laying hens demonstrating that glycometabolism and amino acid metabolism were enriched in the cecal microbiota of the higher-FE group (Yan et al., 2017). Consistent with studies on pigs, “phenylalanine, tyrosine, and tryptophan biosynthesis”and “C5-branched dibasic acid metabolism” pathways were significantly enriched in higher-FE animals (Mccormack et al., 2017). Differences in enriched pathways might be associated with distinct microorganisms between the HFE and LFE groups. In our study, Bacteroides was significantly more abundant in the HFE group than in the LFE group, and the bacterial genes were enriched in pathways related to carbohydrate metabolism. We could thus infer that Bacteroides improves nutrient digestion and absorption of the host through carbohydrate metabolism. The primary carbohydrates available to colon bacteria include resistant starch, non-starch polysaccharides, and oligosaccharides (Flint et al., 2012). Resistant starch refers to dietary starch that escapes digestion from host enzymes and enters into the large intestine; these are estimated to be the largest dietary source of colonic bacteria (Nugent, 2005). Our functional predictions showed that the starch and sucrose pathways were significantly enriched in the HFE group. Perhaps the HFE-specific microorganisms were better host consumers of starch, resulting in different FEs between groups. Interestingly, glycan biosynthesis and metabolism pathways were less abundant in the HFE group than in the LFE group. Microbial studies related to obesity have shown that glycan biosynthesis and metabolism (biosynthesis of various types of N-glycans, glycosphingolipids, lipopolysaccharide, and degradation of glycosaminoglycans and other glycans) are underrepresented in obese children (Hou et al., 2017). Therefore, in this experiment, the metabolism-related pathway was associated with FE, among which starch and sucrose metabolism may be important. Further work is required to clarify these assumptions.
In conclusion, we profiled cecal microbial communities and revealed the compositional differences related to FE. These findings suggested that the cecal microbiota has a possible connection with FE in yellow broilers. Of note, the differentially abundant bacteria, particularly Bacteroides, may potentially be adopted as biomarkers for FE or used to modify dietary strategies for improving commercial poultry performance. Moreover, FE-associated correlation analysis also revealed that there may be some relationship between Bacteroides and FE. However, the development and application of microbial biomarkers are dependent on future improvements in microorganism isolation and cultivation technology. Pearson's correlations suggested that there may be different relationships between genera in HFE and LFE groups. Functional prediction confirmed the differences in metabolic pathways between the HFE and LFE groups owing to different bacterial communities. We expect that the applications of our findings will be further expanded with future studies that use a larger population to verify the reliability of the FE-related microbial taxa identified here. Intervention trials and functional analyses of metagenomics will also help to better interpret our results. Nevertheless, the identification of FE-associated microbial taxa and metagenomic predictions in our study provide valuable insights into the connection between cecal microbiota and FE.
Acknowledgments
The current research was supported in part by funding from the National Natural Science Foundation of China (U1702232-1), Primary Research & Development Plan of Jiangsu Province (BE2017309), and the Programs for Changjiang Scholars and Innovative Research in University (IRT_15R62).
Footnotes
Supplementary data associated with this article can be found in the online version at https://doi.org/10.1016/j.psj.2021.01.019.
Disclosures
The authors declared that they have no conflicts of interest to this work.
Supplementary data
References
- Aggrey S.E., Karnuah A.B., Sebastian B., Anthony N.B. Genetic properties of feed efficiency parameters in meat-type chickens. Genet. Sel Evol. 2010;42:25. doi: 10.1186/1297-9686-42-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Fataftah A.R.A., Abu-Dieyeh Z.H.M. Effect of Chronic heat stress on broiler performance in Jordan. Int. J. Poult. Sci. 2007;6:64–70. [Google Scholar]
- Awad W., Ghareeb K., Abdel-Raheem S.M., Böhm J. Effects of dietary inclusion of probiotic and synbiotic on growth performance, organ weights, and intestinal histomorphology of broiler chickens. 2009;88:49–55. doi: 10.3382/ps.2008-00244. [DOI] [PubMed] [Google Scholar]
- Beckmann L., Simon O., Vahjen W. Isolation and identification of mixed linkedβ -glucan degrading bacteria in the intestine of broiler chickens and partial characterization of respective 1,3-1,4-β -glucanase activities. J. Basic Microbiol. 2006;46:175–185. doi: 10.1002/jobm.200510107. [DOI] [PubMed] [Google Scholar]
- Bervoets L., Van Hoorenbeeck K., Kortleven I., Noten C., Hens N., Vael C., Goossens H., Desager K., Vankerckhoven V. Differences in gut microbiota composition between obese and lean children: a cross-sectional study. Gut Pathog. 2013;5:10. doi: 10.1186/1757-4749-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., Fierer N., Peã A A.G., Goodrich J.K., Gordon J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G., Lauber C.L., Walters W.A., Berg-Lyons D., Huntley J., Fierer N., Owens S.M., Betley J., Fraser L., Bauer M., Gormley N., Gilbert J.A., Smith G., Knight R. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Isme J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplin S.B. Effect of cecectomy on water and nutrient absorption of birds. J. Exp. Zool Suppl. 1989;3:81–86. doi: 10.1002/jez.1402520514. [DOI] [PubMed] [Google Scholar]
- Chen Y., Wang J., Yu L., Xu T., Zhu N. Microbiota and metabolome responses in the cecum and serum of broiler chickens fed with plant essential oils or virginiamycin. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-60135-x. 5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clench M., Mathias J. The avian cecum: a review. Wilson Bull. 1995;107:93–121. [Google Scholar]
- Corrigan A., Horgan K., Clipson N., Murphy R. Effect of dietary Supplementation with a Saccharomyces cerevisiae Mannan oligosaccharide on the bacterial community structure of broiler cecal contents. Appl. Environ. Microb. 2011;77:6653–6662. doi: 10.1128/AEM.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damms Machado A., Mitra S., Schollenberger A., Kramer K., Meile T., Königsrainer A., Huson D., Bischoff S. Effects of Surgical and dietary weight Loss Therapy for obesity on gut microbiota composition and nutrient absorption. Biomed. Res. Int. 2015 doi: 10.1155/2015/806248. Article ID 806248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan B., Macfarlane S., Quigley M.E., Macfarlane G.T. Starch utilization by Bacteroides ovatus isolated from the human large intestine. Curr. Microbiol. 1997;34:290–296. doi: 10.1007/s002849900184. [DOI] [PubMed] [Google Scholar]
- Degolier T., Mahoney S., Duke G. Relationships of avian cecal lengths to food Habits, taxonomic position, and intestinal lengths. Condor. 1999;101:622–634. [Google Scholar]
- DeSantis T., Philip H., Larsen N., Rojas M., Brodie E., Keller K., Huber T., Dalevi D., Hu P., Andersen G. Greengenes, a Chimera-Checked 16S rRNA gene database and Workbench Compatible with ARB. Appl. Environ. Microb. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Sanchez S., Perrotta A.R., Rockafellow I., Alm E.J., Okimoto R., Hawken R., Hanning I. Using fecal microbiota as biomarkers for predictions of performance in the selective breeding process of pedigree broiler breeders. Plos One. 2019;14:e216080. doi: 10.1371/journal.pone.0216080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- Eerden E., Van den Brand H., Parmentier H., De Jong M., Kemp B. Phenotypic selection for residual feed intake and its effect on Humoral immune responses in growing layer hens. Poult. Sci. 2004;83:1602–1609. doi: 10.1093/ps/83.9.1602. [DOI] [PubMed] [Google Scholar]
- Fadrosh D.W., Ma B., Gajer P., Sengamalay N., Ott S., Brotman R.M., Ravel J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2:6. doi: 10.1186/2049-2618-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint H., Scott K., Duncan S., Louis P., Forano E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes. 2012;3:289–306. doi: 10.4161/gmic.19897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G.R., Roberfroid M.B. Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J. Nutr. 2004;17:259–275. doi: 10.1079/NRR200479. [DOI] [PubMed] [Google Scholar]
- Goodrich J., Waters J., Poole A., Sutter J., Koren O., Blekhman R., Beaumont M., Treuren W., Knight R., Bell J., Spector T., Clark A., Ley R. Human genetics Shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havenstein G., Ferket P. Growth, Livability, and feed conversion of 1957 versus 2001 broilers when fed representative 1957 and 2001 broiler diets. Poult. Sci. 2003;82:1500–1508. doi: 10.1093/ps/82.10.1500. [DOI] [PubMed] [Google Scholar]
- Hou Y.P., He Q.Q., Ouyang H.M., Peng H.S., Wang Q., Li J., Lv X.F., Zheng Y.N., Li S.C., Liu H.L., Yin A.H. Human gut microbiota associated with obesity in Chinese children and Adolescents. Biomed. Res. Int. 2017;2017:7585989. doi: 10.1155/2017/7585989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume D.A., Whitelaw B., Archibald A.L. The future of animal production: improving productivity and sustainability. J. Agric. Sci. 2011;149:9–16. [Google Scholar]
- Jamroz D., Jakobsen K., Bach K.K., Wiliczkiewicz A., Orda J. Digestibility and energy value of non-starch polysaccharides in young chickens, ducks and geese, fed diets containing high amounts of barley. Comp. Biochem. Physiol. A. Mol. Integr. Physiol. 2002;131:657–668. doi: 10.1016/s1095-6433(01)00517-7. [DOI] [PubMed] [Google Scholar]
- Johansson K., Sarles W., Shapiro S. The intestinal microflora of hens as influenced by various carbohydrates in a biotin-deficient ration. J. Bacteriol. 1948;56:619–634. doi: 10.1128/jb.56.5.619-634.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.R., Lee T.K., Park J., Fenner K., Helbling D.E. The functional and taxonomic richness of wastewater treatment plant microbial communities are associated with each other and with ambient nitrogen and carbon availability. Environ. Microbiol. 2015;17:4851–4860. doi: 10.1111/1462-2920.12429. [DOI] [PubMed] [Google Scholar]
- Kaakoush N.O., Sodhi N., Chenu J.W., Cox J.M., Riordan S.M., Mitchell H.M. The interplay between Campylobacter and Helicobacter species and other gastrointestinal microbiota of commercial broiler chickens. Gut Pathog. 2014;6:18. doi: 10.1186/1757-4749-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaoutari A.E., Armougom F., Gordon J.I., Raoult D., Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013;11:497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- Kong L., Tap J., Aron-Wisnewsky J., Pelloux V., Basdevant A., Bouillot J.L., Zucker J., Dore J., Clément K. Gut microbiota after gastric bypass in human obesity: increased richness and associations of bacterial genera with adipose tissue genes. Am. J. Clin. Nutr. 2013;98:16–24. doi: 10.3945/ajcn.113.058743. [DOI] [PubMed] [Google Scholar]
- Konikoff T., Gophna U. Oscillospira: a Central, Enigmatic component of the human gut microbiota. Trends Microbiol. 2016;24:523–524. doi: 10.1016/j.tim.2016.02.015. [DOI] [PubMed] [Google Scholar]
- Lan Y., Verstegen M.W.A., Tamminga S., Williams B.A., Boer H. The role of the commensal gut microbial community in broiler chickens. World Poult. Sci. J. 2005;61:95–104. [Google Scholar]
- Langille M.G.I., Zaneveld J., Caporaso J.G., McDonald D., Knights D., Reyes J.A., Clemente J.C., Burkepile D.E., Vega Thurber R.L., Knight R., Beiko R.G., Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Luo C., Wang J., Ma J., Shu D., Lund M.S., Su G., Qu H. Assessment of the genomic prediction accuracy for feed efficiency traits in meat-type chickens. Plos One. 2017;12:e173620. doi: 10.1371/journal.pone.0173620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C.A., Stombaugh J.I., Gordon J.I., Jansson J.K., Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., Idris U., Harmon B., Hofacre C., Maurer J., Lee M. Diversity and Succession of the intestinal bacterial community of the maturing broiler chicken. Appl. Environ. Microb. 2003;69:6816–6824. doi: 10.1128/AEM.69.11.6816-6824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund M., Bjerrum L., Pedersen K. Quantification of Faecalibacterium prausnitzii- and Subdoligranulum variabile-like bacteria in the cecum of chickens by real-time PCR. Poult. Sci. 2010;89:1217–1224. doi: 10.3382/ps.2010-00653. [DOI] [PubMed] [Google Scholar]
- Macy J.M., Probst I. The biology of gastrointestinal bacteroides. Annu. Rev. Microbiol. 1979;33:561. doi: 10.1146/annurev.mi.33.100179.003021. [DOI] [PubMed] [Google Scholar]
- Magoc T., Salzberg S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marounek M., Suchorska O., Savka O. Effect of Substrate and feed antibiotics on in vitro production of volatile fatty acids and methane in caecal contents of chickens. Anim. Feed Sci. Tech. 1999;80:223–230. [Google Scholar]
- Mccormack U., Curiao T., Buzoianu S., Prieto M.L., Ryan T., Varley P., Crispie F., Magowan E., Metzler-Zebeli B., Berry D., O'Sullivan O., Cotter P., Gardiner G., Lawlor P. Exploring a possible link between the intestinal microbiota and feed efficiency in pigs. Appl. Environ. Microb. 2017;83 doi: 10.1128/AEM.00380-17. :e00380-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medinger R., Nolte V., Pandey R., Jost S., Ottenwälder B., Schlötterer C., Boenigk J. Diversity in a hidden world: potential and limitation of next-generation sequencing for surveys of molecular diversity of eukaryotic microorganisms. Mol. Ecol. 2010 doi: 10.1111/j.1365-294X.2009.04478.x. 19 (Suppl 1): 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohd Shaufi M., Sieo C., Chong C., Gan H., Ho Y. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 2015;7:4. doi: 10.1186/s13099-015-0051-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Indias I., Torres M., Sánchez-Alcoholado L., Cardona F., Almendros I., Gozal D., Montserrat J., Queipo Ortuño M.I., Farré R. Normoxic Recovery Mimicking treatment of Sleep Apnea does not reverse intermittent hypoxia-Induced bacterial Dysbiosis and low-grade Endotoxemia in mice. Sleep. 2016;39 doi: 10.5665/sleep.6176. :1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council . 9th rev. ed. Natl. Acad. Press; Washington, DC: 1994. Nutrient Requirements of Poultry. [Google Scholar]
- Nugent A.P. Health properties of resistant starch. Nutr. Bull. 2005;30:27–54. [Google Scholar]
- Oakley B.B., Lillehoj H.S., Kogut M.H., Kim W.K., Maurer J.J., Pedroso A., Lee M.D., Collett S.R., Johnson T.J., Cox N.A. The chicken gastrointestinal microbiome. Fems Microbiol. Lett. 2015;360:100–112. doi: 10.1111/1574-6968.12608. [DOI] [PubMed] [Google Scholar]
- Paradis E., Claude J., Strimmer K. APE: analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Patrone V., Vajana E., Minuti A., Callegari M., Federico A., Loguercio C., Dallio M., Tolone S., Docimo L., Morelli L. Postoperative changes in fecal bacterial communities and fermentation products in obese patients Undergoing bilio-intestinal bypass. Front Microbiol. 2016;7:200. doi: 10.3389/fmicb.2016.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedroso A., Menten J., Lambais M., Racanicci A., Longo F., Sorbara J.O. Intestinal bacterial community and growth performance of chickens fed diets containing antibiotics. Poult. Sci. 2006;85:747–752. doi: 10.1093/ps/85.4.747. [DOI] [PubMed] [Google Scholar]
- Rinttilä T., Apajalahti J. Intestinal microbiota and metabolites—Implications for broiler chicken health and performance. J. Appl. Poult. Res. 2013;22:647–658. [Google Scholar]
- Saxena S., Saxena V., Tomar S., Sapcota D., Gonmei G. Characterisation of caecum and crop microbiota of Indian indigenous chicken targeting multiple hypervariable regions within 16S rRNA gene. Br. Poult. Sci. 2016;57:381–389. doi: 10.1080/00071668.2016.1161728. [DOI] [PubMed] [Google Scholar]
- Schloss P.D., Westcott S.L., Ryabin T., Hall J.R., Hartmann M., Hollister E.B., Lesniewski R.A., Oakley B.B., Parks D.H., Robinson C.J., Sahl J.W., Stres B., Thallinger G.G., Van Horn D.J., Weber C.F. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K.P., Gratz S.W., Sheridan P.O., Flint H.J., Duncan S.H. The influence of diet on the gut microbiota. Pharmacol. Res. 2013;69:52–60. doi: 10.1016/j.phrs.2012.10.020. [DOI] [PubMed] [Google Scholar]
- Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant M.J., Constantinidou C., Cogan T.A., Bedford M.R., Pallen M.J. Extensive microbial and functional diversity within the chicken cecal microbiome. PLoS One. 2014;9:e91941. doi: 10.1371/journal.pone.0091941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegerstetter S., Schmitz-Esser S., Magowan E., Wetzels S., Zebeli Q., Lawlor P., O'Connell N., Metzler-Zebeli B. Intestinal microbiota profiles associated with low and high residual feed intake in chickens across two geographical locations. PLoS One. 2017;12:e187766. doi: 10.1371/journal.pone.0187766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K., Shah T., Deshpande S., Jakhesara S., Koringa P., Rank D.N., Joshi C. High through put 16S rRNA gene-based pyrosequencing analysis of the fecal microbiota of high FCR and low FCR broiler growers. Mol. Biol. Rep. 2012;39:595–602. doi: 10.1007/s11033-012-1947-7. [DOI] [PubMed] [Google Scholar]
- Singh K.M., Shah T.M., Reddy B., Deshpande S., Rank D.N., Joshi C.G. Taxonomic and gene-centric metagenomics of the fecal microbiome of low and high feed conversion ratio (FCR) broilers. J. Appl. Genet. 2014;55:145–154. doi: 10.1007/s13353-013-0179-4. [DOI] [PubMed] [Google Scholar]
- Sohail M.U., Hume M., Byrd J., Nisbet D., Shabbir M.Z., Ahmad I., Rehman H. Molecular analysis of cecal and tracheal microbiome of heat-stressed broilers supplemented with prebiotic and probiotic. Avian Pathol. 2015;44:1–28. doi: 10.1080/03079457.2015.1004622. [DOI] [PubMed] [Google Scholar]
- Stanley D., Denman S., Hughes R.J., Geier M., Crowley T., Chen H., Haring V., Moore R. Intestinal microbiota associated with differential feed conversion efficiency in chickens. Appl. Microbiol. Biot. 2012;96:1361–1369. doi: 10.1007/s00253-011-3847-5. [DOI] [PubMed] [Google Scholar]
- Stanley D., Hughes R.J., Moore R.J. Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. Appl. Microbiol. Biot. 2014;98:4301–4310. doi: 10.1007/s00253-014-5646-2. [DOI] [PubMed] [Google Scholar]
- Stanley D., Hughes R.J., Geier M.S., Moore R.J. Bacteria within the gastrointestinal tract microbiota correlated with improved growth and feed conversion: Challenges presented for the identification of performance enhancing probiotic bacteria. Front Microbiol. 2016;7:187. doi: 10.3389/fmicb.2016.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappenbeck T.S., Hooper L.V., Gordon J.I. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc. Natl. Acad. Sci. U S A. 2002;99:15451–15455. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z., Wang Y., Yang T., Ao H., Chen S., Xing K., Zhang F., Xitong Z., Liu J., Wang C. Differences in gut microbiota composition in finishing Landrace pigs with low and high feed conversion ratios. Antonie Van Leeuwenhoek. 2018;111:1673–1685. doi: 10.1007/s10482-018-1057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tims S., Derom C., Jonkers D., Vlietinck R., Saris W., Kleerebezem M., De Vos W., Zoetendal E. 2013. Microbiota Conservation and BMI Signatures in Adult Monozygotic Twins, ISME J. 7. :707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P., Hamady M., Yatsunenko T., Cantarel B., Duncan A., Ley R., Sogin M., Jones J., Roe B., Affourtit J., Egholm M., Henrissat B., Heath A., Knight R., Gordon J. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wielen P., Keuzenkamp D.A., Lipman L.J.A., Knapen F., Biesterveld S. Spatial and Temporal variation of the intestinal bacterial community in commercially raised broiler chickens during growth. Microb. Ecol. 2002;44:286–293. doi: 10.1007/s00248-002-2015-y. [DOI] [PubMed] [Google Scholar]
- Verdam F., Fuentes S., de Jonge C., Zoetendal E., Erbil R., Greve J.W., Buurman W., De Vos W., Rensen S. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) 2013;21:E607–E615. doi: 10.1002/oby.20466. [DOI] [PubMed] [Google Scholar]
- Wang Q., Garrity G., Tiedje J., Cole J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73 doi: 10.1128/AEM.00062-07. :5264-5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems O.W., Miller S.P., Wood B.J. Aspects of selection for feed efficiency in meat producing poultry. World Poult. Sci. J. 2013;69:275–288. [Google Scholar]
- Yan W., Sun C., Yuan J., Yang N. Gut metagenomic analysis reveals prominent roles of Lactobacillus and cecal microbiota in chicken feed efficiency. Sci. Rep. 2017;7:45308. doi: 10.1038/srep45308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuidhof M.J., Schneider B.L., Carney V.L., Korver D.R., Robinson F.E. Growth, efficiency, and yield of commercial broilers from 1957, 1978, and 2005. Poult. Sci. 2014;93:2970–2982. doi: 10.3382/ps.2014-04291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.