

Abstract
Development of renal fibrosis is a hallmark of renal aging and chronic kidney disease of all etiologies and characterized by extensive renal cell injuries and subsequent myofibroblast transdifferentiations (MTDs), which are significantly influenced by aberrant histone deacetylase (HDAC) activities. However, the key HDAC isoforms and effectors that are causally involved in the processes remain poorly understood. Here, we report that aberrant HDAC3 induction and its inhibition of Klotho, a renal epithelium-enriched aging suppressor, contribute significantly to renal fibrogenesis. HDAC3 was preferentially elevated with concomitant Klotho suppression in fibrotic kidneys incurred by unilateral ureter obstruction (UUO) and aristolochic acid nephropathy (AAN), whereas Hdac3 knockout resisted the fibrotic pathologies. The HDAC3 elevation is substantially blocked by the inhibitors of TGFβ receptor and Smad3 phosphorylation, suggesting that TGFβ/Smad signal activates Hdac3 transcription. Consistently, an HDAC3-selective inhibitor RGFP966 derepressed Klotho and mitigated the renal fibrotic injuries in both UUO and AAN mice. Further, HDAC3 overexpression or inhibition in renal epithelia inversely affected Klotho abundances and HDAC3 was inducibly associated with transcription regulators NCoR and NF-kB and bound to Klotho promoter in fibrotic kidney, supporting that aberrant HDAC3 targets and transcriptionally inhibits Klotho under renal fibrotic conditions. More importantly, the antirenal fibrosis effects of RGFP966 were largely compromised in mice with siRNA-mediated Klotho knockdown. Hence, HDAC3 aberration and the subsequent Klotho suppression constitute an important regulatory loop that promotes MTD and renal fibrosis and uses of HDAC3-selective inhibitors are potentially effective in treating renal fibrotic disorders.
Subject terms: Epigenetics, Pathogenesis
Introduction
Renal fibrosis is an indispensible pathohistological feature of renal aging and chronic kidney disease (CKD) irrespective of etiologies and characterized by myofibroblast transdifferentiation (MTD) of injured renal cells of almost all origins [1]. Renal fibrosis represents an overhealing/repairing process, during which the injured renal cells lose their own phenotypes and transdifferentiate to myofibroblasts, resulting in excessive production and deposition of extracellular matrix (ECM) proteins that progressively deteriorate renal structure and function [2]. At the subcellular level, renal fibrosis is positively regulated by several profibrotic signaling pathways and negatively affected by a number of antifibrotic factors such as Klotho, RAS protein activator like 1, and bone morphogenesis protein 7 (BMP-7), which are extensively studied [3]. Intriguingly, aged people and renal CKD patients of the same primary cause often experience various susceptibility and severity of renal fibrosis with different prognoses and outcomes, which are attributed to epigenetic influences, but poorly understood [4]. Recent studies have showed that the developments of MTD and renal fibrosis are influenced by aberrant epigenetic modifications of numerous fibrogenic genes [5]. More pertinently, various pan- and class-selective inhibitors of histone deacetylase (HDAC) exhibit impressive antirenal fibrosis properties in various animal models [6–14], suggesting that increased HDAC expressions or activities additionally control the development or progression of renal fibrogenesis.
In mammals, the protein acetylations are reversibly regulated by histone acetyltransferases (HATs) and HDACs that consist of four major classes of 18 members. The class I (1, 2, 3, 8), class IIa (4, 5, 7, 9), IIb (6,10), and class IV (11) HDACs require Zn2 for their catalytic activities, whereas class III HDACs (Sirt1–7) are nicotinamide adenine dinucleotide dependent and its modifications of nonhistone proteins tend to beneficially affect various cellular processes [15]. In general, HDACs inhibit gene transcription by deacetylating lysine residues and restoring the positive charges of chromatin histones, leading to a condensed chromatin that blocks the access of transcription factors [16]. Notably, class I HDACs often form a transcription repressor complex consisting of transcription factors/repressors and actively silence gene transcription [17]. Although altered HDAC expressions of various class members have been occasionally reported in animal studies of fibrotic diseases [10, 18, 19], the information regarding the essential HDAC isoforms and the key targets/effectors that are causally involved in renal fibrogenesis is lacking.
Genes whose expressions are affected by either HAT or HDAC aberrations during renal fibrogenesis include profibrotic growth factors, cellular signaling molecules, ECM proteins, and renal antifibrotic factors [5]. Many of them, for example ECM proteins, are upregulated in fibrotic kidney, seemly opposing the inhibitory effects of HDAC. On the other hand, the antifibrosis protein Klotho and BMP-7 are reportedly repressed that can be reversed by HDAC inhibitions [20, 21]. The antifibrosis factors inhibit renal fibrogenesis mainly by interfering with profibrotic signaling and ECM expression [22–24]. However, whether preserving a key antifibrotic mediator by HDAC subtype-selective inhibition provides sufficient antirenal fibrosis functions remain to be established.
In this study, we seek to identify the key HDAC isoforms and mediators critically involved in renal fibrogenesis with both pharmacological and genetic approaches. We provide strong evidence that HDAC3 aberration, likely induced by TGFβ/Smad signaling, and its inhibition of Klotho play essential roles in promoting epithelial to MTD and renal fibrogenesis. Thus, our study reveals an important regulatory pathway in epigenetic renal fibrogenesis with clinical therapeutic implications.
Materials and methods
Animal study
C57BL/6 mice were purchased from the Model Animal Research Center of Nanjing University. Since germline Hdac3 knockout is lethal, a strain of conditional Hdac3 knockout mice (Hdac3fl/fl/Cre-ERT2) was generated by crossing Hdac3 floxed mice (Hdac3fl/fl) [25] with transgenic Cre-ERT2 mice (B6.Cg-Ndor1Tg(UBC-cre/ERT2)1Ejb/1J, Jackson lab, USA) that express a cytoplasm-tethered Cre-ERT2 fusion protein subjected to tamoxifen activation. Mouse genotype was confirmed by polymerase chain reaction (PCR) with mouse tail DNA using the following primers: HD3flF: GCTTGGTAGCCAGCCAGCTTAG, HD3flR: CATGTGACCCCAGACATGACTGG; CreF: ACCAGCCAGCTATCAACTCG, CreR: TTACATTGGTCCAGCCACC. For inducible deletion of Hdac3, mice were injected intraperitoneally with tamoxifen (1 mg per mouse, dissolved in corn oil containing 10% ethanol) for 5 consecutive days and unilateral ureter obstruction (UUO) was performed 7 days after the last injection. Mice are free to diet and water and housed under 22 °C temperature, 50–60% humidity, and regular lighting conditions (12 h light/dark cycles).
Mouse models of renal fibrosis were established by procedures of UUO [26] and aristolochic acid nephropathy (AAN) [27]. The experimental mice, male and 8-10 weeks of age, were randomly assigned to following four groups: (1) control solvent injection or Sham operation; (2) SB431542 (5 mg/kg, T1726, TargetMol, USA, daily intraperitoneal injection) or RGFP966 (10 mg/kg subcutaneous injection starting the first day and following every other day, S7229, Selleck, USA); (3) renal fibrotic mice: UUO (7 days) or AAN (AAI, A5512, Sigma-Aldrich, USA, 5 mg/kg/day daily i.p. injection for 2 weeks); and (4) SB431542 or RGFP966 interventions of the fibrotic mice. After the experiment completion, mice were sacrificed and the kidneys harvested by surgical procedure and stored at −80 °C for further analysis. At least six animals were included in each group and at least three independent cell experiments were performed to ensure the effect size. No animals were excluded from experiments unless the presence of technical issues. The animal experimental procedures were in accordance with the animal use guidelines and approved by the Institutional Animal Care and Use Committee of Nanjing University Medical School.
Klotho suppression by RNA interferences
Klotho knockdown in kidney was performed with small interference RNA (siRNA) as before [28]. A small interfering RNA targeting mouse Klotho gene (5′-GCGACTACCCAGAG AGTAT-3′, 10 nm in 200 μl of PBS each injection) and a scrambled RNA control (CGUACGCGGAAUACUUCGA dTdT) were injected into mouse tail vein one day before UUO operation.
Western blotting
Western blotting assays of renal tissue or cell lysates were performed as described before [29] with following antibodies: HDAC1 (A0238, ABclonal, China), HDAC2 (A2084), HDAC3 (A2139), HDAC8 (A5829), β-catenin (A11932), silencing mediator for retinoid and thyroid hormone receptors (SMRT) (A8388), Ski-related novel protein N (SnoN) (A5844), acetylated histone site-specific antibody H3K4ac (A17019), H3K9ac (A7255), H3K27ac (A7253), H4K5ac (A15233), Klotho (A12028) from ABclonal, China; E-cadherin (GB11082) and collagen 1 (GB11022-2) from Servicebio, China; α-SMA (sc-32251), BMP-7(sc-53917), NCoR (sc-8994), and NF-kB (sc-372) from Santa Cruz, USA; glyceraldehyde-3-phosphate dehydrogenaseGAPDH (GAPDH, 60004-1-Ig, Proteintech, Rosemont, USA); acetyl-Histone 3 (ac-H3, 06-599, Millipore, USA), histone 3 (4499), histone 4(13919), phosphorylated Smad3 (9520) from Cell Signaling Technology, USA; phosphorylated Smad2 (AF3450, Affinity Biosciences, USA); and β-actin (YFMA0052), goat anti-rabbit IgG-HRP (YFSA02), and goat anti-mouse IgG-HRP (YFSA01) from Yifeixue biotech, Nanjing, China. The blots were developed using an ECL plus western blotting detection system (Vazyme, USA). The protein expression levels were assessed by Image J software and expressed as relative levels after adjusted to loading controls.
Histology, immunohistochemistry, and immunofluorescent staining
Kidney section preparations and the staining procedures of Masson’s trichrome, immunohistochemistry, and immunofluorescence for HDAC3 (sc-376957, Santa Cruz, USA) were performed following the protocols established in lab before [30]. Photomicrographs were taken with a DP74 light or a FV3000 confocal Olympus microscope. Renal fibrosis severity was assessed blindly by Image-Pro Plus 6.0 software based on ten randomly selected nonoverlapping areas for each animal and presented as the ratio of blue-stained collagens over the whole examined field.
Cell culture and treatment
Human kidney tubular HK2 and human embryo kidney HEK293 cells (ATCC, USA) were cultured in DMEM/F12 or DMEM medium, respectively, supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Gibco, USA) in a humidified 5% CO2 incubator at 37 °C. HDAC3-selective inhibitor RGFP966, TGFβ (240-B-002, R&D Systems, USA), inhibitor of Smad3 phosphorylation SIS3 (HY-13013, MCE, USA) were added as indicated.
Plasmid construction and cell transfection
The murine Klotho promoter reporter plasmid mKLp-Luc, the positive control plasmid containing three copies of TGFβ/Smad responsive element 3TP-luc, and a plasmid overexpressing flag-tagged HDAC3 have been previously described [31, 32]. Mouse Hdac3 promoter reporter Hd3p-luc was constructed in pGL-3-luc plasmid by inserting a PCR-amplified mouse genomic DNA fragment at XhoI and HindIII sites (forward primer GTACTCGAGAGCACGTGGCAGATCATGAAGC and reverse primer GACAAGCTTGCAAGCACGCAGCCTACTACTG, the cloning sites were underlined). A mutant form mHd3p-luc in which the Smad responsive element CAGACA was mutated to CAGCAC was generated by PCR-based mutagenesis and the mutated sequences were confirmed by sequencing. Cell transfections were performed with Lipofectamine 2000 agents (Invitrogen, USA) following the manufacturer’s instruction.
Luciferase assay
HEK293 cells were transiently transfected with plasmid Hd3p-luc, mHd3p-luc, 3TP-luc, or mKLp-Luc, plus a renilla luciferase reporter as internal control. The transfected cells were treated with TGFβ, and/or SIS3, RGFP966 as indicated. Luciferase activities were assayed using a dual luciferase reporter assay kit (Promega, USA). Luciferase activities were normalized to renilla luciferase levels and expressed as relative fold changes.
Reverse-transcription PCR (RT-PCR)
RT-PCR detections of Hdac1, Hdac2, Hdac3, and Hdac8 mRNAs in renal tissue was performed essentially as before [28] with following primers: Hd1F: ACCGTCCTCACAAAGCCAAT, Hd1R: AAACACCGGACAGTCCTCAC; Hd2F: CTATCCCGCTCTGTGCCCTA, Hd2R: CACACTTTCTTCTTGCCGCC; HD3F: CCCCACCAATATGCAGGGTT, Hd3R: CAGAAGCCAGAGGCCTCAAA; Hd8F: GCCTGTTTCACCAGAACTCC, Hd8R: GGCCACTGACACAAGAAAGAC; Gapdh (GapdhF: TATGTCGTGGAGTCTACTGGTGT, GapdhR: GTCATCATACTTGGCA GGTTTCT) served as internal control. PCR products were resolved on a 1.5% agarose gel and visualized under UV light.
Co-immunoprecipitation (Co-IP)
Co-IP was performed to detect the protein associations reciprocally following a previous protocol [31]. The kidney lysates were first immunoprecipitated with antibody to HDAC3, NCoR, NF-kB, or isoform-matched immunoglobulin (Ig), and then the immunoprecipitants were assayed by western blotting with antibody to NCoR, HDAC3, or NF-kB, respectively.
Chromatin immunoprecipitation (ChIP)
ChIP assay is performed with renal tissues as before [31]. The immunoprecipitation was performed with ChIP quality antibody to acetylated histone 3, HDAC3, NF-kB, and NCoR. The starting (input) and immunoprecipitated DNAs were analyzed by PCR and quantitative real-time PCR (qRT-PCR) using primer set NbF: AGAAGCCTCACACAACCCATC (-594/-574) and NbR: CAGAGAACATCCCAGGAGAGC (-385/-365), which cover a putative NF-KB binding motif (-428/GAATTCCC). Regular PCR products were separated on 1.5% agarose gels and analysis of PCR product densitometry were performed with Image J Software. The qRT-PCR was performed by a ViiA 7 Real-Time PCR System (Applied Biosystems, USA).
Statistical analysis
Data analyses were performed with GraphPad Prism or SPSS Statistics 22. Number of biological replicates is indicated in the figure legends. Data normality and the assumption of homogeneity of variances were determined by Shapiro–Wilk test and Levene’s test, respectively. Statistical analysis and the main/interaction effects were assessed by Student’s t test, analysis of variance (ANOVA), or ANOVA followed by Tukey’s post-hoc tests for multiple group comparisons. Data were expressed as means ± SEM. Statistically significance was defined as P < 0.05.
Results
HDAC3 is preferentially upregulated in fibrotic kidney of UUO mice
Since various class I HDAC (HDAC1, 2, 3, and 8) inhibitors are reportedly protective against renal fibrosis [12, 33], we decided to determine the expression nature of family I HDACs in fibrotic kidney induced by UUO, a typical renal fibrosis model displaying extensive tubular injury and fibrosis as a results of obstructed urine flow. As anticipated, mice subjected to UUO for 7 days showed marked renal tubular damage and fibrotic lesions (Fig. 1a, indicated by arrows). Examination of HDAC1, 2, 3, and 8 mRNAs and proteins revealed a preferential upregulation of HDAC3 (Fig. 1b, c) and its predominant accumulation in nucleus of renal tubular cells (Fig. 1d, indicated by arrows). To gain insight into the role of HDAC3 in renal fibrosis, we generated a strain of tamoxifen-inducible (germline Hdac3 knockout is lethal) Hdac3 knockout mice (see methods). Hdac3KO mice were verified by genotyping (Fig. 1g) and western blotting (Fig. 1h, i, the upper panel) and displayed much less renal fibrotic lesions (9.59 ± 1.34% vs.18.44 ± 0.94% of Hdac3WT UUO mice, P < 0.05, Fig. 1e, f). As expected, the myofibroblast marker α-SMA was markedly induced and the renal-specific antifibrosis protein Klotho was repressed in the wild-type mouse kidney, but not in Hdac3KO mice (Fig. 1h, i). These results indicate that HDAC3 is a critical profibrotic factor whose elevation correlates with Klotho suppression and renal fibrosis.
Fig. 1. HDAC3 is preferentially upregulated in fibrotic kidney of UUO mice.

a Representative photomicrographs of kidney sections (Masson’s trichrome staining) from Sham and UUO mice (7 days, six mice in each group). The dark arrows indicate collagen-stained fibrotic areas. b RT-PCR of renal tissues from Sham and UUO mice for Hdac1 (Hd1), Hd2, Hd3, and Hd8 mRNA levels. Gapdh served as internal control. Two samples from each group were shown. c Western blots of renal tissues for HDAC1 (HD1), HD2, HD3, and HD8 proteins from Sham and UUO mice. Three samples from each group were shown. GAPDH served as loading control. d Representative photomicrographs of kidney sections of Sham and UUO mice stained for HDAC3 by immunohistochemistry (IHC) or immunofluorescence (IF). The arrows indicated positively stained renal tubular cells. The red-circled were glomeruli. e Representative photomicrographs of kidney sections (Masson’s trichrome staining) of Hdac3WT and Hdac3KO mice subjected to Sham or UUO for 7 days. The dark arrows indicate the fibrotic areas. f Quantitation of renal fibrosis in e (n = 6). g Genotyping of Hdac3 wild-type, Hdac3 floxed (Hdac3fl/fl. the upper panel), and the conditional Hdac3 knockout mice (Hdac3fl/fl/CreERT2, the middle and lower panel). h Renal tissue western blots for HDAC3, Klotho, α-SMA and GAPDH from mice in e. Two randomly selected samples from each group were shown. i Quantification of h. The values (f, i) were presented as means ± SEM. *P < 0.05, two-way ANOVA.
TGFβ/Smad signaling upregulates HDAC3 transcription
To search for the possible cause of the HDAC3 upregulation, we analyzed Hdac3 promoter and found a putative Smad binding element (-1035/CAGACA) [29, 34], suggesting that HDAC3 induction is likely regulated by TGFβ, a primary pathological factor known to promotes renal fibrosis [35]. Mice treated with a selective inhibitor of TGFβ receptor I SB431542 were largely resistant to UUO-induced renal fibrosis (8.79 ± 1.01% vs. 19.72 ± 0.86% of UUO mice, P < 0.05, Fig. 2a, b) and the abnormal expressions of α-SMA, epithelia marker E-cadherin, ECM protein collagen 1 and HDAC3 (Fig. 2c, d). In addition, TGFβ preferentially induced HDAC3 in a dose-dependent manner in renal epithelial HK2 cells (Fig. 2e, f), but the effect was blocked by SIS3, a selective inhibitor of Smad3 phosphorylation (Fig. 2g, h). We also made a Hdac3 promoter reporter plasmid Hd3p-luc and a mutant mHd3p-luc in which the Smad binding motif CAGACA was replaced by CAGCAC (Fig. 2i, the upper panel) and found that TGFβ significantly induced the transactivations of a positive control plasmid 3TP-luc and Hd3p-luc, but not the mutant mHd3p-luc. Also the TGFβ-induced transactivation of Hd3p-luc was blocked by SIS3 (Fig. 2i). These results indicate that TGFβ can directly up-regulate HDAC3 via Smad signaling under fibrotic conditions.
Fig. 2. TGFβ/Smad signaling upregulates HDAC3 transcription.

Mice were separated into Sham, SB431542, UUO, and SB431542-treated UUO groups (n = 6, 7 days). a Representative photomicrographs of kidney sections (Masson’s trichrome staining). The dark arrows indicate fibrotic areas. b Quantitation of renal fibrosis in a. c Western blots of renal HDAC3, α-SMA, E-cadherin (E-cad), and collagen 1 (Colla 1). Two samples from each group were shown. d Quantification of c. e Western blots. HK2 cells were treated with increasing doses of TGFβ (1, 2, 5 ng/ml) for 24 h and then assayed for HDAC1, HDAC2, HDAC3, and HDAC8. f Quantification of e. *P < 0.05, **P < 0.01; one-way ANOVA. g Western blot. HK2 cells were treated with TGFβ (5 ng/ml) in presence and absence of SIS3 (10 μM) for 24 h, and then assayed for phosphorylated Smad3 (p-Smad3) and HDAC3 levels. h Quantitations of HDAC3 in g. i Luciferase assay. HEK293 cells were transfected with a positive control plasmid 3TP-luc (P1), the murine Hdac3 promoter reporter Hd3p (P2) or the mutant mHd3p (P3, the replacement of ACA by CAC was confirmed by sequencing shown as the inserts) plus a renilla luciferase plasmid, and then cells were treated with TGFβ (5 ng/ml) and/or SIS3 (10 μM) for 24 h and luciferase activities measured and normalized with renilla luciferase activities. Data were presented as means ± SEM of six mice in each group or three repeated cell assays. *P < 0.05; one- (P1) or two-way (P2) ANOVA.
HDAC3-selective inhibition alleviates Klotho suppression and renal fibrosis
To gain further insights into the functional relevance of the HDAC3 aberration, we explored the effects of HDAC3 inhibition on renal fibrosis in UUO and an additional mouse model—AAN, in which aristolochic acid I overexposure causes extensive renal tubular atrophy and progressive interstitial fibrosis [27, 36]. For specific and effective HDAC3 inhibition, we used a HDAC3-selective inhibitor RGFP966 that reportedly inhibited HDAC3 with IC50 value of 80 nM while had no effective inhibition on other HDACs up to 15 μM [37]. Mice were separately subjected to Sham/Control, RGFP966, UUO/AAN, or RGFP966-treated UUO/AAN, respectively. The results showed that RGFP966 treatments did not significantly affect the normal renal histomorphologies or HDAC3 levels, but effectively reduced renal fibrotic lesions in UUO (9.08 ± 0.98% vs. 18.62 ± 1.62 % of UUO, P < 0.05, Fig. 3a, b, the upper panel) and in AAN mice (7.27 ± 0.51 % vs. 14.24 ± 1.07 % of AAN, P < 0.05, Fig. 3a, b, the lower panel). Consistently, RGFP966 significantly corrected the abnormal expressions of α-SMA, collagen 1, E-cadherin, and BMP-7 in UUO mice (Fig. 3c, d) and similarly normalized the Klotho, α-SMA and collagen 1 in AAN mice (Fig. 3e, f), suggesting that HDAC3 inhibition by RGFP966 prevents renal fibrosis. Since Klotho preservations are known to reduce renal fibrosis [30, 38], these results also suggest that the HDAC3 inhibition might exert the antirenal fibrosis effects at least in part via derepressing Klotho.
Fig. 3. HDAC3-selective inhibition alleviates Klotho suppression and renal fibrosis.

B6 mice were separated into Control/Sham, RGFP966, UUO AAN, or RGFP966-treated UUO/AAN groups (six mice in each group). a Representative photomicrographs of kidney sections of UUO (the upper panel) and AAN (the lower panel) experimental mice (Masson’s trichrome staining). b Quantitation of renal fibrosis in a. c Western blots. The renal tissues of UUO experiments were assayed for α-SMA, collagen 1 (Colla 1), E-cadherin (E-cad), Klotho, BMP-7, and HDAC3. Two samples from each group were shown. d Quantification of c . e Western blots. The renal tissues of AAN experimental mice were assayed for Klotho, HDAC3, α-SMA, and collagen 1 (Colla 1). Two samples from each group were shown. f Quantification of e. Data were presented as means ± SEM; *P < 0.05; two-way (b, d) or one-way (f) ANOVA.
HDAC3 aberration represses Klotho transcription
Klotho is a renal epithelium-enriched protein and HDAC3 was apparently upregulated in renal epithelial cells of fibrotic kidney (Fig. 1d). Their inverse expressions in normal vs. fibrotic kidneys raise a possibility that the HDAC3 aberration is a direct cause of the Klotho suppression. To answer this question, we examined the effects of gain or loss of HDAC3 on Klotho levels in renal cells. The results showed that overexpression of a flag-tagged HDAC3 in HK2 cell reduced the basal levels of Klotho and E-cadherin and induced collagen 1 (Fig. 4a). Conversely, HDAC3 inhibition by RGFP966 reduced TGFβ inductions of α-SMA and collagen 1 and Klotho repression (Fig. 4b, c), which were blocked by HDAC3 overexpression (Fig. 4d, e). Further, we performed Klotho promoter/reporter luciferase assay and found that TGFβ inhibited mouse Klotho promoter transactivation that was significantly blocked by RGFP966 (Fig. 4f). These results indicate that HDAC3 acts upstream of Klotho and its aberrant elevation inhibits Klotho transcription during renal epithelial to MTD.
Fig. 4. HDAC3 aberration represses Klotho transcription.

a Western blots. HK2 cells were transfected with vector or plasmid overexpressing flag-tagged HDAC3 (F-HDAC3). Twenty-four hours later, cell lysates were assayed with antibody to HDAC3, Klotho, E-cadherin, and collagen 1 (Colla 1). b Western blots. HK2 cells were treated with TGFβ (5 ng/ml) in presence or absence of RGFP966 (RG,10 μM) for 24 h, and then cell lysates were assayed for HDAC3, Klotho, α-SMA, or collagen 1 (Colla 1). c Quantification of b. d Western blots. HK2 cells were transfected with vector or plasmid overexpressing flag-tagged HDAC3 for 12 h, and then treated with TGFβ (5 ng/ml) and/or RGFP966 (RG, 10 μM) for 24 h. Cell lysates were assayed with antibody to Klotho or HDAC3. e Quantification of d. f Luciferase assay. HEK293 cells were transfected with murine Klotho promoter reporter mKLp-Luc plus a renilla luciferase reporter control, and then treated with TGFβ (5 ng/ml) with or without RGFP966 (RG, 10 μM) for 24 h. Cell lysates were assayed with a dual luciferase kit and the luciferase activities were normalized with renilla’s. Data information: data were presented as means ± SEM of three repeated experiments. *P < 0.05. Two-way ANOVA.
HDAC3 inhibits Klotho via NCoR and NF-kB-associated transcriptional repression
HDAC3 is known to function exclusively by forming a complex with transcription factors/repressors that guide the complex to a specific gene promoter [39]. We found that HDAC3 started to accumulate as early as on day 3 after UUO that was accompanied by Klotho decline and α-SMA induction, while transcription repressor NCoR (nuclear receptor corepressor) known to associate with HDAC3 under various pathological conditions [40] also increased concomitantly (Fig. 5a). Peroxisome proliferator-activated receptor-gamma (PPARγ) acetylation, which was regulated by HDAC3 in adenine-induced chronic renal injury [31], was not detected in either UUO or RGFP966-treated UUO kidneys (data not shown). Other potential HDAC3 partners such as SMRT or SnoN were not positively affected either; however, NF-kB, a transcription factor reportedly causing inflammatory Klotho repression [41], was substantially increased (Fig. 5b), suggesting that NCoR and NF-kB might participate in the HDAC3-incurred Klotho transcriptional repression.
Fig. 5. HDAC3 inhibits Klotho via NCoR and NF-kB-associated transcriptional repression.

a Western blots. Mice were subjected to UUO for 3, 5, and 7 days, (four mice in each group) and then renal tissues were assayed for HDAC3 (HD3), Klotho, NCoR, and α-SMA . Two samples from each group were shown. b Western blots. Renal tissues from Sham and UUO (7 days) mice were assayed for NCoR, SMRT, SnoN, and NF-kB. Two samples from each group were shown. c Co-IP assay. Renal tissues of Sham and UUO mice (7 days) were tested for NCoR, HDAC3 (HD3) and NF-kB (left panel). The same tissue lysates were immunoprecipitated with isoform-matched immunoglobulin (Ig) or antibody to HDAC3 (HD3), NCoR, or NF-kB, and then immunoprecipitants were assessed for NCoR, NF-kB, or HDAC3 by western blotting. d Quantification of Co-IP in c. Data were presented as means ± SD based on three renal samples from each group. *P < 0.05, Student’s t test. e Western blot assays of Sham, RG, UUO, or RG/UUO renal tissues using site-specific antibody to acetylated histone 3 lysine 4 (H3K4ac), H3K9ac, H3K27ac, or H4K5ac. Total histone 3 (Hist3) and histone 4 (Hist4) served as controls. Two samples from each group were shown. f ChIP assay. The renal tissues as indicated were immunoprecipitated with antibody to isotype-matched immunoglobulin (∇), HDAC3 (HD3), NCoR, NF-kB, or acetylated histone 3 (Ac-H3), respectively, and then the genomic DNA (Input) and the antibody-bound DNA fragments were PCR-amplified with primers covering the NF-kB site on Klotho promoter. The PCR products were analyzed on agarose gel. One (HDAC3, NCoR, and NF-kB) or two representative samples (acetyl-Hinstone 3, Ac-H3) from each group were shown. g The same input and IP-DNAs were analyzed by quantitative real-time PCR (qRT-PCR). The results were normalized with input DNA and presented as fold changes relative to Sham. Data were presented as means ± SEM of six renal samples from each group. *P < 0.05, Student’s t test or two-way ANOVA (Ac-H3).
To test this hypothesis, we first performed immunoprecipitation assays and found that HDAC3 inducibly associated with NCoR and NF-kB in UUO kidney (Fig. 5c, d). In addition, UUO kidney displayed reduced acetylation of histone 3 lysine 4 (H3K4), but increased acetylations of H3K9, H3K27, and H4K5 (Fig. 5e), whereas RG treatment further increased the acetylation levels of H3K4, H3K9, and H4K5, but not that of H3K27 (Fig. 5e), suggesting that H3K4, H3K9, and H4K5 are sensitive to HDAC3 in fibrotic kidney. To obtain the direct evidence that HDAC3 and NF-kB/NCoR inhibit Klotho transcription, we performed ChIP assays and found that HDAC3, NF-kB, and NCoR all accumulated on the Klotho promoter region containing a putative NF-kB binding site (-428/GAATTCCC) in UUO kidney. Also, this locus was acetylated in Sham and RG-treated kidneys, hypoacetylated in UUO kidney, and regained the acetylation after RG treatment, which correlated with the Klotho expression patterns under these conditions (Fig. 5f, g). Altogether, these results strongly support that HDAC3 forms a repressive complex with NCoR and NF-kB that dynamically regulates fibrotic Klotho expressions.
Klotho derepression is essential for the antifibrosis function of HDAC3 inhibition
To explore the critical role of the Klotho derepression by HDAC3 inhibition, we tested whether Klotho knockdown by siRNA affects the antifibrotic effects of RGFP966. Mice receiving either siRNA-control (siCon) or siRNA-Klotho (siKL) were subjected to Sham, RGFP966, UUO, or RGFP966 plus UUO treatments. As expected, Klotho is substantially knocked-down (Fig. 6c, the upper panel) in siKL mice and UUO similarly induced renal fibrosis in siCon mice (Fig. 6a, b, 19.5 ± 0.94%) as before. However, siKL mice displayed a significant increase of renal fibrotic lesions after UUO (Fig. 6a, b, 25.4 ± 1.17%, P < 0.05, comparing groups 7 and 3). RGFP966 effectively reduced the renal fibrosis in siCon mice (8.01 ± 1.27%, P < 0.05, comparing groups 4 and 3), but the effect was largely abrogated in mice lacking Klotho (Fig. 6, comparing groups 8 and 7). We also assessed the effects of siKL treatment, RGFP966 intervention and the interaction between siKL and RGFP966 intervention. The results showed that renal fibrotic lesions were significantly affected by siKL (P1 < 0.000001), RGFP966 intervention (P2 = 0.000047), and the interaction between siKL and the intervention (P3 = 0.015). Similarly, RGFP966 normalized the fibrotic expression of α-SMA, BMP-7, collagen 1, β-catenin, and phosphorylated Smad2 in siCon mice (Fig. 6c–e, comparing groups 3 and 2), but the beneficial effects were largely abolished in siKL mice (Fig. 6c–e, comparing groups 6 and 5). These results strongly indicate that Klotho derepression is essential for the antirenal fibrosis functions of HDAC3 inhibition in fibrotic kidney.
Fig. 6. Klotho derepression is essential for the antifibrosis function of HDAC3 inhibition.

Mice receiving siRNA-Control (siCon) or siRNA-Klotho (siKL) were subgrouped into Sham, RGFP966, UUO, and RGFP966-treated UUO (n = 6). a Representative photomicrographs of kidney sections (Masson’s trichrome staining). The dark arrows indicate fibrotic areas. b Quantitation of renal fibrosis in a. The effects of siRNA-Klotho (P1), RGFP966 intervention (P2), and the interaction between siRNA-Klotho and RGFP966 intervention (P3) were indicated. c Western blots. The renal tissues from experimental mice in a were assayed for Klotho, α-SMA, BMP-7, collagen 1 (Colla 1), and d β-catenin (β-cat) and phosphorylated Smad2 (p-Smd2). Two samples from each group were shown. e Quantification of c and d. Data were presented as means ± SEM. *P < 0.05, three-way ANOVA followed by Tukey’s post-hoc test.
Discussion
In this study, we have made several novel findings towards a better understanding of epigenetic mechanisms of renal fibrotic fibrogenesis. We discover that HDAC3 is preferentially upregulated, likely by TGFβ/Smad2/3 signaling, in renal epithelial cells of fibrotic kidney, which transcriptionally inhibits a major antifibrosis protein Klotho via forming a transcriptional repressive complex with NCoR and NF-kB and promotes renal fibrosis. In turn, HDAC3-selective inhibition by RGFP966 effectively alleviates renal fibrotic pathologies in a Klotho-sensitive manner (Fig. 7). Hence, HDAC3 aberration and its inhibition of Klotho are important epigenetic events that are causally involved in renal fibrogenesis and might serve as targets for antirenal fibrosis therapies.
Fig. 7. A schematic diagram of sequential HDAC3 aberration, Klotho suppression, and renal fibrosis.
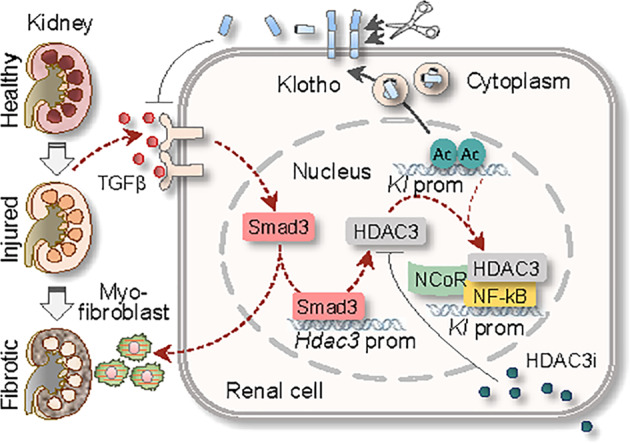
Persistent renal injury releases excessive TGFβ that activates Smad signaling, promotes myofibroblast differentiation and renal fibrosis, and increases Hdac3 transcription. Aberrant HDAC3 is associated with NCoR and NF-kB and binds to and deacetylates Klotho promoter (Kl prom), leading to Klotho transcriptional suppression (dashed line). Selective HDAC3 inhibition (HDAC3i) by RGFP966 blocks HDAC3 activity and preserves Klotho, resulting in inhibition of TGFβ-incurred profibrotic signaling (solid line) and renal fibrosis.
TGFβ is the most important profibrotic cytokine actively involved in tissue repair/remodeling and its dysregulation is a major cause of MTD and renal fibrosis [35]. TGFβ promotes MTD and renal fibrosis by activating canonical Smad and other noncanonical profibrotic signaling pathways and affecting a large number of genes, either increasing or decreasing their expressions [42]. HDAC3 upregulation by TGFβ both in vitro and in vivo and its subsequent repression of Klotho are important discoveries of our study, suggesting that the repressive effects of TGFβ on at least some of its target genes are likely mediated by its transcriptional activation of Hdac3.
The aberrant HDAC isoform expressions and their individual contributions to renal fibrosis are not conformably reported [9–13]. Several class IIa HDAC inhibitors displayed antifibrotic properties by lowing HDAC4 and HDAC5 [10]; however, HDAC4 and HDAC5 reportedly had little to no catalytic activity on canonical HDAC substrates [43]. A number of pan-HDAC inhibitors including TSA, valproic acid, SB939, and CG200745 [9, 11, 14] and class I HDAC-selective inhibitor MS-275 and FK228 [8, 13] similarly attenuated renal fibrotic injuries in animal studies. We further found that HDAC3-selective inhibition and genetic Hdac3 knockout mitigated Klotho repression and renal fibrotic pathologies (Figs. 1 and 3), providing stronger evidence that HDAC3 is a key HDAC subtype. In addition, HDAC3 is highly induced in renal tubular epithelial cells (Fig. 1d), the same cell type in which Klotho is enriched [44]. HDAC3 inhibition of Klotho positively affects myofibroblastic phenotypes in renal epithelial cells (Fig. 4), suggesting that the aberrant HDAC3 and its inhibition of Klotho play critical roles in renal epithelial-MTD during renal fibrogenesis.
HDAC3 is a unique HDAC subtype with a non-conserved C-terminal region and functions exclusively as a component of corepressor complex reportedly containing NCoR, SMRT, Ski, or SnoN [45–48]. The repressor complex often silences gene transcription by interacting with other transcription factors [49, 50]. We initially thought that PPARγ might participate in the fibrotic Klotho suppression since its acetylation by HDAC3 inhibition led to Klotho preservation in adenine-incurred chronic renal injury [31]. However, we could not detect PPARγ’s involvement by either immunoprecipitation or acetylation assay, Instead, we found that increased NCoR and NF-kB, but not PPARγ, SMRT, Ski, or SnoN, were associated with HDAC3 on Klotho promoter in UUO kidney. These results predict a regulatory loop of fibrotic HDAC3 aberration and subsequent Klotho repression: namely, HDAC3 upregulation by TGFβ in fibrotic kidney recruits NCoR and NF-kB to Klotho promoter and reduces the promoter acetylation (Fig. 5), leading to its transcriptional repression. Apparently this working mode reveals the important components of a HDAC3-associated repressive complex that contributes to the fibrotic Klotho suppression and suggests that Klotho suppressions under various pathological processes or disease stages are regulated by alternative signaling pathways.
Klotho is initially identified as a kidney-enriched aging suppressor whose deficiency causes premature aging and short life span in mice [44]. Klotho exists in both membrane and soluble forms. The full-length membrane Klotho mainly acts as an obligatory coreceptor for bone-derived hormone fibroblast growth factor 23 [51], while the soluble Klotho generated by proteolytic cleavage of membrane Klotho or alternative mRNA splicing circulated in blood, urine, and cerebral spinal fluid regulates the functions of a number of cellular membrane receptors and ion transporters through physical interactions or via its intrinsic glycosidase activities [52]. Klotho’s antiaging functions are mainly conferred by its inhibition of the intracellular insulin/insulin-like growth factor-1 signaling cascade [53]. Its FGF23 coreceptor action and deglycosylation of the calcium channel transient receptor potential vallinoid-5 [52] are critical for mineral and vitamin D metabolisms and maintaining kidney homeostasis. Klotho is known to inhibit profibrotic TGFβ/Smad and Wnt/β-catenin signaling by binding to TGFβ receptors and Wnt ligands [22, 23, 54]. In addition, Klotho beneficially regulates other fibrosis-related cellular processes such as oxidative stress, inflammation, and autophagy [55–58] and exerts a great spectrum of renoprotective activities. The strategies of maintaining Klotho levels by exogenous supplementations [59, 60] or endogenous Klotho preservations [30, 38, 61, 62] are protective against renal fibrotic injuries and proven beneficial in treating various renal diseases and the extrarenal complications in animal studies [63, 64]. Our results demonstrate that HDAC3 inhibition effectively derepresses Klotho and Klotho sensitively mitigates the renal fibrotic injuries, the abnormal expressions of fibrotic proteins, the profibrotic signaling molecules, and even another antifibrotic protein BMP-7 (Fig. 6), supporting that Klotho among other renal antifibrotic factors is a major target/effector that mediates the antifibrosis function of HDAC3 inhibition.
In summary, our data suggest that TGFβ-incurred HDAC3 aberration and its transcriptional inhibition of Klotho form an epigenetic regulatory loop that contributes significantly to renal fibrogenesis. Although several pan- or class HDAC inhibitors, namely vorinostat, romidepsin, belinostat, and panobinostat, are approved by US Food and Drug Administration for treating cutaneous and peripheral T-cell lymphomas [65], long term uses of nonspecific HDAC inhibitors have raised safety concerns. Our results provide strong evidence that strategies of Klotho derepression by HDAC3-selective inhibitors possess therapeutic potentials in treating renal fibrotic disorders.
Acknowledgements
This study is supported by research grants from National Nature Science Foundation of China General Program 81970577 and 81670762 (to WC).
Author contributions
Conception and design: WC; data acquisition, analysis, and interpretation: FC and WC; investigation: FC, QG, AW, XC, YS, HW, and WC; article drafting and revising: FC and WC; and article writing: WC. All authors approved the final version of the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by D. Aberdam
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hongwei Wang, Email: hwang@nju.edu.cn.
Wangsen Cao, Email: wangsencao@nju.edu.cn.
References
- 1.Zeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21:1819–34. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]
- 2.Black LM, Lever JM, Agarwal A. Renal inflammation and fibrosis: a double-edged sword. J Histochem Cytochem. 2019;67:663–81. doi: 10.1369/0022155419852932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol. 2010;6:643–56. doi: 10.1038/nrneph.2010.120. [DOI] [PubMed] [Google Scholar]
- 4.Wing MR, Ramezani A, Gill HS, Devaney JM, Raj DS. Epigenetics of progression of chronic kidney disease: fact or fantasy? Semin Nephrol. 2013;33:363–74. doi: 10.1016/j.semnephrol.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tampe B, Zeisberg M. Contribution of genetics and epigenetics to progression of kidney fibrosis. Nephrol Dial Transplant. 2014;29:72–9. doi: 10.1093/ndt/gft025. [DOI] [PubMed] [Google Scholar]
- 6.Chun P. Therapeutic effects of histone deacetylase inhibitors on kidney disease. Arch Pharmacal Res. 2017;41:162–83. doi: 10.1007/s12272-017-0998-7. [DOI] [PubMed] [Google Scholar]
- 7.Brilli LL, Swanhart LM, de Caestecker MP, Hukriede NA. HDAC inhibitors in kidney development and disease. Pediatr Nephrol. 2012;28:1909–21. doi: 10.1007/s00467-012-2320-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine MH, Wang Z, Bhatti TR, Wang Y, Aufhauser DD, McNeal S, et al. Class-specific histone/protein deacetylase inhibition protects against renal ischemia reperfusion injury and fibrosis formation. Am J Transplant. 2015;15:965–73. doi: 10.1111/ajt.13106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi HS, Song JH, Kim IJ, Joo SY, Eom GH, Kim I, et al. Histone deacetylase inhibitor, CG200745 attenuates renal fibrosis in obstructive kidney disease. Sci Rep. 2018;8:11546. doi: 10.1038/s41598-018-30008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiong C, Guan Y, Zhou X, Liu L, Zhuang MA, Zhang W, et al. Selective inhibition of class IIa histone deacetylases alleviates renal fibrosis. FASEB J. 2019;33:8249–62. doi: 10.1096/fj.201801067RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, et al. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol-Ren Physiol. 2009;297:F996–1005. doi: 10.1152/ajprenal.00282.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu N, He S, Ma L, Ponnusamy M, Tang J, Tolbert E, et al. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS ONE. 2013;8:e54001. doi: 10.1371/journal.pone.0054001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang M, Chen G, Zhang X, Guo Y, Yu Y, Tian L, et al. Inhibition of class I HDACs attenuates renal interstitial fibrosis in a murine model. Pharmacol Res. 2019;142:192–204. doi: 10.1016/j.phrs.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Kang SW, Lee SM, Kim JY, Kim SY, Kim YH, Kim TH, et al. Therapeutic activity of the histone deacetylase inhibitor SB939 on renal fibrosis. Int Immunopharmacol. 2017;42:25–31. doi: 10.1016/j.intimp.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Balasubramanian S, Verner E, Buggy JJ. Isoform-specific histone deacetylase inhibitors: the next step? Cancer Lett. 2009;280:211–21. doi: 10.1016/j.canlet.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, et al. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeshi Marumo KH, Yoshikawa Masahiro, Hirahashi Junichi, Kawachi Shoji, Fujita Toshiro. Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am J Physiol Ren Physiol. 2010;298:F133–41. doi: 10.1152/ajprenal.00400.2009. [DOI] [PubMed] [Google Scholar]
- 19.Sen U, Choi SY, Piao ZH, Jin L, Kim JH, Kim GR, et al. Piceatannol attenuates renal fibrosis induced by unilateral ureteral obstruction via downregulation of histone deacetylase 4/5 or p38-MAPK signaling. PloS ONE. 2016;11:e0167340. doi: 10.1371/journal.pone.0167340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Chen F, Wei A, Bi F, Zhu X, Yin S, et al. Klotho recovery by genistein via promoter histone acetylation and DNA demethylation mitigates renal fibrosis in mice. J Mol Med. 2019;97:541–52. doi: 10.1007/s00109-019-01759-z. [DOI] [PubMed] [Google Scholar]
- 21.Manson SR, Song JB, Hruska KA, Austin PF. HDAC dependent transcriptional repression of Bmp-7 potentiates TGF-β mediated renal fibrosis in obstructive uropathy. J Urol. 2014;191:242–52. doi: 10.1016/j.juro.2013.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satoh M, Nagasu H, Morita Y, Yamaguchi TP, Kanwar YS, Kashihara N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am J Physiol Ren Physiol. 2012;303:F1641–51. doi: 10.1152/ajprenal.00460.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doi S, Zou Y, Togao O, Pastor JV, John GB, Wang L, et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. 2011;286:8655–65. doi: 10.1074/jbc.M110.174037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manson SR, Niederhoff RA, Hruska KA, Austin PF. The BMP-7–Smad1/5/8 pathway promotes kidney repair after obstruction induced renal injury. J Urol. 2011;185:2523–30. doi: 10.1016/j.juro.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu X-F, Cao X-Y, Zhu Y-J, Wu Z-R, Zhuang X, Shao M-Y, et al. Histone deacetylase 3 promotes liver regeneration and liver cancer cells proliferation through signal transducer and activator of transcription 3 signaling pathway. Cell Death Dis. 2018;9:398. doi: 10.1038/s41419-018-0428-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin T, Du R, Huang F, Yin S, Yang J, Qin S, et al. Sinomenine activation of Nrf2 signaling prevents hyperactive inflammation and kidney injury in a mouse model of obstructive nephropathy. Free Radic Biol Med. 2016;92:90–9. doi: 10.1016/j.freeradbiomed.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 27.Ye J, Qian Z, Xue M, Liu Y, Zhu S, Li Y, et al. Aristolochic acid I aggravates renal injury by activating the C3a/C3aR complement system. Toxicol Lett. 2019;312:118–24. doi: 10.1016/j.toxlet.2019.04.027. [DOI] [PubMed] [Google Scholar]
- 28.Yin S, Zhang Q, Yang J, Lin W, Li Y, Chen F, et al. TGFβ-incurred epigenetic aberrations of miRNA and DNA methyltransferase suppress Klotho and potentiate renal fibrosis. Biochim et Biophys Acta. 2017;1864:1207–16. doi: 10.1016/j.bbamcr.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Yin S, Bi F, Liu L, Qin T, Wang H, et al. TIMAP repression by TGFβ and HDAC3-associated Smad signaling regulates macrophage M2 phenotypic phagocytosis. J Mol Med. 2016;95:273–85. doi: 10.1007/s00109-016-1479-z. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Q, Yin S, Liu L, Liu Z, Cao W. Rhein reversal of DNA hypermethylation-associated Klotho suppression ameliorates renal fibrosis in mice. Sci Rep. 2016;6:34597. [DOI] [PMC free article] [PubMed]
- 31.Lin W, Zhang Q, Liu L, Yin S, Liu Z, Cao W. Klotho restoration via acetylation of peroxisome proliferation–activated receptor γ reduces the progression of chronic kidney disease. Kidney Int. 2017;92:669–79. doi: 10.1016/j.kint.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 32.Liu L, Lin W, Zhang Q, Cao W, Liu Z. TGF-β induces miR-30d down-regulation and podocyte injury through Smad2/3 and HDAC3-associated transcriptional repression. J Mol Med. 2015;94:291–300. doi: 10.1007/s00109-015-1340-9. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci USA. 2012;109:E2865–74. doi: 10.1073/pnas.1121131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu M-Z, Tsai Y-P, Yang M-H, Huang C-H, Chang S-Y, Chang C-C, et al. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 2011;43:811–22. doi: 10.1016/j.molcel.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 35.Böttinger EP. TGF-β in renal injury and disease. Semin Nephrol. 2007;27:309–20. doi: 10.1016/j.semnephrol.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Baudoux TER, Pozdzik AA, Arlt VM, De Prez EG, Antoine M-H, Quellard N, et al. Probenecid prevents acute tubular necrosis in a mouse model of aristolochic acid nephropathy. Kidney Int. 2012;82:1105–13. doi: 10.1038/ki.2012.264. [DOI] [PubMed] [Google Scholar]
- 37.Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, et al. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci USA. 2013;110:2647–52. doi: 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irifuku T, Doi S, Sasaki K, Doi T, Nakashima A, Ueno T, et al. Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int. 2016;89:147–57. doi: 10.1038/ki.2015.291. [DOI] [PubMed] [Google Scholar]
- 39.McQuown SC, Wood MA. HDAC3 and the molecular brake pad hypothesis. Neurobiol Learn Mem. 2011;96:27–34. doi: 10.1016/j.nlm.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. 2007;26:5439–49. doi: 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- 41.Moreno JA, Izquierdo MC, Sanchez-Nino MD, Suarez-Alvarez B, Lopez-Larrea C, Jakubowski A, et al. The inflammatory cytokines TWEAK and TNFalpha reduce renal klotho expression through NFkappaB. J Am Soc Nephrology. 2011;22:1315–25. doi: 10.1681/ASN.2010101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–34. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 43.Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci USA. 2007;104:17335–40. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 45.Tabata T, Kokura K, ten Dijke P, Ishii S. Ski co-repressor complexes maintain the basal repressed state of the TGF-β target gene,SMAD7, via HDAC3 and PRMT5. Genes Cells. 2009;14:17–28. doi: 10.1111/j.1365-2443.2008.01246.x. [DOI] [PubMed] [Google Scholar]
- 46.Tecalco-Cruz AC, Ríos-López DG, Vázquez-Victorio G, Rosales-Alvarez RE, Macías-Silva M. Transcriptional cofactors Ski and SnoN are major regulators of the TGF-β/Smad signaling pathway in health and disease. Signal Transduct Target Ther. 2018;8:15. doi: 10.1038/s41392-018-0015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wen YD, Perissi V, Staszewski LM, Yang WM, Krones A, Glass CK, et al. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci USA. 2000;97:7202–7. doi: 10.1073/pnas.97.13.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, et al. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000;19:4342–50. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20:182–7. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao Z, Chiao P, Zhang X, Zhang X, Lazar MA, Seto E, et al. Coactivators and corepressors of NF-κB in IκBα gene promoter. J Biol Chem. 2005;280:21091–8. doi: 10.1074/jbc.M500754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–4. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 52.Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro OM, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci USA. 2008;105:9805–10. doi: 10.1073/pnas.0803223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–33. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou L, Li Y, Zhou D, Tan RJ, Liu Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J Am Soc Nephrology. 2013;24:771–85. doi: 10.1681/ASN.2012080865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu YN, Zhou J, Li T, Wu J, Xie SH, Liu H-f, et al. Sulodexide protects renal tubular epithelial cells from oxidative stress-induced injury via upregulating klotho expression at an early stage of diabetic kidney disease. J Diabetes Res. 2017;2017:1–10. doi: 10.1155/2017/4989847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao Y, Banerjee S, Dey N, LeJeune WS, Sarkar PS, Brobey R, et al. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes. 2011;60:1907–16. doi: 10.2337/db10-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu F, Wu S, Ren H, Gu J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat Cell Biol. 2011;13:254–62. doi: 10.1038/ncb2167. [DOI] [PubMed] [Google Scholar]
- 58.Shi M, Flores B, Gillings N, Bian A, Cho HJ, Yan S, et al. Klotho mitigates progression of AKI to CKD through activation of autophagy. J Am Soc Nephrol. 2015;27:2331–45. doi: 10.1681/ASN.2015060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guan X, Nie L, He T, Yang K, Xiao T, Wang S, et al. Klotho suppresses renal tubulo-interstitial fibrosis by controlling basic fibroblast growth factor-2 signalling. J Pathol. 2014;234:560–72. doi: 10.1002/path.4420. [DOI] [PubMed] [Google Scholar]
- 60.Takenaka TKH, Inoue T, Miyazaki T, Suzuki H, Nishiyama A, Ishii N, et al. Klotho supplementation ameliorates blood pressure and renal function in DBA/2-pcy mice, a model of polycystic kidney disease. Am J Physiol Ren Physiol. 2020;318:F557–64. doi: 10.1152/ajprenal.00299.2019. [DOI] [PubMed] [Google Scholar]
- 61.Hu YML, Yang F, Tu H, Lin W. Curcumin attenuates cyclosporine A-induced renal fibrosis by inhibiting hypermethylation of the klotho promoter. Mol Med Rep. 2016;14:3229–36. doi: 10.3892/mmr.2016.5601. [DOI] [PubMed] [Google Scholar]
- 62.Grange C, Papadimitriou E, Dimuccio V, Pastorino C, Molina J, O’Kelly R, et al. Urinary extracellular vesicles carrying klotho improve the recovery of renal function in an acute tubular injury model. Mol Ther. 2020;28:490–502. doi: 10.1016/j.ymthe.2019.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neyra JA, Hu MC, Moe OW. Klotho in clinical nephrology: diagnostic and therapeutic implications. Clin J Am Soc Nephrol. 2020;22:CJN.02840320. [DOI] [PMC free article] [PubMed]
- 64.Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR. Targeting the progression of chronic kidney disease. Nat Rev Nephrol. 2020;16:269–88. doi: 10.1038/s41581-019-0248-y. [DOI] [PubMed] [Google Scholar]
- 65.Shah RR. Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Saf. 2019;42:235–45. doi: 10.1007/s40264-018-0773-9. [DOI] [PubMed] [Google Scholar]