

Abstract
Background
The identification of recent human immunodeficiency virus (HIV) 1 infections among people with new HIV diagnoses is important to both tailoring and assessing the impact of HIV-1 prevention strategies.
Methods
We developed a multiplexed Primer ID–next-generation sequencing approach to identify recent infections by measuring the intrahost viral diversity over multiple regions of the HIV-1 genome, in addition to detecting drug resistance mutations (DRMs) and phylogenetically linked clusters. We summarize the field implementation of this all-in-one platform among persons with newly diagnosed HIV-1 by the North Carolina State Laboratory of Public Health in 2018.
Results
Overall, recent infection was identified in 94 (35%) of 268 patients with new HIV diagnoses. People <30 years old, and people who inject drugs were more likely to have diagnoses of recent infection. The reverse-transcriptase region K103N was the most commonly detected DRM (prevalence, approximately 15%). We found a total of 28 clusters, and persons with recent infection were more likely to be cluster members than were those with chronic infections (P = .03).
Conclusions
We demonstrate the rapid identification of recent infection and pretreatment DRMs coupled with cluster analysis that will allow prioritization of linkage to care, treatment, and prevention interventions to those at highest risk of onward transmission.
Keywords: recency, drug resistance mutations, transmission network, next-generation sequencing
We developed an all-in-one phylodynamics platform to detect recent human immunodeficiency virus (HIV) infection, drug resistance, and transmission networks using next-generation-sequencing from remnant HIV diagnostic tests. Of persons with new diagnoses, 35% were estimated to be recently infected, and clustered infections were frequent.
The human immunodeficiency virus (HIV) 1 epidemic continues to be a significant threat to public health worldwide. In the United States, the Centers for Disease Control and Prevention estimates that 1.1 million people are currently living with HIV-1, with >38 000 cases newly diagnosed in 2017. This number has not declined since 2013, suggesting that novel methods are needed to interrupt onward transmission [1]. In particular, southern states have a disproportionately higher burden of HIV-1, higher than all other regions in the United States combined [2, 3], partially owing to the opioid crisis in this area in recent years [4, 5].
A key component of strategies to limit new infections (including in the context of the End the HIV Epidemic initiative [6]) is to identify recent (incident) HIV-1 infections among persons with new diagnoses, as they represent ongoing transmission events that may be linked in active transmission clusters where a real-time intervention could block further spread locally. However, to achieve this goal, tools are needed that can rapidly and efficiently identify recency and that can be deployed in near real time. A method that provides clinically useful information and identifies transmission clusters in addition to an assessment of recency would amplify the utility of these tools.
We have described a next-generation sequencing (NGS) approach to identify recent HIV-1 infections based on sequence diversity in the reverse-transcriptase (RT) and env V1–V3 regions of the viral genome [7]. To accomplish this, we used a state-of-art multiplexed Primer ID (MPID) sequencing approach to reveal the true genome sampling depth of the viral population in each serum sample, to greatly reduce methodological errors in polymerase chain reaction (PCR) sequencing, and to reconstruct true haplotypes, which is critical for accurately determining sequence diversity [8, 9]. The combination of the variable V1–V3 region of env and the more conserved RT region of pol gave the most accurate estimate of recency.
Based on this approach we established a pilot phylodynamics platform to identify recent HIV-1 infections; document the presence of transmitted drug resistance mutations (DRMs), by including additional amplicons covering the protease (PR) and integrase (IN) coding regions; and define transmission networks from a single NGS reaction using HIV-positive diagnostic specimens. This novel near real-time platform could serve as a key component for monitoring and intervention in individual HIV-1 outbreaks. In this article, we discuss the details of the platform and summarize the data from diagnostic specimens collected across North Carolina for the calendar year 2018.
MATERIALS AND METHODS
HIV-1 Diagnostic Specimens
The North Carolina State Laboratory of Public Health (NC-SLPH), conducts HIV-1 screening assays for public health agencies and for designated counseling and testing sites across the state. Approximately one-third of all new HIV diagnoses in North Carolina are identified by the NC-SLPH [10]. All HIV-positive serum specimens are routinely stored at the NC-SLPH. After confirmation of test results and starting with samples collected in 2018, aliquots of remnant HIV-positive serum were shipped to the University of North Carolina, Chapel Hill, for downstream sequencing and analysis. Individuals were considered new diagnoses and antiretroviral therapy naive if there was ≤30 days between the date of HIV-1 diagnosis recorded by the North Carolina Division of Public Health (NC-DPH) and the date of sample collection for HIV-1 testing by the NC-SLPH. This study was approved by institutional review boards at University of North Carolina, Chapel Hill, and the NC-DPH.
MPID Library Preparation and NGS
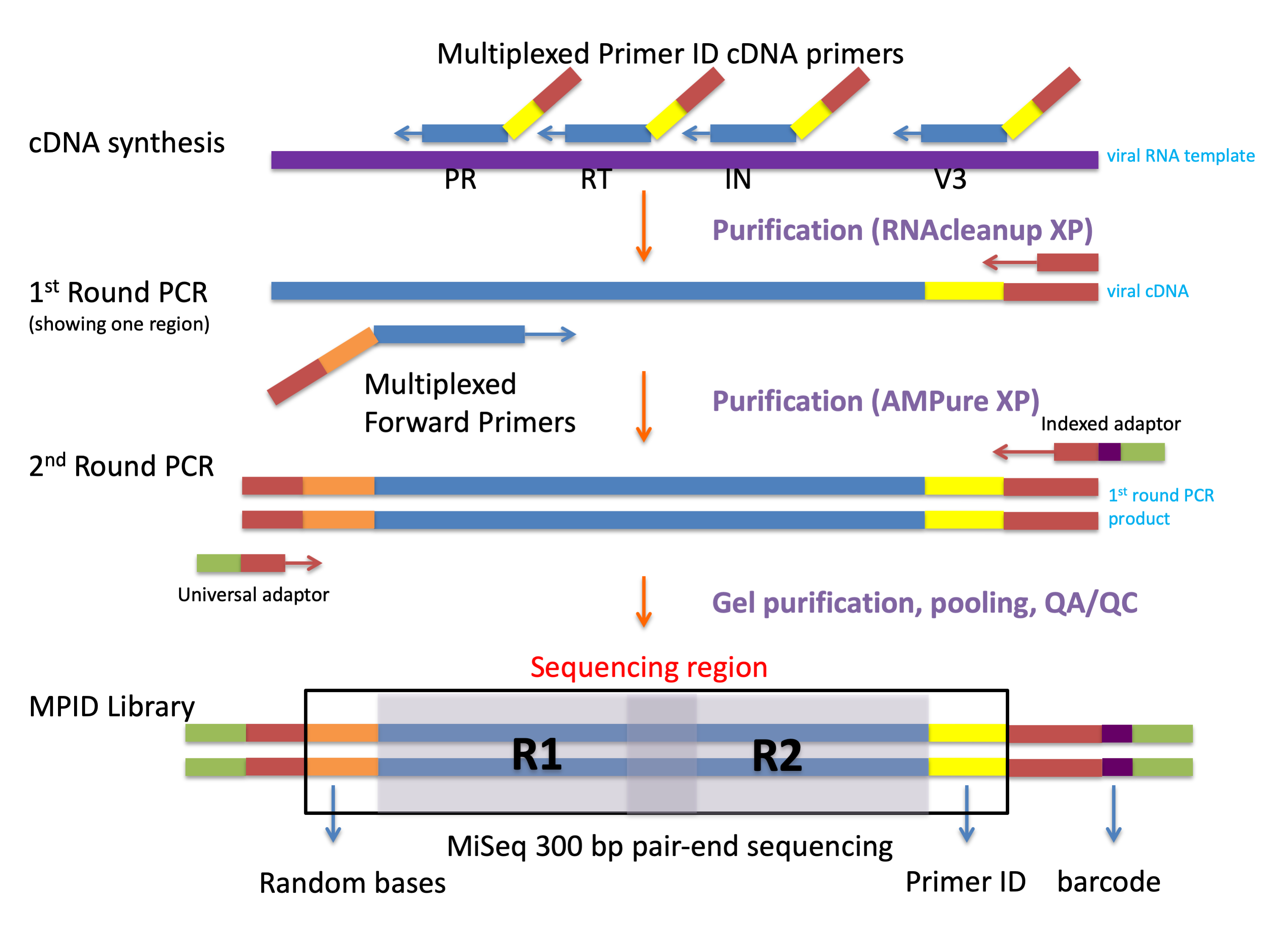
We used a MPID protocol for the sequencing HIV-1 in the diagnostic specimens. Viral particles were first pelleted followed by extraction of viral RNA (Qiagen). Viral RNA was used as template for complementary DNA (cDNA) synthesis, with a mixture of 4 primers, each with a block of random nucleotides (the Primer ID) [8, 9] and a gene-specific region targeting different sites in the HIV-1 genome, and the Superscript III system for reverse-transcription (Thermo Fisher Scientific). All of the bead-purified cDNA (Beckman Coulter) was included to start 2 rounds of PCR amplification. In the first-round PCR we used a mixture of forward primers containing gene-specific regions and a universal reverse primer, and in the second-round PCR we introduced MiSeq adaptors and barcodes. We pooled up to 24 libraries for a single MiSeq 300-base paired-end sequencing run (Illumina). Supplementary Table 1 hows the primer sequences, and Supplementary Figure 1 shows the workflow of MPID library preparation.
This protocol allowed us to amplify multiple regions of the HIV-1 genome in a single cDNA synthesis reaction/PCR amplification. We sequenced regions covering the HIV-1 PR (HXB2 nos. 2164–2593), RT (HXB2 nos. 2648–2914 and 3001–3257, with the 2 sequence fragments linked), IN (HXB2 nos. 4384–4751), and the env V1–V3 region (HXB2 nos. 6585–7208, with a small segment of the C2 region missing); a total of approximately 1.8 kb of the 9-kb HIV-1 genome was sequenced in each multiplexed reaction for each sample.
Bioinformatics Analysis
MiSeq data were initially processed using Illumina bcl2fastq pipeline software. TCS pipeline software, version 1.3.8, was used to sort the reads from the different regions of the genome and construct a template consensus sequence (TCS) for all the reads with the same Primer ID and filter out off-target sequences based on the targeted HXB2 coordinates. The depth of sampling of the viral population was equal to the number of different TCSs, each representing a different viral genome sequenced. We used the parameters identified in our previous work to estimate recent HIV-1 infection based on the sequence diversity (π) and the first quintile of the pairwise comparison [7]. Specimens were classified as recent HIV infection (within 9 months of transmission), chronic infection, or indeterminate (based on criteria in the supplementary text and the algorithm flowchart in Supplementary Figure 2).
We first filtered TCSs for stop codons and APOBEC3G/F hypermutations and generated a cutoff for minority variants using a Poisson model for each sequenced region of each individual specimen. There was no arbitrary cutoff for the minority variants; rather, we generated cutoffs for each individual specimen and region based on the number of TCS to distinguish potential residual methodological errors from true mutations using on a Poisson distribution model [8]. We selected the HIV-1 surveillance DRMs from the Stanford HIV drug resistance database for the PR and RT region mutations, and we used the list of DRMs in the IN region from the International Antiviral Society–USA [11, 12]. We calculated the prevalence and 95% confidence interval (CI) for each mutation. We used the following numbers of TCSs as thresholds to be included in the analysis of the detection of minor variants, based on the upper 95% confidence limits for the binomial proportion when no mutation has been observed, given the number of TCSs: 10 TCS (30% detection limit sensitivity), 34 (10%), 350 (1%), and 1208 (0.3%).
We identified close transmission clusters among the individuals with new diagnoses by first making a consensus sequence from the TCS of the RT region to look for transmission clusters among people with newly diagnosed HIV. We used MUSCLE software version 3.8.1 to alignment the sequences [13, 14], and then used FastTree software version 2.1 [15, 16] to make approximately maximum likelihood phylogenetic trees. Putative transmission clusters were defined as clades with ≤1% genetic difference between sequences.
Statistical Analysis
Demographic characteristics that are routinely collected during HIV-1 surveillance by the NC-DPH were evaluated using descriptive statistics. We fit multivariable logistic regression to identify characteristics associated with diagnosis during recent HIV-1 infection. Adjusted odds ratios (ORs) were calculated, with 95% CIs reported as measures of precision. We calculated the Clopper-Pearson binomial proportion CIs for DRMs identified in each specimen. The detection sensitivity of DRMs was based on the upper 95% confidence limits for the binomial proportion when no mutation has been observed, given the number of TCSs. We used R software, version 3.5.0, for the analysis.
RESULTS
Summary of Samples and Sequencing
We used MPID-NGS to sequence HIV-positive serum specimens from a total of 294 patients with new HIV diagnoses who had specimens submitted to the NC-SLPH for HIV-1 diagnostic testing during the calendar year of 2018; 268 (91%) were successfully sequenced (defined as obtaining ≥10 TCSs in ≥2 genomic regions). The frequency of sequencing success decreased as a function of time since diagnosis, rising to >60% failure if specimen collection was >1 year after recorded diagnosis (Supplementary Table 2). In a sample of specimens from people with HIV diagnosed >1 year before specimen collection, we detected the presence of antiviral drugs in 80% of the specimens that failed to sequence, suggesting that they were obtained from persons receiving suppressive therapy (Supplementary Table 3).
Characteristics of Persons With Newly Diagnosed HIV-1
We examined the demographic characteristics among the 268 individuals whose serum specimens were successfully sequenced. Overall, based on the genetic diversity at the HIV-1 RT and V1–V3 regions by MPID-NGS assay, we found 94 (35.1%) of the specimens were from individuals who were recently infected (estimated within 9 months from transmission), 131 (48.9%) were from people infected for >9 months, and 43 (16%) were indeterminate. Table 1 shows the demographic characteristics of individual with new HIV diagnoses stratified by the sequence-based inference of recency. Overall, most individuals were male (84%) and black/African American (72%); the median age at diagnosis was 27.6 years (interquartile range, 23.7–36.5 years). Most of the individuals (63.4%) identified as men who have sex with men.
Table 1.
Demographic Characteristics in Persons With New Human Immunodeficiency Virus 1 Diagnoses, Stratified by Recency of Infection According to Next-Generation Sequencinga
| Persons With New HIV-1 Diagnoses by Recency of Infection, No. (%) | ||||
|---|---|---|---|---|
| Characteristic | Chronic (n = 131; [48.9]) | Recent (n = 94 [35.1%]) | Indeterminate (n = 43 [16.0%]) | Total (n = 268) |
| Sex | ||||
| Female | 17 (13.0) | 20 (21.3) | 6 (14.0) | 43 (16.0) |
| Male | 114 (87.0) | 74 (78.7) | 37 (86.0) | 225 (84.0) |
| Race | ||||
| Black, non-Hispanic | 94 (71.8) | 68 (72.3) | 30 (69.8) | 192 (71.6) |
| Hispanic | 13 (9.9) | 6 (6.4) | 6 (14.0) | 25 (9.3) |
| White, non-Hispanic | 21 (16.0) | 18 (19.1) | 6 (14.0) | 45 (16.8) |
| Other | 3 (2.3) | 2 (2.1) | 1 (2.3) | 6 (2.2) |
| Age category, y | ||||
| >44 | 22 (16.8) | 12 (12.8) | 7 (16.3) | 41 (15.3) |
| 18–24 | 31 (23.7) | 34 (36.2) | 12 (27.9) | 77 (28.7) |
| 25–34 | 60 (45.8) | 38 (40.4) | 15 (34.9) | 113 (42.2) |
| 35–44 | 18 (13.7) | 10 (10.6) | 9 (20.9) | 37 (13.8) |
| Transmission risk factor | ||||
| MSM | 87 (66.4) | 62 (66.0) | 31 (72.1) | 180 (67.2) |
| Heterosexual transmission | ||||
| Men | 21 (16.0) | 4 (4.3) | 4 (9.3) | 29 (10.8) |
| Women | 10 (7.6) | 16 (17.0) | 4 (9.3) | 30 (11.2) |
| PWID | ||||
| Men | 0 (0) | 4 (4.3) | 1 (2.3) | 5 (1.9) |
| Women | 2 (1.5) | 3 (3.2) | 0 (0) | 5 (1.9) |
| Other/unknown | 11 (8.4) | 5 (5.3) | 3 (7.0) | 19 (7.1) |
| STI coinfection | ||||
| No | 94 (71.8) | 64 (68.1) | 25 (58.1) | 183 (68.3) |
| Yes | 37 (28.2) | 30 (31.9) | 18 (41.9) | 85 (31.7) |
| County of residence | ||||
| Urban | 103 (78.6) | 68 (72.3) | 33 (76.7) | 204 (76.1) |
| Rural | 16 (12.2) | 15 (16.0) | 8 (18.6) | 39 (14.6) |
| Missing | 12 (9.2) | 11 (11.7) | 2 (4.7) | 25 (9.3) |
Abbreviations: HIV, human immunodeficiency virus; MSM, men who have sex with men; PWID, people who inject drugs; STI, sexually transmitted infection.
aNew diagnoses were defined as those made ≤30 days before specimen collection.
Indeterminate Samples
We identified a total of 43 indeterminate samples (16%) that represented either chronic infections with borderline diversity or multivariant infection (either recent dual infection or superinfection). In phylogenetic analysis, we found 7 individuals with ≥2 discrete homogenous intrahost viral lineages, indicating recent multivariant infection (phylogenetic trees of these subjects shown in Supplementary Figure 3).
Recent Versus Chronic Infection
We used bivariable and multivariable logistic regression analysis to explore factors associated with recent HIV-1 infection at diagnosis. For this analysis, we excluded the 43 individuals with specimens of indeterminate chronicity. We found that younger people (<30 years old) were more likely to have recent HIV-1 infection diagnosed (adjusted OR, 2.17; 95% CI, 1.21–3.98; P = .01), as were people who inject drugs (PWID) (5.31; 1.18–37.59; P = .047) (Table 2).
Table 2.
Demographic Characteristics Associated With Diagnosis of Recent Human Immunodeficiency Virus 1 Infectiona
| Characteristic | Patients, No. (%) | OR (95% CI) [P Value] | ||
|---|---|---|---|---|
| Chronic Infection | Recent Infection | Univariable | Multivariable | |
| Sex | ||||
| Male | 114 (60.6) | 74 (39.4) | … | … |
| Female | 17 (45.9) | 20 (54.1) | 1.81 (.89–3.72) [.10] | 1.91 (.89–4.14) [.10] |
| Black race | ||||
| No | 37 (58.7) | 26 (41.3) | … | … |
| Yes | 94 (58.0) | 68 (42.0) | 1.03 (.57–1.87) [.92] | … |
| Age <30 y | ||||
| No | 58 (66.7) | 29 (33.3) | … | … |
| Yes | 73 (52.9) | 65 (47.1) | 1.78 (1.03–3.13) [.04] | 2.17 (1.21–3.98) [.01] |
| MSM | ||||
| No | 44 (57.9) | 32 (42.1) | … | … |
| Yes | 87 (58.4) | 62 (41.6) | 0.98 (.56–1.72) [ .94] | … |
| PWID | ||||
| No | 129 (59.7) | 87 (40.3) | … | … |
| Yes | 2 (22.2) | 7 (77.8) | 5.19 (1.22–35.39) [.04] | 5.31 (1.18–37.59) [.047] |
| Unknown risk | ||||
| No | 120 (57.4) | 89 (42.6) | … | … |
| Yes | 11 (68.8) | 5 (31.2) | 0.61 (.19–1.75) [.38] | - |
| STI coinfection | ||||
| No | 94 (59.5) | 64 (40.5) | … | … |
| Yes | 37 (55.2) | 30 (44.8) | 1.19 (.67–2.12) [.55] | … |
| County of residence | ||||
| Urban | 103 (60.2) | 68 (39.8) | … | … |
| Rural | 16 (51.6) | 15 (48.4) | 1.42 (.65–3.07) [.37] | … |
Abbreviations: CI, confidence interval; MSM, men who have sex with men; OR, odds ratio; PWID, people who inject drugs; STI, sexually transmitted infection.
aRecent defined as within 9 months of transmission.
Sequence Sampling Depth to Validate Sequence Data
Figure 1 shows the distribution of TCS numbers at 4 regions of all individuals with newly diagnosed HIV. These represent the number of viral genomes that were actually sequenced, validating the sampling depth of the viral sequence population and thus defining the sensitivity of detection of minor variants in the population. Overall, we obtained a median of 145, 162, 165, and 81 TCSs for the PR, RT, IN and env V1–V3 regions, respectively. Samples from individuals with recent infections had more TCSs than samples from those with chronic infection, consistent with higher viremia early after infection.
Figure 1.

Distribution of template consensus sequence (TCS) numbers at 4 sequenced regions, with. bars representing the median number and interquartile range for each region. Abbreviations: IN, integrase; PR, protease; RT, reverse-transcriptase.
Identification of DRMs
The sampling depth of the viral sequence population defines the sensitivity for the detection of DRMs that are not fixed (ie, at 100%). Therefore, it is essential to group the specimens based on the sequence sampling depth, and thus the levels of sensitivity of detection of minor variants. We pooled the results from samples that had TCS counts of at least 10, 34, 350, and 1208 TCSs, which provided sampling depth to detect mutations as low as 30%, 10%, 1%, and 0.3% abundance, respectively, with 95% confidence of detection. At a detection sensitivity of 30% abundance, we identified DRMs in 2.4% of the specimens (6 of 254) in PR, in 18.8% (49 of 260) in RT, and in 3.6% (9 of 252) in the IN region. At the sensitivity level of 10% abundance, we found similar percentages of 2.3%, 21.7%, and 4.6% of the specimens had any PR, RT, or IN region DRMs, respectively. However, at the sensitivity level of 1% abundance, 24.4%, 27.7% and 3.3% of the specimens had any PR, RT or IN region DRMs, respectively, a significant increase in PR. Finally, at the sensitivity level of 0.3% abundance, 60.5%, 51.5% and 2.3% of the individuals had any PR, RT, or IN region DRMs, respectively (Supplementary Table 4).
In the PR region, M46I/L was the most frequently identified DRM. In the RT region the nonnucleoside RT inhibitor (NNRTI) mutation K103N mutation was a major mutation in a similar percentage of samples (approximately 15%) at all levels of detection sensitivity, indicating its presence at high abundance in the viral population within an individual when detected. In the IN region, the accessory mutation T97A was the most frequently seen mutation, found in approximately 3% of samples at all levels of detection sensitivity. By contrast, the clinically important DRMs, including RT region K65R and M184V and IN region major mutations, were rarely seen. We compared the detection of DRMs in individuals with recent or chronic infections, based on the NGS recency assay, and we found that recently infected individuals were more likely to have RT region DRMs at the detection sensitivity of 30%, with a borderline significant trend (23.7% vs 13.7%; P = .07), driven by both nucleoside RT inhibitor and NNRTI mutations (Supplementary Table 5).
Figure 2 shows the percentage of individuals identified with the presence of a panel of the most frequently identified DRMs at the different levels of detection sensitivity. We found that there were 2 distinct types of DRMs. The prevalence of RT K103N and IN T97A were similar across all levels of detection sensitivity. In contrast, the prevalence of PR region mutations M46L/I, D30N, V82A, and RT region mutations M41L, D67N, G190E, T215F/I/C/ D/V/E increased greatly (up to 10-fold), with an increase in detection sensitivity from 30% to 0.3%. This demonstrates the frequent presence of these mutations in the viral populations at low abundance, but with their detection strongly dependent on an adequate depth of sampling of the individual viral populations. Identification of X4 viruses and non–subtype B viruses is included in the Supplementary Materials.
Figure 2.

Selected surveillance drug resistance mutations (SDRMs) identified using different sensitivity cutoffs. G190E was plotted at 0.1% instead of zero values on the log scale. A, SDRMs with a trend of increased abundance with an increase in detection sensitivity. B, SDRMs with steady abundance regardless of the increase in detection sensitivity. Abbreviation: IN, integrase; PR, protease; RT, reverse-transcriptase; TCSs, template consensus sequences.
Identification of Genetic Clusters
We identified a total of 28 clusters among the individuals with newly diagnosed HIV, with clusters of 2–4 individuals. The maximum genetic distance between any 2 members of the cluster was within 1%. Persons with recent infection were more likely to be cluster members compared than with chronic infections (32% vs 18% in clusters; P = .03). Figure 3 shows the ML phylogenetic tree of the RT specimen consensus sequences with the transmission clusters highlighted.
Figure 3.

Maximum likelihood tree of reverse-transcriptase (RT) individual consensus sequences for 268 patients with newly diagnosed human immunodeficiency virus infection. Transmission clusters are highlighted in yellow. Recent (incident) infections are represented by red stars, chronic infections by blue triangles, and indeterminate or possible dual infections by purple diamonds. A total of 28 clusters were identified, each including 2–4 members.
DISCUSSION
In the current study, we developed a near real-time phylodynamics platform to detect recent HIV-1 infections, the presence of high and low abundance DRMs, and close transmission clusters, using diagnostic specimens sent to a centralized state testing laboratory. Among persons with new diagnoses, we estimate that >35% had recent infection (within 9 months of specimen collection), and that the most significant DRM was the NNRTI mutation K103N, seen in about 15% of the specimens tested. We also found that diagnosis during recent infection was associated with younger age and PWID status, and that persons with recent infections were more likely than those with chronic infections to be identified in a cluster . Overall, this approach provides near real-time information about features of the ongoing HIV-1 epidemic that could be integrated into public health efforts to reduce transmission.
Several approaches have been used to estimate recency in the setting of HIV-1 testing and diagnosis. Serology-based assays have been developed to predict recency but their accuracy usually varies by geographic location and HIV-1 subtypes. Sanger (bulk) sequencing has been used previously to estimate recency by counting sequence ambiguities or mixtures, but this approach has limited sensitivity and is also susceptible to background noise in the sequencing chromatogram [17]. More recently, NGS has also been used [18–20]. A key limitation in the typical use of deep sequencing is that the number of sequence reads or distinct haplotypes is used as an indirect measure of the unknown number of viral genomes actually sequenced, leading to significant errors in the inferences that can be drawn. By contrast, the use of unique molecular identifiers, in this case called Primer ID, to add a unique sequence tag to each starting genome (via its cDNA primer) allows an exact quantification of the number of genomes that have been sequenced with enhanced accuracy. This has allowed us to use the increase in pairwise diversity of the viral population over time as a molecular clock to infer the time since infection.
Recency is an important concept in monitoring the HIV-1 epidemic, as opposed to less informative “new diagnoses.” Being able to identify incident infections would allow a focus of prevention efforts where transmission events have recently occurred and may be ongoing. In the public health setting, knowing recency over a longer time frame than acute HIV-1 infection is more useful, because this approach will capture more transmission events beyond just the few with distinctive biological features that are present only over a short period of time (eg, antibody negative and/or high viremia). In the current study, we found that younger people (aged <30 years) were more likely than older people to have HIV diagnosed within this 9-month window. We did not find significant differences in recency of diagnosis by sex or known risk factor, apart from PWID status. Although a small number of samples were tested from PWID, and therefore the OR had a very large CI, such data could be used as a justification to increase monitoring and focus prevention efforts. Our platform confirmed a focal outbreak of HIV among PWID that was previously intensely investigated by the NC-DPH [21].
Recent infections in transmission clusters are highly relevant to public health in terms of targeted prevention [22]. We were able to define transmission clusters with recent infections, identifying significantly more recent infections in the clusters than the chronic infections. A previous study using a large set of pol sequences showed that people with acute or recent infections were more likely than those with chronic infections to be clustered together [23]. Although our sequenced regions were relatively short, the 1.2–kilobase pair pol sequence can still be integrated with pol gene databases collected for drug resistance testing [24] to study clusters on a much larger background, with annotated recency information provided by the MPID approach [2, 25, 26].
Transmitted DRMs has been extensively studied owing to clinical resistance testing done by bulk sequencing (ie, Sanger sequencing) to guide antiretroviral drug use when initiating therapy. DRMs are commonly acquired during treatment failure, and rates of transmitted drug resistance have been increasing over the years [27–30]. Deep sequencing has the potential to detect minor variants in the viral population within an individual, with the sensitivity of detection limited by the depth of sampling of viral genomes sequenced, which unfortunately is an uncontrolled variable in most sequencing applications. Efforts have been made in the past decade to study transmitted drug resistance, especially NGS analysis of minority mutations, but effects on treatment outcomes are still not fully understood [27, 31–36].
As noted above, an important advantage of the Primer ID approach is that it can be used to define the depth of sampling of the viral population. Using this approach, we found 2 distinct classes of DRMs. One class, with K103N in RT as an example, was present in a similar number of specimens regardless of the genome sampling depth (in approximately 15% of all specimens tested). This indicates that, when present, these mutations are at high abundance and are likely relatively stable over time. In contrast, there were more DRMs present at low abundance, such that the perception of how often they are present is strongly determined by the depth of sampling of the viral populations in the specimens. Thus, a group of mutations appear to be present in ≤1% of specimens when the sampling depth of the viral population is poor but can be detected in 10 times as many specimens when the true sampling depth allows detection of minor variants present at an abundance of 0.3%–1%. Of note, some of the APOBEC3g/f-related mutations, such as PR D30N, M46I, and RT G190E [37], are among those in this group.
The current study had several limitations and possible sources of bias. The NC-SLPH diagnoses only about one-third of new HIV cases in North Carolina, which might result in sampling bias of the population. Compared with the data on new diagnoses reported by North Carolina in 2018, the specimens tested in the state laboratory were more likely to be from African American/black individuals (72% vs 63%) and slightly less likely to be from women (16% vs 19%) [10]. Another current limitation of our approach is that some specimens (16%) yielded an indeterminate interpretation. Further bioinformatics algorithms need to be developed to make full use of the information in these indeterminate specimens.
In summary, our NGS/phylodynamics platform is a novel application to monitor the key HIV-1 epidemic components of recency, DRMs, and transmission clustering, all from a single sequencing reaction. This monitoring system is particularly important for high-burden countries, to monitor the percentage of recent infection in patients with newly diagnosed HIV over the years as an assessment of HIV control efforts.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
Notes
Acknowledgments. We thank the University of North Carolina Center for AIDS Research Clinical Pharmacology/Analytical Chemistry Core for performing the antiviral drug detection.
Financial support. This work is supported by the National Institutes of Health (grants NIH R01 AI135970 and NIH R01 AI140970, and grant P01 AI050410 to the University of North Carolina Center for AIDS Research Clinical Pharmacology/Analytical Chemistry Core.
Potential conflicts of interest. The University of North Carolina is pursuing intellectual property protection for Primer ID, and R. S. has received nominal royalties. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Centers for Disease Control and Prevention. HIV surveillance report 2017.https://www.cdc.gov/hiv/library/reports/hiv-surveillance.html. Accessed 24 July 2019.
- 2. Samoff E. HIV End the epidemic in the South: the importance of measuring progression to AIDS and death. Am J Public Health 2019; 109:1159–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trujillo L, Chapin-Bardales J, German EJ, Kanny D, Wejnert C;. National HIV Behavioral Surveillance Study Group . Trends in sexual risk behaviors among Hispanic/Latino men who have sex with men—19 urban areas, 2011–2017. MMWR Morb Mortal Wkly Rep 2019; 68:873–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Handel MM, Rose CE, Hallisey EJ, et al. County-level vulnerability assessment for rapid dissemination of HIV or HCV infections among persons who inject drugs, United States. J Acquir Immune Defic Syndr 2016; 73:323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lerner AM, Fauci AS. Opioid injection in rural areas of the United States: a potential obstacle to ending the HIV epidemic. JAMA 2019; 322:1041–2. [DOI] [PubMed] [Google Scholar]
- 6. Fauci AS, Redfield RR, Sigounas G, Weahkee MD, Giroir BP. Ending the HIV epidemic: a plan for the United States. JAMA 2019; 321:844–5. [DOI] [PubMed] [Google Scholar]
- 7. Dennis AM, Zhou S, Sellers CJ, et al. Using Primer-ID deep sequencing to detect recent human immunodeficiency virus type 1 infection. J Infect Dis 2018; 218:1777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou S, Jones C, Mieczkowski P, Swanstrom R. Primer ID validates template sampling depth and greatly reduces the error rate of next-generation sequencing of HIV-1 genomic RNA populations. J Virol 2015; 89:8540–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci U S A 2011; 108:20166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. HIV/STD/Hepatitis Surveillance Unit, Division of Public Health, North Carolina Department of Health and Human Services. 2018 North Carolina HIV surveillance report. 2019. https://epi.dph.ncdhhs.gov/cd/stds/figures/hiv18rpt_02042020.pdf. Accessed 19 March 2020. [Google Scholar]
- 11. Menza TW, Billock R, Samoff E, Eron JJ, Dennis AM. Pretreatment integrase strand transfer inhibitor resistance in North Carolina from 2010–2016. AIDS 2017; 31:2235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wensing AM, Calvez V, Gunthard HF, et al. 2017Update of the drug resistance mutations in HIV-1. Top Antivir Med 2017; 24:132–3. [PMC free article] [PubMed] [Google Scholar]
- 13. Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004; 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu K, Linder CR, Warnow T. RAxML and FastTree: comparing two methods for large-scale maximum likelihood phylogeny estimation. PLoS One 2011; 6:e27731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moyo S, Wilkinson E, Novitsky V, et al. Identifying recent HIV infections: from serological assays to genomics. Viruses 2015; 7:5508–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Puller V, Neher R, Albert J. Estimating time of HIV-1 infection from next-generation sequence diversity. PLoS Comput Biol 2017; 13:e1005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carlisle LA, Turk T, Kusejko K, et al. ; Swiss HIV Cohort Study . Viral diversity based on next-generation sequencing of HIV-1 provides precise estimates of infection recency and time since infection. J Infect Dis 2019; 220:254–65. [DOI] [PubMed] [Google Scholar]
- 20. Kafando A, Fournier E, Serhir B, et al. HIV-1 envelope sequence-based diversity measures for identifying recent infections. PLoS One 2017; 12:e0189999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Samoff E, Mobley V, Hudgins M, et al. HIV outbreak control with effective access to care and harm reduction in North Carolina, 2017–2018. Am J Public Health 2020; 110:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Truong HM, Pipkin S, OʼKeefe KJ, et al. Brief report: recent infection, sexually transmitted infections, and transmission clusters frequently observed among persons newly diagnosed with HIV in San Francisco. J Acquir Immune Defic Syndr 2015; 69:606–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Volz EM, Koopman JS, Ward MJ, Brown AL, Frost SD. Simple epidemiological dynamics explain phylogenetic clustering of HIV from patients with recent infection. PLoS Comput Biol 2012; 8:e1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Billock RM, Powers KA, Pasquale DK, et al. Prediction of HIV transmission cluster growth with statewide surveillance data. J Acquir Immune Defic Syndr 2019; 80:152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Evans ME, Labuda SM, Hogan V, et al. Notes from the field: HIV infection investigation in a rural area—West Virginia, 2017. MMWR Morb Mortal Wkly Rep 2018; 67:257–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peters PJ, Pontones P, Hoover KW, et al. HIV infection linked to injection use of oxymorphone in Indiana, 2014–2015. N Engl J Med 2016; 375:229–39. [DOI] [PubMed] [Google Scholar]
- 27. Clutter DS, Jordan MR, Bertagnolio S, Shafer RW. HIV-1 drug resistance and resistance testing. Infect Genet Evol 2016; 46:292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rhee SY, Blanco JL, Jordan MR, et al. Geographic and temporal trends in the molecular epidemiology and genetic mechanisms of transmitted HIV-1 drug resistance: an individual-patient- and sequence-level meta-analysis. PLoS Med 2015; 12:e1001810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. TenoRes Study Group. Global epidemiology of drug resistance after failure of WHO recommended first-line regimens for adult HIV-1 infection: a multicentre retrospective cohort study. Lancet Infect Dis 2016; 16:565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gupta RK, Gregson J, Parkin N, et al. HIV-1 drug resistance before initiation or re-initiation of first-line antiretroviral therapy in low-income and middle-income countries: a systematic review and meta-regression analysis. Lancet Infect Dis 2018; 18:346–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Keys JR, Zhou S, Anderson JA, et al. Primer ID informs next-generation sequencing platforms and reveals preexisting drug resistance mutations in the HIV-1 reverse transcriptase coding domain. AIDS Res Hum Retroviruses 2015; 31:658–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Telele NF, Kalu AW, Gebre-Selassie S, et al. Pretreatment drug resistance in a large countrywide Ethiopian HIV-1C cohort: a comparison of Sanger and high-throughput sequencing. Sci Rep 2018; 8:7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perrier M, Visseaux B, Landman R, et al. No impact of HIV-1 protease minority resistant variants on the virological response to a first-line PI-based regimen containing darunavir or atazanavir. J Antimicrob Chemother 2018; 73:173–6. [DOI] [PubMed] [Google Scholar]
- 34. Weis JF, Baeten JM, McCoy CO, et al. ; Partners PrEP Study Team . Preexposure prophylaxis-selected drug resistance decays rapidly after drug cessation. AIDS 2016; 30:31–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paredes R, Lalama CM, Ribaudo HJ, et al. ; AIDS Clinical Trials Group (ACTG) A5095 Study Team . Pre-existing minority drug-resistant HIV-1 variants, adherence, and risk of antiretroviral treatment failure. J Infect Dis 2010; 201:662–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li JZ, Paredes R, Ribaudo HJ, et al. Low-frequency HIV-1 drug resistance mutations and risk of NNRTI-based antiretroviral treatment failure: a systematic review and pooled analysis. JAMA 2011; 305:1327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rhee SY, Sankaran K, Varghese V, et al. HIV-1 protease, reverse transcriptase, and integrase variation. J Virol 2016; 90:6058–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.