

Abstract
Autotaxin (ATX, aka. ENPP2) is the main source of the lipid mediator lysophosphatidic acid (LPA) in biological fluids. This study reports on inhibitors of ATX derived by lead optimization of the benzene-sulfonamide in silico hit compound 3. The new analogues provide a comprehensive structure–activity relationship of the benzene-sulfonamide scaffold that yielded a series of highly potent ATX inhibitors. The three most potent analogues (3a, IC50 ~ 32 nM; 3b, IC50 ~ 9 nM; and 14, IC50 ~ 35 nM) inhibit ATX-dependent invasion of A2058 human melanoma cells in vitro. Two of the most potent compounds, 3b and 3f (IC50 ~ 84 nM), lack inhibitory action on ENPP6 and ENPP7 but possess weak antagonist action specific to the LPA1 G protein-coupled receptor. In particular, compound 3b potently reduced in vitro chemotherapeutic resistance of 4T1 breast cancer stem-like cells to paclitaxel and significantly reduced B16 melanoma metastasis in vivo.
Graphical Abstract

INTRODUCTION
Autotaxin (ATX), a member of the ectonucleotide pyrophosphate and phosphatase (ENPP) family, is primarily known to catalyze the hydrolysis of lysophosphatidylcholine (LPC), resulting in the production of the growth-factor-like bioactive phospholipid lysophosphatidic acid (LPA).1–3 LPA activates a set of six G protein-coupled receptors (GPCRs) LPA1–6, ion channels, and the transcription factor PPARγ, which regulate a host of cellular responses, including cell proliferation and migration, vascular tone, and platelet aggregation.4,5 Many of these responses are at the core of human diseases that include fibrotic diseases, neuropathic pain, and rheumatoid arthritis as well as cardiovascular diseases.1,4–6 In recent years, the ATX–LPA signaling pathway has been linked to several aspects of cancer cell biology that include cancer growth, invasion, metastasis, and therapeutic resistance.6–8 Recently, ATX has been found to play an important role in the maintenance and proliferation of ovarian cancer stem cells.9 Because of the explicit role of LPA in these human pathologies, development of drug-like ATX inhibitors has begun.1,3,10–15 These synthetic efforts have resulted in a number of novel ATX inhibitors that can arbitrarily be divided into two categories: lipid-like ATX inhibitors, which mimic either LPC or LPA, and non-lipid ATX inhibitors.1–3,7,11,12,14,16–20 Despite these efforts, limited success has been achieved in preclinical and regulatory development of ATX inhibitor drug candidates.1 In this context, the high partition coefficient (Log P > 5) renders most lipid-like inhibitors unsuitable for potential clinical development.2,7,16 The elucidation of ATX crystal structure with the inhibitor cocrystallized in the active site provided insight into the potential active site surfaces.10 We and others pursued rational discovery that yielded a new generation of small molecule drug-like non-lipid ATX inhibitors with pharmacological properties that meet Lippinski’s rule of five.1,6,16,18 A subgroup of recently developed non-lipid ATX inhibitors, including (Z)-4-[(4-{[3-(4-fluorobenzyl)-2,4-dioxo-1,3-thiazolan-5-yliden]methyl}phenoxy) methyl]phenyl boronic acid (HA 155)10 and 3,5-dichlorobenzyl-[4-[3-oxo-3-(2-oxo-2,3-dihydrobenzoxazol-6-yl)propyl]]-piperazine-1-carboxylate (PF 8380),23 compounds reported by pharmaceutical companies Novartis and PharmAkea, have similar structural features and contain an acid or acid-like moiety (for interacting with one of the Zn2+ ions at the catalytic site), a core spacer, and a hydrophobic tail accommodated within the hydrophobic pocket of ATX.1–3,12,21–24 However, to the best of our knowledge, none of the active site inhibitors has successfully made it through clinical trials yet. Thus, recent approaches have focused on developing small molecule non-lipid ATX inhibitors without an acidic moiety (Figure 1), allosteric modulators of the enzyme,15 and DNA aptamers.25 Our group utilized high-throughput and in silico searching to identify a small molecule ATX inhibitor, 2,4-dichloro-N-(3-fluorophenyl)-5-(morpholinosulfonyl)benzamide [GRI918013 (3), Figure 1A], which exerts its inhibition by binding to the hydrophobic pocket remote from the catalytic site and lacks an acidic moiety.2,16 This chemical entity, even without an acidic head group, shows the same pharmacological and biological effects as catalytic site inhibitors, but inhibition is mediated via interference with the binding of the hydrophobic chain of the lipid substrates, although it lacks sufficient potency and stability in vivo.2,16 Recently, Galapagos Inc. has developed a number of potent compounds that have a major structural similarity to 3, lacking the acidic group (Figure 1).3 One of the Galapagos compounds has been reported to successfully pass the first in-human phase one clinical trial. Thus, the objectives of our synthetic campaign were (1) to improve the potency of ATX inhibition with IC50 values in the low nanomolar range and (2) to obtain compounds that were effective in in vitro and in vivo assays dependent on ATX activity. Our lead optimization set four structural criteria for the novel compounds: (1) lack of an acidic moiety, (2) lack of a chiral center, (3) molecular weight (MW) less than 500 Da, and (4) partition coefficients (Log P) less than 3. Here, we report on the structure–activity relationship (SAR) of several modifications of hit 3 that yielded two novel compounds with potent in vitro and in vivo activities, namely, blocking B16 melanoma invasion and metastasis and reducing chemotherapeutic resistance of 4T1 breast cancer stem-like cells to paclitaxel.
Figure 1.

(A) Examples of non-acidic headgroup non-lipid ATX inhibitors.1–3 Galapagos 2015 is one of the non-carboxylic acid autotaxin inhibitors developed and reported by Galapagos Inc. in 2015.3 (B) Position of top-ranked pose of 3 (space-filling model) in the mouse ATX crystal structure (PDB 3NKM) represented as a ribbon shaded from blue at the N-terminus to red at the C-terminus. The position of 3 is outside of the catalytic core of the ATX active site (enclosed in the dotted magenta circle). Catalytic site metal ions are shown as cyan spheres. The additional outlined areas are the hydrophobic pocket (orange) and the hydrophobic tunnel (green).
RESULTS
Chemical Synthesis.
Modification of Ring A.
We designated the three rings in 3 as rings A, B, and C (Figure 1A). Our previous screening experiments, mutagenesis, and molecular modeling findings suggested that 3 binds into the hydrophobic pocket of ATX without protruding into and blocking substrate access to the catalytic site as shown in Figure 1B. First, we designed and synthesized a series of derivatives with varying substituents on ring A as shown in Scheme 1.
Scheme 1. Design and Synthesis of Ring A-Modified Benzene Sulfonamide Analoguesa.

aPercent yields are shown in parentheses. (i) CISO3H, 12 h reflux; (ii) morpholine, Et3N, RT, 12 h; (iii) (a) SOCI2, DMF (cat), 12 h, RT, (b) substituted-aniline pyridine, 12 h, RT; (iv) LiOH, MeOH/THF.
Compound 3 was synthesized following the strategy described in Scheme 1 with 70% yield. We first increased the number of fluorine substituents on ring A by synthesizing the 3,4-difluoro analogue (3a) and the 3,4,5-trifluoro analogue (3b) depicted in Scheme 1 and tested them for ATX inhibitory activity (Table 1). The simple manipulation of aromatic substituents resulted in 4-fold IC50 improvements in 3a (IC50 = 31 nM) and 13-fold in 3b (IC50 = 9 nM) relative to the IC50= 120 nM parent compound. When 3b was docked in to the ATX crystal structure (PDB 3NKM),22 we found that three fluoro substituents promote a tighter binding of this compound over 3 (Figure 2). Top poses of these two compounds show strong volume overlap except in the angle of the halogenated ring A. 3 makes a largely edge-to-face π interaction with residue W260, whereas 3b has a rotated aromatic ring that allows a hydrogen bond to form between a fluorine substituent and aromatic N–H of W260 (Figure 3A). We continued this series by making a penta-fluoro analogue (3c) that was poorly active. Next, we synthesized and tested a relatively less electron-deficient 3,4,5-trichloro analogue (3d) and an electron-donating 3,4,5-trimethoxy analogue (3e), both of which were predicted to be sterically more congested than trifluoro analogue 3b in modeling studies. These two analogues were poorly active, suggesting increased steric congestion by ring A, which did not allow the molecule to be properly accommodated into the hydrophobic pocket. Compound 3d was also docked into the ATX crystal structure, and a dramatically different orientation was observed in concert with our experimental findings (Figure 3B). Top poses of 3b and 3d showed good volume overlap, but 3d ran in the opposite direction and central aromatic ring B was twisted completely out of conjugation with the amide linker. This represents a very high energy conformation and is unlikely to bind because of the conformational energy penalty. Next, we tested the effects of combining two different kinds of ring A substituents on the inhibitory activity of the molecule. We synthesized the 3,5-difluoro-4-chloro analogue (3f) and the 3,5-dichloro-4-fluoro analogue (3g) and tested them for the ATX inhibition. Both analogues were highly potent ATX inhibitors with IC50 values of 83 and 40 nM, respectively. When docked to ATX, 3f and 3b adopted similar poses, with the electronegative chlorine in 3f and the fluorine in 3b exposed to water in the hydrophobic tunnel (Figure 3C). However, 3g (not shown) adopted a completely different pose in which the halogenated aromatic A ring was in the hydrophobic pocket, not in the hydrophobic tunnel, whereas the polar ends of the molecules overlapped, although they ran in opposite directions. Distances between the chlorine atoms of 3g in the hydrophobic pocket predicted a water-mediated hydrogen bond with backbone atoms of L214 or A218 and a weak hydrogen bond with W276, which can be the reason for the 2-fold higher potency of 3g over 3f.
Table 1.
Structure–Activity Relationship of the New ATX Inhibitorsa
| compd | IC50(nM ± SD) vs FS-3 | pNP-TMP inhibition | mechanism | Log P | MW |
|---|---|---|---|---|---|
| 3 | 117 ± 2 | none | competitive | 2.99 | 433.28 |
| 3a | 32 ± 2 | none | competitive | 3.17 | 451.27 |
| 3b | 9 ± 0.2 | none | competitive | 3.23 | 469.26 |
| 3c | 1034 ± 120 | none | ND | 3.67 | 505.24 |
| 3d | >1000 | none | ND | 4.35 | 518.63 |
| 3e | >1000 | none | ND | 2.91 | 505.36 |
| 3f | 84 ± 0.1 | none | competitive | 3.62 | 485.72 |
| 3g | 40 ± 0.3 | none | competitive | 3.89 | 502.17 |
| 3h | 190 ± 13 | none | ND | 2.46 | 476.28 |
| 3i | 864 ± 52 | none | ND | 2.78 | 509.31 |
| 4 | >1000 | none | ND | 2.26 | 476.83 |
| 5 | 18 ± 2 | none | ND | 3.71 | 548.16 |
| 7 | >1000 | none | ND | 2.74 | 431.26 |
| 8 | >1000 | none | ND | 3.42 | 448.27 |
| 9 | >1000 | none | ND | 2.56 | 447.31 |
| 10 | >1000 | none | ND | 3.92 | 437.29 |
| 11 | 396 ±44 | none | ND | 3.98 | 455.28 |
| 12 | 121 ± 1 | none | ND | 3.57 | 449.35 |
| 13 | 70 ± 1 | none | ND | 3.72 | 467.34 |
| 14 | 35 ± 3 | none | competitive | 3.87 | 485.33 |
| 15 | 98 ± 7 | none | ND | 4.95 | 518.24 |
| 19 | >1000 | none | ND | 1.92 | 469.33 |
| 20 | >1000 | none | ND | 2.65 | 627.48 |
| 22 | >1000 | none | ND | 1.91 | 469.33 |
| 23 | >1000 | none | ND | 2.06 | 505.32 |
| 25 | >1000 | none | ND | 3.49 | 420.21 |
| 26a | >1000 | none | ND | 2.83 | 498.30 |
| 28a | 67 ± 7.1 | none | ND | 3.23 | 482.30 |
| 28b | 56 ±9 | none | ND | 3.53 | 515.21 |
| 29 | 100 ±8 | none | ND | 4.38 | 531.28 |
ND = not determined. Log P was calculated using Schrödinger Molecular Modeling Suite (Schrödinger LLC, New York).
Figure 2.

Binding poses (low energy conformation) of 3 (dark gray carbon atoms) and 3b (light gray carbon atoms) in the ATX crystal structure (PDB 3NKM). Distance measured between a fluorine atom of 3b and the aromatic amine hydrogen of W260 in ATX is shown in green.
Figure 3.

Molecular models of ATX inhibitors docked into the ATX crystal structure (PDB 3NKM). Superpositions of compound 3b (light gray carbons) with 3 (dark gray carbons) (A), 3d (black carbons) (B), and 3f (orange carbons) (C) explain the observed potency differences. Panel A displays the hydrogen bond formed between the 3-fluoro group of trifluorinated compound 3b (magenta dotted line), which is not formed with the 3-fluoro group of the less electron-rich monofluorinated compound 3. The additional hydrogen bond formed by 3b is consistent with the more potent inhibition of ATX observed for 3b relative to the parent compound. Panel B illustrates the inability of trichlorinated compound 3d to adopt a pose similar to that seen for trifluorinated compound 3b. The larger size of the three chlorine substituents of 3d is not accommodated within the hydrophobic tunnel at the lower left corner, and the best-ranked pose has a poorly conjugated linkage between the central aryl group and the amide linker in the region encircled in the magenta dotted line (98° torsion angle relative to preferred angle in isolation of 40°). The pose of 3b exhibits much lower conformational distortion, with a 48° torsion angle for the corresponding bond. The difference in energy between the docked conformation of 3d and the closest local minimum energy (both in the absence of the protein and computed using the MMFF94 force field)39 is over 15 kcal/mol. The difference for 3b is less than 3 kcal/mol. Thus, 3d has a 12 kcal/mol greater conformational energy penalty than 3b. Panel C shows excellent superposition of the trihalogenated aromatic rings of 3b and 3f, with the 4-halo substituent exposed to the aqueous solution that would fill the remainder of the hydrophobic tunnel. The larger and less electronegative chlorine atom has sufficient space in the 4-position to allow adoption of a similar binding pose, but it lowers the potency due to a lower preference for aqueous exposure relative to the 4-fluoro substituent of 3b.
In an attempt to introduce more polar substituents into ring A, we prepared the 3,5-difluoro-4-cyano analogue (3h) and the 3,5-difluoro 4-methyl ester analogue (3i). ATX activity measurements with these compounds showed that 3h, although modestly tolerated, 3i was much less potent than 3 with IC50 values of 190 and 863 nM, respectively. Next, we hydrolyzed the ester bond in 3i to the corresponding carboxylic acid and found that this new compound (4) was inactive.
Modifications of the Linker between Rings A and B.
Subsequent synthetic modifications were aimed at the replacement of the amide bond of 3 to explore the importance of the amide linker in ATX inhibition and to improve biological stability to amidases. To accomplish this, we first tried to synthesize the trifluoro benzoxazole (6) from intermediate 5 (Scheme 2). Although intermediate 5 was obtained in good yield (76%) following the same strategy as shown in Scheme 1, the synthetic procedure failed to turn intermediate 5 into desired oxazole 6. An alternative strategy was applied to generate oxazole 7 and imidazole 8 using commercially available amino-phenol and amino-aniline precursors, respectively (Scheme 2). We also synthesized the N-methyl analogue (9). The four compounds (5, 7, 8, and 9) obtained with the synthetic route shown in Scheme 2 were tested for ATX inhibition (Table 1). Compound 5 showed potent ATX inhibition (IC50 = 18 ± 2 nM), although it was less potent than the trifluoro compound 3b. Compounds 7 and 8 closely mimic the amide framework, yet they failed to inhibit ATX. It is noteworthy that the A and B aromatic rings in compounds 7 and 8 were predicted to be closer to planar than in the compounds with amide linkers. Additionally, compound 9 showed only very weak potency inhibiting ATX. We next examined the role of the carbonyl oxygen/carbonyl bond for ATX inhibitory activity because the SAR of the analogues we synthesized to this point indicated that the orientation of ring A relative to ring B around the amide linkage is critical for the activity. To achieve this, we reduced amide bonds of the two most active ATX inhibitors, 3a and 3b (Scheme 3). Desired amines 10 and 11 were obtained in good yields and were subjected to ATX inhibition analysis to find that the inhibitory activity of the 3,4,5-trifluoro amine (11) was dramatically reduced relative to 3b (IC50 = 395 versus 9 nM; Table 1). Difluoro amine 10 was inactive.
Scheme 2. Design and Synthesis of Amide Bond-Modified Benzene Sulfonamide Analoguesa.

a(i) (a) SOCI2, DMF (cat), (b) substituted aniline, pyridine; (ii) CuI (cat), 1,10-phenanthroline (cat), Cs2C03, 2 h reflux; (iii) (a) SOCI2, DMF (cat), 12 h, RT, (b) substituted aniline, Et3N, (c) p-TSA, H2O, toluene, reflux; (iv) SOCI2, DMF (cat), 12 h, RT, (b) substituted aniline, Et3N, (c) POCI3, 100 min reflux; (v) (a) SOCI2, DMF (cat), (b) N-methyl-substituted aniline, pyridine
Scheme 3. Design and Synthesis of Reduced Amide Benzene Sulfonamide Analoguesa.

a(i) BH3-DMS, THF (sol), reflux 24 to 72 h.
In the next phase of our SAR analysis, we prepared compounds with thioamide linkers because of their higher stability against proteases compared to amides. We selected the four most potent inhibitors and synthesized the corresponding thioamides utilizing the synthetic strategy shown in Scheme 4.26 ATX inhibition assays showed that compounds 12, 13, 14, and 15 retained similar inhibitory activity as the amide analogues (Table 1). We also replaced the amide linker with a sulfonamide (Scheme 5).27–30 Intermediate 18 was obtained in good yield and was converted into two different kinds of sulfonamides. Compounds 19 and 20 have the sulfonamide nitrogen linked to the ring B, whereas in compounds 22 and 23 the sulfonamide nitrogen is linked to ring A. All four compounds lost their ATX inhibitory activity, as shown in Table 1. These results underline the importance of linker geometry for ATX inhibition.
Scheme 4. Design and Synthesis of Thioamide Benzene Sulfonamide Analoguesa.

a(i) Lawesson’s reagent, toluene (sol), reflux.
Scheme 5. Design and Synthesis of Disulfonamide Analoguesa.

a(i) CISO3H, reflux, 12 h; (ii) morpholine, Et3N, RT, 12 h; (iii) iron powder, HCI, reflux, 5 h; (iv) substituted benzenesulfonyl chloride, Et3N, DMAP, 96 h; (v) NaNO2, HCI, 0 °C, SO2, CH3CO2H; (vi) 3-fluoroaniline, Et3N; (vii) 3,4,5-trifluoro aniline, Et3N.
Modifications of the Linker between Rings B and C.
Because these findings identified the importance of the amide bond linkage for the inhibitory activity, we decided to further explore variations of the sulfonamide linkage between rings B and C to assess the role of the sulfonyl group. First, we replaced the sulfonamide linkage with a polar but smaller linkage, keeping the morpholine ring intact. Additionally, we avoided the introduction of chirality in linking ring B to C. We constructed the hydrazine moiety, where the sulfonyl is replaced by a nitrogen atom as shown in Scheme 6. Compound 25 was obtained through intermediate 24 using the Buchwald–Hartwig cross-coupling reaction following the synthetic tools described by Cacchi et al.31 Compound 25 completely lost ATX inhibitory activity (Table 1). This result confirms that the sulfonyl linkage provides a better contact with the polar surface of the ATX substrate binding pocket and forms some electrostatic interactions to promote strong binding. We hypothesized that introduction of a mesyl group (26) on the hydrazine in compound 25 could be as effective as compound 3b in contacting the polar enzyme surface. Many attempts to obtain 26b remained unsuccessful.32–34 However, utilizing excess sodium tert-butoxide and a 1:11 ratio of methane sulfonyl chloride, compound 26a was obtained. Testing revealed that compound 26a was inactive, once again reinforcing the importance of the linker geometry. We also replaced ring C with N-methyl piperazine, as shown in Scheme 7. The rationale behind this modification was that it would be more water soluble than the morpholine analogues. We selected the two most potent morpholine analogues, 3b and 3g, and prepared their N-methyl piperazine analogues, 28a and 28b, following the synthetic strategy shown in Scheme 7. Analogues 28a and 28b were tested for ATX inhibition and found to inhibit ATX with IC50 values of 66 and 55 nM, respectively (Table 1). Finally, we selected the more potent analogue, 28b, and converted it into the corresponding thioamide (29), which showed a potency of IC50 = 100 nM.
Scheme 6. Design and Synthesis of Hydrazine Analoguesa.

a(i) (a) SOCI2, DMF(cat), 12 h, RT, (b) 3,4,5-trifluoro-aniline, pyridine, 12 h, RT; (ii) 4-aminomorpholine, LiCI, Pd2(dba)3, Xantphos, NaOtBu, 12 h, reflux; (iii) NaOtBu, methanesulfonyl chloride, 24 h, RT; (iv) N-morpholinomethanesulfonamide, [Pd(allyl)CI]2, t-BuXphos, K2C03.
Scheme 7. Design and Synthesis of Piperazine Analoguesa.

a(i) ClSO3H, 12 h, reflux; (ii) N-methylpiperazine, Et3N, RT, 12 h; (iii) (a) SOCl2, DMF (cat), 12 h, RT, (b) substituted aniline, pyridine, 12 h, RT; (iv) Lawesson’s reagent.
Pharmacological Characterization of the Benzenesulfonamide ATX Inhibitors.
ATX has dual nucleotide pyrophosphate phosphatase and lysophosholipase D activities that can be selectively measured using the pNP-TMP and FS-3 substrates, respectively.2,13,16 The inhibitory activities of the new compounds against the two different substrates are listed in Table 1. Our primary test was to assess the inhibitory action of the new compounds on the lysophospholipase D activity of purified recombinant human ATX. Compound 3b was found to be the most potent ATX inhibitor, with an IC50 of ~9 nM in this assay. The mechanism of action was competitive; however, the traditional measure of a competitive inhibitor may not be appropriately applied in this context. 3b inhibited hydrolysis of the lipid-like substrate FS-3 while sparing effects on hydrolysis of the nucleotide substrate pNP-TMP. Molecular modeling predicted that 3b does not bind at the active site, in which case both substrates would be affected. However, because the compound is targeted toward the hydrophobic pocket of ATX, it shares space with the binding site for the hydrophobic tail of lipid-like substrates like FS-3. As such, the mechanism of action presents itself as “competitive” in the FS-3 hydrolysis assay even though the binding site of the 3b exists outside the catalytic site of the enzyme. This particular behavior of ATX inhibitors has been noted before.16
Selective Dose-Dependent Inhibition of LPA1 GPCR by Compounds 3b, 3f, and 3g.
The product of ATX (LPA) is ligand to at least six GPCRs, and the benzenesulfonamide compounds we have generated in this study could also possess some ligand activity at the different LPAR subtypes. For this reason, we tested all new chemical entities with useful ATX inhibitory potency (<250 nM) for agonist and antagonist activities at LPA1/2/3/4/5 using cells stably transfected with each individual GPCR.35 None of the compounds tested exhibited agonist activity at any of these LPA receptor subtypes. Additionally, of those screened, only 3b, 3f, and 3g exhibited antagonist activity, and this inhibition was specific to the LPA1 receptor (Table 2, Figure 4). Compounds 3b (ATX IC50 = 9 nM, LPA1 IC50 ~ 14 μM), 3f (ATX IC50 = 83 nM, LPA1 IC50 ~ 6 μM), and 3g (ATX IC50 = 40 nM, LPA1 IC50 = 18 μM) were dual inhibitors of both ATX and the LPA1 receptor.
Table 2.
Effect of ATX Inhibitors on LPA-Mediated Ca2+ Mobilizationa
| LPA1 | LPA2 | LPA3 | LPA4 | LPA5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 or IC50 (μM) | Emax | EC50 or IC50 (μM) | Emax | EC50 or IC50 (μM) | Emax | EC50 or IC50 (μM) | Emax | EC50 or IC50 (μM) | μEmax | |
| LPA 18:1 | 0.08 ± 0.01 | 100.00 ± 0.00 | 0.003 ± 0.00 | 100.00 ± 0.00 | 0.29 ± 0.01 | 100.00 ± 0.00 | 0.57 ± 0.01 | 100.00 ± 0.00 | 0.14 ± 0.00 | 100.00 ± 0.00 |
| LPA + 3b | 14.47 ± 2.75 | 46.14 ± 15.54 | NE | NE | NE | NE | NE | NE | NE | NE |
| LPA + 3f | 6.12 ± 2.22 | 51.03 ± 13.99 | NE | NE | NE | NE | NE | NE | NE | NE |
| LPA + 3g | 18.39 ± 7.44 | 50.04 ± 13.04 | NE | NE | NE | NE | NE | NE | NE | NE |
| LPA + 14 | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE |
| LPA + 5 | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE |
NE = no effect detected.
Figure 4.

Effect of ATX inhibitors on LPA-activated Ca2+ responses mediated by LPA1 RH7777 cells. All measurements were done in quadruplicates, and data represent the mean ± SD.
Effect of Benzenesulfonamide ATX Inhibitors on Other Members of the ENPP Family.
ATX constitutes one of three members of the ENPP family, composed also of ENPP6 and ENPP7. Both ENPP6 and ENPP7 retain similar phosphodiesterase activity to ATX but lack its hydrophobic pocket. To test the inhibitory effects of benzenesulfonamide ATX inhibitors versus purified recombinant ENPP6 and ENPP7, the nucleotide substrate p-nitrophenylphosphorylcholine (pNPPC) was used. None of the compounds tested exhibited inhibitory activity for ENPP6 or ENPP7 (data not shown) as the benzenesulfonamide structures are targeted to the hydrophobic pocket of ATX, which is not found in either of the other ENPP family members.
Inhibition of ATX-Mediated Invasion of A2058 Human Melanoma in Vitro.
We utilized the Boyden chamber assay to assess the effect of benzenesulfonamide analogues 3a, 3b, 3f, and 14 on invasion of A2058 human melanoma cells in vitro. A2058 cells invade the matrigel layer in an LPA-dependent manner. Therefore, we used exogenous LPC and recombinant ATX as a source of LPA. First, we screened the compounds at 10 μM concentration alongside (4-pentadecylbenzyl)phosphonic acid [30 (BMP22)],13 a potent ATX inhibitor that we have previously identified and characterized.13 We found that all four inhibitors were as effective as 30 in inhibiting A2058 cell invasion across the matrigel layer in response to exogenous LPC and recombinant ATX (Figure 5A). Subsequently, we generated dose–response curves for compounds 3a, 3b, 3f, and 14 (Figure 5B). All four inhibitors dose dependently decreased A2058 cell invasion with similar potencies (Table 3).
Figure 5.

Characterization of ATX inhibitors on A2058 human melanoma invasion. (A) Effect of the various ATX inhibitors on A2058 cell invasion. (B) Dose–response curves were generated in the presence of exogenous LPC, recombinant ATX, and increasing concentrations of inhibitors as indicated. All measurements were done in triplicates, and the data represent the mean ± SD of an experiment performed three times. **p < 0.001 and *p < 0.0001 relative to LPC plus ATX treatment based on one-way ANOVA analysis followed by Bonferroni post-test.
Table 3.
IC50 Values of Compounds 3b, 3a, and 14 in the A2058 Cell Invasion Assay
| compd | IC50 (nM) |
|---|---|
| 3b | S28 ± 14 |
| 3a | 1478 ± 12 |
| 3f | 740.4 ± 13 |
| 14 | 403 + 13 |
In Vitro Stability Analysis.
The lead 3, 3b, 3f, and 14 were analyzed for stability in the presence of cells in culture using liquid chromatography mass spectrometry. All analogues tested showed significant stability over the 40 h of exposure to 4T1 cells in serum-free culture medium. The lead, 3, maintained 79.7 ± 7.8% (16 h) and 79.5 ± 12.6% (40 h) of the 0 h control signal, whereas the polyhalogenated amide and thioamide analogues appear to be a bit more stable in this system over this limited time course (3b maintained 102.6 ± 2.6% (16 h) and 105.3 ± 2.7% (40 h), 3f maintained 101.3 ± 3.7% (16 h) and 108.2 ± 1.3%, and thioamide 14 maintained 93.2 ± 20.4% (16 h) and 102.0 ± 14.5% (40 h) of their corresponding 0 h controls).
Inhibition of B16 Murine Melanoma Lung Metastasis by Benzenesulfonamide ATX Inhibitors.
To complement our in vitro invasion studies, we tested the effect of 3b and 14 in the B16 lung metastasis in vivo model. We found that daily administration of compounds 3b and 14 for a total of 10 days significantly reduced the number of metastatic lung nodules formed in mice (Figure 6).
Figure 6.

Effect of the various ATX inhibitors on the lung metastasis of B16F10 melanoma cells in C57BL/6 mice. Mice were treated daily with 1 mg/kg of compound for up to 10 days. All mice were sacrificed at day 21, and the number of metastatic lung nodules was counted. Differences between groups were determined by one-way ANOVA followed by Bonferroni post-test.
Sensitization of Therapy-Resistant 4T1 Murine Breast Carcinoma Cells and Stem Cell-Like Cells (CSC) to Paclitaxel by Benzenesulfonamide ATX Inhibitors.
To further characterize the three most potent compounds, we tested them on the viability and growth of 4T1 murine breast cancer cells (Figure 7). Adherent cultures of 4T1 cells were treated for 24 h with a range of 0–10 μM compounds 3b, 3f, or 14 or reference inhibitor 30 in RPMI with 1% (v/v) charcoal-stripped FBS. After 48 h treatment with daily replenishment of inhibitor, cell number/viability was assessed (Figure 7A). Only 30 inhibited cell growth at 3 μM, whereas all other compounds failed to cause a significant reduction in cell number. However, unlike the other compounds, 14 was toxic at 10 μM, the highest concentration tested.
Figure 7.
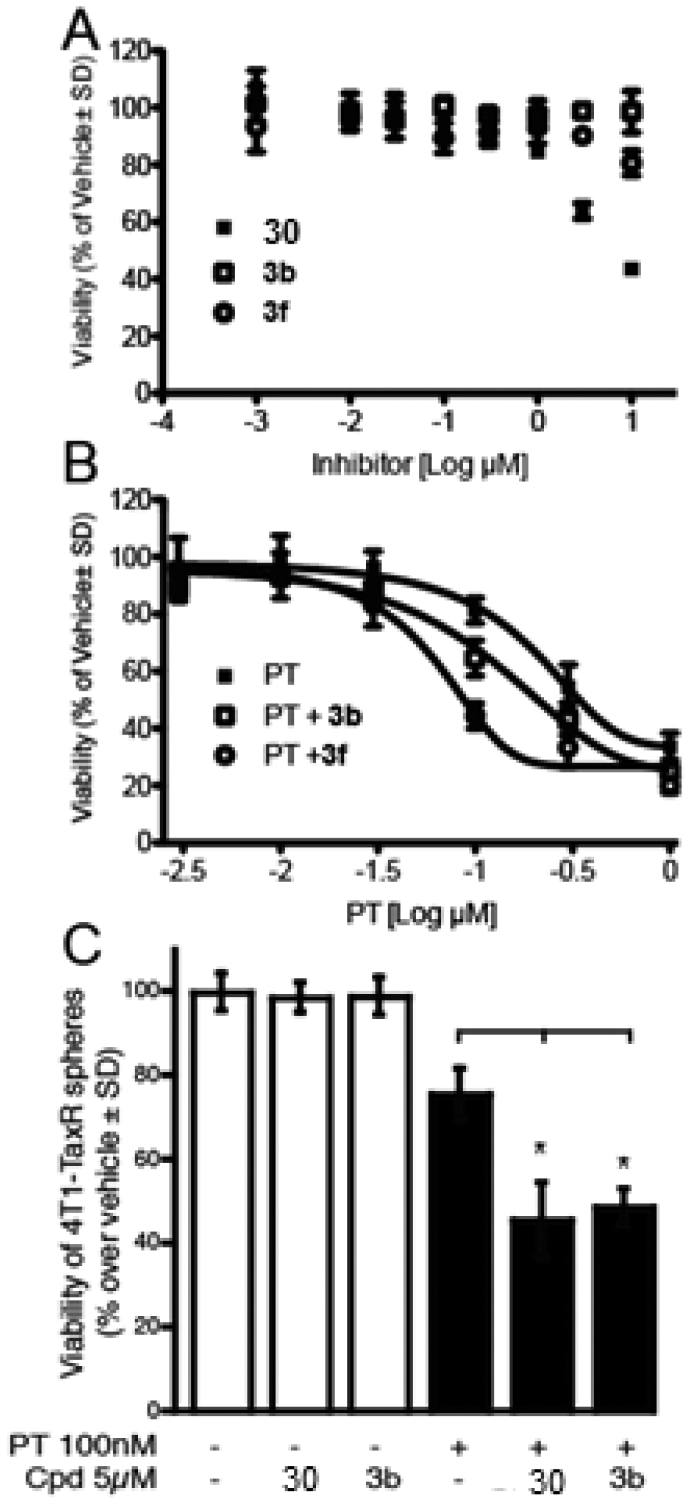
Effect of ATX inhibitors on the therapeutic resistance of 4T1 breast carcinoma cells and cancer stem-like cells. (A) The rate of growth of 4T1 breast carcinoma cells grown in adherent cultures is unaffected by ATX inhibitor 30 and compounds 3b and 3f. (B) ATX inhibitors (3 μM) when added together with PT shift the dose–response curve of cell killing to the left, indicating that these inhibitors make 4T1 cells more sensitive to PT-induced death. (C) Mammospheres were generated from the 4T1-TaxR cell line and treated with PT at 100 nM with or without the presence of 30 and compound 3b at 5 μM, respectively. All measurements were done in quadruplicates, and data represent the mean ± SD of an experiment performed three times. * p < 0.05 relative to PT alone based on one-way ANOVA followed by Bonferroni post-test.
LPA and ATX increase the resistance of cancer cells to chemotherapy and radiation therapy.6 To assess whether the new ATX inhibitors could overcome chemoresistance, the 4T1 breast cancer cell line was exposed to increasing concentrations of paclitaxel (PT) for 48 h with or without a 24 h pretreatment with 3 μM of compound 3b or 3f. Both ATX inhibitors caused a significant shift in the LD50 of PT’s dose–response relationship, indicating that ATX inhibitor treatment made them more sensitive to PT (Figure 7B). The PT LD50 for reducing cell growth by 50% was 202 nM when applied alone. In contrast, 3b and 3f reduced the LD50 to PT to 127 and 66 nM, respectively.
Next, we evaluated if these inhibitors were able to overcome chemoresistance to PT in cancer stem-like cells. To do this, we generated a PT-resistant 4T1 cell line (4T1-TaxR) and grew them in an anchorage-independent environment to induce sphere formation. This culture method has been widely used to enrich for breast CSCs. We found that both 30 and compound 3b were able to further reduce cell viability by ~25–30% compared to PT alone, suggesting that the ATX inhibitors were able to resensitize CSCs to PT treatment (Figure 7C).
DISCUSSION AND CONCLUSIONS
Here, we report on the synthesis of ATX inhibitors that are benzene-sulfonamide analogues derived from hit compound 3, which we identified via in silico screening of the Genome Research Institute library at the University of Cincinnati Drug Discovery Center.2,16 The parent compound 3, although a potent non-lipid ATX inhibitor with an IC50 value ~117 nM, lacked sufficient stability and activity in cellular and in vivo assays. Thus, our study objectives were (1) to improve the potency of ATX inhibition with IC50 values in the low nanomolar range and (2) to obtain compounds that were effective in in vitro cell-based assays and an in vivo metastasis model dependent on ATX activity. We have conducted four different kinds of SAR explorations of the scaffold of 3. The structural features explored included (1) modification of ring A substituents, (2) modification of the amide bond A–B ring linker, (3) modification of sulfonyl B–C ring linker, and (4) modification of ring C. The A ring optimization resulted in a number of substantially more active ATX inhibitors, including inhibitors 3b (IC50 = 9 nM) and 5 (IC50 = 18 nM). When tested for agonist action at LPA1/2/3/4/5 GPCRs, none of the compounds were active up to 10 μM, the highest concentration tested. However, three of the potent ATX inhibitors, 3b, 3f, and 3g, were also effective in antagonizing LPA1-elicited Ca2+ responses. The antagonist potency of these ATX inhibitors was in the low micromolar range, which is not negligible when assessing their cellular actions. Our synthetic improvements to increase the LPA1 antagonist activity of derivatives of compounds 3b and 3f without compromising their low nanomolar ATX inhibitory activity were unsuccessful.
Unexpectedly, inhibition of ATX was highly sensitive to transformations of the amide linkage as any alteration led to a deterioration of potency. Thioamides, including compound 14 (IC50 = 35 nM), were the most potent in this series, with 3-fold better potency than the screening hit 3. This finding underlines the importance of linker geometry for inhibition of ATX. The third optimization was modification of the sulfonyl bond, and this did not yield any compound that was more potent than 3. Finally, replacement of morpholine with piperazine was tolerated, resulting in three more active compounds than the starting scaffold, with the most potent 28b showing an IC50 of 55 nM. It is noteworthy that the piperazine series with log P values around 3.5 are water soluble.
We selected the three most potent amide analogues (3a, 3b, and 3f) and the most potent thioamide analogue (14) in cellular assays using the ATX-dependent invasion of A2058 human melanoma cells in vitro. All four inhibitors showed good in vitro stability and dose dependently decreased A2058 cell invasion with overlapping potencies (Table 3). In addition, two of these inhibitors, 3b and 14, were shown to be effective in reducing the metastasis of B16F10 cells to the lungs of C57BL/6 mice. Compounds 30 and 3b effectively resensitized the resistant 4T1 cell line (4T1-TaxR) to PT treatment. Our results complement the findings of Venkatraman et al., in which activation of the LPA1 receptor promotes chemoresistance in breast cancer cells in part via the stabilization of Nrf2 and the subsequent increase in the transcription of various genes involved in drug resistance.36 Thus, compounds that have both inhibitory actions against ATX and LPA1 might have therapeutic utility as an adjuvant for the treatment of therapy-resistant cancers.
In summary, this series of inhibitors meets the four objectives of our synthetic campaign as they lack carboxylic acid functionality, contain no chiral center, their molecular weight is less than 500 Da, and each has IC50 values less than 85 nM for ATX. During our study, Galapagos Inc. developed a number of potent compounds with structural similarity to 3 (Figure 1A).1 One of the Galapagos compounds has been reported to have successfully completed the first in-human phase one clinical trial,1 giving us hope that our inhibitors will yield similar outcomes with higher potency than the Galapagos compound.
EXPERIMENTAL SECTION
Chemistry.
General Methods.
All nonaqueous reactions were performed in oven-dried glassware under an inert atmosphere of dry nitrogen. All reagents and solvents were purchased from Aldrich (St. Louis, MO), Alfa-Aesar (Ward Hill, MA), Combi-Blocks (San Diego, CA), or Ark Pharm (Libertyville, IL) and used without further purification. Analytical thin-layer chromatography was performed on Silica Gel GHLF 10 × 20 cm Analtech TLC Uniplates (Analtech, Newark, DE) and visualized by fluorescence quenching under UV light. A Biotage SP1 flash chromatography purification system (Charlotte, NC) (Biotage SNAP cartridge, silica, 50 g and 100 g) was used to purify the compounds. 1H and 13C NMR spectra were recorded on a Varian Inova-500 spectrometer (500 MHz) (Agilent Technologies, Santa Clara, CA) or a Bruker Ascend 400 (400 MHz) (Billerica, MA) spectrometer. Chemical shifts are reported in ppm on the δ scale and referenced to the appropriate solvent peak. Mass spectra were collected on a Bruker ESQUIRE electrospray/ion trap instrument in the positive and negative modes. High-resolution mass spectrometry (HRMS) data were acquired on a Waters Xevo G2-S QTOF (Milford, MA) system equipped with an Acquity I-class UPLC system. The purity of all tested compounds was determined to be >95% by 1H NMR and HPLC. The HPLC method used to determine purity is as follows: Compound purity was analyzed using an Agilent 1100 HPLC system (Santa Clara, CA) with a Zorbax SB-C18 column, particle size 3.5 μm, 4.6 × 150 mm, from Agilent. Mobile phases consist of water with 0.1% formic acid (A) and acetonitrile with 0.1% formic acid (B). A flow rate of 1 mL/min was used. The gradient elution started at 50% B. It reached 100% B from 0 to 9 min, was maintained at this from 9 to 12 min, and was then decreased to 50% B from 12 to 15 min and stopped. Compound purity was monitored with a DAD detector set at 254 nm.
Synthesis of 2,4-Dichloro-5-(chlorosulfonyl)benzoic Acid (1).
To cooled (0 °C) chlorosulfonic acid (10.4 mL, 157 mmol) was added 2,4-dichlorobenzoic acid (5 g, 26 mmol) portionwise. The reaction mixture was subsequently heated at 140 °C and stirred for 16 h. The reaction mixture was cooled to room temperature and poured into crushed ice. The formed white precipitates were collected by filtration and dried under vacuum. Yield = 80%. The characterizations are consistent with the literature.37
General Procedure for the Synthesis of Compounds 2 and 27.
To a solution of 1 (3g, 10.3 mmol) in 20 mL of CH2Cl2 was added trimethylamine (3.61 mL, 26 mmol). The reaction mixture was cooled to 0 °C under ice. The corresponding secondary amine (11.4 mmol) was added dropwise, and the reaction mixture was stirred overnight. The reaction mixture was concentrated, and the crude was taken for the next step without further purification.
General Procedure for the Synthesis of Compounds 3 and 3a–3i.
To a solution of compound 2 (0.46g, 1.4 mmol) in 20 mL of CH2Cl2 were added 1 mL of SOCl2 (13.6 mmol) and 2 drops of DMF under an argon atmosphere. The reaction was stirred at room temperature overnight. The reaction mixture was concentrated and dissolved again in 20 mL of CH2Cl2. Pyridine (330 μL, 4.1 mmol) and the corresponding aniline (1.2 mmol) were added under an argon atmosphere. The reaction mixture was allowed to stir overnight at room temperature. The reaction mixture was extracted with 10% HCl, water, and brine solution. The organic layer was dried over MgSO4, concentrated, and purified by column chromatography, leading to the pure product.
2,4-Dichloro-N-(3-fluorophenyl)-5-(morpholinosulfonyl)-benzamide (3).
Compound 3 was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 70%. 1H NMR (400 MHz, DMSO-d6) δ 10.94 (s, 1H), 8.18 (d, J = 3.8 Hz, 2H), 7.77–7.67 (m, 1H), 7.53–7.37 (m, 2H), 7.12–6.95 (m, 1H), 3.69 (t, J = 4.70 Hz, 4H), 3.28 (t, J = 5.0 Hz, 4H). HRMS [C17H16N2O4FSCl2+]: calcd 433.0192, found 433.0182.
2,4-Dichloro-N-(3,4-difluorophenyl)-5-(morpholinosulfonyl)-benzamide (3a).
Compound 3a was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 72%. mp = 221.3 °C. 1H NMR (400 MHz, DMSO) δ 10.90 (s, 1H), 8.13 (s, 2H), 7.85 (ddd, J = 12.8, 7.4, 2.4 Hz, 1H), 7.45 (ddd, J = 27.0, 18.1, 9.0 Hz, 2H), 3.63 (t, J = 4.46, 4H), 3.22 (t, J = 4.77, 4H). 13C NMR (101 MHz, DMSO) δ 162.71, 135.72, 135.29, 134.19, 133.20, 133.10, 131.49, 117.76, 117.58, 116.22, 109.00, 108.79, 65.72, 45.49. HRMS [C17H15N2O4F2SCl2+]: calcd 451.0098, found 451.0099.
2,4-Dichloro-5-(morpholinosulfonyl)-N-(3,4,5-trifluorophenyl)-benzamide (3b).
Compound 3b was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 71.5%. mp = 237.8 °C. 1H NMR (400 MHz, DMSO) δ 11.05 (s, 1H), 8.15 (d, J = 2.1 Hz, 2H), 7.58 (dd, J = 9.9, 6.5 Hz, 2H), 3.63 (t, J = 4.67, 4H), 3.22 (t, J = 4.53, 4H). 13C NMR (101 MHz, DMSO) δ 162.99, 135.70, 134.94, 134.27, 133.42, 133.17, 131.53, 104.39, 104.15, 65.72, 45.49. HRMS [C17H14N2O4F3SCl2+]: calcd 469.0003, found 451.0012.
2,4-Dichloro-5-(morpholinosulfonyl)-N-(perfluorophenyl)-benzamide (3c).
Compound 3c was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 65%. mp = 237.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H), 8.15 (dd, J = 24.5, 19.0 Hz, 2H), 3.70–3.55 (m, 4H), 3.27–3.13 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 166.95, 163.05, 136.00, 134.33, 133.82, 133.46, 131.49, 65.73, 45.43. HRMS [C17H12N2O4F5SCl2+]: calcd 504.9815, found 504.9838.
2,4-Dichloro-5-(morpholinosulfonyl)-N-(3,4,5-trichlorophenyl)-benzamide (3d).
Compound 3d was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 75%. mp = 303.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 8.18 (s, 1H), 8.15 (s, 1H), 7.94 (s, 2H), 3.68–3.57 (t, J = 4.32, 4H), 3.27–3.16 (t, J = 4.84, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.10, 138.36, 135.74, 134.78, 134.30, 133.52, 133.20, 133.01, 131.59, 124.51, 119.89, 65.72, 45.48. HRMS [C17H14N2O4SCl5+]: calcd 516.9117, found 516.9108.
2,4-Dichloro-5-(morpholinosulfonyl)-N-(3,4,5-trimethoxyphenyl)-benzamide (3e).
Compound 3e was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 65%. mp = 281.2 °C. 1H NMR (400 MHz, chloroform-d) δ 8.34 (s, 1H), 7.83 (s, 1H), 7.66 (s, 1H), 6.93 (s, 2H), 3.89 (s, 6H), 3.84 (s, 3H), 3.73 (dd, J = 5.9, 3.7 Hz, 4H), 3.39–3.18 (m, 4H). HRMS [C20H23N2O7SCl2+]: calcd 505.0603, found 505.0606.
2,4-Dichloro-N-(4-chloro-3,5-difluorophenyl)-5-(morpholinosulfonyl)benzamide (3f).
Compound 3f was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 75%. mp = 275.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.17 (s, 1H), 8.16 (d, J = 6.1 Hz, 2H), 7.60 (d, J = 9.1 Hz, 2H), 3.63 (t, J = 4.35, 4H), 3.22 (t, J = 4.93, 4H). 13C NMR (101 MHz, CDCl3) δ 168.38, 140.96, 140.08, 139.55, 138.75, 138.43, 136.82, 109.16, 108.89, 70.97, 50.74. HRMS [C17H14N2O4F2SCl3+]: calcd 484.9708, found 484.9729.
2,4-Dichloro-N-(3,5-dichloro-4-fluorophenyl)-5-(morpholinosulfonyl)benzamide (3g).
Compound 3g was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 76%. mp = 297.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.00 (s, 1H), 8.16 (d, J = 9.9 Hz, 2H), 7.85 (d, J = 6.1 Hz, 2H), 3.63 (t, J = 4.8 Hz, 4H), 3.22 (t, J = 5.22 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.96, 135.74, 134.89, 134.28, 133.45, 133.18, 131.56, 121.11, 120.93, 120.19, 65.72, 45.49. HRMS [C17H14N2O4FSCl4+]: calcd 500.9412, found 500.9409.
2,4-Dichloro-N-(4-cyano-3,5-difluorophenyl)-5-(morpholinosulfonyl)benzamide (3h).
Compound 3h was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 72%. mp = 240.7 °C. 1H NMR (400 MHz, DMSO) δ 11.51 (s, 1H), 8.19 (d, J = 17.4 Hz, 2H), 7.63 (d, J = 10.1 Hz, 2H), 3.63 (t, J = 4.75 Hz, 4H), 3.22 (t, J = 4.68, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.67, 135.69, 134.51, 134.36, 133.73, 133.23, 131.69, 109.75, 103.18, 102.93, 65.72, 45.48. HRMS [C18H14N3O4F2SCl2+]: calcd 476.0050, found 476.0048.
Methyl 4-(2,4-Dichloro-5-(morpholinosulfonyl)benzamido)-2,6-difluorobenzoate (3i).
Compound 3i was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 69%. mp = 216.4.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.29 (s, 1H), 8.17 (d, J = 14.8 Hz, 2H), 7.50 (d, J = 10.6 Hz, 2H), 3.87 (s, 3H), 3.63 (t, J = 4.46 Hz, 4H), 3.23 (t, J = 4.69 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.37, 160.85, 143.00, 135.70, 134.69, 134.34, 133.61, 133.19, 131.63, 103.16, 102.90, 65.72, 52.70, 45.49. HRMS [C19H17N2O6F2SCl2+]: calcd 509.0152, found 509.0146.
4-(2,4-Dichloro-5-(morpholinosulfonyl)benzamido)-2,6-difluoro-benzoic Acid (4).
An amount of 8.5 mg of LiOH (0.35 mmol) was added to a solution of compound 3i (90 mg, 0.17 mmol) in 10 mL of a THF/MeOH (2:1) mixture. The reaction mixture was allowed to stir overnight at room temperature. The reaction mixture was diluted with water, acidified with 10% HCl (pH 4), and extracted with ethyl acetate. The organic layer was dried over MgSO4 and purified via flash chromatography (2% MeOH/CH2Cl2), leading to the pure product as a white solid. Yield = 62%. mp = 256.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.63 (s, 1H), 10.73 (s, 1H), 8.12 (s, 1H), 7.57 (s, 1H), 7.52 (d, J = 10.4 Hz, 2H), 4.01 (s, 3H), 3.62 (t, J = 4.66 Hz, 4H), 3.14 (t, J = 4.51 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.12, 161.79, 159.94, 135.47, 132.99, 126.54, 123.22, 116.17, 103.04, 102.74, 65.67, 57.40, 45.50. HRMS [C19H18N2O7F2SCl+]: calcd 491.0491, found 491.0500.
N-(2-Bromo-3,4,5-trifluorophenyl)-2,4-dichloro-5-(morpholinosulfonyl)benzamide (5).
Compound 5 was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 76%. mp = 196.1 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 8.21 (s, 1H), 8.12 (s, 1H), 7.86 (m, 1H), 3.64 (t, J = 4.7 Hz, 4H), 3.23 (t, J = 4.7 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.37, 135.82, 134.80, 134.02, 133.29, 133.07, 131.77, 111.61, 65.71, 45.46. HRMS [C19H17N2O6F2SCl2+]: calcd 546.9109, found 546.9113.
2-(2,4-Dichloro-5-(morpholinosulfonyl)phenyl)-7-fluorobenzo[d]-oxazole (7).
To a solution of 0.28 g of compound 2 (0.82 mmol) in 20 mL of CH2Cl2 was added 0.6 mL of SOCl2 (8.2 mmol) and a catalytic amount of DMF (2 drops) under an argon atmosphere. The reaction was allowed to sir for 12 h. The reaction mixture was concentrated and dissolved in 20 mL of CH2Cl2. A volume of 0.2 mL of pyridine (2.5 mmol) as well as 0.105 g (0.82 mmol) of 2-amino-6-fluorophenol were added to the solution under an argon atmosphere. The reaction mixture was allowed to stir overnight at room temperature. The reaction mixture was diluted with 20 mL of CH2Cl2 and extracted with 10% HCl. The organic layer was dried over MgSO4 and concentrated. The crude was dissolved in 25 mL of toluene. An amount of of 156 mg p-TSA (0.82 mmol) was added to the solution under an argon atmosphere. The reaction mixture was refluxed over 12 h. The reaction was cooled to room temperature, poured into water, and extracted with ethyl acetate. The organic layer was separated, washed with water and brine, dried (MgSO4), filtered, and concentrated. The crude product was purified by column chromatography (silica gel, 30% ethyl acetate/hexanes) to afford the desired product as an off-white solid. Yield = 77%. mp = 167.4 °C. 1H NMR (400 MHz, chloroform-d) δ 8.87 (s, 1H), 7.80 (s, 1H), 7.66 (dd, J = 8.0, 0.9 Hz, 1H), 7.37 (td, J = 8.2, 4.6 Hz, 1H), 7.21 (ddd, J = 9.9, 8.3, 0.9 Hz, 1H), 3.90–3.62 (m, 4H), 3.49–3.20 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 158.89, 148.30, 145.78, 144.63, 138.44, 137.84, 135.44, 134.80, 125.61, 124.72, 116.62 (d, J = 4.3 Hz), 113.09, 112.93, 66.51, 45.90. HRMS [C17H14N2O4FSCl2+]: calcd 431.0035, found 431.0045.
4-((2,4-Dichloro-5-(5,6-difluoro-1H-benzo[d]imidazol-2-yl)-phenyl)sulfonyl)-morpholine (8).
To a solution of 0.28 g of compound 2 (0.82 mmol) in 20 mL of CH2Cl2, was added 0.6 mL of SOCl2 (8.2 mmol) and a catalytic amount of DMF (2 drops) under an argon atmosphere. The reaction was allowed to sir for 12 h. The reaction mixture was concentrated and dissolved in 20 mL of CH2Cl2. A volume of 0.2 mL of pyridine (2.5 mmol) and 0.118 g (0.82 mmol) of 4,5-difluorobenzene-1,2-diamine were added to the solution under an argon atmosphere. The reaction mixture was allowed to stir overnight at room temperature. The mixture was diluted with dichloromethane (30 mL) and washed with saturated aqueous sodium bicarbonate solution (80 mL). The organic phase was dried over MgSO4, and the solvent was removed to provide the amide as a mixture of isomers. The crude amide was combined with phosphorus oxychloride (10 mL) under an argon atmosphere, heated to reflux for 100 min, and then allowed to cool to room temperature. The mixture was poured into ice water (100 mL) and neutralized with 2 N sodium hydroxide solution. Saturated aqueous sodium bicarbonate solution (60 mL) was added, and the aqueous phase was extracted with ethyl acetate (3 × 70 mL). Water (150 mL) was added to the combined organic phases, and the pH was adjusted to 3 with 2 N aqueous hydrochloric acid. The organic layer was washed with brine (100 mL) and dried over MgSO4, and the solvent was removed to give a brown solid. The crude was purified by flash chromatography (35% EtOAc/hexanes) to afford the desired product as a light brown solid. Yield = 71%. mp = 175.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.51 (s, 1H), 8.19 (s, 1H), 7.83–7.71 (m, 2H), 3.64 (t, J = 4.8 Hz, 5H), 3.24 (dd, J = 5.9, 3.6 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 148.55, 148.41, 146.18, 136.45, 134.30, 134.20, 133.91, 132.88, 128.32, 65.71, 54.87, 45.48. HRMS [C17H14N3O3F2SCl2+]: calcd 448.0101, found 448.0106.
2,4-Dichloro-N-(3-fluorophenyl)-N-methyl-5-(morpholinosulfonyl)benzamide (9).
Compound 9 was prepared following the general procedure for the synthesis of 3 and 3a–3i. The crude was purified in 1% MeOH/CH2Cl2, yielding the pure product as a white solid. Yield = 75%. mp = 128.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.16–7.72 (m, 2H), 7.47–7.24 (m, 2H), 7.22–6.99 (m, 2H), 3.57 (t, J = 4.6 Hz, 4H), 3.40 (s, 3H), 2.95 (bt, J = 5.2 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 164.16, 160.61, 144.17, 135.50, 135.09, 133.32, 133.03, 132.68, 132.05, 131.84, 130.86, 123.31, 114.57, 65.53, 45.47, 36.60. HRMS [C18H18N2O4FSCl2+]: calcd 447.0348, found 447.0369.
N-(2,4-Dichloro-5-(morpholinosulfonyl)benzyl)-3,4-difluoroani-line (10).
To a solution of compound 3a (0.15g, 0.33 mmol) in anhydrous THF (20 mL) was added 47 μL of BH3·DMS (0.5 mmol) under an argon atmosphere. The reaction mixture was allowed to reflux over 72 h, and reaction progress was monitored by TLC. The reaction was quenched with 30 mL of water and extracted with ethyl acetate. The organic layer was extracted with brine and dried over MgSO4. The crude was purified via flash chromatography (silica gel, 40% EtOAc/hexanes), providing access to the pure product as a white solid. Yield = 75%. mp = 154.8 °C. 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1H), 7.53 (s, 1H), 6.96–6.79 (m, 1H), 6.33–6.10 (m, 2H), 4.35 (d, J = 6.0 Hz, 2H), 4.23 (t, J = 5.9 Hz, 1H), 3.58 (t, J = 4.44 Hz, 4H), 3.03 (t, J = 4.54 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 145.36, 137.86, 136.88, 133.19, 132.53, 131.01, 130.11, 117.66, 117.48, 108.10, 100.73, 100.53, 65.50, 45.45, 43.83. HRMS [C17H17N2O3F2SCl2+]: calcd 437.0305, found 437.0307.
N-(2,4-Dichloro-5-(morpholinosulfonyl)benzyl)-3,4,5-trifluoroani-line (11).
To a solution of compound 3b (0.15g, 0.32 mmol) in anhydrous THF (20 mL) was added 46 μL of BH3·DMS (0.48 mmol) under an argon atmosphere. The reaction mixture was allowed to reflux over 24 h, and reaction progress was monitored by TLC. The reaction was quenched with 30 mL of water and extracted with ethyl acetate. The organic layer was extracted with brine and dried over MgSO4. The crude was purified via flash chromatography (silica gel, 40% EtOAc/hexanes), providing access to the pure product as a white solid. Yield = 72%. mp = 149.4 °C. 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1H), 7.54 (s, 1H), 6.16–5.94 (m, 2H), 4.31 (dd, J = 13.5, 5.0 Hz, 3H), 3.61 (t, J = 4.84 Hz, 4H), 3.08 (t, J = 5.0 Hz, 4H). 13C NMR (101 MHz, DMSO) δ 152.26, 149.91, 144.61, 137.99, 136.42, 133.27, 132.60, 131.01, 130.33, 96.08, 95.84, 65.54, 45.47, 43.66. HRMS [C17H16N2O3F3SCl2+]: calcd 455.0211, found 455.0206.
General Procedure for the Synthesis of Compounds 12–15.
To a solution of 0.2 g (1 equiv) of 3, 3a, 3b, or 3g in toluene (20 mL) was added Lawesson’s reagent (0.9 equiv) under an argon atmosphere, as demonstrated in the literature.26 The reaction was allowed to reflux over 12 h. The reaction was cooled to room temperature. The solvent was evaporated, and the crude was purified via flash chromatography (silica gel, 30–35% EtOAc/hexanes), yielding the pure product as a light yellow solid.
2,4-Dichloro-N-(3-fluorophenyl)-5-(morpholinosulfonyl)-benzothioamide (12).
Compound 12 was obtained following the general procedure for the preparation of compounds 12–15. The crude was purified through flash column chromatography (silica gel, 30% EtOAc/hexanes), leading to the pure product as a light yellow solid. Yield = 68%. mp = 181.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.45 (s, 1H), 8.11 (s, 1H), 8.06 (dt, J = 11.4, 2.3 Hz, 1H), 8.00 (s, 1H), 7.71–7.64 (m, 1H), 7.53 (td, J = 8.2, 6.7 Hz, 1H), 7.19 (td, J = 8.5, 1.8 Hz, 1H), 3.65 (t, J = 4.64 Hz, 4H), 3.21 (t, J = 4.72 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 141.97, 133.92, 133.71, 132.78, 131.68, 131.18, 130.68, 130.59, 118.73, 109.48, 109.23, 65.68, 45.51. HRMS [C17H16N2O3FS2Cl2+]: calcd 448.9963, found 448.9956.
2,4-Dichloro-N-(3,4-difluorophenyl)-5-(morpholinosulfonyl)-benzothioamide (13).
Compound 13 was obtained following the general procedure for the preparation of compounds 12–15. The crude was purified through flash column chromatography (silica gel, 32% EtOAc/hexanes), leading to the pure product as a light yellow solid. Yield = 70%. 1H NMR (400 MHz, CDCl3) δ 9.55 (s, 1H), 8.19–7.86 (m, 2H), 7.71–7.40 (m, 2H), 7.31–7.15 (m, 1H), 3.71 (bs, 4H), 3.23 (bs, 4H). 13C NMR (101 MHz, CDCl3) δ 192.37, 151.23 (d, J = 13.4 Hz), 150.07 (d, J = 12.8 Hz), 148.76 (d, J = 13.4 Hz), 147.58 (d, J = 12.7 Hz), 141.70, 134.58, 134.44, 133.99, 133.39, 132.76, 132.39, 119.40 (d, J = 2.4 Hz), 117.69, 117.50, 112.99, 112.78, 66.31, 45.87. HRMS [C17H15N2O3F2S2Cl2+]: calcd 466.9869, found 466.9869.
2,4-Dichloro-5-(morpholinosulfonyl)-N-(3,4,5-trifluorophenyl)-benzothioamide (14).
Compound 14 was obtained following the general procedure for the preparation of compounds 12–15. The crude was purified through flash column chromatography (silica gel, 35% EtOAc/hexanes), leading to the pure product as a light yellow solid. Yield = 71%. mp = 152.1 °C. 1H NMR (400 MHz, CDCl3) δ 9.41 (s, 1H), 8.05 (s, 1H), 7.68 (dd, J = 8.7, 6.2 Hz, 2H), 7.59 (s, 1H), 3.73 (t, J = 5.23 Hz, 4H), 3.25 (t, J = 5.23 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 191.63, 151.41, 140.50, 133.31, 133.21, 132.36, 132.08, 131.50, 106.82, 106.57, 65.33, 44.87. HRMS [C17H14N2O3F3S2Cl2+]: calcd 484.9775, found 484.9778.
2,4-Dichloro-N-(3,5-dichloro-4-fluorophenyl)-5-(morpholinosulfonyl)-benzothioamide (15).
Compound 15 was obtained following the general procedure for the preparation of compounds 12–15. The crude was purified through flash column chromatography (silica gel, 35% EtOAc/hexanes), leading to the pure product as a light yellow solid. Yield = 72%. mp = 175.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.51 (s, 1H), 8.17 (d, J = 6.0 Hz, 2H), 8.11 (s, 1H), 8.05 (s, 1H), 3.65 (bt, J = 4.41 Hz, 4H), 3.21 (bt, J = 4.31 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 192.75, 152.43, 149.96, 141.53, 135.81 (d, J = 4.4 Hz), 134.01, 133.78, 132.83, 131.92, 131.20, 123.89, 121.07, 120.88, 65.68, 45.51. HRMS [C17H14N2O3FS2Cl4+]: calcd 516.9184, found 516.9227.
2,4-Dichloro-5-nitrobenzene-1-sulfonyl Chloride (16).
Intermediate 16 was prepared following the experimental procedure for the preparation of intermediate 1 starting with 5 g of 2,4-dichloro-1-nitrobenzene (26 mmol). An amount of 5.7 g (19.6 mmol, 75%) of pure product was obtained as an off-white solid upon drying under vacuum. 1H NMR (400 MHz, DMSO-d6) δ 8.44 (s, 1H), 7.91 (s, 1H).
4-((2,4-Dichloro-5-nitrophenyl)sulfonyl)morpholine (17).
Intermediate 17 was prepared following the general procedure for the preparation of intermediates 2 and 27 staring with 3.5 g of intermediate 16 (12 mmol). An amount of 3.2 g (9.3 mmol, 78%) of pure product was obtained as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 7.79 (s, 1H), 3.75 (t, J = 4.71 Hz, 4H), 3.35 (t, J = 4.31 Hz, 4H).
2,4-Dichloro-5-(morpholinosulfonyl)aniline (18).
Nitro intermediate 17 (3 g, 8.8 mmol) was dissolved in a mixture of EtOH and H2O (40 mL, 10:1). Powdered iron (2.25 g, 35.2 mmol) and five drops of HCl (12 M) were added. The mixture was refluxed 5 h. The mixture was cooled to room temperature, and the solvent was evaporated. HCl (1 N, 100 mL) was added, and the mixture was extracted with EtOAc (100 mL). The organic phase was extracted with brine, dried over MgSO4, and concentrated, providing the pure product as a light red solid (2.6 g, 8.3 mmol, 95%). 1H NMR (400 MHz, DMSO-d6) δ 7.58 (s, 1H), 7.41 (s, 1H), 6.04 (bs, 2H), 3.54 (t, J = 5.0 Hz, 4H), 3.13 (t, J = 4.86 Hz, 4H).
Synthesis of Compounds 19 and 20.
To a solution of 0.2 g of 18 (0.64 mmol) in 15 mL of CH2Cl2 were added Et3N (267 μL, 1.9 mmol), 3-fluorobenzenesulfonyl chloride (0.149g, 0.77 mmol), and a catalytic amount of DMAP under an argon atmosphere. The reaction was allowed to stir at room temperature over 96 h and monitored by TLC. The reaction led to the formation of two new spots, as determined by TLC. The reaction mixture was diluted with 30 mL of CH2Cl2. The organic layer was extracted with 10% HCl, H2O, and brine and evaporated under reduced pressure. The crude with two spots was isolated by flash chromatography (silica gel, 30–45% EtOAc/hexanes), providing pure compounds 19 (yield = 33%) and 20 (yield = 60%) as white solids. Compound 19. 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.72–7.43 (m, 4H), 7.33 (tdd, J = 8.3, 2.5, 0.9 Hz, 1H), 3.74 (t, J = 5.10 Hz, 4H), 3.28 (t, J = 5.04 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 163.71, 161.19, 140.34 (d, J = 6.8 Hz), 135.81, 132.61, 132.43, 131.42 (d, J = 7.8 Hz), 129.09, 128.41, 123.97, 123.28 (d, J = 3.4 Hz), 121.42, 121.21, 114.94, 114.70, 66.47, 45.93. HRMS [C16H16N2O5FS2Cl2+]: calcd 468.9862, found 467.9870. Compound 20. 1H NMR (400 MHz, CDCl3) δ 7.90–7.37 (m, 10H), 3.73 (t, J = 4.86 Hz, 4H), 3.27 (t, J = 4.69 Hz, 4H). [C22H19N2O7F2S3Cl2+]: calcd 626.9699, found 626.9702.
2,4-Dichloro-5-(morpholinosulfonyl)benzene-1-sulfonyl Chloride (21).
Intermediate 21 was prepared from 1.5 g (4.8 mmol) of intermediate 18 following the literature reported procedure.32 An amount of 1.7 g of intermediate 21 (4.3 mmol, 90%) was obtained as an off-white solid upon drying the precipitate under vacuum. The product was taken for the next step.
General Procedure for the Formation of Compounds 22 and 23.
To a solution of 0.2 g of 21 (0.51 mmol) in 15 mL of CH2Cl2, were added Et3N (1.53 mmol) and the appropriate aniline derivative (1.2 equiv) under an argon atmosphere. The reaction was allowed to stir at room temperature over 12 h. The reaction was diluted with 30 mL of CH2Cl2 and extracted with 10% HCl, H2O, and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The crude was purified via flash column chromatography (silica gel, 2% MeOH/CH2Cl2), yielding the pure product as an off-white solid.
2,4-Dichloro-N-(3-fluorophenyl)-5-(morpholinosulfonyl)-benzenesulfonamide (22).
Yield = 95%. mp = 170.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.28 (s, 1H), 8.41 (s, 1H), 8.22 (s, 1H), 7.33 (dd, J = 15.0, 8.2 Hz, 1H), 7.01–6.77 (m, 3H), 3.59 (t, J = 4.23 Hz, 4H), 3.09 (t, J = 4.64 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.43, 161.0, 159.69, 138.36, 136.84, 136.21, 135.68, 135.42, 134.05, 133.34, 131.33, 131.24, 115.46, 111.25, 111.04, 106.59, 106.34, 65.60, 45.43. HRMS [C16H16N2O5FS2Cl2+]: calcd 468.9861, found 468.9857.
2,4-Dichloro-5-(morpholinosulfonyl)-N-(3,4,5-trifluorophenyl)-benzenesulfonamide (23).
Yield = 94%. mp = 107.8 °C. 1H NMR (400 MHz, chloroform-d) δ 8.61 (s, 1H), 7.73 (s, 1H), 7.18 (bs, 1H), 6.90–6.70 (m, 2H), 3.76–3.65 (m, 4H), 3.31–3.17 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 151.57, 149.06, 137.15, 136.19, 135.81, 135.12, 134.30, 133.34, 104.62, 104.55, 104.38, 65.63, 45.42. HRMS [C16H14N2O5F3S2Cl2+]: calcd 504.9673, found 504.9679.
5-Bromo-2,4-dichloro-N-(3,4,5-trifluorophenyl)benzamide (24).
Intermediate 24 was prepared following the synthetic technique for the preparation of 3a–3i starting with 2 g of 5-bromo-2,4-dichlorobenzoic acid (7.4 mmol). An amount of 2.22 g (5.6%, 75%) of pure 24 was obtained as a white solid upon purification through flash column chromatography (silica gel, 45% EtOAc/hexanes). 1H NMR (400 MHz, chloroform-d) δ 8.15 (s, 1H), 7.58 (s, 1H), 3.92 (s, 3H).
2,4-Dichloro-5-(morpholinoamino)-N-(3,4,5-trifluorophenyl)-benzamide (25).
Compound 25 was prepared from compound 24 (0.3 g, 0.75 mmol) by LiCl-mediated palladium-catalyzed coupling of hydrazine and phenyl bromide.32 An amount of 0.195 g (0.46 mmol, 62%) of pure product was obtained as a light yellowish solid upon purification via flash column chromatography (silica gel, 30% EtOAC/hexanes). mp = 196.1 °C. 1H NMR (400 MHz, chloroform-d) δ 7.95 (s, 1H), 7.65 (s, 1H), 7.51–7.29 (m, 3H), 5.15 (s, 1H), 3.81 (t, J = 4.6 Hz, 4H), 2.78 (bt, 4H). 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 7.77–7.41 (m, 3H), 7.29 (s, 1H), 7.05 (s, 1H), 3.67 (t, J = 4.4 Hz, 4H), 2.79 (t, J = 4.7 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.16, 151.31, 148.88, 142.82, 135.57, 129.64, 117.71, 116.95, 112.40, 103.99, 103.75, 66.28, 54.47. HRMS [C17H15N3O2F3Cl2+]: calcd 420.0493, found 420.0484.
2,4-Dichloro-N-(methylsulfonyl)-5-(morpholinoamino)-N-(3,4,5-trifluorophenyl)-benzamide (26a).
To a solution of 0.1 g of 25 (0.24 mmol) in DMSO was added NaOtBu (0.069 g, 0.72 mmol) at 0 °C under an argon atmosphere. Methanesulfonyl chloride (22 μL, 0.29 mmol) was added, and the reaction was allowed to stir over 24 h at ambient temperature. The reaction was diluted with water and extracted with EtOAc (30 mL). The organic layer was washed with water and brine, dried over MgSO4, and concentrated under reduced pressure. The crude was purified via column chromatography, leading to compound 26a (0.04 g, 0.08 mmol) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 7.18 (s, 1H), 7.12 (s, 1H), 6.99 (dd, J = 7.4, 6.0 Hz, 2H), 5.06 (s, 1H), 3.84 (t, J = 4.46 Hz, 4H), 3.58 (s, 3H), 2.68 (bt, 4H). 13C NMR (101 MHz, CDCl3) δ 167.13, 142.04, 132.69, 130.52, 130.16, 120.12, 118.85, 115.26, 115.03, 112.93, 66.81, 56.42, 42.05. HRMS [C18H17N3O4F3SCl2+]: calcd 498.0269, found 498.0263.
2,4-Dichloro-5-((4-methylpiperazin-1-yl)sulfonyl)-N-(3,4,5-trifluorophenyl)-benzamide (28a).
Compound 28a was prepared from compound 27 (0.2 g, 0.57 mmol) and 3,4,5-trifuluro aniline following the synthetic technique for the preparation of 3a–3i. Pure compound 28a was obtained as a white solid upon purification (silica gel, 10% MeOH/CH2Cl2). Yield = 65%. mp = 201.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.04 (s, 1H), 8.14 (d, J = 6.4 Hz, 2H), 7.58 (dd, J = 9.9, 6.5 Hz, 2H), 3.24 (t, J = 4.49 Hz, 4H), 2.36 (t, J = 4.74 Hz, 4H), 2.18 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.01, 135.55, 134.90, 134.72, 133.32, 133.09, 131.42, 104.42, 104.17, 53.91, 45.24. HRMS [C18H17N3O3F3SCl2+]: calcd 482.0320, found 482.0340.
2,4-Dichloro-N-(3,5-dichloro-4-fluorophenyl)-5-((4-methylpiperazin-1-yl)sulfonyl)benzamide (28b).
Compound 28b was prepared from compound 27 (0.2 g, 0.57 mmol) and 3,5-dichloro-4-fluoro aniline following the synthetic technique for the preparation of 3a–3i. Pure compound 28b was obtained as a white solid upon purification (silica gel, 10% MeOH/CH2Cl2). Yield = 64%. mp = 232.7 °C. 1H NMR (400 MHz, DMSO) δ 11.00 (s, 1H), 8.15 (d, J = 14.8 Hz, 2H), 7.85 (d, J = 6.1 Hz, 2H), 3.23 (t, J = 4.38 Hz, 4H), 2.35 (t, J = 4.38 Hz, 4H), 2.18 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 162.98, 135.57, 134.86, 134.73, 133.33, 133.10, 131.45, 121.10, 120.92, 120.20, 53.94, 45.41, 45.35. HRMS [C18H17N3O3F3SCl2+]: calcd 513.9729, found = 513.9743.
2,4-Dichloro-N-(3,5-dichloro-4-fluorophenyl)-5-((4-methylpipera-zin-1-yl)sulfonyl)-benzothioamide (29).
Compound 29 was prepared from 28b following the synthetic procedure for the preparation of 12–15. Pure compound 29 was obtained as a light purple solid upon purification (silica gel, 3% MeOH/CH2Cl2). Yield = 69%. 1H NMR (400 MHz, CDCl3) δ 9.60 (s, 1H), 8.04 (s, 1H), 7.94 (d, J = 5.8 Hz, 2H), 7.57 (s, 1H), 3.30 (bm, 4H), 2.47 (bm, 4H), 2.31 (s, 3H). 13C NMR (101 MHz, DMSO) δ 141.62, 134.36, 133.63, 132.75, 131.74, 131.08, 123.93, 121.04, 120.85, 53.79, 45.18. HRMS [C18H17N3O2FS2Cl4+]: calcd 529.9500, found = 529.9503.
Molecular Modeling.
Molecular modeling studies were performed using the induced fit docking protocol within MOE software, as in prior studies.2,38 Briefly, hydrogen atoms and partial charges were added to the crystal structure (PDB 3NKM), and crystallographic water molecules were removed to produce the docking target.22 Ligands were constructed in th e protonated and tautomeric states expected to predominate at pH 7.0. The docking target site was based on residues that fall within 5 Å of crystallographic LPA molecules found in crystal structures 3NKN–3NKR.22
Testing of ATX Inhibitors on ATX and LPAR.
ATX Generation.
Human recombinant ATX was expressed as published previously, using Sf9 Spodoptera frugiperda ovary cells (Invitrogen, Carlsbad, CA).16 Suspension cells were grown to a 1 L quantity at a concentration of 1 × 106 cells/mL in Sf-900 III serum-free medium (Invitrogen) supplemented with 50 U/mL penicillin and 50 μg/mL streptomycin at 27 °C with agitation. Cells were then infected with high-titer baculovirus generated via the Bac-to-Bac baculovirus expression system (Invitrogen) using the pCMV5 mammalian expression vector containing a C-terminal FLAG-tagged human ATX sequence (a generous gift from Dr. Junken Aoki, Tohoku University, Sendai, Japan) subcloned into the pFastBac1 transfer vector. Expression was allowed to proceed for 72 h, and secreted protein was harvested by centrifugation and filtration of the culture medium followed by affinity chromatography using anti-FLAG M2 agarose beads (Sigma-Aldrich, St. Louis, MO) and competitive elution with 50 μg/mL FLAG peptide (Sigma-Aldrich). Resultant ATX was then concentrated via centrifugation in Amicon Ultra 30 000 molecular weight cut off filter units (Millipore, Billerica, MA) and subsequent buffer exchange into storage buffer composed of 50 mM Tris, pH 7.4, with 20% (v/v) ethylene glycol. Protein was held at −80 °C for long-term storage.
ATX, ENPP6, and ENPP7 Inhibition.
ATX activity was assessed via hydrolysis of the synthetic lipid-like FRET-based substrate FS-3 (Echelon Biosciences, Salt Lake City, UT) or via hydrolysis of the nucleotide substrate p-nitrophenyl thymidine 5′-monophosphate (pNP-TMP) as described previously.2 ENPP6 and ENPP7 activities were assessed by hydrolysis of the nucleotide substrate p-nitrophenylphos-phorylcholine (pNPPC) as described previously.13 Reaction wells were loaded in 60 μL volumes in triplicate wells of black-wall 96-well plates in assay buffer consisting of 50 mM Tris, 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 μM BSA, pH 8.0 for ATX; 100 mM Tris, 500 mM NaCl, 0.05% Triton, pH 9.0 for ENPP6; or 50 mM Tris, 150 mM NaCl, 10 mM taurocholate, pH 8.5 for ENPP7. For dose–response and IC50 generation, final concentrations per reaction well were composed of 1 μM FS-3 (or 1 mM pNP-TMP), 0 or 4 nM human recombinant ATX, and test compound concentrations ranging from 0 to 1 μM. For ENPP6 and ENPP7 screening, wells were composed of 10 μM pNPPC, 0 or 10 nM ENPP6 or ENPP7, and a range of test compound concentrations from 0 to 10 μM. To determine the mechanism of action, triplicate wells were loaded with assay buffer with FS-3 concentrations ranging from 0 to 10 μM, 0 or 4 nM ATX, and inhibitor concentrations of 0, 0.5 × IC50, or 2 × IC50. Fluorescence was read every 2 min for 1 h at excitation/emission wavelengths of 485/528 nm for FS-3 hydrolysis or 405 nm absorbance for pNP-TMP or pNPPC hydrolysis using a FlexStation 3 microplate reader (Molecular Devices, Sunnyvale, CA). Data (relative fluorescence or absorbance) were then recorded as a mean value of the triplicates for each sample versus time. Percent ATX, ENPP6, or ENPP7 activity (±SD) were calculated from the 1 h time point data for each inhibitor concentration, and GraphPad Prism version 5.0a (GraphPad Software, San Diego, CA) was then used to fit nonlinear regression curves, fitting the Hill slope from the data in a variable slope model and interpolating from the curve to determine the IC50 (±SD) for each compound. In the case of mechanism determination, the linear fluorescence data from 10–30 min were then transformed using a carboxyfluorescein standard curve to determine product concentration and plotted separately. Linear trend lines were inserted using Microsoft Excel, the slope of which represents the rates of reaction for each substrate concentration. This reaction rate data for each inhibitor concentration was then plotted against substrate (FS-3) concentration and simultaneously fitted via nonlinear regression in the Michaelis–Menten equations for competitive, noncompetitive, uncompetitive, and mixed-mode inhibition using GraphPad Prism, version 5.0a. The mechanism of inhibition was identified by determining which curve had the best global fit (R2 value).
Selective Dose-Dependent Inhibition of LPA1 Receptor by 3b, 3f, and 3g.
Cell Culture.
LPA1 and LPA3 RH7777 rat hepatoma cells were generated in-house as described previously39 and maintained in DMEM supplemented with 10% FBS and 2 mM l-glutamine with 250 μg/mL G418. LPA2 mouse embryonic fibroblast (MEF) cells were also derived from LPA1 and LPA2 double knockout mouse embryos and maintained in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. LPA4 CHO chinese hamster ovary cells were a generous gift from Dr. Takao Shimizu (Tokyo University, Tokyo, Japan) and were maintained in Ham’s F12 medium supplemented with 10% FBS, 2 mM l-glutamine, and 350 μg/mL G418. LPA5 B103 rat neuroblastoma cells were derived in-house via lentiviral transduction of FLAG-tagged LPA5 and puromycin selection and maintained in DMEM supplemented with 10% FBS and 0.4 μg/mL puromycin. All cells were maintained at 37 °C with 5% CO2 in a humidified atmosphere.
Calcium Mobilization Assay.
As published previously, LPAR activation leads to transient calcium mobilization. In order to assess receptor activation or antagonism, compounds were tested in stable transfectant cell lines engineered to overexpress a single LPAR subtype.35 At the level of each receptor, calcium mobilization was assessed via fluorescence in Fura-2AM-loaded cells treated with a dose range of test compound both in the absence and presence of the EC50 concentration of LPA 18:1 corresponding to the appropriate receptor subtype and cell line. All cells were plated in triplicate in 96-well, black-wall plates at a density of 5 × 104 cells per well and allowed to adhere overnight. LPA1 and LPA3 RH7777 cells were plated in poly-l-lysine coated plates. LPA5 B103 cells were plated in Matrigel-coated plates. After adherence, cells were serum-deprived in Krebs buffer consisting of 10 mM HEPES, pH 7.4, with 120 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.62 mM MgSO4, and 6 mM d-glucose. LPA1 RH7777, LPA3 RH7777, and LPA5 B103 cells were serum-deprived for 4 h, LPA2 MEF cells were serum-deprived for 1 h, and LPA4 CHO cells were not serum deprived. Fura-2AM was then loaded for 30 min at a concentration of 4.5 μg/mL in Krebs buffer with 0.45% (v/v) Pluronic F-127 (and additionally 0.1% BSA and 2.5 mM Probenicid for LPA4 CHO cells), after which cells were switched to fresh Krebs buffer. Finally, a FlexStation 3 microplate reader was used to apply a dose range of LPA 18:1 (in a 1:1 molecular complex with lipid-depleted BSA) or test compounds ranging from 0 to 100 μM in the presence and absence of the EC50 concentration of LPA 18:1 for each LPA receptor subtype. Fluorescence corresponding to calcium mobilization was immediately monitored upon addition every 3.42 s over a span of 70 s at excitation/emission wavelengths of 340/510 and 380/510 nm. Data (relative fluorescence) were then recorded as a mean fluorescence ratio value of the triplicates for each concentration and normalized to percentage of LPA 18:1 Emax. GraphPad Prism, version 5.0a, was then used to plot the data and fit nonlinear regression curves in a variable slope model in order to determine the pharmacodynamics (EC50 or IC50) of the compounds in Ca2+ mobilization.
In Vitro Invasion Assay.
The A2058 human melanoma cell line (gift from Dr. Timothy Clair, NCI, National Institutes of Health) was cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (V/V) FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine (Life Technologies). Cell invasion was performed using the 24-well BD Biocoat tumor invasion system (BD Biosciences, 8 μm-pore size). Briefly, the matrigel coating was rehydrated with PBS at 37 °C for 2 h. After removal of PBS, 5 × 104 A2058 cells in serum-free DMEM supplemented with 0.1% BSA were plated in each upper chamber. Serum-free DMEM/0.1% BSA (0.75 mL) containing chemoattractant (recombinant ATX plus 1 μM LPC) with or without the respective ATX inhibitors was added to the bottom chamber. Cells were allowed to invade the matrigel for 20 h at 37 °C. To stain the invaded cells, the media in the upper chamber was first removed and the inset was transferred to a new 24-well plate containing 4 μg/mL Calcein AM (Molecular Probes, Life Technologies) in Hank’s balanced salt solution (HBSS) and incubated for 1 h at 37 °C. The fluorescent invaded cells were measured using the FLEXstation II plate reader (Molecular Devices) at excitation and emission wavelengths of 485 and 530 nm, respectively.
Cell Growth Assay for Cytotoxicity and Chemoresistance of 4T1 Cells.
To assess cytotoxicity of benzenesulfonamide ATX inhibitors, wild-type 4T1 cells were plated in 96-well plates at a density of 7500 cells per well in RPMI with 1% (v/v) charcoal-stripped FBS and incubated for 4 h at 37 °C with 5% CO2. Cells were then treated for 48 h with 0–10 μM benzenesulfonamide ATX inhibitors or the reference ATX inhibitor 30 with daily refreshment of compound treatment. Viability was assessed via Promega CellTiter Blue reagent as per the manufacturer’s instructions and subsequently normalized to % vehicle control. For chemoresistance studies, wild-type 4T1 cells were plated in 96-well plates at a density of 5000 cells per well in RPMI with 1% (v/v) charcoal-stripped FBS and incubated overnight at 37 °C with 5% CO2. Cells were then treated for 24 h with 3 μM ATX or ATX/LPA1 inhibitors followed by 48 h treatment with a dose range of 0–1 μM PT in RPMI with 1% (v/v) charcoal-stripped FBS. After 48 h of incubation, cells were allowed to recover in RPMI with 10% FBS for an additional 72 h. Viability was then assessed via Promega CellTiter Blue following the manufacturer’s instructions and normalized to % vehicle control samples. All viability data were plotted using GraphPad Prism, and nonlinear regression analysis was performed in a variable slope model in order to determine LD50 concentrations for ATX inhibitors alone or PT in the presence and absence of ATX inhibitors. The 4T1-TaxR cell line was generated by treating wild-type 4T1 at increasing doses of PT from 40, 100, to 200 nM. Briefly, 1.5 × 105 cells per 10 cm dish were treated at each PT dose for 7 days with daily replenishment and allowed to recover for 7 days before the start of the next increment dose of PT treatment. To generate mammospheres, 3.5 × 103 4T1-TaxR cells were plated in each well of a 24-well plate in serum-free RPMI supplemented with 1× N-2 supplement, 20 ng/mL FGF, and 20 ng/mL EGF. Cells were cultured at 37 °C with 5% CO2 for 4 days to allow sphere formation. At day 4, spheres were treated with or without 100 nM PT and the various ATX inhibitors at 5 μM, as indicated, for 3 days. Viability was then assessed using the Promega CellTiter Blue reagent.
In Vivo Lung Metastasis Study.
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Tennessee Health Science Center. B16F10 murine melanoma cells were cultured in minimum essential medium (MEM) supplemented with 5% (V/V) heat-inactivated FBS, 2 mM l-glutamine (Life Technologies), 1% (v/v) MEM-vitamin solution, 1 mM sodium pyruvate, and 1% (v/v) nonessential amino acids (NEAA). Briefly, 1 × 105 cells were injected into the tail vein of 8–12 week old C57BL/6 mice purchased from Jackson Laboratory (Bar Harbor, ME). Mice were treated either with vehicle (PBS containing 1% DMSO and 50 μM charcoal-stripped mouse serum albumin (MSA)) or the ATX inhibitors as indicated. Treatment began at day 0 (prior to tumor inoculation) via intraperitoneal injection of 1 mg/kg of compound and continued daily up to day 10. All mice were sacrificed at day 21. The number of metastatic tumor nodules on the lungs was counted.
In Vitro Stability Determination by LC-MS.
To assess stability of amide (and thioamide) analogues in short-term in vitro assays, 4T1 mouse mammary carcinoma cells (1 × 104 cells per well) were plated in 48-well plates (a concentration equivalent to that used in the cytotoxicity/viability assay described). The cells were allowed to attach and recover overnight before 550 μL of fresh DMEM containing 1% charcoal-stripped FBS and 10 μM 3, 3b, 3f, or 14 was added (with a final concentration of 0.1% DMSO). Quadruplicate samples were taken at 0, 16 and 40 h post treatment for analysis by LC-MS/MS. All samples were centrifuged at 16 000g for 3 min at 4 °C to remove cell debris, and 500 μL aliquots were extracted twice with 500 μL of diethyl ether. The pooled organic extracts were dried under N2 before analysis by LC-MS/MS (for amide analogues 3, 3b, and 3f) or LC-MS for thioamide analogue 14) using a Thermo LTQ-XL mass spectrometer. Target compounds were isolated using a C18 column with a solvent gradient starting with 60% solvent A for 1 min with a linear gradient to 100% solvent B by 4 min. This composition was maintained for an additional 2 min before returning to 100% solvent A for a total run time of 8 min (solvent A: water + 0.025% NH4OH; solvent B: methanol + 0.025% NH4OH). The following parent to product ion MS/MS transitions were monitored for 3 (430.9 to 293.4 m/z), 3b (469.9 to 430.8 m/z), and 3f (484.8 to 448.8 m/z). Since MS/MS of 14 resulted in very low signals, this compound was analyzed following its parent ion at 482.7 m/z. Peak areas for each MS/MS transitions (or parent ion) were determined and normalized to the 0 h control.
ACKNOWLEDGMENTS
This work was supported by NIH/NCI grants CA-092160 to Drs. Gabor G. Tigyi and Duane D. Miller. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH/NCI.
ABBREVIATIONS USED
- ATX
autotaxin
- LPA
lysophosphatidic acid
- LPC
lysophosphatidylcholine
- LPAR
lysophosphatidic acid receptor
- GPCR
G protein-coupled receptor
- ENPP
ectonucleotide phosphodiesterase
- pNPPC
p-nitrophenylphophorylcholine
- IC50
half-maximum inhibitory concentration
- SAR
structure–activity relationship
- MOE
molecular operating environment
- pNP-TMP
p-nitrophenyl thymidylate
- DMEM
Dulbecco’s modified Eagle’s medium
- HBSS
Hank’s balanced salt solution
- IACUC
Institutional Animal Care and Use Committee
- NEAA
nonessential amino acids
- MSA
mouse serum albumin
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01270.
Molecular formula strings (CSV)
1H and 13C NMR for target compounds (PDF)
Accession Codes
3 (PDB 3NKM) and 3b (PDB 3NKM).
The authors declare no competing financial interest.
REFERENCES
- (1).Castagna D; Budd DC; Macdonald SJ; Jamieson C; Watson AJ Development of autotaxin inhibitors: an overview of the patent and primary literature. J. Med. Chem 2016, 59, 5604–5621. [DOI] [PubMed] [Google Scholar]
- (2).Fells JI; Lee SC; Norman DD; Tsukahara R; Kirby JR; Nelson S; Seibel W; Papoian R; Patil R; Miller DD; Parrill AL; Pham TC; Baker DL; Bittman R; Tigyi G Targeting the hydrophobic pocket of autotaxin with virtual screening of inhibitors identifies a common aromatic sulfonamide structural motif. FEBS J. 2014, 281, 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Albers HM; Ovaa H Chemical evolution of autotaxin inhibitors. Chem. Rev 2012, 112, 2593–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kihara Y; Mizuno H; Chun J Lysophospholipid receptors in drug discovery. Exp. Cell Res 2015, 333, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tigyi G Aiming drug discovery at lysophosphatidic acid targets. Br. J. Pharmacol 2010, 161, 241–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Brindley DN; Lin FT; Tigyi GJ Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2013, 1831, 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Baker DL; Fujiwara Y; Pigg KR; Tsukahara R; Kobayashi S; Murofushi H; Uchiyama A; Murakami-Murofushi K; Koh E; Bandle RW; Byun HS; Bittman R; Fan D; Murph M; Mills GB; Tigyi G Carba analogs of cyclic phosphatidic acid are selective inhibitors of autotaxin and cancer cell invasion and metastasis. J. Biol. Chem 2006, 281, 22786–22793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lee S-C; Fujiwara Y; Liu J; Yue J; Shimizu Y; Norman DD; Wang Y; Tsukahara R; Szabo E; Patil R; Banerjee S; Miller DD; Balazs L; Ghosh MC; Waters CM; Oravecz T; Tigyi GJ Autotaxin, lysophosphatidic acid receptors 1 and 5 exert different functions in tumor cells versus the host tissue microenvironment in melanoma invasion and metastasis. Mol. Cancer Res 2015, 13, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Seo EJ; Kwon YW; Jang IH; Kim DK; Lee SI; Choi EJ; Kim KH; Suh DS; Lee JH; Choi KU; Lee JW; Mok HJ; Kim KP; Matsumoto H; Aoki J; Kim JH Autotaxin regulates maintenance of ovarian cancer stem cells through lysophosphatidic acid-mediated autocrine mechanism. Stem Cells 2016, 34, 551–564. [DOI] [PubMed] [Google Scholar]
- (10).Albers HM; Hendrickx LJ; van Tol RJ; Hausmann J; Perrakis A; Ovaa H Structure-based design of novel boronic acid-based inhibitors of autotaxin. J. Med. Chem 2011, 54, 4619–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Albers HM; van Meeteren LA; Egan DA; van Tilburg EW; Moolenaar WH; Ovaa H Discovery and optimization of boronic acid based inhibitors of autotaxin. J. Med. Chem 2010, 53, 4958–4967. [DOI] [PubMed] [Google Scholar]
- (12).Castagna D; Duffy EL; Semaan D; Young LC; Pritchard JM; Macdonald SJF; Budd DC; Jamieson C; Watson AJB Identification of a novel class of autotaxin inhibitors through cross-screening. MedChemComm 2015, 6, 1149–1155. [Google Scholar]
- (13).Gupte R; Patil R; Liu J; Wang Y; Lee SC; Fujiwara Y; Fells J; Bolen AL; Emmons-Thompson K; Yates CR; Siddam A; Panupinthu N; Pham TC; Baker DL; Parrill AL; Mills GB; Tigyi G; Miller DD Benzyl and naphthalene methylphosphonic acid inhibitors of autotaxin with anti-invasive and anti-metastatic activity. ChemMedChem 2011, 6, 922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hoeglund AB; Bostic HE; Howard AL; Wanjala IW; Best MD; Baker DL; Parrill AL Optimization of a pipemidic acid autotaxin inhibitor. J. Med. Chem 2010, 53, 1056–1066. [DOI] [PubMed] [Google Scholar]
- (15).Stein AJ; Bain G; Prodanovich P; Santini AM; Darlington J; Stelzer NM; Sidhu RS; Schaub J; Goulet L; Lonergan D; Calderon I; Evans JF; Hutchinson JH Structural basis for inhibition of human autotaxin by four potent compounds with distinct modes of binding. Mol. Pharmacol 2015, 88, 982–992. [DOI] [PubMed] [Google Scholar]
- (16).Fells JI; Lee SC; Fujiwara Y; Norman DD; Lim KG; Tsukahara R; Liu J; Patil R; Miller DD; Kirby RJ; Nelson S; Seibel W; Papoian R; Parrill AL; Baker DL; Bittman R; Tigyi G Hits of a high-throughput screen identify the hydrophobic pocket of autotaxin/lysophospholipase D as an inhibitory surface. Mol. Pharmacol 2013, 84, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mize CD; Abbott AM; Gacasan SB; Parrill AL; Baker DL Ligand-based autotaxin pharmacophore models reflect structure-based docking results. J. Mol. Graphics Modell 2011, 31, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Barbayianni E; Magrioti V; Moutevelis-Minakakis P; Kokotos G Autotaxin inhibitors: a patent review. Expert Opin. Ther. Pat 2013, 23, 1123–1132. [DOI] [PubMed] [Google Scholar]
- (19).Katsamakas S; Bermperoglou E; Hadjipavlou-Litina D Considering autotaxin inhibitors in terms of 2D-QSAR and 3D-mapping-review and evaluation. Curr. Med. Chem 2015, 22, 1428–1461. [DOI] [PubMed] [Google Scholar]
- (20).Saga H; Ohhata A; Hayashi A; Katoh M; Maeda T; Mizuno H; Takada Y; Komichi Y; Ota H; Matsumura N; Shibaya M; Sugiyama T; Nakade S; Kishikawa K A novel highly potent autotaxin/ENPP2 inhibitor produces prolonged decreases in plasma lysophosphatidic acid formation in vivo and regulates urethral tension. PLoS One 2014, 9, e93230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hausmann J; Kamtekar S; Christodoulou E; Day JE; Wu T; Fulkerson Z; Albers HM; van Meeteren LA; Houben AJ; van Zeijl L; Jansen S; Andries M; Hall T; Pegg LE; Benson TE; Kasiem M; Harlos K; Kooi CW; Smyth SS; Ovaa H; Bollen M; Morris AJ; Moolenaar WH; Perrakis A Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol 2011, 18, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Nishimasu H; Okudaira S; Hama K; Mihara E; Dohmae N; Inoue A; Ishitani R; Takagi J; Aoki J; Nureki O Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol 2011, 18, 205–U271. [DOI] [PubMed] [Google Scholar]
- (23).Gierse J; Thorarensen A; Beltey K; Bradshaw-Pierce E; Cortes-Burgos L; Hall T; Johnston A; Murphy M; Nemirovskiy O; Ogawa S; Pegg L; Pelc M; Prinsen M; Schnute M; Wendling J; Wene S; Weinberg R; Wittwer A; Zweifel B; Masferrer J A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J. Pharmacol. Exp. Ther 2010, 334, 310–317. [DOI] [PubMed] [Google Scholar]
- (24).Furminger V; Hughes OR; Legrand DM; Stanley E; Thomson C Preparation of N-substituted nonaryl-heterocyclo carboxylate as autotaxin inhibitors. U.S. Pat. Appl. 2014/0171404 A1, 2014. [Google Scholar]
- (25).Kato K; Ikeda H; Miyakawa S; Futakawa S; Nonaka Y; Fujiwara M; Okudaira S; Kano K; Aoki J; Morita J; Ishitani R; Nishimasu H; Nakamura Y; Nureki O Structural basis for specific inhibition of autotaxin by a DNA aptamer. Nat. Struct. Mol. Biol 2016, 23, 395–401. [DOI] [PubMed] [Google Scholar]
- (26).Banerjee S; Smith J; Smith J; Faulkner C; Masterson DS A stereoselective cyclization strategy for the preparation of gamma-lactams and their use in the synthesis of alpha-methyl-beta-proline. J. Org. Chem 2012, 77, 10925–10930. [DOI] [PubMed] [Google Scholar]
- (27).Pelcman B; Olofsson K; Suna E; Kalvins I; Ozola V; Andrianov V Disulfonamides useful in the treatment of inflammation. Int. Pat. Appl. WO2008129276 A1, 2008. [Google Scholar]
- (28).Rashad AA; Jones AJ; Avery VM; Baell J; Keller PA Facile synthesis and preliminary structure-activity analysis of new sulfonamides against trypanosoma brucei. ACS Med. Chem. Lett 2014, 5, 496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cholody WM; Zang Y; Zuck K; Watthey JWH; Ohler Z; Strovel J; Ohler NE; Chellappan S; Padia J Derivatives of multi-ring aromatic compounds and uses as anti-tumor agents. Int. Pat. Appl. WO 2010082912 A1, 2010. [Google Scholar]
- (30).Alyar S;Özbek N; Kuzukıran K; Karacan N Quantitative structure–activity relationships studies for prediction of antimicrobial activity of synthesized disulfonamide derivatives. Med. Chem. Res 2011, 20, 175–183. [Google Scholar]
- (31).Cacchi S; Fabrizi G; Goggiamani A; Licandro E; Maiorana S; Perdicchia D Synthesis of N,N-Dialkyl-N’-arylhydrazines via palladium-catalyzed N-arylation by using N,N-dialkylhydrazines/2LiCl adducts. Org. Lett 2005, 7, 1497–1500. [DOI] [PubMed] [Google Scholar]
- (32).De Luca L; Giacomelli G An easy microwave-assisted synthesis of sulfonamides directly from sulfonic acids. J. Org. Chem 2008, 73, 3967–3969. [DOI] [PubMed] [Google Scholar]
- (33).Kim JG; Jang DO Mild and efficient indium metal catalyzed synthesis of sulfonamides and sulfonic esters. Synlett 2007, 2007, 2501–2504. [Google Scholar]
- (34).Rosen BR; Ruble JC; Beauchamp TJ; Navarro A Mild Pd-catalyzed N-arylation of methanesulfonamide and related nucleophiles: avoiding potentially genotoxic reagents and byproducts. Org. Lett 2011, 13, 2564–2567. [DOI] [PubMed] [Google Scholar]
- (35).Zhang H; Xu X; Gajewiak J; Tsukahara R; Fujiwara Y; Liu J; Fells JI; Perygin D; Parrill AL; Tigyi G; Prestwich GD Dual activity lysophosphatidic acid receptor pan-antagonist/autotaxin inhibitor reduces breast cancer cell migration in vitro and causes tumor regression in vivo. Cancer Res. 2009, 69, 5441–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Venkatraman G; Benesch MG; Tang X; Dewald J; McMullen TP; Brindley DN Lysophosphatidate signaling stabilizes Nrf2 and increases the expression of genes involved in drug resistance and oxidative stress responses: implications for cancer treatment. FASEB J. 2015, 29, 772–785. [DOI] [PubMed] [Google Scholar]
- (37).Hrast M; Turk S; Sosic I; Knez D; Randall CP; Barreteau H; Contreras-Martel C; Dessen A; O’Neill AJ; Mengin-Lecreulx D; Blanot D; Gobec S Structure-activity relationships of new cyanothiophene inhibitors of the essential peptidoglycan biosynthesis enzyme MurF. Eur. J. Med. Chem 2013, 66, 32–45. [DOI] [PubMed] [Google Scholar]
- (38).Norman DD; Ibezim A; Scott WE; White S; Parrill AL; Baker DL Autotaxin inhibition: development and application of computational tools to identify site-selective lead compounds. Bioorg. Med. Chem 2013, 21, 5548–5560. [DOI] [PubMed] [Google Scholar]
- (39).Halgren TA MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem 1999, 20, 720–729. [DOI] [PubMed] [Google Scholar]