

Abstract
Background
Acquired von Willebrand syndrome (AVWS) has been associated with monoclonal gammopathy of undetermined significance (MGUS), with limited data on its management.
Methods
We conducted a systematic literature search in Medline (Ovid), Embase, and Scopus up to September 11, 2019, for studies reporting on the management of AVWS associated with MGUS (AVWS‐MGUS). Data on patient characteristics, laboratory parameters at presentation, and clinical and laboratory outcomes were extracted.
Objectives
To describe the clinical presentation and outcomes of different therapeutic approaches.
Results
Seventy‐five studies were included in the final review, for a total of 137 patients. Most patients had von Willebrand factor ristocetin cofactor activity <30 IU/dL (86.6%) and factor VIII levels <50 IU/dL (91.8%). Bleeding severity ranged from no bleeding (16.1%) to minor bleeding (46.4%) and major bleeding (37.5%). The overall clinical success rates for 1‐deamino‐8‐D‐arginine vasopressin (DDAVP), factor replacement therapy, and intravenous immunoglobulin (IVIG) were 43.8%, 33.3%, and 85.4%, respectively. The laboratory response rates for DDAVP, factor replacement therapy, and IVIG were 39.0%, 62.9%, and 88.6%, respectively. Several other treatments were also reported in small numbers, out of which myeloma‐directed therapies, plasma exchange, recombinant factor VIIa, and antifibrinolytics appeared most successful, while immunosuppressive agents were largely ineffective.
Conclusion
IVIG appears to be an effective treatment for AVWS‐MGUS bleeding, conferring a high clinical success rate with measurable laboratory outcomes; albeit temporary. DDAVP and factor replacement therapy may be partially successful in controlling minor bleeds, but not major bleeds. Other less commonly used agents may be effective in certain cases, although data are limited.
Keywords: DDAVP, IVIG, monoclonal gammopathy of undetermined significance, treatment, von Willebrand disease
Essentials.
Acquired von Willebrand syndrome is a rare complication of monoclonal gammopathy of undetermined significance with limited data on management.
It can present with spontaneous bleeding, with severity ranging from mild to life‐threatening.
A scoping review of the literature suggests that intravenous immunoglobulin may be the most effective hemostatic treatment.
1‐Deamino‐8‐D‐arginine vasopressin (DDAVP) may be successful in controlling minor bleeds but not major bleeds.
1. INTRODUCTION
Acquired von Willebrand syndrome (AVWS) is a rare bleeding disorder that presents with laboratory findings and clinical features similar to that of congenital von Willebrand disease (VWD). However, unlike congenital VWD, AVWS arises in individuals with no prior or family history of bleeding. AVWS has been reported to occur in association with various underlying disorders, most frequently with lymphoproliferative disorders, myeloproliferative neoplasms, and cardiovascular diseases. 1 Among the lymphoproliferative disorders, monoclonal gammopathy of undetermined significance (MGUS) is the most frequent association 2 and is considered as one of the main disorders related to the evolving concept of “monoclonal gammopathy of clinical significance.” 3
MGUS, in itself, does not require treatment. However, AVWS associated with MGUS (AVWS‐MGUS) can present with severe spontaneous bleeding, with a severity that ranges from mild to life‐threatening. Diagnosing this disorder requires a high index of suspicion and a thorough clinical and laboratory evaluation before treatment. Similar to congenital VWD, treatment with 1‐deamino‐8‐D‐arginine vasopressin (DDAVP) and von Willebrand factor (VWF)‐containing concentrate has been used. However, unlike the response in congenital VWD, the response to treatment for AVWS‐MGUS can be highly variable and is often limited in duration. As such, adjunct therapies have been used, which include intravenous immunoglobulin (IVIG), plasmapheresis, and immunomodulatory agents such as lenalidomide, with the goal to induce the bleeding symptoms of AVWS into remission. The rationale behind the use of these treatment modalities is based on the current understanding of the pathophysiology of this disease.
The pathophysiology of AVWS‐MGUS likely involves an accelerated immunologic clearance of circulating VWF as a result of direct binding to the monoclonal antibody. 4 , 5 , 6 , 7 , 8 Evidence from a few studies suggests that monoclonal IgG in patients with IgG MGUS binds to VWF/factor VIII (FVIII) complexes in vivo, through a binding site located on the VWF molecule. 9 The complexes are subsequently rapidly removed by the reticuloendothelial system through an Fc‐receptor–dependent mechanism. 4 , 5 , 6 , 7 , 8 Studies by Gan et al 8 , 10 and van Genderen et al 8 , 10 demonstrated a preferential binding of the monoclonal antibody to high‐ and intermediate‐molecular‐weight VWF multimers, leading to the preferential clearance of large VWF multimers, a pattern similar to type 2A VWD.
While several reviews in the literature highlight AVWS as a general overview, comprehensive reviews in the literature summarizing the evidence for the management of AVWS‐MGUS are lacking. 1 , 11 , 12 This presents a real challenge in the field, since currently available data do not provide sufficient information to guide evidence‐based management of this challenging disorder. Currently, relevant data are limited to case reports and small case series—literature that is highly subjected to publication bias. Furthermore, these cases have additional significant limitations, including inconsistent reporting of key information related to diagnosis and a lack of standardized reporting of laboratory findings and outcomes. In addition, the treatment approaches reported are highly variable, highlighting the lack of a standard of care approach in management.
Given the significant limitations of current data, as well as the variable treatment approaches used, we conducted a scoping review of the medical literature using the framework as recommended by Arksey and O’Malley. 13 This method has become increasingly common in recent years, since scoping reviews allow for inclusion of a greater, more flexible range of study designs and standards in comparison to systematic reviews. 14 Systematic reviews, on the other hand, require specific study types, such as randomized control trials, that must meet certain quality standards in order to be included. 13 Scoping reviews have been useful in providing clinical guidance in the management of disorders on which individual studies may be insufficient to suggest therapeutic approaches, while a summation of available data may be used to provide useful recommendations. 15 , 16 , 17 For a rare disorder such as AVWS‐MGUS where there is a paucity of large studies, and data are limited to case reports or small series of different designs, a scoping review was the more appropriate approach for our study. This allowed us to provide an overview of the existing evidence regardless of methodological quality or risk of bias.
The purpose of this study was to describe the clinical presentation, laboratory assessments of MGUS and AVWS, and outcomes of therapeutic approaches for AVWS‐MGUS, from a clinical and laboratory perspective.
2. METHODS
2.1. Protocol and registration
The review was conducted and reported in line with the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses extension for Scoping Reviews guidelines scoping reviews. 18 The protocol for this scoping review was submitted to an internal registry hosted at the University of Utah Eccles Health Sciences Library.
2.2. Eligibility criteria
To determine study eligibility, the following criteria were applied to the studies identified in the initial search: Patients had a diagnosis of MGUS with no other concomitant lymphoproliferative disorder that would have warranted treatment; patients had a diagnosis of AVWS based on clinical presentation and laboratory parameters; and the study reported on therapeutic agents used in the management of AVWS and its outcomes. All studies, including conference abstracts, case reports, case series, and retrospective studies, containing a patient population of n ≥ 1 were included. Only studies published in English were considered. Review papers, editorials, and commentaries were excluded from this scoping review.
2.3. Search strategy
We conducted a comprehensive search in Medline (Ovid), Embase (embase.com) and Scopus (scopus.org) from 1946 to September 11, 2019. The search strategies used for each database are detailed in Supporting Information S1. The results of all searches were entered into Covidence software (Covidence, Melbourne, Australia) for analyses.
2.4. Study selection
After duplicates were removed, all titles and abstracts of the literature search results were screened by two authors (YAI and MYL) to the eligibility criteria to determine whether the study should receive a more in‐depth review. All potentially eligible studies were then independently reviewed by both authors. Disagreements about the inclusion of studies were resolved, where necessary, by consultation with a third author (GR). There were no conflicts.
2.5. Data collection and outcomes
Data extraction was undertaken independently by two authors (YAI and MYL) using a standardized data extraction form, which was designed beforehand. Any discrepancies in interpretation between the two reviewers were resolved through a discussion of the text of the original articles. The following data were extracted from all included studies: author; year of publication; patient demographics, type of monoclonal gammopathy (IgG, IgA, or IgM), monoclonal spike (in grams per deciliter), type of bleeding presentation, partial thromboplastin time (PTT), baseline factor FVIII/VWF parameters, including FVIII coagulant activity (FVIII:C), VWF antigen (VWF:Ag) level, VWF ristocetin cofactor activity (vWF:RCo), VWF propeptide (VWFpp), VWF multimers; and type and outcome of therapeutic interventions used, including FVIII/VWF parameters in response to the intervention (if reported). The baseline FVIII/VWF parameters were not reported in every study; if not reported, this was documented as “not documented.” For VWF activity, we opted to document only vWF:RCo, as this was the most commonly used laboratory assay used in the studies. The bleeding presentation was categorized on the basis of location and severity. For severity of bleeding, the International Society on Thrombosis and Haemostasis definition for major and minor bleeding were used. 19 , 20 We did not use the definition of clinically relevant nonmajor bleeding, as this definition requires that either a medical intervention or a hospitalization occurred, which was not helpful for classification given that this study was looking at outcomes of therapeutic interventions. 21 As almost all patients presented with multiple types of bleed, we reported the highest severity of bleed for each patient.
2.6. Synthesis of results
Due to the heterogeneity of outcomes of therapeutic agents reported in the studies, findings were aggregated to report a clinically meaningful account of the included literature.
Outcomes for the three most common therapeutic agents—DDAVP, factor replacement therapy, and IVIG—were reported via two methods: clinical outcomes and laboratory outcomes.
For other therapeutic agents, when used either on its own or in combination with factor replacement therapy and/or IVIG, outcomes were reported on the basis of clinical outcomes only.
2.6.1. Clinical outcomes
Clinical outcomes were defined as a clinical success if there was cessation of bleeding symptoms or prevention of bleeding complications for surgical procedures, regardless of laboratory parameters. If the therapeutic intervention resulted in no cessation of bleeding, or did not prevent bleeding complications after a surgical procedure, this was defined as a clinical failure.
2.6.2. Laboratory outcomes
FVIII/VWF parameters and timing of the parameters after therapeutic interventions were used to determine the laboratory outcomes. The type of FVIII/VWF parameters performed after therapeutic interventions were not reported consistently in all studies. In addition, the format through which the results were reported differed between studies and used either numerical values, graphical representations, or verbal descriptions (ie, showed good response, observed a minimal rise, had no response, reached normal level, total correction). Only studies that included numerical values or interpretable graphical representations for FVIII/VWF parameters were used to determine laboratory outcomes. Responders to a therapeutic agent were defined as those in whom (i) VWF:RCo increased to ≥50 IU/dL and (ii) ≥2‐fold increase above their baseline levels at any time points after the therapeutic intervention (whichever is highest, if multiple time points available). We used VWF:RCo, as this was the most commonly reported assay used in laboratory measures. In selected studies where VWF:RCo was not used, we then used other VWF/FVIII parameters to determine laboratory outcomes and are reported separately. We excluded studies that used verbal descriptions to document response to the intervention due to the variability in interpretation among studies on what constitutes a response or normal levels. If a response was achieved for either DDAVP or factor replacement therapy, we then determined duration of response, which was categorized into <6 or ≥6 hours. We selected 6 hours as the cutoff as most studies collected serial VWF/FVIII parameters at either 4 or 6 hours. In selected studies, duration of response was not given; instead, the half‐life of the response was given, which we reported separately. If a response was achieved for IVIG or in combination with factor replacement therapy, we report the duration of the response in days (if available).
3. RESULTS
3.1. Study characteristics
A total of 1164 citations were identified (Figure 1). After duplicate removal, 1157 references were screened by title and abstract review. All studies that did not meet the inclusion criteria based on the study title or abstract were excluded first. Subsequently, there were 324 full‐text articles and conference abstracts, which were then reviewed in‐depth for eligibility, out of which 75 were identified as meeting the inclusion criteria. This included 10 conference abstracts that were not published as full‐text articles elsewhere. A list of all included studies is available in Supporting Information S2. The flowchart for the studies included in the scoping review and the primary reasons for study exclusion are described in Figure 1.
FIGURE 1.
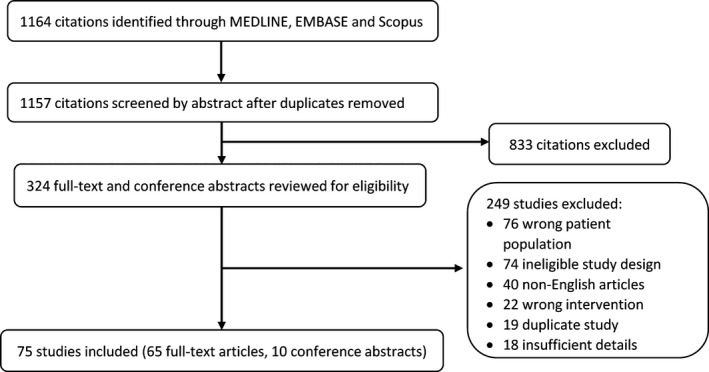
Flowchart for the studies included in the scoping review
3.2. Patient and disease characteristics
Seventy‐five case reports and case series, consisting of 65 full‐text articles and 10 conference abstracts, which included 137 patients, reported the outcomes of therapeutic approaches for AVWS‐MGUS, from either a clinical and/or laboratory perspective.
Of the total cohort of 137 patients, 112 had data linked to individual patients, which were used for the baseline patient characteristics analysis (Table 1). The median age of onset of AVWS‐MGUS was 61.5 years (interquartile range, 52‐72), and the mean age of onset was 60.6 years (standard deviation,14.5). Almost two‐thirds of patients were men (n = 73; 65.2%). The most common MGUS subtype was IgG (n = 90; 87%), followed by IgM (n = 10; 10%). There were three patients presenting with both IgG and IgM subtype, one with IgA, and eight were not documented. Forty patients had a quantitative monoclonal band (M‐spike) reported; of which 95% (n = 38) had an M‐spike < 1 g/dL. The lower limit of detectable VWF:Ag and VWF:RCo varied notably in the studies in which they were reported. The majority of patients presented with VWF:Ag <30 IU/dL (86.6%) and VWF:RCo < 30 IU/dL (94.4%). One hundred patients (91.8%) presented with FVIII:C levels <50 IU/dL. Fifty‐eight patients had a multimeric pattern of VWF reported—absent or relative decrease of the high‐molecular‐weight and/or intermediate‐molecular‐weight multimers (n = 32; 56.1%), normal multimers (n = 16; 28.1%), decrease of all multimers (n = 4; 7.0%), absence of all multimers (n = 2, 3.5%), and abnormal multimeric pattern (n = 3; 5.3%). We were not able to clinically aggregate data on PTT and VWFpp across studies due to data heterogeneity.
TABLE 1.
Baseline characteristics of 112 patients with AVWS‐MGUS
| Characteristics | n | % |
|---|---|---|
| Age at onset, y | ||
| Mean | 60.6 | |
| Median | 61.5 | (IQR, 52‐72) |
| Sex | ||
| Men | 73 | 65.2 |
| Women | 39 | 34.8 |
| MGUS subtype | ||
| IgG | 90 | 86.5 |
| IgM | 10 | 9.6 |
| IgA | 1 | 1.0 |
| IgG and IgM | 3 | 2.9 |
| Not documented | 8 | |
| M‐spike | ||
| <1 g/dL | 38 | 95.0 |
| ≥1 g/dL | 2 | 5.0 |
| Not documented | 72 | |
| VWF:Ag | ||
| <15 IU/dL | 57 | 64.1 |
| 15‐29 IU/dL | 20 a | 22.5 |
| 30‐50 IU/dL | 5 | 5.6 |
| >50 IU/dL | 4 | 4.5 |
| Decrease | 1 | 1.1 |
| Absent/undetectable | 2 | 2.2 |
| Not documented | 23 | |
| VWF activity | ||
| <15 IU/dL | 81 | 76.4 |
| 15 ‐ 29 IU/dL | 19 b | 18.0 |
| 30 ‐ 50 IU/dL | 3 | 2.8 |
| Decrease | 1 | 0.9 |
| Absent/undetectable | 2 | 1.9 |
| Not documented | 6 | |
| FVIII | ||
| <50 IU/dL | 100 | 91.8 |
| ≥50 IU/dL | 8 | 7.3 |
| Decrease | 1 | 0.9 |
| Not documented | 3 | |
| Multimeric pattern | ||
| Absent or relative decrease of the HMW and/or IMW | 32 | 56.1 |
| Normal | 16 | 28.1 |
| Decrease of all multimers | 4 | 7.0 |
| Absent of all multimers | 2 | 3.5 |
| Abnormal multimeric patterns | 3 | 5.3 |
| Not documented | 55 | |
Abbreviations: AVWS, acquired von Willebrand syndrome; FVIII, factor VIII; HMW, high molecular weight; IMW, intermediate molecular weight; MGUS, monoclonal gammopathy of undetermined significance; VWF, von Willebrand factor; VWF:Ag, von Willebrand factor antigen; VWF:RCo, von Willebrand factor ristocetin cofactor.
This includes two patients with VWF:Ag levels documented as 26‐37 IU/dL and three patients with VWF:Ag levels documented as 9‐23 IU/dL.
This includes four patients with VWF:RCo documented as < 20 IU/dL and three patients with vWF:RCo documented as < 10‐20 IU/dL.
The most common bleeding type reported was epistaxis/gum bleeding (n = 36; 32.1%), followed by gastrointestinal bleed/angiodysplasia (n = 34; 30.4%) (Table 2). In terms of severity, 37.5% (n = 42) of patients had major bleeding, 46.4% (n = 52) had minor bleeding, and 16.1% (n = 18) reported no bleeding. For the “no bleeding” group, the diagnosis of AVWS was discovered fortuitously as a result of routine coagulation screening or during preoperative coagulation testing. The confirmation of an AVWS diagnosis, as opposed to a laboratory artefactual error, was based on the authors’ interpretation of these assays. The “no bleeding” group of patients received therapeutic interventions to determine their laboratory outcome or for hemostatic support peri‐ and postoperatively.
TABLE 2.
Location and types of bleeding events
| Location/type of bleed | na | % |
|---|---|---|
| Epistaxis/gum bleeding | 36 | 32.1 |
| Gastrointestinal bleed/angiodysplasia | 34 | 30.4 |
| None/not documented | 19 | 17.0 |
| Ecchymoses/bruising | 17 | 15.2 |
| Postsurgery complications | 17 | 15.2 |
| Hematoma | 16 | 14.3 |
| Menorrhagia | 9 | 8.0 |
| Post‐dental complications | 7 | 6.3 |
| Hematuria/genitourinary bleed | 3 | 2.7 |
| Prolonged bleeding after minor cuts | 3 | 2.7 |
| Intracerebral hemorrhage | 2 | 1.8 |
N = 112. Majority of patients presented with > 1 type of bleed.
3.3. Treatment‐related outcomes
We summarize the clinical and laboratory outcomes for the three most common therapeutic interventions in Tables 3 and 4, respectively.
TABLE 3.
Clinical outcomes for therapeutic agents used
| Therapeutic Agent | Reason for using each therapeutic agent | ||||
|---|---|---|---|---|---|
| Major bleeding | Minor bleeding | Perioperative | Unclear | Total | |
|
DDAVP, n = 16 (%) |
0/5 (0) | 4/6 (66.7) | 3/4 (75) | 0/1 b (0) | 7/16 (43.8) |
| Factor replacement therapy, n= 33, (%) | 2/15 (13.3) | 4/9 (44.4) | 5/9 (55.6) | 0 | 11/33 (33.3) |
|
IVIG, n= 48 (%) |
13/17 (76.5 | 6/6 (100) | 13/14 (92.9) | 9/11 a (81.8) | 41/48 (85.4) |
| Factor replacement therapy + IVIG, n= 6 (%) | 1/1 (100) | 0 | 3/5 (60) | 0 | 4/6 (66.7) |
TABLE 4.
Laboratory outcomes for therapeutic agents used
| Therapeutic agent | Response rate |
|---|---|
|
DDAVP, n= 41 (%) |
16 a (39.0) |
| Factor replacement therapy, n= 35 (%) | 22(62.9) |
|
IVIG, n= 44 (%) |
39 (88.6) |
| Factor replacement therapy + IVIG, n= 3 (%) | 2 (66.7) |
Abbreviations: DDAVP, desmopressin; IVIG, intravenous immunoglobulin
In one patient, the results of von Willebrand factor GP1b activity was used.
3.3.1. DDAVP
Thirty‐three studies, which included 59 patients, reported the use of DDAVP in AVWS‐MGUS.
3.3.1.1. Clinical outcome
Sixteen patients received DDAVP only for clinical management of bleeding symptoms (major, n = 5; minor, n = 6), for perioperative hemostatic support (n = 4) and for unclear indications (n = 1). DDAVP was clinically effective as a hemostatic agent in seven cases (Table 3). Four (66.7%) cases were for minor bleeds, and three (75%) cases were for perioperative hemostatic support, corresponding to an overall success rate of 43.8%.
3.3.1.2. Laboratory outcome
Forty‐one patients had numerical values or interpretable graphical representations for the FVIII/VWF parameters that were used to determine laboratory outcomes (Table 4). Only 16 patients (39.0%) satisfied the predetermined criteria for response. The majority of responses were short‐lived with a return to baseline within 6 hours (n = 12) or had a short half‐life reported (n = 2). Two patients had a documented duration of response of ≥6 hours.
3.3.2. Factor replacement therapy
Fifty‐five studies were identified in which a total of 78 patients with AVWS‐MGUS received factor replacement therapy, including VWF/FVIII concentrate, cryoprecipitate, or fresh‐frozen plasma.
3.3.2.1. Clinical outcome
For patients where only factor replacement therapy was used (n = 33), the treatment was effective in 11 cases (major, n = 2; minor, n = 4; perioperative, n = 5), corresponding to a success rate of 33.3% (Table 3).
3.3.2.2. Laboratory outcome
Laboratory measures when factor replacement therapy only was used were available for 35 patients, of which 22 (62.9%) satisfied the predetermined criteria for response (Table 4). Similar to the duration of response with DDAVP, the majority of responses to factor replacement therapy were short‐lived with a return to baseline within 6 hours (n = 18) or had a short half‐life reported (n = 3). One patient had a documented duration of response lasting 24 hours.
3.3.3. IVIG
Forty‐nine studies were identified in which a total of 89 patients with AVWS‐MGUS received IVIG.
3.3.3.1. Clinical outcome
For patients in whom only IVIG was used (n = 48), the treatment was effective in 41 cases (major, n = 13; minor, n = 6; perioperative, n = 13; unclear indication, n = 9), corresponding to a success rate of 85.4%.
3.3.3.2. Laboratory outcome
Laboratory measures after IVIG were interpretable for 44 patients, of which 39 (88.6%) satisfied the predetermined criteria, with the duration of response ranging between 10 and 35 days.
3.3.4. Combination of factor replacement and IVIG
3.3.4.1. Clinical outcome
Six patients received both factor replacement therapy and IVIG for major bleed (n = 1) and perioperative hemostatic support (n = 5). This combination was effective in four cases (66.7%).
3.3.4.2. Laboratory outcome
Three patients who received both factor replacement therapy and IVIG had numerical values or interpretable graphical representations for the FVIII/VWF parameters, of which two (66.7%) responded to the combined treatment. The duration of response was 4 days and 9 days in the two responders.
3.3.5. Other treatments
Various other treatments were attempted in different studies. The outcomes of these results are summarized in Table 5. Immunosuppressive agents such as rituximab, cyclophosphamide, and azathioprine were ineffective in all cases except for one, where rituximab was used in conjunction with steroids. Steroids used individually were successful in only two of nine cases; however, the agents, doses, and methods of administration varied in those studies. Myeloma‐directed therapies, including lenalidomide, thalidomide, or bortezomib, were successful in four of five cases. Plasmapheresis was effective in four of six cases. In all four successful cases, either factor replacement therapy and/or IVIG was infused after plasmapheresis. Antifibrinolytic agents and recombinant FVIIa (rFVIIa) were successful in 8 of 13 and 6 of 6 patients, respectively, and often used in conjunction with factor replacement therapy and/or IVIG.
TABLE 5.
Outcomes of other treatment options
| Treatment options | Success rate | Concomitant therapy |
|---|---|---|
| a) Immunosuppressive therapies | ||
| Azathioprine | 0/2 | |
| Melphalan | 0/1 | |
| Rituximab | 0/6 | |
| Rituximab + bendamustine | 0/1 | |
| Rituximab + steroids | 1/2 | |
| Chlorambucil + steroids | 0/1 | |
| Dapsone + steroids | 0/1 | |
| MMF + steroids | 1/1 | 1 case with IVIG |
| Cyclophosphamide + steroids | 0/2 | |
| Cyclophosphamide | 0/1 | |
| Steroids | 2/9 | |
| b) Myeloma‐directed therapies | ||
| Thalidomide | 1/1 | |
| Lenalidomide | 2/2 | 1 case with factor replacement therapy |
| Bortezomib + steroids | 1/2 | |
| c) Hemostatic agents | ||
| Antifibrinolytics | 8/13 |
Success 1 case with rFVIIa 3 cases with factor replacement therapy 1 case with factor replacement therapy and/or IVIG 1 case with DDAVP 1 case with estrogen Failure 2 cases with factor replacement therapy 1 case with steroids |
| rFVIIa | 6/6 |
1 case with IVIG 1 case with factor replacement therapy and antifibrinolytic agent 1 case with factor replacement therapy, IVIG, and antifibrinolytic agent |
| d) Other | ||
| Plasmapheresis | 4/6 |
Success 2 cases with factor replacement therapy and IVIG 1 case with factor replacement therapy 1 case with IVIG Failure 1 case with factor replacement therapy |
| Romiplostim a | 1/1 | |
Abbreviations: DDAVP, desmopressin; IVIG, intravenous immunoglobulin; MMF, mycophenolate mofetil; rFVIIa, recombinant factor VIIa.
Presented with type 2B‐like acquired von Willebrand syndrome with thrombocytopenia.
4. DISCUSSION
Our review indicates that while the data on AVWS‐MGUS are scarce, several relevant observations can be drawn from it collectively. When we compared the baseline characteristics of our cohort to overall MGUS epidemiologic data, a similar sex difference is noted, where men were twice as likely as women to be diagnosed. 22 , 23 However, some differences were observed. AVWS‐MGUS patients were likely to be younger at diagnosis than MGUS patients (60.8 years vs 70 years). 22 This may be caused by symptomatic bleeding with AVWS, leading to an earlier diagnosis. There was an overrepresentation of IgG subtype in our cohort, higher than the general incidence of IgG subtype in MGUS overall (87% vs 69%). 24 It has previously been demonstrated in a few studies that monoclonal IgG binds to the VWF directly on a binding site on the VWF molecule. 9 Our observation of higher prevalence of IgG MGUS suggests that monoclonal IgG may perhaps bind more readily to VWF than non‐IgG monoclonal antibodies, although such a conclusion would require further studies on the interaction of these other subtypes with VWF.
In patients in whom multimeric analysis was reported, a majority had decreased intermediate‐molecular‐weight or high‐molecular‐weight multimers (56.1%), resembling a type 2A or type 2B VWD pattern, and 28.1% had normal multimers suggestive of a type 1 pattern. This observation is consistent with findings by Gan et al 8 , 10 and van Genderen et al 8 , 10 that demonstrated a preferential binding of the monoclonal antibody to high‐ and intermediate‐molecular‐weight VWF multimers, leading to increased clearance of these multimers. 8 , 10 The bleeding phenotype of AVWS‐MGUS appears to vary in severity, with the majority of patients in our study having mild or no bleeding (total, 62.5%). However, the remaining patients had a severe and often life‐threatening bleed.
There have been many different management approaches for the bleeding phenotype of this disorder in the literature. In general, our scoping review shows that both DDAVP, which promotes increased endogenous release of VWF/FVIII, and factor replacement therapy offer limited treatment success in the hemostatic management of AVWS‐MGUS. The subsequent elevations of VWF/FVIII were transient and short‐lived in laboratory studies. Clinically, both agents were poorly effective in controlling major bleeds, but had better success in controlling minor bleeds (DDAVP, 66.7%; factor replacement therapy, 55.6%). Thus, we suggest the use of DDAVP as first line for minor bleeds in AVWS‐MGUS, factoring into consideration its cost, availability, and side effects in comparison to factor replacement therapy (Figure 2).
FIGURE 2.
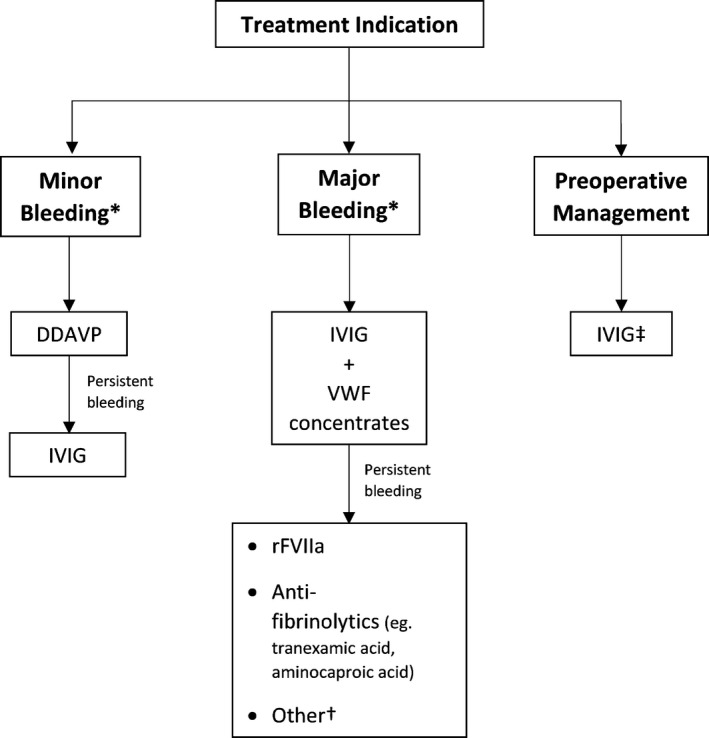
Proposed algorithm for choice of hemostatic agent for AVWS‐MGUS based on treatment indication. *Per ISTH criteria. †Other considerations include plasmapheresis and myeloma‐directed therapies. ‡ IVIG should be initiated a day prior to surgery, with repeat laboratory testing the day of surgery. Proceed only if VWF and FVIII levels are above target levels; otherwise, addition of VWF‐containing concentrate to IVIG may be necessary. Postoperative bleeding can be managed similarly to major bleeding in the above algorithm. FVIII, factor VIII; ISTH, International Society on Thrombosis and Haemostasis; IVIG, intravenous immunoglobulin; AVWS‐MGUS, acquired von Willebrand syndrome associated with monoclonal gammopathy of undetermined significance; VWF, von Willebrand factor
The limited response of DDAVP and factor replacement therapy is expected, given that the pathophysiology of AVWS‐MGUS likely involves an accelerated immunologic clearance of circulating VWF as a result of direct binding to the monoclonal antibody. 4 , 5 , 6 , 7 , 8 Thus, it would also be expected that treatments that eradicate or neutralize the antibody would be successful, which was a finding that we observed in our review. One such treatment is IVIG, which has been shown to be beneficial in various immune‐mediated disorders due to various mechanisms including neutralizing the autoantibodies through the effect of anti‐idiotypic antibodies, as well as modulation of B‐cell and T‐cell effects. 25 In our study, IVIG had an overall success rate of 85.4% in managing all bleeding or perioperative cases, including major bleeds, where its success rate was 76.5%—a rate noticeably higher than that of DDAVP or factor replacement therapy. Plasmapheresis, which leads to temporary clearance of the autoantibodies, when given in conjunction with factor replacement therapy and/or IVIG, also appears to offer clinical success, as it was effective in four of six cases. It is worth noting that the laboratory effect of IVIG is temporary and lasted between 10 and 21 days on average. However, from a management perspective, the high rate of success of IVIG as a temporizing measure for symptomatic bleeds or surgical procedures bears more clinical relevance than the duration of its laboratory effect, offering a rationale for its use for these indications. In patients with recurrent major bleeds, an alternative approach to be considered is the administration of IVIG at regular time intervals (eg, every 3 weeks); however, this has not been well studied in this setting and may not be cost‐effective. Such patients may be indicated for myeloma‐directed therapies, which is further discussed below. We suggest the use of IVIG, in combination with VWF/FVIII concentrate, as first‐line treatment for all acute major bleeds. In patients with or without bleeding symptoms who are undergoing surgery, we recommend IVIG for preoperative management (Figure 2). We suggest an IVIG dose of 400 mg/kg for 5 days or 1 g/kg for 2 days, with modifications as needed for renal dysfunction.
We also reviewed the effect of other less commonly used treatment approaches in our review. Immunosuppressive agents such as rituximab, cyclophosphamide, and azathioprine were largely ineffective. This provides further evidence that the cause of AVWS in MGUS is likely a direct result of monoclonal antibodies binding to VWF and leading to its accelerated immunologic clearance, since these immunosuppressive treatments do not affect the clonal plasma cells that produce the monoclonal antibodies. Myeloma‐directed therapies, including lenalidomide, thalidomide or bortezomib, were successful in four of five cases. However, it remains unclear whether they should be used to treat AVWS associated with MGUS, which is considered one of the main disorders now recognized as monoclonal gammopathy of clinical significance. 3 From a safety perspective, both proteasome inhibitors and immunomodulatory imide drugs (IMiDs) can lead to significant side effects, and the high incidence of thrombocytopenia with bortezomib could complicate the management of a bleeding disorder. 26 , 27 The optimal management of the prothrombotic effects of IMiDs may be challenging in the setting of an acquired bleeding disorder, especially in patients who would otherwise be considered to receive prophylaxis anticoagulation along with IMiD therapy. Nevertheless, from an efficacy standpoint, myeloma‐directed therapy may theoretically offer the advantage of a durable remission of the AVWS due to its long‐lasting effects on clonal plasma cells that cause the disease. This is in contrast to IVIG, which serves as only a temporizing measure and may require frequent administration in patients with recurrent severe, life‐threatening bleeding episodes. Such patients may be candidates for more aggressive therapy aimed at long‐term remission, including myeloma‐directed therapy or hematopoietic stem cell transplant. Ultimately, these therapeutic options need to be considered on an individual, case‐by‐case basis, after taking into account the benefit‐risk ratio.
In our review, adjunct hemostatic agents such as rFVIIa and antifibrinolytics appeared to also be successful in managing AVWS‐MGUS, although the numbers of studies were limited. These agents do not directly impact the pathophysiological mechanism of this disorder. Their success is likely related to their effect in rebalancing the hemostatic pathways, leading to a shift toward a prothrombotic state. Both rFVIIa and tranexamic acid have been successfully used in the management of congenital VWD. 28 , 29 We consider these agents to be useful under circumstances where IVIG is unavailable or was ineffective.
As described above, there are significant limitations to this study based on the quality of available literature. Current data are limited to case reports or small case series, with a small number of eligible patients and inconsistent reporting of key information. The retrospective aspect of analysis, the heterogeneity of study design and therapeutic approaches in the studies, and publication bias are also significant limiting factors. This poses a real challenge in managing this serious disorder in an evidence‐based fashion, since these studies do not provide sufficient information individually to guide treatment approaches. As with all scoping reviews, we collated currently available published literature in a meaningful manner that would be beneficial to clinicians and used this information to make recommendations based on our expert opinion, which we summarized in Figure 2. While larger, prospective studies are needed to more accurately assess management outcomes, this would confer a major challenge given the rarity of this disease.
5. CONCLUSION
AVWS is a complication of MGUS that leads to a bleeding disorder of a variable degree of severity, and is likely caused by monoclonal autoantibodies against VWF. Limited data in the literature suggest that the hemostatic management of this disorder should be focused on neutralizing or eliminating the autoantibody. IVIG appears to be an effective treatment of bleeding symptoms, and it is our agent of choice for major bleeds or preoperative management. Agents such as DDAVP or factor replacement therapy offer limited success, but given the general availability of DDAVP, it may be useful in controlling minor bleeds. While other hemostatic agents such as rFVIIa do not neutralize the antibodies, their prothrombotic effects could offer success in managing major bleeds and may be considered in emergent situations or cases of drug shortage, although the data are limited.
AUTHOR CONTRIBUTIONS
YAI and MYL designed the study, analyzed the data, and wrote the manuscript. GMR and PFB reviewed and edited the manuscript; all authors approved the final version of the manuscript. All the authors agree to be accountable for all aspects of the work, thereby ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
RELATIONSHIP DISCLOSURE
GMR reports consulting and clinical trial funding from Sanofi, Octapharma, and Baxter, and consulting funding from Bayer and Genentech, outside the submitted work. MYL reports receiving honoraria from the American Society of Hematology, outside the submitted work. PFB and YAI declare no conflicts of interest.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mary M. McFarland, Information Specialist at the Eccles Health Sciences Library, University of Utah, for her assistance in performing the literature search.
Abou‐Ismail MY, Rodgers GM, Bray PF, Lim MY. Acquired von Willebrand syndrome in monoclonal gammopathy – A scoping review on hemostatic management. Res Pract Thromb Haemost. 2021;5:356–365. 10.1002/rth2.12481
Handling Editor: Dr Cihan Ay.
Contributor Information
Mouhamed Yazan Abou‐Ismail, DoctorYazanA.
Ming Y. Lim, Email: ming.lim@hsc.utah.edu, @MingLim.
REFERENCES
- 1. Tiede A. Diagnosis and treatment of acquired von Willebrand syndrome. Thromb Res. 2012;130(suppl 2):S2‐6. [DOI] [PubMed] [Google Scholar]
- 2. Federici AB, Budde U, Rand JH. Acquired von Willebrand syndrome 2004: International Registry–diagnosis and management from online to bedside. Hamostaseologie. 2004;24:50‐5. [DOI] [PubMed] [Google Scholar]
- 3. Fermand J‐P, Bridoux F, Dispenzieri A, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. 2018;132:1478‐85. [DOI] [PubMed] [Google Scholar]
- 4. Michiels JJ, Budde U, van der Planken M, et al. Acquired von Willebrand syndromes: clinical features, aetiology, pathophysiology, classification and management. Best Pract Res Clin Haematol. 2001;14:401‐36. [DOI] [PubMed] [Google Scholar]
- 5. Dicke C, Schneppenheim S, Holstein K, et al. Distinct mechanisms account for acquired von Willebrand syndrome in plasma cell dyscrasias. Ann Hematol. 2016;95:945–57. [DOI] [PubMed] [Google Scholar]
- 6. Michiels JJ, Berneman Z, Gadisseur A, et al. Immune‐mediated etiology of acquired von Willebrand syndrome in systemic lupus erythematosus and in benign monoclonal gammopathy: therapeutic implications. Semin Thromb Hemost. 2006;32:577‐88. [DOI] [PubMed] [Google Scholar]
- 7. Federici AB, Stabile F, Castaman G, et al. Treatment of acquired von Willebrand syndrome in patients with monoclonal gammopathy of uncertain significance: comparison of three different therapeutic approaches. Blood. 1998;92:2707‐11. [PubMed] [Google Scholar]
- 8. Gan TE, Sawers RJ, Koutts J. Pathogenesis of antibody‐induced acquired von Willebrand syndrome. Am J Hematol. 1980;9:363‐71. [DOI] [PubMed] [Google Scholar]
- 9. Fricke WA, Brinkhous KM, Garris JB, Roberts HR. Comparison of inhibitory and binding characteristics of an antibody causing acquired von Willebrand syndrome: an assay for von Willebrand factor binding by antibody. Blood. 1985;66:562‐9. [PubMed] [Google Scholar]
- 10. van Genderen PJ, Terpstra W, Michiels JJ, et al. High‐dose intravenous immunoglobulin delays clearance of von Willebrand factor in acquired von Willebrand disease. Thromb Haemost. 1995;73:891‐2. [PubMed] [Google Scholar]
- 11. Federici AB, Budde U, Castaman G, et al. Current diagnostic and therapeutic approaches to patients with acquired von Willebrand syndrome: a 2013 update. Semin Thromb Hemost. 2013;39:191‐201. [DOI] [PubMed] [Google Scholar]
- 12. Tiede A, Rand JH, Budde U, et al. How I treat the acquired von Willebrand syndrome. Blood. 2011;117:6777‐85. [DOI] [PubMed] [Google Scholar]
- 13. Arksey H, O'Malley L. Scoping studies: towards a methodological framework. Int J Soc Res Methodol. 2005;8:19‐32. [Google Scholar]
- 14. Anderson S, Allen P, Peckham S, Goodwin N. Asking the right questions: scoping studies in the commissioning of research on the organisation and delivery of health services. Health Res Policy Syst. 2008;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rizwan I, Minuk L, Jackson S, Iorio A. Cardiovascular disease prevalence and relevance in haemophilia: a scoping review. Haemophilia. 2015;21:e156‐166. [DOI] [PubMed] [Google Scholar]
- 16. Yu JK, Iorio A, Edginton AN. Using pharmacokinetics for tailoring prophylaxis in people with hemophilia switching between clotting factor products: a scoping review. Res Pract Thromb Haemost. 2019;3:528‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Al‐Ani F, Chehade S, Lazo‐Langner A. Thrombosis risk associated with COVID‐19 infection: a scoping review. Thromb Res. 2020;152‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tricco AC, Lillie E, Zarin W, et al. PRISMA Extension for Scoping Reviews (PRISMA‐ScR): checklist and explanation. Ann Intern Med. 2018;169:467‐73. [DOI] [PubMed] [Google Scholar]
- 19. Schulman S, Angeras U, Bergqvist D, et al. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in surgical patients. J Thromb Haemost. 2010;8:202‐4. [DOI] [PubMed] [Google Scholar]
- 20. Schulman S, Kearon C, Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis . Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non‐surgical patients. J Thromb Haemost. 2005;3:692‐4. [DOI] [PubMed] [Google Scholar]
- 21. Kaatz S, Ahmad D, Spyropoulos AC, et al. Definition of clinically relevant non‐major bleeding in studies of anticoagulants in atrial fibrillation and venous thromboembolic disease in non‐surgical patients: communication from the SSC of the ISTH. J Thromb Haemost. 2015;13:2119‐26. [DOI] [PubMed] [Google Scholar]
- 22. Wadhera RK, Rajkumar SV. Prevalence of monoclonal gammopathy of undetermined significance: a systematic review. Mayo Clin Proc. 2010;85:933‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Therneau TM, Kyle RA, Melton LJ 3rd, et al. Incidence of monoclonal gammopathy of undetermined significance and estimation of duration before first clinical recognition. Mayo Clin Proc. 2012;87:1071‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354:1362‐9. [DOI] [PubMed] [Google Scholar]
- 25. Boros P, Gondolesi G, Bromberg JS. High dose intravenous immunoglobulin treatment: mechanisms of action. Liver Transplant. 2005;11:1469‐80. [DOI] [PubMed] [Google Scholar]
- 26. Cengiz Seval G, Beksac M. The safety of bortezomib for the treatment of multiple myeloma. Expert Opin Drug Saf. 2018;17:953‐62. [DOI] [PubMed] [Google Scholar]
- 27. Raza S, Safyan RA, Lentzsch S. Immunomodulatory drugs (IMiDs) in multiple myeloma. Curr Cancer Drug Targets. 2017;17:846‐57. [DOI] [PubMed] [Google Scholar]
- 28. Sucker C, Scharf RE, Zotz RB. Use of recombinant factor VIIa in inherited and acquired von Willebrand disease. Clin Appl Thromb Hemost. 2009;15:27‐31. [DOI] [PubMed] [Google Scholar]
- 29. Lavin M, O'Donnell JS. New treatment approaches to von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2016;2016:683‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bertolino J. Intravenous immunoglobulin in patients with acquired Von Willebrand syndrome: A single referral centre experience. Haemophilia. 2019;25(1):e42–e45. [DOI] [PubMed] [Google Scholar]
- 31. Macik B. G. The use of high‐dose intravenous gamma‐globulin in acquired von Willebrand syndrome. Arch Pathol Lab Med. 1988;112(2):143–146. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.