

Abstract
Epigenetic processes play a crucial role in the regulation of plant stress responses, but their role in plant–pathogen interactions remains poorly understood. Although histone‐modifying enzymes have been observed to be deregulated in galls induced by root‐knot nematodes (RKN, Meloidogyne graminicola) in rice, their influence on plant defence and their genome‐wide impact has not been comprehensively investigated. First, the role of histone modifications in plant–nematode interactions was confirmed by pharmacological inhibition of histone‐modifying enzymes, which all significantly affected rice susceptibility to RKN. For a more specific view, three histone marks, H3K9ac, H3K9me2, and H3K27me3, were subsequently studied by chromatin‐immunoprecipitation‐sequencing on RKN‐induced galls at 3 days postinoculation. While levels of H3K9ac and H3K27me3 were strongly enriched, H3K9me2 was generally depleted in galls versus control root tips. Differential histone peaks were generally associated with plant defence‐related genes. Transcriptome analysis using RNA‐Seq and RT‐qPCR‐based validation revealed that genes marked with H3K9ac or H3K9me2 showed the expected activation or repression gene expression pattern, but this was not the case for H3K27me3 marks. Our results indicate that histone modifications respond dynamically to RKN infection, and that posttranslational modifications mainly at H3K9 specifically target plant defence‐related genes.
Keywords: ChIP‐Seq, epigenetics, gene expression, histone modifications, nematode, Oryza sativa
Histone modifications respond dynamically to root‐knot nematode infection in rice, with levels of H3K9ac and H3K27me3 enriched, and H3K9me2 generally depleted, in galls versus control root tips.
1. INTRODUCTION
In eukaryotes, DNA is wrapped around histone proteins and the resulting DNA–histone complex or nucleosome is the basic repeating unit of chromatin. The histone tails protruding from the nucleosome core can be modified by addition of chemical groups, mainly acetyl and methyl groups, affecting the physical accessibility of DNA to the transcriptional machinery of the cell (Berger, 2007; Lawrence et al., 2016). Fine‐tuning of gene expression is obtained by an interplay between different types and levels of posttranslational histone modifications (Schones & Zhao, 2008; Zhou et al., 2011). Some histone marks are associated with transcriptional activation, for example acetylation of lysine residues on histone H3 is correlated with gene activation and in some cases DNA repair. In contrast, depending on the residue, H3 methylation can be associated with transcriptional activation (lysine 4 and 36) or repression (lysine 9 and 27) (Armstrong, 2014; Berger, 2007; Fuchs et al., 2006). Both lysine and arginine can be methylated and up to three methyl groups can bind to each residue. The histone acetylation level is balanced by the activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs or HDAs). The relatively small genome of rice contains eight HATs and 19 HDACs (Liu et al., 2012; Pandey et al., 2002; Zhou et al., 2013). The rice genome also contains 37 Su(var)3‐9/Enhancer of Zeste/Trithorax (SET)‐domain containing histone methyltransferases also named the SET domain‐containing group (SDG), eight protein arginine methyltransferases (PRMTs), and 24 Jumonji C domain‐containing histone‐lysine demethylases (JmjC‐KDMs), which regulate the histone methylome (Zhou et al., 2013). The genome‐wide distribution of histone marks has been mainly investigated in unstressed plants. Du et al. (2013) found H3K9ac to be mainly present (73.4%) in genic regions of the rice genome. H3K9ac was associated with 781 transposable‐element genes (TE genes) and 19,616 non‐TE genes (Du et al., 2013). In another study, the distribution patterns of H3K4me3, H3K9ac, and H3K27me3 showed overall enrichment in genic and euchromatic regions. Furthermore, out of 41,043 non‐TE genes in the TIGR database, 25,207 (61.4%), 26,623 (64.9%), and 17,211 (41.9%) were associated with H3K4me3, H3K9ac, and H3K27me3, respectively. In contrast, these histone marks were associated with less than 5.2% of TE‐related genes in the same database (He et al., 2010). The role of histone modifications in plant responses to biotic stress has been described. On infection of Paulownia fortunei with phytoplasma, 1,788 and 939 genomic regions were hyper‐ and hypoacetylated, respectively, for H3K9ac (Yan et al., 2019). Application of salicylic acid (SA)‐analogue benzothiadiazole (BTH) induced H3K9ac enrichment in the promoter of pathogenesis‐related gene 1 (PR1) (López et al., 2011). Pathogen infection or treatment with agents that induce plant resistance tends to lead to higher expression levels of HATs, as was, for example, observed on application of abscisic acid and SA (Liu et al., 2012). Expression of HDA19, the best‐studied HDAC in Arabidopsis, is induced by wounding, infection by the fungal pathogen Alternaria brassicicola, and application of jasmonic acid (JA) and ethylene (ET) (Zhou et al., 2005). Overexpression of HDA19 confers higher plant resistance against A. brassicicola, whereas the hda19‐deficient mutant shows increased susceptibility through differential regulation of genes involved in the JA and ET pathways (Wang et al., 2017). Similarly, Kim et al. (2008) showed that overexpression of HDA19 results in enhanced resistance against Pseudomonas syringae, while the deficient mutant was more susceptible to infection through compromised activation of WRKY38 and WRKY62, which are negative regulators of plant defence (Kim et al., 2008). However, in another study it was demonstrated that HDA19 represses the SA‐mediated defence response in Arabidopsis, and the hda19‐mutant showed enhanced resistance against P. syringae pv. tomato DC3000 (Pst DC3000), correlated with enhanced expression of PR genes (Choi et al., 2012). Similarly, the hda6 mutant showed increased levels of acetylation, which led to enhanced resistance to Pst DC3000 by increased activation of PR1, PR2, and PR4 (Wang et al., 2017). Given its clear role in plant defence, it is not unexpected that pathogens interfere with histone acetylation. An effector of the sugar beet cyst nematode Heterodera schachtii alters acetylation levels in Arabidopsis by inhibiting HDACs, leading to rRNA gene acetylation and activation (Vijayapalani et al., 2018). Next to histone acetylation, other marks are also involved in plant defence. In bean plants infected with Uromyces appendiculatus, next to H4K12ac also H3K9me2 marks were associated with several plant defence genes, including WRKY, bZIP, MYB transcription factors, chitinase, calmodulin, and leucine‐rich repeat (LRR) genes. However, at the genome‐wide level, peaks for both histone marks were mainly located in intergenic regions (Ayyappan et al., 2015). Similarly, JmjC DOMAIN‐CONTAINING PROTEIN 27 (JMJ27), an H3K9me1/2 demethylase, was shown to be a positive regulator of plant defence against Pst DC3000 in Arabidopsis. JMJ27 negatively regulates WRKY25 (a repressor of defence) and positively regulates PR genes (Dutta et al., 2017). JMJ705 overexpression in rice confers enhanced resistance against Xanthomonas oryzae pv. oryzae by reduction in H3K27me2/3 levels, correlated with higher expression of peroxidases, JA‐related genes, and PR‐proteins (Li et al., 2013). The use of chemical inhibitors of histone‐modifying enzymes to counteract epigenetic malfunctions in human oncogenes has been studied extensively (Heerboth et al., 2014). Recent studies have also shown the potential of chemical reagents in influencing epigenetic mechanisms in plants (Bond et al., 2009; Miwa et al., 2017; Tanaka et al., 2008; Zhang et al., 2012, 2013). As HDAC‐inhibitor, nicotinamide application induces the expression of VERNALIZATION INSENSITIVE 3 (VIN3) in Arabidopsis, causing flowering and repression of flowering locus C (Bond et al., 2009). Its derivative, nicotinamide mononucleotide, accumulates in Fusarium‐resistant barley cultivars to Fusarium while pretreatment with nicotinamide mononucleotide confers resistance to Fusarium graminearum in Arabidopsis leaves and flowers and barley spikes (Miwa et al., 2017). Sulfamethazine (SMZ) belongs to the family of antibacterial sulphonamides that impair folate synthesis and thereby methyl supply in plants. Application of sulfamethazine on Arabidopsis reduces the level of H3K9me2 methylation and DNA methylation, and consequently derepresses epigenetic silencing (Zhang et al., 2012). Fumarate inhibits histone demethylation at sites of DNA damage (Lees‐Miller, 2015) and enhances resistance against Pst DC3000 in Arabidopsis without direct antimicrobial effect (Balmer et al., 2018).
In this study, we aimed to assess the role of histone modifications in the interaction between rice and root‐knot nematodes (RKN). Rice is the staple food of half the world's population. With a relatively small and fully sequenced genome, it is an excellent model system for genomic studies in monocotyledonous plants. The RKN Meloidogyne graminicola is one of the most damaging nematodes attacking rice and other monocots (Bridge et al., 1990). After penetration of second‐stage juveniles of M. graminicola into the rice roots, they establish a feeding site that consists of giant cells. Giant cell establishment causes root deformation, leading to symptoms called galls, which in the case of M. graminicola are typically located at the root tip and are macroscopically visible from about 3 days postinoculation (dpi) (Mantelin et al., 2017). Studies on epigenetic changes in plant–nematode interactions have thus far focused on DNA methylation and small RNA expression. In the compatible interaction between soybean and Arabidopsis with cyst nematodes genome‐wide DNA hypomethylation was observed (Hewezi et al., 2017; Rambani et al., 2015) and this was recently confirmed in the rice–RKN interaction (Atighi et al., 2020). Concerning smRNAs, the majority of differentially expressed microRNAs were down‐regulated (Cabrera et al., 2016) in the RKN–Arabidopsis interaction, while an up‐regulation of repeat‐associated small interfering RNAs was observed (Ruiz‐Ferrer et al., 2018). A number of miRNAs have been functionally characterized, with most of them being identified as negative regulators of plant immunity (Hewezi, 2020).
Despite the fact that previous transcriptome studies revealed that expression of many histone modification enzymes was significantly altered in galls and giant cells induced by M. graminicola in comparison with control tissues/cells in rice (Ji et al., 2013; Kyndt et al., 2012), no study has yet been published focusing on changes in genome‐wide histone modification patterns during nematode infection in host plants. After first assessing whether interference with histone marks affects rice susceptibility to M. graminicola, we subsequently studied how histone modification patterns are affected in galls at very early time points after infection with M. graminicola using chromatin‐immunoprecipitation (ChIP‐Seq).
2. RESULTS
2.1. Effects of chemical inhibitors of histone‐modifying enzymes on rice susceptibility to RKN
After M. graminicola infection in rice, a strong transcriptional deregulation occurs in many histone‐modifying genes at the early stage of infection, 3 dpi (Kyndt et al., 2012). At this moment, RKN are initiating a feeding site at the rice root tips, which is correlated with strong effects on plant defence (Kyndt et al., 2012; Mantelin et al., 2017). About 80 genes encoding enzymes involved in histone lysine acetylation and methylation are differentially expressed after nematode infection (Figure S1), indicating changes at the epigenomic level.
In a first experiment, chemical inhibitors of histone‐modifying enzymes were applied to evaluate if interference with histone marks would affect rice susceptibility to RKN. Three previously reported chemical inhibitors of histone‐modifying enzymes, namely nicotinamide (HDAC inhibitor), sulfamethazine (histone and DNA methyltransferase inhibitor), and fumaric acid (histone demethylase inhibitor), were applied on rice plants. The activity of these chemical inhibitors and their effect on plant development and defence were described in previous studies, using the same concentrations as applied here (Bond et al., 2009; Miwa et al., 2017; Tanaka et al., 2008; Zhang et al., 2012, 2013). All chemicals were sprayed (Dawood et al., 2019; Latzel et al., 2016; Miwa et al., 2017; Puy et al., 2018) on plants 24 hr before nematode inoculation. To rule out direct effects on nematodes, we executed foliar application including a surfactant to allow adequate uptake and systemic effects in the plants (Nahar et al., 2011). The effects of application of these chemical inhibitors on histone‐modifying enzymes in rice was confirmed by western blot at 24 hr after spray treatment (Figure S2). As expected, application of fumaric acid and sulfamethazine increased and decreased H3K27me3 levels, respectively, while H3K9ac levels increased after treatment with nicotinamide. Nematodes were inoculated at 24 hr after the spray treatment and 13 days later numbers of galls and nematodes per plants were counted. Data were also expressed per milligram of dry root to correct for potential growth retardation effects induced by these chemicals in treated plants. Two different concentrations of fumaric acid, 2.5 and 5 mM, were applied, showing no observable developmental or growth retardation effects in plants (Figure S3a). However, the number of galls and nematodes decreased significantly and in a concentration‐dependent manner, whether expressed per plant or per milligram of dry root (Figure S3b,c). Sulfamethazine application caused negative effects on plant growth at 2 mM but not at 200 µM (Figure S3d). Numbers of galls and nematodes were significantly lower in sulfamethazine‐treated plants in comparison with mock‐treated control plants, whether expressed per plant (Figure S3e) or per milligram of root (Figure S3f). We applied two concentrations of nicotinamide, 1 and 10 mM. The lower concentration slightly promoted root development, as previously observed in wheat roots and shoots (Mohamed et al., 1989). However, this application had no significant effect on the number of galls or nematodes compared to untreated control plants (Figure S3g–i). The 10 mM concentration caused a significant retardation in root and shoot development compared to the 1 mM treated and control plants (Figure S3g). The number of nematodes was significantly lower in treated plants, per plant and per milligram of root, but no significant differences were observed for the number of galls (Figure S3h,i). Despite the fact that a lack of specificity of these compounds precludes us from drawing specific conclusions on the role of histone modifications in this interaction, these results indicate that histone modifications play a role in the parasitic interaction between M. graminicola and rice. That is why we decided to execute ChIP‐Seq analyses for specific monitoring of histone modifications in nematode‐induced gall tissue. Three histone marks with previously described roles in plant–pathogen interactions were selected: H3K9ac, H3K9me2, and H3K27me3 (Ayyappan et al., 2015; Yan et al., 2019, 2020).
ChIP‐Seq was performed for three biological replicates of gall tissue, in comparison with three biological replicates of root tips sampled on mock‐inoculated control plants. Root tips are the ideal control material for studies on plant responses to M. graminicola, as this nematode preferentially induces galls close to the root apical meristem (Mantelin et al., 2017). Galls at 3 dpi and corresponding root tips of mock‐inoculated plants were collected and used for chromatin immunoprecipitation following DNA sequencing.
2.2. Nematode‐induced galls have increased levels of H3K9ac
In total 63,994 H3K9ac peak regions were called, of which 1,062 were significantly differentially modified in galls compared to control root tips (File S1). We observed H3K9 hyperacetylation in galls compared to root tips (Figure 1a). A majority of the peaks overlap with a promoter region less than 1 kb from the transcription start site (TSS), while most other peaks are intergenic (Figure 1b). H3K9ac peaks were significantly associated with TE class DTM, while TE classes RLG and RLX overlapped significantly less with H3K9ac peaks than expected (Figure 2a). In total, 631 and 262 protein‐coding genes were hyperacetylated in their gene body or promoter, respectively (File S1), significantly more than randomly expected (Figure 2a). When focusing on genes with H3K9ac peaks in their promoters no significant Gene Ontology (GO) terms were found and the WRS test (Benjamini Hochberg‐corrected) in MapMan also showed no enriched pathways. However, GO analysis on genes with H3K9ac peaks in their gene body revealed over‐representation of genes related to protein modification, more specifically phosphorylation, response to organic substance, and sequence‐specific DNA binding (Figure 3). MapMan confirmed significant enrichment in Bin 30.2, containing “signalling receptor kinases” (number of genes: 23, p = .02). Interestingly, a set of SET domain‐containing proteins and HATs were enriched for H3K9ac in their promoters or gene bodies. Many of the differentially modified peaks covered genes that are involved in plant defence, for example encoding PR proteins, transcription factors of the WRKY, MYB, and ERF families, receptor kinases, MAP kinases, and hormone‐related genes especially abscisic acid, auxin, and ET (Figure 4 and File S1).
FIGURE 1.
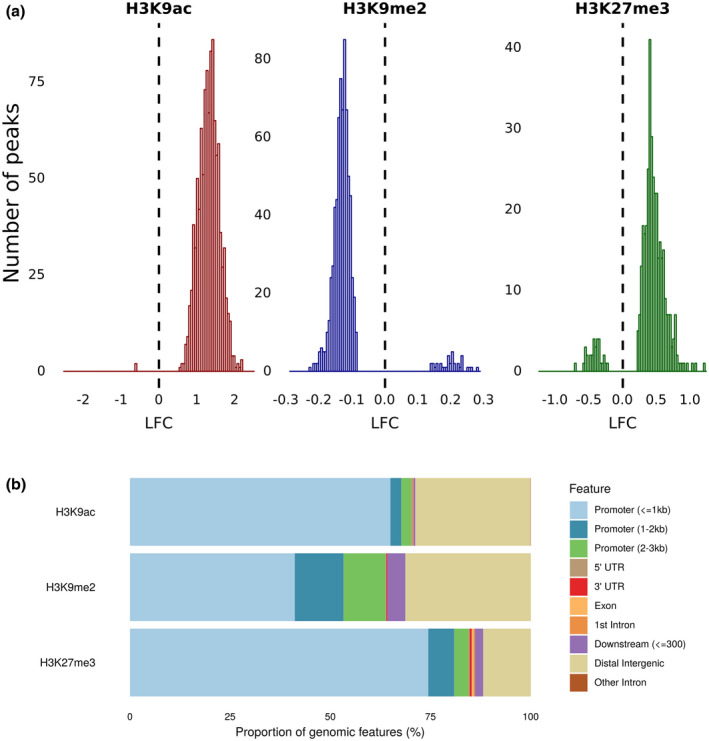
Analysis of differentially modified histone peaks in 3 days postinoculation Meloidogyne graminicola‐induced galls versus uninfected root tips of rice plants. (a) Log2 fold change (LFC, infected versus control) of significantly differentially enriched peaks for the three studied histone marks. (b) Peak distribution over different genomic regions for the three studied histone marks
FIGURE 2.
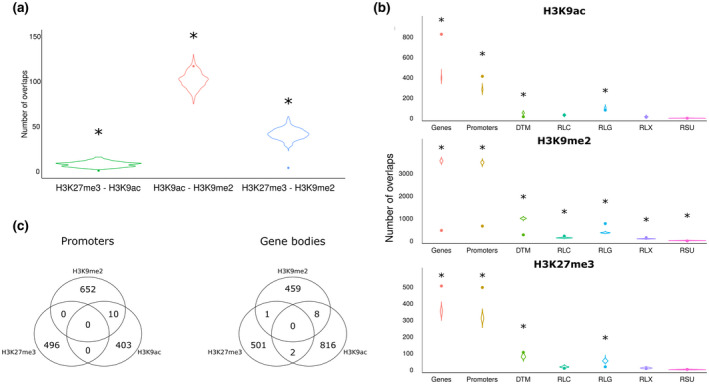
Concurrence of histone marks detected in 3 days postinoculation Meloidogyne graminicola‐induced galls and uninfected root tips of rice plants. (a) Significance of overlap between histone peaks and different genic regions or transposable elements (TEs). The violin plots show the distribution of the number of overlapping histone peaks and randomly scattered TE or gene regions (1,000 simulations) and the dots the observed number. Asterisks indicate significant differences between the observed number and the random distribution (p < .05). (b) Similar to (a), but for the overlap between the different histone peaks. (c) Venn diagrams of the number of promoter regions and gene body regions shared between the three studied histone marks
FIGURE 3.
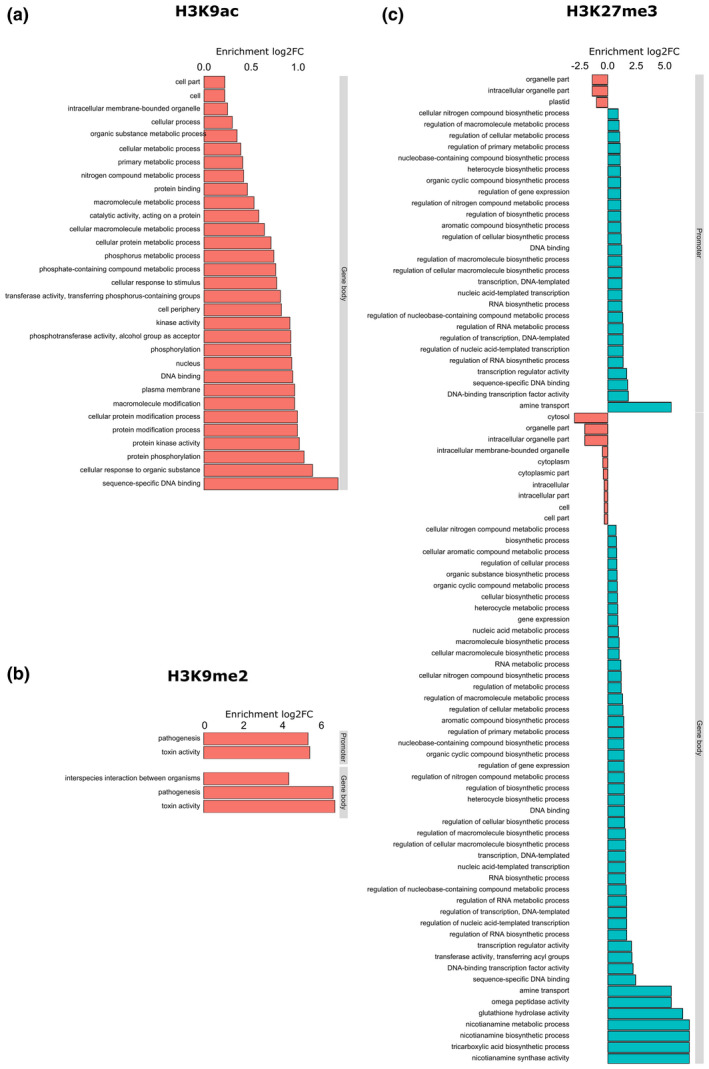
Gene ontology (GO) analysis of the genes associated with differentially modified histone peaks in 3 days postinoculation Meloidogyne graminicola‐induced galls versus uninfected root tips of rice plants: (a) H3K9ac, (b) H3K9me2, and (c) H3K27me3. The graphs show the significantly enriched (log2 fold change [FC] > 0) or depleted (log2 FC < 0) GO terms detected among the genes overlapping with the detected peaks in either gene body or gene promoter. Note that no significant GO terms were found in the set of genes that overlap with H3K9ac in their promoters
FIGURE 4.

MapMan visualization of genes that are associated with significantly affected histone peaks in 3 days postinoculation (dpi) galls versus uninfected root tips in rice. (a) The visualization shows the pattern of the histone mark in Meloidogyne graminicola‐induced galls compared to uninfected control root tips. Red and blue denote enrichment and depletion, respectively, of this mark in infected roots versus uninfected root tips. (b) Expression profile of these genes in 3 dpi galls versus uninfected root tips in rice (based on data published in Kyndt et al., 2012). Red and blue denote gene up‐ and down‐regulation in galls versus uninfected root tips, respectively
2.3. Nematode‐induced galls feature depleted levels of H3K9me2
A total of 11,162 H3K9me2 peaks were called. When comparing the presence of this mark in galls versus root tips we found a total of 732 genomic regions to be differentially methylated (File S2). A majority of these peaks showed hypomethylation in the galls (Figure 1a). Compared to other histone peaks, H3K9me2 peaks showed the least overlap with gene promoters (Figure 1b). Only 13 genes were found to have a hypermethylated promoter versus 246 genes with a hypomethylated promoter (File S2). When focusing on the gene body, eight genes were observed to be hypermethylated in H3K9me2 versus 100 genes hypomethylated (File S2). Next to the significant depletion of this mark in gene bodies and promoters, H3K9me2 depletion was also seen for TE classes DTM and RSU. Moreover, positive associations were found between H3K9me2 peaks and TE classes RLC, RLG, and RLX (Figure 2a). The WRS test analyses in MapMan showed no enrichment for any pathway/bin among the genes with H3K9me2 in their promoter or gene bodies regions. However, H3K9me2 peak‐containing genes were strongly enriched for GO terms related to pathogenesis and toxin activity (Figure 3). More specifically, we detected this mark to be associated with several genes involved in plant defence, such as cell wall synthesis, PR genes, thionin genes, transcription factors of the MYB family, receptor kinases, as well as hormone‐related genes mainly involved in the ET pathway (Figure 4 and File S2).
2.4. Nematode‐induced galls have increased levels of H3K27me3
Galls and corresponding uninfected root tips were also used for ChIP‐Seq analyses targeting H3K27me3. Data analysis of the sequenced DNA resulted in 10,836 called H3K27me3 peaks of which 496 peak regions were significantly differentially methylated on RKN infection (File S3). The H3K27me3 peaks mainly showed hypermethylation in galls compared to control root tips (Figure 1a) and more than 75% of the peaks were located in a 1 kb region preceding the TSS of genes (Figure 1b). Hypermethylation and hypomethylation of promoters occurred in 241 and 12 genes, respectively. Similarly, hypermethylation and hypomethylation of gene bodies occurred in 241 and 14 genes, respectively (File S3). Next to the generally positive association between H3K27me3 and gene bodies as well as promoters, this was also the case for TE class DTM. A negative association was found for the presence of H3K27me3 peaks in TE classes RLC and RLG (Figure 2a).
The WRS test analyses in MapMan showed that no specific pathway was enriched among the genes with differential H3K27me3 peaks in promoters or gene bodies. However, GO analyses revealed a large set of significant GO terms related to transcription (factors), glutathione hydrolase, and nicotianamine biosynthesis (Figure 3). The list of genes associated with hypermethylated H3K27me3‐patterns in galls contains many well‐known players in plant defence, including transcription factors from the MYB, ERF, and WRKY families, heat shock proteins, pectinesterase, and thaumatin (Figure 4 and File S3).
2.5. Concurrence of studied histone marks
We evaluated whether the investigated histone marks concurred on genomic regions. The presence of H3K27me3 marks showed a significantly negative association with H3K9ac peaks and H3K9me2 peaks. H3K9ac and H3K9me2 peaks featured a moderate but significantly positive overlap, while exhibiting opposite levels of modification changes between galls and roots, that is, an increase in H3K9 acetylation for a decrease in methylation and vice versa (Figure 2b). When evaluating solely gene bodies or promoters (Figure 2c and File S4), concurrence between marks was less clear, most probably due to the smaller part of the genome considered.
2.6. Only minor correlation between differentially modified histones and genome‐wide gene expression profiles in galls induced by M. graminicola
H3K9ac has been typically associated with transcriptional activation while H3K9me2 and H3K27me3 are mainly associated with transcriptional repression. Here, we created a total RNA‐Seq data set to see whether there is a genome‐wide correlation between differentially modified histones and gene expression profiles in M. graminicola‐induced galls at 3 dpi. Differential expression analysis of total RNA sequencing samples of galls at 3 dpi versus uninfected rice roots showed 18,629 protein‐coding genes to be differentially expressed of which 9,306 were down‐regulated and 9,323 were up‐regulated (File S5).
Although some correlations between histone marks and gene expression profiles were found to be statistically significant, the generally low correlation values and the appearance of the correlation plots make it unlikely that these correlations are biologically relevant (Figure 5). A significantly lower than randomly expected number of differentially expressed genes was found among the genes with differentially modified H3K9me peaks in promoter or gene bodies (Figure 6).
FIGURE 5.
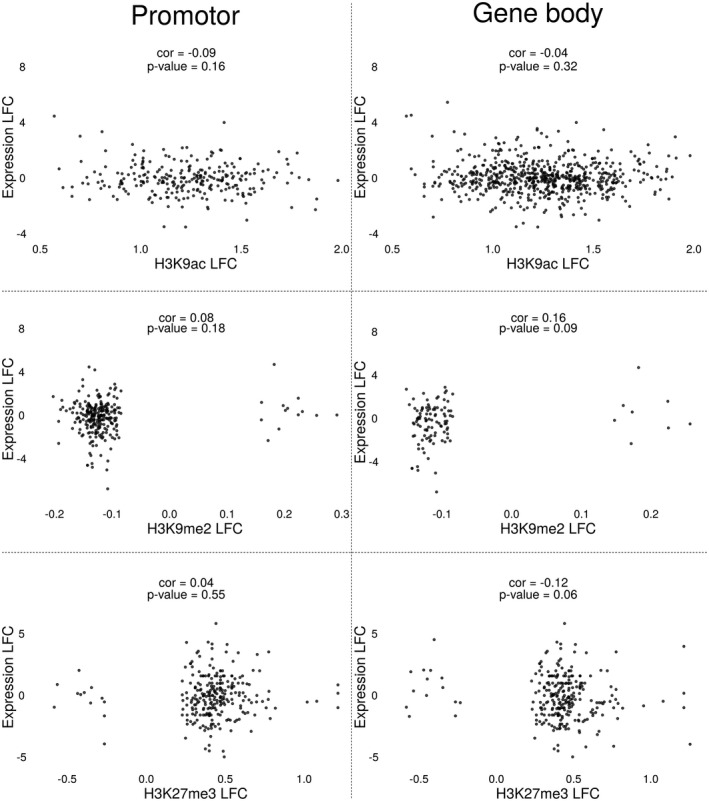
Correlation analyses between differentially modified genes and gene expression in Meloidogyne graminicola‐induced galls in rice. Correlation analyses between differentially modified H3K9ac, H3K9me2, and H3K27me3 peaks between 3 days postinoculation galls and uninfected root tips, in comparison with the gene expression profile of the associated genes at the same time point. These analyses were performed separately for peaks detected to be associated with gene promoters (left) or with gene bodies (right). LFC, log2 fold change
FIGURE 6.
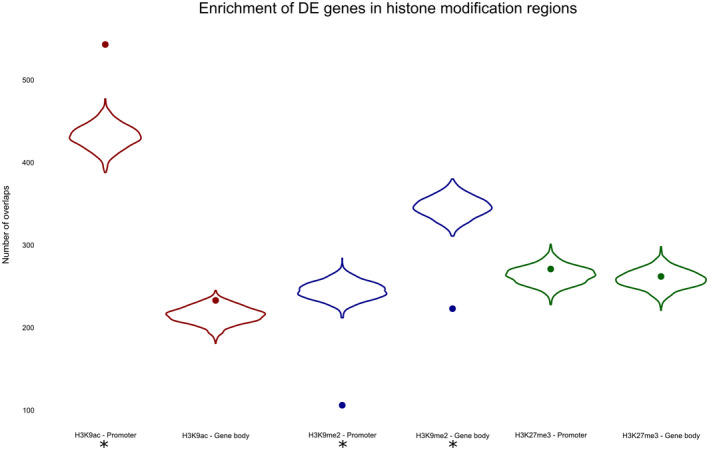
Association between differentially expressed (DE) genes and differentially modified histone peaks. The violin plots show the background distribution of overlaps between differentially modified peaks and a randomly sampled group of genes/promoters. The dots show the here‐observed number of overlaps between differentially modified peaks and DE genes, promoters of DE genes in Meloidogyne graminicola‐induced galls versus uninfected rice root tips. Asterisks indicate significant associations (p < .05)
Although an enrichment for differentially expressed genes was found among the genes with differentially modified H3K9ac peaks in their promoter (Figure 6), no tendency towards up‐regulation was detected, as would be expected if the H3K9ac mark were an activating histone mark. Of the 233 differentially expressed genes whose promoter overlaps with a differentially modified H3K9ac peak, 106 were up‐regulated (p = .93). Differentially expressed genes with a differentially modified H3K9ac peak in their gene body did show a near‐significant trend towards up‐regulation: of the 543 differentially expressed genes whose gene body overlaps with a differentially modified H3K9ac peak, 291 were up‐regulated (p = .051).
Details about differentially expressed genes and their overlap with differentially modified peaks can be found in File S5.
2.7. Association between histone marks and plant defence genes
Despite the lack of a clear genome‐wide correlation between histone peaks and differentially expressed genes, the GO analyses and manual screening of the lists of genes indicated an enrichment for plant defence‐related genes. Fifteen genes were selected for quantitative reverse transcription PCR (RT‐qPCR)‐based validation of the expression patterns in independently collected M. graminicola‐induced gall samples at 3 dpi versus uninfected root tips. For five defence‐related genes associated with significant H3K9 hyperacetylation and gene activation, activation patterns were generally confirmed by RT‐qPCR (Table 1a), validating the general robustness of our data and the correlation between this mark and defence gene activation.
TABLE 1.
Quantitative reverse transcription PCR (RT‐qPCR)‐based confirmation of the expression pattern of a subset of genes that are associated with differential histone marks as well as differential gene expression levels in 3 days postinoculation galls induced by Meloidogyne graminicola in rice in comparison with uninfected control root tips
| Modification | Locus number | Annotation | Log2FC (galls/root tips) | |||
|---|---|---|---|---|---|---|
| RT‐qPCR | RNA‐Seq | ChIP‐Seq | ||||
| a | H3K9ac | Os04g0618700 | OsFLS2, leucine‐rich repeat receptor protein kinase | 2.23 | 1.69 | 0.77 |
| Os04g0493100 | OsbHLH065, ethylene responsive | 0.59 | 0.33 | 1.27 | ||
| Os06g0662200 | bZIP transcription factor, OsbZIP52 | 1.48 | 0.27 | 1.77 | ||
| Os04g0531400 | Lectin‐like receptor kinase | 0.47 | 0.32 | 0.9 | ||
| Os03g0116700 | OsFBX76: F‐box domain containing protein | 1.13 | 0.24 | 1.28 | ||
| b | H3K9me2 | Os01g0278600 | OsDegp1: putative Deg protease homologue | 0.42 | −0.07 | −0.12 |
| Os04g0316200 | Cysteine‐rich receptor‐like protein kinase | −2.25 | −3.30 | −0.1 | ||
| Os09g0272600 | Expressed protein | 3.01 | 4.06 | −0.09 | ||
| Os11g0303800 | Transposon protein, putative, Pong subclass, | 1.57 | 0.03 | −0.13 | ||
| Os06g0513050 | THION3: plant thionin family protein | 0.87 | 1.27 | −0.14 | ||
| c | H3K27me3 | Os04g0580700 | OsMADS17, MADS‐box family gene | 8.78 | 7.21 | 0.46 |
| Os02g0178100 | OsBBX3, B‐box‐containing protein 3 | −1.14 | −0.73 | 0.49 | ||
| Os01g0164300 | POEI43: pollen Ole e I allergen and extensin family | 1.93 | 1.27 | 0.25 | ||
| Os03g0132900 | CHIT16: chitinase family protein | −0.38 | −0.47 | 0.39 | ||
| Os06g0624900 | Zinc‐binding protein | −2.98 | −3.38 | 1.25 | ||
The values indicate the mean of three biological replicates. Each biological replicate consists of a pool of galls obtained from about 20 plants. ChIP, chromatin immunoprecipitation; FC, fold change.
For five defence‐related genes that were significantly associated with H3K9me2 hypomethylation and that showed gene activation, RT‐qPCR generally corresponded with the transcriptome results, except for one gene (Table 1b). This reveals that depletion of this mark is slightly correlated with activation of these defence‐related genes.
Five defence‐related genes associated with significant H3K27me3 hypermethylation were selected. These genes showed diverging expression profiles in the RNA‐Seq data, and this pattern was here independently confirmed by RT‐qPCR on gall samples (Table 1c). These data reveal no apparent correlation between the presence of this mark and gene expression.
3. DISCUSSION
Infection of rice plants by root‐knot nematode M. graminicola was previously reported to cause a striking modulation in the expression of genes involved in histone modifications (Figure S1; Ji et al., 2013; Kyndt et al., 2012). Here, we provide the first genome‐wide data of histone modification patterns in the interaction between rice plants and parasitic nematodes. At the investigated time point, 3 dpi, early giant cell formation is observable in rice roots and galls become macroscopically visible, allowing specific sampling of the local infection site (Mantelin et al., 2017). Our experiments with pharmacological inhibition revealed that these chemicals generally decrease plant susceptibility for M. graminicola infection. This confirms their previously reported activity as defence priming agents (Azooz et al., 2013; Balmer et al., 2018; Noutoshi et al., 2012). Noteworthy, application of these chemicals could have widely affected histone marks across the genome and this could hence affect the expression of a multitude of genes involved in defence and development. We assume that these chemicals generally affect the primary metabolic flux and pool of precursors for many development as well as defence‐related processes in plants, leading to reduced susceptibility for M. graminicola in rice. For example, application of sulfamethazine reduces the plant folate pool and consequently the levels of S‐adenosyl methionine, the donor of methyl groups for many substrates (Shen et al., 2016). These data hence precluded us from drawing specific conclusions about the role of histone modifications in rice responses to nematode infection, although they do suggest that histone modifications play a role in the interaction between M. graminicola and rice.
To gain more robust and specific insights into the changed profiles on nematode infection, we studied the genome‐wide pattern of three histone marks, namely, H3K9ac, H3K9me2, and H3K27me3, with the first one generally considered as a gene activation mark and the two latter as gene repression marks (Armstrong, 2014). Confirming its role in gene activation the generally hyperacetylated H3K9ac peaks were mainly detected around the TSS site. Changes of the repressive mark H3K9me2 were mainly detected in non‐genic regions, such as TEs, but were significantly under‐represented in genes, promoters, and around the TSS (Figures 1b and 2a). These observations are in line with previous research. An in‐depth analysis of chromosome 4 of the rice genome determined that H3K9ac is mainly present in gene‐rich euchromatic regions, whereas H3K9me2 tends to occur in heterochromatic regions (Yin et al., 2008). In dragon trees under phytoplasma stress, differentially modified H3K9ac peaks overlapped with the largest number of genes (2,577) compared to H3K4me3 (1,738) and H3K36me3 (986) peaks (Yan et al., 2019). However, in common bean infected by Uromyces appendiculatus (rust) most differentially modified H3K9me2 as well as H4K12ac peaks were mainly located in intergenic regions (Ayyappan et al., 2015). In the case of H3K27me3, the generally hypermethylated peaks in galls were mostly detected around the TSS. Only a small number of genes showed significant changes for more than one of the here‐investigated histone marks (Figures 1b and 2a).
Our data revealed a strong genome‐wide quasi‐unilateral shift in these histone marks early after M. graminicola infection, with general enrichment of genome‐wide H3K9ac and H3K27me3, but depletion of H3K9me2 in young gall tissue compared to uninfected root tips. Strong genome‐wide modification of histone marks in plants after pathogen infection has been described before, but only after aboveground infection by phytoplasmas and fungi. For example, in rust‐infected bean plants, H3K9me2 and H4K12ac showed strong unilateral modification and, similar to our data, transcription factors from the WRKY, MYB, and bZIP families were affected (Ayyappan et al., 2015). In another study on phytoplasma‐infected P. fortunei, 1,788 and 939 genomic regions were hyper‐ and hypoacetylated at H3K9 (Yan et al., 2019). Although a sharp hyperacetylation was not observed, the peaks reported in that study were also related with the GO terms metabolic process, cellular process, and response to stimulus, as seen in our data (Figure 3) (Yan et al., 2019). In 3 dpi galls compared to control tissue, many defence‐related genes were marked by differential histone marks at H3K9 (Figure 4). These genes were strongly hyperacetylated at H3K9 in galls, which is correlated with activated expression of these modified genes (Table 1a). In contrast to H3K9me2, which was mainly detected in distal promoter regions and intergenic regions, H3K9ac and H3K27me3 peaks were strongly associated with proximal gene promoters and gene bodies (Figure 1b). Although histone modifications are generally believed to affect expression of the genes that they are associated with, no clear genome‐wide correlation between the here‐studied histone marks and RNA‐Seq data were observed in 3 dpi galls (Figure 5). Such a lack of general correlation between gene expression and specific histone modifications at the same time point has been reported before. Our observations confirm a previous study where no general correlation between loss or gain of H3K27me3 and gene expression was detected in the rice inflorescence meristem (Liu et al., 2015). Similarly, a report of Yan et al., who studied dragon trees, revealed that only 16.8% (292 of 1,738), 18.1% (178 of 986), and 16.8% (434 of 2,577) of the genes differentially modified by H3K4me3, H3K36me3, and H3K9ac showed differential gene expression under phytoplasma stress (Yan et al., 2019). We therefore hypothesize that there is a sophisticated cross‐talk between multiple epigenetic marks in the regulation of gene expression in plants. Very probably the here‐observed histone changes are offset by changes in other histone marks, which can enhance or reduce the effects of a particular histone mark, depending on the genomic location. Moreover, gene expression could also be affected by distant histone modifications that do not necessarily overlap with a gene or its promoter (Lawrence et al., 2016) or there could be a time‐lag between histone modification and effects on gene expression.
According to our observations there are strong modulations in the histone acetylome and methylome of rice after M. graminicola infection, although this is not associated with genome‐wide changes in expression profiles at the same time point. Further studies using other histone marks and multiple time points will be required to elucidate if and how the network of histone‐modifying genes, resulting marks, and their interactions can modulate the genome‐wide expression of the targeted genes. For example, it is possible that genes are primed for activation at later time points. Nevertheless, we have detected specific changes in histone marks around a subset of plant‐defence‐related genes for each investigated mark. Also, the unilateral change in histone dynamics for each of the here‐studied marks indicates that histone modifications play a role in the plant response to M. graminicola infection. The question arises of how the genome is able to target histone changes specifically towards a subset of defence‐related genes, and whether the parasitic nematode interferes with this response to attain susceptibility. We speculate that each histone‐modifying enzyme has the specific task to control a certain group of plant defence genes, such as, for example, shown for HDAC19 in controlling JA/ET‐based defence responses (Zhou et al., 2005). Further detailed functional investigations on specific HDAC, HMTs, and SDGs will be needed to prove this hypothesis.
In summary, we present the first genome‐wide study of three histone marks in young galls induced by M. graminicola infection in rice roots. We reveal that each of these histone marks showed a different unilateral shift (hypo‐ or hypermethylation or acetylation) and many of the genes marked with changes at H3K9 are involved in important plant defence pathways. Additional research targeting other histone marks and the elucidation of the exact underlying defence mechanism activated on chemical modulation of specific histone modification pathways are needed to depict a complete picture of epigenetic mechanisms after infection of plants with M. graminicola and potentially other pathogens.
4. EXPERIMENTAL PROCEDURES
4.1. Plant growth, nematode culture, and inoculation
Rice (Oryza sativa 'Nipponbare', GSOR‐100, USDA) seeds were germinated for 5 days in darkness at 30 °C, after which they were transferred to synthetic absorbent polymer (SAP) substrate in polyvinylchloride tubes (Reversat et al., 1999) and grown at 28 °C (16 hr light/8 hr darkness). Plants were watered by Hoagland solution every other day.
A pure culture of M. graminicola was originally obtained from the Philippines (kindly provided by Professor Dirk De Waele, Catholic University Leuven). Nematodes were cultured on susceptible rice and grasses (Echinochloa crus‐galli). Nematodes were extracted from 3–4‐month‐old cultures using the tray method (Whitehead & Hemming, 1965). After 2–3 days, the nematode suspension was collected and concentrated using 20 µm sieves. Two‐week‐old plants were inoculated with about 300 second‐stage juveniles per plant or mock inoculated with water.
4.2. Application of chemical inhibitors and evaluation of plant susceptibility
Thirteen‐day‐old rice plants were sprayed with nicotinamide, sulfamethazine, or fumaric acid, while control plants were mock‐sprayed with water. The surfactant Tween 20 was added to all spraying solutions at 0.02% (vol/vol). Twenty‐four hours later, plants were inoculated. For infection assays, the level of infection was evaluated 2 weeks after inoculation. Root and shoot lengths and weights were measured. Root systems were harvested and stained by boiling in 0.013% acid fuchsin for 5 min. After destaining in acid glycerol, the total numbers of galls and nematodes were counted under a stereomicroscope SMZ1500 (Nikon).
4.3. Western blot analysis
Thirteen‐day‐old rice plants were sprayed with 10 mM nicotinamide, 200 µM sulfamethazine, or 5 mM fumaric acid or mock‐sprayed with water. For each treatment, two biological replicates, each consisting of pooled whole‐plant material of 10 individual plants, was collected 24 hr after treatment. Protein extraction was done as described by Tariq et al. (2003), and concentrations were measured using Bradford reagent. Proteins were separated on 15% sodium dodecyl sulphate (SDS)‐polyacrylamide gel electrophoresis (PAGE) and blotted onto a FluoroTrans PVDF membrane (Pall Laboratory). The membrane was blocked overnight with Tris‐buffered saline (TBS; 10 mM Tris, 150 mM NaCl, 0.1% vol/vol Triton X‐100, pH 7.6) containing 5% (wt/vol) nonfat milk powder, after which it was washed three times for 5 min with TBS and incubated for 1 hr with primary antibodies anti‐H3K27me3 (1:1,000, C15410195; Diagenode), anti‐H3K9ac (1:1,000, C15410004; Diagenode), or anti‐H3 (1:4,000, ab1791; Abcam) as positive control. After three more 5 min washes with TBS, the membrane was incubated for 1 hr with goat antirabbit antibody (1:10,000, ab205718; Abcam) conjugated with horseradish peroxidase. After two washes with TBS and one wash with 0.1 M Tris (pH 7.6), detection was performed using 700 μM 3,3′‐diaminobenzidine tetrahydrochloride (Sigma‐Aldrich) and 0.03% (vol/vol) hydrogen peroxide in the same buffer. The reaction was stopped after 30 s by rinsing the membrane with distilled water. All washes and incubations were performed at room temperature, while shaking gently.
4.4. Data analysis
For statistical analyses software SPSS v. 25 (IBM) was used. Data normality was checked using a Kolmogorov–Smirnov test (α = 0.05). Homoscedasticity was verified using the Levene test (α = 0.05). If confirmed, parametric tests, that is, t test, analysis of variance (ANOVA), and Duncan's test were used. Otherwise, nonparametric Mann–Whitney or Kruskal–Wallis tests were applied.
4.5. Synchronization of infection and ChIP
Two‐week‐old rice plants were grown and inoculated as described above. After 36 hr, when the majority of nematodes had entered the root system (Mantelin et al., 2017), they were transferred to 50% Hoagland solution in glass tubes to synchronize the infection process. At 3 dpi, galls of infected plants and uninfected root tips of mock‐inoculated plants were harvested. For each sample, three independent biological replicates were sampled.
Chromatin extraction and shearing was performed according to the Diagenode Chromatin Shearing Optimization Kit for Universal Plant ChIP‐Seq Kit (cat. no. C01020014) and precipitation according to the Plant ChIP‐Seq kit (cat. no. C011010150) with some modifications. Briefly, collected galls and root tips were cross‐linked in 1% formaldehyde for 15 min on ice in a desiccator under vacuum. Glycine was added to stop crosslinking under an additional 5 min of vacuum. Root materials were ground and chromatin was extracted using three extraction buffers as in the manufacturer's guidelines and collected and dissolved in 600 µl of sonication buffer supplemented with Protease Inhibitor Cocktail (Sigma). Sonication was performed using a Covaris M220 Focused‐Ultrasonicator: peak power of 75, duty factor of 10, and cycle/burst of 200 for 15 min. Chromatin immunoprecipitation was performed on the supernatant using three antibodies (Diagenode), H3K9ac, H3K9me2, and H3K27me3. Magnetic beads (DiaMag protein A‐coated magnetic beads; Diagenode) were incubated with each antibody overnight, after which they were washed with ChIP dilution buffer to remove unbound antibodies. Beads were incubated for 10 hr with 50 µl sheared chromatin. One tenth of the diluted chromatin was collected and kept aside as input sample before incubation with antibody. After incubation, beads were washed to remove unbound DNA fragments and DNA was eluted. Input and precipitated DNA were washed in absolute ethanol and the pellet was dissolved in 20 µl DNase‐free water.
4.6. Library preparation and sequencing
Library preparation was done using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs). To prepare samples, 34 µl of DNase‐free water was added to 16 µl of precipitated DNA and adaptors were ligated to DNA fragments (NEBNext Multiplex Oligos for Illumina kit; NEB). After ligation, the DNA samples were cleaned up using AMPure XP beads (0.9×) (Beckman Coulter). Resulting fragments were amplified for 14 cycles using NEBNext Ultra II PCR protocol (New England Biolabs) and quality was assessed with the Agilent High sensitivity DNA kit. Amplicons were excised from a 2% agarose gel (200–800 bp fragments were excised) and purified using the Gel DNA recovery kit (Zymo Research). Library concentrations were measured with quantitative PCR (qPCR) according to Illumina's Sequencing Library qPCR Quantification Guide. Sequencing was done on a NextSeq500 using single reads (76 bp). A 2.3 pM library was loaded on the flow cell with 2% PhiX spike‐in.
4.7. Data analysis of ChIP‐sequencing data
Reads were trimmed using Trim Galore v. 0.4.0 with default settings. Mapping was done with Bowtie v. 1.1.1. Unmapped reads were filtered out and multiplexed subsamples were merged with samtools v. 1.9. Redundant reads were filtered out using Picard v. 2.18.27. For peak calling, the MUSIC (multiscale enrichment calling) software (Harmanci et al., 2014) was used. A mappability profile was generated using the O. sativa subsp. japonica (build MSU7.0) genome with a read length parameter of 76. For peak calling, all samples were merged per histone modification (excluding the input sample, which was used as a control for the genome background). For H3K9me2 and H3K27me3 the broad peak subroutine was used while for H3K9ac punctate peaks were called, following the classification of the ENCODE project (https://www.encodeproject.org/chip‐seq/histone/). Read quality was checked using FastQC v. 0.11.8. Sample quality was evaluated by calculating the nonredundant fraction and PCR bottleneck coefficient. Successful enrichment of genomic regions was checked using R package ChipQC v. 1.18.2 (Carroll et al., 2014) and calculating the fraction of reads in peaks as well as the standardized standard deviation. Normalized strand cross‐correlation coefficient and relative strand cross‐correlation coefficients were also calculated using phantompeakqualtools (Kharchenko et al., 2008; Landt et al., 2012). Read counts and quality control metrics can be found in File S6. For H3K27me3 and H3K9me2 three biological replicates were used per condition, for H3K9ac two biological replicates were used because a third replicate was of insufficient quality. To assess significant changes in peak regions between galls and roots, count tables of read counts in peak regions were generated by the summarizeOverlaps function in the GenomicAlignments R package v. 1.16.0. For differential peak analysis, DESeq2 package v. 1.22.2 with an false discovery rate (FDR) cut‐off of 0.05 was applied on nondeduplicated samples. The p values were corrected for multiple testing using Benjamini–Hochberg correction (Love et al., 2014). Peak annotation of differential peaks and TSS analysis was done using ChIPseeker v. 1.18.0 (Yu et al., 2015). Gene and TE annotations were obtained from Ensembl (Oryza_sativa.IRGSP‐1.0.42.gff3) and Rice Transposable Element databases. Five TE classes were used: DNA Transposon Mutator (DTM), long tandem repeat Copia (RLC), long tandem repeat Gypsi (RLG), long tandem repeat unknown (RLX), and short interspersed nuclear elements (RSU). Overlaps between genomic features and histone peaks was assessed using ChIPseeker. Promoter regions were defined as regions 2,000 bp upstream of the TSS.
4.8. Association between gene sets
Significance of association between sets of genomic regions was evaluated using permutation tests using the regioneR package v. 1.14.0 (Catoni et al., 2017; Gel et al., 2016; Zhao et al., 2019). First, 1,000 permutations were performed to create a null distribution. Per permutation, a set of genes of equal size to the set of genomic regions was randomly selected from the total set of rice genes and the number of overlaps was counted. Subsequently, the number of true overlaps between the two gene sets was compared to this null distribution (cut‐off: p < .05).
4.9. RNA sequencing
Rice was grown and inoculated as described above. For each treatment (3 dpi galls or root tips), three independent biological replicates were sampled. RNA was extracted using the ZR Plant RNA Miniprep kit (Zymo Research) and DNase‐treated (Thermo Fisher). RNA quality was verified with an RNA 6000 nanochip (Agilent Technologies) and concentration was measured with a Quant‐it Ribogreen RNA assay (Life Technologies). For rRNA depletion, 1.5 µg of RNA was treated with the Ribo Zero Plant Seed/Root kit (Illumina). The Truseq stranded total RNA library prep (Illumina) was used for library preparation. The cDNA was used for enrichment PCR (13 cycles), purified with the double AMPure XP cleanup (1:1) (Beckman Coulter), and checked with a high‐sensitivity DNA chip (Agilent Technologies). Quantification of the libraries was done with a qPCR assay according to Illumina's protocol to enable equimolar pooling of libraries. Finally, sequencing was performed on a NextSeq 500 using 2% Phix spike‐in (single‐end reads, 76 bp). Read quality was verified with FastQC v. 0.11.8, and reads were trimmed using Trimmomatic v. 0.38 with following parameters: ILLUMINACLIP:3:30:10, MAXINFO:23:1, SLIDINGWINDOW:5:30, MINLEN:17. STAR v. 2.6.1d was used for mapping on the O. sativa subsp. japonica (build MSU7.0) genome with the following parameters: readFilesCommand zcat, outFilterMultimapNmax 1, and outSAMtype BAM SortedByCoordinate. Samtools v. 1.10 was used to merge multiplexed samples. A count table was made using rtracklayer v. 1.44.4 to convert the GTF (Ensembl release 42) file into a Granges object, Rsamtools v. 2.0.3 to create a BamFileList object, and GenomicAlignments v. 1.20.1 for the summarizeOverlaps function to create the count table. To find differentially expressed genes, DESeq2 v. 1.24.0 was used with an FDR cut‐off of 0.05. The p values were corrected for multiple testing using the Benjamini–Hochberg correction.
Correlation between changes in histone modifications and gene expression was evaluated by comparing log2 fold changes of gene expression level between galls and control samples with the log2 fold changes of their overlapping histone peaks using the Pearson correlation coefficient. Association between differentially expressed genes and differentially modified histone peaks—associated with either gene bodies or promoters—was tested with the regioneR package as described above.
4.10. Gene ontology and pathway enrichment
For GO analyses, PLAZA v. 4.5 with default parameters was used (with Bonferroni correction; p < .05,) (Van Bel et al., 2018). The Wilcoxon rank sum (WRS) test (with Benjamini–Hochberg correction, p < .05) was used to evaluate statistical enrichment of pathways in MapMan (Thimm et al., 2004).
4.11. RNA extraction, reverse transcription, and RT‐qPCR analysis
RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) and cDNA was synthesized using Tetro reverse transcriptase (Bioline). RT‐qPCRs were performed with three technical and three biological replicates. The qPCR conditions consisted of initial denaturation at 95 °C for 10 min followed by 50 cycles of 95 °C for 25 s, 58 °C for 25 s, and 72 °C for 20 s. Expression data were normalized using two reference genes (all primers are listed in File S7) and analysed using REST2009.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interests.
Supporting information
FIGURE S1 Expression profile of genes involved in histone acetylation, deacetylation, and lysine methylation and demethylation in nematode‐induced galls at 3 days postinoculation, as observed in our previously published transcriptome analysis of the rice–Meloidogyne graminicola interaction (Kyndt et al., 2012). The gene annotation is based on (Zhou et al., 2013)
FIGURE S2 Western blotting analysis of histone modifications H3K27me3 and H3K9ac after treatment with fumaric acid, sulfamethazine, and nicotinamide. Two biological replicates are shown per condition
FIGURE S3 Effect of chemical of histone‐modifying enzymes on rice growth and susceptibility for root‐knot nematode infection. Plants were treated with different chemicals at 24 hr before inoculation with 300 J2 of Meloidogyne graminicola per plant. Root and shoot length as well as number of galls were evaluated 14 days later. (a–c) Effect of fumaric acid on rice growth and susceptibility to nematodes. (d–f) Effect of sulfamethazine on plant growth and susceptibility to nematodes. (g–i) Effect of nicotinamide on plant growth and susceptibility to nematodes (n = 20). For all subpanels different letters denote significant differences (p < .05). Error bars indicate SEM
TABLE S1 List of all called H3K9ac peaks, the subset of differentially modified H3K9ac peaks, and all genes associated with differentially modified H3K9ac peaks in their promoter or gene bodies at 3 days postinoculation on nematode infection in rice roots
TABLE S2 List of all called H3K9me2 peaks, the subset of differentially modified H3K9me2 peaks, and all genes associated with differentially modified H3K9me2 peaks in their promoter or gene bodies at 3 days postinoculation on nematode infection in rice roots
TABLE S3 List of all called H3K27me3 peaks, the subset of differentially modified H3K27me3 peaks, and all genes associated with differentially modified H3K27me3 peaks in their promoter or gene bodies at 3 days postinoculation on nematode infection in rice roots
TABLE S4 List of genes associated with multiple detected histone marks in their promoter or gene bodies at 3 days postinoculation on nematode infection in rice roots
TABLE S5 Read count statistics of RNA sequencing samples, differentially expressed genes after Meloidogyne graminicola infection 3 days postinoculation, and their overlap with differentially modified histones
TABLE S6 Read counts per sample and quality control metrics of ChIP sequencing samples
TABLE S7 Primers used in this study
ACKNOWLEDGEMENTS
This work was supported by Fonds Wetenschappelijk Onderzoek (FWO)‐Vlaanderen (G007417N) and Bijzonder Onderzoeksfonds UGent (BOF‐starting grant).
Atighi MR, Verstraeten B, De Meyer T, Kyndt T. Genome‐wide shifts in histone modifications at early stage of rice infection with Meloidogyne graminicola . Mol Plant Pathol. 2021;22:440–455. 10.1111/mpp.13037
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in the Gene Expression Omnibus at https://www.ncbi.nlm.nih.gov/geo/, accession numbers GSE145501 (Chip‐Seq) and GSE152783 (RNA‐Seq).
REFERENCES
- Armstrong, L. (2014) Epigenetics. Garland Science New York. [Google Scholar]
- Atighi, M.R. , Verstraeten, B. , De Meyer, T. & Kyndt, T. (2020) Genome‐wide DNA hypomethylation shapes nematode pattern‐triggered immunity in plants. New Phytologist, 227, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyappan, V. , Kalavacharla, V. , Thimmapuram, J. , Bhide, K.P. , Sripathi, V.R. , Smolinski, T.G. et al. (2015) Genome‐wide profiling of histone modifications (H3K9me2 and H4K12ac) and gene expression in rust (Uromyces appendiculatus) inoculated common bean (Phaseolus vulgaris L.). PLoS One, 10, e0132176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azooz, M.M. , Alzahrani, A.M. & Youssef, M.M. (2013) The potential role of seed priming with ascorbic acid and nicotinamide and their interactions to enhance salt tolerance in broad bean (Vicia faba L.). Australian Journal of Crop Science, 7, 2091–2100. [Google Scholar]
- Balmer, A. , Pastor, V. , Glauser, G. & Mauch‐Mani, B. (2018) Tricarboxylates induce defense priming against bacteria in Arabidopsis thaliana . Frontiers in Plant Science, 9, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, S.L. (2007) The complex language of chromatin regulation during transcription. Nature, 447, 407–412. [DOI] [PubMed] [Google Scholar]
- Bond, D.M. , Dennis, E.S. , Pogson, B.J. & Finnegan, E.J. (2009) Histone acetylation, VERNALIZATION INSENSITIVE 3, FLOWERING LOCUS C, and the vernalization response. Molecular Plant, 2, 724–737. [DOI] [PubMed] [Google Scholar]
- Bridge, J. , Michel, L. & Sikora, R. (1990) Nematode parasites of rice. In: Sikora, R. , Coyne, D. , Hallmann, J. and Timper, P. (Eds.) Plant Parasitic Nematodes in Subtropical and Tropical Agriculture. Wallingford, UK: CABI, pp. 9–108. [Google Scholar]
- Cabrera, J. , Barcala, M. , García, A. , Rio‐Machín, A. , Medina, C. , Jaubert‐Possamai, S. et al. (2016) Differentially expressed small RNAs in Arabidopsis galls formed by Meloidogyne javanica: A functional role for miR390 and its TAS3‐derived tasiRNAs. New Phytologist, 209, 1625–1640. [DOI] [PubMed] [Google Scholar]
- Carroll, T.S. , Liang, Z. , Salama, R. , Stark, R. & de Santiago, I. (2014) Impact of artifact removal on ChIP quality metrics in ChIP‐seq and ChIP‐exo data. Frontiers in Genetics, 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catoni, M. , Griffiths, J. , Becker, C. , Zabet, N.R. , Bayon, C. , Dapp, M. et al. (2017) DNA sequence properties that predict susceptibility to epiallelic switching. The EMBO Journal, 36, 617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S.‐M. , Song, H.‐R. , Han, S.‐K. , Han, M. , Kim, C.‐Y. , Park, J. et al. (2012) HDA19 is required for the repression of salicylic acid biosynthesis and salicylic acid‐mediated defense responses in Arabidopsis. The Plant Journal, 71, 135–146. [DOI] [PubMed] [Google Scholar]
- Dawood, M.G. , Abdel‐Baky, Y.R. , El‐Awadi, M.‐E.‐S. & Bakhoum, G.S. (2019) Enhancement quality and quantity of faba bean plants grown under sandy soil conditions by nicotinamide and/or humic acid application. Bulletin of the National Research Centre, 43, 28. [Google Scholar]
- Du, Z. , Li, H. , Wei, Q. , Zhao, X. , Wang, C. , Zhu, Q. et al. (2013) Genome‐wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. japonica . Molecular Plant, 6, 1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta, A. , Choudhary, P. , Caruana, J. & Raina, R. (2017) JMJ27, an Arabidopsis H3K9 histone demethylase, modulates defense against Pseudomonas syringae and flowering time. The Plant Journal, 91, 1015–1028. [DOI] [PubMed] [Google Scholar]
- Fuchs, J. , Demidov, D. , Houben, A. & Schubert, I. (2006) Chromosomal histone modification patterns–from conservation to diversity. Trends in Plant Science, 11, 199–208. [DOI] [PubMed] [Google Scholar]
- Gel, B. , Díez‐Villanueva, A. , Serra, E. , Buschbeck, M. , Peinado, M.A. & Malinverni, R. (2016) regioneR: An R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics, 32, 289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmanci, A. , Rozowsky, J. & Gerstein, M. (2014) MUSIC: Identification of enriched regions in ChIP‐Seq experiments using a mappability‐corrected multiscale signal processing framework. Genome Biology, 15, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, G. , Zhu, X. , Elling, A.A. , Chen, L. , Wang, X. , Guo, L. et al. (2010) Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. The Plant Cell, 22, 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerboth, S. , Lapinska, K. , Snyder, N. , Leary, M. , Rollinson, S. & Sarkar, S. (2014) Use of epigenetic drugs in disease: An overview. Genetics & Epigenetics, 6, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewezi, T. (2020) Epigenetic mechanisms in nematode–plant interactions. Annual Review of Phytopathology, 58, 119–138. [DOI] [PubMed] [Google Scholar]
- Hewezi, T. , Lane, T. , Piya, S. , Rambani, A. , Rice, J.H. & Staton, M. (2017) Cyst nematode parasitism induces dynamic changes in the root epigenome. Plant Physiology, 174, 405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, H. , Gheysen, G. , Denil, S. , Lindsey, K. , Topping, J.F. , Nahar, K. et al. (2013) Transcriptional analysis through RNA sequencing of giant cells induced by Meloidogyne graminicola in rice roots. Journal of Experimental Botany, 64, 3885–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko, P.V. , Tolstorukov, M.Y. & Park, P.J. (2008) Design and analysis of ChIP‐seq experiments for DNA‐binding proteins. Nature Biotechnology, 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.‐C. , Lai, Z. , Fan, B. & Chen, Z. (2008) Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone deacetylase 19 in basal defense. The Plant Cell, 20, 2357–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyndt, T. , Denil, S. , Haegeman, A. , Trooskens, G. , Bauters, L. , Van Criekinge, W. et al. (2012) Transcriptional reprogramming by root knot and migratory nematode infection in rice. New Phytologist, 196, 887–900. [DOI] [PubMed] [Google Scholar]
- Landt, S.G. , Marinov, G.K. , Kundaje, A. , Kheradpour, P. , Pauli, F. , Batzoglou, S. et al. (2012) ChIP‐seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research, 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latzel, V. , Rendina González, A.P. & Rosenthal, J. (2016) Epigenetic memory as a basis for intelligent behavior in clonal plants. Frontiers in Plant Science, 7, 1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, M. , Daujat, S. & Schneider, R. (2016) Lateral thinking: How histone modifications regulate gene expression. Trends in Genetics, 32, 42–56. [DOI] [PubMed] [Google Scholar]
- Lees‐Miller, S.P. (2015) Fumarate in DNA repair. Nature Cell Biology, 17, 1096–1097. [DOI] [PubMed] [Google Scholar]
- Li, T. , Chen, X. , Zhong, X. , Zhao, Y. , Liu, X. , Zhou, S. et al. (2013) Jumonji C domain protein JMJ705‐mediated removal of histone H3 lysine 27 trimethylation is involved in defense‐related gene activation in rice. The Plant Cell, 25, 4725–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Luo, M. , Zhang, W. , Zhao, J. , Zhang, J. , Wu, K. et al. (2012) Histone acetyltransferases in rice (Oryza sativa L.): Phylogenetic analysis, subcellular localization and expression. BMC Plant Biology, 12, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Zhou, S. , Wang, W. , Ye, Y. , Zhao, Y.U. , Xu, Q. et al. (2015) Regulation of histone methylation and reprogramming of gene expression in the rice inflorescence meristem. The Plant Cell, 27, 1428–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López, A. , Ramírez, V. , García‐Andrade, J. , Flors, V. & Vera, P. (2011) The RNA silencing enzyme RNA polymerase V is required for plant immunity. PLoS Genetics, 7, e1002434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. & Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantelin, S. , Bellafiore, S. & Kyndt, T. (2017) Meloidogyne graminicola: A major threat to rice agriculture. Molecular Plant Pathology, 18, 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa, A. , Sawada, Y. , Tamaoki, D. , Yokota Hirai, M. , Kimura, M. , Sato, K. et al. (2017) Nicotinamide mononucleotide and related metabolites induce disease resistance against fungal phytopathogens in Arabidopsis and barley. Scientific Reports, 7, 6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed, Y.A.H. , El‐Din, A.S. & Foda, E. (1989) Role of nicotinamide and salicylaldehyde on some growth parameters in wheat. Phyton, 29, 33–40. [Google Scholar]
- Nahar, K. , Kyndt, T. , de Vleesschauwer, D. , Höfte, M. & Gheysen, G. (2011) The jasmonate pathway is a key player in systemically induced defense against root knot nematodes in rice. Plant Physiology, 157, 305–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noutoshi, Y. , Ikeda, M. , Saito, T. , Osada, H. & Shirasu, K. (2012) Sulfonamides identified as plant immune‐priming compounds in high‐throughput chemical screening increase disease resistance in Arabidopsis thaliana . Frontiers in Plant Science, 3, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey, R. , Müller, A. , Napoli, C.A. , Selinger, D.A. , Pikaard, C.S. , Richards, E.J. et al. (2002) Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Research, 30, 5036–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puy, J. , Dvořáková, H. , Carmona, C.P. , de Bello, F. , Hiiesalu, I. & Latzel, V. (2018) Improved demethylation in ecological epigenetic experiments: Testing a simple and harmless foliar demethylation application. Methods in Ecology and Evolution, 9, 744–753. [Google Scholar]
- Rambani, A. , Rice, J.H. , Liu, J. , Lane, T. , Ranjan, P. , Mazarei, M. et al. (2015) The methylome of soybean roots during the compatible interaction with the soybean cyst nematode. Plant Physiology, 168, 1364–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reversat, G. , Boyer, J. , Sannier, C. & Pando‐Bahuon, A. (1999) Use of a mixture of sand and water‐absorbent synthetic polymer as substrate for the xenic culturing of plant‐parasitic nematodes in the laboratory. Nematology, 1, 209–212. [Google Scholar]
- Ruiz‐Ferrer, V. , Cabrera, J. , Martinez‐Argudo, I. , Artaza, H. , Fenoll, C. & Escobar, C. (2018) Silenced retrotransposons are major rasiRNAs targets in Arabidopsis galls induced by Meloidogyne javanica . Molecular Plant Pathology, 19, 2431–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones, D.E. & Zhao, K. (2008) Genome‐wide approaches to studying chromatin modifications. Nature Reviews Genetics, 9, 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y. , Issakidis‐Bourguet, E. & Zhou, D.‐X. (2016) Perspectives on the interactions between metabolism, redox, and epigenetics in plants. Journal of Experimental Botany, 67, 5291–5300. [DOI] [PubMed] [Google Scholar]
- Tanaka, M. , Kikuchi, A. & Kamada, H. (2008) The Arabidopsis histone deacetylases HDA6 and HDA19 contribute to the repression of embryonic properties after germination. Plant Physiology, 146, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq, M. , Saze, H. , Probst, A.V. , Lichota, J. , Habu, Y. & Paszkowski, J. (2003) Erasure of CpG methylation in Arabidopsis; alters patterns of histone H3 methylation in heterochromatin. Proceedings of the National Academy of Sciences of the United States of America, 100, 8823–8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm, O. , Bläsing, O. , Gibon, Y. , Nagel, A. , Meyer, S. , Krüger, P. et al. (2004) MAPMAN: A user‐driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. The Plant Journal, 37, 914–939. [DOI] [PubMed] [Google Scholar]
- Van Bel, M. , Diels, T. , Vancaester, E. , Kreft, L. , Botzki, A. , Van de Peer, Y. et al. (2018) PLAZA 4.0: An integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Research, 46, D1190–D1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayapalani, P. , Hewezi, T. , Pontvianne, F. & Baum, T.J. (2018) An effector from the cyst nematode Heterodera schachtii derepresses host rRNA genes by altering histone acetylation. The Plant Cell, 30, 2795–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Hu, Q. , Wu, Z. , Wang, H. , Han, S. , Jin, Y. et al. (2017) HISTONE DEACETYLASE 6 represses pathogen defence responses in Arabidopsis thaliana . Plant, Cell & Environment, 40, 2972–2986. [DOI] [PubMed] [Google Scholar]
- Whitehead, A.G. & Hemming, J.R. (1965) A comparison of some quantitative methods of extracting small vermiform nematodes from soil. Annals of Applied Biology, 55, 25–38. [Google Scholar]
- Yan, L. , Fan, G. & Li, X. (2019) Genome‐wide analysis of three histone marks and gene expression in Paulownia fortunei with phytoplasma infection. BMC Genomics, 20, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, L. , Zhai, X. , Zhao, Z. & Fan, G. (2020) Whole‐genome landscape of H3K4me3, H3K36me3 and H3K9ac and their association with gene expression during Paulownia witches’ broom disease infection and recovery processes. 3 Biotech, 10, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, B.‐L. , Guo, L. , Zhang, D.‐F. , Terzaghi, W. , Wang, X.‐F. , Liu, T.‐T. et al. (2008) Integration of Cytological features with molecular and epigenetic properties of rice chromosome 4. Molecular Plant, 1, 816–829. [DOI] [PubMed] [Google Scholar]
- Yu, G. , Wang, L.‐G. & He, Q.‐Y. (2015) ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics, 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Deng, X. , Miki, D. , Cutler, S. , La, H. , Hou, Y.‐J. et al. (2012) Sulfamethazine suppresses epigenetic silencing in Arabidopsis by impairing folate synthesis. The Plant Cell, 24, 1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Wang, B. , Duan, C.‐G. & Zhu, J.‐K. (2013) Chemical probes in plant epigenetics studies. Plant Signaling & Behavior, 8, e25364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, S. , Cheng, L. , Gao, Y. , Zhang, B. , Zheng, X. , Wang, L. et al. (2019) Plant HP1 protein ADCP1 links multivalent H3K9 methylation readout to heterochromatin formation. Cell Research, 29, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, C. , Zhang, L. , Duan, J. , Miki, B. & Wu, K. (2005) HISTONE DEACETYLASE19 is involved in jasmonic acid and ethylene signaling of pathogen response in Arabidopsis . The Plant Cell, 17, 1196–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, D.X. , Hu, Y. & Zhao, Y. (2013) Epigenomics. In: Zhang, Q. and Wing, R.A. (Eds.) Genetics and Genomics of Rice. Springer, pp. 129–144. [Google Scholar]
- Zhou, V.W. , Goren, A. & Bernstein, B.E. (2011) Charting histone modifications and the functional organization of mammalian genomes. Nature Reviews Genetics, 12, 7–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Expression profile of genes involved in histone acetylation, deacetylation, and lysine methylation and demethylation in nematode‐induced galls at 3 days postinoculation, as observed in our previously published transcriptome analysis of the rice–Meloidogyne graminicola interaction (Kyndt et al., 2012). The gene annotation is based on (Zhou et al., 2013)
FIGURE S2 Western blotting analysis of histone modifications H3K27me3 and H3K9ac after treatment with fumaric acid, sulfamethazine, and nicotinamide. Two biological replicates are shown per condition
FIGURE S3 Effect of chemical of histone‐modifying enzymes on rice growth and susceptibility for root‐knot nematode infection. Plants were treated with different chemicals at 24 hr before inoculation with 300 J2 of Meloidogyne graminicola per plant. Root and shoot length as well as number of galls were evaluated 14 days later. (a–c) Effect of fumaric acid on rice growth and susceptibility to nematodes. (d–f) Effect of sulfamethazine on plant growth and susceptibility to nematodes. (g–i) Effect of nicotinamide on plant growth and susceptibility to nematodes (n = 20). For all subpanels different letters denote significant differences (p < .05). Error bars indicate SEM
TABLE S1 List of all called H3K9ac peaks, the subset of differentially modified H3K9ac peaks, and all genes associated with differentially modified H3K9ac peaks in their promoter or gene bodies at 3 days postinoculation on nematode infection in rice roots
TABLE S2 List of all called H3K9me2 peaks, the subset of differentially modified H3K9me2 peaks, and all genes associated with differentially modified H3K9me2 peaks in their promoter or gene bodies at 3 days postinoculation on nematode infection in rice roots
TABLE S3 List of all called H3K27me3 peaks, the subset of differentially modified H3K27me3 peaks, and all genes associated with differentially modified H3K27me3 peaks in their promoter or gene bodies at 3 days postinoculation on nematode infection in rice roots
TABLE S4 List of genes associated with multiple detected histone marks in their promoter or gene bodies at 3 days postinoculation on nematode infection in rice roots
TABLE S5 Read count statistics of RNA sequencing samples, differentially expressed genes after Meloidogyne graminicola infection 3 days postinoculation, and their overlap with differentially modified histones
TABLE S6 Read counts per sample and quality control metrics of ChIP sequencing samples
TABLE S7 Primers used in this study
Data Availability Statement
The data that support the findings of this study are openly available in the Gene Expression Omnibus at https://www.ncbi.nlm.nih.gov/geo/, accession numbers GSE145501 (Chip‐Seq) and GSE152783 (RNA‐Seq).