

SUMMARY
Connectivity webs mediate the unique biology of the mammalian brain. Yet while cell circuit maps are increasingly available, knowledge of their underlying molecular networks remains limited. Here we applied multi-dimensional biochemical fractionation with mass spectrometry and machine learning to survey endogenous macromolecules across the adult mouse brain. We defined a global ‘interactome’ comprised of over one thousand multi-protein complexes. These include hundreds of brain-selective assemblies that have distinct physical and functional attributes, show regional and cell-type specificity, and have links to core neurological processes and disorders. Using reciprocal pulldowns and a transgenic model, we validated a putative 28-member RNA-binding protein complex associated with amyotrophic lateral sclerosis, suggesting a coordinated function in alternative splicing in disease progression. This Brain Interaction Map (BraInMap) resource facilitates mechanistic exploration of the unique molecular machinery driving core cellular processes of the central nervous system. It is publicly available and can be explored here https://www.bu.edu/dbin/cnsb/mousebrain/.
Graphical Abstract
eTOC Blurb
In this ground-breaking work, Pourhaghighi et al. have carried out a survey of over one thousand multi-protein complex interactions in the mouse brain using a platform they have named BraInMap (for Brain Interaction Map). This approach uses computer learning to reconstruct protein interactions from brain tissues that have been extensively purified. This important resource will allow neuroscientists to explore important neurobiological questions and identify pathways that are adversely affected in disease.
INTRODUCTION
The mammalian brain consists of intricate physical and functional protein interaction networks whose compositions are largely uncharacterized. These circuits support essential functions of a vast interconnected array of neurons, glial, oligodendrocytes and other cell types (Elmer and McAllister, 2012; Grant and O’Dell, 2001; Migaud et al., 1998; Sherman and Brophy, 2005; Slepnev and De Camilli, 2000; Small and Petsko, 2015; Zhu et al., 2016). Proper synaptic formation and activity resulting from these networks is essential for core brain functions, such as neurotransmission, synaptic plasticity, and memory. These molecular circuits are perturbed in neurological syndromes by genetic variants and environmental factors, resulting in behavioral, cognitive and neurodegenerative impairments. For example, abnormal protein-protein interactions among tau and α-synuclein lead to pathological accumulation preceding neurodegeneration (Forman et al., 2004; Ross and Poirier, 2004; Vanderweyde et al., 2016). Disease-causing disruptions in macromolecular assemblies have also been documented in amyotrophic lateral sclerosis (ALS) and Frontotemporal Dementia (FTD) (Dormann et al., 2010) as well as Parkinson’s disease (PD) (Carrion et al., 2017; Malty et al., 2017). Hence, mapping the global physical cartography of brain protein interaction networks is essential to understanding normal neuronal functions, the causal mechanisms driving disease, and for discovery of new targets as a basis for more effective and selective clinical therapies. Whereas large-scale physical interaction maps have been reported for transformed human cell lines (Havugimana et al., 2012; Hein et al., 2015; Huttlin et al., 2017; Wan et al., 2015), to our knowledge, no direct large-scale experimental study of the mammalian brain regional protein circuity, or ‘interactome’, has ever been reported, thwarting clinically actionable mechanistic understanding of neuronal processes and dysfunction.
To fill this gap, we systematically isolated and characterize endogenous protein assemblies on a global scale from mammalian brain lysates. Given its experimental tractability, widespread use in the neurobiology field, and the short post-mortem intervals enabled by animal studies, we opted to study mouse as a model. Multi-protein complexes were biochemically resolved and their cognate components identified by mass spectrometry based on their reproducible co-fractionation over orthogonal separations. Using an integrative co-complex scoring pipeline, we then generated a high-resolution survey, termed the BraInMap, representing the largest experiment-based protein interaction network for the central nervous system (CNS) to date and to the best of our knowledge. BraInMap comprises hundreds of putative macromolecular assemblies, most of which are conserved in human and expressed in a regional and cell-type specific manner.
To illustrate the utility of BraInMap, we explore the functional and biophysical properties of brain-specific assemblies with significant associations to core neurological functions and disorders in humans. We provide evidence that disease-associated processes and genetic variants disrupt the physical interfaces between components of neuronal protein assemblages critical for normal brain physiological homeostasis, suggesting a common causal basis for diverse neuropathies. Particularly prevalent were assemblies enriched for RNA-binding proteins (RBPs) whose physical associations are fundamentally linked to the etiology and pathogenesis of progressive neurological disorders such as Alzheimer’s disease (AD), ALS and FTD. While toxic gain-of-function and loss-of-function mutations in certain RBPs have previously been reported to elicit deleterious effects on splicing and RNA homeostasis (Arnold et al., 2013; Fratta et al., 2018), BraInMap describes their normal physical interactions in healthy adult brain, and therefore serves as a useful resource to bridge the gap between macro-level cell-cell connectivity studies, neuronal cell biology and epidemiological genetics, opening up new research avenues in molecular systems neuroscience.
RESULTS
Biochemical fractionation and precision mass spectrometry reveals brain selective protein assemblies
As illustrated schematically in Figure 1A, soluble protein extracts were prepared from homogenized adult whole brain CD1 mice using gentle isolation procedures (STAR Methods). The lysates were subject to extensive non-denaturing biochemical fractionation followed by mass spectrometry to identify and quantify stably associated proteins reproducibly co-eluting together. To maximize resolution and coverage, we deployed multiple orthogonal workflows to separate native macromolecules from functionally unrelated constituents (i.e., to mitigate ‘chance’ co-elution). This included two-dimensional separations combining isoelectric focusing (IEF) with mixed-bed ion exchange high performance liquid chromatography (IEX-HPLC). In parallel, we performed repeat IEX-HPLC-based separations using alternate chromatography procedures to selectively enrich for cytoskeletal, nuclear, membrane-bound and synaptic protein assemblies (STAR Methods).
FIGURE 1 – Integrative workflow used to generate the mammalian Brain Interactome Map (BraInMap).
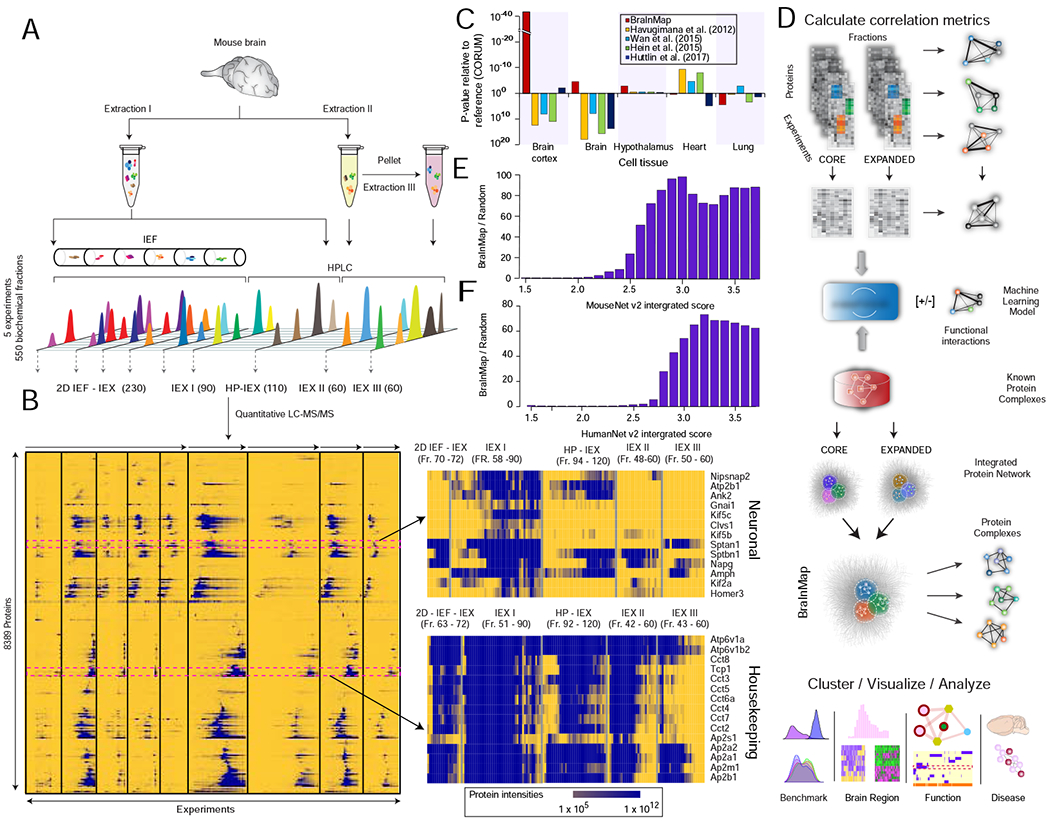
A Multi-pronged biochemical fractionation (high performance ion exchange chromatography, HPLC-IEX; isoelectric focusing, IEF; fraction numbers in brackets) of soluble macromolecular assemblies from mouse brain extracts.
B Hierarchical clustering of protein co-fractionation intensity profiles recorded by precision liquid chromatography-tandem mass spectrometry (LC-MS/MS); (right) neuronal (top) and housekeeping (bottom) components highlighted.
C Enrichment analysis (DAVID (Huang da et al., 2009)) of representative tissue annotations (UniProt) for proteins detected in this work relative to previously published interactome studies.
D Schematic depicting steps in the integrative BraInMap computational scoring pipeline: calculation of protein similarity (correlation) metrics, integrative classifier training (EPIC machine learning; (Hu et al., 2019)) and scoring of co-fractionation data (this study) and supporting (public) evidence to predict high-confidence co-complex interactions, followed by network partitioning, benchmarking and meta-analysis (pathobiological relevance) of the predicted complexes.
E Enrichment of interacting (co-eluting) brain proteins relative to random pairs for high functional similarity based on association scores reported in MouseNet (v2) (Kim et al., 2016)
F Enrichment of orthologs of interacting mouse brain proteins relative to random pairs for high functional association scores in HumanNet (v2) (Hwang et al., 2019).
Altogether, 550 biochemical fractions were collected in total across nine different fractionation experiments, which included replicate runs as a test for reproducibility. After trypsinization, each fraction was analyzed by nanoflow liquid chromatography coupled to quantitative (Orbitrap) mass spectrometry. The spectra were subject to stringent database searching and filtering (false discovery rate <1% at both the peptide- and protein-level) using multiple search algorithms, which on integration (STAR Methods) resulted in 8,389 high-confidence protein identifications (Table S1). Hierarchical clustering of the recorded protein profiles, covering two-thirds (5505 of 9121) of previously reported mouse brain tissue annotations (The UniProt Consortium, 2017), demonstrated the characteristic elution patterns of both neuronal and ubiquitous (housekeeping) protein assemblies (Figure 1B). In comparison to previously reported large-scale interactome studies of cultured cell lines (Havugimana et al., 2012; Huttlin et al., 2017; Wan et al., 2015), BraInMap was significantly enriched for annotated proteins known to be selectively expressed in mammalian cortex (FDR = 7.2 x 10−56), brain (FDR = 1.34 x 10−41) and other brain regions relative to other mouse tissues (Figure 1C; Table S6).
Scoring high-confidence co-complex associations
We devised a quantitation-based computational pipeline to tally the likelihood of co-complex interactions based on the pairwise similarity of the measured protein co-elution patterns. The premise is that stably associated components (subunits) of a multi-protein complex exhibit correlated profiles (i.e., components reproducibly co-purify together). Proteomic precursor ion (MS1) intensity profiles across all the fractions were acquired using both MaxQuant (CORE) and 3 additional search engines (X!Tandem, MSGF+, Comet) (EXPANDED) to derive preliminary protein co-complex associations. We calculated five established similarity measures (APEX, Jaccard, Bayes, Euclidean Distance, Mutual Information; see STAR Methods) that evaluate different features recorded in each experimental profile separately.
In a subsequent step (Figure 1D), protein pairs from both the CORE and EXPANDED datasets showing high similarity were input into a supervised machine-learning model (random forest classifier; STAR Methods). Two models were trained to predict high confidence co-complex associations based on the co-fractionation patterns we observed alone, or together with other publicly available supporting functional association evidence, with reference to curated ‘gold standard’ brain associated mammalian macromolecules (Table S2). Positive examples were obtained from public curated databases – namely CORUM (Ruepp et al., 2010), IntAct (Orchard et al., 2014), Gene Ontology (Ashburner et al., 2000), while negatives were created from randomized combinations of components assigned to distinct clusters. To minimize classifier bias, known mouse exemplars were supplemented with annotated human protein assemblies based on strict one-to-one orthology projections (InParanoid) (Sonnhammer and Ostlund, 2015). Moreover, complexes in the training set sharing a majority of subunits were merged (fractional overlap >0.8), while those with more than 50 members (e.g. ribosome) were excluded.
High concordance was evident when comparing both sets of co-fractionation patterns to probabilistic functional associations previously predicted based on protein domain co-occurrence, co-expression and co-citation in both mouse (MouseNet v2 database) (Kim et al., 2016) (Figure 1E) and human (HumanNet v2) (Hwang et al., 2019) (Figure 1F; conversion of human to mouse identifiers was done through one-to-one orthology mapping via InParanoid (Sonnhammer and Ostlund, 2015) and wherever applicable human orthologs of mouse proteins are named using uppercase letters e.g. the human ortholog of mouse protein Tdp-43 is referred to as TDP-43. These observations establish the broad physiological relevance of our initial interactome data.
The trained classifiers were then used to generate probabilistic co-complex relationships from both the CORE and EXPANDED datasets (STAR Methods). We evaluated all possible feature combinations to optimize precision and recall. The models were merged (average) into a single final high confidence protein-protein interaction (PPI) network, the BraInMap, consisting of 27,043 co-complex interactions (Table S3). We benchmarked the model prediction performance (precision and recall) by two-fold cross validation, using a fully independent set (i.e. exclusive of the training set) of manually curated complexes (from CORUM) for evaluation. These tests established a stringent False Discovery rate (FDR) of 11% with a precision-recall area-under-the-curve AUC of 0.92 (Figure 2A).
FIGURE 2 – Benchmarking reveals diverse, evolutionarily conserved brain complexes.
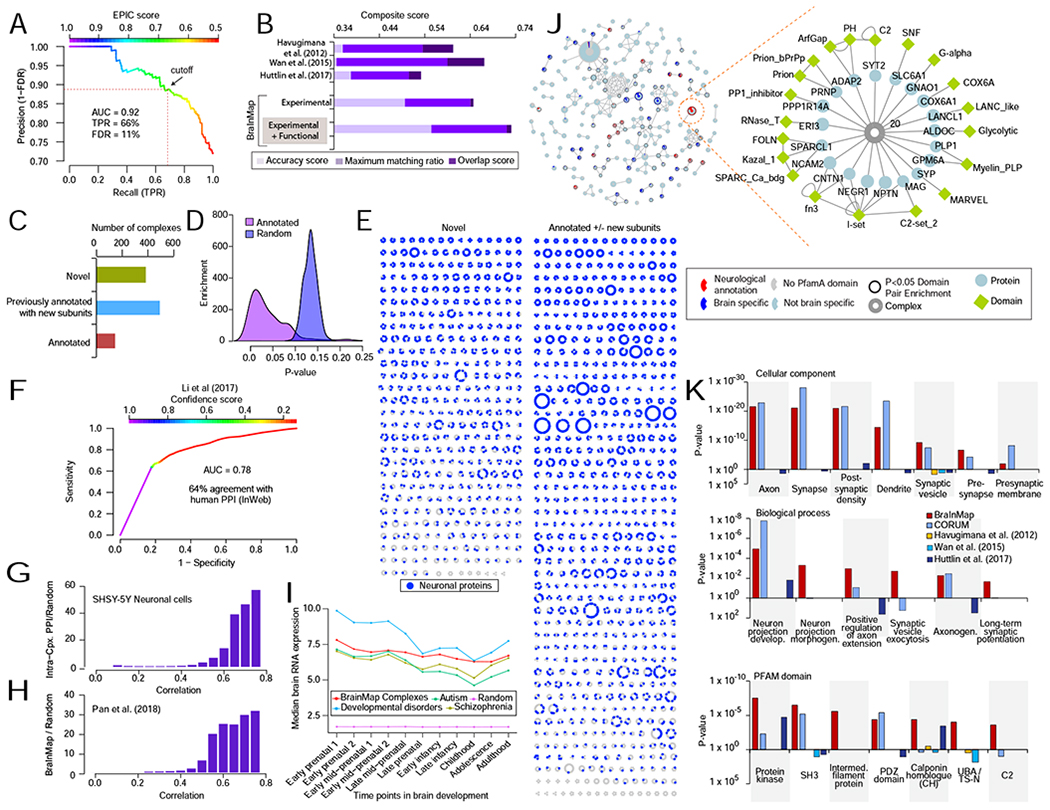
A Precision Recall (PR) analysis of predicted (EPIC score) co-complex interactions (CORE + EXPANDED) benchmarked against an independent (holdout) set of brain-derived reference assemblies establishes a false discovery rate (FDR) of 11%.
B Benchmark quality metrics of putative complexes (this work) versus other interactome maps. Bar length reflects total composite score, calculated as the sum of complex maximal matching ratio, overlap, and accuracy (see STAR methods) relative to select reference curated brain macromolecules.
C Bar chart of categorized complexes (partial or complete match to annotated assemblies vs novel).
D Highly significant (hypergeometric p-values) overlap of predicted complexes with annotated assemblies compared to randomized protein sets.
E Schematic of protein assemblies in BraInMap, sorted according to novelty, showing the distribution of neuron-associated components (purple).
F ROC analysis of predicted co-complex interactions showing high agreement with previously reported high confidence orthologous human protein interactions in the InWeb database (Li et al., 2017).
G Enrichment of human orthologs of BraInMap complex subunits relative to randomized protein pairs for highly correlated co-fractionation profiles of SHSY5Y neuronal cell extracts.
H Enrichment of human orthologs of interacting proteins in BraInMap relative to random pairs for high functional ‘co-fitness’ scores (Pan et al., 2018).
I Median expression of orthologs of BraInMap components during development of the human cortex; lines indicate levels of all interacting components (red) versus the subset associated with risk for schizophrenia (olive)(Schizophrenia Working Group of the Psychiatric Genomics et al., 2014), autism (green)(Sanders et al., 2015), or other neurodevelopmental disorders (cyan)(Deciphering Developmental Disorders et al., 2017), as compared to random proteins (magenta).
J Schematic of protein domains enriched in BraInMap. Complexes (nodes) sharing two or more domains are joined according to overlap (Jaccard Index). Colors reflect the proportion of domains restricted to brain (blue) or linked to neuropathology (red). Highlighted bipartite subnetwork shows relationship between subunits (ellipses) and domains (diamonds) of a representative assembly (complex 20).
K Annotation enrichment (DAVID; (Huang da et al., 2009) in BraInMap relative to previous interactome studies: Gene Ontology (i) cellular component or (ii) biological process terms, or (iii) PFAM domains (Finn et al., 2016).
We portioned the integrated network using the ClusterONE (Nepusz et al., 2012) clustering algorithm which revealed 1030 putative brain protein assemblies (Table S4). Based on their degree of connectivity and the initial source network from which a particular subunit within a given assembly was derived, each complex can be deconstructed into a ‘core’ and ‘extended’ set of interacting components (Figure S1A). To rigorously evaluate classifier performance at the protein complex level (rather than PPI as before), we calculated three stringent evaluation metrics (maximum matching ratio, accuracy, overlap score) (Nepusz et al., 2012) and combined the results into a single summary ‘composite’ quality score (F-measure) (STAR Methods).
As seen in Figure 2B, complexes based on our brain co-fractionation data alone (i.e. built without external data) produced a comparable or higher total composite score than other recently reported cell line-based interactomes, establishing the overall reliability of our scoring pipeline. We boosted classifier performance further by incorporating additional supporting functional association evidence (see STAR Methods) from MouseNet (Kim et al., 2016) and other public sources. We emphasize that the external data was used primarily as a filter to reinforce the primary findings of our proteomics data and that none (zero) of the protein assemblies in BraInMap are based solely on external sources (all macromolecular complexes are derived from replicate co-fractionation data).
To establish the degree of agreement with previously known complexes, we systematically examined the overlap of BraInMap complexes with annotated assemblies using multiple similarity metrics (Figure S1B). Of the 6 metrics tested, we settled on average matching index (AMI) and hypergeometric score as the most inclusive and stringent criteria to define macromolecules not reported in public databases (Figure S1C). We calculated the AMI as the average fraction of subunits matched between a known and predicted complex (STAR Methods).
As shown in Figure 2C & D, just over half (638, or 62%) of our complexes overlapped (AMI ≥ 0.25, hypergeometric p-value ≤ 0.05) significantly with one or more previously reported complexes (Havugimana et al., 2012; Huttlin et al., 2017; Ruepp et al., 2010; Wan et al., 2015); of these, 146 were considered as fully annotated (AMI ≥ 0.5) while the others (492) were deemed to have additional subunits not previously reported. Using this rigorous definition, the remaining (392) complexes appear to be reported here for the first time (Table S4). Consistent with the source tissue, over half (57%) of all the assemblies recovered by our survey consisted predominantly (≥50%) of components annotated as neuronal according to the Gene Ontology (STAR Methods), whereas only 33 assemblies (3%) lacked neuronal constituents (Figure 2E).
Brain complexes exhibit recent evolutionary adaptations that extend to human
To assess the human physiological relevance of BraInMap, we compared the underlying co-complex interactions against a fully independent curated public database of high quality human PPI (‘InWeb’, pooled from the InWeb3 and InWeb_IM resources) (Lage et al., 2007; Li et al., 2017) and found an overall agreement of 64% (Figure 2F). Consistent with this high apparent conservation, our own independent validation experiments showed that human orthologs of putatively interacting mouse components also tend to co-elute together (i.e., have correlated co-elution profiles) in independent chromatographic fractionation experiments performed on protein extracts from cultured human neuroblastoma SH-SY5Y cells (Figure 2G; Table S5). Likewise, human orthologs of BraInMap components strikingly showed higher correlated co-fitness profiles upon mRNA knockdown in human cell culture (Pan et al., 2018) as compared to random target pairs (Figure 2H), implying functional conservation of these complexes in human brain as well. Further support for the apparent conserved roles of these putative complexes in the CNS was demonstrated by the observation that these same orthologs are highly expressed during human brain development (embryogenesis through adulthood; Figure 2I) based on messenger RNA expression data obtained from the BrainSpan Atlas (Miller et al., 2014).
We examined the domain architecture of brain specific assemblies by assessing their corresponding Pfam A domain and family assignments (STAR Methods). By definition, domains are highly conserved sequence patterns that are presumed to represent independent folding units, while domain pairs in multi-domain proteins represent combinations of units operating in tandem (Cromar et al., 2016). In general, while rarely in the majority, brain specific protein domains and domain pairs were found to occur widely across the BraInMap (Figure 2J), suggesting they confer brain-specific functions. For some complexes, the occurrence of brain specific domains was a dominant feature. The presence of unique folds, both independently and in combination, reinforces the concept that the complexes we found in brain differ markedly from those revealed in previous interactome studies of cell lines. These observations are also consistent with specialized roles in processes linked to neurodevelopment and brain physiology. Consistent with this, many of the conserved complexes showed broad functional annotation diversity (Table S6) and enrichment for associations with specialized neuronal compartments (e.g. synapse, axon, and dendrite), processes (e.g. neurogenesis) and particular protein domains (Figure 2K).
Regionalization and cell-type specificity of brain protein assemblies
The mechanisms underlying the regional specification of the vertebrate CNS are of broad interest. This specification may be driven in part, by differences in the abundance (expression) and composition of different macromolecular complexes. To directly examine the potential regionalization of the protein assemblies in BraInMap, we performed independent biochemical fractionations (quadruplicate IEX-HPLC runs) and mass spectrometric profiling on 10 distinct brain regions (Figure 3A; frontal cortex, parietal cortex, occipital cortex, hippocampus, striatum, thalamus, midbrain, hindbrain, cerebellum, and cervical spinal cord) isolated from age and gender matched CD1 mice (4x males, 16 weeks) (STAR Methods). To accurately quantify differences in relative abundance, we used a two-pronged multiplexing procedure based on stable isotope labeling (tandem mass tags) to measure both the regional expression patterns and the corresponding regional co-elution profiles of most of the BraInMap assemblies in parallel (Figure 3B). We found that complexes that were significantly enriched (hypergeometric p-value ≤ 0.05) in the brain total protein measurements (Figure 3C) were likewise enriched for subunit pairs showing significantly (hypergeometric p-value ≤ 0.05) and reproducibly correlated co-fractionation profiles (as compared to random pairs) across the same regions (Figure 3D), allowing us to infer the regional selectivity of most of the assemblies in BraInMap (Table S7).
Figure 3 – Regional- and cell-type selective macromolecules.
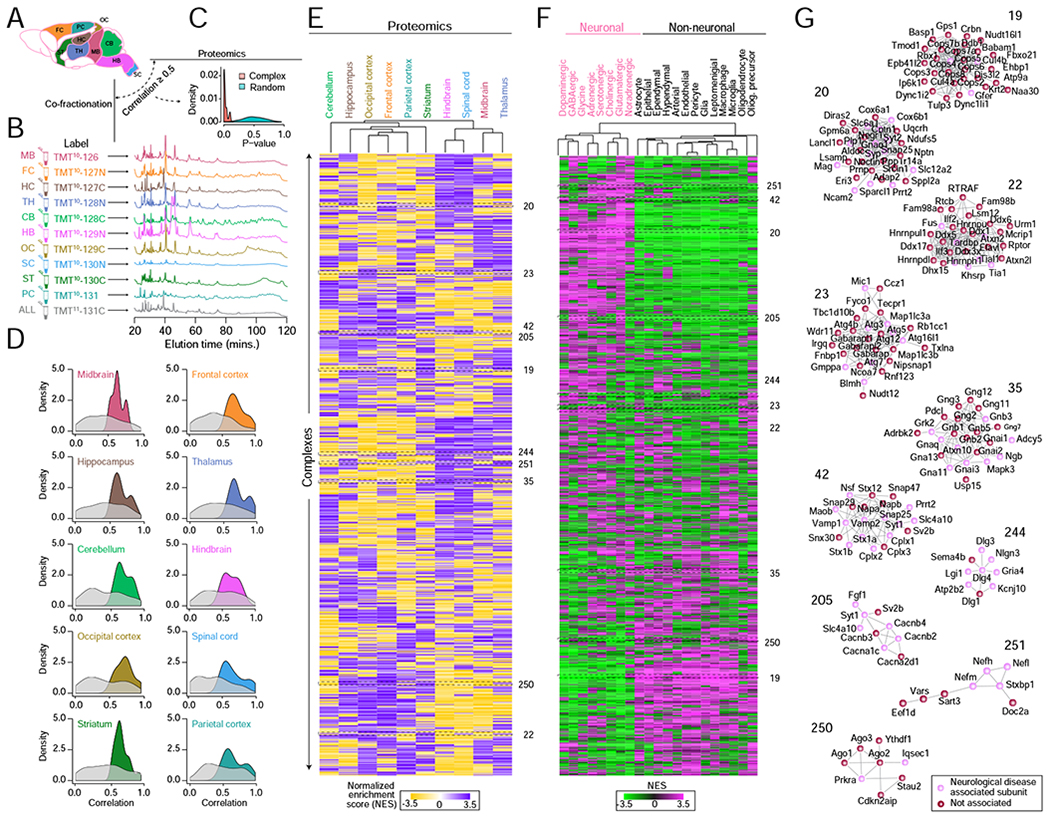
A Schematic of 10 mouse brain regions subjected to quantitative proteomic profiling and biochemical (HPLC-IEX) fractionation in parallel.
B Representative chromatograms and isobaric (TMT) labeling of fractionated regional assemblies.
C Highly significant (hypergeometric) agreement between the regional abundance patterns recorded by quantitative profiling versus co-fractionation of BraInMap components (derived by whole tissue analysis) as compared to randomized protein sets.
D Complex subunits with highly correlated regional co-fractionation profiles also show significantly co-enrichment (hypergeometric p-value ≤ 0.05, relative to randomized protein pairs) in the same brain compartments as determined by quantitative proteomics (E).
E Heatmap clustergram showing complex regional specificity (enrichment P-value ≤ 0.01 by Kolmogorov–Smirnov test) as measured by quantitative proteomics.
F Heatmap clustergram of complexes showing preferential (P ≤ 0.01 by KS test after normalization) component mRNA expression in neuronal versus non-neuronal cell classes based on recently published mouse brain scRNA-seq data (Zeisel et al., 2018).
G Representative complexes displaying regional (proteomic) and neuronal cell-type (scRNAseq) specificity. Highlighted (red) nodes represent subunits associated with neurological disorders.
To further examine the extent of specification, we overlaid BraInMap with recent mouse single-cell RNA (scRNA Seq) data (Zeisel et al., 2018). After collapsing the cell taxonomy from Zeisel et al. into 21 broad neuronal and non-neuronal cell-types, we observed widespread evidence of selective cognate gene expression (Figure 3F; Table S7). For example, complex 20, implicated in adhesion and signaling of axons with the myelin sheath, was enriched in neurons as well as abundantly expressed in the hippocampus and spinal cord (Figure 3F & G). Functional annotations of its membrane-associated subunits corroborate regional specificity in hippocampus (Ntpn, Prrt2, Slc6a1), and spinal cord (Lancl1, Prrt2, Srcin1), reflecting roles supporting and maintaining axon growth signals (Gpm6a, Negr1, Nptn), and vesicle targeting and release (Snap25, Syp, Syt2). Another component, Slc6a1, terminates GABAergic signal through sodium-dependent reuptake to presynaptic terminals, leading to myoclonic-atonic seizures when mutated (Carvill et al., 2015), while Plp1, a key constituent of compact myelin, along with Mag and Cntn1, mediates adhesion of the insulating sheath to axons in the internodes and paranodes, respectively (Jahn et al., 2009). PLP1 mutations cause a spectrum of neuronal disorders from Pelizaeus-Merzbacher disease to spastic paraplegia 2 (Hobson and Kamholz, 1993), while variants in CNTN1 cause lethal congenital myopathy (Compton et al., 2008), which may reflect an adhesion role at the neuromuscular junction. Mouse prion protein (Prnp) is also present in complex 20 and its interaction with PLP1, MAG, CNTN1, DPP6, ERI3, and SPARCL1 has previously been described (Schmitt-Ulms, Hansen et al. 2004). In addition to affecting transmissible neurodegenerative disease, neuronal expression of Prnp is essential for maintaining myelination (Bremer et al., 2010). Taken as a whole, this transmembrane assembly is likely critical to formation of myelin sheaths around GABAergic axons.
Likewise, complex 251, which contains neuron-specific neurofilament light, medium and heavy chain (Nefl, Nefm, Nefh) axoskeletal components, showed enriched expression in neuronal cells, as well as higher abundance in cortex and hippocampus (and lower expression in midbrain, hindbrain and spinal cord), while complex 42, comprised of SNARE protein components necessary for neurotransmitter release, was enriched in neurons (Chen et al., 2002) as well as in cerebellum and spinal cord. Conversely, complex 35, which contains alpha, beta and gamma subunits of Guanine nucleotide-binding protein (G-protein), showed high abundance in midbrain and thalamus as well as broad expression in both neurons and non-neurons (Figure 3G & F). Upon extracellular ligand binding to G-protein coupled receptors (GPCRs), G-proteins are activated by GDP to GTP replacement, facilitating one of the most prevalent signaling systems in diverse cell types through downstream effectors. Notably, this assembly included β-adrenergic receptor kinases 1 and 2 (Grk2, Adrbk2), mitogen-activated protein kinase 3 (Mapk3), as well as Ataxin 10 (Atxn10), in which a repeat expansion mutation is associated with spinocerebellar ataxia type 10 (Matsuura et al., 2000). In support of these findings, Atxn10 has previously been shown to interact with Gbeta2 (Gnb2) to potently activate the Ras/Mapk/Elk-1 signaling cascade (Waragai et al., 2006).
BraInMap assemblies showing regional enrichment in thalamus and striatum and preferential enrichment in non-neuronal cells include complex 19, which contains subunits 1 to 8 of the COP9 signalosome (responsible for deneddylation of cullin-RING ubiquitin E3 ligases), cullin4A-RING (Cul4a, Cul4b, Crbn, Ddb1) and cullin2-RING (Rbx1) E3 ubiquitin ligases (Cavadini et al., 2016; Dubiel et al., 2015). Likewise, complex 250 is enriched in non-neurons and hippocampus and is composed of argonaute proteins 1-3 (Ago1-3) necessary for RNA silencing and other double-stranded RNA interacting proteins (Stau2, Prkra). It also contains RNA binding protein Ytfdh1 needed to facilitate learning and memory formation in the hippocampus (Shi et al., 2018).
Subcellular compartmentalization
BraInMap identifies an array of complexes associated with neuronal subcellular compartments such as the axon, dendritic spine and synapse (Figure 4A; Table S8). The latter include assemblies that form a higher order molecular architecture on outer cell membranes as well as the synaptic vesicles involved in neurotransmission (Figure 4B; Table S8). For example, complex 42 (Figure 3F) and 51 share 14 components that encompass SNARE proteins (including Syt1, Snap25, Syntaxins 1a/1b/12, Complexins 1/2/3, Vamp1/2) necessary for synaptic vesicle docking (Chen et al., 2002). While complex 42 is characterized by the inclusion of additional synaptic-vesicle transmembrane factors (Sv2b, Slc4a10, Prrt2), complex 51 is differentiated by the presence of factors mediating ER-Golgi vesicle transport and fusion (Vcp, Sec22b, Scfd1, Arfgap2). Likewise, complex 234 and 267 share components required for Glutamatergic neurotransmission such as Gad1/2 (Glutamate decarboxylases) and Slc17a7 (Vesicular glutamate transporter 1). Complex 234 differs by exhibiting additional interaction with components of excitatory synapses (Vdac1, Nlgn2, Slc17a6), whereas complex 267 contains endosomal trafficking components (Rab21, Itgb1). These observations highlight compositional variations relevant to core neuronal activities.
FIGURE 4 – Compartmentalized brain protein assemblies.
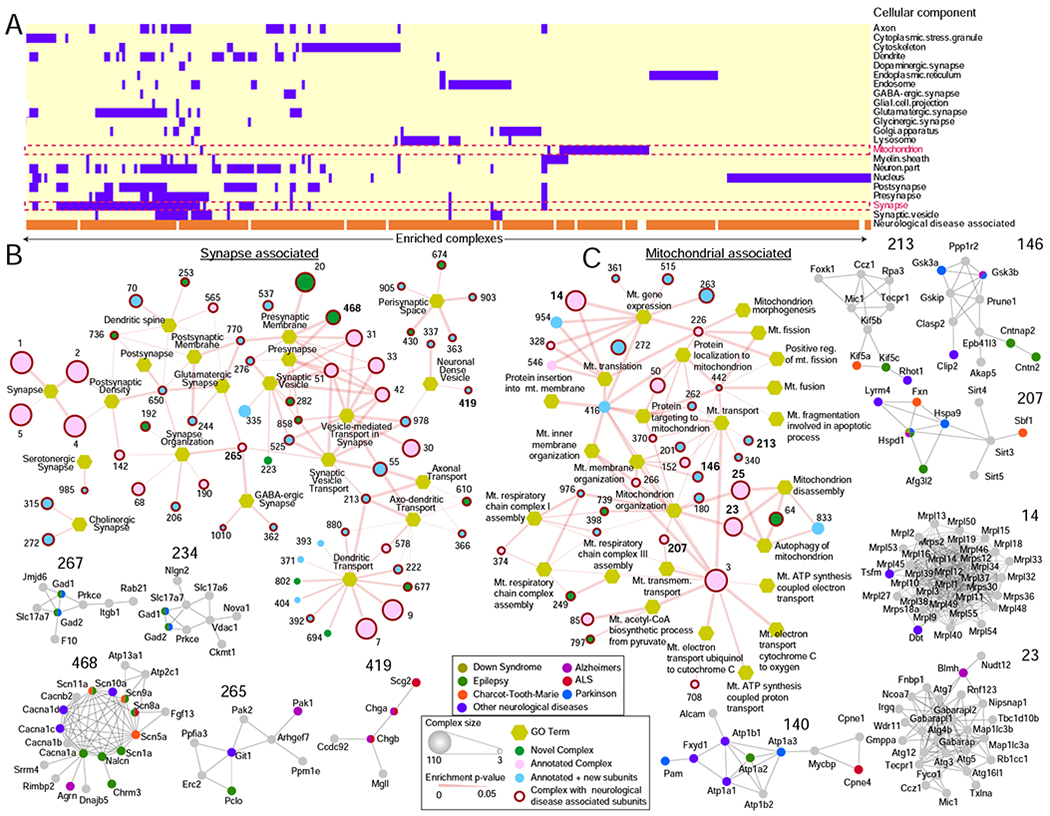
A BraInMap assemblies enriched for select neuronal functions (GO annotation terms).
B Protein complexes (circles; size proportional to subunit number) enriched for synaptic functions (hexagons). Red outlines indicate links to neurological disorders (examplars shown at bottom).
C Protein complexes enriched for mitochondrial (Mt.) functions.
Manifold other complexes in BraInMap are linked to mitochondrial function (Figure 4C; Table S8), which plays a crucial role in meeting the elevated energetic demands required for neuronal homeostasis. These include complex 14, which consists of mitochondrial ribosomal proteins (Mrpl/s), and the related complexes 23 and 25, which contain autophagosomal proteins involved in mitochondrial turnover. Conversely, complex 226, comprised of factors involved in mitochondrial fission (Dnm1l, Mff), has links to neurological disorders through Scg3, which is involved in secretion of neuropeptides and hormones such as pre-opiomelanocortin from the CNS (Tanabe et al., 2007) as well as neurotoxin-induced apoptosis of Dopaminergic neurons in a PD model (Li et al., 2012).
BraInMap identifies manifold RNA binding assemblies
Previously unreported complexes in BraInMap are significantly enriched for involvement in RNA metabolism (Figure 5A), including messenger RNA processing (FDR P = 1.6 x 10−2) and binding (FDR P = 2.7 x 10−2). These assemblies typically comprise RNA binding proteins (RBPs) (Figure 5B; Table S8), which mediate the biogenesis, distribution, and metabolism of both coding and non-coding RNAs (Hentze et al., 2018). BraInMap identifies assemblies ranging in size from 8 interacting RBPs, such as complex 250, which includes Ago1/2/3 and Stau2 (Figure 3F) to larger complexes with over a dozen subunits. For instance, complex 22 (Figure 5C) contains 28 RBPs (Atxn2/2l, Ddx1/3x/5/6/17, Dhx15, Elavl 1, Fam98a/b, Fus, Hnrnpdl/h1/u/ul1, Ilf2/3, Khsrp, Lsm12, Mcrip1, Rptor, Rtcb, Rtraf, Tdp-43, Tia1, Tial1, Urm1).
Figure 5 – BraInMap identifies complexes with diverse functions.
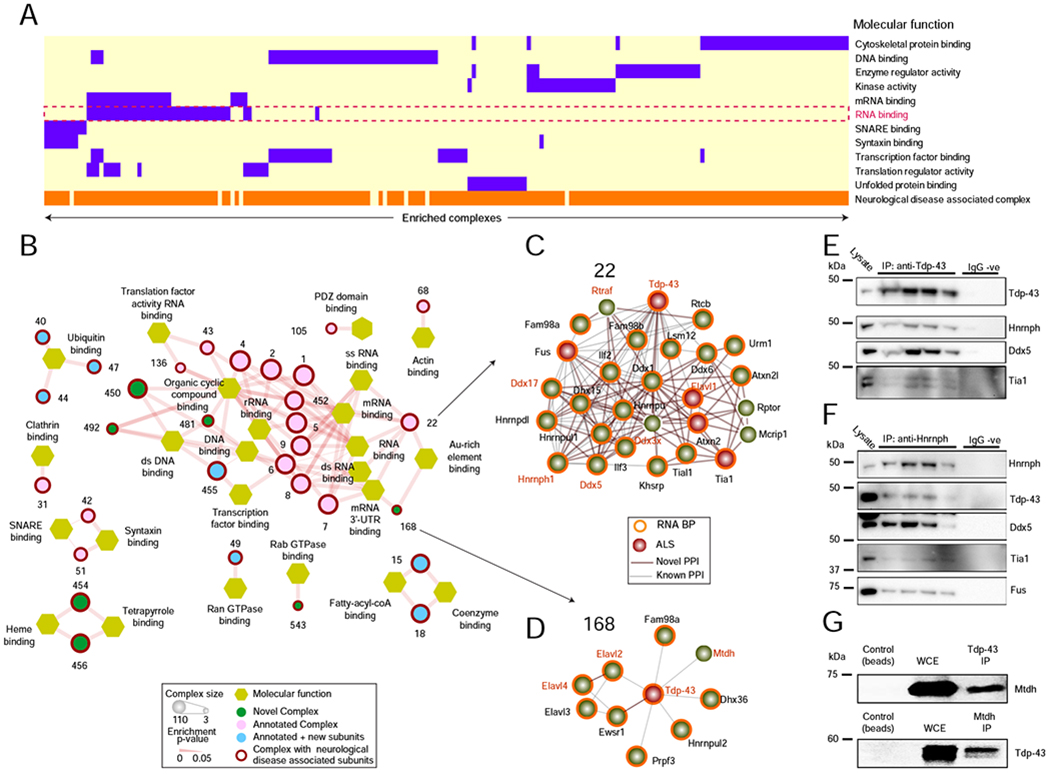
A BraInMap complexes enriched for RNA-binding (dashed box), other annotation terms (purple), and disease associations (orange).
B Sub-network of RNA-related complexes (olive); outline (red) indicates a link to neurological disorder.
C Putative module (complex 22), composed of 28 RBPs (orange) with links to ALS (red).
D Co-Immunoprecipitation (Western blot) analysis of endogenous Tdp-43 confirms physical associations with Hnrnph, Ddx5, and Tia1 (doublet). Lysate and replicate pulldowns provided; no non-specific signal observed using rabbit or mouse IgG (IgG −ve).
E Co-IP analysis of endogenous Hnrnph confirms interactions with Tdp-43, Ddx5, Tia1, and FUS/TLS.
F Complex 168 (Tdp-43 co-complexed with Elavl2/3/4, Ewsr1, Fam98a, Dhx36, Hnrnpul2,Mdth, Prpf3).
G Reciprocal co-IP analysis confirms the association of Mtdh with Tdp-43 in the mouse brain.
We confirmed the interaction between mouse Tdp-43, Hnrnph1, Ddx5, Tia1 and Fus, key members of complex 22, by co-immunoprecipitation (co-IP) of either endogenous mouse Tdp-43 or Hnrnph1 from brain cortices of wild-type C57BL/6J mice (n=4). Whereas RBP components were absent from control IPs using either rabbit or mouse IgG, IP of Tdp-43 co-precipitated endogenous Hnrnph, Ddx5 and Tia1 (Figure 5D; Figure S2A). Likewise, IP of endogenous Hnrnph1 reciprocally pulled-down Tdp-43, Ddx5, Tia1 and Fus (Figure 5E; Figure S2B). Tdp-43 was also detected as a component of complex 168 that contains Dhx36, Elavl2 (HuB), Elavl3 (HuC), Elavl4 (HuD), Ewsr1, Fam98a, Hnrnpul2, Mtdh (Aeg-1), and Prpf3 (Figure 5F; Table S4). In this other RBP complex we confirmed the previously unreported association of Mtdh (AEG-1) with Tdp-43 in mouse brain lysates by reciprocal co-IP (Figure 5G).
RBP-containing complexes with relevance to ALS/FTD are affected by disease state
Complexes 22 and 168 are of particular interest since they contain multiple RBPs genetically linked to ALS and FTD. Mutations in (TARDBP) (Kabashi et al., 2008; Rutherford et al., 2008; Sreedharan et al., 2008), FUS/TLS (Kwiatkowski et al., 2009; Vance et al., 2009) and TIA1 (Mackenzie et al., 2017) lead to the accumulation of pathological insoluble cytoplasmic inclusions in motor and cortical neurons (Mackenzie et al., 2010; Sreedharan et al., 2008). ATXN2 is a common genetic modifier of ALS, in addition to its role in spinal cerebellar ataxia (Elden et al., 2010), and EWSR1 mutations are associated with the disease (Couthouis et al., 2012).
Given the multiple links of complex 22 to neurodegeneration, we examined a mouse model of ALS to explore the relationship of the components of this complex to disease progression. Overexpression of human TDP-43 (TDP-43WT/WT) in mice results in rapid degeneration of motor neurons with associated pathological aggregates (Wils et al., 2010), whereas depletion of Atxn2 (a component of complex 22) reduces aggregation of the transgenic TDP-43, increasing motor neuron survival and extending lifespan (Becker et al., 2017). We, therefore, performed co-IP experiments of exogenous human TDP-43 from brain cortices in both disease prone TDP-43WT/WTAtxn2[+/+] and protected TDP-43WT/WTAtxn2[+/−] mice (n=4/group), and used quantitative mass spectrometry to explore changes in complex 22 components linked to neuroprotection (STAR Methods)
Immunoprecipitated TDP-43 pulled down complex 22 RBPs Ddx1/3x/5/17, Elavl4, Fam98b, Fus, Hnrnpdl/h1/u, Khsrp, Rtcb and Rtraf from the brains of susceptible TDP-43WT/WTAtxn2[+/+] mice (Figure 6A), confirming the interactions detected by co-fractionation. Interestingly, however, in the protected TDP-43WT/WTAtxn2[+/−] mice these interactions were all reduced, with the exception of Ddx1 (fold change of 1.00) (Figure 6A). This finding is highlighted in the reproducible reductions observed in TDP-43 binding to Hnrnph1, Ddx3x, Ddx5, Ddx17 and Rtraf (Hnrnph1: −1.83 fold-change, −Log10 P value = 1.02; Ddx3x: −1.82 FC, −Log10 P value = 1.28; Ddx5: −1.95 FC, −Log10 P value = 1.09; Ddx17: −1.93 FC, −Log10 P value = 1.06; Rtraf: −1.52 FC, −Log10 P value = 1.10; n= 4/group) (red text labels; Figure 6A). Reduced co-IP of Hnrnph1 with exogenous TDP-43 was further confirmed by immunoblot (Figure S2E, F).
FIGURE 6 – RBP complexes are affected in ALS models.
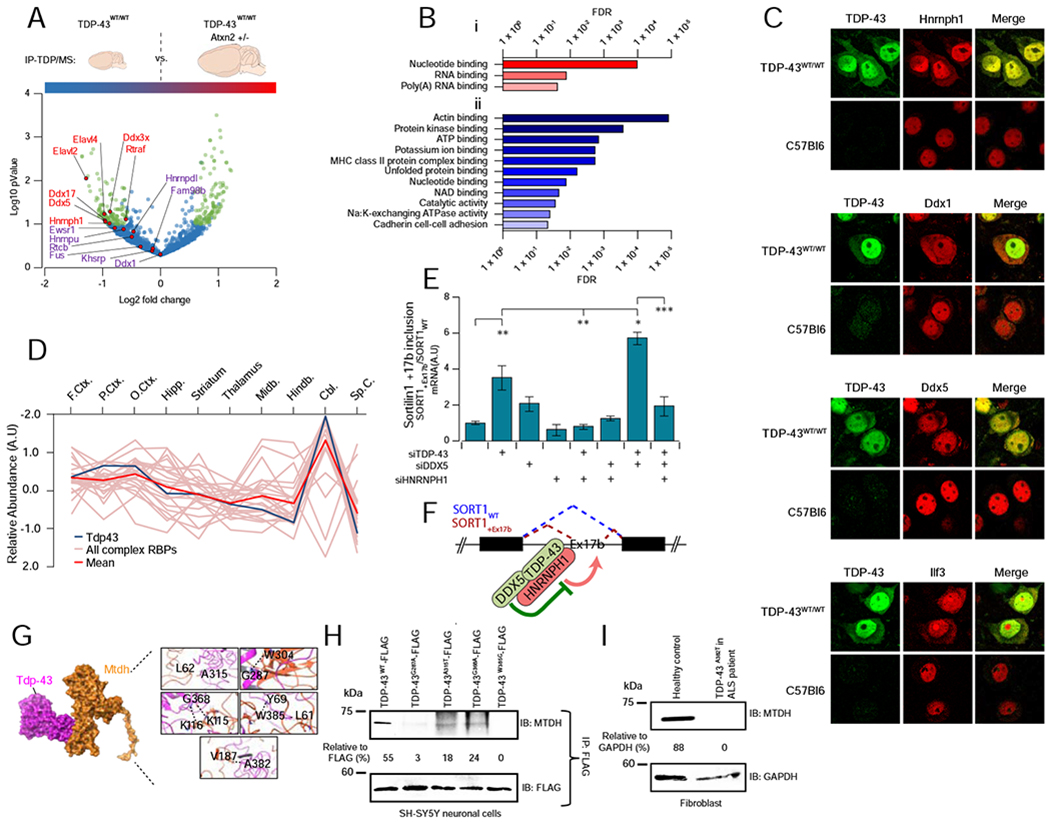
A Complex 22 is responsive to neuropathology. Volcano plot summarizing results from co-IP pulldowns of exogenous TDP-43 from cortical lysates from diseased (TDP-43WT/WT) versus protected (TDP-43WT/WTAtxn2[+/−]) transgenic mice. Precipitates were subject to quantitative mass spectrometry to define differential binding to pathogenic TDP-43 (> ±0.50x Log2-fold, −Log10 P < 1, highlighted in green). Interaction of Hnrnph1, Ddx5 and Ddx17 significantly reduced in protected animals (n = 4 per group, students t-test P ≤ 0.05).
B Gene ontology molecular function annotations of proteins showing (i) decreased interaction and (ii) increased interaction with transgenic TDP-43 in protected TDP-43WT/WTAtxn2[+/−] murine brain. Shown are terms with FDR−1 >20.
C Confocal immunofluorescent microscopy showing a redistribution of Complex 22 RBPs (Hnrnph1, Ddx1, Ddx5, Ilf3) into human TDP-43 positive cytoplasmic accumulations (arrows) in affected cortical neurons of transgenic TDP-43WT/WT mice, which is not seen in wild type animals. Scale bar = 20μm.
D The relative brain region expression pattern of Tardbp (TDP-43; dark blue line) closely mirrors the mean expression complex 22 expression (red line). Other RBP components are traced in pink.
E Knockdown (siRNA) of TDP-43 or TDP-43/DDX5 together results in the inclusion of Exon 17b of sortilin1 (SORT1) in SH-SY5Y cells (quantified by qPCR), whereas knockdown of interacting partner HNRNPH1 blocks this effect. Graphs show ratio (mean ± SEM) of SORT1 transcripts with/without exon17b (SORT1+Ex17b vs SORT1WT); n = 3 per group (ANOVA with Tukey’s multiple comparison between all groups: * P < 0.05, ** P< 0.01, *** P < 0.001).
F Model of TDP-43 and DDX5 interaction illustrates coordinate inhibition of SORT1 Ex17b inclusion, dependent upon joint association with HNRNPH1.
G Structural model of mutations in residues of TDP-43 linked to familial ALS (A315T, G287A, G368A, W385G, A382T) that map to the interaction interface with MTDH.
H Co-IP analysis showing a reduced association of MTDH in SH-SY5Y cells expressing FLAG-tagged TDP-43 with ALS-relevant mutations at the predicted interaction interface.
I TDP-43 interaction with MTDH is abrogated in ALS-patient-derived fibroblasts carrying a pathogenic mutation (A382T), as compared to fibroblasts from a healthy control.
Elavl2 and Elavl4 of complex 168 were also depleted in the co-IP pulldowns from disease resistant TDP-43wT/wTAtxn2[+/−] mice as compared to the susceptible strain (Elavl2: −2.43 FC, −Log10 P value = 2.05; Elavl4: −1.96 FC, −Log10, P value = 1.23; n = 4/group) (red text labels; Figure 6A). A similar trend was observed with Ewsr1, but did not reach statistical significance (Ewsr1: −1.74 FC, −Log10, P value = 0.91) (purple text labels; Figure 6A). Elavl proteins are cytosolic RBPs, which suggests that Atxn2 modulates the interaction of TDP-43 with cytoplasmic RNP granules, thereby decreasing pathologic insoluble inclusions in TDP-43WT/WTAtxn2[+/−] mice. These results point to selective dissociation of pathologic TDP-43 from different RBP assemblies upon Atxn2 reduction, consistent with reduced recruitment of TDP-43 to cytoplasmic SGs resulting in fewer inclusions in the disease resistant strain (Becker et al., 2017; Elden et al., 2010).
In the protected TDP-43WT/WTAtxn2[+/−] mice, TDP-43 showed decreased interaction with proteins associated with RNA binding functional terms (Figure 6Bi), this is exemplified by the volcano plot distribution of heterogeneous nuclear ribonucleoproteins (HNRNPs; Figure S2C). Interestingly, TDP-43 in the protected TDP-43WT/WTAtxn2[+/−] mice showed increased interaction with proteins clustering with functional categories such as protein folding, ATP binding, and sodium/potassium ion homeostasis (Figure 6Bii); this is exemplified by the volcano plot distribution of heat shock proteins and protein isomerases that form the unfolded protein response (Figure S2D). These data indicate that the interaction of pathologic TDP-43 with Hnrnphl, Ddx3x, 5 and 17, Rtraf and other RBPs involved in RNA processing is responsive to neuropathophysiological states.
Our observation that TDP-43 shows increased interaction with complex 22 components in disease-affected mice led us to predict that these RBP components would be dysregulated in neurons affected with TDP-43 pathology. We, therefore, investigated distribution of RBPs in the cortices of transgenic TDP-43WT/WTAtxn2[+/+] mice by immunofluorescent confocal microscopy.
As shown previously, neurons in the TDP-43WT/WTAtxn2[+/+] mice exhibited increased levels of cytoplasmic TDP-43 and pTDP-43 (Becker et al., 2017). Neurons showing cytoplasmic distribution of transgenic TDP-43 (also immuno-positive for phosphorylation at S409/410; Figure S2Hi) exhibited increased cytoplasmic distribution of complex 22 RBPs Hnrnph1, Ddx5, Ddx1, and Ilf3 (Figure 6C). The cytoplasmic distribution of these RBPs mirrored that of TDP-43; for instance, neurons showing focal accumulations of TDP-43 also showed co-localized accumulations of complex 11 RBPs (Figure 6C; Figure S2Gi–iv). These RBPs are restricted to the nuclei of neurons and non-neurons in wild type C57Bl6 mice. The RBP U2af, which is not a component, does not redistribute out of neuronal nuclei in TDP-43WT/WTAtxn2[+/+] mice, nor does the nuclear protein Histone H3 (Figure S2Hii–iii). This finding emphasizes how TDP-43 pathology may specifically disrupt complex function through subunit sequestration and aberrant cellular relocalization.
By examining our regional proteomic data (Figure 3E), we examined the distribution of complex 22 RBPs (Figure 6D; Figure S2I). Cortical regions show a small relative increase in complex 22 expression compared to normalized whole brain expression (mean ±SEM, F.Ctx 0.27 ±0.09, P.Ctx 0.19 ±0.08, O.Ctx 0.36 ±0.10), while the thalamus, midbrain, and hindbrain exhibited a moderate decrease (mean ±SEM, Thalamus −0.46 ±0.07, Midbrain (Midb.) −0.25 ±0.13, Hindbrain (Hindb.) −0.46 ±0.13). The region displaying highest expression is the cerebellum (an unaffected area in ALS) while the region showing the lowest expression is the spinal cord (mean ±SEM, Cerebellum (Cbl) 1.32 ±0.23, Spinal Cord (Sp.C) −0.73 ±0.15). The regional expression of Tdp-43 (Figure 6D) and Fus clearly correlate with the pooled expression pattern of complex 22 (Pearson correlation of region mean to Tdp-43 r =0.977, P <0.001, to Fus r =0.951, P <0.001). As pathological inclusions of TDP-43 are almost universally detected in sporadic ALS-affected spinal cord and FTD-affected cortical neurons (but rarely in cerebellar neurons) (Brettschneider et al., 2014; Brettschneider et al., 2013), it appears that total regional expression of TDP-43 (or complex 22) does not directly correlate with distribution of pathology, suggesting that relative cellular expression or complexation levels may be more relevant. Notably, as indicated above, while the expression of many RBPs is ubiquitous, complex 22 RBPs are enriched in neurons (Dopaminergic, GABAergic, Glycinergic, Adrenergic, and Serotonergic) as compared to non-neuronal cells (except for Oligodendrocytes; Figure 3G). Hence, factors other than absolute level of expression of TDP-43 (or complex 22) may drive motor neuron susceptibility.
HNRNPH1 function is antagonistic to that of TDP-43 and DDX5
We investigated the functional relationships of RBPs associated with TDP-43. We assayed alternative splicing upon depletion of one or more co-complex members based on the previously reported observation that a reduction in TDP-43 leads to increase inclusion of exon 17b of SORT1 (Sortilin 1) (Polymenidou and Cleveland, 2011; Prudencio et al., 2012). Expression of TDP-43, HNRNPH1 and DDX5 was reduced in SH-SY5Y cells through transfection of siRNA (Figure S2J, K). As previously reported (Prudencio et al., 2012), knockdown of TDP-43 (siTDP-43) significantly increased the ratio of SORT1+17b transcript (inclusion of exon 17b) to SORT1WT (predominant transcript lacking exon 17b) (ANOVA with Tukey’s multiple comparison between all groups: siTDP-43 = 3.54 A.U. ± 0.082, P < 0.01 vs control/siCtrl non-targeting siRNA) (Figure 6E; Figure S2L). The knockdown of DDX5 slightly increased SORT1 exon 17b inclusion, but the effect was not statistically significant. Strikingly, however, coordinate knockdown of TDP-43 and DDX5 together led to a dramatic increase in inclusion of SORT1 exon 17b (siTDP-43/DDX5 ratio = 5.74 ± 0.36, P < 0.05 vs siTDP-43 alone). Conversely, while knockdown of HNRNPH1 alone had no significant effect on alternate splicing of SORT1, it exerted a profound antagonizing effect on the splicing functions of both TDP-43 and DDX5. Knockdown of HNRNPH1 blocked inclusion of SORT1 exon 17b resulting from depletion of either TDP-43 (siTDP-43/HNRNPH1 = 0.84 ± 0.15, P < 0.01 vs siTDP-43) or TDP-43 and DDX5 together (siTDP-43/DDX5/HNRNPH1 = 1.97 ± 0.51, P < 0.001 vs siTDP-43/DDX5) (Figure 6E).
These data imply that TDP-43 and DDX5 exert cooperative (synergistic) functions in repressing HNRNPH1-mediated inclusion of alternatively spliced exons (Figure 6F). TDP-43 was known to participate with RBPs to facilitate splicing (Mohagheghi et al., 2016), and HNRNPH1 was known to bind the intron upstream of SORT1 exon 17b (i.e. same binding region as TDP-43), but no significant effect on splicing was recorded. Our study establishes the interaction of specific RBPs as a functional module in which members exert antithetical effects on exon usage.
ALS-mutations disrupt TDP-43-MTDH association within an RBP complex
TDP-43 co-purified with other factors linked to ALS, including MTDH (metadherin, also known as Astrocyte-elevated gene-1). Molecular docking using the crystal structures of human MTDH and TDP-43 revealed that five residues of TDP-43, which when mutated cause familial ALS (A315, G287, G368, W385, A382), are located in the predicted interaction interface (Figure 6G). Functional studies support the association of TDP-43 with MTDH: TDP-43 plays a role in microRNA (miRNA) biogenesis and activity via Microprocessor and RNA-induced silencing complex (RISC) (Kawahara and Mieda-Sato, 2012) and regulates miRNA loading to RISC (King et al., 2014). MTDH, an RBP, has also been shown to be physically and functionally associated with RISC activity (Yoo et al., 2011).
To independently assess the effect of these ALS-associated variants on MTDH and TDP-43 interaction, we generated four mutant TDP-43-FLAG variants (A315T, G287A, G368A, W385G), and found that G287A and W385G variants disrupted the association in differentiated SH-SY5Y neuronal cells, while two other variants partially impaired the interaction (Figure 6H). In addition, we demonstrated a perturbed interaction in fibroblasts isolated from an ALS patient carrying a pathogenic mutation (A382T) in TDP-43 as compared to healthy control (Figure 6I). Taken together, our data suggest that the disruption of miRNA regulation observed in ALS pathobiology (Eitan and Hornstein, 2016) may reflect disruption of interaction between TDP-43 and factors such as MTDH.
Modules connected to human neurological conditions
Conserved components of multiple BraInMap assemblies had strong links to diverse human neurological disorders (Figure 7A). These include macromolecules showing significant enrichment (Figure 7B) for components associated with psychiatric conditions such as schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics et al., 2014), neurodevelopmental disorders such as autism (Sanders et al., 2015) and neurodegenerative diseases such as AD, PD and ALS (Dormann et al., 2010) and for associated genetic variants (Table S9). Relative to other diseases, neurological dysfunction annotations (DisGeNet) (Piñero et al., 2017) (Figure 7C) were often associated with several subunits of certain novel brain complexes. This suggests that disruption of complex function by multiple avenues can lead to similar mechanistic and phenotypic outcomes.
FIGURE 7 – Macromolecular links to neurological disorders.
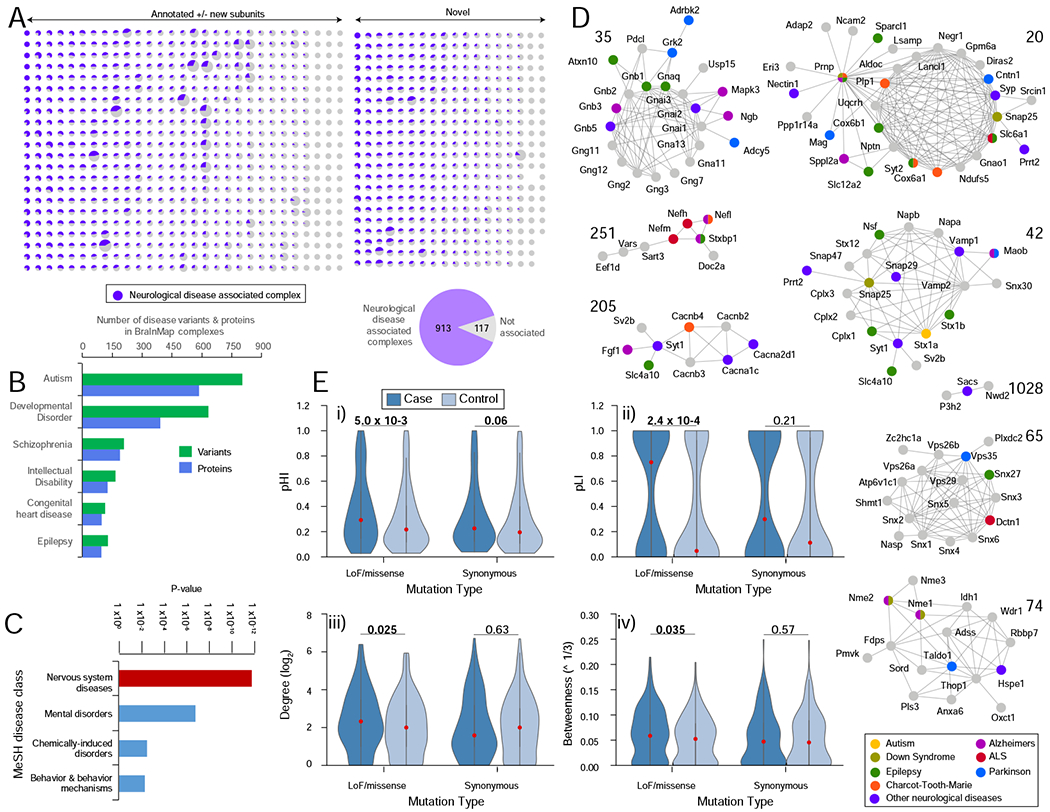
A Putative pathophysiological relevance of complexes in BraInMap. Proportion (purple) of subunits of each assembly linked to neurological impairment (see Table S6 for details).
B Number of BraInMap components (orthologs) and corresponding human genetic variants associated with specific neuropathologies (see Table S5).
C Enrichment (hypergeometric p-value) of complex subunits with links to neuropathology as annotated in DisGeNET (Pinero et al., 2017).
D Representative complexes associated with Alzheimer’s (magenta), autism (yellow), amyotrophic lateral sclerosis (red), epilepsy (green), Down syndrome (olive), Charcot-Toothe-Marie syndrome (orange), Parkinson’s (blue), or other neurological disorders (purple).
E Enrichment of genes encoding BraInMap components harboring de novo variants for (i) haploinsufficiency (pHI) and (ii) pLI (probability a gene is intolerant to loss of function (LoF) mutations) versus synonymous variants in affected individuals in comparison to unaffected controls; (iii) network degree and (iv) betweeness of genes with de novo LoF/missense or synonymous mutations in neurodevelopmental disorder afflicted individuals or unaffected controls. Violin plot width proportional to protein abundance (red dot, median); P-values (one-tailed U-test; P< 0.05 in bold) shown at the top.
An illustrative example, highlighted in Figure 7D, is complex 42, which includes SNARE protein components necessary for synaptic vesicle fusion in neurotransmitter release (Chen et al., 2002) that are deficient in neurodegenerative impairments such as in PD (Burre et al., 2010) and Huntington’s disease (HD)(Smith et al., 2007). Alterations in SNARE component SNAP25 have also been associated with psychiatric disorders, particularly attention-deficit hyperactivity disorder, in both mice and humans (Brophy et al., 2002; Bruno et al., 2007). In a similar vein, BraInMap complex 35 (discussed further above) contains the ortholog of ATXN10 in which repeat expansion mutations have been shown to cause spinocerebellar ataxia type 10 (Matsuura et al., 2000). Likewise, complex 20 (discussed above) contains a number of subunits associated with neurological disorders, including PLP1 (Hobson and Kamholz, 1993), CNTN1 (Compton et al., 2008) and PRNP. In complex 205, the human ortholog of the voltage-gated L-type calcium channel subunit Cacnb3 has been linked to bipolar disorder (Psychiatric Gwas Consortium Bipolar Disorder Working Group et al., 2011) while a rare mutation in the ortholog of the subunit synaptogamin1 SYT1 results in severe juvenile motor deficits and cognitive impairment (Baker et al., 2015). Complex 251 neurofilaments (Nefl, Nefm, Nefh) accumulate in certain neurodegenerative diseases and are associated genetically with ALS (Campos-Melo et al., 2018). NEFL mRNA stability is regulated by direct binding to the 3‘UTR by TDP-43 and mutant SOD1, the major disease proteins of ALS, potentially dysregulating the stoichiometry of neurofilament polymerization (Ge et al., 2005; Strong et al., 2007).
Another example is complex 65 (Figure 7D), which consists of a number of retromer complex vacuolar protein sorting-associated components (Vps26a/26b/29/35) and sorting nexins (Snx1-6/27), implicating this assembly in endosomal delivery. Mutations in VPS35 impair vacuole dynamics leading to defects in macroautophagy, mitochondrial turnover and AMPA receptor trafficking (Williams et al., 2017) resulting in PD (Vilarino-Guell et al., 2011). Also present in this complex is dynactin 1 (DCNT1), important for retrograde transport of vesicles and autophagic clearance (Laird et al., 2008) and in which mutations cause ALS and Perry’s syndrome (Farrer et al., 2009; Munch et al., 2004). These observations mesh with accumulating evidence that autophagic deficiencies underlie the neurological dysfunction seen in diverse clinical disorders, potentially explaining their heterogeneous etiology.
BraInMap assemblies are frequent targets of disruptive mutations impairing neurodevelopment
To evaluate whether brain complexes had an elevated rate of disease-linked mutations, we investigated the correspondence of ~21,000 de novo variants previously detected in neurodevelopmental disorder-affected individuals as compared to unaffected controls. The variant data was compiled from 40 different published studies (listed in denovo-db v.1.5), including the Deciphering Developmental Disorders project and genetic studies of Autism, Schizophrenia, Epilepsy, and Intellectual Disability (Turner et al., 2017). To control for differences in abundance in the enrichment analyses, we compared subsets of proteins (2,298 per group) from BraInMap and background (detectable) proteome with matched abundance distributions for the enrichment analyses (one-tail Fisher’s exact test). The same procedures were applied in Figure 7E analyses (calculating node degree, betweenness, pLI, pHI).
As summarized in Table S9, BraInMap was significantly enriched for gene products harboring de novo loss-of-function (LoF) mutations in neurologically impaired individuals (LoF, expected:observed ratio = 1.41, P value = 3.4 x 10−4 by a two-tail binomial test; see STAR Methods), but not for synonymous mutations or variants seen in unaffected controls (Figure 7E). Starting with a curated list of 1,007 known Autism-associated gene products (Basu et al., 2009), we again observed significant overlap with BraInMap (expected:observed = 1.50, P value = 3.2×10−4; Table S9). In contrast, gene products with rare synonymous variants from the National Heart Lung Blood Institute Exome Sequencing Project (NHLBI ESP) study exhibited the background rate (expected:observed = 0.86, P value = 1.0).
De novo variants typically occur on one copy of a gene; hence, to confer risk, should arise more frequently in genes susceptible to haploinsufficiency. In neurodevelopmental disorder-affected individuals, orthologs of BraInMap components harboring de novo LoF and missense mutations had, on average, a significantly higher probability of being haploinsufficient (pHI) (Huang et al., 2010) than unaffected controls (median 0.33 versus 0.23, binomial test P value = 5.0 x 10−3; Figure 7E, panel i). Components with disruptive de novo mutations were less tolerant to genetic variation, with a higher average probability of loss-of-function intolerance (pLI)(Lek et al., 2016) compared to controls (median 0.78 versus 0.05, P value = 2.4 x 10−4; Figure 7E panel ii) or synonymous variants (median 0.33 versus 0.1, P value = 0.21). Taken together, the analyses in Table S9 show that BraInMap assemblies are frequent targets of disease-related variants that potentially impair neurodevelopment.
Disruptive missense mutations in autistic individuals reportedly impact highly connected network (hub) components (Chen et al., 2018). Consistent with this, proteins with loss of function (LoF) and missense mutations in affected individuals exhibited, on average, significantly higher network connectivity (‘degree’) relative to unaffected controls (median 0.24 versus 0.2, P value = 0.025 by one-tail U-test; Figure 7E, panel iii). In contrast, no significant difference was observed for synonymous variants (median 0.15 versus 0.2, P = 0.63). Similar trends were evident with other measures of network centrality such as shortest paths or ‘betweenness’ (median 0.06 versus 0.05, P value = 0.035; Figure 7E, panel iv). BraInMap, therefore, offers a potential mechanistic framework for determining how genetic variants confer clinical risk through interaction perturbation.
DISCUSSION
Tissue, regional and cell-type enriched macromolecules drive brain function, physiology, and disease. However, direct mapping of molecular connections in the CNS is challenging. While a number of experimental methods have been devised to study protein interactions that occur in neurons (see for example (Zhu et al., 2018) and (Ganapathiraju et al., 2016)), most studies have typically been executed in a piecemeal manner that does not allow for a comprehensive interrogation of the brain interactome. To address this gap, we applied a systematic, data-driven functional proteomic approach. Using deep biochemical fractionation, we identified endogenous protein complexes in murine brain in a near native context, avoiding artifacts due to epitope tagging or ectopic over-expression. Moreover, it is complementary to existing genetic surveys (e.g. GWAS), cell connectivity maps (i.e. NIH BRAIN initiative), and single cell transcriptome data.
In doing so, we establish an approach to identifying macromolecular protein complexes in post-mortem tissues, which could be highly useful in studying human brain samples. Rather than viewing ALS/FTD as a TDP-43 proteinopathy, a growing consensus is to consider the condition as resulting from insolubility and splicing defects of a number of RBPs. Our discovery that ALS-associated RBPs natively assemble as a functional splicing module raises the possibility that a more accurate descriptor of ALS/FTD is as an RBP ‘complexopathy’ that results in part from splicing defects due to insolubility of a subnetwork of RBPs. Therefore, BraInMap represents an important new tool to interrogate the composition, distribution, and function of the macromolecules of the CNS and their role in normal and diseased brain physiology.
Comprehensive characterization of the multi-protein architecture of the mammalian brain, therefore, represents a milestone for neurobiology. It also provides access to unusual classes of questions. For example, of the 5,677 proteins to a complex, 2,836 (or just under 50%) were assigned to multiple complexes. These ‘promiscuous’ interactors (or “moonlighting proteins”), which are members of multiple complexes, show an enrichment for functional roles linked to protein transport (Benjamini Hochberg FDR p = 9.8 x 10−32), mRNA processing (Benjamini Hochberg FDR p = 1.7 x 10−27) and translation (Benjamini Hochberg FDR p = 1.6 x 10−17), as well as a higher (1.8-fold) average abundance in brain according to PAXdb (students T test P = 4.7 x 10−3)(Wang et al., 2015) as compared to proteins assigned only to one complex (Figure S1D, Table S8).
Given the rapid evolution of mammalian brain, we could also examine the evolutionary trajectories of these macromolecular assemblies by assigning individual proteins an ‘age’ that represents their phylogenetic origin based on ortholog projections (see STAR Methods). In this preliminary investigation, most brain complexes exhibited a mixture of component ages (Figure S1G & H; Table S4), suggesting that younger, possibly less tightly bound components represent more recent evolutionary adaptations relative to more ancient assemblies. The previously unreported complexes in our network were also enriched for mammalian proteins, whereas documented assemblies exhibited a higher fraction of components of lower eukaryotic origin (Figure S1F). Taken together, these observations are consistent with the notion that mammal restricted macromolecules are more likely to mediate brain-specific functions, whereas previously described complexes are more likely associated with house-keeping roles common to multiple lineages. Further in depth analysis of the BraInMap resource is needed to refine and rigorously test this notion.
To support such follow up studies, BraInMap can be accessed via a dedicated web portal (https://www.bu.edu/dbin/cnsb/mousebrain/) that supports search queries, network visualization, and biological inference. The resource is currently a static representation of neuronal circuits, however, whereas cellular interaction networks are dynamic. The mammalian brain is impacted by changing developmental and physiological cues and contextual signaling cascades. In principle, our interactome profiling technology can be used to study these network fluxes to reveal interactions underlying particular neurological and pathological states. Thus, follow up studies using this platform together with sophisticated functional manipulation tools, such as optogenetic control of protein interactions and complex assembly, data science methods based on newer deep learning classifiers, as well as more precise information about macromolecular localization and topology, may define the mechanistic principles driving neurological processes central to healthy and impaired brain function.
STAR ★ METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the lead contact, Dr. Andrew Emili, by email at aemili@bu.edu. Plasmids are available upon request; this study did not generate any other new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Tissue Harvest and Protein Extraction
12 week old male CD1 mice were euthanized and the brains excised, washed several times in ice-cold phosphate-buffered saline (PBS) to remove blood, snap-frozen and stored in −80°C. For protein isolation, tissues were homogenized in ice-cold lysis buffer (10mM Tris-HCl pH7.4, 10% glycerol, 50mM NaCl, 1mM Dithiothreitol (DTT), 1mM Ethylenediaminetetraacetic acid (EDTA), 1% Triton X-114 and complete Mini EDTA-free Protease Inhibitor (Roche)) using a dounce tissue grinder on ice. The lysates were kept on ice for 30 minutes and centrifuged at 20,000rcf for 10 minutes to pellet cellular debris. Prior to fractionation, lysates were treated with 100 units/ml Benzonase (Sigma) to remove nucleic acids and further clarified by centrifugation to remove debris. A Bradford assay was performed to determine protein concentrations. The lysate saved as protein extract I.
The detergent-free protein extract was prepared by homogenization of the brain tissue in lysis buffer II (containing 10mM Tris-HCl pH7.4, 20% glycerol, 50mM NaCl, 1mM DTT, 1mM EDTA and complete Mini EDTA-free Protease Inhibitor (Roche)). The suspension was incubated on ice for 30 minutes and then centrifuged at 4,000rcf for 10 minutes. The supernatant was saved as protein extract II while the pellet was resuspended in detergent containing lysis buffer (10mM Tris-HCl pH7.4, 20% glycerol, 50mM NaCl, 1mM DTT, 1mM EDTA, 1% Triton X-114 and protease inhibitors). The sample was incubated with gentle shaking for 45 min at 4°C and centrifuged at 6,000rcf for 10 min. The supernatant was saved as protein extract III. 100 units/ml Benzonase was then added to both protein extracts II and III and protein concentrations measured by Bradford assay.
METHOD DETAILS
2-D Biochemical Fractionation
Isoelectric Focusing (IEF) Fractionation
The protein extract was fractionated by isoelectric focusing using a MicroRotofor IEF cell (Bio-Rad) set up. 3mg of total protein were added to IEF running buffer (20% glycerol, 2% IPG buffer pH5-8) and an electric field at a constant power of 1W was applied to the focusing cell while the voltage and current were limited to 350V and 2500 mA, respectively. The separation was stopped after 150 min once the voltage held constant for about 45 min. Five fractions per sample were collected across a pH range of 5-8.
Ion Exchange (IEX) Fractionation of IEF fractions
Each IEF fraction was subjected to IEX-HPLC separation using mixed-bed PolyCATWAX chromatography columns (200 × 2.1mm i.d., 5μm, 1000-Å) purchased from PolyLC Inc (MD, USA) without any preparation step. Depending on the pH of IEF fractions collected, an IEX buffer system of Tris pH8 or MES pH6 was used. IEX buffers were always freshly prepared with HPLC grade H2O and comprised of a low salt buffer A (10mM Tris-HCl pH8 (or 10mM MES pH6), 0.01%-NaN3, 5%-Glycerol) and high salt buffer B (buffer A + 1.5M NaCl). All HPLC fractionations were performed using an Agilent 1260 infinity HPLC system (Agilent Technologies, ON, Canada). The PolyCATWAX IEX column was equilibrated with running low salt buffer A for 30 minutes immediately before loading protein samples. Bound proteins were eluted from the column using a linear gradient to 30% buffer B from 5 to 95 min, followed by a gradient to 100% buffer B from 95 to 105 min and an isocratic hold at 100% B until 120 min. Protein elution was monitored by absorption at 260 and 280 nm. The gradient was run at a flow rate of 0.2ml/min and 60x 0.4ml fractions were collected (the first and last fractions with no peak at 280 nm were discarded). Fractions (i.e. 46 fractions per IEX run, 230 fractions for entire 2-D IEF-IEX fractionation experiment) were prepared and by LC-MS/MS as described.
Ion Exchange Fractionation of Protein Extract I
A total of 2mg soluble protein of protein extract I was loaded to a PolyCATWAX column (200 × 2.1mm i.d., 5μm, 1000-Å). A MES pH6 buffer system (described above) was employed and elution of bound proteins was achieved through application of a linear gradient to 15% buffer B from 2 to 80 min, followed by a gradient to 50% buffer B from until 140 min and a final 20 min long gradient of 50%--100% buffer B. An isocratic hold at 100% B applied until 180 min to elute tightly bound proteins. A total of 90x 0.4ml fractions were collected using a flow rate of 0.2ml/min.
Ion Exchange Fractionation of Protein Extracts II and III
1.2 to 1.5mg total proteins in cytoplasmic and membrane extracts were fractionated on a PolyCATWAX column (200 × 2.1mm i.d., 5μm, 1000-Å) using the MES pH6 buffer system. Protein extracts were resolved using a 120 min gradient program as follows: A linear gradient to 20% buffer B from 2 to 60 min, a gradient to 60% buffer B from 60 to 90 min followed by final 10 min gradient to 100% buffer B and 20 min run with 100% buffer B. 60 fractions by 2 min intervals and using a flow rate of 0.2 ml/min were collected.
Dual-Phase Heparin-Ion Exchange Fractionation of Protein Extract I
In order to enrich low abundance nuclear proteins, a TSKgel Heparin-5PW affinity column (75 × 7.5mm i.d., 10μm, 1000-Å) hyphenated with a PolyCATWAX mixed-bed ion exchange column (200 × 4.6mm i.d., 5μm, 1000-Å). 4mg of protein was loaded to columns and the MES pH6 buffer system was used to resolve multi-proteins complexes in protein extracts. A 240 min elution program consisting of a 10 min gradient with 100% buffer A, followed by a 120 min gradient from 0 to 15% buffer B, a 60 min gradient from 15 to 50% buffer B and a 30 min gradient to 100% buffer B followed by 30 min isocratic hold at 100% buffer B was applied to resolve and fractionate proteins. A total of 120x 0.5ml fractions were collected using a flow rate of 0.25ml/min.
Sample Preparation and Trypsin Digestion
HPLC protein fractions were precipitated overnight at 4°C by adding 10% v/v Trichloroacetic acid (TCA). The fractions then precipitated at 20,000rcf for 30 min and the pellets washed twice with 300μl ice-cold acetone. The pellets were air dried then dissolved in 90μl 50mM NH4HCO3. The samples were reduced by adding DTT (Thermo) to a final concentration of 5mM and incubated for 20 min at 50°C with gentle agitation. The samples were cooled to room temperature and alkylated by adding 10mM lodoacetamide (Sigma) and incubation in the dark for 20 min. To quench excess of lodoacetamide, 5mM DTT was added to each sample. The protein fractions were then digested by adding ĝ of mass spectrometry grade trypsin gold (Promega) and incubated overnight at 37°C with gentle agitation. The digestion was quenched by adding formic acid (FA) to 1% v/v final concentration and the peptide mixture was subjected to purifying using ziptip C18 tips (Millipore). The ziptip C18 tips were first conditioned with 10μ! acetonitrile and then equilibrated with 2x 10μl of 0.1% trifluoroacetic acid (TFA). After loading the peptide mixture to ziptip C18 tips, the samples were washed three times with 0.1 v/v TFA and eluted with 2x 10μl elution buffer (80% acetonitrile, 0.1% TFA). The desalted peptides then lyophilized by using Speed-Vac (Thermo Scientific) and dissolved in 1% FA prior to LC-MS/MS analysis.
LC-MS/MS analysis
All LC-MS/MS analyses performed on an EASY nLC 1200 system (Thermo Scientific) coupled to a Q Exactive HF mass spectrometer equipped with an EASY-Spray ion source (all from Thermo Scientific). A C18 Acclaim PepMap 100 pre-column (3μm, 100 Å, 75μm × 2cm) hyphenated to a PepMap RSLC C18 analytical column (2μm, 100 Å, 75μm × 50cm) (all from Thermo Scientific) was used to separate peptide mixtures prior injection into the mass spectrometer. Depending on sample complexity in each fractionation experiment, 60 or 90-min gradients were used to elute peptides from columns. The quality of LC-MS/MS analysis was repeatedly controlled for by running Trypsin-digested BSA MS Standard (BioLabs) between sample runs.
Regional sample preparation for quantitative mass spectrometry
Tissue from 10 brain regions were dissected from four 12 week old male CD1 mice: 1) Frontal Cortex, 2) Parietal Cortex, 3) Occipital Cortex, 4) Hippocampus, 5) Striatum, 6) Thalamus and Hypothalamus, 7) Midbrain (including Substantia Nigra), 8) Hindbrain (including Pons and Medulla), 9) Cerebellum and 10) Spinal Cord. Each was individually placed in 8M urea with phosphatase (PhosSTOP™, Roche) and protease (cOmplete™, Roche) inhibitors, then sonicated (1 minute, in 2 second pulses) on ice. Sonicated samples were snap-frozen in liquid N2 and then sonicated again as described above. Proteins were reduced for 1 hour with 5mM dithiothreitol (DTT) and alkylated for 30 minutes with 15mM iodoacetamide in the dark. Protein concentration was estimated using a BCA kit (Pierce™ BCA Protein Assay Kit, Thermo) and about 1mg of each sample was allocated for trypsin digestion. Prior to digestion, the 8M urea solution was diluted to 1M with 50mM ammonium bicarbonate. Each sample was digested overnight at 37°C with 10μg sequencing grade trypsin (Pierce™ Trypsin Protease, MS Grade, Thermo)
Prior to TMT (Tandem Mass Tag) labeling, peptides were extracted from each digested sample using c18 Sep-Pak (Waters, 50mg cartridge) and peptide concentrations were measured with a peptide quantification assay (Pierce™ Quantitative Colorimetric Peptide Assay, Thermo). Sample peptide concentrations were adjusted to 1μg/μL in 100mM triethylammonium bicarbonate (TEAB), and 100μg of the sample was aliquoted for labelling. A common pool of samples was generated by combining equal parts of each of the 40 samples. Peptides from the samples and the pool were labelled with 0.4mg and 1.6mg of TMT label, respectively (TMT11plex™ Isobaric Label Reagent Set, 1 x 0.8mg, Thermo). Ten labelled regions from each mouse along with an aliquot of the common pool were pooled and fractionated by high pH reverse-phase HPLC into 12 fractions. Mobile phase A was 0.1% ammonium hydroxide and 2% acetonitrile, mobile phase B was 0.1% ammonium hydroxide and 98% acetonitrile. Fractions were collected over a 48 minute gradient.
Mass spectrometry
Samples were analyzed by a Q Exactive HFX mass spectrometer connected to Easy nLC 1200 reverse-phase chromatography system (Thermo Scientific). Mobile phase A was 0.1% formic acid and 2% acetonitrile, mobile phase B was 0.1% formic acid and 80% acetonitrile. Peptides were resuspended in 0.1% formic acid for loading. The samples were loaded onto a nano-trap column with mobile phase A, (75μm i.d. × 2 cm, Acclaim PepMap100 C18 3μm, 100Å, Thermo Scientific) and were separated over an EASY-Spray column, (50 cm × 75 μm ID, PepMap RSLC C18, Thermo Scientific) by an increasing mobile phase B gradient over 180 minutes at a flow rate of 250 nL/min. The mass spectrometer was operated in positive ion mode with a capillary temperature of 300°C, and with a potential of 2100V applied to the frit. All data were acquired with the mass spectrometer operating in automatic data dependent switching mode. A high resolution (60,000) MS scan (350-1500 m/z) was performed using the Q Exactive to select the 12 most intense ions prior to MS/MS analysis using HCD (NCE 33, 45,000 resolution).
MaxQuant search and data analysis
Raw files were searched in MaxQuant Version 1.6.0.16 against the Mus musculus canonical Swiss-Prot proteome downloaded from UniProt on January 24, 2019. Two missed cleavage events were allowed and carbamidomethylation of cysteine was set as a fixed modification while variable modifications were oxidation of methionine and acetylation of protein N-termini. Reporter ion MS2 was used for quantification with 11plex TMT and a reporter mass tolerance of 0.003 Da. Peptide search tolerance was set to 4.5ppm for MS1, and MS2 fragment tolerance was set to 10ppm. Match between runs was active with an alignment window of 20 min and a match window of 0.7 min. The obtained protein intensities of each sample were first normalized to its median for each of the 10 brain regions, the replicates summed and then normalized to the intensities of the reference pool. The summed normalized intensities were used for enrichment analysis to detect regional specificity of BraInMap complexes.
Regional sample preparation for co-fractionation analysis
Snap-frozen mouse brain tissues (frontal cortex, parietal cortex, occipital cortex, hippocampus, striatum, thalamus, midbrain, hindbrain, and spinal cord) were transferred to 2ml microcentrifuge tubes and homogenized using 2x5mm stainless steel grinding beads. The tissues were homogenized for four 0.5 min cycles in Mixer Mill (MM400, Retsch Technology) at 25 Hz. Ground tissues were solubilized in a mild-detergent buffer (10mM Tris-HCl, 250mM Sucrose, 5mM MgCl2, 1mM DTT, 5mM ATP, 1% DDM) containing protease and phosphatase inhibitors (Roche) and treated with Benzonase at 100 units/ml for 30 min at 4°C. The crude lysates were centrifuged at 18,000rcf for 10 mins at 4°C and the clarified supernatant collected. Protein concentration in the clarified lysates was estimated with Brad-Ford assay (Bio-Rad). The protein extracts were further clarified at 14,000rcf for 30 min at 4°C and fractionated using an optimized volatile-salt based IEX-HPLC fractionation approach (manuscript in preparation). We deployed a previously described dual IEX-HPLC elution gradient (Havugimana et al., 2007), comprising PolyWAX LP and PolyCAT A (200 x4.6 mm i.d., 5μm, 1000-A; PolyLC Inc) column in series, to generate a total of 960-IEX protein fractions (i.e., 96 fractions per mouse regional tissue). The fractions were dried in a speed vac, digested, and each set of 96 fractions was labeled with a unique Tandem Mass Tag (TMT) using the TMT-10plex kit (ThermoFisher Scientific). The TMT-labeled fractions were pooled and desalted. The desalted samples were then analyzed via LC-MS/MS using a Q Exactive Orbitrap HF mass spectrometer (ThermoFisher Scientific) (Havugimana et al., 2007).
Database search and data analysis
Raw file for each fraction was searched against the Mus musculus canonical Swiss-Prot proteome downloaded from UniProt on January 24, 2019, using 3 search algorithms (X!Tandem, MSGF+, and Comet). MS1 intensities were extracted from the results using the utilities developed in-house as described above. The obtained protein intensities of each fraction were normalized to its median for each of the 10 brain regions and then normalized to the intensities of the reference pool. The protein-protein correlation was calculated for each of the four replicates using the co-elution profile of each protein across all fractions. Protein pairs in BraInMap complexes that showed high correlation (≥ 0.5) in their co-elution profiles for two or more replicates and that also exhibited a high concordance in terms of their corresponding proteomic expression profiles were selected for further analysis.
Co-immunoprecipitation of complexed RBPs from C57BI/6J mice
The right cortices from four 5 months old C57Bl/6J wild type mice were homogenized in lysis buffer (50mM Tris pH7.4, 150mM NaCl, 2mM EDTA, 0.2% NP-40, 0.05% SDS, 1mM PMSF, 1x HALT PIC (Pierce), PhosSTOP (Roche) and 40U/ml RNasin (Promega)) using a motorized homogenizer. Protein concentration was determined by BCA assay. Co-immunoprecipitation was performed using Direct-IP kits (Pierce) according to the manufacturer’s protocol. Briefly, 5μg of either (mouse) monoclonal anti-Tdp-43 (Ling et al., 2010) (FL4; a gift from Ling Shuo-Chien) or (rabbit) polyclonal anti-hnRNP-H (Bethyl Labs; A300-511A) was conjugated to AminoLink resin, blocked (1 hr at RT) with 1% BSA in lysis buffer then washed with lysis buffer. Negative controls were performed using normal mouse IgG (Santa Cruz) and rabbit control IgG (Proteintech). 1mg of sample lysates was precleared by incubation (1 hr at 4°C) with control agarose resin, before incubating overnight at 4°C in IP columns. The following day, the flow-throughs were collected then the columns washed 3 times with lysis buffer, once with lysis buffer containing 0.1% SDS then eluted by incubating at 98°C for 10 mins in TBS with 2× LDS and 1× reducing agent (Life Tech.). Samples were resolved in 4-12% BisTris Bolt gels (Thermo) with 10μg lysates, transferred to 0.45μM PVDF membrane, blocked in 5% non-fat dry milk in TBSt and probed overnight at 4°C with the antibodies as follows. IP-Tdp-43 immunoblot: 1. (rabbit) anti-hnRNP-H (Bethyl Labs; A300-511A; 1:2000), 2. (rabbit) anti-DDX5 (Abcam; ab21696; 1:2000), 3. (goat) anti-TIA-1 (Santa Cruz; sc-1751; 1:300), 4. (rabbit) anti-TDP-43 (Proteintech; 12892-1-AP; 1:2000). IP-Hnrnph immunoblot: 1. (mouse) anti-Tdp-43 (FL4; 1:4000), 2. (rabbit) anti-DDX5-Biotin (ab21696) 3. (goat) anti-TIA-1 (sc-1751), 5. (rabbit) anti-FUS/TLS-Biotin (11570-1-AP), 5. (goat) anti-hnRNP H (Santa Cruz; sc-10042; 1:2000). Where indicated, 10μg primary antibodies were conjugated to Biotin using One-Step Antibody Biotinylation kit (Miltenyi Biotec). Blots were probed with secondary (donkey) anti-mouse-HRP, (donkey) anti-rabbit-HRP (Jackson; 1:5000) or Strepavidin-HRP (Jackson; 0.2μg/ml) 1 hour at RT before activating with SuperSignal HRP substrate (Thermo) and imaging with a ChemiDoc XRS+ (Bio-Rad). Between antibodies, blots were stripped with Restore PLUS (Thermo), blocked, washed and re-probed.
TDP-43 immunoprecipitation from transgenic TDP-43 murine brain and proteomic analysis
Cortical sections from 21 day old wild type (gait score 0/4), TDP-43WT/WT; Atxn2[+/+] (gait score 3.75/4) and TDP-43WT/WT; Atxn2[+/−] (gait score 2.75/4) mice (Becker et al., 2017) (n=4 per group) were lysed in 50mM Tris-HCl pH7.4, 150mM NaCl, 0.2% NP-40 with 1mM PMSF, cOmplete PIC, PhosSTOP and 40U/ml RNasin by motorized pestle. Samples were spun at 1,000rcf for 5 mins at 4°C and the supernatants collected and assessed for concentration. Direct-IP columns were generated with 10ug each of (mouse) anti-TARDBP (Abnova; H00023435-M01) and immunoprecipitations, using 500μg lysate per column, were bound, washed and eluted as above. Negative control experiments were performed using anti-TARDBP bound columns and hippocampal lysate from a conditional Tardbp knockout mouse (gift from Phillip Wong (Chiang et al., 2010)) and using TDP-43WT/WTAtxn2[+/+] lysates in columns bound with normal mouse IgG. Quantitative proteomic analysis of TDP-43 interactions was performed, as previously described (Vanderweyde et al., 2016), using LC-MS/MS, less nonspecific interactions identified in negative controls and normalized to the iBAQ levels of TDP-43 in each sample. Protein interactors were excluded if not identified in duplicate or more per group. Equal amounts of TDP-43 immunoprecipitated material from TDP-43WT/WTAtxn2[+/+] and TDP-43WT/WTAtxn2[+/−] mice (n=3) was immunoblotted and probe for hnRNP-H (Bethyl) and DDX5 (Abcam) as detailed above.
TDP-43 immunofluorescence imaging from transgenic TDP-43 murine brain
Hemispheres from TDP-43WT/WTAtxn2[+/+] and wild type litter mates were drop fixed in 4% PFA for 48 hours before washing in PBS and storing in 30% sucrose in PBS. Hemispheres were sliced into 30μm sagittal sections, treated 20 mins at room temperature in 1mg/ml sodium borohydrate to block aldehydes, washed in water then mounted to slides. After drying, slides were washed in PBS, incubated 1 hour at 95°C in citrate buffer (Vector Labs; H-3300) then cooled in PBS. Tissue was permeabilized in 0.2% Tween-20 in PBS, blocked in 5% normal donkey serum, 0.05% Tween-20 in PBS then incubated overnight at 4°C with primary antibodies in PBS with 0.5% NDS and 0.05% Tween-20. Primary antibodies used were (mouse) anti-TARDBP (Abnova; H00023435-M01; 1:500), (rabbit) anti-TDP-43 phosph-S409/410 (a gift from Leonard Petrucelli; Rb3655; 1:250), (rabbit) anti-DDX1 (ProteinTech; 11357-1-AP; 1:500), (rabbit) anti-DDX5 (Abcam; ab21696; 1:1000), (rabbit) anti-hnRNP-H (Bethyl; A300-511A; 1:500), (rabbit) anti-ILF3 (Bethyl; NF90/NF110, A303-121A), (rabbit) anti-U2AF2 (Novus; NBP2-04138), (rabbit) anti-Histone H3 (Abcam; ab18521), (chicken) anti-MAP2 (Aves; MAP2) and (chicken) anti-NeuN (EMD; ABN91; 1:500). Slides were then washed with PBS and fluorescently immunolabelled as appropriate with the (donkey) anti-mouse, rabbit, or chicken with Alexafluor conjugates (Jackson Immuno; 1:750). Slides were washed again in PBS, counterstained with DAPI then autofluorescence was quenched by incubating 10 mins at RT in 0.1% Sudan Black in 50% Ethanol. The sections were coverslipped in Prolong Gold antifade reagent (ThermoFisher). Sections were then imaged at 63x on a Zeiss AxioObserver LSM700 confocal with standardized exposures given additional gain to observe cytoplasmic distribution.
Human cell culture and differentiation:
SH-SY5Y cells (ECACC, 94030304) were maintained in high-glucose DMEM (Millipore Sigma, D5671) medium supplemented with 10% fetal bovine serum (FBS, Lifetchnologies; 12483020), penicillin (50 u/ml), streptomycin (50μg/ml) (Lifetechnologies; 15070-063), L-glutamine (2mM) (Lifetechnologies; 25030-081). Cells were incubated at 37°C and 5% CO2/95% air with saturated humidity. SH-SY5Y cells were differentiated by all-trans retinoic (at-RA, Millipore Sigma; R2625) and BDNF (eBiosciences; 14-8366-80) as described before (Encinas et al., 2000). For differentiation, cells were plated at the density of 4 X 104 cells/cm2 in complete DMEM medium containing 5% FBS and at-RA acid was added to cells and the medium was changed daily for a total of 5 days. From days 6-12, cells were incubated with DMEM supplemented with penicillin (50μg/ml), streptomycin (50μg/ml), L-glutamine (2mM) and BDNF (20ng/ml) but no FBS and medium was changed every 2-3 days.
Human anti-FLAG/MDTH immunoprecipitation, Western blotting, and affinity-purification/mass spectrometry:
Differentiated live cells were cross linked using the cell membrane-permeable bifunctional cross linking reagent dithiobis[succinimidyl propionate] (DSP, Lomant’s reagent, ThermoScientific; 22585). DSP solution was made in DMSO at the concentration of 0.25M and diluted in PBS to a final concentration of 1mM immediately prior to incubation with cells at room temperature. Excess DSP was quenched by reacting with 100mM Tris-HCl pH 7.5. Cells were then washed in 1X PBS twice, detached by incubating with versine at 37°C for 10 minutes, pelleted and lysed in RIPA buffer. Protein concentration was assayed and approximately 10mg protein was incubated in each reaction with protein-specific or control antibodies for 1 hr at 4°C with tumbling. After that, 50μl protein G magnetic microbeads were added and the mixture is incubated for an additional 4 hr at 4°C with tumbling. Subsequently, samples are purified using magnetic columns and washed using detergent-free buffers. For mass spectrometric analysis, purified proteins and their associated partners are eluted and proteolytically-digested overnight at room temperature. Samples are subsequently desalted and purified using ZipTip and analyzed using nLC-MS. For Western blotting, elution was done in Laemlli buffer and a fraction was run on SDS-PAGE, transferred to PVDF membranes and subsequently blotted with primary protein specific antibodies and subsequently with HRP-conjugated secondary antibodies. Visualization was done using luminescence.
Mouse brain co-Immunoprecipitation (anti-Tdp-43/anti-MTDH):
The cell lysate was adjusted to 1ml with RIPA buffer containing PIC and 3μl of each antibody (TDP-43 or MTDH) were added. After 1 hr agitation at 4°C, 100μl of μMACS protein A or G magnetic microbeads (Miltenyi) was added with continued agitation overnight at 4°C. Microbeads suspension was passed through μMACS columns (Miltenyi) equilibrated with RIPA and PIC, and the retained microbeads were washed 2 times with 1ml of RIPA buffer containing 0.1% of detergents and PIC followed by another one wash with 1ml detergent-free RIPA buffer. Proteins bound to the microbeads were released by addition of 100μl Laemmli loading buffer 2X and heated at 95°C. Eluate was analyzed using western blot and visualized using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher, 34095).
Structure modeling and docking:
The I-TASSER (Iterative Threading ASSEmbly Refinement) server was used to predict non-resolved full-length structures of human MTDH and TDP-43. The structure of the lowest energy was selected, which was then refined by a fragment-guided molecular dynamic procedure, with the purpose of optimizing the hydrogenbinding network and removing steric clashes. The docking studies were carried out using PIPER. We produced 7,000 structural conformations between two structure chains. For the highest scoring docked structure, we determined residues at the complex interface using Schrodinger to measure the change in solvent accessible surface area between bound and unbound states of this complex. Residues with a minimum 15% solvent accessible surface area in the unbound state whose absolute solvent accessible surface area changes decreased by ≥1.0Å squared were considered to be at the interface of the bound structure.
Generation of human TDP-43 mutants and mutagenesis
Sequence confirmed TDP-43 cDNA clone (HsCD00079870) from the human ORFeome collection (Dana Farber/Harvard Cancer Center DNA Resource Core) was used to generate the mutations, following the PCR-driven overlap extension method (PMID: 29128334). All PCR reactions were performed using the high-fidelity Phusion polymerase. A set of forward and reverse primers were designed to flank the desired mutation sites and the two unique restriction sites at NsbI and EcoRV. At each mutation site, another set of forward and reverse primers that bind at the mutation sites was used, ensuring that their Tm was within ± 5 °C of the outer flanking primers. For each mutation, two first-stage PCR reactions were performed with the outer flanking forward primer and the mutation site reverse primer, and vice versa. After a successful PCR, the reaction products were cleaned up using a PCR cleanup kit. For each mutation, a single second-stage overlap-extension PCR was carried out in which an equimolar mixture of two PCR products of the first-stage reactions and the outer forward and reverse flanking primers were used. Overlap extension was verified using agarose gel. Both TDP-43 plasmid (HsCD00079870) and the final PCR products were cut with restriction enzymes, NsbI and EcoRV, following manufacturer’s recommendations. Gel purified products were ligated using T4-DNA ligase, transformed into competent DH5α cells, and the successful mutagenesis was verified using Sanger sequencing.
To clone TDP-43 into mammalian expression vectors, we used Gateway LR Clonase II (ThermoFisher) according to the manufacturer’s instructions. Briefly, a mixture of TDP-43 wild type or mutant entry vector was mixed with the enzyme mixture and appropriate amount of the destination vector pLD-puro-Cc-VA (Addgene) containing a C-terminal versatile affinity tag containing 3× Flag, 6× histidine and 2× Streptactin epitopes (Flag and His separated by dual tobacco etch virus protease cleavage sites). After the incubation and enzyme inactivation, cloning mixture was transformed into NEB stable competent bacteria (to avoid recombination of the repetitive lentiviral sequences in the destination vector). Successful cloning was verified using Sanger sequencing at The Centre for Applied Genomics (TCAG), Toronto Hospital for Sick Children sequencing facility.
siRNA knockdown of complexed neuronal RBPs
SH-SY5Y human neuroblastoma cells were maintained in 50:50 DMEM/F12, 10% FBS, Pen/Strep, NEAA and L-glutamine using standard culturing techniques. Cells were plated (DIV0) to 6 well plates (2.0x105 cells/well) or 12 well plates (1.0x105 cells/well). The following day (DIV1), knockdown in triplicate was achieved using PepMute (SignaGen; according to manufacturer’s protocol) with 50nM of the following siRNA SMARTpools (Dharmacon): siGENOME Human TDP-43 (siTDP-43, 23435), siGENOME Human HNRNPH1 (siH1, 3187), siGENOME Human DDX5 (siDDX5, 1655) and siGENOME Non-Targeting siRNA Pool #1 (siCtrl). Where appropriate, siCtrl was included so as equimolar siRNA was added to each well. After 24 hours (DIV2), the media and knockdown reagents were removed and replaced for a total of 72 hours knockdown before collection (on DIV4). SH-SY5Ys from 12 well plates were collected and lysed in RIPA buffer (50mM Tris pH7.4, 150mM NaCl, 2mM EDTA, 1% NP-40, 0.1% SDS, 0.1% sodium deoxycholate, 1mM PMSF, cOmplete PIC (Roche)) and the concentrations determined by BCA reagent. Samples were immunoblotted as above.
qPCR analysis of SORT1 Exon17b mis-splicing
Total RNA from siRNA treated SH-SY5Ys from 6 well plates was collected using the RNeasy Minikit (Qiagen). Random hexamer primed cDNA was generated using the High Capacity cDNA Reverse Transcription Kit (Thermo). qPCR was performed using iQ SYBR Green Supermix (Bio-Rad) to detect ACTB (NM_001101.3), total Sortilin 1 (Prudencio et al., 2012) (NM_002959.6; SORT1total; Ex15_F 5’-TCCATCTGCCTCTGTTCCCTG, Ex16_ 5’-GGTGTTCTTCTCTTCCGTACAGACAA), SORT1WT (omitting exon 17b; Ex17_F 5’-TGGGGTAAATCCAGTTCGAG, Ex17-18_R 5’-GACTTGGAATTCTGTTTTTCCGGAC) and SORT1+17b (including exon 17b; Ex17b_F 5’-AATCCAGCTCTGCCTCCTCT, Ex18_R 5’-TCCCACGATGGCCAGGATAA). Sample transcripts were normalized to ACTB levels then to the mean of the siCtrl treated group. Plotting and ANOVA with Tukey’s multiple comparison posthoc statistical analysis was performed using GraphPad.
QUANTIFICATION AND STASTICAL ANALYSIS
Data analysis
MS1 intensity elution profiles of 550 fractions from 5 experiments were determined by searching the spectra with MaxQuant version 1.6.0.16 (Tyanova et al., 2015) against the UniProt reviewed Mus musculus proteome protein sequence database (version: Feb 21, 2017, (The UniProt Consortium, 2017), number of sequences: 50,915). Searches were performed with fragment ion mass tolerance of 20 ppm, maximum missed cleavage of 2. Oxidation of methionine was considered as variable modification. The false discovery was controlled using a target/decoy approach with false discovery level set to 1%. Only protein groups identified with at least two or more peptides (sum of razor and unique) in more than one fraction were carried forward in the analysis. This resulted in the detection of 4,134 proteins. To increase protein coverage we ran additional database searches using 3 more search algorithms (X!Tandem, MSGF+, and Comet) subject to the same parameters as given above. The results of these 3 search engines were integrated using the MSblender integration tool which led to the identification of 8,075 proteins. Since the results were obtained in the form of MS2 spectral counts, an in-house script was developed to extract MS1 intensities. Three utilities were developed to extract MS1 intensity data. The first pair of programs scanned the X!Tandem, MSGF+ and Comet search results, producing a list of peptides identified for each fraction by a given search engine along with a range of scan numbers for each combination of identified peptide and precursor ion charge state. The range of scan numbers consisted of the lowest and highest scan numbers of MS2 spectra for which the peptide was considered identified. The third program read each list and scanned the associated spectra file, extracting and reporting the highest MS1 peak intensity within plus and minus 10 ppm of the precursor peak m/z for (1) the MS1 spectrum immediately preceding the range, (2) the first MS1 spectrum following the range, and (3) each MS1 spectrum between.
Both sets of MS1 intensities were run through the EPIC prediction tool (Hu et al., 2019) to predict PPI and complexes. Correlation scores were calculated for each experiment using 5 different methods (Euclidean, Bayes, Jaccard, Apex and Mutual information) and 15 additional functional annotation features were included to boost performance and PPI prediction. 678 complexes from GO, IntAct and CORUM (Table S2) were used as the reference set for training the data through machine learning to predict PPI. Complexes were predicted using ClusterOne (Nepusz et al., 2012)and benchmarked against a set of 78 brain specific reference complexes obtained from CORUM for mouse and other orthologs (Human, Rat, Bovine, Rabbit, and Pig; Table S2).
Given that subunits of a complex should reproducibly co-elute, chromatographic profile similarity is taken as a proxy for physical association. We applied machine-learning procedures to capture and weigh different features from the biochemical data (Hu et al., 2019). The results from each fractionation were processed by random forest classifier trained on experimentally-verified co-complexes PPIs from public curated databases to assign PPI confidence scores (CORUM (Ruepp et al., 2010), IntAct (Orchard, Ammari et al. 2014), GO (Ashburner et al., 2000). To maximize coverage and accuracy, we integrate supporting functional association evidence in the random forest step. After generating a high confident co-elution network we used ClusterONE to generate a set of stable protein complexes from the co-elution network. See below for a more detailed description of each set of the data analysis pipeline and how the model evaluation was conducted (Nepusz et al., 2012).
Removing low scoring proteins
To ensure proper quality across different co-elution experiments we integrated two preprocessing steps: a) removing low scoring proteins, and b) normalizing peptide counts. Both of the filtering steps treated each coelution experiment as an individual entity and we merged all experiments at a later stage. First, we removed all proteins for which peptides were observed in one fraction only. For example, if protein A was only observed in fraction 21 in a co-elution experiment that protein was discarded. We justify this filtering based on the fact that calculating any kind of co-elution is impossible for a protein with exactly one observation. We observed that some fractions contain more peptide than others, to minimize this fraction bias we performed a column-wise normalization followed by a row-wise normalization. In the column-wise normalization, we divided the number of identified peptides for each protein for each fraction by the total number of peptides in that fraction. For row wise normalization, we divided the number of peptides of a protein in a specific fraction by the total number of identified peptides of that protein.
Co-elution scores
We expect proteins that are physically interacting will co-elute in our fractionation experiments and thus the elution profile of interaction proteins should be similar. To measure this relationship we deploy several methods that capture elution profile similarity. At its core, these methods are different correlation metrics that are tuned to measure different aspects of correlation. In the formulas for each correlation metric: pa,pb denote protein a and protein b in the same co-fractionation experiment, N denotes the total number of proteins and M is the total number of fractions.
Euclidean Distance
The Euclidean distance denotes the distance of two vectors (or two points) in a high-dimensional space (also known as 2-norm). The two points, for which the distance is calculated, represent the protein pair and the number of fractions is the dimension of the space to which the Euclidean distance applies. The Euclidean distance feature uses normalized counts and lies between 0 and the square root of 2, where identical elution profiles have a distance of 0 and elution profiles that differ greatly have a distance close to square root of 2.
Bayes correlation
In this work, we integrated a novel method (Sanchez-Taltavull et al., 2016) that utilizes a Bayesian framework for calculating correlation scores between two MS2 spectral counts based vectors. Originally, this method was proposed to process RNA-Seq gene expression data that is based on sequence counts for various genes under various conditions. Here we propose to use the same method for protein peptide counts for various proteins across various biochemical fractions. The main advantage of the Bayesian correlation over Pearson correlation is that it considers both measured signal magnitudes and associated uncertainties in those magnitudes. Thus, Bayesian correlation will retain high correlation values if the measurement confidence is high and will prevent high correlation values when the measurement confidence is low. Moreover, it was shown that the Bayesian correlation could be used as a kernel in any kernel based machine-learning method, such as support vector machines, which makes Bayesian correlation a useful feature for our co-fractionation pipeline. To integrate Bayesian correlation we downloaded the R script (http://www.perkinslab.ca/sites/perkinslab.ca/files/Bayes_Corr.R) and integrated into the python pipeline using the rpy python package that allows the import of R code into python. Bayesian correlation calculation scores support three different assumptions of how the priors distributed: uniform, Dirichlet-marginalized and zero count-motivated. Zero-count was used here, as it performed better than the others (unpublished data).
Mutual information (MI)
Mutual information (MI), unlike linear correlation metrics such as PCC, considers information about both linear and nonlinear dependencies. The initial step in calculating MI is to binarize the spectral count vector elements into ‘with protein’ and ‘without protein’, since mutual information measures statistical dependence between the two given proteins based on their relative co-elution frequency (% co-eluted fractions) and each protein’s individual relative frequency (% fractions containing the respective protein). We binarize the elution matrix by temporarily changing each protein peptide count to 1 if there were spectral counts observed in the fraction and to 0 if not. Thus, P(pa =1) denotes the individual relative frequency of pa, which is calculated by dividing the total number of fractions with value 1 for protein pa by the total number of fractions in the corresponding cofractionation experiment. Whereas, the joint relative co-elution frequency of protein pa and pb named P(pa =1, pb =1) is calculated by counting the total number of fractions that contain both pa and pb and dividing this number by the total number of fractions. MI is calculated as follows:
In the formula above, H(pa) denotes the entropy of protein a and H(pa, pb) the joint entropy with the following formulas:
Jaccard score
Jaccard score computes the ratio of how often proteins are eluted in the same fractions and how often proteins are eluted in different fractions. Thus the Jaccard score between two proteins is calculated by counting the number of fractions that contain both proteins and dividing by the number of fractions that have at least one of the two proteins.
Apex
Most proteins tend to elute only at a specific time, and thus the fraction that contains the largest amount of a particular protein is also the most critical fraction for the given protein. Thus, two proteins are considered to be more likely to interact with each other if the fractions that have the largest amount of proteins across all fractions are the same. Based on this assumption, the previous co-fractionation experiments utilized the co-apex score, which scores protein co-elution profiles highly if their respective peak fraction is the same (apex score = 1) or not (apex score = 0).
Functional evidence
We enriched our experimental data with high quality functional evidence and other brain-related experiments taken from various sources. In order to prevent circular reasoning, we removed all evidences that used information derived from protein complexes. Adding functional evidence only slightly increased the composite score.
MouseNetV2
MouseNetV2 is a functional gene network for the laboratory mouse that combines various functional evidence from both mouse and other model organisms mapped to mouse (Kim et al., 2016).
Allen brain Atlas
The Allen brain atlas is a gene expression database for mouse brain that contains exhaustive in situ experimental data for various mouse brain regions. We extracted expression values for each gene for all available brain regions that are: Isocortex, Olfactory areas, Hippocampal formation, cortical subplate, striatum, pallidum, thalamus, hypothalamus, midbrain, pons, medulla, cerebellum. The expression is measured in expression energy, which is calculated as follows: Within a given area A (voxel or structure), expression energy = (sum of intensity of expressing pixels in A) / (sum of all pixels in A). The final interaction score is derived by calculating Pearson correlation for all protein pairs based on their expression energy.
Published brain networks
We also integrated brain data from various other sources. A recently published work on mouse brain proteasome that contains MS expression analysis for 12934 proteins across major brain regions and cell types was integrated. Additionally, we mapped a study of the human subcellular location to their respective mouse orthologs via InParanoid. We calculated Pearson correlation for each protein pair in each experiment respectively.
RNA-Seq data
RNA-Seq data was extracted from the Gene expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/gds) using their R library, and selected using several criteria to extract high quality brain data. Only transcriptomic RNA-Seq data for adult mouse generated using Illumina HiSeq 2000 and 2500 and having as source tissue one of the following descriptors: brain, cortex, thalamus, striatum, cerebellum, cerebellum dentate, or olfactory bulb was selected. We then use the Sequence read archive (SRA) tool to map each GEO to their respective raw data set on the SRA. In accordance with a recently published Nature protocol (Pertea et al., 2016), we processed the raw RNA-Seq read data by using StringTie (Pertea et al., 2015) and HISAT (Kim et al., 2015) with the Ensembl (Zerbino et al., 2018) mouse reference genome (Mus_musculus.GRCm38.84.gtf) as an annotation source. Once we quantified RNA expression in each experiment, we calculated co-expression based on those experiments using Pearson correlation.
Reference complexes
We created a comprehensive set of 678 mouse protein complexes by extracting known protein complexes from CORUM, IntAct, and GO. We downloaded a recent set of complexes from CORUM and only kept those complexes annotated with biochemical evidence. We further expanded this set by adding experimentally verified mouse protein complexes from IntAct and protein complexes that we constructed by using GO annotation. GO complexes were derived by taking all genes that are annotated with a complex specific GO annotation. We identified them by taking all experimentally validated GO cellular component annotations that are a leaf annotation (lowest level, i.e. most specific) and are a descendent of the protein complex GO term. Genes with the same GO annotation are grouped together in the same complex. In the next step, we repeated the same procedure for Human protein complexes, followed by a strict one-to-one mapping of human proteins to mouse proteins using only human-mouse pairs that have a 100% InParanoid score (highest confidence score). After obtaining this set of complexes, we performed several preprocessing steps. In the first step, we removed all proteins, for which we have no elution profile, followed by removing all assemblies that have more than 50 members. In an effort to eliminate redundancy we merged all protein complexes that have an overlap coefficient of 0.8 or more. In addition, for complex benchmarking we used a distinct set (i.e. not a part of the reference complexes) of 78 brain associated complexes from CORUM.
Positive and negative protein complexes
As in previous work (Havugimana et al., 2012; Wan et al., 2015), we generated a set of positive co-occurring protein pairs by taking all possible protein pairs that were observed in the same protein complex. Negative protein pairs are all possible protein pairs that are never observed in the same protein complex. For example, we observed protein A and B are members of the same complex we consider them to be part of the positive training set, and if we never observe protein A and C together in any of our reference complexes we would consider them to be part of the negative training set. Furthermore, previous studies showed that co-elution prediction works best when having a ratio of one to five between positive and negative protein pairs. We created that ratio of positive to negative training data points by under sampling negative protein pairs since there are considerably more negative PPIs than positives.
Model evaluation
Protein complexes consist of multiple proteins and determining if two complexes are matching is a non-trivial problem. The most common way of measuring it is using the overlap coefficient. The overlap between two protein complexes A and B is calculated as follows (note that |A| denotes the number of proteins in complex A):
We defined two protein complexes as matching when the overlap score between them is larger than 0.25 since two clusters of the same size would have this score if the intersection set is half of the complex size.
Additionally, we calculated prediction sensitivity, accuracy, positive predictive value, and cluster separation (Brohee and van Helden, 2006). For the following scores, we considered a1,...,ai,..,am predicted complexes, which we compared to a set of b1,..,bj,..,bn reference complexes, and Ti,j denotes the number of proteins that were found in both complexes i and j.
Sensitivity (Sn): the fraction of proteins in predicted complexes that were found in reference complexes.
Positive predictive value (PPV): indicates how specific and complete the predicted complexes match the reference complexes. A score of 1 indicates that each predicted complex only overlaps with exactly one reference complex, and a low score indicates low or redundant overlap with the reference.
Accuracy (Acc): shows the trade-off between PPV and Sn.
Maximum matching ratio (MMR): The MMR was developed to cope with some of the limitations of the PPV. PPV tends to be lower if there is substantial overlap in the reference data (Nepusz et al., 2012), but those overlaps are common in biological data sets such as CORUM. Our merging step only removes highly overlapping clusters, but smaller overlaps are still present. Thus, even if EPIC perfectly predicts the reference complexes it will not achieve a score of 1 for PPV and Sep (clustering-wise separation score suggested previously by (Brohee and van Helden, 2006)). MMR addresses this problem:
As established by others (Nepusz et al., 2012), we summarized MMR, overlap score, and accuracy to create the composite score, and we considered the parameter combination with the highest composite score to be the best combination.
Cross fold evaluation
Our primary goal was to accurately infer stable protein complexes from the experimental data in order to properly evaluate our performance. Therefore, we measured how well we could reconstruct known reference complexes from our experimental data. We performed a two-fold cross validation to ensure that we have the same amount of complexes for training and validation. To train the model we first split our set of reference complexes 50:50 and then generated positive and negative PPIs for one set and then trained a random forest model to distinguish them. Next, we predicted all PPIs for which we have elution data and retained all PPIs pairs with a random forest score greater than 50%. We then generated protein clusters from these interactions using ClusterOne with default parameters. The performance was evaluated using overlap score, MMR, and accuracy of those predicted clusters against the separate set of brain specific reference complexes. To perform an extensive benchmark, we tried out all possible combinations of co-elution scores and found the best result using Apex, Jaccard, Bayes, Euclidean distance, and mutual information. We performed a global optimization to select the elution profile correlation metrics that generated the highest composite score. We also noted that adding functional evidence considerably increased the composite score.
Scored protein co-fractionation networks were calculated by correlation analysis (Apex, Jaccard, Bayes, Euclidean distance, and mutual information) based on the protein intensities recorded across each set of fractions (STAR Methods). Weighted networks were constructed based on functional evidence reported in MouseNet v2 (Kim et al., 2016) omitting mammalian protein interaction data to minimize circularity that might bias our association predictions. For the machine-learning classifier, we used the Fast Random Forest implementation (STAR Methods) to integrate all generated networks. Cross-validated decision trees were learned and benchmarked using independent training and test sets of reference complexes (Ruepp et al., 2010) (STAR Methods). Clusters were defined using ClusterONE parameter settings maximizing the bipartite matching ratio between the predicted complexes and set of cluster-training complexes (STAR Methods).
Random forest cut-off
The final output of the random forest returns a confidence score on how likely two proteins are interacting based on their functional evidence and their co-elution. This score ranges between 0 and 1, and we would only consider two proteins to be interacting if they have a score of at least 0.5. Higher cut-off results in better composite scores and better-predicted complexes, but at a cost of reducing the number of complexes predicted. To explore the effects of this parameter, we evaluated prediction performance for each random forest score cutoff between 0.5 and 1 for two fold evaluation. We observed a steady increase in scores for cut-off scores 0.5 to 0.683, with a drop in MMR and accuracy for higher cut-offs. At the same time, we see an increase in overlap score, which in turn causes a significant increase in composite score. We see that the number of PPIs and predicted clusters declines for higher cut-off and the jump in overlap score is most likely caused by over fitting. Thus, we select a random forest confidence score cut-off of 0.683.
Classification of BraInMap complexes
There are many metrics for measuring cluster agreement (overlap), but none is universally accepted in the field. To define novelty in a stringent and transparent manner, we applied 6 independent similarity measures reported in previous interactome publications. These include the Jaccard, Sorensen-Dice, Anderberg, Ochai (Meyer et al., 2004), and Overlap scores (Nepusz et al., 2012), and the hypergeometric distribution, to define the overlap between our predicted protein complexes and known assemblies in CORUM. Though these established similarity metrics gave generally similar results (Figure S1B), they did not account for instances wherein the subunits of a small (known) complex were found as part of a larger predicted assembly. Hence, to address this shortcoming, we then calculated an average matching index (AMI) that looked at overlaps with respect to both the vantage of the annotated and the predicted complex as follows:
where p and k represent the number of subunits in predicted and known complexes respectively and (p ∩ k) the number of subunits present in both.
As both a pragmatic and stringent solution, we classified putative complexes with an average matching index ≥ 0.5 as “annotated”, those between ≥ 0.25 & < 0.5 as “previously reported assemblies with new subunits”, and finally only those complexes with < 0.25 average matching index that are also not statistically significant (p-value > 0.05) by hypergeometric test as “novel” (Figure S1C).
Selection of neurological and other disease annotation
Neurological and other disease associations for BraInMap complexes were compiled from disease annotations in DisGeNET 5.0 (Pinero et al., 2017). We used high quality curated associations obtained by applying stringent filtering to exclude associations with EI (Evidence Index) < 0.9 and DisGeNET score < 0.005. (Figure S1E), to map 1710 members of BraInMap complexes to various neurological diseases (Table S8).
Enrichment analysis
Enrichment analysis was carried out with Gene Ontology (GO) version 1.2 (downloaded on 2019-03-07), and mouse gene associations downloaded from Gene Ontology (Ashburner et al., 2000). A subset of the gene ontology comprising ~3,221 GO terms were defined using goslim synapse and selected neuronal terms for enrichment. Interaction space was constrained to only those interactions between pairs of proteins that were observed both in our high-confidence PPIs and in the target annotated dataset.
Over-representation analysis of gene ontology terms was performed using the Cytoscape app BiNGO Version 3.0.3 (Maere et al., 2005). Enrichment for each annotated term among genes in each of the 1030 complexes was calculated using the hypergeometric test (p < 0.05) with Benjamini-Hochberg FDR correction, using genes in our high confidence network as the reference set.
Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) of brain regions and cell types was performed to determine brain-specific and cell type specific complexes in BraInMap. In each case, our 1030 complexes were used as gene sets. To determine brain-region specificity we used normalized protein intensity data from regional mouse brain co-fractionations performed in our lab (see STAR Methods below) restricted to proteins in our high confidence network, while for cell type specificity we used the sc-RNA-Seq gene expression data from mouse brain (Zeisel et al., 2018). Average normalized CPM values were computed using the edgeR package for R (Robinson et al., 2010) and grouped into representative neuronal and non-neuronal cell types. The gene expression data was again constrained to genes present in our high confidence network. Results were visualized using the Cytoscape Enrichment map app and hierarchical clustering.
Enrichment of Neurodevelopmental disorder-related genes in BraInMap PPI network
The overlap between a given gene set and our network genes was evaluated using a binomial model
Where:
n is the number of genes in the gene set being examined
p is the probability of observing a random protein-coding gene in our brain PPI network, which is calculated as the fraction of 2,304 genes in the network over all 20,210 mouse protein-coding genes (The UniProt Consortium, 2017).
Domain enrichment
Domain architectures for all mouse proteins were obtained from PhyloPro 2.0 (Cromar et al., 2016) for the longest peptide associated with each gene. Domain predictions are based on Hidden Markov Models of curated seed alignments comprising Pfam A Domains and Families. To avoid frequency biases, all architectures were stripped of domain repeats using a custom Perl script (e.g. AABBAAA becomes ABA). The resulting architectures were then used to determine domain pairs as follows. Domain architectures within proteins were determined by ordering domains by sequence start site and creating adjacent pairs. These were used to define brain specific pairs as seen in the overlap analysis (see below). Domain architectures within complexes were compared to produce all possible combinations of cross-protein domain pairs, ignoring adjacent domains. For example, comparing ABC to DEF would yield AD, AE, AF, BD, BE, BF, CD, CE, CF but not AB, BC, DE or DF). We did this because, at the complex level, we were interested to discover domain associations’ particular to the complex rather than the proteins themselves. Neurologically associated domains are defined as those appearing in proteins that are annotated to one or more neurological diseases. To determine unique brain and neurologically associated domains, an overlap analysis was performed as follows. A list of domains in each complex was obtained by pooling the domain architectures of proteins in each complex. This was also done for complexes in the assembly CORUM, Havugimana et al. (2012), and Wan, Borgeson et al. 2015 data sets. These lists were compared using (http://bioinformatics.psb.ugent.be/webtools/Venn/) to determine domain overlaps between the four data sets and identify domains unique to the brain. Brain specific and neurologically associated proteins were determined similarly. To determine statistical significance of features, we constructed 10,000 random data sets consisting of complexes of the same size as the real data set by selecting random genes (and their associated domain architectures) from a list comprising all mouse proteins with domain predictions. Custom Perl scripts were used to calculate the frequency of specific proteins, domains and domain pairs for the real data set and compare them with the sum of frequency of occurrences in the random networks, counting a score of 1 for each random network in which the protein, domain or domain pair was present as frequently or more frequently than in the real network. For the domain similarity network (Main Figure), domains and domain pairs were classified as either brain specific (b), neurologically associated (n) or non-brain specific (nb) and enrichments were determined by category. The p-value is the ratio of the real frequency to the score of the random frequencies. Network construction and visualization was done in Cytoscape.
Phylogenetic conservation of complexes
Ortholog predictions for all proteins were obtained from PhyloPro 2.0 (Cromar et al., 2016) and clustered using Cluster 3.0 (City Block, Complete Linkage) to group proteins with similar phylogenetic conservation patterns across 164 taxa. Taxa were phylogenetically arranged and grouped into: Eukaryotes, Opisthokonts, Metazoans, Vertebrates, and Mammals. Within each group, we scored the presence or absence of an ortholog prediction for each gene and used an unbiased, consensus approach to predict gene origin. To account for gene losses in some clades within a group we defined an arbitrary cutoff of 30% representation as a requirement to score a gene as being present within a group. To determine whether the group comprising novel complexes was enriched for proteins of a particular age category versus the group comprising non-novel complexes, the assignment of complexes to novel or non-novel groups was randomly shuffled and the frequency of proteins of different ages was compared between real groups versus 10,000 random assignments. A tally was kept in which the frequency of proteins in an age category equaled or exceeded the frequency in the real group.
DATA AVAILABILITY
All raw proteomic (co-fractionation) data from this work is submitted to the PRIDE repository (Accession: PXD011304) at the European Bioinformatics Institute, in accordance with the data sharing policy. Codes used in generating the results are described above in detail.
Supplementary Material
Table S1. Protein intensities of 4,314 and 8075 high-confidence protein identifications across 550 biochemical fractions from five experiments performed using adult mice. Related to Figure 1 and STAR Methods.
Table S2. List of 564 gold standard reference complexes used for PPI prediction and 78 brain specific reference complexes used for cluster evaluation. Related to Figures 1–2 and STAR Methods.
Table S3. 27,403 high confidence protein-protein interactions in BraInMap. Related to Figure 1–2 and STAR Methods.
Table S4. BraInMap complexes showing core and expanded members, human orthologs, neuronal and non-neuronal members, membrane components, similarity metrics, and subunit ages. Related to Figure 2–7 and STAR Methods.
Table S5. BraInMap interactions mapped to human SHSY-5Y neuronal cell line interactions. Related to Figure 2 and STAR Methods.
Table S6. Tissue enrichment of protein detected in BraInMap compared to studies in mammalian cell culture models based on annotation from UniProt Tissue. Enrichment of proteins BraInMap complexes compared to studies in mammalian cell culture models based on annotation from GO cellular component, molecular function, and PFAM domains. Related to Figure 1–2 and STAR Methods.
Table S7. Brain region and cell type specific enrichment of BraInMap complexes. Related to Figure 3 and STAR Methods.
Table S8. Complexes enriched for GO cellular component and molecular function annotations. Enrichment of proteins belonging to more than one complex. Related to Figure 4–6, S1 and STAR Methods.
Table S9. Disease annotations of proteins in BraInMap complexes based on curated annotations DisGeNET database and neurodevelopmental disorders enriched in BraInMap. Related to Figure 7 and STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| (mouse) anti-Tdp-43 | Ling Shuo-Chien | FL4 |
| (rabbit) anti-hnRNP-H | Bethyl Labs | A300-511A |
| normal mouse IgG | Santa Cruz | sc-20-25 |
| rabbit control IgG | Proteintech | 30000-0-AP |
| (rabbit) anti-DDX5 | Abcam | ab21696 |
| (rabbit) anti-FUS/TLS | Proteintech | 11570-1-AP |
| (goat) anti-TIA-1 | Santa Cruz | sc-1751 |
| (rabbit) anti-TDP-43 | Proteintech | 12892-1-AP |
| (goat) anti-hnRNP H | Santa Cruz | sc-10042 |
| (donkey) anti-mouse-HRP | Jackson Immuno | 715-035-150 |
| (donkey) anti-rabbit-HRP | Jackson Immuno | 711-035-152 |
| Streptavidin-HRP | Jackson Immuno | 016-030-084 |
| (mouse) anti-TARDBP | Abnova | H00023435-M01 |
| (rabbit) anti-TDP-43 phosph-S409/410 | Lab of Leonard Petrucelli | |
| (rabbit) anti-DDX1 | ProteinTech | 11357-1-AP |
| (rabbit) anti-ILF3 | Bethyl Labs | NF90/NF110 Antibody, A303-121A |
| (rabbit) anti-U2AF2 | Novus Biologicals | NBP2-04138 |
| (rabbit) anti-Histone H3 | Abcam | ab18521 |
| (chicken) anti-MAP2 | Aves | MAP2 |
| (chicken) anti-NeuN | EMD | ABN91 |
| Anti-TARDBP | Santa Cruz | Cat# sc-376532 |
| Anti-MTDH (LYRIC) | Abcam | Cat# ab227981 |
| TMT10plex Isobaric Label Reagent Set plus TMT11-131C Label Reagent | Thermo Fisher | Cat# A34808 |
| Pierce™ Trypsin Protease, MS Grade | Thermo Fisher | Cat# 90057 |
| Sep-Pak C18 1 cc Vac Cartridge, 50 mg Sorbent per Cartridge, 55-105 μm Particle Size | Waters | Cat# WAT054955 |
| Thermo Scientific™ EASY-Spray™ HPLC Column | Thermo Fisher | Cat# ES805 |
| Bacterial and Virus Strains | ||
| DH5α competent cells | Thermo Fisher Scientific | Cat# 18255017 |
| E. coli NEB stable | New England Biolabs | Cat# C3040I |
| Biological Samples | ||
| Control fibroblast (code: pz2) | I IRCCS, Neurology and Laboratory of Neuroscience | Female; Mutation: none; age at biopsy-45 yrs; Healthy |
| Mutant TARDBP fibroblasts (code: A577 MF) | IRCCS, Neurology and Laboratory of Neuroscience | Male; Mutation TARDBP p.A382T; age at biopsy-56 yrs. |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DL-Dithiothreitol | Millipore Sigma | Cat# D0632 |
| Iodoacetamide | Millipore Sigma | Cat# I6125 |
| Urea | Fisher | Cat# BP169 |
| PhosSTOP™ | Millipore Sigma | Cat# PHOSS-RO |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Millipore Sigma | Cat# 11836170001 |
| Triethylammonium bicarbonate buffer | Millipore Sigma | Cat# T7408 |
| Formic Acid, 99.0+%, Optima™ LC/MS Grade, Fisher Chemical | Fisher | Cat# A117 |
| Acetonitrile, Optima™ LC/MS Grade, Fisher Chemical | Fisher | Cat# A955 |
| Ammonium Hydroxide, ACS Reagent Grade, 28.0-30.0% as NH3 | Fisher | Cat# RABA0020500 |
| Critical Commercial Assays | ||
| Pierce™ Quantitative Colorimetric Peptide Assay | Thermo Scientific | Cat# 23275 |
| Pierce™ BCA Protein Assay Kit | Thermo Scientific | Cat# 23225 |
| Deposited Data | ||
| Protein sequences | UniProt Consortium | PMID: 29425356 |
| Reference complexes | CORUM | PMID: 19884131 |
| Reference complexes | IntAct | PMID:24234451 |
| Reference complexes | GO | PMID: 10802651; PMID: 27899567 |
| Reference complexes | Metazoan | PMID: 26344197; PMID: 26870755 |
| Reference complexes | Human soluble | PMID: 22939629 |
| Functional evidence | Mouse Net v2.0 | PMID: 26527726 |
| Functional evidence | Human Subcellular localization | PMID:28495876 |
| Functional evidence | Cell type and brain region – Mouse brain Proteome | PMID:26523646 |
| Functional evidence | Allen Brain Atlas – Mouse brain | PMID: 17151600 |
| Functional evidence | RNA-Seq from SRA | PMID: 21062823 |
| Functional evidence | GeneCards | PMID: 27322403 |
| Protein abundances | PAXdb | PMID: 25656970 |
| Disease association | DisGenNET | PMID: 27924018 |
| Experimental PPIs | AP/MS, BF/MS, Y2H | PMID: 22939629; PMID: 26344197; PMID: 26496610; PMID: 28514442 |
| Gene expression | Sc-RNA-Seq | PMID: 30096314; PMID: 30096299 |
| RNA binding protein assignment | Census of RNA binding proteins | PMID: 25365966 |
| Functional PPIs | Human Net v2.0, Mouse Net v.2 | PMID: 30418591; PMID: 26527726 |
| Domains and Orthologues | PhyloPro 2.0 | PMID: 26980519 |
| RNA-Seq database | Gene expression Omnibus | PMID: 27008011 |
| De novo variants | denovo-db v.1.5 | PMID: 27907889 |
| Autism genes | SFARI (Mar 5, 2018) | PMID: 19015121 |
| Population variants | NHLBI ESP exome-sequencing study | PMID: 23201682 |
| Protein interaction interface | Interactome INSIDER | PMID: 29355848 |
| Mouse to human gene mapping | Ensemble 92 | PMID: 29155950 |
| Human PPI databases | Inweb3, InWeb_IM | PMID: 17344885, PMID: 27892958 |
| Human prenatal gene expression | BrainSpan | PMID: 24695229 |
| Schizophrenia risk genes | PMID: 25056061 | |
| Autism risk genes | PMID: 26402605 | |
| Developmental disorder associated genes | PMID: 28135719 | |
| Experimental Models: Cell Lines | ||
| SH-SY5Y | ATCC | CRL-2266 |
| Experimental Models: Organisms/Strains | ||
| CD1 wild type mice | ||
| C57BI/6J | JAX labs | 000664 |
| TDP-43WT/WT | JAX via AD. Gitler | 012836; B6;SJL-Tg(Thy1-TARDBP)4Singh/J |
| Atxn2[+/−] | JAX via AD. Gitler | 101043; B6129SF1/J |
| CamKCreER TDP-43 KO | Gift from P. Wong | Chiang et al., 2010. PMID: 20660762 |
| Oligonucleotides | ||
| siGENOME Human TARDBP | Dharmacon | siTDP, 23435 |
| siGENOME Human HNRNPH1 | Dharmacon | siH1, 3187 |
| siGENOME Human DDX5 | Dharmacon | siDDX5, 1655 |
| siGENOME Non-Targeting siRNA Pool #1 | Dharmacon | siCtrl |
| SORT1total_Ex15_F | Life Tech. | 5’-TCCATCTGCCTCTGTTCCCTG |
| SORT1total_Ex16_R | Life Tech. | 5‘-GGTGTTCTTCTCTTCCGTACAGACAA |
| SORT1WT_Ex17_F | Life Tech. | 5‘-TGGGGTAAATCCAGTTCGAG |
| SORT1WT_Ex17-18_R | Life Tech. | 5‘-GACTTGGAATTCTGTTTTTCCGGAC |
| SORT1+17b_Ex17b_F | Life Tech. | 5‘-AATCCAGCTCTGCCTCCTCT |
| SORT1+17b_Ex18_R | Life Tech. | 5‘-TCCCACGATGGCCAGGATAA |
| ACTB_931F | Life Tech. | 5‘-GACAGGATGCAGAAGGAGAT |
| ACTB_1011R | Life Tech. | 5‘-GTACTTGCGCTCAGGAGGA |
| TARDBP-outer_Forward | Millipore-Sigma | CAAGATGAGCCTTTGAGAAGC |
| TARDBP-outer_Reverse | Millipore-Sigma | AGAGCTGCCAGGAAACAGC |
| TARDBP-G287A_ Forward | Millipore-Sigma | AATCAGGCTGGATTTGGTAATAGCAGAGGG |
| TARDBP-G287A_ Reverse | Millipore-Sigma | AAATCCAGCCTGATTCCCAAAGC |
| TARDBP-A315T_ Forward | Millipore-Sigma | TTGGTACGTTCAGCATTAATCCAGCC |
| TARDBP-A315T_Reverse | Millipore-Sigma | GAACGTACCAAAGTTCATCCCACC |
| TARDBP-G368A_ Forward | Millipore-Sigma | GCCTTCGCTTCTGGAAATAACTCTTATAGTGG |
| TARDBP-G368A_ Reverse | Millipore-Sigma | CCAGAAGCGAAGGCCTGG |
| TARDBP-W385G_ Forward | Millipore-Sigma | AATTGGTGGCGGATCAGCATCCAATGC |
| TARDBP-W385G_ Reverse | Millipore-Sigma | ATCCGCCACCAATTGCTGCACC |
| Recombinant DNA | ||
| pENTR-TARDBP-G287A | ||
| pENTR-TARDBP-A315T | ||
| pENTR-TARDBP-G368A | ||
| pENTR-TARDBP-W385A | ||
| pLD-puro-Cc-TARDBP-WT-VA | ||
| pLD-puro-Cc-TARDBP-G287A-VA | ||
| pLD-puro-Cc-TARDBP-A315T-VA | ||
| pLD-puro-Cc-TARDBP-G368A-VA | ||
| pLD-puro-Cc-TARDBP-W387G-VA | ||
| Software and Algorithms | ||
| Sequence database searching | MaxQuant 1.5.5.1 & 1.6.0.16 | PMID: 19029910 |
| Ortholog mapping | InParanoid8 | PMID: 25429972 |
| PPI Prediction | EPIC | PMID: 31308550 |
| Complex prediction | ClusterONE | PMID: 22426491 |
| Network visualization | Cytoscape v. 3.5.1 | PMID: 14597658 |
| Gene Set Enrichment Analysis | GSEA | PMID: 16199517; PMID: 12808457 |
| Enrichment analysis | BinGO 3.0 Cytoscape App | PMID: 15972284 |
| Enrichment analysis | DAVID Bioinformatics resource 6.8 | PMID: 19131956 |
| Hierarchical clustering | Cluster 3.0 | PMID: 14871861 |
| Cluster visualization | Java TreeView v 1.1.6r4 | PMID: 15180930 |
| Hypergeometric test | R function | Stats: R package |
| RNA-Seq data analysis | R function | edgeR: R Package |
| RNA-SEQ analysis | StringTie | PMID:25690850 |
| RNA-SEQ analysis | HiSAT | PMID:25751142 |
| Binomial test | Scipy function | Python package |
| Mann-Whitney U test | Scipy function | Python package |
| Network analysis | NetworkX | Python package |
| PIPER | Schrödinger, LLC | Protein-protein docking |
| ITASSER | Protein structure and function prediction | PMID: 25549265 |
| Plots | R function | ggplot2: R package |
| Venn diagram | R function | VennDiagram: R package |
| Overlap analysis | Venn Draw Tool | http://bioinformatics.psb.ugent.be/webtools/Venn/ |
| Surrogate Variable Analysis | R function | sva: R package |
| Quantile Normalization | R function | preprocessCore: R package |
| Heatmap | R function | ComplexHeatmap: R package |
| R | R version 3.5 | R Foundation for Statistical Computing |
| IsobaricAnalyzer | C++ Library | OpenMS v2.4 |
| Other | ||
| Proteomics data deposition | PRIDE | PXD011304 |
| Proteomics data deposition | BioGRID | To be deposited |
| Nano-HPLC | Thermo Scientific | EASY-nLC™ 1200 System |
| HPLC | Agilent | Agilent 1100 Series |
| Mass Spectrometer | Thermo Scientific | Q Exactive™ HF-X Hybrid Quadrupole-Orbitrap™ Mass Spectrometer |
Highlights.
BraInMap is a global proteomic survey of over 1000 multi-protein brain complexes.
Near native complex identification by CF/MS and reconstruction by computer learning.
Technique interrogates complexes in normal and pathophysiological context.
Allows study of functional modules that are adversely affected in neurological diseases.
ACKNOWLEDGEMENTS
We thank R. Isserlin and A. Alpert for technical assistance and L. Shuo-Chien (National University of Singapore) for the generous gift of FL4 (mouse) monoclonal anti-Tdp-43 antibody. The authors thank Boston University’s Research Computing Services who administer the Shared Computing Cluster. This work was supported in part by funds from the ALS Society of Canada, Canadian Institutes of Health Research (FDN-154318), and the U.S. National Institutes of Health (R01GM106019) to M.B.; Boston University and the CIHR (FDN-148399) to A.E., the U.S. NIH GM103504 / GM070743 / HG006623 to G.D.B.; and AG050471 / AG056318 / NS089544 / AG059925 / ES020395 to B.W.), and the BrightFocus Foundation and Alzheimer Association to B.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Arnold ES, Ling S-C, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, et al. (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proceedings of the National Academy of Sciences of the United States of America 110, E736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker K, Gordon SL, Grozeva D, van Kogelenberg M, Roberts NY, Pike M, Blair E, Hurles ME, Chong WK, Baldeweg T, et al. (2015). Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling. The Journal of clinical investigation 125, 1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basu SN, Kollu R, and Banerjee-Basu S (2009). AutDB: a gene reference resource for autism research. Nucleic acids research 37, D832–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, et al. (2017). Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave K-A, Weis J, et al. (2010). Axonal prion protein is required for peripheral myelin maintenance. Nature neuroscience 13, 310–318. [DOI] [PubMed] [Google Scholar]
- 7.Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB, Fang L, Van Deerlin VM, Ludolph AC, Lee VM, et al. (2014). Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta neuropathologica 127, 423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y, et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Annals of neurology 74, 20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brohée S, and van Helden J (2006). Evaluation of clustering algorithms for protein-protein interaction networks. BMC bioinformatics 7, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brophy K, Hawi Z, Kirley A, Fitzgerald M, and Gill M (2002). Synaptosomal-associated protein 25 (SNAP-25) and attention deficit hyperactivity disorder (ADHD): evidence of linkage and association in the Irish population. Molecular psychiatry 7, 913–917. [DOI] [PubMed] [Google Scholar]
- 11.Bruno KJ, Freet CS, Twining RC, Egami K, Grigson PS, and Hess EJ (2007). Abnormal latent inhibition and impulsivity in coloboma mice, a model of ADHD. Neurobiology of disease 25, 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, and Sudhof TC (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campos-Melo D, Hawley ZCE, and Strong MJ (2018). Dysregulation of human NEFM and NEFH mRNA stability by ALS-linked miRNAs. Molecular brain 11, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrion MDP, Marsicano S, Daniele F, Marte A, Pischedda F, Di Cairano E, Piovesana E, von Zweydorf F, Kremmer E, Gloeckner CJ, et al. (2017). The LRRK2 G2385R variant is a partial loss-of-function mutation that affects synaptic vesicle trafficking through altered protein interactions. Scientific reports 7, 5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carvill GL, McMahon JM, Schneider A, Zemel M, Myers CT, Saykally J, Nguyen J, Robbiano A, Zara F, Specchio N, et al. (2015). Mutations in the GABA Transporter SLC6A1 Cause Epilepsy with Myoclonic-Atonic Seizures. American journal of human genetics 96, 808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavadini S, Fischer ES, Bunker RD, Potenza A, Lingaraju GM, Goldie KN, Mohamed WI, Faty M, Petzold G, Beckwith REJ, et al. (2016). Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 531, 598–603. [DOI] [PubMed] [Google Scholar]
- 17.Chen S, Fragoza R, Klei L, Liu Y, Wang J, Roeder K, Devlin B, and Yu H (2018). An interactome perturbation framework prioritizes damaging missense mutations for developmental disorders. Nature genetics 50, 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Tomchick DR, Kovrigin E, Araç D, Machius M, Südhof TC, and Rizo J (2002). Three-dimensional structure of the complexin/SNARE complex. Neuron 33, 397–409. [DOI] [PubMed] [Google Scholar]
- 19.Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, and Wong PC (2010). Deletion of TDP-43 downregulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A 107, 16320–16324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Compton AG, Albrecht DE, Seto JT, Cooper ST, Ilkovski B, Jones KJ, Challis D, Mowat D, Ranscht B, Bahlo M, et al. (2008). Mutations in contactin-1, a neural adhesion and neuromuscular junction protein, cause a familial form of lethal congenital myopathy. Am J Hum Genet 83, 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S, et al. (2012). Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Human molecular genetics 21, 2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cromar GL, Zhao A, Xiong X, Swapna LS, Loughran N, Song H, and Parkinson J (2016). PhyloPro2.0: a database for the dynamic exploration of phylogenetically conserved proteins and their domain architectures across the Eukarya. Database : the journal of biological databases and curation 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deciphering Developmental Disorders S, McRae JF, Clayton S, Fitzgerald TW, Kaplanis J, Prigmore E, Rajan D, Sifrim A, Aitken S, Akawi N, et al. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IRA, Capell A, Schmid B, et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. The EMBO journal 29, 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubiel D, Rockel B, Naumann M, and Dubiel W (2015). Diversity of COP9 signalosome structures and functional consequences. FEBS letters 589, 2507–2513. [DOI] [PubMed] [Google Scholar]
- 26.Eitan C, and Hornstein E (2016). Vulnerability of microRNA biogenesis in FTD-ALS. Brain research 1647, 105–111. [DOI] [PubMed] [Google Scholar]
- 27.Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elmer BM, and McAllister AK (2012). Major histocompatibility complex class I proteins in brain development and plasticity. Trends in neurosciences 35, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Ceña V, Gallego C, and Comella JX (2000). Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. Journal of neurochemistry 75, 991–1003. [DOI] [PubMed] [Google Scholar]
- 30.Farrer MJ, Hulihan MM, Kachergus JM, Dachsel JC, Stoessl AJ, Grantier LL, Calne S, Calne DB, Lechevalier B, Chapon F, et al. (2009). DCTN1 mutations in Perry syndrome. Nat Genet 41, 163–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44, D279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forman MS, Trojanowski JQ, and Lee VMY (2004). Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nature medicine 10, 1055–1063. [DOI] [PubMed] [Google Scholar]
- 33.Fratta P, Sivakumar P, Humphrey J, Lo K, Ricketts T, Oliveira H, Brito-Armas JM, Kalmar B, Ule A, Yu Y, et al. (2018). Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. Embo j 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ganapathiraju MK, Thahir M, Handen A, Sarkar SN, Sweet RA, Nimgaonkar VL, Loscher CE, Bauer EM, and Chaparala S (2016). Schizophrenia interactome with 504 novel protein-protein interactions. NPJ schizophrenia 2, 16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ge WW, Wen W, Strong W, Leystra-Lantz C, and Strong MJ (2005). Mutant copper-zinc superoxide dismutase binds to and destabilizes human low molecular weight neurofilament mRNA. The Journal of biological chemistry 280, 118–124. [DOI] [PubMed] [Google Scholar]
- 36.Grant SG, and O’Dell TJ (2001). Multiprotein complex signaling and the plasticity problem. Current opinion in neurobiology 11, 363–368. [DOI] [PubMed] [Google Scholar]
- 37.Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al. (2012). A census of human soluble protein complexes. Cell 150, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Havugimana PC, Wong P, and Emili A (2007). Improved proteomic discovery by sample pre-fractionation using dual-column ion-exchange high performance liquid chromatography. Journal of chromatography B, Analytical technologies in the biomedical and life sciences 847, 54–61. [DOI] [PubMed] [Google Scholar]
- 39.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. (2015). A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723. [DOI] [PubMed] [Google Scholar]
- 40.Hentze MW, Castello A, Schwarzl T, and Preiss T (2018). A brave new world of RNA-binding proteins. Nature reviews Molecular cell biology 19, 327–341. [DOI] [PubMed] [Google Scholar]
- 41.Hobson GM, and Kamholz J (1993). PLP1-Related Disorders., G. [Internet]. ed. (Seattle (WA): University of Washington, Seattle;: University of Washington). [Google Scholar]
- 42.Hu LZ, Goebels F, Tan JH, Wolf E, Kuzmanov U, Wan C, Phanse S, Xu C, Schertzberg M, Fraser AG, et al. (2019). EPIC: software toolkit for elution profile-based inference of protein complexes. Nature Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang da W, Sherman BT, and Lempicki RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hwang S, Kim CY, Yang S, Kim E, Hart T, Marcotte EM, and Lee I (2019). HumanNet v2: human gene networks for disease research. Nucleic acids research 47, D573–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jahn O, Tenzer S, and Werner HB (2009). Myelin proteomics: molecular anatomy of an insulating sheath. Molecular neurobiology 40, 55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard J-P, Lacomblez L, Pochigaeva K, Salachas F, et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nature genetics 40, 572–574. [DOI] [PubMed] [Google Scholar]
- 48.Kawahara Y, and Mieda-Sato A (2012). TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci U S A 109, 3347–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim E, Hwang S, Kim H, Shim H, Kang B, Yang S, Shim JH, Shin SY, Marcotte EM, and Lee I (2016). MouseNet v2: a database of gene networks for studying the laboratory mouse and eight other model vertebrates. Nucleic acids research 44, D848–D854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.King IN, Yartseva V, Salas D, Kumar A, Heidersbach A, Ando DM, Stallings NR, Elliott JL, Srivastava D, and Ivey KN (2014). The RNA-binding protein TDP-43 selectively disrupts microRNA-1/206 incorporation into the RNA-induced silencing complex. The Journal of biological chemistry 289, 14263–14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (New York, NY) 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- 53.Lage K, Karlberg EO, Størling ZM, Ólason PÍ, Pedersen AG, Rigina O, Hinsby AM, Tümer Z, Pociot F, Tommerup N, et al. (2007). A human phenome-interactome network of protein complexes implicated in genetic disorders. Nature Biotechnology 25, 309. [DOI] [PubMed] [Google Scholar]
- 54.Laird FM, Farah MH, Ackerley S, Hoke A, Maragakis N, Rothstein JD, Griffin J, Price DL, Martin LJ, and Wong PC (2008). Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 1997–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li F, Tian X, Zhou Y, Zhu L, Wang B, Ding M, and Pang H (2012). Dysregulated expression of secretogranin III is involved in neurotoxin-induced dopaminergic neuron apoptosis. Journal of neuroscience research 90, 2237–2246. [DOI] [PubMed] [Google Scholar]
- 56.Li T, Wernersson R, Hansen RB, Horn H, Mercer J, Slodkowicz G, Workman CT, Rigina O, Rapacki K, Staerfeldt HH, et al. (2017). A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods 14, 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ling SC, Albuquerque CP, Han JS, Lagier-Tourenne C, Tokunaga S, Zhou H, and Cleveland DW (2010). ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci U S A 107, 13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, et al. (2017). TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 95, 808–816.e809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mackenzie IR, Rademakers R, and Neumann M (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. The Lancet Neurology 9, 995–1007. [DOI] [PubMed] [Google Scholar]
- 60.Maere S, Heymans K, and Kuiper M (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics (Oxford, England) 21, 3448–3449. [DOI] [PubMed] [Google Scholar]
- 61.Malty RH, Aoki H, Kumar A, Phanse S, Amin S, Zhang Q, Minic Z, Goebels F, Musso G, Wu Z, et al. (2017). A Map of Human Mitochondrial Protein Interactions Linked to Neurodegeneration Reveals New Mechanisms of Redox Homeostasis and NF-kappaB Signaling. Cell systems 5, 564–577.e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, et al. (2000). Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 26, 191–194. [DOI] [PubMed] [Google Scholar]
- 63.Meyer A.d.S., Garcia AAF, Souza A.P.d., and Souza C.L.d Jr.. (2004). Comparison of similarity coefficients used for cluster analysis with dominant markers in maize (Zea mays L). Genetics and Molecular Biology 27, 83–91. [Google Scholar]
- 64.Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, et al. (1998). Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396, 433–439. [DOI] [PubMed] [Google Scholar]
- 65.Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohagheghi F, Prudencio M, Stuani C, Cook C, Jansen-West K, Dickson DW, Petrucelli L, and Buratti E (2016). TDP-43 functions within a network of hnRNP proteins to inhibit the production of a truncated human SORT1 receptor. Human molecular genetics 25, 534–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. [DOI] [PubMed] [Google Scholar]
- 68.Nepusz T, Yu H, and Paccanaro A (2012). Detecting overlapping protein complexes in protein-protein interaction networks. Nature Methods 9, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, et al. (2014). The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42, D358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pan J, Meyers RM, Michel BC, Mashtalir N, Sizemore AE, Wells JN, Cassel SH, Vazquez F, Weir BA, Hahn WC, et al. (2018). Interrogation of Mammalian Protein Complex Structure, Function, and Membership Using Genome-Scale Fitness Screens. Cell systems 6, 555–568.e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pertea M, Kim D, Pertea GM, Leek JT, and Salzberg SL (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature protocols 11, 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pinero J, Bravo A, Queralt-Rosinach N, Gutierrez-Sacristan A, Deu-Pons J, Centeno E, Garcia-Garcia J, Sanz F, and Furlong LI (2017). DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res 45, D833–d839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz F, and Furlong LI (2017). DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Research 45, D833–D839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Polymenidou M, and Cleveland DW (2011). The seeds of neurodegeneration: prion-like spreading in ALS. Cell 147, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Prudencio M, Jansen-West KR, Lee WC, Gendron TF, Zhang YJ, Xu YF, Gass J, Stuani C, Stetler C, Rademakers R, et al. (2012). Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc Natl Acad Sci U S A 109, 21510–21515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Psychiatric Gwas Consortium Bipolar Disorder Working Group, Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, Edenberg HJ, Nurnberger JI Jr, Rietschel M, et al. (2011). Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nature Genetics 43, 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ross CA, and Poirier MA (2004). Protein aggregation and neurodegenerative disease. Nat Med 10 Suppl, S10–17. [DOI] [PubMed] [Google Scholar]
- 80.Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Mewes HW (2010). CORUM: the comprehensive resource of mammalian protein complexes--2009. Nucleic Acids Res 38, D497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rutherford NJ, Zhang Y-J, Baker M, Gass JM, Finch NA, Xu Y-F, Stewart H, Kelley BJ, Kuntz K, Crook RJP, et al. (2008). Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS genetics 4, e1000193–e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sanchez-Taltavull D, Ramachandran P, Lau N, and Perkins TJ (2016). Bayesian Correlation Analysis for Sequence Count Data. PloS one 11, e0163595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schizophrenia Working Group of the Psychiatric Genomics, C., Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, Lee P, Bulik-Sullivan B, Collier DA, et al. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sherman DL, and Brophy PJ (2005). Mechanisms of axon ensheathment and myelin growth. Nature reviews Neuroscience 6, 683–690. [DOI] [PubMed] [Google Scholar]
- 86.Shi H, Zhang X, Weng YL, Lu Z, Liu Y, Lu Z, Li J, Hao P, Zhang Y, Zhang F, et al. (2018). m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Slepnev VI, and De Camilli P (2000). Accessory factors in clathrin-dependent synaptic vesicle endocytosis. Nature reviews Neuroscience 1, 161–172. [DOI] [PubMed] [Google Scholar]
- 88.Small SA, and Petsko GA (2015). Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nature reviews Neuroscience 16, 126–132. [DOI] [PubMed] [Google Scholar]
- 89.Smith R, Klein P, Koc-Schmitz Y, Waldvogel HJ, Faull RL, Brundin P, Plomann M, and Li JY (2007). Loss of SNAP-25 and rabphilin 3a in sensory-motor cortex in Huntington’s disease. Journal of neurochemistry 103, 115–123. [DOI] [PubMed] [Google Scholar]
- 90.Sonnhammer EL, and Ostlund G (2015). InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res 43, D234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, and Shoesmith C (2007). TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci 35, 320–327. [DOI] [PubMed] [Google Scholar]
- 93.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tanabe A, Yanagiya T, Iida A, Saito S, Sekine A, Takahashi A, Nakamura T, Tsunoda T, Kamohara S, Nakata Y, et al. (2007). Functional single-nucleotide polymorphisms in the secretogranin III (SCG3) gene that form secretory granules with appetite-related neuropeptides are associated with obesity. The Journal of clinical endocrinology and metabolism 92, 1145–1154. [DOI] [PubMed] [Google Scholar]
- 95.The UniProt Consortium (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Research 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Turner TN, Yi Q, Krumm N, Huddleston J, Hoekzema K, HA FS, Doebley AL, Bernier RA, Nickerson DA, and Eichler EE (2017). denovo-db: a compendium of human de novo variants. Nucleic Acids Res 45, D804–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tyanova S, Temu T, Carlson A, Sinitcyn P, Mann M, and Cox J (2015). Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics 15, 1453–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, et al. (2016). Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell reports 15, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, et al. (2011). VPS35 mutations in Parkinson disease. Am J Hum Genet 89, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, et al. (2015). Panorama of ancient metazoan macromolecular complexes. Nature 525, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang M, Herrmann CJ, Simonovic M, Szklarczyk D, and von Mering C (2015). Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 15, 3163–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Waragai M, Nagamitsu S, Xu W, Li YJ, Lin X, and Ashizawa T (2006). Ataxin 10 induces neuritogenesis via interaction with G-protein beta2 subunit. Journal of neuroscience research 83, 1170–1178. [DOI] [PubMed] [Google Scholar]
- 104.Williams ET, Chen X, and Moore DJ (2017). VPS35, the Retromer Complex and Parkinson’s Disease. Journal of Parkinson’s disease 7, 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, and Kumar-Singh S (2010). TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 107, 3858–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yoo BK, Santhekadur PK, Gredler R, Chen D, Emdad L, Bhutia S, Pannell L, Fisher PB, and Sarkar D (2011). Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology (Baltimore, Md) 53, 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, et al. (2018). Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014.e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Giron CG, et al. (2018). Ensembl 2018. Nucleic Acids Res 46, D754–d761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhu F, Cizeron M, Qiu Z, Benavides-Piccione R, Kopanitsa MV, Skene NG, Koniaris B, DeFelipe J, Fransen E, Komiyama NH, et al. (2018). Architecture of the Mouse Brain Synaptome. Neuron 99, 781–799.e710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhu J, Shang Y, and Zhang M (2016). Mechanistic basis of MAGUK-organized complexes in synaptic development and signalling. Nature reviews Neuroscience 17, 209–223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Protein intensities of 4,314 and 8075 high-confidence protein identifications across 550 biochemical fractions from five experiments performed using adult mice. Related to Figure 1 and STAR Methods.
Table S2. List of 564 gold standard reference complexes used for PPI prediction and 78 brain specific reference complexes used for cluster evaluation. Related to Figures 1–2 and STAR Methods.
Table S3. 27,403 high confidence protein-protein interactions in BraInMap. Related to Figure 1–2 and STAR Methods.
Table S4. BraInMap complexes showing core and expanded members, human orthologs, neuronal and non-neuronal members, membrane components, similarity metrics, and subunit ages. Related to Figure 2–7 and STAR Methods.
Table S5. BraInMap interactions mapped to human SHSY-5Y neuronal cell line interactions. Related to Figure 2 and STAR Methods.
Table S6. Tissue enrichment of protein detected in BraInMap compared to studies in mammalian cell culture models based on annotation from UniProt Tissue. Enrichment of proteins BraInMap complexes compared to studies in mammalian cell culture models based on annotation from GO cellular component, molecular function, and PFAM domains. Related to Figure 1–2 and STAR Methods.
Table S7. Brain region and cell type specific enrichment of BraInMap complexes. Related to Figure 3 and STAR Methods.
Table S8. Complexes enriched for GO cellular component and molecular function annotations. Enrichment of proteins belonging to more than one complex. Related to Figure 4–6, S1 and STAR Methods.
Table S9. Disease annotations of proteins in BraInMap complexes based on curated annotations DisGeNET database and neurodevelopmental disorders enriched in BraInMap. Related to Figure 7 and STAR Methods.
Data Availability Statement
All raw proteomic (co-fractionation) data from this work is submitted to the PRIDE repository (Accession: PXD011304) at the European Bioinformatics Institute, in accordance with the data sharing policy. Codes used in generating the results are described above in detail.