

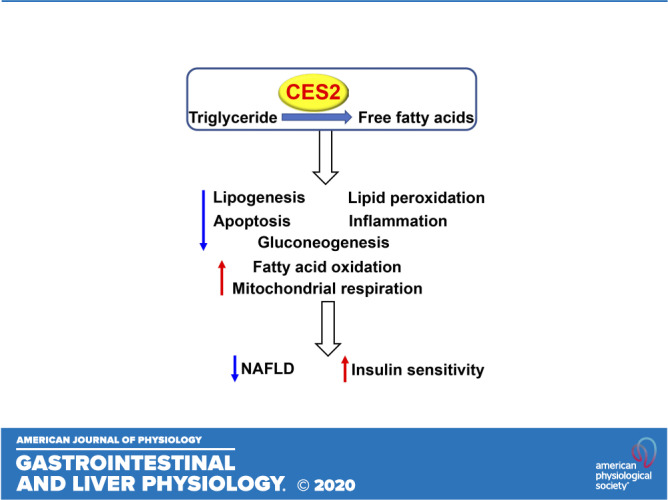
Keywords: CES2, fatty acid oxidation, lipolysis, lipotoxicity, steatohepatitis
Abstract
Human carboxylesterase 2 (CES2) has triacylglycerol hydrolase (TGH) activities and plays an important role in lipolysis. In this study, we aim to determine the role of human CES2 in the progression or reversal of steatohepatitis in diet-induced or genetically obese mice. High-fat/high-cholesterol/high-fructose (HFCF) diet-fed C57BL/6 mice or db/db mice were intravenously injected with an adeno-associated virus expressing human CES2 under the control of an albumin promoter. Human CES2 protected against HFCF diet-induced nonalcoholic fatty liver disease (NAFLD) in C57BL/6J mice and reversed steatohepatitis in db/db mice. Human CES2 also improved glucose tolerance and insulin sensitivity. Mechanistically, human CES2 reduced hepatic triglyceride (T) and free fatty acid (FFA) levels by inducing lipolysis and fatty acid oxidation and inhibiting lipogenesis via suppression of sterol regulatory element-binding protein 1. Furthermore, human CES2 overexpression improved mitochondrial respiration and glycolytic function, and inhibited gluconeogenesis, lipid peroxidation, apoptosis, and inflammation. Our data suggest that hepatocyte-specific expression of human CES2 prevents and reverses steatohepatitis. Targeting hepatic CES2 may be an attractive strategy for treatment of NAFLD.
NEW & NOTEWORTHY Human CES2 attenuates high-fat/cholesterol/fructose diet-induced steatohepatitis and reverses steatohepatitis in db/db mice. Mechanistically, human CES2 induces lipolysis, fatty acid and glucose oxidation, and inhibits hepatic glucose production, inflammation, lipid oxidation, and apoptosis. Our data suggest that human CES2 may be targeted for treatment of non-alcoholic steatohepatitis (NASH).
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a very common chronic disorder around the world. It initiates from simple steatosis, which may further progress to nonalcoholic steatohepatitis (NASH), cirrhosis, and hepatocellular carcinoma. Unfortunately, the mechanisms underlying the pathogenesis of NAFLD are still poorly understood, and no FDA-approved therapies are available for treatment of NAFLD.
Carboxylesterase 2 (CES2) is an esterase that can catalyze the hydrolysis of ester- and amide bonds-containing drugs or compounds (1). The mouse Ces2 family has eight members (Ces2a–Ces2h) that are present in a wide variety of cell types and tissues (1). Among the eight mouse Ces2 genes, the function of Ces2c is relatively well-characterized, which shares 71% homology in amino acids with human CES2. Mouse Ces2c has been shown to display both triacylglycerol hydrolase (TGH) and diacylglycerol hydrolase (DAG) activity (2–4). Hepatic deficiency of mouse Ces2c causes liver steatosis (2). On the contrary, gain of Ces2c function in liver improves hepatic steatosis, energy metabolism, and insulin signaling (2). In addition, intestine-specific overexpression of mouse Ces2c is shown to protect against high-fat diet-induced liver steatosis and obesity (3).
Human CES2 is abundantly expressed in liver. Given that so many rodent Ces2 isoforms exist, it is important to determine the role of human CES2 in metabolism. So far, only one report showed limited role of human CES2 in regulation of obesity-related metabolic disturbances (4). The role of human CES2 in regulating the development or reversal of NAFLD remains to be determined. In this report, we show that adeno-associated virus serotype 8 (AAV8)-mediated overexpression of human CES2 in hepatocytes protects against high-fat/high-cholesterol/high-fructose (HFCF) diet-induced steatohepatitis or reverses steatohepatitis in genetically obese db/db mice. We also investigated the underlying mechanisms. Our data demonstrate that human CES2 in hepatocytes is protective against NAFLD.
MATERIALS AND METHODS
Mice and Diets
C57BL/6J mice and db/db mice were purchased from the Jackson Laboratories (Bar Harbor, ME). C57BL/6J mice were fed for 16 wk a high-fat/cholesterol/fructose diet containing 40% fat/0.2% cholesterol (AIN-76A; TestDiet, St. Louis, MO) and 4.2% fructose (in drinking water). Unless otherwise stated, male mice were used and were fasted for 5–6 h before euthanasia. All the animal experiments were approved by the Institutional Animal Care and Use Committee at Northeast Ohio Medical University.
Adeno-Associated Viruses
The coding sequence of human CES2 was amplified by polymerase chain reaction (PCR) and cloned into an AAV vector under the control of a mouse albumin promoter to generate AAV-ALB-CES2. AAV8-ALB-Null (control) or AAV8-ALB-CES2 was produced and titrated by Vector BioLabs. Each mouse was intravenously injected with 3 × 1011 genome copies (GCs) AAVs and then fed either a chow diet for 12 wk or an HFCF diet for up to 16 wk.
Real-Time PCR
Total RNA was isolated using TRIzol Reagent (Thermo Fisher). mRNA levels were quantified by quantitative real-time PCR (qRT-PCR) using PowerUP SYBR Green Master Mix (ThermoFisher) on a 7500 Real-Time PCR machine (Applied Biosystems). Results were calculated using Ct values and normalized to 36B4 mRNA level.
Western Blot and Immunostaining Assays
Western blot assays were performed using total liver lysates or nuclear extracts of the liver samples as described previously (5). CES2 (Cat. No. ab56528) and Tubulin (Cat. No. ab4074) antibodies were purchased from Abcam. Antibodies against acetyl-CoA carboxylase (ACC) (Cat. No. 3662), cleaved caspase 3 (Cat. No. 9661), total caspase 3 (Cat. No. 9662), phospho-Smad2/3 (Cat. No. 8828), total Smad2/3 (Cat. No. 5678), phospho serine/threonine kinase 1 (AKT)-AKT (Cat. No. 4051), or total AKT (Cat. No. 9272) were purchased from Cell Signaling Technology (Boston, MA). The sterol regulatory element-binding protein 1 (SREBP-1) antibody was purchased from Santa Cruz Biotechnology (Cat. No. SC-8984). Immunostaining was performed using an ABC-HRP kit from Vector Laboratories (Cat. No. PK-4001) and an F4/80 antibody from Abcam (Cat. No. ab6640).
Analysis of Plasma or Hepatic Lipids, Glucose, Hydroxyproline, Malondialdehyde, ROS, AST, ALT, β-Hydroxybutyrate, and Apoptosis
Approximately 100 mg liver tissue was homogenized in methanol, and lipids were extracted in chloroform/methanol (2:1 vol/vol) as described previously (6). Triglycerides (TGs) and cholesterol in the livers were then quantified using Infinity reagents from Thermo Scientific (Waltham, MA). Plasma glucose, alanine aminotransferase (ALT), and aspartate aminotransferase (AST) levels were measured using Infinity reagents. Hydroxyproline level in liver was quantified using a kit from Cell BioLabs (Cat. No. STA675). Hepatic malondialdehyde (MDA) levels were determined using a thiobarbituric acid reactive substances (TBARS) assay kit (Cat. No. STA-330), and reactive oxygen species (ROS) were measured using an OxiSelect in Vitro ROS/RNS Assay kit (Cat. No. STA-347) from Cell Biolabs as described. Plasma β-hydroxybutyrate level was determined using a kit from Pointe Scientific (Fisher; Cat. No. 23-666-471). Liver apoptosis was determined using a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) kit from Abcam (Cat. No. ab206386).
Liver Histology
Livers were fixed in 10% formalin and then embedded in optimal cutting temperature (OCT) compound or paraffin. Hepatic neutral lipid accumulation, morphology, and fibrosis were determined by Oil Red O staining, hematoxylin and eosin staining, and picrosirius red staining, respectively, and images were acquired using an Olympus microscope.
TGH Activity Assays
Hepatic microsome proteins were isolated, and TGH activity was measured using [3H]triolein as the substrate as described previously (7).
Fatty Acid Oxidation
Primary hepatocytes were isolated from C57BL/6J mice injected with AAV8-ALB-Null or AAV8-ALB-CES2, and then cultured in DMEM containing 10% FBS in 12-well dishes. Fatty acid oxidation (FAO) was determined using [3H]palmitic acid as substrates as previously described (7).
Analysis of Fatty Acid Levels and Fatty Acid Composition
Hepatic or plasma total free fatty acids (FFAs) were measured using a kit from Wako Chemicals (Richmond, VA). Hepatic fatty acid composition was analyzed by GC-MS as described previously (2, 8).
De Novo Lipogenesis
Mice were injected intraperitoneally with 2H2O. After 4 h, de novo lipogenesis (DNL) was performed by GC-MS as described previously (7).
Glucose, Insulin, or Pyruvate Tolerance Tests
Mice were fasted overnight before glucose or pyruvate tolerance tests were performed, or fasted for 6 h before insulin tolerance test was performed. Glucose, insulin, or pyruvate tolerance tests were performed by intraperitoneally injecting mice with 2 g/kg glucose, 1 U/kg insulin, or 1.5 g/kg pyruvate, respectively. Blood glucose concentrations were measured with a glucometer using blood taken from tail tips at the designated time points.
Metabolic Flux Analysis
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using the XF24 Extracellular Flux analyzer and a Cell Energy Phenotype test kit (Seahorse Bioscience, North Billerica, MA). Briefly, mouse primary hepatocytes were isolated from mice infected with AAV8-ALB-Null or AAV8-ALB-CES2. They were seeded on an XF24 plate at a density of 120,000 cells/well and cultured in DMEM. The cells were incubated for 24 h in a humidified 37°C incubator with 5% CO2. Before an assay was performed, growth medium in the wells of an XF cell plate was exchanged with the appropriate assay medium. About 500 µL of the assay medium was added to cells for an XF assay. Although sensor cartridges were calibrated, cell plates were cultured in a 37°C/non-CO2 incubator for 1 h before the start of an assay. All experiments were performed at 37°C. Each measurement cycle consisted of a mixing time of 5 min and a data acquisition period of 5 min. OCR and ECAR data points refer to the average rates during the measurement cycles. All compounds were prepared at appropriate concentrations in desired assay medium and adjusted to pH 7.4. About 75 µL of the compounds (inhibitors of electron transfer chain, such as oligomycin and FCCP) were added to each injection port. OCR and ECAR were reported as absolute rates (pmole/min for OCR and mpH/min for ECAR) and normalized to total proteins of cell lysates.
Body Fat Content Measurement
Body fat content was detected using Echo-MRI (Echo-MRI, LLC, Houston, TX).
Statistical Analysis
All data are expressed as means ± SE. Statistical significance was analyzed using unpaired Student’s t test or analysis of variance (ANOVA; for more than two groups; GraphPad Prism, CA). Differences were considered statistically significant at P < 0.05.
RESULTS
Hepatocyte-Specific Expression of Human CES2 Prevents HFCF Diet-Induced Hepatosteatosis by Promoting Lipolysis and FAO and Inhibiting Lipogenesis
Our previous studies have shown that hepatic Ces2c/CES2 expression is markedly reduced in genetically obese db/db mice, high-fat diet (HFD)-fed mice, or patients with NASH (2). Given that 8 Ces2 genes exist in mice (1) and that the role of human CES2 in the development of NAFLD remains unclear, we generated an AAV expressing human CES2 under the control of an albumin promoter (AAV8-ALB-CES2) and used it for subsequent studies. AAV8-ALB-CES2 and AAV8-ALB-Null (control) were injected intravenously into C57BL/6J mice, which were then fed an HFCF diet for 16 wk. Overexpression of human CES2 in hepatocytes reduced the body and liver weight (Fig. 1A), fat content (Supplemental Fig. S1A; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.12925847), and plasma ALT levels (Fig. 1B), but it had no effect on hepatic or plasma total cholesterol (TC) levels (Fig. 1C and Supplemental Fig. S1B). Hepatocyte-specific expression of human CES2 also significantly decreased plasma TG and FFA levels (Supplemental Fig. 1, C and D) as well as hepatic TG and FFA levels (Fig. 1, D and E). Analysis of hepatic fatty acid composition using GC-MS showed that human CES2 expression reduced C14:0, C16:0, C16:1, C18:0, C18:1, and C18:2 fatty acids by 47%, 58%, 60%, 67%, 54%, and 67%, respectively, in HFCF-fed C57BL/6 mice (Fig. 1F). In addition, histological staining studies showed that human CES2 overexpression reduced neutral lipid accumulation and improved liver histology (Fig. 1G).
Figure 1.

Hepatocyte-specific expression of human carboxylesterase 2 (CES2) prevents high-fat/high-cholesterol/high-fructose (HFCF) diet-induced hepatosteatosis by inducing lipolysis and fatty acid oxidation (FAO) and inhibiting lipogenesis. C57BL/6J mice were intravenously injected with AAV8-ALB-Null or AAV8-ALB-CES2 at a dose of 3 × 1011 genomic copies/mouse and fed an HFCF diet for 16 wk (n = 8 mice per group). A: body or liver weight (A). B: plasma alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels. C: hepatic total cholesterol (TC) levels. D: hepatic triglyceride (TG) levels. E: hepatic free fatty acid (FFA) levels. F: hepatic fatty acid (FA) composition analyzed by GC-MS (n = 6–8 mice). G: representative images of liver sections stained with Oil Red O (ORO) (top) or hematoxylin and eosin (H&E) (bottom). H: liver microsome proteins were isolated and triacylglycerol hydrolase (TGH) activity was measured using [3H]triolein as substrates. I: mouse primary hepatocytes were isolated, and FAO was determined using [3H]palmitic acid as substrates (n = 5). J: newly synthesized TG levels were quantified by GC-MS. K: hepatic mRNA levels were quantified by quantitative real-time PCR. L: hepatic proteins were analyzed by Western blot assays. M: hepatic protein levels were quantified. *P < 0.05, **P < 0.01. AAV, adeno-associated virus; ACC, acetyl-CoA carboxylase.
To investigate how human CES2 regulates TG metabolism, we used liver lysates and [3H]triolein as substrates to determine whether human CES2 affected TGH activity in vivo. Our data showed that overexpression of human CES2 increased hydrolysis of [3H]triolein by 1.9-fold (Fig. 1H), indicating that human CES2 promotes TG hydrolysis. Newly released FFAs from lipolysis are known to function as a signaling molecule by activating peroxisome proliferator-activated receptor α (PPARα) and fatty acid oxidation (FAO) and/or inhibiting SREBP-1c via antagonizing liver X receptor activity (2, 9, 10). Consistent with these findings, human CES2 overexpression increased hepatocyte FAO (Fig. 1I) and plasma levels of β-hydroxybutyrate (β-HB) (Supplemental Fig. S1E). β-HB is an indicator of hepatic FAO. Human CES2 also reduced newly synthesized TG levels in liver (Fig. 1J). Consistent with the functional changes, overexpression of human CES2 in hepatocytes significantly induced hepatic mRNA levels of genes implicated in FAO (Cpt1 and Pdk4), but inhibited genes involved in lipogenesis (Srebp1c, Acc1, Fasn; Fig. 1K). There was no change in genes involved in lipolysis (Ces1, Atgl) or VLDL secretion (Apob, Mttp) (Fig. 1K). We also confirmed the expression of some of the genes by Western blotting assays. AAV-mediated expression of human CES2 increased hepatic CES2 protein levels by 3.2-fold, but it repressed hepatic protein levels of sterol regulatory element-binding protein 1 (SREBP1) and ACC, the key molecules in lipogenesis, by 51% and 79%, respectively (Fig. 1, L and M). Taken together, the data of Fig. 1 suggest that human CES2 prevents HFCF diet-induced hepatosteatosis likely by inducing lipolysis and FAO and suppressing lipogenesis.
Human CES2 Improves Glucose Homeostasis and Inhibits Glucose-Stimulated Lipogenesis via SREBP-1C
Although circulation-derived FFA is one of the main mechanisms for hepatic lipid loading, glucose-mediated de novo lipogenesis (DNL) also plays an important role in the pathogenesis of NAFLD (11). To determine whether human CES2 plays a role in glucose metabolism, we analyzed plasma glucose levels in the HFCF diet-fed mice. Interestingly, overexpression of human CES2 in hepatocytes markedly reduced both postprandial (251.9 ± 12.27 vs. 204 ± 8.1 mg/dL) and fasting (129.3 ± 6.6 vs. 101.6 ± 4.16 mg/dL) plasma glucose levels (Fig. 2A), and it improved glucose tolerance (Fig. 2B) and insulin sensitivity (Fig. 2C). Analysis of metabolic flux by a Seahorse XF Analyzer showed that human CES2 overexpression increased mitochondrial respiration (Fig. 2D, left) and glycolytic function (Fig. 2D, right). Consistent with improved glucose homeostasis, human CES2 overexpression increased hepatic phospho-AKT level (Fig. 2E), and inhibited hepatic genes involved in gluconeogenesis, including Pgc1α, Pepck, and G6pc (Fig. 2F).
Figure 2.

Human carboxylesterase 2 (CES2) improves glucose homeostasis and inhibits glucose-stimulated lipogenesis via sterol regulatory element-binding protein 1 (SREBP-1c). A–F: C57BL/6J mice were intravenously injected with AAV8-ALB-Null or AAV8-ALB-CES2 and fed a high-fat/high-cholesterol/high-fructose (HFCF) diet for 16 wk (n = 8 mice per group). Postprandial glucose and fasting glucose levels were measured (A). Glucose-tolerance test (GTT) (B) and insulin-tolerance test (ITT) (C) were performed. Oxygen consumption rate (OCR) (D, left) and extracellular acidification rate (ECAR) (D, right) were measured using an XF24 Extracellular Flux analyzer. Hepatic protein (E) and mRNA (F) levels were determined. G and H: mouse primary hepatocytes were isolated from C57BL/6J mice that were intravenously injected with AAV8-ALB-Null or AAV8-ALB-CES2 and were then infected with adenoviruses expressing SREBP-1c (Ad-SREBP1c) or control adenoviruses (Ad-Empty). The cells were cultured with 5 mM or 25 mM glucose for 48 h. Cellular triglyceride (TG) levels were determined (G) (n = 5 per group). H: Western blot assays were performed. Overexpression of SREBP-1c normalized SREBP-1C levels in CES2-expressing hepatocytes. I and J: mouse primary hepatocytes were isolated from Srebp1c+/+ mice or Srebp1c−/− mice that were intravenously injected with AAV8-ALB-Null or AAV8-ALB-CES2 and cultured in the presence of 5 mM or 25 mM glucose for 48 h. Cellular TG levels were quantified (I) (n = 5 per group) and protein levels were determined (J). *P < 0.05, **P < 0.01. AAV, adeno-associated virus.
The data of Fig. 1 show that human CES2 inhibits hepatic SREBP-1C expression. To determine whether SREBP-1C is required for glucose-stimulated lipogenesis in the context of CES2 overexpression, we isolated primary hepatocytes from C57BL/6J mice infected with AAV8-ALB-Null or AAV8-ALB-CES2, and these hepatocytes were then infected with Ad-Empty (control) or Ad-SREBP-1c. Compared with 5 mM glucose, 25 mM glucose increased TG levels (Fig. 5G). Overexpression of human CES2 reduced TG levels in hepatocytes treated with 5 or 25 mM glucose, which was blunted when SREBP-1C levels were normalized (Fig. 2, G and H). We also isolated primary hepatocytes from Srebp-1c+/+ or Srebp-1c−/− mice infected with AAV8-ALB-Null or AAV8-ALB-CES2. Overexpression of human CES2 lowered TG levels in Srebp-1c+/+ hepatocytes but not in Srebp-1c−/− hepatocytes (Fig. 2, I and J). These data suggest that CES2 inhibits glucose-stimulated lipogenesis via SREBP-1C.
Figure 5.

Human carboxylesterase 2 (CES2) improves triglyceride (TG) and glucose homeostasis in db/db mice. Twelve-week-old male db/db mice were intravenously injected with AAV8-ALB-Null or AAV8-ALB-CES2 and fed a chow diet for 12 wk (n = 7–8 mice per group). Hepatic triacylglycerol hydrolase (TGH) activity (A) and newly synthesized TG (B) were determined. Hepatic levels of mRNA (C) and proteins (D and E) related to lipid metabolism were analyzed. Postprandial and fasting glucose levels were measured (F). GTT (G) and pyruvate-tolerance test (PTT) (H) were performed. Hepatic levels of mRNA (I) and proteins (J and K) related to glucose metabolism were determined. *P < 0.05, **P < 0.01. AAV, adeno-associated virus.
Taken together, the data of Fig. 2 show that human CES2 improves glucose homeostasis and reduces glucose-stimulated lipogenesis via SREBP-1C.
Hepatocyte-Specific Expression of Human CES2 Protects Against HFCF Diet-Induced Liver Inflammation, Apoptosis, and Fibrosis
Excessive FFAs, high level of glucose, or insulin resistance can cause hepatic endoplasmic reticulum stress, mitochondrial dysfunction, apoptosis, and inflammation, which play a key role in the progression of NAFLD from simple steatosis to steatohepatitis. The data of Figs. 1 and 2 show that human CES2 reduces FFA or glucose levels and improves insulin resistance. Therefore, we investigated whether human CES2-regulated apoptosis, inflammation, and liver injury. Hepatic overexpression of human CES2 inhibited a number of genes involved in inflammation (Tnfα, Il-1β, Mcp, F4/80) and fibrogenesis (Timp1, α-Sma, Col1α1, Col1α2), and reduced protein levels of phospho-Smad2/3 and cleaved Caspase 3 (Fig. 3, B and C). Consistent with the changes in gene expression, human CES2 reduced hepatic levels of apoptosis (Fig. 3, D and E), malondialdehyde (MDA; Fig. 3F), reactive oxygen species (ROS; Fig. 3G), macrophage infiltration (Fig. 3H), and collagen accumulation (Fig. 3, I and J), as indicated by picrosirius red staining (Fig. 3I) and hepatic hydroxyproline levels (Fig. 3J). Together, these data suggest that liver-specific expression of human CES2 attenuates HFCF diet-induced steatohepatitis likely by inhibiting ROS production, lipid peroxidation, apoptosis, and inflammation.
Figure 3.

Liver-specific expression of human carboxylesterase 2 (CES2) protects against high-fat/high-cholesterol/high-fructose (HFCF) diet-induced steatohepatitis. The mice have been described in the legend of Fig. 1. Hepatic mRNA (A) and protein (B and C) levels were determined. Hepatic apoptosis was analyzed by TUNEL assays (D) and then quantified (E). Hepatic malondialdehyde (MDA) (F) and reactive oxygen species (ROS) (G) levels were analyzed. Liver sections were immunostained with an antibody against F4/80 (H, left) or stained with picrosirius red (I, left). F4/80-positve area (%) (H, right) or picrosirius red-positive area (%) (I, right) was quantified. Hepatic hydroxyproline levels were determined (J). In (D), (H), and (I), arrows point to apoptotic cells, monocytes/macrophages, and fibrosis, respectively. *P < 0.05, **P < 0.01. AAV, adeno-associated virus.
Hepatocyte-Specific Expression of Human CES2 Reverses Hepatosteatosis in db/db Mice
The data from Figs. 1–3 show that human CES2 can prevent the development of diet-induced NAFLD. To explore whether human CES2 could also reverse NAFLD, we overexpressed human CES2 in genetically obese and diabetic db/db mice for 12 wk. Hepatic-specific expression of human CES2 reduced body and liver weight (Fig. 4A), fat content (Supplemental Fig. S2A), and plasma ALT and AST levels (Fig. 4B), but it had no effect on hepatic or plasma TC levels (Fig. 4C and Supplemental Fig. S2B). Overexpression of human CES2 in hepatocytes also reduced plasma TG or FFA levels (Supplemental Fig. S2, C and D) and hepatic TG or FFA levels (Fig. 4, D and E). Analysis of hepatic fatty acid composition by GC-MS shows that human CES2 expression reduced a number of fatty acyl-CoA levels, with C16:0 and C18:1 fatty acids being reduced by 68% and 55%, respectively (Fig. 4F). Consistent with these data, hematoxylin and eosin (H&E) or Oil Red O (ORO) staining showed that overexpression of human CES2 improved liver histology and reduced hepatic neutral lipid accumulation (Fig. 4G). Thus, the data of Fig. 4 demonstrate that hepatic-specific expression of human CES2 reverses hepatosteatosis in db/db mice.
Figure 4.

Liver-specific expression of human carboxylesterase 2 (CES2) reverses hepatosteatosis in db/db mice. Twelve-week-old male db/db mice were intravenously injected with AAV8-ALB-Null or AAV8-ALB-CES2 and fed a chow diet for 12 wk (n = 7–8 mice per group). A: body or liver weight. B: plasma alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels. C: hepatic cholesterol levels. D: hepatic triglyceride (TG) levels. E: hepatic free fatty acid (FFA) levels. F: hepatic fatty acid composition. G: representative images of liver sections stained with Oil Red O (top) or hematoxylin and eosin (bottom). *P < 0.05, **P < 0.01. AAV, adeno-associated virus.
Hepatocyte-Specific Expression of Human CES2 Induces TGH Activity and Inhibits TG Synthesis and Glucose Production in db/db Mice
To investigate how human CES2 regulated hepatic TG levels in db/db mice, we analyzed hepatic TGH activity and DNL. Overexpression of human CES2 increased hepatic TGH activity by 1.5-fold (Fig. 5A) and inhibited newly synthesized TG levels by 37% in the liver (Fig. 5B). Overexpression of human CES2 also increased plasma β-HB levels (Supplemental Fig. 2E), suggesting that human CES2 increases hepatic FAO. Consistent with these data, overexpression of human CES2-induced genes involved in FAO (Cpt1, Pdk4) and inhibited lipogenic genes (Srebp1c, Acc1, Fasn; Fig. 5, C–E). There was no change in other genes involved in lipolysis (Ces1, Atgl) or genes involved in VLDL secretion (Apob, Mttp; Fig. 5C).
In addition, overexpression of human CES2 reduced both postprandial (380.5 ± 43.43 vs. 264 ± 22.56 mg/dL) and fasting (184.9 ± 8.19 vs. 133.4 ± 6.32 mg/dL) glucose levels (Fig. 5F). We then performed glucose-tolerance test (GTT) and pyruvate-tolerance test (PTT) to assess global glucose disposal and hepatic glucose production, respectively. Overexpression of human CES2 improved glucose tolerance (Fig. 5G) and reduced hepatic glucose production (Fig. 5H). In line with these data, human CES2 significantly inhibited hepatic genes involved in gluconeogenesis (Pgc1α, Pepck, and G6pc) or glucose uptake (Glut2; Fig. 5I), and increased hepatic phospho-AKT level by greater than threefold (Fig. 5K). Together, these data demonstrate that human CES2 increases TGH activity and suppresses DNL and glucose production in db/db mice.
Hepatic Overexpression of Human CES2 Attenuates Steatohepatitis by Inhibiting Inflammation and Apoptosis in db/db Mice
Next, we investigated whether and how human CES2 regulated steatohepatitis in db/db mice. Overexpression of human CES2 in hepatocytes suppressed genes involved in inflammation (Tnfα, Il-6, Il-1β, Mcp1, F4/80) or fibrogenesis (Tgfβ, Timp1, α-Sma, Col1α1, and Col1α2; Fig. 6A), and reduced protein levels of phospho-Smad2/3 and cleaved Caspase 3 (Fig. 6B) that were involved in fibrogenesis and apoptosis. Further studies showed that human CES2 inhibited hepatic apoptosis (Fig. 6C), as well as hepatic MDA and ROS levels (Fig. 6, D and E). In addition, human CES2 inhibited hepatic macrophage infiltration (Fig. 6F) and fibrogenesis (Fig. 6, G and H). Thus, hepatic expression of human CES2 reverses steatohepatitis likely by attenuating inflammation, ROS production, lipid peroxidation, and apoptosis.
Figure 6.

Human carboxylesterase 2 (CES2) attenuates steatohepatitis in db/db mice. Mice have been described in the legend of Figs. 4 and 5 (n = 7–8 mice). Hepatic mRNA (A) and protein (B) levels were determined. Hepatic apoptosis was analyzed by TUNEL assays (C). Hepatic malondialdehyde (MDA) (D) and reactive oxygen species (ROS) (E) levels were analyzed. Liver sections were immunostained with an antibody against F4/80 (F, left) or stained with picrosirius red (G, left). F4/80-positve area (%) (F, right) or picrosirius red-positive area (%) (G, right) was quantified. Hepatic hydroxyproline levels were determined (H). In (C), (F), and (G), arrows point to apoptotic cells, monocytes/macrophages, and fibrosis, respectively. *P < 0.05, **P < 0.01. AAV, adeno-associated virus.
DISCUSSION
We and others (2, 4) have shown that hepatic CES2 levels or activity are decreased in mice or humans with obesity or NAFLD. Yet, the role of human CES2 in the development of NAFLD remains to be determined. In this study, we used AAVs overexpressing human CES2 specifically in mouse hepatocytes to investigate whether and how human CES2 regulates the development or reversal of NAFLD. Our data show that human CES2 protects against HFCF diet-induced NAFLD and reverses steatohepatitis in db/db mice. Mechanistically, we demonstrate that human CES2 promotes lipolysis, FAO, and glucose oxidation and inhibits DNL, gluconeogenesis, lipid peroxidation, and apoptosis.
Mice have eight Ces2 genes. The role of many of mouse Ces2 genes in lipid metabolism remains unknown. In addition, it is unclear which mouse Ces2 gene is the homologue of human CES2 gene. Thus, it is necessary to investigate the function of human CES2. So far, only one report shows that adenovirus-mediated expression of human CES2 for eight days reduces hepatic TG levels in mice that have been fed a high-fat diet (4). Given that adenoviruses can affect different cell types, cause significant inflammation, and express exogenous genes only for a very short time in the liver, it is impossible to investigate whether human CES2 regulates the development of NAFLD using adenoviruses-mediated gene transfer. Therefore, we utilized AAVs that overexpressed human CES2 only in the liver, which overcame the shortcomings noted with the use of adenoviruses. Nonetheless, overexpression of a human gene in mice remains an artificial system, which may not completely mimic what happens in humans.
NAFLD is often initiated from simple steatosis. Increased DNL and defective lipolysis or VLDL secretion all contribute to hepatic TG accumulation. We show that human CES2 has TGH activity but does not regulate genes involved in VLDL secretion. Increased TGH activity can release more FFAs which can in turn promote FAO (2, 7, 9). The newly released FFAs may also inhibit SREBP-1C via antagonizing liver X receptor (LXR) activity (2, 12). Glucose is one of the major sources for DNL. Our data also show that human CES2 inhibits glucose-stimulated DNL at least partly through SREBP-1C. Moreover, CES2 inhibits hepatic glucose production. Thus, human CES2 lowers hepatic TG and FFA levels likely by inhibiting DNL and increasing lipolysis and FAO.
ACC inhibition has been shown to reduce hepatic TG levels in rodents and humans, but also induces hypertriglyceridemia (13, 14). The underlying mechanism may include an increase in LXR/SREBP1 and a reduction in PPARα activation (13, 14). Interestingly, human CES2 also reduces hepatic ACC expression. However, human CES2 inhibits SREBP1 expression and promotes fatty acid oxidation, and reduces plasma TG levels. In addition, human CES2 inhibits NASH development. Therefore, human CES2 may be translatable to a novel NASH therapy.
NAFLD may progress from simple steatosis to NASH in the presence of elevated inflammation, ROS, lipid peroxidation, ER stress, and apoptosis, which may be caused by insulin resistance and lipotoxicity (15). The finding that human CES2 improves insulin resistance and reduces hepatic lipid accumulation may underlie the protective role of human CES2 in steatohepatitis. In addition, induction of phosphor-AKT by CES2 may induce production of nitric oxide (NO) via phosphorylation and activation of endothelial nitric oxide synthase (eNOS), which may in turn inhibit fibrogenesis as NO signaling has been shown to prevent fibrogenesis (16, 17).
In summary, we have demonstrated that overexpression of human CES2 prevents diet-induced development of NAFLD. Reversal of NAFLD is desirable clinically. We also show that long-term expression of human CES2 reverses steatohepatitis in db/db mice. Mechanistically, CES2 exerts its beneficial effects via induction of lipolysis and subsequent metabolic changes. A more global profiling of changes in gene expression or metabolites may shed more light on the underlying mechanisms. Given that hepatic CES2 expression or activity is reduced in patients with NAFLD or obesity (2, 4), our data suggest that targeting hepatic CES2 may be a new strategy for treatment of NAFLD.
GRANTS
This work is supported by National Institutes of Health Grants R01DK102619 and R01DK118941.
DISCLOSURE
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.X., X.P., S.H., and Y.Z. conceived and designed research; Y.X., X.P., S.H., Y.Z., F.C., Y.L., and L.Y. performed experiments; Y.X., X.P., S.H., and Y.Z. analyzed data; Y.X., X.P., S.H., and Y.Z. interpreted results of experiments; Y.X., X.P., S.H., and Y.Z. prepared figures; Y.X., X.P., and S.H. drafted manuscript; Y.Z. edited and revised manuscript; Y.Z. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of Y. Li: Zhongshan Institute for Drug Discovery, the Institutes of Drug Discovery and Development, Chinese Academy of Sciences, Zhongshan, Guangdong Province, China; Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China. Future address of X. Pan: Divison of Gastroenterology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, China.
Footnotes
Human carboxylesterase 2 (CES2) has triacylglycerol hydrolase (TGH) activities and plays an important role in lipolysis. In this study, we aim to determine the role of human CES2 in the progression or reversal of steatohepatitis in diet-induced or genetically obese mice. High-fat/high-cholesterol/high-fructose (HFCF) diet-fed C57BL/6 mice or db/db mice were intravenously injected with an adeno-associated virus expressing human CES2 under the control of an albumin promoter. Human CES2 protected against HFCF diet-induced nonalcoholic fatty liver disease (NAFLD) in C57BL/6J mice and reversed steatohepatitis in db/db mice. Human CES2 also improved glucose tolerance and insulin sensitivity. Mechanistically, human CES2 reduced hepatic triglyceride (T) and free fatty acid (FFA) levels by inducing lipolysis and fatty acid oxidation and inhibiting lipogenesis via suppression of sterol regulatory element-binding protein 1. Furthermore, human CES2 overexpression improved mitochondrial respiration and glycolytic function, and inhibited gluconeogenesis, lipid peroxidation, apoptosis, and inflammation. Our data suggest that hepatocyte-specific expression of human CES2 prevents and reverses steatohepatitis. Targeting hepatic CES2 may be an attractive strategy for treatment of NAFLD.
REFERENCES
- 1.Holmes RS, Wright MW, Laulederkind SJ, Cox LA, Hosokawa M, Imai T, Ishibashi S, Lehner R, Miyazaki M, Perkins EJ, Potter PM, Redinbo MR, Robert J, Satoh T, Yamashita T, Yan B, Yokoi T, Zechner R, Maltais LJ. Recommended nomenclature for five mammalian carboxylesterase gene families: human, mouse, and rat genes and proteins. Mamm Genome 21: 427–441, 2010. doi: 10.1007/s00335-010-9284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, Zalzala M, Jadhav K, Xu Y, Kasumov T, Yin L, Zhang Y. Carboxylesterase 2 prevents liver steatosis by modulating lipolysis, endoplasmic reticulum stress, and lipogenesis and is regulated by hepatocyte nuclear factor 4 alpha in mice. Hepatology 63: 1860–1874, 2016. doi: 10.1002/hep.28472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maresch LK, Benedikt P, Feiler U, Eder S, Zierler KA, Taschler U, Kolleritsch S, Eichmann TO, Schoiswohl G, Leopold C, Wieser BI, Lackner C, Rulicke T, van Klinken J, Kratky D, Moustafa T, Hoefler G, Haemmerle G. Intestine-specific overexpression of carboxylesterase 2c protects mice from diet-induced liver steatosis and obesity. Hepatol Commun 3: 227–245, 2019. doi: 10.1002/hep4.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruby MA, Massart J, Hunerdosse DM, Schonke M, Correia JC, Louie SM, Ruas JL, Naslund E, Nomura DK, Zierath JR. Human carboxylesterase 2 reverses obesity-induced diacylglycerol accumulation and glucose intolerance. Cell Rep 18: 636–646, 2017. doi: 10.1016/j.celrep.2016.12.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4alpha pathway regulates lipid and lipoprotein metabolism. Nat Commun 6: 7466, 2015. doi: 10.1038/ncomms8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917, 1959. doi: 10.1139/y59-099, . doi: 10.1139/y59-099. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Li Y, Chen WD, Xu Y, Yin L, Ge X, Jadhav K, Adorini L, Zhang Y. Hepatic carboxylesterase 1 is essential for both normal and farnesoid X receptor-controlled lipid homeostasis. Hepatology 59: 1761–1771, 2014. doi: 10.1002/hep.26714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Y, Zhu Y, Jadhav K, Li Y, Sun H, Yin L, Kasumov T, Chen X, Zhang Y. Lipocalin-2 protects against diet-induced nonalcoholic fatty liver disease by targeting hepatocytes. Hepatol Commun 3: 763–775, 2019. doi: 10.1002/hep4.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 53: 116–126, 2011. doi: 10.1002/hep.24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, Goldstein JL, Brown MS. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci USA 98: 6027–6032, 2001. doi: 10.1073/pnas.111138698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, Nakagawa T, Kuwabara M, Sato Y, Kang DH, Tolan DR, Sanchez-Lozada LG, Rosen HR, Lanaspa MA, Diehl AM, Johnson RJ. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J Hepatol 68: 1063–1075, 2018. doi: 10.1016/j.jhep.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pawar A, Botolin D, Mangelsdorf DJ, Jump DB. The role of liver X receptor-alpha in the fatty acid regulation of hepatic gene expression. J Biol Chem 278: 40736–40743, 2003. doi: 10.1074/jbc.M307973200. [DOI] [PubMed] [Google Scholar]
- 13.Alkhouri N, Lawitz E, Noureddin M, DeFronzo R, Shulman GI. GS-0976 (Firsocostat): an investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin Investig Drugs 29: 135–141, 2020. doi: 10.1080/13543784.2020.1668374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goedeke L, Bates J, Vatner DF, Perry RJ, Wang T, Ramirez R, Li L, Ellis MW, Zhang D, Wong KE, Beysen C, Cline GW, Ray AS, Shulman GI. Acetyl-CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology 68: 2197–2211, 2018. doi: 10.1002/hep.30097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arab JP, Arrese M, Trauner M. Recent Insights into the pathogenesis of nonalcoholic fatty liver disease. Annu Rev Pathol Mech Dis 13: 321–350, 2018. doi: 10.1146/annurev-pathol-020117-043617. [DOI] [PubMed] [Google Scholar]
- 16.Dong Z, Su L, Esmaili S, Iseli TJ, Ramezani-Moghadam M, Hu L, Xu A, George J, Wang J. Adiponectin attenuates liver fibrosis by inducing nitric oxide production of hepatic stellate cells. J Mol Med 93: 1327–1339, 2015. doi: 10.1007/s00109-015-1313-z. [DOI] [PubMed] [Google Scholar]
- 17.Nozaki Y, Fujita K, Wada K, Yoneda M, Kessoku T, Shinohara Y, Imajo K, Ogawa Y, Nakamuta M, Saito S, Masaki N, Nagashima Y, Terauchi Y, Nakajima A. Deficiency of iNOS-derived NO accelerates lipid accumulation-independent liver fibrosis in non-alcoholic steatohepatitis mouse model. BMC Gastroenterol 15: 42, 2015. doi: 10.1186/s12876-015-0269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]