

Abstract
Rewarming the intact heart after a period of hypothermia is associated with reduced myocardial contractility, decreased Ca2+ sensitivity, and increased cardiac troponin-I (cTnI) phosphorylation. We hypothesized that hypothermia/rewarming (H/R) induces left ventricular (LV) contractile dysfunction due to phosphorylation of cTnI at Ser23/24. To test this hypothesis, the response of wild-type mice (n = 7) to H/R was compared with transgenic (TG) mice expressing slow skeletal TnI (TG-ssTnI; n = 7) that lacks the Ser23/24 phosphorylation sites. Hypothermia was induced by surface cooling and maintained at 23–25°C for 3 h. Subsequently, the animals were rewarmed to 37°C. LV systolic and diastolic function was assessed using a 1.4 F pressure-volume Millar catheter introduced via the right carotid artery. At baseline conditions, there were no significant differences in LV systolic function between wild-type and TG-ssTnI mice, whereas measurements of diastolic function [isovolumic relaxation constant (τ) and end-diastolic pressure-volume relationship (EDPVR)] were significantly (P < 0.05) reduced in TG-ssTnI animals. Immediately after rewarming, significant differences between groups were found in cardiac output (CO; wild-type 6.6 ± 0.7 vs. TG-ssTnI 8.8 ± 0.7 mL/min), stroke work (SW; wild-type 796 ± 112 vs. TG-ssTnI 1208 ± 67 mmHg/μL), and the preload recruited stroke work (PRSW; wild-type 38.3 ± 4.9 vs. TG-ssTnI 68.8 ± 8.2 mmHg). However, EDPVR and τ returned to control levels within 1 h in both groups. We conclude that H/R-induced LV systolic dysfunction results from phosphorylation of cTnI at Ser23/24.
NEW & NOTEWORTHY Rewarming following a period of accidental hypothermia leads to a form of acute cardiac failure (rewarming shock), which is in part due to reduced sensitivity to Ca2+ activation of myocardial contraction. The results of the present study support the hypothesis that rewarming shock is due to phosphorylation of cardiac troponin I.
Keywords: hypothermia, rewarming, transgenic slow skeletal Tnl, troponin I
INTRODUCTION
Rewarming patients following profound accidental hypothermia (<28°C) results in the clinically well-recognized combination of inadequate cardiac output (CO) and hypotension (“rewarming shock”) (2, 13–16, 18, 33). Epidemiological studies have reported a 30–80% overall survival of hypothermic patients after rewarming (16, 17, 22, 36). To further increase survival in these hypothermic patients, the mechanisms responsible for the intrinsic depression of cardiac contractility must be elucidated (1, 11, 15, 16, 32).
Phosphorylation of cardiac troponin I (cTnI) plays an important role in cardiac contractility by enhancing the relaxation rate and decreasing the contraction-relaxation cycle time during adrenergic stimulation (25, 26). The NH2-terminal region of cTnI contains serines at positions 23 and 24 (Ser23/24) which are substrates for protein kinase A (PKA), PKG, PKD, and PKCδ (25). PKA-induced phosphorylation of the NH2-terminal extension of cTnI at Ser23/24 alters the cTnI structure diminishing the cTnI NH2-terminal interaction with troponin C (TnC), resulting in a decrease in Ca2+ sensitivity of cardiac muscle force generation (30). Phosphorylation of cTnI at Ser23/24 also increases the rate of myocardial relaxation and thus helps to maintain adequate diastolic filling during increased heart rate induced by β-adrenergic stimulation to meet increased cardiac demand (24–26).
The significance of cTnI phosphorylation and its impact on cardiac function have been previously studied in a genetically altered murine model, where slow skeletal troponin I (ssTnI) is expressed replacing cTnI (5). The ssTnI isoform lacks the last 27–33 amino acids of the NH2-terminal region of cTnI; hence, the phosphorylation sites at Ser23/24 are absent. This transgenic (TG-ssTnI) model has a normal phenotype and with no cardiac pathology. However, significant differences in cardiac contractile function are present in TG-ssTnI mice, including an increase in myofibrillar Ca2+ sensitivity and altered cardiac relaxation (12, 37).
Previously, using a rat papillary muscle preparation, we (11) showed that H/R induces a decrease in peak contractile force and a reduction in Ca2+ sensitivity of force generation. This H/R-induced contractile dysfunction in papillary muscle is associated with an increase in PKA-specific phosphorylation of cTnI at Ser23/24. These results were further supported in experiments using isolated rat cardiomyocytes, in which we found that H/R-induced contractile dysfunction and reduced Ca2+ sensitivity are associated with an increase in cTnI phosphorylation (23). The aim of the present study was to examine the impact of H/R on left ventricular (LV) contractile function in TG-ssTnI compared with wild-type mice. We hypothesized that H/R-induced phosphorylation of cTnI at Ser23/24 underlies reduced LV contractile function.
MATERIALS AND METHODS
The Institutional Animal Care and Use Committee of the Mayo Clinic approved experimental protocols and the care of the animals. Adult female TG-ssTnI (on a CD1 background) mice and wild-type controls ranging from 14 to 27 wk of age (35–44 g) were used in this study. Animals were genotyped by PCR using specific primers directed against the inserted transgene (5). Hearts from TG-ssTnI mice demonstrated full stoichiometric replacement of cTnI with ssTnI. These TG-ssTnI mice were fertile and viable and showed no signs of increased mortality or cardiovascular pathology up to 18 mo of age (5).
Anesthesia.
Anesthesia was induced by intraperitoneal injections with pentobarbital sodium and ketamine (respectively, 50 + 45 mg/kg). A tracheostomy was performed and a small-size metal tube (19 G) was inserted to create a free airway. was set to 0.3, and spontaneous respiration was maintained throughout experiments. Respiratory rate was monitored during experiments. Anesthesia was continued by infusing pentobarbital sodium (9 mg·kg−1·h−1) through a left jugular venous catheter. The infusion was discontinued at temperatures below 25°C, since hypothermia has anesthetic effects and decreases drug metabolism. The animals were monitored for any sign of discomfort during H/R so that additional anesthesia might be provided if necessary.
Core cooling and rewarming.
Animals were cooled and rewarmed by circulating warm or cold water through a double-layered aluminum operating table circulated by temperature-adjusted water. Core temperature was continuously monitored using a thermocouple wire with the sensor tip positioned in the lower third of the esophagus and connected to a digital thermometer (Fluke 52II; Fluke, Everett, WA). Animals were cooled to 25°C over a 30-min period, maintained at 25°C for 1 h, thereafter cooled to 23°C, maintained at that temperature for 2 h, and subsequently rewarmed to 37°C over a 30-min period. Time-matched controls were maintained at 37°C for 4 h.
Hemodynamic measurements, data acquisition, and analysis.
A microtip pressure-volume catheter (SPR-839, 1.4 Fr; Millar Instruments, Houston, TX) was inserted into the right carotid artery and advanced into the LV under pressure control. LV pressure and volume signals were digitized at 1 kHz and recorded using ADInstruments Chart DAQ software. Acquired data included heart rate, maximum LV systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), maximum slopes of the LV systolic pressure increment (dP/dtmax), and the diastolic pressure decrement (dP/dtmin), time constant of LV pressure decay (τ), stroke volume (SV), end-diastolic volume (EDV), cardiac output (CO), and stroke work (SW). Data were analyzed off-line using a cardiac pressure/volume analysis program (PVAN 3.2, Millar Instruments). Data were collected during steady-state baseline conditions and during transient inferior vena cava occlusions performed to vary LV preload for determination of various load-independent indexes of systolic function, e.g., preload recruitable stroke work (PRSW), which is a robust index of LV contractility in the intact heart (6, 7) also at low-core temperatures (34).
Specific adjustments related to use of conductance catheter measurements at low-core temperatures.
The methods for using the Millar catheter system under these low-temperature conditions have been previously described in detail (10). Briefly, the measured conductance needs to be corrected for parallel conductance induced by the alternating current passing from the catheter through the blood and into the surrounding LV wall or interventricular septum. Parallel conductance is usually measured by saline bolus injection at the end of the experiment (19). However, difficulties were anticipated when applying this method to our experimental protocol, which required measurements of LV volume at low-core temperatures (37, 34, 32, 30, 28, 25, and 23°C). In light of temperature-dependent changes in viscosity of blood, the bolus injection performed at the end of experiment (37°C) cannot represent the true parallel conductance at lower-core temperatures, and repeated injections of hypertonic saline at each temperature would eventually intoxicate the animal. For this reason, parallel conductance measurements were not included in our volume measurements.
The cuvette calibration method was used to adjust for temperature-dependent changes in viscosity of blood. Insulator-type cuvettes of known diameter (1.5, 2.0, 2.5, 3.0, 3.5, and 4.0 mm) were filled with heparinized blood from mice and immersed in a temperature-controlled water bath to adjust blood temperature during calibration. Slopes and y-intercepts obtained at the above temperatures were applied to the analysis software to convert conductance units to true volume units (μL).
Blood gas measurements.
At the end of the experiment, during removal of the conductance catheter from the carotid artery, arterial blood was sampled, and oxygen and acid/base status were analyzed using a blood gas analyzer (i-Stat portable clinical analyzer, Heska, Loveland, CO).
Experimental protocol.
Following surgery and introduction of the pressure/volume catheter, animals were allowed to rest for 60 min to obtain a stable steady-state condition before hemodynamic measurements. At each temperature, hemodynamic functional variables were recorded. After experiments were completed, hearts were removed through a thoracotomy, freeze clamped in liquid N2, and stored at −80°C for subsequent analyses of myofilament TnI phosphorylation.
Western blot analysis.
Western immunoblotting techniques used for detection and estimation of relative amounts of phospho-TnI and total TnI have been previously described in detail (9–11). Briefly, cardiac homogenates (25 µg of protein) were first subjected to SDS-PAGE in 4–15% gels, and fractionated proteins were transblotted to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Membranes were blocked for 1 h with 5% milk in Tris-buffered saline containing 0.1% Tween, incubated with the appropriate primary antibody overnight at 4°C, washed with Tris-buffered saline containing 0.1% Tween, detected with horseradish peroxidase-conjugated secondary antibodies, and expressed with Supersignal West Pico Chemiluminescent Substrate (Pierce Chemical, Rockford, IL) and then analyzed using a Kodak Imaging System.
Chemicals.
Reagents for electrophoresis were obtained from Bio-Rad; anti-phospho (Ser23/24)-TnI polyclonal antibody was from Cell Signaling; anti-TnI monoclonal antibody (Clone 5) antibody was from Fitzgerald. All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Statistical analysis.
Hemodynamic data were assessed by two-way ANOVA for repeated measurements. If the F value was greater than critical, further analysis was performed using a Holm-Sidak method for multiple comparisons including comparisons to prehypothermic baseline values for each group and comparisons of values at corresponding temperatures between groups. Differences were considered to be significant at P < 0.05.
RESULTS
There were no significant differences in body weight between wild-type (36 ± 4 g, n = 10) and TG-ssTnI mice (40 ± 5 g, n = 10). Stability of the model with respect to hemodynamic function was confirmed with no significant changes across the 4 h period at 37°C in time-matched control groups (wild-type and TG-ssTnI; n = 3 each). These animals underwent identical anesthesia and instrumentation as those animals subjected to the H/R protocol.
In the wild-type group, two animals died during the last phase of rewarming due to falling SV and an abrupt drop in blood pressure at temperatures between 30 and 37°C. Data from these animals were not included. In surviving animals, except for single ectopic ventricular beats, there were no episodes of other types of arrhythmias during H/R.
Prehypothermic baseline hemodynamic values.
In TG-ssTnI mice, baseline normothermic measures of diastolic function, dP/dtmin, and EDPVR revealed a reduced diastolic function compared with wild-type mice (−dP/dtmin: wild-type −7,591 ± 1,216 vs. TG-ssTnI −5,358 ± 433 mmHg/s; and EDPVR: wild-type 0.03 ± 0.01 vs. TG-ssTnI 0.07 ± 0.02; Table 1, and Figs. 1 and 2). At baseline, LVEDV was significantly lower in TG-ssTnI compared with wild-type animals.
Table 1.
Hemodynamic function of wild-type and TG-ssTnI mice before cooling and after rewarming
| Variable | Prehypothermic | Posthypothermic |
|---|---|---|
| HR, beats/min | 472 ± 34 487 ± 24 |
541 ± 47† 597 ± 27† |
| LVESP, mmHg | 93 ± 8 87 ± 4 |
88 ± 11 104 ± 5 |
| LVESV, µl | 47 ± 1 43 ± 3 |
46 ± 2 40 ± 1* |
| LVEDP, mmHg | 6 ± 1 5 ± 0.5 |
6 ± 0.8 9 ± 1.6† |
| LVEDV, µl | 63 ± 2 57 ± 2* |
58 ± 2 55 ± 2 |
| SV, µl | 15.6 ± 0.8 14.0 ± 1.2 |
12.2 ± 0.7 14.8 ± 0.6 |
| CO, ml/min | 7.4 ± 0.7 6.9 ± 0.8 |
6.6 ± 0.7 8.8 ± 0.7†* |
| SW, mmHg/ml | 1.1 ± 0.1 1.0 ± 0.1 |
0.8 ± 0.1† 1.2 ± 0.1* |
| dP/dtmax, mmHg/s |
8,444 ± 1,631 9,705 ± 1,277 |
8,485 ± 2,569 12,983 ± 454†* |
| PRSW, mmHg | 78 ± 7 62 ± 7 |
38 ± 5† 79 ± 8†* |
| V0, µl | 25.6 ± 3.6 20.0 ± 3.9 |
23.0 ± 4.1 23.3 ± 3.3 |
| τ (W), ms | 7.0 ± 0.5 8.6 ± 0.4 |
7.2 ± 0.6 8.6 ± 0.9 |
| τ (G), ms | 11.6 ± 1.0 14.8 ± 1.0 |
10.8 ± 1.3 13.0 ± 1.1 |
| dP/dtmin, mmHg/s | −7,591 ± 1,216 −5,358 ± 433* |
−7,672 ± 1,715 −6,896 ± 778 |
| EDPVR | 0.03 ± 0.01 0.07 ± 0.02* |
0.03 ± 0.02 0.06 ± 0.05* |
Values are means ± SE (n = 7 female mice, both groups). Values in boldface are data from transgenic mice expressing slow skeletal troponin I (TG-ssTnI). HR, heart rate; LVESP, left ventricular end-systolic pressure; LVESV, end-diastolic volume; LVEDP, end-diastolic pressure; LVEDV, end-diastolic volume; SV, stroke volume; CO, cardiac output; TPR, total peripheral resistance MAP/CO; SW, stroke work; dP/dtmax, maximum rate of left ventricular pressure rise; PRSW, preload recruited stroke work (slope of stroke work – EDV relationship); V0, volume axis intercept; τ (W), relaxation time constant calculated by Weiss method (regression of log pressure); τ (G), relaxation time constant calculated by Glantz method (regression of dP/dtmin vs. pressure); EDPVR, end-diastolic pressure-volume relationship. Results were analyzed by 2-way ANOVA for repeated measurements. If the F value was greater than critical, further analysis was performed using the Holm-Sidak method for multiple comparisons including comparisons to prehypothermic baseline values for each group and comparisons of values at corresponding temperatures between groups.
P < 0.05 vs. prehypothermic baseline value within group;
P < 0.05 comparing values at corresponding temperatures between TG-ssTnI and WT groups.
Fig. 1.

Heart rate, cardiac output, maximum rate of left ventricular (LV) pressure rise (dP/dtmax), and stroke work expressed as a function of temperature during cooling, 3 h at 25–23°C, and rewarming in transgenic mice expressing slow skeletal troponin I (TG-ssTnI) and wild-type mice. Values are means ± SE (each group, n = 7 female mice). Data are presented as median and 25th and 75th percentiles box plot, 10th and 90th percentiles whisker (each group, n = 7 female mice). †P < 0.05 vs. prehypothermic baseline value within group (Holm-Sidak method); *P < 0.05 comparing values at corresponding temperatures between TG-ssTnI and wild-type groups (Holm-Sidak method).
Fig. 2.

Changes in left ventricular (LV) stroke volume (SV) during hypothermia/rewarming as the difference between end-diastolic and end-systolic volume (SV = EDV – ESV) in wild-type mice (A) and transgenic mice expressing slow skeletal troponin-I (TG-ssTnI; B) groups at each temperature. Values are means ± SE (each group, n = 7 female mice). †P < 0.05 vs. prehypothermic baseline value within group (Holm-Sidak method); *P < 0.05 comparing values at corresponding temperature between TG-ssTnI and wild-type groups (Holm-Sidak method).
Cooling and stable hypothermia (3 h at 25.22°C).
When compared with baseline prehypothermic conditions (37°C), cooling to 25°C (over a 30-min period), maintained hypothermia (25–23°C for 3 h), and rewarming to 37°C (over a 30-min period), induced dynamic changes in LV volumes in both groups (Figs. 1 and 2). Already at 34°C during cooling, LVESV was reduced to a greater extent in TG-ssTnI compared with wild-type mice, leading to a significant increase in SV compared with baseline between 34 and 28°C, and after 1 h at 23°C in the TG-ssTnI group. Furthermore, these changes were accompanied by a significant increase in CO, dP/dtmax, and SW at the same temperatures in TG-ssTnI animals. No significant elevation in any of these variables took place in wild-type animals at the same temperatures. A decrease in respiratory rate was also observed during cooling, but no differences in respiratory rates were observed between groups at any temperature.
Rewarming.
During rewarming, heart rate returned to prehypothermic control levels at 32°C in wild-type mice and at 28°C in TG-ssTnI animals, whereas in both groups heart rate was significantly increased immediately after rewarming (Table 1 and Figs. 1 and 2). In TG-ssTnI animals, CO was significantly elevated compared with prehypothermic levels during rewarming (34–37°C), such that CO was significantly higher after rewarming compared with CO in wild-type animals. Simultaneously, in TG-ssTnI animals, both dP/dtmax, PRSW (Fig. 3), and SW were significantly elevated compared with wild-type animals at 37°C. In wild-type animals, there was a significant reduction in SW and PRSW after rewarming, with no change in dP/dtmax.
Fig. 3.
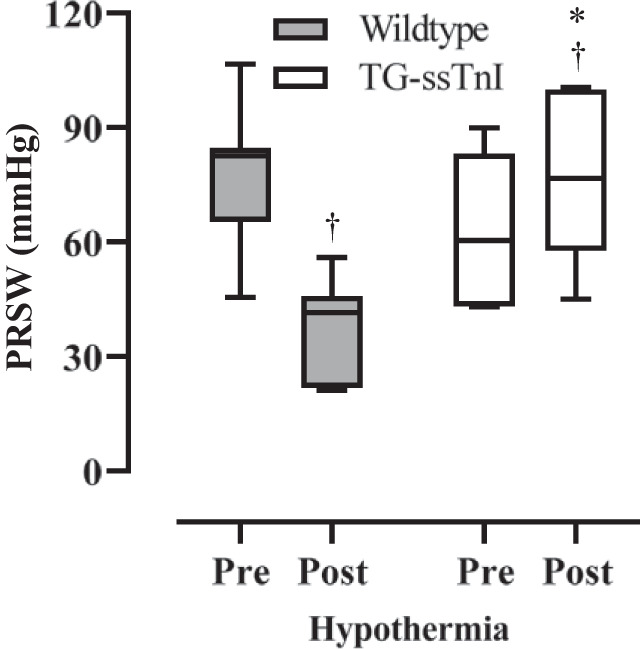
Changes in left ventricular preload recruited stroke work (PRSW) in wild-type mice and transgenic mice expressing slow skeletal troponin I (TG-ssTnI). Data are presented as median and 25th and 75th percentiles box plot, 10th and 90th percentiles whisker (each group, n = 7 female mice). †P < 0.05 comparing prehypothermic baseline value with posthypothermic value within group; *P < 0.05 comparing corresponding values in TG-ssTnI and wild-type groups (Holm-Sidak method).
The LV catheter was backed out into the ascending aorta at each temperature to confirm stable aortic pressure. Differences between peak systolic pressure in the aorta and LV end-systolic pressure (LVESP) never exceeded ± 7%. No within- or intergroup differences in LVESP were found (Table 1) throughout the H/R protocol, confirming that LV afterload was comparable between groups.
dP/dtmin and EDPVR were significantly higher in wild-type than in TG-ssTnI mice. After rewarming, the diastolic functional variable EDPVR was still higher in wild-type animals, whereas the difference in −dP/dtmin between groups no longer existed, which may have been due to the significant increase in heart rate that occurred in both groups.
Blood gases and lactate level.
No significant differences in arterial blood gases or lactate levels were found between time-matched (4 h) normothermic control, TG-ssTnI, and wild-type mice after rewarming (Table 2). These values were all within physiological range for anesthetized mice (16a, 32).
Table 2.
Arterial blood gases and lactate measured after rewarming in TG-ssTnI (TG) and WT mice, and after 4 h in normothermic time-matched controls
| TG | WT | Control | |
|---|---|---|---|
| n | 6 | 7 | 6 |
| pH | 7.31 ± 0.06 | 7.31 ± 0.02 | 7.27 ± 0.02 |
| Pco2, mmHg | 28.9 ± 10.1 | 30.7 ± 3.6 | 38.6 ± 3.3 |
| Po2, mmHg | 142.3 ± 30.1 | 149.6 ± 32.1 | 106.8 ± 27.9 |
| , mmol/L | 14.4 ± 3.2 | 14.9 ± 1.2 | 18.3 ± 1.3 |
| BE, mmol/L | −11.7 ± 2.7 | −11.5 ± 1.0 | −8.5 ± 1.4 |
| Sat O2, % | 99.5 ± 1.3 | 99.6 ± 0.8 | 98 ± 1 |
| Lactate, mmol/L | 2.23 ± 0.30 | 1.99 ± 0.21 | 1.65 ± 0.20 |
Values are means ± SE. WT, wild type; TG-ssTnI, transgenic mice expressing slow skeletal troponin I; BE, base excess; Sat, saturated.
TnI phosphorylation.
The extent of TnI phosphorylation was analyzed in cardiac homogenates from wild-type and TG-ssTnI hearts subjected to H/R. Following H/R, the extent of cTnI phosphorylation at Ser23/24 increased significantly in wild-type hearts compared with time-matched control hearts. In TG-ssTnI mice exposed to H/R and in time-matched controls, no phosphorylation of ssTnI was detected (Fig. 4).
Fig. 4.
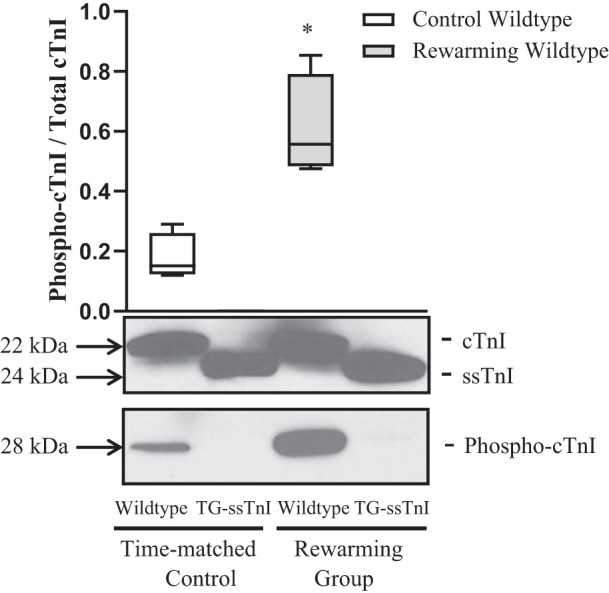
Phosphorylation status of troponin I (TnI) in experimental groups. Cardiac homogenate preparations obtained from wild-type mice and transgenic mice expressing slow skeletal TnI (TG-ssTnI) subjected to hypothermia/rewarming showing significant expression of TnI when probed with clone 5 of TnI-specific antibody. TnI showed a significantly increased level of phosphorylation at Ser23/24 in wild-type rewarmed hearts vs. time-matched control. We observed no phosphorylation of ssTnI under these experimental conditions. Values are means ± SE (each group, n = 6). Results were analyzed by unpaired 2-tailed Student’s t test. *P < 0.05 vs. control wild-type.
DISCUSSION
The results of the present study demonstrate that H/R-induced cardiac dysfunction (rewarming shock) depends on TnI phosphorylation, most likely phosphorylation of cTnI at Ser23/24. Complete restoration of cardiac function was observed after rewarming in TG-ssTnI mice, which completely lack the Ser23/24 phosphorylatable site on TnI. In contrast, wild-type mice expressing cTnI showed significant reduction of LV contractility after rewarming in concert with increased phosphorylation of cTnI at Ser23/24. After rewarming, diastolic function returned to prehypothermic baseline levels in both animal groups.
Differences between groups in response to cooling became distinct already at 34°C (Fig. 2) as LVESV was significantly reduced in TG-ssTnI mice, leading to a significant increase in SV, which persisted throughout the period of stable hypothermia accompanied by a significant elevation of CO, dP/dtmax and SW. In contrast, wild-type mice showed no change in LVESV, SV, CO, dP/dtmax, or SW in response to cooling. No within- or intergroup differences in LVESP were found (Table 1) throughout the H/R protocol, confirming that LV afterload was comparable between groups.
It is known that temperature change is one of the major factors that alter cardiac myofilament Ca2+ sensitivity (8). Cooling was reported to diminish Ca2+ sensitivity of TnC in chemically skinned rabbit ventricular muscle, but in skeletal muscle, exhibiting the ssTnI, Ca2+ sensitivity of TnC is increased by cooling (8, 27–29). These results indicate a thermodynamic difference between cardiac and skeletal muscle regulation at the level of TnC. The underlying mechanism for the increase in Ca2+ sensitivity of TnC in skeletal muscle is not fully understood, but the increase in Ca2+ sensitivity and contractile force of skeletal muscle ssTn induced by cooling was reported to be completely reversed by rewarming (8). Therefore, in contrast to the effects of cooling and rewarming in TG-ssTnI mice, the hypothermia-induced decrease in Ca2+ sensitivity of cTnC in wild-type mice was not reversed by rewarming and likely contributed to the posthypothermic reduction in myocardial contractility.
A role of cTnI phosphorylation at Ser23/24 in mediating H/R-induced cardiac dysfunction is supported by previous studies from our group using rat papillary muscle or isolated cardiomyocytes (9, 10, 23). In those studies, we found that H/R-induced reductions in force/contractility (sarcomere length shortening) and myofilament Ca2+ sensitivity are associated with increased PKA-specific phosphorylations of cTnI. Importantly, in the present study, we found that CO, SW, and indexes of cardiac contractility (dP/dtmax and PRSW) were all significantly reduced in wild-type mice after rewarming in conjunction with cTnI phosphorylation compared with TG-ssTnI mice in which ssTnI was not phosphorylated. Thus, TG-ssTnI mice tolerate H/R better than wild-type mice. The significant increase in the cardiac contractility index dP/dtmax in TG-ssTnI mice during cooling may be related to hypothermia-dependent elevation of Ca2+ sensitivity. However, any increase in Ca2+ sensitivity in TG-ssTnI mice was apparently reversed by rewarming.
Previous H/R experiments from our group had documented that by applying levosimendan, a myofilament Ca2+ sensitizer used to support LV cardiac contractility, hypothermia-induced cardiac dysfunction was abolished. Importantly, if given before rewarming, levosimendan elevates dP/dtmax and PRSW in concert with a significant increase in SV and CO (3). These findings are consistent with the results of the present study indicating that the lower tolerance of wild-type hearts to H/R is related to reduced Ca2+ sensitivity due to cTnI phosphorylation.
The rate of cardiac relaxation is influenced by both myofilament properties and sequestration of cytosolic Ca2+. In the present study, both dP/dtmin and EDPVR were significantly lower in TG-ssTnI compared with wild-type mice, indicating differences in diastolic function. After rewarming, the difference in EDPVR between groups remained, whereas the difference in dP/dtmin was not present. However, the elevated dP/dtmin after rewarming can be ascribed to the significant increase in heart rate in both groups, which influences this diastolic variable.
In cardiac muscle, the ssTnI isoform is expressed during embryonic life and later replaced by cTnI expression during the early postnatal period. As a consequence, myofilaments of neonatal hearts expressing ssTnI are more sensitive to cytosolic Ca2+ than the adult heart expressing cTnI. In this respect, it has long been known that newborn mammals display greater cardiac tolerance to hypothermia than adults (4, 5).
In the present study, the level and duration of the hypothermic exposure was relatively modest compared with temperature interventions in previous studies (10, 14, 20, 21, 35). The 23–25°C temperature was selected to maintain spontaneous circulation and respiration throughout experiment. In pilot experiments, we found that spontaneous cardiorespiratory function in mice halted at temperatures below 20°C. The fact that two wild-type mice died due to circulatory collapse during the late phase of rewarming indicated that the actual temperature exposure protocol used in this study was adequate for testing temperature tolerance between groups.
In conclusion, by comparing the effects of H/R on LV contractile function between wild-type and TG-ssTnI mice, we showed that the absence of the PKA-specific cTnI phosphorylation prevents rewarming shock and reduced cardiac contractility. Thus, treatment of accidental hypothermia patients should target improvement of cardiac contractility during rewarming via mechanisms that avoid increase of PKA activity (and cTnI phosphorylation) while increasing Ca2+ sensitivity of cTnC.
GRANTS
This study was supported by the Mayo Foundation and Norwegian Air Ambulance Foundation (Y. S. Han).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.T., G.M.A., and G.C.S. conceived and designed research; T.T. performed experiments; T.T. analyzed data; T.T., G.M.A., and G.C.S. interpreted results of experiments; T.T. and Y.-S.H. prepared figures; T.T. and G.M.A. drafted manuscript; T.T., Y.-S.H., and G.C.S. edited and revised manuscript; T.T., G.M.A., Y.-S.H., and G.C.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to Dr. Solaro for providing us with the knockout mice and to Dr. Venkatachalem for assistance with biochemical measurements.
REFERENCES
- 1.Blair E, Montgomery AV, Swan H. Posthypothermic circulatory failure. I. Physiologic observations on the circulation. Circulation 13: 909–915, 1956. doi: 10.1161/01.CIR.13.6.909. [DOI] [PubMed] [Google Scholar]
- 2.Connolly E, Worthley LI. Induced and accidental hypothermia. Crit Care Resusc 2: 22–29, 2000. [PubMed] [Google Scholar]
- 3.Dietrichs ES, Håheim B, Kondratiev T, Sieck GC, Tveita T. Cardiovascular effects of levosimendan during rewarming from hypothermia in rat. Cryobiology 69: 402–410, 2014. doi: 10.1016/j.cryobiol.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Edwards W. Influence des Agnes Physiques Sur La Vie. Paris: Crochard, 1824, p. 650. [Google Scholar]
- 5.Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol 517: 143–157, 1999. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glower DD, Spratt JA, Kabas JS, Davis JW, Rankin JS. Quantification of regional myocardial dysfunction after acute ischemic injury. Am J Physiol 255: H85–H93, 1988. doi: 10.1152/ajpheart.1988.255.1.H85. [DOI] [PubMed] [Google Scholar]
- 7.Glower DD, Spratt JA, Snow ND, Kabas JS, Davis JW, Olsen CO, Tyson GS, Sabiston DC Jr, Rankin JS. Linearity of the Frank-Starling relationship in the intact heart: the concept of preload recruitable stroke work. Circulation 71: 994–1009, 1985. doi: 10.1161/01.CIR.71.5.994. [DOI] [PubMed] [Google Scholar]
- 8.Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J Gen Physiol 80: 279–297, 1982. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han YS, Schaible N, Tveita T, Sieck G. Discontinued stimulation of cardiomyocytes provides protection against hypothermia-rewarming-induced disruption of excitation-contraction coupling. Exp Physiol 103: 819–826, 2018. doi: 10.1113/EP086774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han YS, Tveita T, Kondratiev TV, Prakash YS, Sieck GC. Changes in cardiovascular beta-adrenoceptor responses during hypothermia. Cryobiology 57: 246–250, 2008. doi: 10.1016/j.cryobiol.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Han YS, Tveita T, Prakash YS, Sieck GC. Mechanisms underlying hypothermia-induced cardiac contractile dysfunction. Am J Physiol Heart Circ Physiol 298: H890–H897, 2010. doi: 10.1152/ajpheart.00805.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res 88: 1059–1065, 2001. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- 13.Kirkpatrick AW, Chun R, Brown R, Simons RK. Hypothermia and the trauma patient. Can J Surg 42: 333–343, 1999. [PMC free article] [PubMed] [Google Scholar]
- 14.Kondratiev TV, Flemming K, Myhre ES, Sovershaev MA, Tveita T. Is oxygen supply a limiting factor for survival during rewarming from profound hypothermia? Am J Physiol Heart Circ Physiol 291: H441–H450, 2006. doi: 10.1152/ajpheart.01229.2005. [DOI] [PubMed] [Google Scholar]
- 15.Maclean D. Emergency management of accidental hypothermia: a review. J R Soc Med 79: 528–531, 1986. doi: 10.1177/014107688607900909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacLean D, Emslie-Smith D. Accidental Hypothermia. Melbourne, Australia: Blackwell Scientific, 1977. [Google Scholar]
- 16a.Meek TH, Lonquich BP, Hannon RM, Garland T Jr. Endurance capacity of mice selectively bred for high voluntary wheel running. J Exp Biol 212: 2908–2917, 2009. doi: 10.1242/jeb.028886. [DOI] [PubMed] [Google Scholar]
- 17.Mégarbane B, Axler O, Chary I, Pompier R, Brivet FG. Hypothermia with indoor occurrence is associated with a worse outcome. Intensive Care Med 26: 1843–1849, 2000. doi: 10.1007/s001340000702. [DOI] [PubMed] [Google Scholar]
- 18.Mizushima Y, Wang P, Cioffi WG, Bland KI, Chaudry IH. Restoration of body temperature to normothermia during resuscitation following trauma-hemorrhage improves the depressed cardiovascular and hepatocellular functions. Arch Surg 135: 175–181, 2000. doi: 10.1001/archsurg.135.2.175. [DOI] [PubMed] [Google Scholar]
- 19.Nielsen JM, Kristiansen SB, Ringgaard S, Nielsen TT, Flyvbjerg A, Redington AN, Bøtker HE. Left ventricular volume measurement in mice by conductance catheter: evaluation and optimization of calibration. Am J Physiol Heart Circ Physiol 293: H534–H540, 2007. doi: 10.1152/ajpheart.01268.2006. [DOI] [PubMed] [Google Scholar]
- 20.Popovic V. Physiological characteristics of rats and ground squirrels during prolonged lethargic hypothermia. Am J Physiol 199: 467–471, 1960. doi: 10.1152/ajplegacy.1960.199.3.467. [DOI] [PubMed] [Google Scholar]
- 21.Popovic V. Survival time of hypothermic white rats (15°C) and ground squirrels (10°C). Am J Physiol 199: 463–466, 1960. doi: 10.1152/ajplegacy.1960.199.3.463. [DOI] [PubMed] [Google Scholar]
- 22.Roggela M, Wagner A, Hoedl W, Michalek A, Roeggla G. Successful resuscitation of a child with severe hypothermia after cardiac arrest. Prehosp Disaster Med 10: 286–287, 1995. doi: 10.1017/S1049023X00052250. [DOI] [PubMed] [Google Scholar]
- 23.Schaible N, Han YS, Hoang T, Arteaga G, Tveita T, Sieck G. Hypothermia/rewarming disrupts excitation-contraction coupling in cardiomyocytes. Am J Physiol Heart Circ Physiol 310: H1533–H1540, 2016. doi: 10.1152/ajpheart.00840.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solaro RJ, Rarick HM. Troponin and tropomyosin: proteins that switch on and tune in the activity of cardiac myofilaments. Circ Res 83: 471–480, 1998. doi: 10.1161/01.RES.83.5.471. [DOI] [PubMed] [Google Scholar]
- 25.Solaro RJ, Rosevear P, Kobayashi T. The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochem Biophys Res Commun 369: 82–87, 2008. doi: 10.1016/j.bbrc.2007.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solaro RJ, Sheehan KA, Lei M, Ke Y. The curious role of sarcomeric proteins in control of diverse processes in cardiac myocytes. J Gen Physiol 136: 13–19, 2010. doi: 10.1085/jgp.201010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephenson DG, Wendt IR. Length dependence of changes in sarcoplasmic calcium concentration and myofibrillar calcium sensitivity in striated muscle fibres. J Muscle Res Cell Motil 5: 243–272, 1984. doi: 10.1007/BF00713107. [DOI] [PubMed] [Google Scholar]
- 28.Stephenson DG, Williams DA. Calcium-activated force responses in fast- and slow-twitch skinned muscle fibres of the rat at different temperatures. J Physiol 317: 281–302, 1981. doi: 10.1113/jphysiol.1981.sp013825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stephenson DG, Williams DA. Temperature-dependent calcium sensitivity changes in skinned muscle fibres of rat and toad. J Physiol 360: 1–12, 1985. doi: 10.1113/jphysiol.1985.sp015600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sumandea MP, Burkart EM, Kobayashi T, De Tombe PP, Solaro RJ. Molecular and integrated biology of thin filament protein phosphorylation in heart muscle. Ann N Y Acad Sci 1015: 39–52, 2004. doi: 10.1196/annals.1302.004. [DOI] [PubMed] [Google Scholar]
- 32.Thal SC, Plesnila N. Non-invasive intraoperative monitoring of blood pressure and arterial pCO2 during surgical anesthesia in mice. J Neurosci Methods 159: 261–267, 2007. doi: 10.1016/j.jneumeth.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 33.Tisherman SA. Hypothermia and injury. Curr Opin Crit Care 10: 512–519, 2004. doi: 10.1097/01.ccx.0000145096.28446.f7. [DOI] [PubMed] [Google Scholar]
- 34.Tveita T, Ytrehus K, Myhre ES, Hevrøy O. Left ventricular dysfunction following rewarming from experimental hypothermia. J Appl Physiol (1985) 85: 2135–2139, 1998. doi: 10.1152/jappl.1998.85.6.2135. [DOI] [PubMed] [Google Scholar]
- 35.Tveita T, Ytrehus K, Skandfer M, Oian P, Helset E, Myhre ES, Larsen TS. Changes in blood flow distribution and capillary function after deep hypothermia in rat. Can J Physiol Pharmacol 74: 376–381, 1996. doi: 10.1139/y96-028. [DOI] [PubMed] [Google Scholar]
- 36.Vassal T, Benoit-Gonin B, Carrat F, Guidet B, Maury E, Offenstadt G. Severe accidental hypothermia treated in an ICU: prognosis and outcome. Chest 120: 1998–2003, 2001. doi: 10.1378/chest.120.6.1998. [DOI] [PubMed] [Google Scholar]
- 37.Yasuda S, Coutu P, Sadayappan S, Robbins J, Metzger JM. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ Res 101: 377–386, 2007. doi: 10.1161/CIRCRESAHA.106.145557. [DOI] [PubMed] [Google Scholar]