

Abstract
Objectives
We aimed to highlight the utility of novel dissolving microneedle (MN)-based delivery systems for enhanced transdermal protein delivery. Vaccination remains the most accepted and effective approach in offering protection from infectious diseases. In recent years, much interest has focused on the possibility of using minimally invasive MN technologies to replace conventional hypodermic vaccine injections.
Methods
The focus of this study was exploitation of dissolving MN array devices fabricated from 20% w/w poly(methyl vinyl ether/maleic acid) using a micromoulding technique, for the facilitated delivery of a model antigen, ovalbumin (OVA).
Key findings
A series of in-vitro and in-vivo experiments were designed to demonstrate that MN arrays loaded with OVA penetrated the stratum corneum and delivered their payload systemically. The latter was evidenced by the activation of both humoral and cellular inflammatory responses in mice, indicated by the production of immunoglobulins (IgG, IgG1, IgG2a) and inflammatory cytokines, specifically interferon-gamma and interleukin-4. Importantly, the structural integrity of the OVA following incorporation into the MN arrays was maintained.
Conclusion
While enhanced manufacturing strategies are required to improve delivery efficiency and reduce waste, dissolving MN are a promising candidate for ‘reduced-risk’ vaccination and protein delivery strategies.
Keywords: laser-engineering, microneedles, protein delivery, vaccination
Introduction
Microneedle (MN) arrays are micron scale, minimally invasive devices that painlessly bypass the skin's stratum corneum, which is the principal barrier to topically applied drugs. MN arrays have been extensively investigated in recent years as a means to enhance transdermal drug and vaccine delivery. The current trend in MN-based research has involved recognition of the dubious biocompatibility of silicon and the potential for inappropriate reuse of silicon or metal MNs, which remain fully intact after removal from a patient's skin. Consequently, much recent effort has focused on MN arrays prepared from drug-loaded gels of FDA-approved biocompatible polymers. Such systems typically dissolve in skin interstitial fluid to release their drug payload.[1]
Dissolving MN arrays have been shown to enhance transdermal and intradermal delivery of numerous substances, including insulin,[2,3] 5-aminolevulinic acid,[4] sulforhodamine B,[5] low molecular weight heparin,[6] ovalbumin (OVA),[7,8] adenovirus vector[8] and a variety of vaccine antigens.[9,10,11,12]
We have previously described dissolving polymeric MN arrays prepared using an innovative laser engineering method for enhanced transdermal and intradermal delivery of low molecular weight drugs,[6] peptide-based therapeutics,[13] as well as nanoparticles.[14,15] In the this study, we investigated, for the first time, the feasibility of loading these novel systems with a model protein and the subsequent use of the formed MN arrays in in-vivo delivery and vaccination experiments. The ultimate aim was to study protein delivery from our dissolving MN arrays and to make conclusions as to their potential real-world utility in therapeutics and preventative healthcare strategies. Once fully optimised and manufactured to standards of good manufacturing practice, such technology could prove extremely effective in minimising the risk associated with vaccination with conventional hypodermic needles, particularly in the developing world, since the MN are self-disabling. By dissolving in skin, accidental or intentional reuse is prevented and specialist sharps disposal is not necessary.
Materials and Methods
Chemicals
Gantrez AN-139, a copolymer of methylvinylether and maleic anhydride (PMVE/MA), was provided by Ashland, Kidderminster, UK. Isoflurane inhalation anaesthetic was obtained from Abbott Laboratories Ltd, Kent, UK. Chicken OVA, mouse monoclonal anti-chicken OVA antibody, fish gelatine tetramethylbenzidine substrate (TMP), phosphate-buffered saline (PBS) pH 7.3, Tween 20, diaminobenzidine and concanavalin A (Con A) from Canavalia ensiformis were all purchased from Sigma-Aldrich, St. Louis, MO, USA. Imiquimod (IMI) was purchased from Topharman Ltd, Shanghai, China. Fluorescein isothiocyanate-labelled OVA was purchased from Bioresearch, Redmond, WA, USA. Rabbit anti-OVA horseradish peroxidase (HRP)-conjugated polyclonal antibody was purchased from Gene Tex Inc Alton Pkwy, Irvine, CA, USA. SuperBlock T20 was purchased from Thermo Scientific, Rockford, IL, USA. Hyamine was purchased from JT Baker, Phillipsburg, NJ, USA. The mouse anti-OVA immunoglobulin ELISA kits (total IgG, IgG1 and IgG2a) were obtained from Alpha Diagnostic International, San Antonio, TX, USA. The interleukin-4 (IL-4) and interferon-gamma (IFN-γ) ELISA kits were purchased from Abcam, Cambridge, UK. Roswell Park Memorial Institute medium 1640 (RPMI) medium was obtained from Life Technology Ltd, Paisley, UK. All other chemicals used were of analytical reagent grade.
Fabrication of OVA-loaded dissolving MN arrays
Dissolving MN arrays were prepared from aqueous blends of 20% w/w PMVE/MA loaded with OVA by using laser-engineered silicone micromould templates, as described previously.[16] MN arrays devices used in this study consisted of 0.5 cm2 19 × 19 arrays with needle lengths of 600 μm, base width of 300 μm and a base-to-base distance of 50 μm.
Aqueous blends (500 mg) were loaded with 2.5 mg OVA, 2.5 mg fluorescently labelled OVA (FITC-OVA) or 2.5 mg of both OVA and IMI. Notably, we did not adjust the pH of the blends, which were approximately 2.4. The formulations were poured into the silicone micromoulds, centrifuged for 15 min at 1372 g (Jouan C312 laboratory centrifuge; DJB Labcare, Buckinghamshire, UK) and dried at ambient temperature for 24 h. FITC-OVA-loaded MN arrays were kept out of direct sunlight. The ‘sidewalls’ formed upon drying of the MN arrays were removed using a heated scalpel blade, as depicted in (Figure 1a).
Figure 1.
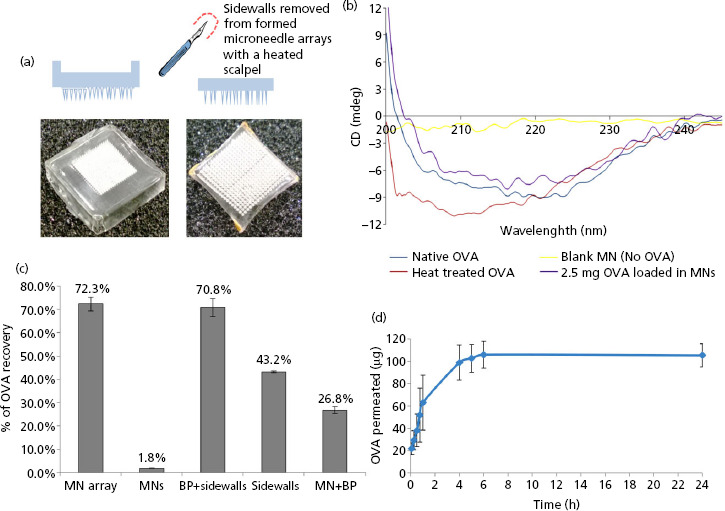
(a) Digital photograph image of a microneedle (MN) array with sidewalls (i) and (ii) after removal of the sidewalls using a hot scalpel blade. (b) Circular dichroism (CD) spectra of ovalbumin (OVA). OVA which had been encapsulated into and released from MN prepared from 20% w/v poly(methyl vinyl ether/maleic acid) (PMVE/MA) at loading of 2.5 mg/array (violet) and CD spectra of OVA standard solution (dark blue), heat-treated OVA solution (red) and non-OVA loaded MN (control, blank MN) (yellow) are presented here. (c) Determination of the loading capacity of the MN device and distribution of OVA within the MN array. MN arrays were loaded with OVA at a concentration of 2.5 mg OVA per device. The content of OVA was measured in either the whole MN array (MN array) or within isolated parts of the MN device, that is, the needles of the array only (MNs), the baseplate of the array including the sidewalls (BP + sidewalls), the sidewalls only (sidewalls) and the MN array including the baseplate but lacking the sidewalls (MN + BP). The results are expressed as the % mean ± SD, (n = 5 MN arrays) of the nominal OVA content recovered from the MN array. (d) Cumulative amount (μg) of OVA which permeated across dermatomed neonatal porcine skin after release from PMVE/MA MN arrays loaded with OVA at a concentration of 2.5 mg per array (means ± SD, n = 7).
Determination of OVA secondary structure by circular dichroism
Circular dichroism (CD) was employed to examine the secondary structure of OVA after incorporation into and subsequent release from dissolving MN arrays. CD spectra were obtained with a Jasco J-185 spectropolarimeter (Jasco, Easton, MD, USA) equipped with a temperature controller. CD spectra were collected at 25°C using a 1 cm quartz cell over the wavelength range of 200–400 nm. A resolution of 0.1 nm and scanning speed of 200 nm/min were employed. Samples for CD analysis were prepared by dissolving OVA-loaded MN arrays (n = 5) in PBS (pH 7.4) to an OVA concentration of 10 μg/ml. The MN arrays-derived OVA sample spectra were compared with that of native fresh OVA solution and heat treated OVA solution (OVA 10 μg/ml in PBS heated at 80°C for 2 h). Negative controls consisting of blank MN arrays dissolved in PBS were also employed in the study.
Determination of MN loading efficiency and distribution of protein within MN arrays
MN arrays with nominal loadings of 2.5 mg per device were fabricated as detailed above. The arrays (n = 3) were dissolved in PBS (pH 7.4) and the OVA content of the device was determined by the bicinchoninic acid (BCA) assay (Micro BCA Protein assay reagent kit, Thermo Scientific, Rockford, IL, USA) using spectroscopic detection at 562 nm (Powerwave XS, Bio-Tek Instruments Inc., Winooski, VT, USA). The amount of OVA in the MN array baseplates (the array devoid of needles), the sidewalls which are routinely removed from the arrays at the end of the fabrication process and the actual needles of the array were also determined. Needles, which had been cut off the array using a hot scalpel blade and baseplate alone, were both, independently, dissolved in PBS (pH 7.4) and protein content was deduced using the BCA assay.
In-vitro OVA permeation studies
Diffusion of OVA from polymeric MN arrays across neonatal porcine skin was investigated in vitro using modified Franz diffusion cells (FDC-400 flat flange, 15 mm orifice diameter, mounted on an Franz Diffusion Cell Driver (FDCD) diffusion drive console providing synchronous stirring at 600 rpm and thermostated at 37 ± 1°C; Crown Glass Co. Inc., Somerville, NJ, USA). Neonatal porcine skin is a good model for human skin in terms of hair sparseness and physical properties.[17,18] The skin was immediately (<24.0 h after birth) excised and trimmed to a thickness of 350 μm using an electric dermatome (Padgett Instruments, Plainsboro, NJ, USA). Skin samples were then covered with aluminium foil and stored at −20°C until further use. Neonatal porcine skin specimens were shaved carefully so as not to damage the skin and pre-equilibrated in PBS (pH 7.4) for 15 min before beginning the experiments. A circular specimen of the skin was secured to the donor compartment of the diffusion cell using cyanoacrylate glue (Loctite Ltd, Dublin, Ireland) with the stratum corneum side facing the donor compartment. This was then placed on top of dental wax, to give the skin support and MN arrays were inserted into the centre of the skin section, using a spring-activated applicator at a force of 11.0 N/array. A circular steel weight (diameter 11.0 mm, mass 3.5 g) was placed on top of each MN array so as to simulate the constant contact between skin and MN array which would be achieved in vivo. A non-adhesive putty material (BluTac, Bostik Ltd, Leicester, UK) was placed on top of the weight and a piece of laboratory film (Parafilm) placed over the Franz cell lid. With MN arrays thus held in place, donor compartments were mounted onto the receptor compartments of the Franz cells. The receptor medium was PBS, degassed before use by sonication. Using a long needle, samples (200 μl) were removed from the receptor compartment at defined time intervals (0.25, 0.5, 1, 2, 3, 4, 5, 6, 7 and 24 h respectively) and replaced with an equal volume of pre-warmed degassed PBS. The concentration of OVA in the receptor medium was determined using a sandwich ELISA methodology adapted from Rolland et al.[19] and optimised to improve sensitivity. Briefly, 50 μl aliquots of OVA monoclonal antibody solution (2.5 μg/ml in 0.1 M bicarbonate buffer, pH 9.6) were dispensed into the wells of a Costar 96 well flat-bottom EIA/RIA plate (Sigma-Aldrich) and incubated overnight at 4°C. Excess antibody was removed by washing the plate 5 times with Tween PBS 0.05% v/v. The blocking step was performed by incubation of the wheels with SuperBlock T20 buffer (150 μl/well) for 2 h at room temperature. OVA samples obtained from the in-vitro study, in addition to freshly prepared OVA standard solutions (concentration range: 0.001–1 μg/ml in PBS) were incubated in triplicate (50 μl/well) for 1 h at room temperature. Samples were then washed as outlined previously and were then incubated for 1 h, at room temperature with 50 μl of rabbit anti-OVA HRP-conjugated polyclonal antibody (5 μg/ml in SuperBlock T20 buffer). The plate was then washed as outlined to remove excess antibody and samples were incubated with the enzymatic substrate tetramethylbenzidine for 10 min (50 μl/well). The reaction was then stopped by the addition of 4.0 M HCl (50 μl/well). Released OVA was thus measured indirectly at 450 nm using a microplate spectrophotometer (Powerwave XS, Bio-Tek Instruments Inc.). Results were expressed as means ± SD of seven replicates.
Dissolution kinetics of dissolving MN arrays
The dissolution profile of the polymeric MN arrays was assessed in mice using optical coherence tomography (OCT) (Vivosight high-resolution OCT scanner, Michelson Diagnostics Ltd, Kent, UK), as described previously.[20] C57Bl/6 female mice were used in all in-vivo experiments. The animals were acclimatised to laboratory conditions for 7 days before the commencement of experimentation. Animals were anaesthetised following intraperitoneal injection of 150 mg/kg ketamine and 10 mg/kg xylazine 24 h before experimentation. OVA-loaded MN arrays were manually applied to the dorsal region of each mouse ear and maintained in place for 1 min by the application of firm thumb pressure. The treated areas, with the MN arrays secured in place using transparent adhesive tape (Sellotape, Sellotape Ltd, Cheshire, UK), were visualised at specific intervals (i.e. 1, 5, 15, 30 and 60 min respectively) by OCT. The swept-source Fourier domain OCT system has a laser centre wavelength of 1305 ± 15 nm, facilitating real-time high-resolution imaging of the upper skin layers (7.5 μm lateral and 10 μm vertical resolution). The skin was scanned at a frame rate of up to 15 B-scans (two-dimensional (2D) cross-sectional scans) per second (scan width = 2 mm). 2D images were converted into a three-dimensional representation using the imaging software ImageJ (National Institutes of Health, Bethesda, MD, USA). The scale of the image files obtained was 1.0 pixel = 4 μm, thus allowing accurate measurements to be made.
Approval for animal experiments was obtained from the Committee of the Biological Research Unit, Queen's University Belfast. The work was carried out under Project Licence PPL 2678 and Personal Licence PIL 1466. All animal experiments throughout this study were conducted according to the policy of the federation of European Laboratory Animal Science Associations and the European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes, with implementation of the principle of the 3Rs (replacement, reduction, refinement). This research also adhered to the ‘Principles of Laboratory Animal Care’ (NIH publication 85–23, revised in 1985).
Quantification of FITC-OVA delivered into mouse skin
Blank MN arrays (n = 4), FITC-OVA-loaded MN arrays (2.5 mg nominal loading) (n = 4) and FITC-OVA-loaded arrays (2.5 mg nominal loading) devoid of MN arrays (baseplate only) (n = 4) were manually applied to the dorsal region of mouse ears and held in situ for 1 min using gentle thumb pressure. The MN arrays were thereafter maintained in place for 24 h with the aid of medical tape (Micropore, 3M, Dublin, Ireland) before being removed. The animals were subsequently sacrificed by carbon dioxide and the ears excised. Each ear was cut into smaller portions, cleaned with 1.0% w/v sodium dodecyl sulphate solution to remove non-delivered FITC-OVA on the stratum corneum and then incubated in hyamine hydroxide aqueous solution consisting of a 1 : 9 mixture of deionised water and hyamine hydroxide solution (1.0 M in methanol). Sample solutions obtained were analysed for FITC-OVA content using a FLUOstar Optima calibrated spectrofluorimetry (BMG Labtech Ltd, Aylesbury, UK).
In-vivo evaluation of MN mediated transdermal delivery of OVA
This experiment was carried out to study the capability of OVA-loaded dissolving MN arrays to elicit immune responses in mice in comparison to inoculation via conventional hypodermic needle delivery. To investigate the effect of the adjuvant IMI on the immune responses induced, MN arrays encapsulating OVA with or without IMI were also employed. MN arrays employed in this study contained OVA at nominal loadings of 2.5 mg; OVA plus IMI, both at nominal loadings of 2.5 mg; IMI only at a nominal loading of 2.5 mg or blank MN arrays containing no OVA or IMI. Mice were divided into eight groups, each group containing four animals. Four groups would be immunised using MN arrays and four groups would be immunised by conventional intradermal injections (26 gauge needles, BD Plastipak, BD, Plymouth, UK). Animals were anaesthetised by intraperitoneal injection of 150 mg/kg ketamine and 10 mg/kg xylazine. Before MN array application, 10 μl of PBS was applied to the dorsal region of the mouse ear and spread thoroughly to initiate bioadhesion of the MN array. MN arrays were subsequently applied to the ears of four animals (two MN arrays per animal) at specific times with 2-week intervals between each application (day 0, 14, 28 and 56 respectively). Each MN array was inserted manually using thumb pressure for 1 min and was then maintained in situ over the course of the 24 h experimental period using medical tape (Micropore).
With regards to OVA administration using conventional needle and syringe, hair was removed from the lower back of the animal using an electric shaver, to facilitate visualisation of the hypodermic needle during injection. Each group of four animals was injected intradermally with 100 μl each of the following solutions: OVA 100 μg/100 μl, OVA plus IMI (each at a concentration of 100 μg/100 μl), IMI (100 μg/100 μl) and PBS as a negative control. The OVA dose injected intradermally was matched to that delivered by the MN over 24 h in the in-vitro skin permeation experiments (Figure 1d). As with the MN-mediated OVA delivery, conventional needle- and syringe-mediated OVA administration was repeated at specific time points (day 0, 14, 28 and 56 respectively) with 2-week intervals between administrations. The presence of a definite pale bleb at the injection site was used as an indication that the intradermal injection was performed correctly.
All the animals receiving the treatments (either via MN arrays or conventional needle and syringe) were observed every 2 weeks to monitor for changes in body weight. Blood samples were collected from the tail vein on day 14 and 47. Mice were sacrificed 2 weeks after the last treatment (day 56) using carbon dioxide gas and blood was immediately collected from the mice via heart puncture with a heparinised syringe into ethylenediaminetetraacetic acid (EDTA)-coated tube. Serum was extracted from blood samples following centrifugation at 10 000 g for 15 min and the samples obtained were stored at −80°C until required.
Detection of specific anti-OVA antibody subclasses (IgG, IgG1 and IgG2a) in serum
Sera obtained from immunised mice were analysed for anti-OVA-specific antibodies. In particular, total IgG, as well as specific IgG1 and IgG2a antibody subclasses were determined using commercially available ELISA kits (Alpha Diagnostic International). In each case, ELISA analysis was carried out according to the manufacturer's instructions. Antibody titres were determined by calibrated spectroscopy (Powerwave XS, Bio-Tek Instruments Inc.) at a wavelength of 450 nm.
Extraction of spleens from mice and preparation of splenocyte cell suspensions
Spleens were removed from the animals immediately after sacrifice and incubated in RPMI media supplemented with 10% foetal calf serum (FCS), 50 μM β-mercaptoethanol and 500 U/L penicillin/streptomycin under sterile conditions. Each spleen was positioned between two sheets of sterilised nylon filter paper and then grasped with forceps and carefully crushed. The resulting cell suspension was centrifuged at 400 g for 5 min at 4°C. The supernatant obtained was discarded and the pellet washed twice in PBS, pH 7.4. The splenocytes were suspended in red blood cell lysing buffer (155 mM ammonium chloride, 12 mM sodium bicarbonate, 0.1 M EDTA) and incubated for 5 min to eliminate erythrocytes before being re-suspended in RPMI to stop the reaction. The samples were centrifuged (400 g, 5 min at 4°C) and the pellets re-suspended in RPMI 1640. The cells were counted using a C-Chip disposable haemocytometer (Cronus Technologies, Surrey, UK). The splenocyte suspension was added to the wells of a 96-well round bottomed microtitre plate at a density of 2 × 105 cells/well together with test antigen (200 μg/ml OVA in a final volume of 200 μl per well). Negative controls (wells containing cell suspension without stimulation with OVA) and positive controls (wells containing cells stimulated with 5 μg/ml Con A in a final volume of 200 μl per well) were prepared. After incubation for 96 h at 37°C in a humidified atmosphere containing 5% carbon dioxide, the culture supernatants were collected for cytokine assays. The supernatants were frozen at −80°C until required for testing. IFN-γ and IL-4 levels in cultured splenocytes were determined using commercially available ELISA kits according to the manufacturer's instructions (AbCam).
Protection against OVA-induced airway inflammation in mice
This section of the study was carried out to evaluate the protection offered to mice by dissolving MN arrays loaded with OVA against anaphylactic shock and airway inflammation induced by the injection of high doses of OVA systemically. Comparisons were made with conventional intradermal administration of the same antigen. T helper 1 (Th1) and T helper 2 (Th2) biomarkers were determined before and after challenge to detect any modulation in the immune responses that occurred during the study.
Mice were divided into four groups containing five mice each. Three groups were immunised via MN arrays, while the final group contained non-immunised mice (i.e. no MN arrays applied). The applied MN arrays were again loaded with OVA at nominal loadings of 2.5 mg/array, or contained no OVA (blank MN arrays). MN arrays were applied to both ears of each mouse as described above and were removed after 24 h. Intradermal dosing by conventional injection with OVA (100 μg/100 μl) was also performed, as described above.
All four groups were immunised twice, with a 2-week interval, on days 0 and 14. On day 34, all mice were challenged by intraperitoneal injection of a solution of 2 mg OVA in 100 μl PBS (pH 7.4), except the untreated group, which was challenged intraperitoneally with 100 μl PBS (pH 7.4) only. Mice were observed for 1 h following challenge to detect any signs of shock. On day 35, mice were sacrificed and blood samples collected by heart puncture with a heparinised syringe into EDTA-coated tubes. The blood samples were centrifuged for 15 min at 10 000 g to obtain cell-free serum. The sera were stored at −80°C until required for use. The titre of anti-OVA-specific antibodies was determined by ELISA, as described previously. The spleens of four mice from each group were removed immediately following sacrifice and were kept in cold RPMI containing 10% FCS, 50 μM β-mercaptoethanol and 500 U/L penicillin/streptomycin for tissue culture production according to the protocol described above. The samples were subsequently analysed for IL-4 quantities by ELISA methodology, as before.
Collection of bronchoalveolar lavage (BAL) fluid
In the same experiment, before sacrifice, BAL fluid was collected from three mice, derived from each of the four groups outlined in the previous section, under anaesthesia. The fluid was collected by lung lavage with 900 μl PBS (pH 7.4) via the trachea using a sterile catheter. After three lavages, approximately 700 μl of BAL fluid was recovered and centrifuged for 10 min at 400 g and 4°C. The supernatant was stored at −80°C for the measurement of IL-4 cytokines by ELISA analysis as previously described.
Statistical analysis
All results were expressed as the mean and the associated standard deviation. Where appropriate, the Kruskal–Wallis test was used to compare different groups. Individual differences between treatments were identified using Dunn's test. In all cases, P < 0.05 denoted significance.
Results and Discussion
Determination of OVA secondary structure by CD
Any conformational changes in the OVA secondary structure upon encapsulation within the PMVE/MA matrix were investigated using CD spectroscopy. The results of the CD determination for various OVA preparations are presented in Figure 1b. The CD spectrum of OVA standard solution is depicted, showing two strong negative minima at 210 and 222 nm. These bands are indicative of a typical alpha-helical structure.[21,22] The OVA spectrum after incorporation of the protein into, and release, from MN arrays prepared from aqueous blends of 20% w/w PMVE/MA is also presented. The CD spectrum of the OVA which had been incorporated into the MN array matrix was comparable to that of the OVA standard solution, indicating that no major changes in the secondary structure of the protein had occurred as a result of encapsulation into the MN arrays. Conversely, prolonged exposure of OVA to high temperature (80°C for 2 h) resulted in the degradation of the protein, as indicated by the considerable variation of the CD spectrum in comparison to the standard OVA solution.
The results highlight the suitability of the MN devices employed in this study as a vehicle for the incorporation of therapeutic proteins or protein-based vaccine antigens. The micromoulding approach, based on casting of aqueous PMVE/MA formulations, does not require harsh conditions, such as elevated temperatures, which would inevitably affect the native structure of the OVA. Other MN array fabrication approaches, reported elsewhere, have also been found to maintain the stability of a variety of macromolecules, such as proteins,[23,24] vaccines[25] and hormones[26] following incorporation and storage within the MN arrays device. For example, in a study conducted by Lee et al.,[27] MN arrays were prepared by casting aqueous solutions of carboxymethylcellulose (CMC) and amylopectin into micromoulds. The process was employed to prepare CMC MN arrays encapsulating the model protein lysozyme. The study indicated that the structural and functional integrity of the protein were retained within the MN arrays device after 2 months storage at room temperature. Similarly, Sun et al.[28] reported intradermal delivery of intact antigen proteins from dissolving poly(vinylpyrrolidone) MN arrays prepared by casting protein formulations into silicone moulds at room temperature. Within the same study, the authors indicated that MN arrays manufacturing by photochemical cross-linking, resulted in alteration of the quaternary structure of the proteins and subsequent loss of their binding activity. Possible detrimental effects of MN manufacture on the stability of fragile biomolecules have also been reported by Park et al.[29] MN arrays incorporating the model protein bovine serum albumin (BSA) were prepared from molten poly-(lactide-co-glycolic acid) at 135°C. Exposure of the BSA to the molten polymers resulted in significant loss of protein. Similarly, a study previously carried out by our group highlighted complete degradation of BSA incorporated in galactose MN arrays, which were also prepared at elevated temperatures.[4]
Maintenance of the structural integrity of a protein is essential to ensure its functionality.[30] The CD results indicated that the secondary structure of OVA was not significantly altered after incorporation into and release from the MN array matrix. The data, therefore, indicates that the manufacturing process outlined here can be successfully employed to produce MN arrays incorporating fragile molecules, such as proteins and potentially other biomolecules, such as nucleic acids and DNA.
Quantification of OVA in the MN arrays
The quantity of protein recovered from distinct parts of the MN device was assessed by calibrated spectroscopy. Complete (100%) recovery of protein from the MN arrays was not achieved, most likely due to electrostatic interactions between OVA and the polymeric matrix constituting the MN arrays. Approximately 70% (1785.3 ± 74.9 μg) of the nominal OVA content was recovered for MN arrays loaded with OVA at a concentration of 2.5 mg per array.
As a consequence of the miniaturised dimensions of the MN arrays, only a very small proportion of OVA was recovered from the individual needles of the MN arrays in comparison to the entire array. Specifically, only 2% (50 ± 3.8 μg) of the loaded protein was extracted from the needles of the MN arrays loaded with OVA at nominal concentrations of 2.5 mg per device. Figure 1c depicts the distribution of OVA throughout the MN arrays, expressed as a percentage of the total OVA loaded into the arrays (2.5 mg). As indicated, the majority of OVA was localised within the baseplate and sidewalls of the MN arrays. This result is not unexpected, as these represent the parts of the array with the greatest volume and, hence, mass. This fact certainly impacts the quantity of OVA which can be successfully delivered to the skin. In fact, as the sidewalls of the MN arrays are removed before MN array application, this implies that a considerable amount of OVA is removed from the MN arrays before they are even applied to the skin. Work carried out in our group previously has shown that the release of biomolecules which have been loaded into dissolving MN arrays is largely confined to that which is encapsulated within the individual needles themselves.[31] Moving this research forwards, it may prove beneficial to localise the active agent, in this case OVA, to the actual needles of the array, thus limiting the wastage illustrated in the current study. Indeed, our group has recently employed such an approach for dissolving MN-mediated delivery of nanoparticle-encapsulated antigen.[32]
In-vitro OVA permeation studies
The permeation profile of OVA released from MN arrays prepared from aqueous blends of 20% w/v PMVE/MA across dermatomed neonatal porcine skin (300–350 μm thickness) is presented in Figure 1d. The delivery of OVA increased with increasing duration of MN application and peaked at the 4 h timepoint. These results confirm the capability of these MN arrays to perforate the stratum corneum and thus facilitate passage of hydrophilic macromolecules across the skin. The importance of MN insertion in enhancing skin permeability was further confirmed by experiments carried out with baseplates (devoid of MN). In these experiments, no permeation of OVA was detected across the dermatomed neonatal porcine skin, thus highlighting that OVA could not be delivered via passive diffusion in detectable amounts (data not shown).
As illustrated in Figure 1d, OVA release from the MN matrix exhibited a burst effect within the first 2 h of the study, with detectable quantities of OVA detected in the receiver compartment even after the shortest application time (5 min). This suggests that the MN dissolved rapidly upon contact with the aqueous environment in the skin interstitial fluid, thus releasing the encapsulated OVA. However, the lack of appreciably increasing protein diffusion after 4 h suggests that blockage of the created microchannels by the dissolved polymeric matrix may have occurred. A reduction of the size of pores created in the stratum corneum following MN dissolution has been previously reported by Verbaan et al.[33] The authors thus identify this phenomenon as a possible cause for the plateaued diffusion of OVA across the skin following 4 h application of dissolvable MN arrays.
The amount of OVA detected in the receptor compartment after 4 h was 98.8 ± 15.7 μg, approximately 4% of the nominal 2.5 mg loading. Quantitative analysis of MN array loadings indicated that approximately 27% of the nominal OVA loading (2.5 mg) could be recovered from both the MN and the baseplate of the device and that approximately 2% was recoverable from the MN only (Figure 1c). This suggests that approximately half of the OVA detected in our in-vitro experiments was delivered from the needles of the MN arrays, with only a very restricted quantity of protein being released from the baseplate of the device. This consolidates the observation discussed above, and made previously by us, for insulin,[30] that it is the reduction of the diameter of the pores created by MN arrays over time, as well as their blockage by the dissolved polymeric matrix, which is likely to prevent further diffusion of the protein in skin at the later time points. Moreover, OVA molecules are known to have a tendency to self-aggregate.[34] This fact, together with the possible presence of electrostatic and steric interactions between extracellular skin components and OVA molecules, might combine to affect the mobility of the protein through the skin. Moreover, it is also possible that OVA may have been enzymatically degraded by residual skin-derived proteases, including skin esterases and serine proteases,[35,36] while diffusing through the skin and this may have further contributed to its reduced detection. The lytic activity of skin proteases has been reported for biomolecules, such as insulin, delivered into the skin by iontophoresis.[37] Further studies are needed to investigate the contribution of OVA enzymatic degradation to its permeation across the skin in the in-vitro scenario, thus addressing the question of whether enzymes residing in the skin are still active post freeze-thaw and are, therefore, capable of degrading the protein target. To this end, evidence from the food industry suggests that enzyme levels are increased in those meat samples which have been freeze-thawed,[38] thus suggesting that the freeze-thaw cycle undergone by the neonatal porcine skin used in these experiments may in fact have resulted in increased enzyme activity in the skin, leading to an increased propensity for degradation of the protein.
MN array dissolution profile in vivo
Real-time visualisation of MN arrays penetration and dissolution in vivo was performed by OCT. OCT analysis revealed that MN arrays penetrated to a depth of 473 ± 34 μm and created pores of surface diameters 133 ± 14 μm when manually applied to the dorsal region of the mouse ear. Increasing MN application times (i.e. 1, 5, 15, 30 and 60 min) resulted in gradual reduction of the MN heights, due to MN array dissolution (Figure 2a, A–D). The major portion of the needle shafts appeared to have dissolved 30 min after application (Figure 2a, D), while both the needles of the array and the baseplate of the same dissolved entirely after 60 min (Figure 2a, E). The results indicate that the MN arrays employed in this study had sufficient mechanical strength to successfully penetrate the outmost layer of the skin, the stratum corneum. These MN arrays then dissolved in skin interstitial fluid, delivering the model antigen OVA to the epidermal and dermal layers of the skin, where the majority of antigen-presenting cells (APCs) are located.[39] In addition, the results indicate that the MN arrays completely dissolved after a relatively short application period (60 min). The dissolution profile of the MN arrays (calculated as reduction of individual MN heights) is presented in Figure 2b. From a clinical viewpoint, it may be desirable for complete dissolution to occur more quickly than this to minimise the time a patient undergoing vaccination would need to remain in the clinical setting to monitor for possible anaphylactic reactions. Dissolution time could be reduced by incorporating a lower content of polymer in the arrays. In this case, care would be required to avoid reducing the inherent mechanical strength of the MN arrays, concomitant with reverberations for stratum corneum penetration.
Figure 2.
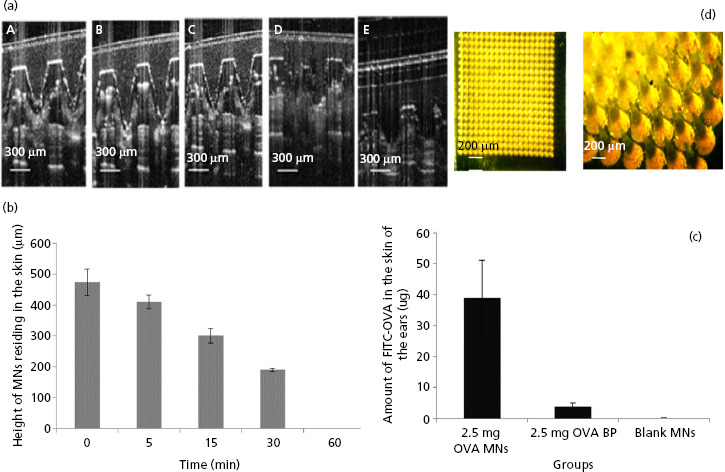
(a) Optical coherence tomography (OCT) real-time in-vivo visualisation of microneedle (MN), fabricated from 20% w/v poly(methyl vinyl ether/maleic acid) (PMVE/MA) and loaded with 2.5 mg ovalbumin (OVA), inserted into the ear of mice for increasing application times: i.e. 1 min (A), 5 min (B), 15 min (C), 30 min (D) and (E) 60 min. (b) Dissolution profile of MN arrays (calculated as reduction of MN height) inserted into mouse skin at increasing application times (mean ± SD, n = 15). (c) The amount (μg) of fluorescently labelled OVA (FITC-OVA) quantified in the excised ear skin of mice that were treated with MN arrays loaded with 2.5 mg FITC-OVA (2.5 mg OVA MNs); baseplates (arrays lacking needles) loaded with 2.5 mg FITC-OVA (2.5 mg OVA BP) or blank MN arrays containing no FITC-OVA (blank MNs). Data are presented as mean ± standard deviation (SD), n = 4. (d) A representative light micrograph of a portion of a MN array prepared from aqueous blends of 20% w/v PMVE/MA and loaded with 2.5 mg of FITC-OVA. The scale bar represents 200 μm.
Quantitative analysis of OVA delivered to mice
Immunisation of mice by OVA-loaded arrays was performed by inserting the MN arrays into the ear skin of the animals. This experiment was performed with the aim of investigating the quantity of antigen delivered to the skin upon MN application, following excision of the ear from the animal. FITC-OVA was used to enable detection of OVA within the cutaneous tissue. The quantity of OVA delivered by OVA-loaded MN arrays was compared with the amount of OVA delivered using OVA-loaded baseplates (arrays devoid of needles). The data indicate that the quantity of OVA recovered from excised ears treated with MN arrays was approximately 10 times higher than the amount extracted from those treated with OVA-loaded baseplates (38.8 ± 12.1 μg and 3.7 ± 1.2 μg were delivered by the MN arrays and baseplates respectively; Figure 2c). A representative light micrograph of a MN array prepared from aqueous blends of 20% w/v PMVE/MA and loaded with 2.5 mg FITC-OVA is presented in Figure 2d. The results clearly indicate the importance of MN arrays in enhancing delivery of the protein into the skin through the creation of micro-channels in the stratum corneum. A small quantity of protein was determined when the baseplate only was applied to the skin. Passive diffusion of OVA through intact stratum corneum would not be expected, due to the protein's high hydrophilicity and molecular weight.[40] As a consequence, the small amount of OVA detected in the skin sample may simply have been due to some retention of OVA on the skin surface following removal of the array. OVA delivered systemically and absorbed by the dermal microcirculation or already taken to the draining lymph nodes by interstitial fluid flow or APCs could, obviously, not be measured here, but may have a role to play in immune responses.
In-vivo evaluation of the efficacy of dissolving MN arrays for transdermal delivery of OVA
The levels of anti-OVA-specific IgG induced in mice immunised by MN arrays fabricated from aqueous blends of 20% w/v PMVE/MA and loaded with OVA were compared with those induced for OVA delivered intradermally using conventional needle and syringe. To investigate the effect of adjuvants on the immune response, the compound IMI, commonly used in vaccine formulations to improve antigen immunogenicity,[41] was added to OVA and delivered intradermally by means of the MN arrays or conventional needle and syringe. The results obtained when delivering OVA (with or without IMI) via MN arrays and conventional hypodermic needles are presented in Figure 3a.
Figure 3.
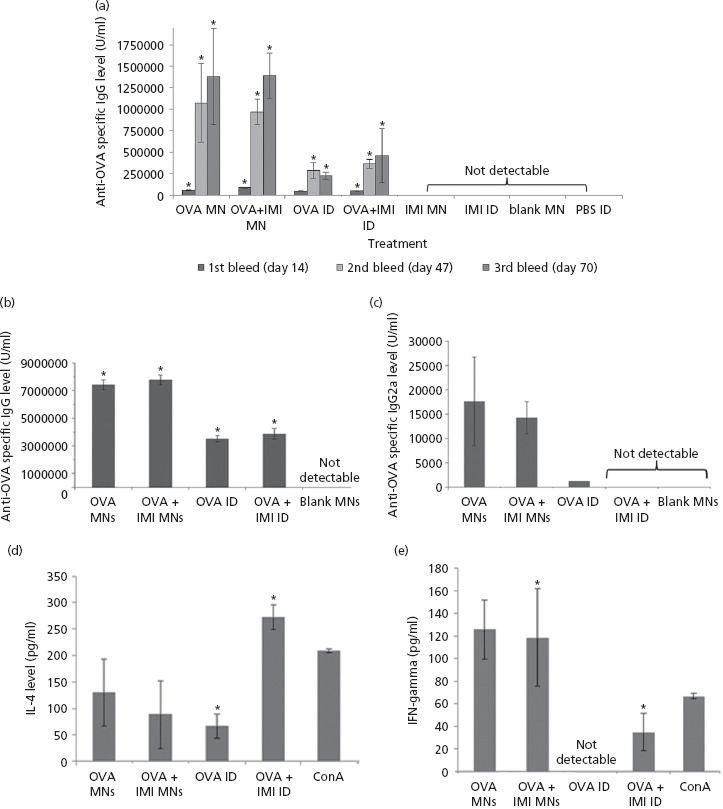
(a) Total anti-ovalbumin (OVA) serum IgG levels after immunisation using either microneedle (MN) or conventional intradermal administration (ID). Mice were treated with OVA-loaded MN, with or without IMI (OVA MN and OVA + IMI), IMI plus MN (IMI MN) or blank MN (blank MN). Mice treated by conventional ID injections received OVA (OVA ID), OVA plus IMI (OVA + IMI ID), IMI (IMI ID) and PBS (PBS ID). Animals were bled on days 14, 47 and 70. Data are presented as mean ± standard deviation (SD), n = 4. (For reference to the statistical significances, please refer to the main text.) Humoral and cellular immune responses elicited in mice following immunisation via MN or ID was determined in sera (b, c) or in cultured splenocytes (d, e). The titres of total anti-OVA IgG subclasses (b, c), as well as IL-4 (d) and IFN- γ (e) cytokines are presented. Mice were treated with OVA-loaded MN, with or without IMI (OVA MN and OVA + IMI), IMI plus MN (IMI MN) or blank MN (blank MN). Mice treated by ID were injected with solutions of OVA (OVA ID) or OVA plus IMI (OVA IMI). Data are presented as means ± SD. In the case of IgG1, n = 4; IgG2a, n = 2; IFN- γ, n = 2; IL-4, n = 4.
The results indicate that, 14 days post-immunisation, the level of IgG induced upon immunisation of OVA plus IMI via MN arrays was greater than those animals immunised with OVA only via MN arrays (P = 0.03). Forty-seven days after immunisation, MN arrays loaded with OVA only induced higher immune responses in comparison to arrays incorporating both OVA and IMI. A further increase was elicited after 70 days, although the differences in antibody levels were not statistically significant (P = 0.85). Similarly, when OVA was administered by conventional needle and syringe, antibody production increased proportionally with time. In contrast to that observed for MN-mediated OVA delivery, the inclusion of IMI in hypodermic needle delivery of OVA, at all three time points, resulted in enhanced production of anti-OVA-specific IgG. However, in all cases, the differences in antibody production between the two groups (OVA only vs OVA plus IMI) at different post-immunisation times were not statistically significant (i.e. P = 0.7 at days 14, P = 0.3 at day 47 and P = 0.3 at day 70 respectively). The results also indicated that OVA-loaded MN arrays were able to induce stronger immune responses in comparison to conventional hypodermic needle. The differences in antibody levels induced by the two administration routes reached statistical significance at days 47 (P = 0.04) and 70 (P = 0.01), while the results were not statistically significant at day 14. The addition of IMI also seemed to increase the degree of antibody production.
The antibody levels induced by OVA plus IMI delivered by MN arrays were significantly higher than those induced by OVA plus IMI delivered by conventional needle and syringe at each of the post-immunisation time points investigated (P = 0.01 at day 14, P = 0.009 at day 47 and P = 0.001 at day 70 respectively). As anticipated, no anti-OVA antibodies were detected in those samples taken from mice immunised with blank MN arrays or conventional syringe and needle. The results also evidenced differences in kinetics of antibody production between MN arrays and conventional needle and syringe, indicating that MN arrays were able to induce greater immune response in a shorter period of time. This may be due to the physical/geometrical differences between the two types of device. The MN arrays consist of 361 micro-projections protruding from a baseplate. This implies that, when the device is applied to the skin, several discrete volumes of antigen are delivered from the needles on the array, as well as from the baseplate of the array. Conversely, in the case of conventional hypodermic needle, a larger volume of antigen is delivered to a defined region of the tissue only. MN array application may therefore result in activation of a greater number of APCs (i.e. Langerhans cells, dendritic cells (DCs), skin homing T cells, macrophages and mast cells) in comparison to hypodermic needle, thus providing a more effective immune response.[32,42]
Other factors may have contributed to the robust MN array-mediated immune responses. In particular, it may be hypothesised that the polymers in the MN array matrix may act as an adjuvant, stimulating antibody production. Polymers have, in fact, been used in several immunological applications, for example in designing new generation vaccines and adjuvants[43,44] and have been reported to efficiently enhance the induced immune responses to purified recombinant or weak antigens.[45] In particular, studies conducted on the immunogenicity of PMVE/MA revealed good adjuvant properties of the copolymer, which was attributed to the presence of reactive anhydride groups.[46,47] This may not be the case here, however, due to hydrolysis of the vast majority of anhydride groups during dissolution. Moreover, Sullivan et al.,[48] in the first published study carried out using dissolving MN arrays fabricated from poly(vinylpyrrolidone), deduced that the robust immune responses generated in the study were possibly attributable to the adjuvant property of the enlisted polymer.
The acidic nature (pH = 2.4) of the aqueous blend of PMVE/MA used to formulate the MN arrays may play a key role in enhancing the adjuvant properties of the MN arrays. A study conducted by Vermeulen et al.[49] revealed that uptake of FITC-OVA by DCs was considerably enhanced by an increase in extracellular acidosis. According to the authors, a reduction in pH in the extracellular space enhanced the capability of immature DCs to endocytose the antigen. Application of polymeric MN arrays and subsequent dissolution of the PMVE/MA acidic matrix could result in reduced pH of peripheral tissues and may, therefore, stimulate antigen engulfment by immune cells. Moreover, the instigation of transient damage to the skin, upon MN array application, may also contribute to the initiation and enhancement of immune responses through release of pro-inflammatory signals, thus leading to subsequent increased recruitment of APC to the site of inflammation.[50] This observation was recently confirmed in a study conducted by Weldon et al.,[51] when it was reported that insertion of solid MN arrays led to enhanced release of pro-inflammatory cytokines in the skin. In the current study, the skin has been punctured 361 times in a single treatment, rather than once only by hypodermic needle. This fact, together with the evidence reported by Weldon et al.,[51] emphasises the potential additive effect of multiple MN punctures on the initiated inflammatory response and suggests that the use of MN arrays, specifically, to deliver vaccines may serve to potentiate the immune response initiated in the patient. In addition to this, the data presented here concur with previous reports regarding the potential adjuvant properties of polymeric MN arrays[48] and thus suggest that MN arrays may act in a two-pronged manner to enhance pro-inflammatory cytokine release in vivo.[52]
Although IMI did, in some instances, potentiate the immunogenic functionality of OVA, this effect cannot be considered to be essential, as comparable immune responses were produced by OVA-loaded MN arrays with or without IMI. The results presented here corroborate the findings of Weldon et al.,[51] where similar total IgG responses were reported in mice immunised by MN arrays previously coated with vaccine formulations with or without adjuvant. The use of MN arrays as a vaccination tool could, therefore, potentially avoid the need for the addition of adjuvant to vaccine formulations, thus leading to reduced production costs. More importantly however, the use of unadjuvanted vaccines, via delivery modes such as MN device, could potentially reduce incidences of immunisation-mediated inflammatory myopathies.[53] In a recent paper, the autoimmune-like clinical syndrome induced by adjuvants (Shoenfeld's syndrome) was discussed, but the author conceded that the development and clinical evaluation of new, alternative adjuvants would prove costly and time-consuming and, for this reason, would prove an unlikely endpoint.[52] The potential future use of unadjuvanted MN devices as vaccine delivery vehicles would circumvent these issues entirely. Specifically, the use of IMI-free vaccine formulations would be particularly desirable from a clinical point of view, as IMI has been associated with the occurrence of adverse side-effects in some clinical studies.[54,55] In addition, the exclusion of IMI would allow for maximisation of antigen loading within the MN arrays. In terms of the potential cost savings and the avoidance of potentially harmful side-effects, the omission of adjuvants from dissolving MN devices used for facilitated delivery of vaccines may prove to be of great benefit to patients.
Determination of anti-OVA-specific antibody subclasses and specific cytokine production
With the aim of identifying the specific immune response elements evoked by the two classes of device used in this study, the concentrations of IgG subclasses, IgG2a and IgG1, associated to thymus helper 1 (Th1) or thymus helper 2 (Th2) cells, respectively, were determined. Additionally, the concentration levels of two pro-inflammatory cytokines, IFN-γ and IL-4, associated with Th1 and Th2 immune responses, respectively, were analysed in animals immunised with either MN arrays or conventional needle and syringe. As outlined in the previous experiment, the influence of the IMI adjuvant was also investigated. Figure 3b and 3c illustrate the levels of specific anti-OVA IgG1 (B) and IgG2a (C) detected in the serum of animals following immunisation.
The results indicate that IgG1 is the predominant IgG subtype expressed after immunisation with both classes of device. Similar to the observations made regarding total IgG production, MN arrays incorporating OVA plus IMI (OVA + IMI MNs) produced slightly greater immune responses than those MN arrays loaded with OVA only (OVA MN), although this difference was not statistically significant (P = 0.1). The same was observed for mice immunised by conventional intradermal injection (OVA ID) (P = 0.4 when comparing mice injected with either OVA only (OVA ID) or OVA plus IMI (OVA + IMI ID)). In line with the results highlighted previously, MN arrays were capable of inducing significantly higher anti-OVA-specific IgG1 levels in comparison to antigen delivery via conventional hypodermic needle, regardless of whether IMI was loaded with the antigen. Statistical analysis deduced that there were significant differences (P = 0.0001 in both cases) when comparing IgG1 levels produced by MN array vs conventional hypodermic needle without or with IMI (OVA MN vs OVA ID and OVA + IMI MNs vs OVA + IMI ID). As expected, the application of blank MN arrays resulted in no induction of IgG1 expression.
Assessing the levels of IgG2a in the mice post-immunisation was more challenging, as only a limited number of animals developed detectable IgG2a quantities over the course of the experimental period. In particular, for MN array-mediated immunisation (OVA MN), IgG2a could be measured in the sera of only two animals out of five (data not shown). Similarly to those results outlined for IgG1 detection, the addition of IMI into the MN (OVA + IMI MN) did not result in a significant increase in antibody production (P = 0.6). With regards to immunisation mediated by conventional intradermal injections, IgG2a levels registered were even lower than those produced in animals treated with MN arrays. IgG2a was undetectable in all animals injected intradermally by conventional needle with OVA plus IMI (OVA + IMI ID), while it was detected in only one of two animals treated with the OVA only (OVA ID). Once again, the application of blank MN arrays resulted in no induction of IgG2a expression.
As deduced in previous studies, the ratios of IgG1 : IgG2a levels provides important information on the type of immune response induced, that is whether the response is Th1 (IgG2a) or Th2 (IgG1) biased.[56,57] Our results indicate higher levels of IgG1, in comparison to IgG2a, induced in all immunised groups, thus indicating that the immune responses favour a Th2-mediated mechanism. Despite the low titres of IgG2a produced, the results indicate that MN arrays are capable of inducing higher levels of IgG2a expression, in comparison to conventional needle-based administration of the antigen. This, therefore, suggests that MN arrays may be able to enhance the production of different subclasses of antibodies and thus induce both Th1- and Th2-mediated immune responses.
The levels of IFN-γ detected in culture supernatants from isolated spleen cells were used as indicators of the Th1 cellular immune response (Figure 3e). The pattern of IFN-γ expression, as a consequence of the different antigen administration approaches, is in agreement with the expression trends for total anti-OVA-specific IgG and IgG subclasses reported here. These data indicate that, upon administration of the antigen via MN arrays, and irrespective of the inclusion of IMI, the overall immune response induced is significantly more pronounced than those achieved via conventional immunisation route. The induced IFN-γ expression was significantly greater when OVA and IMI were delivered by MN rather than injected intradermally by syringe (P = 0.01). Delivery of OVA alone by conventional needle (OVA ID) did not result in any detectable cytokine production, thus indicating the influence of different delivery devices on both the pattern of immune response, as well the strength of immune protection induced. While immunisation by conventional needle but including the addition of an adjuvant (OVA + IMI ID) did elicit the induction of IFN-γ expression, this was to a much lesser degree than that determined using MN delivery vehicles. The addition of IMI to the MN array formulation (OVA + IMI MN) did not significantly increase the concentration of cytokine produced as comparable results were obtained when OVA was delivered by MN arrays with or without the adjuvant (P = 0.7). This further underpins the capability of the MN array delivery system to elicit effective immune responses, even in absence of adjuvant.
In an extension of this cytokine study, IL-4 levels in the splenocytes of OVA-immunised animals were measured to evaluate the extent of Th2-mediated cellular immune response (Figure 3d). In terms of cytokine expression in response to administration in the presence of adjuvant, a similar pattern to that reported for IFN-γ emerged. The addition of IMI to MN arrays (OVA + IMI MN) did not significantly increase the levels of IL-4 produced (P = 0.6). Conversely, delivery of OVA plus IMI via conventional syringe and needle administration (OVA + IMI ID) did result in considerably higher cytokine levels when compared with OVA administration in the absence of the adjuvant (P = 0.01). In contrast with the IFN-γ data, however, cytokine levels induced by MN array application were not significantly higher than those produced following immunisation with conventional hypodermic needle, irrespective of the addition of IMI to the antigen. In particular, delivery of OVA plus IMI by conventional hypodermic needle (OVA + IMI ID) induced IL-4 expression levels which were higher than those produced by MN arrays and adjuvant (OVA + IMI MN), although these differences were not statistically significant (P = 0.5 when comparing cytokine levels produced by OVA delivered by MN arrays (OVA MN) vs conventional hypodermic needle (OVA ID) and P = 0.1 when comparing the levels produced by the two devices in the presence of IMI (OVA + IMI MN vs OVA + IMI ID)). In both instances, Con A stimulation of non-immunised spleen cells was carried out as a positive control.
The results of this study suggest that MN arrays may be able to elicit more balanced immune responses in comparison to conventional hypodermic needles. Both Th1- and Th2-mediated immune responses were, in fact, induced by MN arrays, while conventional hypodermic needle seemed to favour a Th2-biased mechanism, as suggested by the complete absence of expression of specific anti-OVA IgG2a antibodies. In addition, MN arrays were able to elicit cellular immune responses as indicated by the detected cytokine production following MN array application. Stimulation of balanced immune responses would be particularly desirable from a clinical point of view. Unbalanced stimulation of Th1-Th2 mechanisms can, in fact, determine excessive production of pro-inflammatory mediators (e.g. abnormal production of IFN-γ, the main Th1-cytokine) which can cause damage to tissues and immunopathologies.[58] The initiation of a balanced immune response would, therefore, be ideal in a clinical scenario, as it would result in safer and more controlled vaccination.
OVA-induced airway inflammation in mice
Following on from these studies, experiments were then carried out to investigate whether MN array-mediated immunisation would be adequate to protect animals post-challenge. OVA is considered a good model for challenge studies and has frequently been employed to investigate allergic lung disease in guinea pigs and rats,[43,59,60,61,62] where animals develop acute respiratory responses and airway inflammation following challenge with OVA aerosols. Animals have also experienced anaphylactic shock after injection of OVA systemically.[63,64] To this end, mice in the current study were challenged intraperitoneally with high doses of OVA. Initial experiments were performed with the aim of evaluating the immune response of mice immunised with MN arrays at different OVA loadings (1 and 2.5 mg per array) following OVA challenge. The results indicate that mice immunised with MN arrays do not show any sign of anaphylactic shock up to 1 h post-challenge. In addition, no significant changes in body weight occurred and no mortality was reported for any of the treated animals.
Samples collected in this arm of the study were subjected to the same panel of quantitative ELISA methodologies as previously outlined (anti-OVA-specific IGg1; anti-OVA-specific Igg2a; IFN-γ; IL-4). To examine the effect of the MN arrays on the production of anti-OVA-specific antibodies, mice were immunised as described and were bled 34 days after OVA administration, then challenged with OVA and subsequently bled 24 h after challenge (day 35). Anti-OVA-specific total IgG (Figure 4a) and the subclasses IgG1 (Figure 4b) and IgG2a (data not shown) were therefore determined in the sera of the treated animals to assess the level of humoral response initiated. Quantities of the Th2-mediated cytokine, IL-4, were determined in BAL fluids (Figure 4c) and in the supernatants of spleen cells cultured from immunised mice (Figure 4d), to determine the level of cellular immune responses produced in the animals. With regards to the levels of antibody produced (humoral response) following immunisation, IgG2a levels were detected only before OVA challenge and in only two of five mice immunised via MN arrays at OVA loading of 2.5 mg/array (data not shown). IgG2a was not detected in any of the groups immunised with MN arrays at OVA loading of 1 mg or in any of the challenged animals. This in agreement with the results reported previously, indicating that MN arrays induce a weak Th1-mediated immune response. Conversely, total IgG (Figure 4a), in addition to specific IgG1 (Figure 4b), subtype levels were determined in all treated mice.
Figure 4.
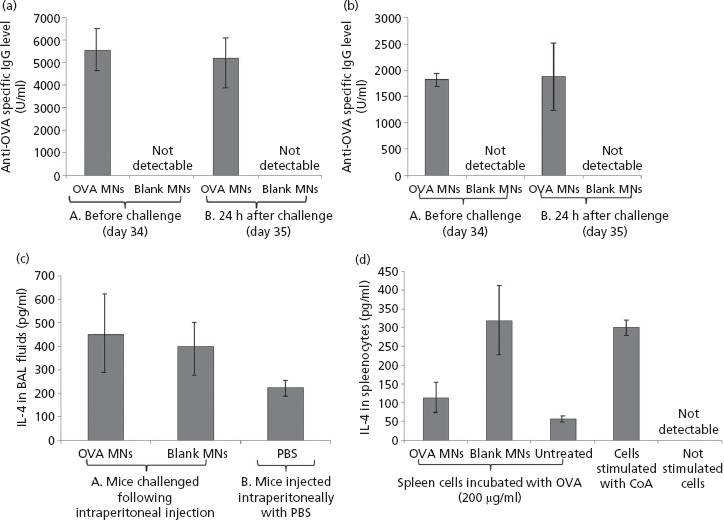
Levels of anti- ovalbumin (OVA)-specific total IgG (a) and IgG1 (b) of C57BL/6 mice before and after intraperitoneal challenge with OVA. Mice were immunised twice (at day 0 and day 14) with microneedle (MN) arrays loaded with 2.5 mg OVA/array or MN arrays devoid of OVA (blank MNs) as a control group. At day 34 before challenge, mice were bled to determine the levels of antibodies in their systems. Mice were then challenged intraperitoneal with 2 mg OVA in a volume of 100 μl phosphate-buffered saline (PBS) and blood samples were collected 24 h after challenge to monitor the levels of antibodies produced in response to the challenge. Brace A indicates the levels pre-challenge while Brace B indicates the total IgG levels and the IgG1 levels post-challenge. Data are presented as mean ± SD, n = 3. Detection of the Th2 cytokine, IL-4, in BAL fluids (c) and in cultured splenocytes (d). C57BL/6 mice were immunised twice with OVA loaded MN arrays at 2.5 mg/array, then challenged intraperitoneal with OVA (2 mg in 100 μl PBS). The control group consisted of mice treated with control MN arrays (blank MNs) before challenge. A second control group were injected with 100 μl PBS (PBS) only. Data are represented as mean ± standard deviation (SD), n = 3.
Figure 4d indicates the levels of Th2 cytokine, IL-4, measured in BAL and splenocytes of immunised animals after OVA challenge respectively. Interestingly, the results indicate that the levels of IL-4 obtained in BAL fluid from animals treated with OVA-loaded MN arrays were comparable to those produced in animals treated with blank MN arrays (not containing OVA). As expected, no IL-4 was detected in the supernatants of cells that were cultured without OVA or control stimulation with the lectin, Con A. Surprisingly, the levels of IL-4 produced by those splenocytes isolated from immunised animals and challenged with OVA were higher in the case of those immunised with blank MN arrays compared with OVA MN arrays, although this difference was not statistically significant. The data confirm the capability of MN arrays to stimulate Th2 cytokine production in animals immunised with OVA. In addition, the increment in IL-4 levels observed in animals treated with blank MN arrays strongly suggests that the MN arrays employed in our study have immunogenic/adjuvant properties as previously suggested.
Conclusions
The work carried out in this study shows that the described dissolving MN arrays are suitable vehicles for incorporation of fragile biomolecules, such as proteins, and their subsequent transdermal delivery. Due to the absence of harsh manufacturing conditions employed during the MN fabrication process, the secondary structure of the model protein, OVA, was maintained post-release from the MN arrays, thus promoting macromolecule integrity. Both in-vitro and in-vivo experimentation confirmed delivery of OVA via MN, thus highlighting these devices as a viable potential means of enhanced transdermal protein delivery. Studies are now warranted to investigate the long-term stability of proteins loaded within the polymeric matrix of these MN arrays. The development of temperature-stable MN arrays incorporating such compounds could, in time, eliminate the requirement for cold chain maintenance. If, for example, MN arrays were engaged as vaccine delivery devices, as suggested in this study, the subsequent improvements and reductions in vaccination costs, vaccine infrastructure and distribution in developing countries particularly would be extensive.
Specifically, this study demonstrates the capability of our laser-engineered dissolving MN system to successfully induce pronounced, and in some instances, enhanced, immune responses in an in-vivo mouse model, when compared with conventional intradermal injection of the same antigen. This was achieved without the necessity for adjuvant inclusion, a fact that in itself has far-reaching benefits in terms of the avoidance of harmful adjuvant-mediated autoimmune reactions. Furthermore, MN-mediated immunisation enabled efficient protection of animals challenged post-immunisation with the model antigen, OVA. Finally, the study highlights the potential immunological/adjuvant properties of the MN devices described.
A shortcoming of the described MN array fabrication process is the loss of OVA upon sidewall removal, in addition to the limited delivery of protein from the baseplates of the arrays. Further studies are, therefore, warranted to optimise the MN loading process, with particular emphasis on the potential for loading of protein into the needles of the MN arrays only. This approach would prevent waste of biomaterials and reduce the inherent loss of active compound which is currently suffered when sidewalls are removed. The development and validation of such an approach would require that back migration of protein into the baseplate be prevented. The means to achieve this may involve the casting of needles and baseplates in a two-step process where the baseplate would potentially be cast from a higher content polymeric gel, so as to reduce back-diffusion. Any MN arrays produced using this approach would then, obviously, be required to undergo similar characterisation experiments to those outlined in the current study. Indeed, we have developed a two-stage casting method that forms the MN and baseplate/sidewalls separately, thus reducing wastage of macromolecules during initial processing and subsequent release. We have now obtained significant funding from the UK's Biotechnology & Biological Research Council to scale up this method to enable production of clinical trial supplies in the first instance.
Declarations
Acknowledgements
This study was supported by BBSRC grant numbers BB/K020234/1, BB/FOF/287 and BB/E020534/1 and Wellcome Trust grant number WT094085MA.
References
- Donnelly RF, et al. Microneedle-Mediated Transdermal and Intradermal Drug Delivery. New Jersey: John Wiley & Sons, 2012. [Google Scholar]
- Ito Y, et al. Transdermal insulin application system with dissolving microneedles. Diabetes Technol Ther 2012; 14: 891–899. [DOI] [PubMed] [Google Scholar]
- Liu S, et al. The development and characteristics of novel microneedle arrays fabricated from hyaluronic acid, and their application in the transdermal delivery of insulin. J Control Release 2012; 161: 933–941. [DOI] [PubMed] [Google Scholar]
- Donnelly RF, et al. Processing difficulties and instability of carbohydrate microneedle arrays. Drug Dev Ind Pharm 2009; 35: 1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, et al. Dissolving microneedles for transdermal drug delivery. Biomaterials 2008; 29: 2113–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomaa YA, et al. Laser-engineered dissolving microneedles for active transdermal delivery of nadroparin calcium. Eur J Pharm Biopharm 2012; 82: 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K, et al. A low-invasive and effective transcutaneous immunization system using a novel dissolving microneedle array for soluble and particulate antigens. J Control Release 2012; 161: 10–17. [DOI] [PubMed] [Google Scholar]
- Naito S, et al. Antigen-loaded dissolving microneedle array as a novel tool for percutaneous vaccination. Vaccine 2012; 30: 1191–1197. [DOI] [PubMed] [Google Scholar]
- Matsuo K, et al. Transcutaneous immunization using a dissolving microneedle array protects against tetanus, diphtheria, malaria, and influenza. J Control Release 2012; 160: 495–501. [DOI] [PubMed] [Google Scholar]
- Sullivan SP, et al. Dissolving polymer microneedle patches for influenza vaccination. Nat Med 2010; 16: 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattani A, et al. Microneedle mediated intradermal delivery of adjuvanted recombinant HIV-1 CN54gp140 effectively primes mucosal boost inoculations. J Control Release 2012; 162: 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraishi Y, et al. Performance and characteristics evaluation of a sodium hyaluronate-based microneedle patch for a transcutaneous drug delivery system. Int J Pharm 2013; 441: 570–579. [DOI] [PubMed] [Google Scholar]
- Garland MJ, et al. Dissolving polymeric microneedle arrays for electrically assisted transdermal drug delivery. J Control Release 2012; 159: 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaric M, et al. Skin dendritic cell targeting via microneedle arrays laden with antigen-encapsulated poly-D,L-lactide-co-glycolide nanoparticles induces efficient antitumor and antiviral immune responses. Photodiagnosis Photodyn Ther 2010; 7: 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly RF, et al. Microneedle-mediated intradermal nanoparticle delivery: potential for enhanced local administration of hydrophobic pre-formed photosensitisers. Anal Biochem 2001; 290: 179–185. [DOI] [PubMed] [Google Scholar]
- Donnelly RF, et al. Design, optimization and characterization of polymeric microneedle arrays prepared by a novel laser-based micromoulding technique. Pharm Res 2011; 28: 41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson AD, et al. Moisture-activated, electrically conducting bioadhesive hydrogels as interfaces for bioelectrodes: effect of film hydration on cutaneous adherence in wet environments. J Appl Polym Sci 1995; 58: 1291–1296. [Google Scholar]
- Fourtanier A, Berrebi C. Miniature pig as an animal model to study photoaging. Photochem Photobiol 1989; 50: 771–784. [DOI] [PubMed] [Google Scholar]
- Rolland JM, et al. Specific and sensitive enzyme-linked immunosorbent assays for analysis of residual allergenic food proteins in commercial bottled wine fined with egg white, milk, and nongrape-derived tannins. J Agric Food Chem 2007; 56: 349–354. [DOI] [PubMed] [Google Scholar]
- Donnelly RF, et al. Optical coherence tomography is a valuable tool in the study of the effects of microneedle geometry on skin penetration characteristics and in-skin dissolution. J Control Release 2010; 147: 333–341. [DOI] [PubMed] [Google Scholar]
- Bouchard M, et al. Formation of insulin amyloid fibrils followed by FTIR and simultaneously with CD and electron microscopy. Protein Sci 2000; 9: 1960–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henricus MM, et al. Investigation of insulin loaded self-assembled microtubulus for drug release. Bioconjug Chem 2008; 19: 2394–2400. [DOI] [PubMed] [Google Scholar]
- Chen MC, et al. Chitosan microneedle patches for sustained transdermal delivery of macromolecules. Biomacromolecules 2012; 13: 4022–4031. [DOI] [PubMed] [Google Scholar]
- Sullivan SP, et al. Minimally invasive protein delivery with rapidly dissolving polymer microneedles. Adv Mater 2008; 20: 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kommareddy S, et al. Dissolvable microneedle patches for the delivery of cell-culture-derived influenza vaccine antigens. J Pharm Sci 2012; 101: 1021–1027. [DOI] [PubMed] [Google Scholar]
- Lee JW, et al. Dissolving microneedle patch for transdermal delivery of human growth hormone. Small 2011; 7: 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, et al. Dissolving microneedles for transdermal drug delivery. Biomaterials 2008; 29: 2113–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, et al. Polyvinylpyrrolidone microneedles enable delivery of intact proteins for diagnostic and therapeutic applications. Acta Biomater 2013; 9: 7767–7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, et al. Polymer microneedles for controlled-release drug delivery. Pharm Res 2006; 23: 1008–1019. [DOI] [PubMed] [Google Scholar]
- Wangoo N, et al. Interaction of gold nanoparticles with protein: a spectroscopic study to monitor protein conformational changes. App Phys Lett 2008; 92: 133104-1–133104-3. [Google Scholar]
- Migalska K, et al. Laser-engineered dissolving microneedle arrays for transdermal macromolecular drug delivery. Pharm Res 2011; 28: 1919–1930. [DOI] [PubMed] [Google Scholar]
- Zaric M, et al. Skin dendritic cell targeting via microneedle arrays laden with antigen-encapsulated poly-D,L-lactide-co-glycolide nanoparticles induces efficient antitumor and antiviral immune responses. ACS Nano 2013; 7: 2042–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbaan FJ, et al. Assembled microneedle arrays enhance the transport of compounds varying over a large range of molecular weight across human dermatomed skin. J Controlled Release 2007; 117: 238–245. [DOI] [PubMed] [Google Scholar]
- Zhao W, Yang R. Pulsed electric field induced aggregation of food proteins: ovalbumin and bovine serum albumin. Food Bioprocess Technol 2012; 5: 1706–1714. [Google Scholar]
- Beisson F, et al. Use of the tape stripping technique for directly quantifying esterase activities in human stratum corneum. Anal Biochem 2001; 290: 179–185. [DOI] [PubMed] [Google Scholar]
- Engelrud T. Stratum corneum chymotryptic enzyme: evidence of its location in the stratum corneum intercellular space. Eur J Dermatol 1992; 2: 50–55. [Google Scholar]
- Huang Y, Wu S. Stability of peptides during iontophoretic transdermal delivery. Int J Pharm 1996; 131: 19–23. [Google Scholar]
- Ballin NZ, Lametsch R. Analytical methods for authentication of fresh vs thawed meat – A review. Meat Sci. 2008; 80: 151–158. [DOI] [PubMed] [Google Scholar]
- Mathers AR, Larregina AT. Professional antigen-presenting cells of the skin. Immunol Res 2006; 36: 127–136. [DOI] [PubMed] [Google Scholar]
- Andrews SN, et al. Transdermal delivery of molecules is limited by full epidermis, not just stratum corneum. Pharm Res 2013; 30: 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauder DN. Immunomodulatory and pharmacologic properties of imiquimod. J Am Acad Dermatol 2000; 43: S6–S11. [DOI] [PubMed] [Google Scholar]
- Koutsonanos DG, et al. Transdermal influenza immunization with vaccine-coated microneedle arrays. PLoS ONE 2009; 4: e4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani N, et al. Targeting of antigens to skin dendritic cells: possibilities to enhance vaccine efficacy. Immunol Cell Biol 2010; 88: 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya AK. Polymers as immunological adjuvants: an update on recent developments. J BioSci Biotech 2012; 3: 199–210. [Google Scholar]
- Wang W, Singh M. Selection of adjuvants for enhanced vaccine potency. World Journal of Vaccines 2011; 1: 33–78. [Google Scholar]
- De Souza Rebouças J, et al. Nanoparticulate adjuvants and delivery systems for allergen immunotherapy. J Biomed Biotechnol 2012, in press. ID 474605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebouças JDS, et al. Development of poly(anhydride) nanoparticles loaded with peanut proteins: the influence of preparation method on the immunogenic properties. Eur J Pharm Biopharm 2012; 82: 241–249. [DOI] [PubMed] [Google Scholar]
- Sullivan S, et al. Dissolving polymer microneedle patches for influenza vaccination. Nat Med 2010; 16: 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, et al. Acidosis improves uptake of antigens and MHC class I-restricted presentation by dendritic cells. J Immunol 2004; 172: 3196–3204. [DOI] [PubMed] [Google Scholar]
- Bal SM, et al. Advances in transcutaneous vaccine delivery: do all ways lead to Rome? J Control Release 2010; 148: 266–282. [DOI] [PubMed] [Google Scholar]
- Weldon WC, et al. Effect of adjuvants on responses to skin immunization by microneedles coated with influenza subunit vaccine. PLoS ONE 2012; 7: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovsky N, Aguilar JC. Vaccine adjuvants: current state and future trends. Immunol Cell Biol 2004; 82: 488–496. [DOI] [PubMed] [Google Scholar]
- Stübgen JP. A review on the association between inflammatory myopathies and vaccination. Autoimmun Rev 2014; 13: 31–39. doi: 10.1016/j.autrev.2013.08.005; [Epub 2013 Aug 31]. [DOI] [PubMed] [Google Scholar]
- Goldstein D, et al. Administration of imiquimod, an interferon inducer, in asymptomatic human immunodeficiency virus-infected persons to determine safety and biologic response modification. J Infect Dis 1998; 178: 858–861. [DOI] [PubMed] [Google Scholar]
- Smits ELJM, et al. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist 2008; 13: 859–875. [DOI] [PubMed] [Google Scholar]
- Schulte S, et al. Genetically programmed biases in Th1 and Th2 immune responses modulate atherogenesis. Am J Pathol 2008; 172: 1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JM, et al. Effects of a Th1- vs a Th2-biased immune response in protection against Helicobacter pylori challenge in mice. Microb Pathog 2008; 44: 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger A. Science commentary: Th1 and Th2 responses: what are they? BMJ 2000; 321: 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy K, et al. Induction of mucosal and systemic immune responses by immunization with ovalbumin entrapped in poly (lactide-co-glycolide) microparticles. Immunology 1994; 81: 661–667. [PMC free article] [PubMed] [Google Scholar]
- Banner KH, et al. Ovalbumin challenge following immunization elicits recruitment of eosinophils but not bronchial hyperresponsiveness in guinea-pigs: time course and relationship to eosinophil activation status. Pulm Pharmacol 1996; 9: 179–187. [DOI] [PubMed] [Google Scholar]
- Wegmann M. Animal models of chronic experimental asthma-strategies for the identification of new therapeutic targets. J Occup Med Toxicol 2008; 3: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JR, et al. Apigenin protects ovalbumin-induced asthma through the regulation of GATA-3 gene. Int Immunopharmacol 2009; 9: 918–924. [DOI] [PubMed] [Google Scholar]
- McCaskill A, et al. Anaphylaxis following intranasal challenge of mice sensitized with ovalbumin. Immunology 1984; 51: 669–677. [PMC free article] [PubMed] [Google Scholar]
- Roman B, et al. Intradermal immunization with ovalbumin-loaded poly-ε-caprolactone microparticles conferred protection in ovalbumin-sensitized allergic mice. Clin Exp Allergy 2007; 37: 287–295. [DOI] [PubMed] [Google Scholar]