

Abstract
Objective
The bioequivalence study was conducted to compare the developed paediatric fixed-dose combination (FDC) zidovudine/lamivudine/nevirapine (60/30/50 mg) tablet – the test formulation – with the combined mixture of single-entity innovator products (reference product).
Methods
A single-dose open-label randomized two-way crossover study was conducted in healthy adult African volunteers after an informed consent was obtained. The 24 volunteers, divided into two groups, were administered the products after an overnight fast on two treatment days with 14 days of washout period. Blood samples were collected for 96 h and analysed using a validated RP-HPLC-UV assay method. Pharmacokinetic (PK) parameters (non-compartmental model) were assessed with WinNonlin® software. Analysis of variance (ANOVA) and FDA bioequivalence statistical criterion of 90% CI or 80% to 125% range (set at P < 0.05) of least square geometric means (LSGM) ratios of test: reference product for Cmax, AUC0–t, and AUC0–∞ were determined.
Results
ANOVA indicated that the period, sequence and formulation had no significant effect on the PK parameters (P > 0.05). The 90% CIs for all the drugs were within the 80% to 125% range.
Conclusion
The developed FDC tablet is bioequivalent to the reference product.
Keywords: bioequivalence, lamivudine, zidovudine, nevirapine, paediatric, tablet, two-way crossover, study
Introduction
The paediatric population infected with human immunodeficiency virus (HIV) is in dire need of suitable treatment regimens of antiretroviral drugs. Transmission of HIV from mother to child remains the main source of acquiring AIDS in children despite the scaling up of prevention of mother-to-child transmission (MTCT) under the Global Plan to eliminate MTCT and new HIV infections in children by 2015.[1] UNAIDS estimates that the number of children under 15 years of age living with AIDS in 2012 was 3.3 million (3.0–3.7 million), and nearly 260 000 (230 000–320 000) children of this age were newly infected, with 210 000 (190 000–250 000) deaths related to the AIDS.[2] Most HIV infections occur in sub-Saharan Africa, with 90% of infected children resident in the region.
One of the strategies to mitigate this compounded problem in treating HIV-infected paediatric patients is to develop a suitable combination formulation with the active drugs that demonstrated safety and efficacy with less toxicity. Combination antiretroviral (ARV) therapy, generally regarded as highly active antiretroviral therapy (ART), has been more effective against suppressing the HIV virus replication and in improving immunological response in terms of CD4 cell count.[3] ART regimen comprises combination of at least three different ARV drugs from at least two classes as follows: (1) combination of two nucleotide reverse-transcriptase inhibitors (NRTIs) including nucleotide reverse-transcriptase inhibitors (NtRTIs), (2) combination of two NRTIs including a protease inhibitor (PI) and (3) combination of two NRTIs including a PI plus and non-nucleotide reverse-transcriptase inhibitors (NNRTI) or others. Although ART has not been able to cure the disease totally, the combination regimen has definitely helped to reduce the morbidity and mortality rates of HIV-infected patients.[4] It is pertinent to mention that the combination dose regimen for HIV treatment is dynamic and continuously evolving as new data on safety and efficacy are being established in humans. Among the various ARV combinations, combination of two NRTIs and a NNRTI (AZT/3TC/NVP) has been extensively used in HIV-infected ARV treatment-naive adults and children below 3 years.[4–6] Recent World Health Organization recommendations for preferred first-line and alternative first-line regimens for paediatrics are as follows. For children under 3 years old, the preferred combination is abacavir (ABC) or AZT + 3TC + lopinavir/ritonavir (LPV/r), while the alternative is ABC or AZT + 3TC + NVP. For children 3–9 years and adolescent (<35 kg), the preferred first-line is ABC + 3TC + efavirenz (EFV); and the alternative is ABC or AZT or tenofovir (TDF) + 3TC (or emtricitabine – FTC) + NVP (or EFV). For adolescents (10–19 years) ≥35 kg, the preferred first-line regimen is TDF + 3TC + (or FTC) + EFV; the alternative is ABC or AZT or TDF + 3TC or EFV.[7] Despite the WHO recommendations, a significant portion of children with AIDS is not receiving treatment. However, according to 2014 WHO Brief on optimization of treatment options and improving access to paediatric ARVs, only 23% of the 3.2 million children estimated to be living with HIV were receiving ART and in 2013 alone, 190 000 [170 000–220 000] children died of HIV-related causes.[8]
To contribute to affordability and availability of the drugs for children with AIDS, an initiative in developing a fixed-dose combination (FDC) formulation with any of the ARV drugs was undertaken. The alternative first-line AZT/3TC/NVP as a particular combination for children up to 9 years has demonstrated suitable efficacy and safety in paediatric population.[7–11] Furthermore, the major aims with FDC formulations are to (1) achieve synergistic therapeutic effects, (2) reduce dose and related toxicity, and (3) minimize or delay the induction of drug resistance. This particular combination of drugs has been indicated to have a synergistic effect making the combination as a choice of therapy.[9]
To achieve the unmet clinical need in the treatment of the paediatric populations and ensure that children are not left behind in the UNAIDS and WHO global efforts to reach the new global targets, it is crucial to develop first-line combination products and expand use of less toxic, simplified and more robust drug regimens tested using respective bioavailability/bioequivalence protocols.[8] The International Conference on Harmonization (ICH) guideline E11 and its evolving revision (E11(R1) on clinical investigation of medicinal products in paediatric population suggest that for relative bioavailability comparison of paediatric formulations, adult subjects can be used to establish the similarity between the formulations.[10,11] The FDA Pediatric Study Plan[12] and the European policies are consistent with the ICH guidelines. Union Regulation on Medicinal Products for Children[13]. Therefore, the main objective of this study was to assess the bioequivalence of a recently developed FDC proprietary tablet formulation comprising of AZT/3TC/NVP (60/30/50 mg) with the individual branded liquid formulations. The successful outcome of this study would be a progress towards availability and successful AIDS treatment and management in paediatric population, especially in resource-limited countries.
Methods
Ethics
In accordance with the Declaration of Helsinki,[14] good clinical practice (GCP) guidelines[15] and relevant regulatory guidelines, an Institutional Review Board (IRB) protocol entailing the purpose and justification of the study design was submitted to and approved before the clinical trial, by the following institutions: (1) Duquesne University, Pittsburgh, PA, 15282 (Protocol ID 09-56), (2) Roosevelt University, Schaumburg, IL, 60173 (Protocol ID 2010-73), Elim Pediatric Pharmaceuticals under Cooperative Research Agreement (CRDA) with Roosevelt University, and Bowen University Teaching Hospital (BUTH), Ogbomoso, Nigeria. BUTH's IRB is registered by Nigeria's Health Research Ethics Committees (NHREC). All the subjects were informed of all the procedures, duration of the study, anticipated risks and possible discomfort. Informed consent was obtained from each subject before the study initiation. The clinical protocol was approved by Nigeria's National Agency for Food and Drug Administration Control (NAFDAC) and the FDA under the Investigation New Drug Application Submission (IND Number: 108,518) through Elim Pediatric Pharmaceuticals (EPPI).
Study design
This clinical trial was a single-centre, randomized, single-dose, open-label, two-period [I, II], two-sequence (test/reference (TR, RT)), non-replicated crossover design under fasted conditions (http://ClinicalTrials.gov Identifier: NCT01469520). The washout period was set for 14 days equivalent to seven half -lives of NVP, owing to its long elimination half -life.[16] All the subjects went through the pre-enrolment evaluation, which was carried out in BUTH, Ogbomoso, Nigeria. Those who met the set inclusion criteria discussed below were enrolled for the clinical study. Subjects were coded and then randomly assigned to each treatment group based on the computer-generated random numbers.
Subjects and treatment groups
Subjects were recruited from the communities of Ogbomoso and surrounding regions. Healthy adults, including both male and female (all of Black race) in a range of 22–55 years of age, were considered. The eligibility criteria included subjects who were: (1) healthy on the basis of a pretrial physical examination, medical history, clinical laboratory tests including blood chemistry and haematology tests, (2) competent and willing to sign informed consent and (3) willing to be hospitalized for 24-hour intensive sampling period.
Subjects were excluded from the study based on the following: (1) history of hypersensitivity to drugs, (2) alcohol or tobacco usage within 48 h of study, (3) abnormal laboratory biochemistry values, (4) any clinically significant disease if diagnosed during the screening period and (5) subjects who were treated with experimental drugs within 30 days before the study entry and (6) pregnancy. Subjects were considered eligible for the study if the medical and medication histories, and demographics and clinical data parameters at the time of the screening were within the clinically acceptable range.
To minimize variances due to subject characteristics such as weight, age and gender, block randomization of the subjects was performed.[17] The block randomization was performed to reduce bias and obtain as much as possible balance in the assigning of subjects to the treatment groups. The process also increases the probability that equal number of subjects will be in each group or block. Part of the aim is also to ensure that variability within the block is much less than between the blocks. For the randomization, each treatment group was formed based on two specified ranges of weight and age characteristics of the subjects. Of total 44 subjects that were screened, 33 subjects were selected as eligible. Twenty-four subjects were then randomly selected and grouped into the two homogenous treatment groups (block randomization). Twenty-four subjects were chosen, as this sample size provides sufficient power of 80% to detect 20% difference at a 5% level of significance.[18]
Adverse events were monitored throughout the study. Subjects were instructed to inform the clinical investigators in case of any adverse events that occurred during the study.
Treatment drugs and administration
The reference drug regimen comprised of a mixture of (1) 24 ml of Retrovir® (AZT), 10 mg/ml syrup (manufactured by GlaxoSmithKline, Lot No. 0B001); (2) 12 ml of Epivir® (3TC), 10 mg/ml syrup (manufactured by GlaxoSmithKline, Lot No. 0F003); and (3) 20 ml of Viramune® (NVP), 10 mg/ml suspension (manufactured by Boehringer Ingelheim Pharmaceuticals Inc, Lot No. 059864J). The test drug comprised of a proprietary formula of AZT/3TC/NVP (60/30/50 mg) orally disintegrating tablet (n = 4). The tablet target weight was 450 mg and the mean per cent drug released (%CV) at 100 rpm in phosphate buffer (pH 2.0) after 60 min for AZT/3TC/NVP is 99%(1.74)/99.01%(1.25)/93.14%(0.39), respectively. Both regimens (test and reference drugs) have equivalent adult doses of AZT/3TC/NVP: 240/120/200 mg.
The clinical batch of the test product was manufactured in a cGMP-certified facility in Norristown, PA, in compliance with the current GCP standards. The product was shipped to the clinical site after all the criteria of end-product release tests were met.
The subjects, after an overnight fast, were administered with either the reference or the test drug regimen. The reference regimen was administered via an oral syringe, while the test product was allowed to be placed in the mouth and allowed to disperse. A total of 240 ml of water was given to ensure the entire drug was swallowed. Additionally, the clinical investigator performed mouth check to assess the administration compliance.
Blood sample collection
Subjects were housed in the clinical facility of BUTH before the dosing started and maintained for 24 h postdosing. A venipuncture was made just before dosing and an in-dwelling catheter was inserted and maintained in the subjects for blood sample collection purpose. Blood samples, 5 ml each, were collected at predetermined intervals of 0, 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 12, 24, 48, 72 and 96 h postdosing. Blood samples after 24 h were collected on an ambulatory basis. Fresh venipuncture was made for each ambulatory blood sample collection. A total of 16 blood samples were collected into labelled heparinized tubes and centrifuged and the plasma samples thus collected were kept frozen at −80 °C until assayed using a validated isocratic HPLC-UV method.
Bioanalytical assay
The sample analysts were blinded to the randomization. Bioanalysis of the samples were performed under the guidelines for good laboratory practice utilizing a validated HPLC-UV method according to the ICH guidelines for validation parameters such as selectivity, linearity, accuracy and precision and lower limit of detection (LOD) and quantitation (LOQ).[19] The plasma samples were equilibrated at room temperature and drugs were extracted using liquid–liquid extraction procedure. A total of 376 μl of equilibrated plasma was transferred to the microcentrifuge tubes to which 200 μl of chilled acetonitrile was added to denature the protein. To this mixture, 24 μl of aprobarbital solution was added as an internal standard and additional 576 μl of chilled acetonitrile was added to extract the drug. The tube was vortexed for 3 min and centrifuged at 1200g for 12 min at −4 °C. A total of 800 μl of clear supernatant liquid was evaporated under nitrogen at 37 °C, and the solid residue thus obtained was dissolved in 25 mm potassium phosphate buffer and analysed by HPLC. The detailed method including the drug extraction and validation is reported elsewhere.[20,21] The chromatograms were extracted for each sample, and ratio of the peak area of drug to the internal standard was evaluated against the calibration samples to estimate the drug concentrations.
Pharmacokinetic and statistical analysis
The plasma concentration profiles of all the analytes for all the subjects were obtained after HPLC analysis. Maximum concentration (Cmax) and time to reach the maximum concentration (Tmax) were evaluated from the concentration–time profiles. A non-compartmental model analysis was performed using Phoenix WinNonlin® 6.1 PK software (Pharsight Corporation, Sunnyvale, CA, USA) to determine (1) area under the curve from time 0 to last measurable concentration (AUC0−t), (2) area under the curve extrapolated to infinity (AUC0−∞), using linear trapezoidal rule and (3) terminal elimination half-life (t1/2) and rate constant (Ke). Arithmetic mean and range of Tmax were also evaluated.
Analysis of variance (ANOVA) were performed on natural-log-transformed AUC0−t, AUC0−∞ and Cmax using the following model (Equation 1) including sequence, subjects nested within sequence, period and drug formulations.
| 1 |
where Yijk = pharmacokinetic response of the kth subject in the ith sequence at the jth period, μ = overall mean, Gi = fixed effect of sequence (i = 1, 2), Sik = random effect of subject k in sequence i (k = 1, 2, 3…N), Pj = fixed effect of period (j = 1, 2), Tt(ij) = fixed effect of treatment (t = 1, 2) in sequence i and period j and Eijk is the residual error. The main objective was to assess whether these model parameters had any effect on the PK parameters. In addition, analysis of variance (ANOVA) was carried out using log-transformed data and 90% CI of least square geometric means (LSGM) ratios Cmax, AUC0−t, and AUC0−inf. Two one-sided t-tests were used for the determination of the 90% CI to assess whether (1) the test product is significantly less bioavailable compared to the reference product, or (2) the reference product is significantly less bioavailable compared to the test product.[18] If the CIs for the mean ratios of test: reference product falls within 80–125% range as per FDA guidance, the products were considered bioequivalent.
Results and Discussion
Demographics and safety results
The study population consisted of 24 healthy subjects (eighteen men, six women) with a mean age of 32.6 years (22–48 years) and mean weight of 62.7 kg (46–73 kg), all of Black race. Each treatment group had comparable groups of subjects with aforementioned characteristics. Of 24 subjects, all of them completed the study. No deaths or serious adverse events occurred during the conduct of study. Both treatments were well tolerated with a few effects (not due to the study drugs) reported by the subjects such as common cold and throat infections (two cases), which were managed by the clinicians at BUTH.
Bioanalytical assay limit of detection and quantitation
The LOQ and LOD were calculated for 3TC, AZT and NVP using the slope and intercept of the standard calibration curves. The LOD for 3TC, AZT and NVP was 6.55, 16.13 and 17.0 ng/ml, respectively. Similarly, LOQ for 3TC, AZT and NVP was observed to be 19.85, 48.88 and 50.00 ng/ml, respectively.
Pharmacokinetics parameters
The mean plasma concentration–time profiles for all the drugs 3TC, AZT and NVP are, respectively, shown in Figures 1–3. The profiles for both the test and reference for each drug are similar. The pharmacokinetic parameters such as Tmax for 3TC, AZT and NVP in both test and reference drugs are represented in Tables 1–3, which are comparable to the data presented in the prescribing information of individual innovator products.[22–24]
Figure 1.
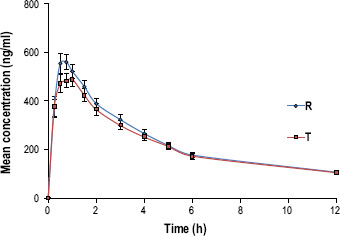
Mean (±SE) plasma concentration–time profile of test product AZT vs reference after a single-dose administration..
Figure 3.
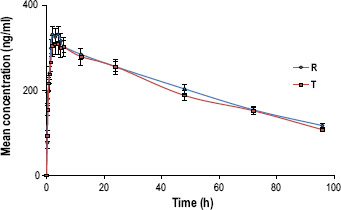
Mean (±SE) plasma concentration–time profile of test product NVP vs reference after a single-dose administration..
Table 1.
Pharmacokinetic parameters data of AZT
| Parameter | T | R | LMS’ ratio (T/R) | 90% CI |
|---|---|---|---|---|
| T max (h)**,a | 0.59 (0.25–1.5) | 0.5 (0.25–1.5) | ||
| C max (ng/ml)b | 605 ± 19 | 623 ± 26 | 97.10 | 86.57–108.90 |
| AUC0−t (ng h/ml)b | 2569 ± 23 | 2734 ± 24 | 93.97 | 82.43–107.12 |
| AUC0−∞ (ng h/ml)b | 3541 ± 24 | 3637 ± 23 | 97.36 | 84.69–111.91 |
CI, confidence interval. **Arithmetic mean (n = 24). aMedian (range). bGeometric least square means.
Table 3.
Pharmacokinetic parameters for NVP
| Parameter | T | R | LMS ratio (T/R) | 90% CI |
| T max (h)**,a | 3.4 (0.75–6.0) | 3.0 (1.5–6.0) | ||
| C max (ng/ml)b | 391 ± 26 | 363 ± 23 | 107.77 | 98.86–117.46 |
| AUC0−t (ng h/ml)b | 19 704 ± 26 | 19 056 ± 21 | 103.40 | 92.70–115.32 |
| AUC0−∞ (ng h/ml)b | 31 122 ± 25 | 30 615 ± 22 | 101.65 | 89.95–114.88 |
CI, confidence interval. **Arithmetic mean (n = 24). aMedian (range). bGeometric least square means.
Figure 2.

Mean (±SE) plasma concentration–time profile of test product 3TC vs reference after a single-dose administration..
Table 2.
Pharmacokinetic parameters data of 3TC
| Parameter | T | R | LMS ratio (T/R) | 90% CI |
|---|---|---|---|---|
| T max (h)**,a | 0.75 (0.5–1.5) | 0.67 (0.5–1.5) | ||
| C max (ng/ml)b | 937.69 ± 15 | 892 ± 20 | 105.04 | 96.53–114.37 |
| AUC0−t (ng h/ml)b | 2949.50 ± 29 | 2796 ± 19 | 105.47 | 94.62–117.56 |
| AUC0−∞ (ng h/ml)b | 3859.42 ± 27 | 3961 ± 30 | 97.42 | 84.54–112.26 |
CI, Confidence interval. **Arithmetic mean (n = 24). aMedian (range). bGeometric least square means.
Considering the AZT, 3TC and NVP disposition based on the values obtained, the maximum concentration reached (Cmax) for AZT and 3TC was relatively lower than the data published in similar other studies utilizing these drugs.[11] This can be rationalized on the basis of the dose used for this particular clinical study, which was lower (3TC/AZT: 120/240 mg) than the dose (3TC/AZT: 150/300 mg) used in other study. This dose ratio was based on the ratio of the doses of AZT and 3TC fixed for the FDC formulation as indicated by the Pediatric Antiretroviral Working Group (PAWG) of World Health Organization as an ideal dosing strength for urgently needed antiretroviral product for paediatrics.[9,25] Pharmacological activity of 3TC and AZT (NRTIs) is dependent on their rate and extent of intracellular phosphorylation to their active intracellular triphosphate metabolites. Their relationships between the plasma drug concentrations and virological/immunological outcomes are not well correlated. Unfortunately, the measurement of these active metabolites is expensive and labour-intensive and requires larger blood volumes than typical plasma samples.[26] Despite this void in correlating the plasma and intracellular concentration, the relevance of using AZT plasma drug concentration as a surrogate has been reported illustrating higher dose of AZT resulted in higher levels of its triphosphate moiety.[27]
The elimination half -life (T1/2) as calculated from the terminal elimination rate constant (λ) was found to be as follows: (1) AZT (test/reference): 5.69/5.56 h, (2) 3TC (test/reference): 5.03/5.94 h and (3) NVP (test/reference): 60.06/62.82 h. The results were comparable with the results reported elsewhere except for NVP.[11] This may be attributed to the truncation of the blood sampling at 96 h not effectively capturing three half-lives of NVP elimination (T1/2 NVP = 45 h). Hence, the data extrapolated from the terminal linear portion of the concentration–time profile might have underestimated the values of elimination rate constant that would have affected the half-life calculation as the two are inversely related.
Statistical analysis
ANOVA of the AUC showed that the period, sequence and formulation did not have any significant effect on the PK parameters (P > 0.05), indicating that the observed differences in PK parameters are independent of these parameters. ANOVA of AUC0−t for AZT is represented below where all the model parameters were found to be not statistically significant (Table 4). ANOVA results for lamivudine and NVP similarly showed no significant model effects.
Table 4.
ANOVA of the model parameters of AUC (AZT)
| Source | DF | SS | MS | F-stat | P-value |
| Sequence | 1 | 0.000733 | 0.000733 | 0.015384 | 0.902415 |
| Sequence × subject | 22 | 1.048417 | 0.047655 | 0.768257 | 0.729206 |
| Formulation | 1 | 0.026469 | 0.026469 | 0.426713 | 0.520375 |
| Period | 1 | 0.02224 | 0.02224 | 0.358526 | 0.555442 |
| Error | 22 | 1.36467 | 0.06203 |
The LSGM ratios (90% CIs) of test: reference product for Cmax, AUC0−t and AUC0−inf, for AZT were found to be 97.1 (86.6% to 108.9%), 94.0 (82.4% to 107.1%) and 97.4 (84.7% to 111.9%); for 3TC were 105.0 (96.5% to 114.4%), 105.5 (94.6% to 117.6%) and 97.4 (84.5% to 112.3%); and for NVP were 107.8 (98.9% to 117.5%), 103.4 (92.7% to 115.3%) and 101.7 (89.9% to 114.9%) respectively. All 90% CIs were within the range of 80% to 125%. These results suggest that the developed fixed-dose combination fast-disintegrating tablet, the test product and the co-administered individual reference products were bioequivalent as indicated by the similar rate and extent of bioavailability.
The power analysis for the PK parameters was calculated by the Phoenix software utilizing Anderson–Hauck test statistic (tAH).[18] Based on this analysis, the powers to detect difference between the various PK parameters represented in Table 5 were greater than 80%.
Table 5.
Power of analysis
| PK parameters | 3TC | AZT | NVP |
| C max | 0.9948 | 0.9407 | 0.9939 |
| AUC0−t | 0.9583 | 0.8802 | 0.9567 |
| AUC0−∞ | 0.8326 | 0.8437 | 0.9145 |
Power = 1 − (probability of type II error).
The current sample size of 24 subjects provided more than 80% power to demonstrate bioequivalence between the test and the reference products. Also, the two treatments were shown to be equally safe and well tolerated, with a few minor adverse effect reported. As a result, the developed FDC product is expected to provide a similar safety/efficacy profile as the individual reference products. Successful management of HIV treatment depends on long-term suppression of viral replication. This can be achieved by the use of potent antiretroviral regimens offering adherence/compliance to the patients. Hence, the developed FDC product is anticipated to fulfil that need.
Conclusions
The average bioequivalence assessment of the test product compared with the reference products conducted in 24 healthy human adult volunteers was successful. Based on the 90% CIs of PK parameters falling in the range of 80% to 125%, as obtained from PK analysis, it can be concluded that the data met the regulatory requirements to assume bioequivalence between the products. This indicated the developed product can be used as a pharmaceutical equivalent product to treat HIV-infected paediatric population. Although the developed product is intended for use in paediatrics, the clinical study was conducted in healthy adults following the ICH guidelines intended for investigations in medicinal products for paediatrics. The results obtained from the crossover bioequivalence studies cannot be extrapolated to the paediatric population, but the assessment allows establishing the similarity between the developed product and its counter single-entity products as marketed by the innovator companies. Detail studies on the pharmacokinetics and pharmacodynamics of the developed product in paediatric HIV-infected population need to be established. This study is a milestone in fulfilling the overarching goal to cater to the unmet clinical need of HIV-infected paediatric populations. Phase II clinical trial is being planned for the future.
Declarations
Conflict of interest
The Author(s) declare that they have no conflict of interest.
Acknowledgements
The authors would like to acknowledge the sponsors of the study: Roosevelt University and Elim Pediatric Pharmaceuticals; Duquesne University for funding very early phase of the product development through the Hunkele Dreaded Disease Grant, and National Institute of Health (NIH) grant 1 R03 HD059540-01 for the initial product development. We also acknowledge Bowen University Teaching Hospital, Nigeria, the clinical site and the clinical trial team members for their contributions.
References
- 2013 Progress report on the Global Plan towards the elimination of new HIV infections among children by 2015 and keeping their mothers alive. Geneva: UNAIDS, 2013. http://www.unaids.org/en/media/unaids/contentassets/documents/unaidspublication/2013/20130625_progress_global_plan_en.pdf (accessed 30 April 2016). [Google Scholar]
- 2013 Report on the global AIDS epidemic. Geneva: UNAIDS, 2013. http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf (accessed 30 April 2016). [Google Scholar]
- Volberding PA, Deeks SG. Antiretroviral therapy and management of HIV infection. Lancet 2010; 376: 49–62. [DOI] [PubMed] [Google Scholar]
- Violari A et al. Early antiretroviral therapy and mortality among HIV-infected infants. N Engl J Med 2008; 359: 2233–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Plan towards the elimination of new HIV infections. Downloaded from http://aidsinfo.nih.gov/guidelines on 9/28/2016.
- Samji H et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS One 2013; 8: e81355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach. Geneva: World Health Organization, 2013. http://www.who.int/hiv/pub/guidelines/arv2013/en (accessed 30 April 2016). [PubMed] [Google Scholar]
- WHO Brief: HIV Treatment: Optimizing Treatment Options and Improving Access to Priority Products for Children Living With HIV. WHO Brief. Geneva, Switzerland. December 2014. Accessed September 28, 2016
- International Conference on Harmonisation (ICH) Guidance for Industry E11 Clinical Investigation of Medicinal Products in the Pediatric Population, 2000. [PubMed]
- Final Concept Paper: ICH E11(R1): Clinical Investigation of Medicinal Products in the Pediatric Population dated 17 July 2014. Endorsed by the ICH Steering Committee on 14 August 2014.
- Chou TC. The median-effect principle and the combination index for quantitation of synergism and antagonism. In: Chou TC, Rideout DC, ed. Synergism and Antagonism in Chemotherapy. San Diego, CA: Academic Press, 1991: 61–102. [Google Scholar]
- FDA's Pediatric Study Plans: Content and Processes for Submitting Initial Pediatric Study Plans and Amended Initial Pediatric Study Plans Guidance for Industry. March 2016 Procedural Revision 1.
- Regulation (EC) No 1901/2006 of the European Parliament and of the Council on medicinal products for paediatric use and amending Regulation (EEC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. 12 December 2006.
- World Medical A . The Helsinki declaration. Orv Hetil 1965; 106: 1715–1716. [PubMed] [Google Scholar]
- ICH Harmonised Guideline: Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2). Current Step 2 Version. June 2015.
- Laurence L. Brunton, John S. Lazo, Keith L. Parker, Eds. Goodman & Gilman's the Pharmacological Basis of Therapeutics, 12th edn. China: McGraw-Hill. 2011. [Google Scholar]
- Kleinbaum DG et al. Applied Regression Analysis and Other Multivariable Methods. CA, USA: Duxbury Press, 2007. [Google Scholar]
- Chow SC, Liu J. Design and Analysis of Bioavailability and Bioequivalence Studies, Vol. 27. Taylor & Francis, USA: Chapman & Hall/CRC Series, 2008. [Google Scholar]
- ICH . Guidelines: Validation of Analytical Procedures: Text and Methodology Q2 (R1). ICH Harmonised Tripartite Guideline, ICH Expert Working Group. 2005. [Google Scholar]
- Joshi A et al. Post marketing in vitro/in vivo assessment of fixed dose combination products of first line antiretrovirals. J Pharm Sci 2010; 99: 2655–2663. [DOI] [PubMed] [Google Scholar]
- Esseku F et al. A randomized Phase I bioequivalence clinical trial of a pediatric fixed-dose combination antiretroviral reconstitutable suspension in healthy adult volunteers. Antivir Ther 2012. doi: 10.3851/IMP2310. http://www.intmedpress.com/journals/avt/abstract.cfm?id=2310&pid=48. [DOI] [PubMed]
- Epivir: Prescribing Information, Manufactured Under Agreement from Shire Pharmaceuticals Group, plc, Basingstoke, UK Glaxosmithkline Inc.
- Retrovir: Prescribing Information, Manufactured Under Agreement from Shire Pharmaceuticals Group, plc, Basingstoke, UK Glaxosmithkline Inc.
- Viramune: Prescribing Information, Boehringer Ingelheim Inc. CT 06877 USA.
- World Health Organization . Antiretroviral Therapy of HIV Infection in Infants and Children: Towards Universal Access: Recommendations for a Public Health Approach. WHO Library Cataloguing-in-Publication Data, 2010. [PubMed] [Google Scholar]
- Neely MN, Rakhmanina NY. Pharmacokinetic optimization of antiretroviral therapy in children and adolescents. Clin Pharmacokinet 2011; 50: 143–189. [DOI] [PubMed] [Google Scholar]
- Fletcher CV et al. Concentration-controlled zidovudine therapy. Clin Pharmacol Ther 1998; 64: 331–338. [DOI] [PubMed] [Google Scholar]