

Abstract
The Major Facilitator Superfamily (MFS) is currently the largest characterized superfamily of transmembrane secondary transport proteins. Its diverse members are found in essentially all organisms in the biosphere and function by uniport, symport, and/or antiport mechanisms. In 1993 we first named and described the MFS which then consisted of 5 previously known families that had not been known to be related, and by 2012 we had identified a total of 74 families, classified phylogenetically within the MFS, all of which included only transport proteins. This superfamily has since expanded to 89 families, all included under TC# 2.A.1, and a few transporter families outside of TC# 2.A.1 were identified as members of the MFS. In this study, we assign nine previously unclassified protein families in the Transporter Classification Database (TCDB; http://www.tcdb.org) to the MFS based on multiple criteria and bioinformatic methodologies. In addition, we find integral membrane domains distantly related to partial or full-length MFS permeases in Lysyl tRNA Synthases (TC# 9.B.111), Lysylphosphatidyl Glycerol Synthases (TC# 4.H.1), and cytochrome b561 transmembrane electron carriers (TC# 5.B.2). Sequence alignments, overlap of hydropathy plots, compatibility of repeat units, similarity of complexity profiles of transmembrane segments, shared protein domains and 3D structural similarities between transport proteins were analyzed to assist in inferring homology. The MFS now includes 105 families.
Keywords: Membrane transport, major facilitator, MFS, Lysyl tRNA ligase, Lysyl phosphatidylglycerol synthase, Cytochrome b561
1.1. Introduction
TCDB implements a system of classification, adopted by the International Union of Biochemistry and Molecular Biology (IUBMB), for representative, recognized and hypothetical transport proteins found in all living organisms on Earth [1–3]. Using functional and phylogenetic information derived from publications on transport systems, TCDB classifies over 14,000 transport systems and potential transporters into over 1,400 families, referencing over 17,000 publications [4–10]. Ongoing efforts are focused on the identification of distant relationships between transport protein families, allowing categorization of both pre-existing and novel families into superfamilies. Currently, TCDB contains over 80 Superfamilies (see http://tcdb.org/superfamily.php) [3, 11].
The largest and most diverse superfamily of secondary carriers characterized to date is the Major Facilitator Superfamily (MFS) [4, 5, 12]. It may, in fact, be the largest superfamily of all types of transporters, since its only “competitor” is the ABC superfamily which has been shown to consist of at least three evolutionarily independently arising families of transport proteins, all of which use homologous ATPases for energy coupling [13]. Structural and sequence analyses of representative members of the MFS show 12 TMSs surrounding a central cavity, forming a semi-symmetrical structure [14]. Evidence suggests that MFS members arose via an intragenic duplication event in which a gene encoding a 6 TMS protein yielded the 12 TMS structure of current MFS proteins [12]. Furthermore, it is believed that each of the 6 TMS domains arose from a primordial 3 TMS ancestor [14, 15]. Crystallographic studies performed on bacterial members of the MFS provided further evidence of a 3+3 inverted structure within the 6 TMS repeat unit [16]. Thus, TMSs 1–3 are related to the TMSs 4–6 by a 180° rotation about the axis running parallel to the lipid bilayer, as are TMSs 7–9 and TMSs 10–12 [16].
Currently recognized members of the MFS bind their substrates stereospecifically and utilize a carrier-mediated process to catalyze transport across biological membranes [17]. MFS secondary carriers transport by (1) uniport, where single molecular entities are transported by facilitated diffusion, or by potential-driven processes for charged solutes, (2) symport, where two or more solutes are transported in the same direction, driven by chemiosmotic energy, in this case, often the electrochemical gradient of protons, called the proton motive force (pmf), or (3) antiport, where two or more solutes are transported in opposite directions, again using chemiosmotic energy to drive the vectorial process. Most members of the MFS share a three dimensional structure that consists of two domains surrounding a central substrate binding site [18, 19]. These transporters operate by an alternating access mechanism where the two halves of the protein move, relative to each other, like a rocker switch, mediated in part by salt bridge formation and breakage during the transport cycle [20, 21].
MFS porters, or permeases, are known to exhibit specificity for sugars, drugs, neurotransmitters, amino acids, vitamins, organic and inorganic ions, as well as many other small compounds, depending on the specific porter, but not macromolecules such as proteins, polysaccharides and nucleic acids [22]. Typical transporters of the MFS are of 400–600 amino acyl residues (aas) in length, and with few exceptions, possess either a 12 or 14 trans-membrane α-helical segment (TMS) topology.
Aside from the MFS families listed under TC# 2.A.1, when the studies reported here were initiated, there were 6 additional transport protein families in TCDB with evidence supporting their membership to the MFS [12]. These families and those reported here are characterized within the MFS superfamily in TCDB (see Superfamily hyperlink in TCDB).
The Glycoside-Pentoside-Hexuronide:Cation Symporter (GPH) Family (TC# 2.A.2), as its name suggests, consists of symporters that usually catalyze uptake of glycosides in conjunction with a monovalent ion, usually H+. The functionally characterized proteins of this family are from bacteria, archaea, and eukaryotes. Members of the GPH family are usually around 500 aas in length and possess the characteristic 12 TMS topology [12].
The ATP:ADP Antiporter (AAA) Family (TC# 2.A.12) contains transporters that appear to be obligate exchange translocases with specificity frequently for ATP and ADP [23]. They take up ATP into the cell in exchange for ADP, but can also transport inorganic phosphate and other phosphorylated nucleosides [24]. These proteins have 12 putative TMSs and are most commonly found in intracellular pathogenic bacteria.
The Proton-dependent Oligopeptide Transporter (POT/PTR) Family (TC# 2.A.17) is another ubiquitous family that catalyzes peptide uptake. Members usually exhibit 12 putative TMSs. It has been suggested that pairs of salt bridge interactions between the transmembrane α-helical structures work together to provide the alternating access transport mechanism [25]. Mammalian members of this transporter family, PepT1 and PepT2, are responsible for the uptake of pharmaceutically important drug molecules such as antibiotics and antiviral agents [25, 26].
The Reduced Folate Carrier (RFC) Family (TC# 2.A.48) includes uptake porters for folates, reduced folates, folate analogues, biotin and thiamine. Folates, also known as vitamin B9, are essential vitamins for humans, and folate deficiency contributes to a variety of health problems. These RFC members mediate the intestinal absorption of the anti-cancer drugs, methotrexate and pralatrexate [27]. Amino acid replacement experiments have shown that the region between TMSs 1 and 2 forms a substrate-binding pocket [28]. Like other MFS family porters, RFC members are typically between 500–600 aas in size and possess 12 putative or established TMSs. The Organo Anion Transporter (OAT) Family (TC# 2.A.60) contains proteins that catalyze facilitated transport of large amphipathic organic ions such as prostaglandins, bile acids, steroid conjugates, thyroid hormones, oligopeptides, drugs, toxins, and various xenobiotics [29, 30]. Human OAT transporters play important roles in drug and metabolite transport across the blood-brain barrier and in the kidneys [31, 32]. These transporters have 12 putative TMSs. Their evolutionary histories and functional diversification have been examined [32].
Members of the Folate-Biopterin Transporter (FBT) Family (TC# 2.A.71) transport folate and biopterin across the cell membrane and are believed to function by H+ symport [33]. Most functionally characterized members of the FBT family are from mammals and protozoa, but homologs exist in bacteria, plants, and algae [34].
In this paper, we incorporate 9 additional TC families into the MFS. We believe that these proteins, or parts of them, all share a common evolutionary origin. In addition to the above described families, currently established constituents of the MFS, we provide evidence that members of the following families share a common origin with recognized MFS superfamily proteins: (1) the Equilibrative Nucleoside Transporter (ENT; TC# 2.A.57) Family, (2) the Aromatic Acid Exporter (ArAE; TC# 2.A.85) Family, (3) the Ferroportin (Fpn; TC# 2.A.100) Family, (4) the Eukaryotic Riboflavin Transporter (E-RFT; TC# 2.A.125) Family, (5) the Lysyl phosphatidylglycerol synthase (MprF; TC# 4.H.1) Family, (6) the Eukaryotic Cytochrome b561 (Cytb561; TC# 5.B.2) Family, (7) the Conidiation and Conidial Germination Protein (CCGP; TC# 9.B.57) Family, (8) the 6 TMS Lysyl tRNA Synthase (LysS; TC# 9.B.111) Family, and (9) the 6 TMS DUF1275 (DUF1275; TC# 9.B.143) Family. Sufficient evidence using our bioinformatic methodologies suggests that members of these families, or domains within these proteins, derive from a common MFS ancestor via pathways similar to those described previously for MFS permeases [35]. These relationships were inferred based on (1) the quality of sequence alignments, (2) overlap of hydrophobic peaks in hydropathy curves plus compatibility of repeat units, (3) compatibility of the locations of simple/complex TMSs based on the Transmembrane helix: simple or complex (TMSOC) classification [36, 37], (4) Pfam [38] domain content, and (5) 3D structural similarity when available. The families included in this study are tabulated with their properties in Table 1, and their average hydropathy characteristics are plotted in Fig 1.
Table 1. Summary of the 9 proposed family additions to the MFS superfamily.
The protein’s family name, abbreviation, TC identifier, typical number of TMSs, typical protein size range (in amino acyl residues), typical topology, proposed evolutionary pathway used to generate this topology, and organismal domain distribution are shown.
| Family name | Short name | TC# | Typical No. of TMS | Typical protein size range | Typical Topology | Topology description | Domain distribution |
|---|---|---|---|---|---|---|---|
| Equilibrative Nucleoside Transporter | ENT | 2.A.57 | 11 | 350–500 | 6 + 5 | 11 TMSs arose from 12 TMSs by loss of the C-terminal TMS | Eukaryota |
| Aromatic Acid Exporter | ArAE | 2.A.85 | 12 | 600–1300 | 6 + 6 | 12 TMSs arose from duplication of 6 TMSs | Archaea, Bacteria, Eukaryota |
| Ferroportin | Fpn | 2.A.100 | 12 | 400–800 | 6 + 6 | 12 TMSs arose from duplication of 6 TMSs | Eukaryota |
| Eukaryotic Riboflavin Transporter | E-RFT | 2.A.125 | 11 | 430–500 | 6 + 5 | 11 TMSs arose from 12 TMSs by loss of the C-terminal TMS | Eukaryota |
| Lysyl-phosphatidylg lycerol synthase | MprF | 4.H.1 | 14 or 15 | 800–910 | 8 or 9+ 6 | Last 6 TMSs are related to MFS | Bacteria |
| 6 TMS Lysyl-tRNA Synthetase | LysS | 9.B.111 | 6 | 550–630 | 6 | Basic 6 TMS repeat unit | Bacteria |
| Eukaryotic Cytochrome b561 | Cytb561 | 5.B.2 | 6 | 222–647 | 6 | Basic 6 TMS repeat unit | Eukaryota |
| Conidiation and Conidial Germination Protein | CCGP | 9.B.57 | 12 | 459–602 | 6+6 | 12 TMSs arose by duplication of 6 TMSs | Eukaryota |
| 6 TMS DUF1275 | DUF1275 | 9.B.143 | 6 | 200–300 | 6 | Basic 6 TMS repeat unit | Archaea Bacteria, Eukaryota |
Fig 1. Average hydropathy plots of the 9 new families added to the MFS in this study.
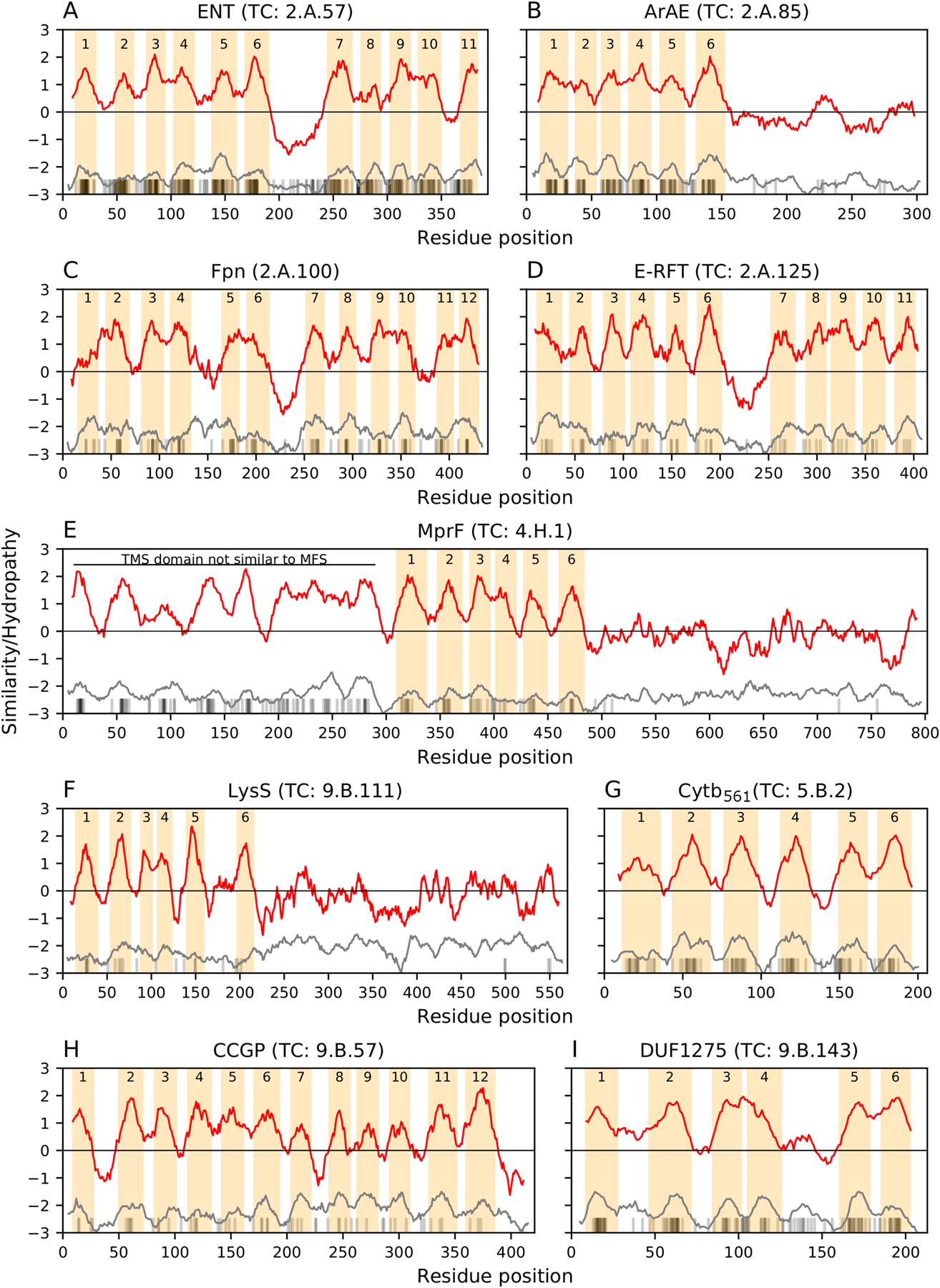
The title of each panel indicates the corresponding family. Red curves indicate average hydropathy, gray curves indicate average similarity, and vertical thin black bars on the x-axis indicate residues predicted to be part of TMSs with HMMTOP. Hydrophobic peaks (i.e. inferred TMSs) are enumerated and highlighted with tan colored bars. Plots were generated with the AveHAS program [39] as described in Methods.
1.2. Methods
1.2.1. Obtaining candidate homologs
The program famXpander [40] was used to retrieve candidate homologs for all members of each family. This program searches the National Center for Biotechnology Information (NCBI) NR protein database using PSI-Blast [41] with a default cut off e-value of 0.0001 and retrieves up to 10,000 sequences. Redundant sequences showing more than 85% sequence identity are then removed using the CD-HIT program [42]. The sequences extracted by famXpander are considered to be candidate homologs of each protein family [3].
1.2.2. Inference of homology
Once candidate homologous sequences for each family are retrieved, they are used to compare each family against all other existing Major Facilitator Superfamily members. The program Protocol2 [6] performs this comparison. Protocol2 takes the sets of proteins returned by famXpander for two query families, and compares them using the Smith-Waterman algorithm, as implemented in the SSEARCH program and correcting E-values for compositional bias using 1000 shuffles [43]. The program reports the top hits between the two families, including the alignments, the TMSs involved in each alignment, and the GSAT score of the alignment [6]. Because unrelated membrane proteins may yield alignment scores beyond significance cutoffs [36, 44], in addition to significant sequence similarity, all candidate homologs identified by Protocol2 must pass four additional criteria: (1) aligned TMSs must be compatible with the repeat units of the families involved in the analysis, and the hydropathy curves of the corresponding alignment should show good overlap of hydrophobic peaks; (2) the locations of simple/complex TMSs should be compatible; (3) Pfam domains of both families should overlap in the aligned region; and (4) if 3D structures are available, significant alignments should further support the relationship.
Although convergent sequence evolution is possible, it has only been shown for short motifs and never for large segments of proteins, such as entire domains [3]. By using a minimal length of 100 amino acyl residues to infer homology, and the above-mentioned criteria, we consider it unlikely that unrelated proteins will pass all of our criteria.
1.2.3. Multiple Alignments and Topological Analyses
MAFFT [45] was used to create multiple alignments of proteins retrieved by famXpander using the L-INS-i algorithm. Residue positions showing >30% gaps in the alignments were removed with trimAL [46]. A few sequences that introduced large gaps into the alignment, usually a result of fragmentation, incorrect sequences, or the inclusion of introns, were removed [3]. Multiple alignments were then inputted into the Average Hydropathy, Amphipathicity, and Similarity (AveHAS) program [47] using a sliding window size of 19 residues and an angle of 100° for α-helical structures. The resulting graphs (Fig 1) allowed us to consider the topological maps of multiple proteins in the same family.
The Web-based Hydropathy, Amphipathicity, and Topology (WHAT) program uses a sliding window to generate hydropathy curves for single protein sequences [47]. HMMTOP was used to determine the transmembrane segments of protein sequences used in this study [48].
In addition to WHAT and HMMTOP, the web based TOPCONS (http://topcons.cbr.su.se) program was used for topological predictions. This program’s algorithm combines a number of topology prediction algorithms into one consensus [49]. TOPCONS proved to be useful as a tool for comparative analyses as it sometimes allowed us to correct for mis-labeled transmembrane segments predicted by WHAT. However it should be noted that the most accurate program to be used for topological predictions is family specific [50].
1.2.4. 3D structural analyses
When full protein 3D structures do not produce meaningful alignments, evidence of homology can still be revealed by cutting structures of transporters into helical bundles of transmembrane α-helices (α-TMSs) and searching for alignments of basic repeat units. Although, the smallest repeat unit in MFS is 3 α-TMS (3HBs), we focused on 4-helix bundles (4HBs) because 3HBs may produce marginally scoring superpositions of the 3 helices between unrelated structures. We extracted α-TMSs from OPM [51] as well as PDBTM [52]. If TMSs corresponded to less than one full α-helix, they were extended to full helices using secondary structure assignments from STRIDE [53]. Helix bundles were compared with the CCP4 [54] implementation of the SSM superpose algorithm [55] or the TM-align program [56]. Alignments were ranked based on RMSD values, coverage, and TM-scores. When significant superpositions of 4HBs were identified, we extended the analysis to 6HBs or the full proteins to investigate the extent of the structural similarity.
1.2.5. Network of relationships within the MFS and their relative confidence levels
Unfortunately, not all of the families we added to the MFS currently have 3D structures available in PDB; thus, most of our inferences were evaluated based on the sequence-based criteria in our strategy. Given that all inferences show significant alignment scores (E-value < 10−7), the hydropathies of the alignments overlap well, the aligned TMSs are congruent with the repeat units of the families, the profile of TMS complexity is compatible between families, and Pfam domains are shared, we rationalized that the degree of confidence of a given inference can be quantified based on the E-values of the B-C alignments, the numbers of TMSs involved in the alignments and, if available, the qualities of the 3D structural superpositions. The contributions of these three factors can be written as
| (1) |
where Nb,c is the number of TMSs in the sequence alignment between proteins b and c, Evalueb,c is the corresponding E-value of the alignment, and CA,D is a function that assesses the contribution of 3D structural alignments. Due to the scarcity of family members with characterized 3D structures in PDB, we regarded the top-scoring superposition of 3D structures between families A and D as representative of the structural similarity between the 2 families. Note that b is a homolog of family A and c is a homolog of family D. We relied on the following empirical assumptions to define the function CA,D: 1) less than 3 α-helices aligning between two 4-helix bundles decreases our confidence in the homology inference; 2) with 3 helices aligned, we are neutral regarding the possibility of homology because 3 helices can also be aligned with marginal scores by chance; 3) more than 3 helices aligned increases our confidence, and 4) the contribution of the structural analysis increases proportionally with higher alignment coverage and is inversely related to the RMSD. This can be modeled as
| (2) |
where cov indicates the coverage of the highest scoring superposition between families A and D, calculated as the number of residues superposed divided by the length of the shorter 4HB. Then, this is divided by the corresponding RMSD value of the alignment. K is an empirical scaling constant to control the magnitude of CA,D relative to the first added term in equation (1). We used K=100, which is equivalent to expressing the coverage as a percentage. WA,D is a function that controls the sign and weight of the contribution of each 3D alignment by following the aforementioned assumptions; it is expressed as:
| (3) |
Three data points are critical in equation 3: cov=0.5 (~2 aligned helices) producing the maximal penalty (WA,D = −1); cov=0.75 (~3 aligned helices) corresponding to a neutral contribution (WA,D = 0); and cov=0.9 (~4 helices aligned) yielding the maximal contribution (WA,D = 1). For simplicity, intermediate coverages are interpolated linearly on the two lines connecting these three points as shown in equation 3.
The score was normalized to 1.0 based on the highest scoring inference, and three arbitrary levels of confidence were defined: high confidence (Score ≥ 0.6), medium confidence score (0.6 > Score ≥ 0.2), and low (Score < 0.2). Table S2 provides the Score and the Confidence level assigned to each inferred relationship within the MFS. All the relationships identified and their confidence levels were plotted in a network layout using the program Gephi 0.9.2 (https://gephi.org/).
1.2.6. The Major Facilitator Superfamily protein tree
We extracted from TCDB all protein sequences that belong to the families considered in this study. However, given that there are more than 900 proteins under TC# 2.A.1, we selected 87 sequences that correspond to the first system listed for each subfamily. In total, we obtained 378 sequences (File S1). Sequences were clustered with the program mkProteinClusters [40], which uses the statistical computing environment R (https://www.R-project.org/) to perform a hierarchical clustering based on a distance matrix calculated from bit scores generated by local Smith-Waterman alignments as implemented in SSEARCH [43]. This method has shown excellent agreement with phylogenetic trees for grouping TCDB families [40]. Clusters were generated using the Ward method (agglomerative coefficient 0.983). The printed version of the tree was generated with the GNU software GIMP 2.10 (https://www.gimp.org/). The original tree file in Nexus format is available in File S2.
1.3. Results and discussion
1.3.1. Distribution of complex and simple TMSs within MFS
In order to generate a reference to test the relationships between MFS families, we generated the distribution of simple and complex TMSs in MFS members using the program TMSOC [36, 37]. In MFS members with 12 TMSs, all TMSs are predominantly complex (Fig 2A). However, a clear trend can be observed where TMSs 3, 6, 9 and 12 have higher frequencies of simple TMSs (Fig 2B). This is in agreement with MFS members evolving from a 3-TMS precursor that duplicated to form a 3+3 topology, which in turn duplicated to generate the 6+6 topology. The fact that TMSs 3, 6, 9, and 12 are more frequently simple than the other TMSs correlates with their locations outside the pore in the 3D structures of MFS permeases. This indicates that they are not directly involved in transport activity [16]. Examining the 14-TMS proteins in the MFS, we found that the same pattern held, except that the first and the two central TMSs, 7 and 8, also showed higher frequencies of simple TMSs (Fig S1). Note that TMSs 7–8 are not part of the two 6-TMS repeat units.
Fig 2. Distribution of complex/simple TMSs in MFS members with 12 TMSs.
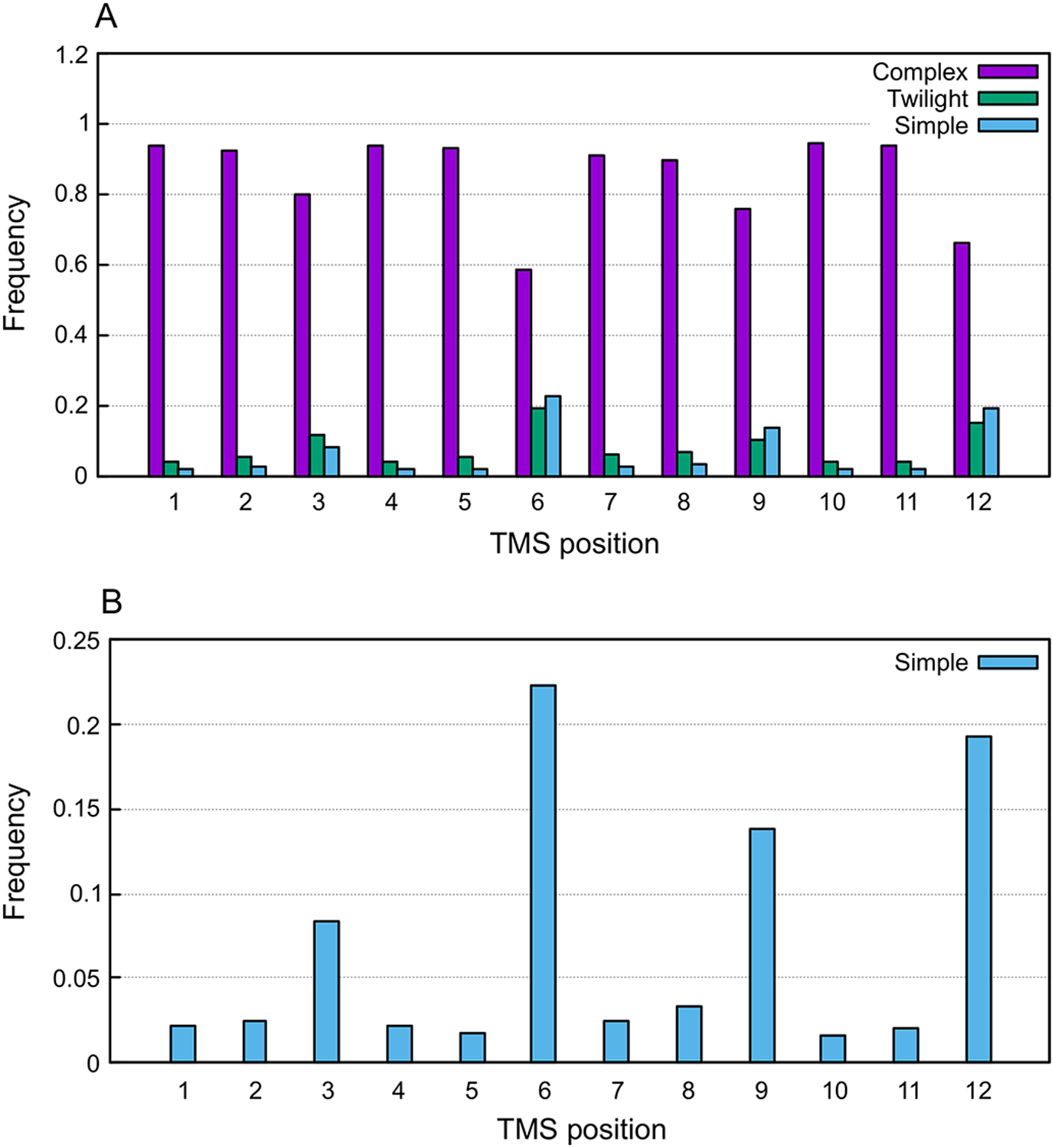
The program TMSOC was used to classify TMSs into three types: complex, twilight and simple. A) Frequency of the type of TMS per TMS position as reported by TMSOC across 658 MFS members with 12 TMSs. B) Frequency of simple TMSs per TMS position. Note that 1) all TMSs are predominantly complex, 2) all TMSs have a small frequency of simple TMSs, and 3) the frequencies of simple TMSs in positions 3, 6, 9, and 12 is larger than TMSs in positions 1–2, 4–5, 7–9, and 10–11.
1.3.2. Inference of evolutionary relationships between pairs of families
Our strategy to infer distant relationships between pairs of families begins with the application of the transitivity property of homology [3, 40]: two proteins, A and D, with no obvious sequence similarity, are considered evolutionarily related if two additional proteins B (a homolog of A) and C (a homolog of D) exist such that a path of significant sequence similarity can be identified connecting proteins A and D (A→B→C→D). The relationship is then deduced by association between the two families to which proteins A and D belong, as long as four additional conditions are met: 1) aligned TMSs must be compatible with the repeat units of both families, that is, there must be a common evolutionary pathway that gave rise to the TMS topology of both families, and the hydropathy of the alignment between proteins B and C must show clear overlap of hydrophobic peaks (i.e., putative TMSs); 2) the presence of complex/simple TMSs should be compatible based on the TMSOC classification, 3) the characteristic Pfam domains of both families must overlap significantly in the B-C alignment; and 4) when 3D structures are available, significant superpositions provide additional evidence of homology (see Methods). For the following discussion, E-values are calculated with the Smith-Waterman algorithm as implemented in SSEARCH [43] unless otherwise specified (See Methods).
As mentioned in the Introduction, we have added nine families to the MFS for a total of fifteen families outside of TC# 2.A.1. Initially, four families (ENT, Fpn, E-RFT, and DUF1275) were identified as candidate members of the MFS using our methodology as previously reported [2, 3, 6, 9, 12, 57]. These results strongly suggested that these families are members of the MFS. These conclusions were further substantiated by incorporating the criteria of TMS complexity and Pfam domain agreement. As a result, 5 more families were added to the MFS (ArAE, CCGP, LysS, MprF and Cytb561). The details of the inferences for each relationship between established MFS families and the new families are discussed below and summarized in Table 2.
Table 2. Evidence for homology of the new families with the MFS.
Summary of the analysis across the transitivity path inferring the relationship between pairs of families. All A-B and C-D alignments had E-values < 10−17 and satisfied all criteria described in our strategy. All B-C alignments have E-value < 10−7, were congruent with the alignment of hydrophobic peaks, and showed agreement of repeat units. Thus, results are summarized based on the number of TMSs in the alignment (N), and the agreement of Pfam domains (D). Domains are shared between families because (1) they are direct hits (dh) using hmmscan [58], (2) the domains belong to the same clan (sc), and/or (3) the domains can be projected (prj) from one family to the other (see Methods). B-C descriptions relating families A and D are shaded.
| Homology transitivity path | Alignment E-values | |||||
|---|---|---|---|---|---|---|
| Family A | Homolog B | Homolog C | Family D | A-B | B-C | C-D |
| 2.A.57.1.10 (ENT) | KVI06040 | WP_056965629 | 2.A.1.2.20 (MFS) | N: 11 | N: 11 | N: 12 |
| D: dh | D: prj | D: dh | ||||
| 2.A.85.3.5 (ArAE) | KII87451 | EIE83441 | 2.A.1.1.43 (MFS) | N: 12 | N: 6 | N:12 |
| D: dh | D: prj | D: dh | ||||
| 2.A.100.2.1 (Fpn) | WP_015091118 | KLE28886 | 2.A.2.3.2 (GPH) | N: 11 | N: 10 | N: 12 |
| D: dh | D: sc | D: dh | ||||
| 2.A.125.1.5 (E-RFT) | XP_004334153 | XP_005604017 | 2.A.57.1.1 (ENT) | N: 10 | N: 10 | N: 11 |
| D: prj | D: prj | D: dh | ||||
| 4.H.1.1.6 (MprF) | Q3M879 | WP_047224658 | 9.B.111.1.4 (LysS) | N: - | N: 7* | N: - |
| D: - | D: prj | D: - | ||||
| 9.B.111.1.2 (LysS) | WP_030772239 | EYC21101 | 2.A.1.2.56 (MFS) | N: 6 | N: 6 | N: 8 |
| D: dh | D: prj | D: dh | ||||
| 5.B.2.1.3 (Cytb561) | XP_005393290 | XP_002320578 | 2.A.17.3.19 (POT) | N: 6 | N: 6 | N: 12 |
| D: dh | D: prj | D: dh | ||||
| 9.B.57.1.1 (CCGP) | KUL88187 | OAK96959 | 5.B.2.3.1 (Cytb561) | N: 11 | N: 5 | N: 5 |
| D: dh | D: dh | D: dh | ||||
| 9.B.143.2.3 (DUF1275) | WP_003499261 | WP_015325006 | 2.A.1.2.75 (MFS) | N: 5 | N: 5 | N:12 |
| D: dh | D: prj | D: dh | ||||
In this comparison, A=B and C=D because the relationship was evident by directly comparing proteins A and D. For simplicity, the summary of the alignment is provided in column B-C, and comparisons A-B and C-D were omitted.
1.3.2.1. The Equilibrative Nucleoside Transporter (ENT) Family (TC# 2.A.57)
Members of the ENT family are typically 350–500 aas in length and possess 11 putative TMS (Fig 1A). ENT family members catalyze nucleoside transport and have homologs in fungi, protozoa, nematodes, and mammals. Members of the human ENT family, SLC29, are known to import drugs used in cancer, AIDS, and parasitic disease treatments [59]. Representative ENT family members have been experimentally shown to have a topology with a cytoplasmic N-terminus and an extracellular C-terminus, suggesting that TMS 12 in the 12 TMS precursor was lost [60]. Site directed mutagenesis experiments provided evidence for structural commonality and a common evolutionary origin between established members of the MFS and the ENT family, implying similar packing of TMSs around a solvent accessible binding site [59].
Fig 3 compares TMSs 1–11 of the 12 TMS MFS homolog WP_056965629 with TMSs 1–11 of the 11 TMS ENT homolog KVI06040 (E-value 9.7×10−10; Fig 3G). According to the TMSOC classification, none of the TMSs in these proteins is simple, which is compatible with the distribution observed in MFS (Fig 2A), thus increasing the reliability of the alignment. The Pfam domain characteristic of the ENT family (PF01733) can be projected to the MFS homolog WP_056965629 (E-value: 6.4×10−8; see Methods), further supporting the relationship between the two families. These results indicate that the last TMS of the original 12-TMS precursor was lost in family ENT, in agreement with the fact that this TMS is more frequently simple in MFS permeases, and not involved in pore formation.
Fig 3. Evidence of homology between families ENT and MFS.
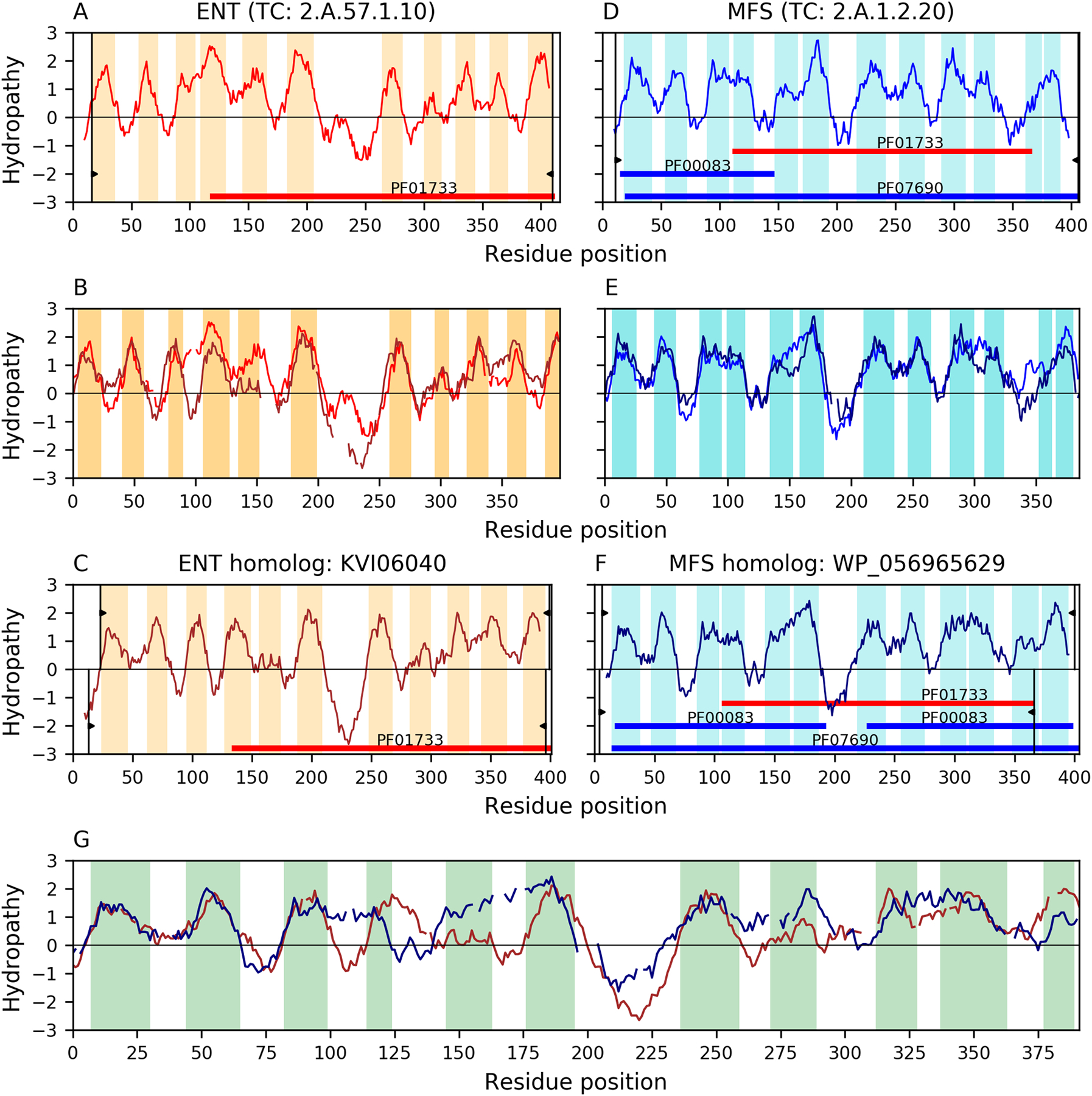
Hydropathy plots are presented across the homology transitivity path between families ENT and MFS. Panels A-C depict relationships within the ENT family, panels D-F depict relationships within the MFS, and panel G presents the evidence supporting the relationship between the two families. Orange (ENT) and Cyan (MFS) bars denote hydrophobic peaks (i.e., putative TMSs). Pfam domains are shown as colored horizontal bars. Different domain Pfam accessions within the same clan have the same color. Thin vertical black lines with wedges delimit the region of a protein involved in an alignment. The wedges in panels A and D delimit the regions covered by the alignments in panels B and E relative to the full-length proteins in panels A and D, respectively. Proteins in Panels C and F have two sets of delimiting wedges (top and bottom of the figures). Upper wedges delimit regions covered by the alignments in panels B and E relative to the full-length proteins in panels C and F, respectively. Panel G presents the alignment between the lower delimited region in panel C and the lower delimited region in panel F. Interruptions in the hydropathy plots of panels B, E, and G, indicate gaps in the corresponding sequence alignments. A. Hydropathy plot of ENT member Q944P0 (TC# 2.A.57.1.10). B. Hydropathy plot of the alignment (E-value: 7.8×10−34) between ENT member Q944P0 and its homolog KVI06040. C. Hydropathy plot of ENT homolog KVI06040. D. Hydropathy plot of MFS member P25744 (TC# 2.A.1.2.20). E. Hydropathy plot of the alignment (E-value: 6.6×10−45) between MFS member P25744 and its homolog WP_056965629. F. Hydropathy plot of MFS homolog WP_056965629. G. Hydropathy plot of the 11 TMS alignment (E-value: 9.7×10−10) between ENT homolog KVI06040 and MFS homolog WP_056965629. Only the regions where hydrophobic peaks overlap are highlighted in the alignments. According to the TMSOC classification [36, 37], none of the TMSs in these proteins is simple, thus increasing the reliability of the alignment. Pfam domain PF01733 in family ENT can be projected to MFS homolog WP_056965629 (E-value: 6.4×10−8; See Methods). The presentation format for supplementary Figures S2-S15 is the same as shown here.
1.3.2.2. The Aromatic Acid Exporter (ArAE) Family (TC# 2.A.85)
The ArAE family is ubiquitous, including members from bacteria, archaea and eukaryotes. These proteins are about 650–750 aas and usually exhibit a repeat sequence due to an internal gene duplication event, typical of MFS proteins. There are five full-length E. coli homologs. At least two of these ArAE family members are encoded within operons that also encode membrane fusion proteins (MFP family; TC# 8.A.1). This implies that these proteins catalyze efflux [61, 62]. The plant proteins, like a homolog in B. subtillis, are of 500–560 residues and exhibit only 6 putative TMSs followed by a long hydrophilic domain. None of the eukaryotic proteins is functionally characterized. A single bacterial member of the ArAE family has been functionally characterized [63]. This protein is AaeB of E. coli which depends on a membrane fusion protein (MFP family; TC# 8.A.1), AaeA, for activity. AaeB proved to be a proton motive force (pmf)-dependent para-hydroxybenzoic acid efflux pump. Only a few aromatic carboxylic acids of hundreds of compounds tested proved to be substrates of the AaeAB efflux pump [63]. It may function as a ‘metabolic relief valve’ to relieve the toxic effects of unbalanced metabolism.
As noted above, members of the ArAE family have either 6 or 12 putative TMSs, each followed by a C-terminal hydrophilic domain (Fig 1B). For example, subfamilies 1, 3, 6, 8, 10 and 11 have 12 TMSs, while subfamilies 2,4, 5, 7 and 9 have six TMSs. According to Pfam, the domains containing 6 TMSs in the 12 TMS proteins (PF10337 and PF13515, respectively) belong to the same clan (CL0307), suggesting that they are likely repeats of each other. Note that the AveHAS plot shown in Fig 1B only depicts a single repeat unit.
Fig S2 presents a comparison of an established ArAE family member (TC# 2.A.85.3.5) with an established member of the MFS (TC# 2.A.1.1.43) using the transitivity property of homology. Comparing TMSs 7–12 of the ArAE homolog, KII87451, with TMSs 7–12 of the MFS homolog, EIE83441, using the same approach as described above for the ENT family, we see that all 6 TMSs of the second repeat unit of both proteins align well (E-value: 9.5×10−11; Fig S2G). According to the TMSOC classification, only TMS 6 of KII87451 and TMS 12 of EIE83441 are simple. This is compatible with the MFS distribution of simple TMSs observed in Fig 2. The Pfam domain characteristic of the MFS family (PF00083) can be projected to the ArAE homolog KII87451 (E-value: 10−6; see Methods). Altogether, this evidence supports the relationship between families ArAE and ENT.
1.3.2.3. The Ferroportin (Fpn) Family (TC# 2.A.100)
Proteins of the Fpn family are required for the export of iron and manganese from animal cells into the systemic circulation [64, 65]. These iron regulated transport proteins are found in the basolateral membranes of mammalian intestinal epithelial cells [66]. Fpn members are essential for iron homeostasis; studies have shown that mice lacking these proteins die during embryonic development [67]. Members of the Fpn family are between 400 and 800 aas in length, and studies with antisera have suggested a topology of 12 TMSs, with the C-termini exposed to the inside of the cell [64] (Fig 1C and Table 1). In Fig 1C, the hydropathy plot has been interpreted in terms of the 12 TMS topology shown in the crystal structure, where the first hydrophobic peak corresponds to two TMSs [68].
Fig S3 shows the comparison of homologs within families Fpn and GPH (TC# 2.A.2), equivalent to the comparison described above for the ENT family (Fig 3). We identified an alignment encompassing TMSs 2–11 of both the Fpn homolog WP_015091118 and the GPH homolog KLE28886 (E-value: 2.8×10−10; Fig S3G), in agreement with the repeat unit of MFS families. According to TMSOC, and compatible with the MFS pattern (Fig 2), only the twelfth TMS in both proteins is simple and Fig S3 shows that it is not part of the alignment. In addition, the Pfam domains of both families belong to the same clan (CL0015). When we compared the 3D structure of Fpn member Q6MLJ0 (TC# 2.A.100.2.1; PDB ID: 5AYN) with the structure of the GPH homolog P30878 (PDB ID: 4M64), the 6 TMSs of the first repeat units in both proteins produced a reasonable alignment (RMSD: 2.93 Å; Coverage: 93%; Fig S4). In addition, the Fpn structure 5AYN also aligns well with other MFS structures. For example, Fig S5 shows a 12 TMS alignment (RMSD: 3.0 Å; Coverage: 98%) with MFS member P11551 (TC# 2.A.1.7.1; PDB ID: 3O7P). This is relevant, particularly between different families, because even within the same MFS family we observe lower quality alignments of 12 TMSs due to the conformational flexibility in MFS proteins afforded by the loops connecting TMSs 6 and 7, responsible for mechanistically important conformational changes [69]. This loop may even contain additional TMSs. Consequently, we were able to improve the quality of the Fpn-MFS alignment (RMSD: 2.27 Å; Coverage: 97%) when only the 6 TMSs of the first repeat unit in both proteins were considered (see Fig S6).
1.3.2.4. The Eukaryotic Riboflavin Transporter (E-RFT) Family (TC# 2.A.125)
Members of the E-RFT family are typically of 430–500 aas in length and possess 11 putative TMS (Fig 1D, Table 1). As its name suggests, the E-RFT family transports riboflavin (vitamin B2). Riboflavin in the forms of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) act as cofactors in biological oxidation-reduction reactions [70]. Deficiencies in riboflavin can lead to developmental abnormalities in mammalian adolescence and is a risk factor for anemia, cancer, and cardiovascular disease [71]. Studies performed on the rat riboflavin transporter 2 (rRFT2) showed that it is inhibited by the presence of lumiflavin, FMN, and FAD [72], suggesting that these transporters are able to transport various riboflavin derivatives.
Fig S7 compares TMSs 2–11 of the E-RFT homolog XP_004334153 with TMSs 2–11 of the ENT homolog XP_005604017 (E-value: 9.3×10−12; Fig S7G), in agreement with the repeat unit of MFS families. According to the TMSOC classification, only protein XP_005604017 has simple TMSs (TMSs 3 and 9) in agreement with the MFS distribution observed in Fig 2. Furthermore, the characteristic Pfam domain of the ENT family (PF01733) can be projected to the E-RFT homolog (E-value: 3.2×10−9; see Methods), further supporting the proposed relationship between these families.
1.3.2.5. The Lysyl Phosphatidylglycerol Transferase (MprF) Family (TC# 4.H.1) and the 6 TMS Lysyl-tRNA Synthetase (LysS) Family (TC# 9.B.111)
The bacterial lysyl phosphatidylglycerol (LPG) transferases in family MprF contain 6 to 15 TMSs (Table 1 and Fig 1E) and have dual functions: derivatization of PG with lysine or alanine to modulate the membrane surface charge, and flipping of the derivatized phospholipid from the inner leaflet to the outer surface of the cytoplasmic membrane in order to provide resistance to cationic antimicrobial peptides [73]. These two functions are reported to be catalyzed by two different domains within these multidomain proteins [74, 75]. Families LysS and MprF of lysyl transferases show high sequence similarity in both the hydrophobic MFS-like and the hydrophilic domains. Fig S8 shows a highly significant alignment (E-value: 1.3×10−48; Fig S8C) between TMSs 1–6 of the MprF member Q3M879 (TC# 4.H.1.1.6) and TMSs 1–6 of the LysS member WP_047224658 (TC# 9.B.111.1.4), in addition to the hydrophilic domain of both proteins. Such a significant level of sequence similarity may seem to warrant membership to the same family; however, given that their functions are completely different, they have been assigned to different TC families. Notice in Fig S8B that the sixth hydrophobic peak of the LysS member is not highlighted; this is because when this protein is aligned with other 6-TMS LysS homologs (e.g., WP_030772239), the sixth peak aligns with the loop connecting TMSs 5 and 6 (compare LysS members in Figs S8 and S10), thus, this is most likely not a TMS. Also note that the MprF member is missing the characteristic N-terminal 8–9 TMS domain, possibly involved in flippase activity [75]. According to the TMSOC classification, only TMS 3 in MprF member Q3M879 is simple, and in LysS member WP_047224658, TMSs 4–5 are simple. While at first glance the simple TMSs in WP_047224658 seem atypical relative to MFS porters, the global distribution of simple/complex TMSs across LysS members with 6 TMSs does show the characteristic MFS pattern where TMSs 3 and 6 have the highest frequencies of simple TMSs (Fig S9). However, in addition, TMSs 4 and 5 also show higher frequencies of simple TMSs relative to TMSs 1–2. Thus, the distributions of simple TMSs are compatible between those two families. Notwithstanding the difference in simple TMSs in WP_047224658, the high significance of the pairwise alignment fully covering both proteins (E-value: 1.3×10−48; Fig S8C) leaves little doubt that they are related. The relationship is further supported by the highly significant projection of the transmembranal Pfam domain characteristic of the LysS family (PF16995) to the MprF member (E-value: 6.9×10−16).
Lysyl-tRNA synthetases are highly conserved enzymes that function in mRNA translation [76]. These synthetases have gained several functions in addition to protein synthesis, playing roles in HIV replication, cytokine-like signaling, and transport of proteins [76]. LysS members are around 600 aas in length and possess a hydrophobic N-terminal domain with 6–7 putative TMSs and an uncharacterized hydrophilic domain at their C-termini (Table 1 and Fig 1F). Fig S10 relates family LysS with the MFS showing that TMSs 1–6 of the LysS homolog WP_030772239 align well (E-value: 4.2×10−8; Fig S10G) with TMSs 1–6 of the MFS homolog EYC21101, in agreement with the repeat units of MFS families. According to the TMSOC classification, TMSs 3 and 6 of both proteins are simple in agreement with the patterns observed in MFS (Fig 2) and LysS (Fig S9). Furthermore, the characteristic Pfam domain of the LysS family (PF016995) can be projected to the MFS homolog (E-value: 2.2×10−5; see Methods), further supporting the proposed relationship.
1.3.2.6. The Eukaryotic Cytochrome b561 (Cytb561) Family
The homodimeric cytochrome b561 proteins contain 6 TMSs and two heme prosthetic groups per subunit (Table 1 and Fig 1G). These hemes are coordinated with His residues from different TMSs [77–79]. Ascorbate and monodehydroascorbate can be enclosed in positively charged pockets on both sides of the membrane. Two highly conserved residues, Lys81 and His 106, play an essential role in substrate recognition and catalysis [80].
Cytochromes b561, residing in chromaffin vesicles, are known to play roles in neuroendocrine-specific transmembrane electron transfer from extravesicular ascorbate to an intravesicular monodehydroascorbate radical to regenerate ascorbate. Some members of the family lack the sequence for putative ascorbate-binding and exhibit a transmembrane ferrireductase activity. Nakanishi et al. [78] proposed that cytochrome b561 has a specific function facilitating the concerted proton/electron transfer from ascorbate by exploiting a cycle of deprotonated and protonated states of the N(delta1) atom of the axial His residue at the extravesicular haem center, as an initial step of transmembrane electron transfer. This mechanism utilizes the well-known electrochemistry of ascorbate for biological transmembrane electron transfer and might be operative for other types of electron transfer reactions from organic reductants [78].
Fig S11 shows the evidence supporting our inference that the Cytb561 family is a member of the MFS. Here we align the 6 TMSs of the Cytb561 homolog XP_005393290 with TMSs 7–12 of the POT homolog XP_002320578 (E-value: 2.5×10−9; Fig S11G). Except for the fourth TMS, the alignment shows excellent correspondence of hydrophobic peaks. According to the TMSOC classification, only the first TMS in Cytb561 homolog XP_005393290 is simple. This is compatible with MFS because there are established MFS families that show a tendency to have the first TMS simple (Fig S1), and because the global distribution of simple/complex TMSs in Cytb561 members with 6 TMSs also shows that TMSs 3 and 6 have higher frequencies of simple TMSs (Fig S12), which is compatible with the pattern observed in MFS proteins (Fig 2).
Furthermore, the characteristic Pfam domain of family POT (Pfam PF00854) projects well to the Cytb561 homolog (E-value: 9.6×10−6).
1.3.2.7. The Conidiation and Conidial Germination Protein (CCGP) Family
The MTP1 gene, encoding a type III integral transmembrane protein, was isolated from the rice blast fungus Magnaporthe oryzae. The Mtp1 protein is 520 aas long and is homologous to the Ytp1 protein of Saccharomyces cerevisiae. Mtp1-GFP (green fluorescent protein) fusion expression results indicated that Mtp1 resides in several membranes exposed to the cytoplasm. The mtp1 gene is primarily expressed in the hyphal and conidial stages and is necessary for conidiation and conidial germination; however, it is not required for pathogenicity. The Δmtp1 mutant grew more efficiently than the wild type strain on non-fermentable carbon sources, implying that Mtp1 has a role in respiratory growth and carbon source utilization [81]. Proteins in this family are around 460 aa long and typically have 12 TMSs (Table 1, Fig 1H).
Fig S13 shows that TMSs 1–5 of the 12 TMS CCGP homolog KUL88187 align with TMSs 1–5 of the Cytb561 homolog OAK96959 (E-value: 7.1×10−11; Fig S13G). By comparing panels A and C in Fig S13, and their corresponding alignment in panel B (E-value: 9.6×10−103), it is evident that the CCPG homolog KUL88187 is missing the first TMS relative to the CCPG member G4MKH1 (TC# 9.B.57.1.1). Worthy of note is that Cytb561 homologs can have either 5 or 6 TMSs, where the missing TMS in the 5 TMS homologs is TMS 1 relative to the 6 TMS homologs. According to the TMSOC classification, only the third TMS of KUL88187 (TMS 4 of the ancestral 6-TMS precursor) and fifth TMS of OAK96959 (TMS 6 of the ancestral 6-TMS precursor) are simple. It may seem that the simple TMS in KUL88187 does not conform with the global MFS distribution observed in Fig 2. However, note 1) that there are a few proteins in the MFS where the fourth TMS is simple, and 2) that in KUL88187 the difference in complexity between TMSs 2 and TMS 3 (0.25) is considerably smaller than the average difference of complexity between TMS 3 and all other complex TMSs (0.84). More importantly, the Pfam domain PF10348 directly hits members of both families (hmmscan E-value: 3.2×10−7) without the need of projection, and the other Pfam domain (PF03188) in the Cytb561 homolog OAK96959 is in the same clan (CL0328) as domain PF10348.
1.3.2.8. The 6 TMS DUF1275/PF06912 (DUF1275) Family (TC# 9.B.143)
Members of this family are ubiquitous, being present in the three domains of life. It is a large family with members having a fairly uniform topology of 6 or sometimes 7 TMSs as expected for half-sized protein members of the MFS (Fig 1I and Table 1). Some members are encoded by genes adjacent to a probable YtcJ-like metalo-amido-hydrolase, suggesting a role in uptake of peptides or other amido compounds or efflux of their hydrolysis products. None of these proteins is functionally characterized, and therefore, these putative transport proteins have not been mechanistically classified.
Fig S14 shows that TMSs 2–6 of the DUF1275 homolog WP_003499261 align with TMSs 2–6 of the MFS homolog WP_015325006 (E-value: 1.3×10−8; Fig S14G). The alignment does not include the first TMS due to the long loop between the first two TMSs in DUF1275 homolog WP_003499261. According to the TMSOC classification, none of the TMSs in both proteins is simple, which is compatible with the distribution observed in MFS (Fig 2A). The Pfam domain characteristic of family DUF1275 (PF06912) can be projected onto the MFS homolog WP_015325006 (E-value: 7.7×10−7).
1.3.3. The MFS network of relationsips
Fig 4 shows the interrelationships that allowed us to conclude that all of the families discussed in this paper are related and therefore members of the MFS. In this plot, the lengths of the lines are meaningless, while the thickness of the lines reflects the three levels of confidence in the homology inferences between pairs of families (see Methods). Thus, the thickest lines indicate the highest level of confidence while the thinnest lines indicate a lowest level of confidence. Families above the MFS node (see the vertical guide line at the right-hand side of the plot) separates the established MFS families, identified in previous reports [12], from those identified in this paper. The two novel highest scoring families with established MFS members are Fpn and ENT; the connection between E-RFT (RFT) and ENT is also significant (see Fig 4). In addition, the plot shows that candidate families CCGP, LysS, Cytb561 (Cytb) and E-RFT (RFT) are directly linked to MFS and other well-established superfamily members. Although the MFS-like domains in families LysS and MprF are highly related in sequence (Fig S8), only the LysS family could be shown to be related to the GPH family and the MFS. This is due, at least in part, to the lower level of conservation of the MFS-like transmembranal domains relative to the large hydrophilic domain in families LysS and MprF (see Fig 1 E–F). Finally, the DUF1275 (DUF) and ArAE families are connected only to the MFS (Fig 4).
Fig 4. Network of relationships within the MFS.

Nodes represent all families in the MFS used in this study. The darker the color of a node, the more connections that family has to other families. The relative confidence level (i.e., high, medium or low) of the homology inference between two families is expressed with three levels of thickness of the edges connecting pairs of nodes; the thickest lines correspond to the connections of highest confidence. The length of the edges connecting nodes is irrelevant (see Methods).
1.3.3.1. The Major Facilitator Superfamily tree
The tree showing the relationships among all established and new families, added to the MFS in this study, is shown in Fig 5, where different colors represent different families. The tree was generated based on pairwise Smith-Waterman bit scores with our program mkProteinClusters [40] as described in Methods. In general, we observed a correlation between the levels of confidence depicted in Fig 4 and the branching pattern in the tree (Fig 5). With two exceptions (MFS and ENT), the grouping of each family is coherent, showing all members of any particular family clustering together. We shall start at the bottom of the tree which includes representative members of the MFS (TC# 2.A.1). Sandwiched between two large groups of MFS proteins is the RFC family, illustrating its close relationship to the MFS proteins included under TC number 2.A.1. MFS vs RFC comparisons show alignments of 11 TMSs (E-value: 5.1×10−14); both families share the Pfam domain PF07690, and the rest of the domains belong to the MFS clan (CL0015). Most closely related to the MFS cluster is an adjacent cluster including Fpn (a novel MFS family) and FBT (an established MFS family), which are followed by the GPH family. A major group (right-hand side) branching from the MFS cluster contains 7 of the 9 new families, clustering adjacent to family AAA (an established MFS member). Families LysS, MprF, Cytb561, and DUF1275 in this group have 6 MFS-like TMSs, and no typical solute transport function has been assigned to them. As noted above for RFC, family RFT is sandwiched between two groups of ENT members. This is explained by the quality of the alignments observed between members of these families. For example, see the 10 TMS alignment in Fig S7. Families OAT, ArAE and POT are the most distant from the MFS cluster, indicating that ArAE (a new MFS family) clusters similarly to two previously established families, relative to the MFS cluster.
Fig 5. Radial tree of protein sequence similarities within the MFS superfamily.
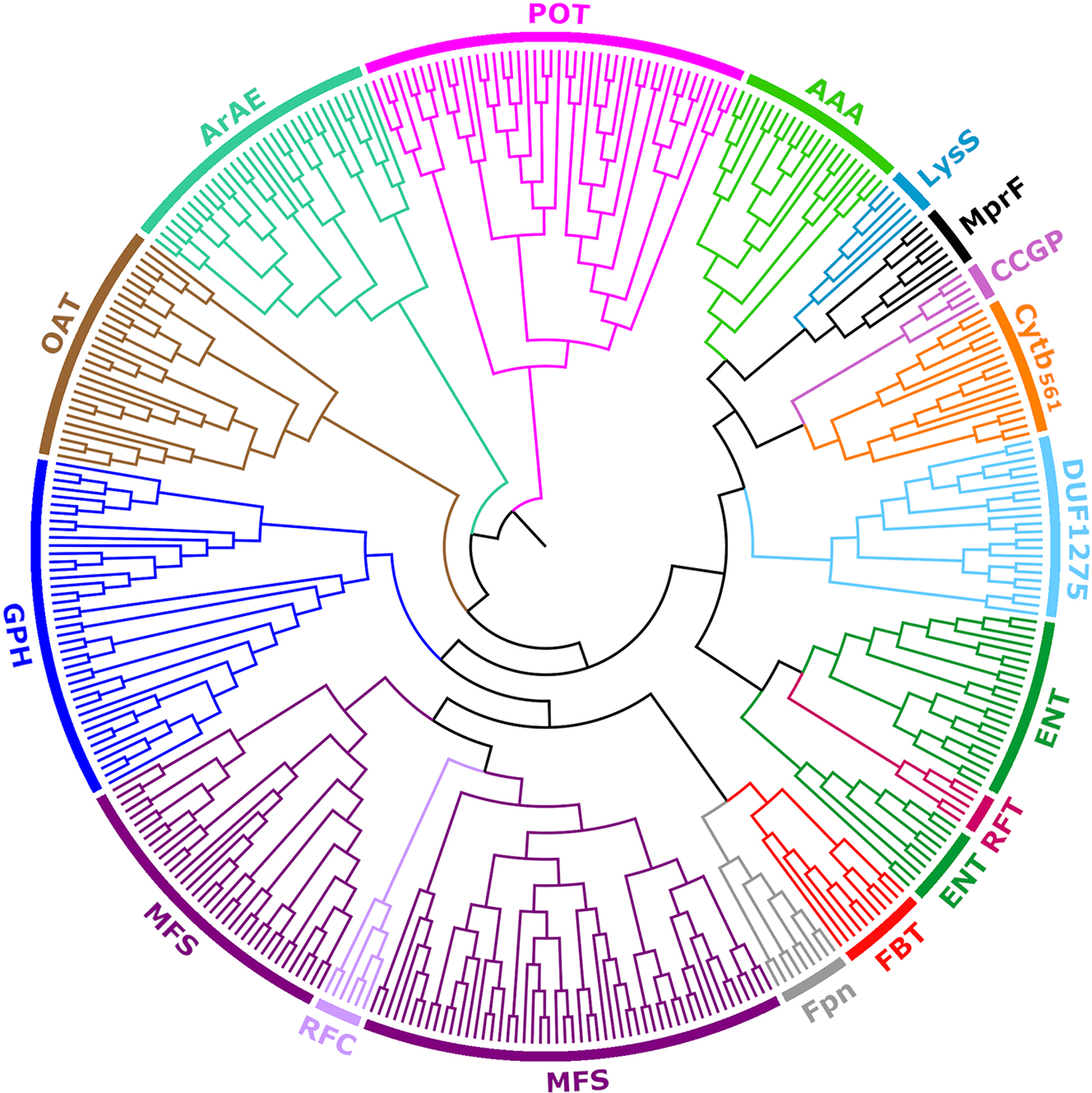
Different families are represented with different colors. For simplicity, family Cytb561 is displayed as Cytb. The tree was generated with the program mkProteinClusters [40] based on Smith-Waterman bit scores of pairwise alignments (agglomerative coefficient: 0.98; see Methods). Note that the tree is shown as a cladogram using FigTree (http://tree.bio.ed.ac.uk/software/figtree/). Thus, only the topology is meaningful and the scale bar was disregarded (See discussion in Section 1.3.3.1). The sequences (in Fasta format) and the tree (in Nexus format) used to generate the figure are available as supplementary files S1 and S2, respectively.
We were able to confirm previously assigned MFS families outside of 2.A.1. For example, the POT family (the most distant family from the MFS cluster in the tree) shows low but significant sequence similarity with MFS; when comparing POT homolog WP_019240224 and MFS member O34546 (TC# 2.A.1.2.69), the alignment covers 11 TMSs with 24% identity (E-value: 7.4×10−8), and both proteins hit the Pfam domain PF07690 while the other domains share the same MFS clan (CL0015). Our confidence in this relationship increases due to the high structural similarity between members of these two families. When TMSs 1–6 of the structure 4UVM of POT member Q8EKT7 (TC# 2.A.17.4.7) are superposed with TMSs 7–12 of the structure 3O7Q of MFS member P11551 (TC# 2.A.1.7.1), an excellent RMSD value of 1.85 Å with 99% coverage of the 6-helix bundle was obtained. This is reflected in the high confidence level observed in Fig 4.
1.3.4. Marginal sequence similarity between the Major Intrinsic Protein (MIP) Superfamily (TC# 1.A.8) and the MFS superfamily.
Unlike the established secondary carriers in families of the MFS, the MIP family includes aquaporins and glycerol facilitator channel proteins [82]. Proteins of the MIP families can be essential for normal cellular function, and they appear to be ancient, including highly diversified members with key conserved structural domains [83]. Members are typically 200–300 aas in length and possess 6 TMS that, like MFS proteins, arose by intragenic duplication of a 3 TMS precursor. The aquaporins transport water, urea, carbon dioxide, ions, and short, neutral straight chain carbohydrates such as glycerol and propanediol by an energy-independent mechanism [83].
The MIP family exhibits a well conserved asparagine-proline-alanine (NPA) motif between TMSs 2–3 and TMSs 5–6, consistent with the proposal that each polypeptide chain of this family arose by tandem duplication of a 3 TMS-encoding primordial genetic element [82]. Because of the high degree of conservation between the two halves, it is presumed that duplication of MIP proteins occurred independently of the 3 TMS duplications that gave rise to the 6 TMS units in most MFS porters. This suggests that the basic unit of sequence similarity is the 3 TMS repeat unit (see Discussion).
Comparing TMSs 1–6 of the MIP homolog CAX48992 (6 TMS) with TMSs 7–12 of the POT homolog EEH20305 (12 TMSs) yielded an E-value of 5.0×10−8 (Fig S15). This alignment shows the second 6 TMS repeat unit of the POT family member aligned with the 6 TMSs of the MIP family member. However, there is poor overlap of hydrophobic peaks 1, 3 and 5; the MIP Pfam domain PF00230 projects with marginal significance (E-value: 1.4×10−4) to the POT homolog EEH20305, and we could not identify meaningful similarity between 3D structures of these two superfamilies. All in all this is not sufficient evidence to infer homology between MIP and the MFS superfamily.
1.3.5. Negative Controls: The Mitochondrial Carrier (MC) (TC# 2.A.29) and the ATP-Binding Cassette-1 (ABC1) Superfamilies (TC# 3.A.1)
The MC superfamily currently contains 32 families. MC member proteins are usually about 300 aas in length and are involved in the transport of amino acids, nucleotides, co-factors, inorganic ions, keto acids, and mono, di, and tri-carboxylic acids [3, 84]. They possess a 6 TMS topology which arose from tandem intragenic triplication of a 2 TMS element [84]. X-ray crystallography was used to solve the 3D structure of a human homolog of the MC family and showed striking similarity between the folds of the three repeat elements [85].
The ABC1 superfamily currently contains 24 families. ABC1 members are primary active transporters and also possess a 6 TMS topology which arose by triplication of a primordial 2 TMS element [13]. The ABC1 family contains efflux transport systems with transport driven by ATP hydrolysis without protein phosphorylation.
Both the MC and ABC1 superfamilies are believed to have arisen via an evolutionary pathway that was different from that of members of the MFS. Additionally, while both the MC and MFS superfamilies are secondary carriers, ABC1 includes primary active transporters. Thus, these two superfamilies, MC and ABC1, represent suitable negative controls for evaluating the relationship among MFS-related proteins.
Table S1 shows the comparison of established MFS members plus the 9 new families against the negative controls. With the exception of one 3 TMS alignment (GPH vs MC), none of the alignments showed good hydropathy overlap. Not surprisingly, none of these comparisons passed the criterion of repeat unit compatibility, despite all alignments having E-values ≤ 10−6. Even though unrelated membrane proteins may artifactually produce alignment scores above thresholds of significance due to biases introduced by physicochemical constraints [36, 44], here we show that they fail our additional criteria for inferring distant relationships: (1) compatibility of repeat units and overlap of hydrophobic peaks, (2) similar Pfam domains within aligned regions, and (3) if available, 3D structural similarity. The footnotes in supplementary Table S1 detail each comparison against the negative control and how they failed to satisfy our criteria for homology.
1.4. Conclussions
In this study, we have expanded the largest superfamily of secondary carriers currently recognized, the Major Facilitator Superfamily, MFS. Prior to this study, 96 families were known to comprise the MFS. However, with the exception of a few receptors, virtually all of these proteins were known or assumed to be transport proteins. Our present efforts have allowed us to include several additional transport protein families, members of which are believed to function as secondary carriers. These include the Equilibrative Nucleoside Transporters (ENT; TC# 2.A.57) with 11 TMS, the Ferroportins (Fpn; TC# 2.A.100) with 12 TMS, and the Eukaryotic Riboflavin Transporters (E-RFT; TC# 2.A.125) with 11 TMS. All of the proteins that comprise these families were shown to have the basic 6 TMS repeat unit, giving rise to 12 TMS, although the predominant members of two of these three families appear to have lost a single TMS at their C-termini. A surprising result was the observation, documented here, that a number of non-transport protein families appear to be at least in part, related to MFS permeases. One of these involved a 6 TMS MFS-like domain linked to Lysyl-tRNALys synthases (lysine tRNA ligases) (LysS) (TC# 9.B.111). The function(s) of this half MFS permease domain is/are not known, but its identification provides incentive for future investigation. Still, another apparent fusion protein type involves a well-characterized phosphatidyl glycerol transferase/synthase (TC# 4.H.1) which has a 14–15 TMS topology, where the last 6 TMSs are related to the MFS. In this case, it has been demonstrated that the protein (MrpF) has a dual function: The C-terminus may catalyze transfer of the lysyl (or alanyl) moiety from lysyl-tRNALys to phosphatidyl glycerol or cardiolipin, while the N-terminal domain translocates the product, the derivatized phospholipid, to the outer membrane [74]. This second process renders the enzyme a flippase. It is possible, but not established, that these two processes both require the entire protein and are tightly coupled, rendering MprF a group translocator [74, 86–89]. Finally, the cytochrome b561 proteins, which catalyze transmembrane election flow, and possibly H+ transport, also have characteristic MFS domains [90–92].
In all of these cases, the detailed functions of the MFS domains or the full-length proteins are not known or fully understood, but one can surmise that many of these domains may function in transmembrane transport. For example, the enzymes that synthesize lysyl-tRNALys could additionally provide a lysine uptake function following dimerization, coupled to esterification with its cognate tRNA. However, such suggestions require experimental verification to elucidate the extent of their functions.
Supplementary Material
The program TMSOC was used to classify TMSs into three types: complex, twilight and simple. A) Frequency of the type of TMS per TMS position as reported by TMSOC across 69 MFS members with 14 TMSs. B) Frequency of simple TMSs per TMS position. Note that 1) all TMSs are predominantly complex in the 2 repeat units (TMSs 1–6 and 9–12); 2) the two central TMSs 7–8, which are not part of the 6-TMS repeat units, have higher frequencies of simple TMSs; 3) the first repeat unit has more simple TMSs in the first TMS compared to the third TMS, but the third TMS still has more simple TMSs compared to TMSs in positions 2, 4 and 5; and 4) the frequencies of simple TMSs in the second repeat unit follows the same pattern as the 12-TMS MFS members in Fig 2.
Sequences were renamed to facilitate visualization of the tree. The family name is followed by the 2 last digits of the TC numbers, separated by underscores. Families with names larger than 4 characters were shortened following the convension: Cytb561 (Cytb), DUF1275 (DUF), POT/PTR (POT), and E-RFT (RFT).
Table S1. Comparison of 9 new MFS families with families in the negative control set.
Hydropathy plots are presented across the homology transitivity path between family ArAE and the MFS. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of ArAE member S7PTY2 (TC# 2.A.85.3.5). B. Hydropathy plot of the alignment (E-value: 1.9×10−184) between ArAE member S7PTY2 and its homolog KII87451. C. Hydropathy plot of ArAE homolog KII87451. D. Hydropathy plot of MFS member A0ZXK6 (TC# 2.A.1.1.43). E. Hydropathy plot of the alignment (E-value: 2.6×10−79) between MFS member A0ZXK6 and its homolog EIE83441. F. Hydropathy plot of MFS homolog EIE83441. G. Hydropathy plot of the 6-TMS alignment (E-value: 9.5×10−11) between ArAE homolog KII87451 and MFS homolog EIE83441. According to the TMSOC classification, only TMS 6 in KII87451and TMS 12 in EIE83441 are simple. This is compatible with the MFS distribution of simple TMSs observed in Fig 2. The Pfam domain characteristic of family MFS (PF00083) can be projected to ArAE homolog KII87451 (E-value: 10−6), further supporting the relationship between these two families.
The leaves are labeled following the same format as for File S1.
Table S2. Relative confidence scores of homology inferences.
Hydropathy plots are presented across the homology transitivity path between families Fpn and GPH. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of Fpn member Q6MLJ0 (TC# 2.A.100.2.1). B. Hydropathy plot of the alignment (E-value: 5.0×10−110) between Fpn member Q6MLJ0 and its homolog WP_015091118. C. Hydropathy plot of Fpn homolog WP_015091118. D. Hydropathy plot of GPH member P94488 (TC# 2.A.2.3.2). E. Hydropathy plot of the alignment (E-value: 2.5×10−54) between GPH member P94488 and its homolog KLE28886. F. Hydropathy plot of GPH homolog KLE28886. G. Hydropathy plot of the 10-TMS alignment (E-value: 2.8×10−10) between Fpn homolog WP_015091118 and GPH homolog KLE28886. According to TMSOC, and compatible with MFS (Fig 2), only the twelfth TMS in both proteins is simple and it is not part of the alignment. The Pfam domains characteristic of families Fpn (PF06963) and GPH (PF03083) are included in the alignment and belong to the same clan (CL0015), further supporting the relationship between these two families.
Ribbon representation of the structural superposition between TMSs 1–6 of Fpn member Q6MLJ0 (TC# 2.A.100.2.1; PDB ID: 5AYN; green) and TMSs 1–6 of the GPH homolog P30878 (PDB ID: 4M64; cyan). The alignment has an RMSD value of 2.93 Å with 93% coverage of the smallest 6-helix bundle. A) Top view and B) side view.
Ribbon representation of the structural superposition between TMSs 1–12 of Fpn member Q6MLJ0 (TC# 2.A.100.2.1; PDB ID: 5AYN; cyan) and TMSs 1–12 of the MFS member P11551 (TC# 2.A.1.7.1; PDB ID: 3O7P; green). The alignment has an RMSD value of 3.0 Å with 98% coverage of the smallest structure. A) Top view and B) side view.
Ribbon representation of the structural superposition between TMSs 1–6 of Fpn member Q6MLJ0 (TC# 2.A.100.2.1; PDB ID: 5AYN; cyan) and TMSs 1–6 of the MFS member P11551 (TC# 2.A.1.7.1; PDB ID: 3O7P; green). The alignment has an RMSD value of 2.27 Å with 97% coverage of the smallest 6-helix bundle. A) Top view and B) side view.
Hydropathy plots are presented across the homology transitivity path between families E-RFT and ENT. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of E-RFT member D2VY17 (TC# 2.A.125.1.5). B. Hydropathy plot of the alignment (E-value: 1.2×10−20) between E-RFT member D2VY17 and its homolog XP_004334153. C. Hydropathy plot of E-RFT homolog XP_004334153. D. Hydropathy plot of ENT member Q99808 (TC# 2.A.57.1.1). E. Hydropathy plot of the alignment (E-value: 1.5×10−135) between ENT member Q99808 and its homolog XP_005604017. F. Hydropathy plot of ENT homolog XP_005604017. G. Hydropathy plot of the 10-TMS alignment (E-value: 9.3×10−12) between E-RTF homolog XP_004334153 and ENT homolog XP_005604017. According to the TMSOC classification, only protein XP_005604017 has simple TMSs (TMSs 3 and 9) in agreement with the MFS distribution observed in Fig 2. The Pfam domain characteristic of family ENT (PF01733) can be projected to E-RFT homolog XP_004334153 (E-value: 2.2×10−7), further supporting the relationship between these two families.
Hydropathy plots are presented comparing families MprF and LysS. Vertical black lines with wedges denote the regions of the proteins in panels A and B involved in the alignment shown in panel C. A. Hydropathy plot of MprF member Q3M879 (TC# 4.H.1.1.6). Hydrophobic peaks (inferred TMSs) are highlighted in tan colored bars. B. Hydropathy plot of LysS member WP_047224658 (TC# 9.B.111.1.4). Inferred TMSs are highlighted with cyan bars. C. Hydropathy plot of the highly significant 6-TMS alignment (E-value: 1.3×10−48) between MprF member Q3M879 and LysS member WP_047224658. Overlapping TMSs are highlighted with green bars. According to the TMSOC classification, only TMS 3 in MprF member Q3M879 is simple, and in LysS member WP_047224658 TMSs 4–5 are simple. See Section 1.3.2.5 for the discussion explaining how these differences are compatible with MFS. Notwithstanding the two simple TMS in WP_047224658, the high significance of the pairwise alignment fully covering both proteins (E-value: 1.3×10−48; Fig S8C) leaves little doubt that they are related. The relationship is further supported by the highly significant projection of the Pfam domain of family LysS (PF16995) to MprF member Q3M879 (E-value: 6.9×10−16). The C-terminal TMS in both proteins is not highlighted because it is not part of the MFS-like repeat unit.
The program TMSOC was used to classify TMSs into three types: complex, twilight and simple. A) Frequency of the type of TMS per TMS position across 124 LysS members with 6 TMSs. B) Frequency of simple TMSs per TMS position. Note that 1) TMSs 3 and 6 have the highest frequencies of simple TMSs in agreement the reference distribution of the 12-TMS MFS members in Fig 2, and 2) TMSs 4 and 5 show higher frequencies of simple TMSs compared to positions 1 and 2.
Hydropathy plots are presented across the homology transitivity path between families LysS and MFS. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of LysS member O69916 (TC: 9.B.111.1.2). B. Hydropathy plot of the alignment (E-value: 5.3×10−209) between LysS member O69916 and its homolog WP_030772239. C. Hydropathy plot of LysS homolog WP_030772239. D. Hydropathy plot of MFS member Q8NHS3 (TC: 2.A.1.2.56). E. Hydropathy plot of the alignment (E-value: 3.7×10−32) between MFS member Q8NHS3 and its homolog EYC21101. F. Hydropathy plot of MFS homolog EYC21101. G. Hydropathy plot of the 6-TMS alignment (E-value: 4.2×10−8) between LysS homolog WP_030772239 and MFS homolog EYC21101. According to the TMSOC classification, TMSs 3 and 6 of both proteins are simple in agreement with the patterns observed for MFS (Fig 2) and LysS (Fig S9). The Pfam domain characteristic of family LysS (PF16995) can be projected to MFS homolog EYC21101 (E-value: 2×10−5), further supporting the relationship between these two families.
Hydropathy plots are presented across the homology transitivity path between families Cytb561 and POT. To save space, Cytb561 is displayed as Cytb in the plots. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of Cytb561 member Q53TN4 (TC: 5.B.2.1.3). B. Hydropathy plot of the alignment (E-value: 2.3×10−77) between Cytb561 member Q53TN4 and its homolog XP_005393290. C. Hydropathy plot of Cytb561 homolog XP_005393290. D. Hydropathy plot of POT member Q9LQL2 (TC: 2.A.17.3.19). E. Hydropathy plot of the alignment (E-value: 2.8×10−181) between POT member Q9LQL2 and its homolog XP_002320578. F. Hydropathy plot of POT homolog XP_002320578. G. Hydropathy plot of the 6-TMS alignment (E-value: 2.5×10−9) between Cytb561 homolog XP_005393290 and POT homolog XP_002320578. According to the TMSOC classification, only the first TMS in Cytb561 homolog XP_005393290 is simple. This is compatible with MFS because there are established MFS families that show a tendency to have the first TMS simple (Fig S1), and because the global distribution of simple/complex TMSs in Cytb561 members with 6 TMSs also shows that TMSs 3 and 6 have higher frequencies of simple TMSs (Fig S12), which is compatible with MFS (Fig 2). The Pfam domain characteristic of family POT (PF00854) can be projected to Cytb561 homolog XP_005393290 (E-value: 9.6×10−6), further supporting the relationship between these two families.
The program TMSOC was used to classify TMSs into three types: complex, twilight or simple. A) Frequency of the type of TMS per TMS position across 693 Cytb561 members with 6 TMSs. B) Frequency of simple TMSs per TMS position. Note that although the first TMS shows high frequency of simple TMSs, TMSs 3 and 6 have considerably higher frequencies of simple TMSs relative positions 2, 4 and 5, which is in agreement with the reference distribution of the 12- and 14-TMS MFS members in Figures 2, and S1.
Hydropathy plots are presented across the homology transitivity path between families CCPG and Cytb561. To save space, family Cytb561 is abbreviated as Cytb in the titles of panels D and F. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of CCPG member G4MKH1 (TC: 9.B.57.1.1). B. Hydropathy plot of the alignment (E-value: 9.6×10−103) between CCGP member G4MKH1 and its homolog KUL88187. C. Hydropathy plot of CCGP homolog KUL88187. D. Hydropathy plot of Cytb561 member R0K6W2 (TC: 5.B.2.3.1). E. Hydropathy plot of the alignment (E-value: 2.4×10−63) between Cytb561 member R0K6W2 and its homolog OAK96959. F. Hydropathy plot of Cytb561 homolog OAK96959. G. Hydropathy plot of the 5-TMS alignment (E-value: 7.1×10−11) between CCGP homolog KUL88187 and Cytb561 homolog OAK96959. According to the TMSOC classification, only the third TMS of KUL88187 (TMS 4 of the ancestral 12-TMS precursor) and the fifth TMS of OAK96959 (TMS 6 of the ancestral 6-TMS precursor) are simple. See Section 1.3.2.7 for the discussion explaining how these differences are compatible with MFS. More importantly, the Pfam domain PF10348 is present (hmmscan E-value: 3.2×10−7) in both families, and the domain characteristic of family Cytb561 (PF03188) belongs to the same clan as domain PF10348 (CL0328), which further supports the relationship between these two families.
Hydropathy plots are presented across the homology transitivity path between families DUF1275 and the MFS. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of DUF1275 member Q8PUP5 (TC: 9.B.143.2.3). B. Hydropathy plot of the alignment (E-value: 4.8×10−35) between DUF1275 member Q8PUP5 and its homolog WP_003499261. C. Hydropathy plot of DUF1275 homolog WP_003499261. D. Hydropathy plot of MFS member Q07282 (TC: 2.A.1.2.75). E. Hydropathy plot of the alignment (E-value: 3.2×10−18) between MFS member Q07282 and its homolog WP_015325006. F. Hydropathy plot of MFS homolog WP_015325006. G. Hydropathy plot of the 5-TMS alignment (E-value: 1.3×10−9) between DUF1275 homolog WP_003499261 and MFS homolog WP_015325006. According to the TMSOC classification, none of the TMSs in both proteins is simple, which is compatible with the distribution observed for MFS (Fig 2A) The Pfam domain characteristic of family DUF1275 (PF06912) can be projected to MFS homolog WP_015325006 (E-value: 7.7×10−7), further supporting the relationship between these two families.
Hydropathy plots are presented across the homology transitivity path between families MIP and POT. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of MIP member P55087 (TC: 1.A.8.8.5). B. Hydropathy plot of the alignment (E-value: 7.6×10−40) between MIP member P55087and its homolog CAX48992. C. Hydropathy plot of MIP homolog CAX48992. D. Hydropathy plot of POT member Q59ST2 (TC: 2.A.17.2.5). E. Hydropathy plot of the alignment (E-value: 9.9×10−104) between POT member Q59ST2 and its homolog EEH20305. F. Hydropathy plot of POT homolog EEH20305. G. Hydropathy plot of the alignment (E-value: 5.0×10−8) between MIP homolog CAX48992 and POT homolog EEH20305. Notice in panel G that the hydropathies of TMSs 1, 3 and 5 are not properly aligned. In addition, the Pfam domain of family MIP (PF00230) is projected to POT homolog EEH20305 with marginal significance (E-value: 1.4×10−4), thus decreasing our confidence in this relationship.
Highlights.
The Major Facilitator Superfamily (MFS) is the largest superfamily of secondary transporters currently known.
Here we expand this superfamily with nine more families, bringing the total to over 100 families.
Among these new families, three are integral membrane proteins not currently recognized as transporters.
These include: (1) The Lysyl Phosphatidyl glycerol synthase (MrpF; TC# 4.H.1), (2) The Eukaryotic Cytochrome b561 (Cytb561; TC# 5.B.2) Family, and (3) The 6 TMS Lysyl tRNA Synthetase (LysS) Family.
The results reported expand the scope and significance of the MFS and reveal novel topological types within the MFS fold.
1.5. Acknowledgements
This work was supported by the National Institutes of Health grant number GM077402. We thank Cali Myers for her assistance in preparing this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
1.6 References
- [1].Saier MH Jr., Tran CV, Barabote RD, TCDB: the Transporter Classification Database for membrane transport protein analyses and information, Nucleic acids research, 34 (2006) D181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Saier MH Jr., Yen MR, Noto K, Tamang DG, Elkan C, The Transporter Classification Database: recent advances, Nucleic acids research, 37 (2009) D274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yee DC, Shlykov MA, Vastermark A, Reddy VS, Arora S, Sun EI, Saier MH Jr., The transporter-opsin-G protein-coupled receptor (TOG) superfamily, FEBS J, 280 (2013) 5780–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marger MD, Saier MH Jr., A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport, Trends Biochem Sci, 18 (1993) 13–20. [DOI] [PubMed] [Google Scholar]
- [5].Pao SS, Paulsen IT, Saier MH Jr., Major facilitator superfamily, Microbiol Mol Biol Rev, 62 (1998) 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reddy VS, Saier MH Jr., BioV Suite--a collection of programs for the study of transport protein evolution, Febs j, 279 (2012) 2036–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Park JH, Saier MH Jr., Phylogenetic characterization of the MIP family of transmembrane channel proteins, J Membr Biol, 153 (1996) 171–180. [DOI] [PubMed] [Google Scholar]
- [8].Morgan JL, Strumillo J, Zimmer J, Crystallographic snapshot of cellulose synthesis and membrane translocation, Nature, 493 (2013) 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Saier MH Jr., Computer-aided analyses of transport protein sequences: gleaning evidence concerning function, structure, biogenesis, and evolution, Microbiol Rev, 58 (1994) 71–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Saier MH Jr., A functional-phylogenetic classification system for transmembrane solute transporters, Microbiol Mol Biol Rev, 64 (2000) 354–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Saier MH Jr., Reddy VS, Tsu BV, Ahmed MS, Li C, Moreno-Hagelsieb G, The Transporter Classification Database (TCDB): recent advances, Nucleic acids research, 44 (2016) D372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Reddy VS, Shlykov MA, Castillo R, Sun EI, Saier MH Jr., The major facilitator superfamily (MFS) revisited, FEBS J, 279 (2012) 2022–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang B, Dukarevich M, Sun EI, Yen MR, Saier MH Jr., Membrane porters of ATP-binding cassette transport systems are polyphyletic, J Membr Biol, 231 (2009) 1–10. [DOI] [PubMed] [Google Scholar]
- [14].Hirai T, Heymann JA, Maloney PC, Subramaniam S, Structural model for 12-helix transporters belonging to the major facilitator superfamily, Journal of bacteriology, 185 (2003) 1712–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Vastermark A, Saier MH, Major Facilitator Superfamily (MFS) evolved without 3-transmembrane segment unit rearrangements, Proceedings of the National Academy of Sciences of the United States of America, 111 (2014) E1162–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yan N, Structural advances for the major facilitator superfamily (MFS) transporters, Trends Biochem Sci, 38 (2013) 151–159. [DOI] [PubMed] [Google Scholar]
- [17].Moraes TF, Reithmeier RA, Membrane transport metabolons, Biochimica et biophysica acta, 1818 (2012) 2687–2706. [DOI] [PubMed] [Google Scholar]
- [18].Yan N, Structural Biology of the Major Facilitator Superfamily Transporters, Annual review of biophysics, 44 (2015) 257–283. [DOI] [PubMed] [Google Scholar]
- [19].Kaback HR, A chemiosmotic mechanism of symport, Proceedings of the National Academy of Sciences of the United States of America, 112 (2015) 1259–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang XC, Zhao Y, Heng J, Jiang D, Energy coupling mechanisms of MFS transporters, Protein science : a publication of the Protein Society, 24 (2015) 1560–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wisedchaisri G, Park MS, Iadanza MG, Zheng H, Gonen T, Proton-coupled sugar transport in the prototypical major facilitator superfamily protein XylE, Nat Commun, 5 (2014) 4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Saier MH Jr., Beatty JT, Goffeau A, Harley KT, Heijne WH, Huang SC, Jack DL, Jahn PS, Lew K, Liu J, Pao SS, Paulsen IT, Tseng TT, Virk PS, The major facilitator superfamily, Journal of molecular microbiology and biotechnology, 1 (1999) 257–279. [PubMed] [Google Scholar]
- [23].Winkler HH, Neuhaus HE, Non-mitochondrial ATP transport, Trends Biochem Sci, 24 (1999) 64–68. [DOI] [PubMed] [Google Scholar]
- [24].Trentmann O, Jung B, Neuhaus HE, Haferkamp I, Nonmitochondrial ATP/ADP transporters accept phosphate as third substrate, The Journal of biological chemistry, 283 (2008) 36486–36493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Newstead S, Molecular insights into proton coupled peptide transport in the PTR family of oligopeptide transporters, Biochimica et biophysica acta, 1850 (2015) 488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Newstead S, Towards a structural understanding of drug and peptide transport within the proton-dependent oligopeptide transporter (POT) family, Biochem Soc Trans, 39 (2011) 1353–1358. [DOI] [PubMed] [Google Scholar]
- [27].Matherly LH, Wilson MR, Hou Z, The major facilitative folate transporters solute carrier 19A1 and solute carrier 46A1: biology and role in antifolate chemotherapy of cancer, Drug metabolism and disposition: the biological fate of chemicals, 42 (2014) 632–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Flintoff WF, Williams FM, Sadlish H, The region between transmembrane domains 1 and 2 of the reduced folate carrier forms part of the substrate-binding pocket, The Journal of biological chemistry, 278 (2003) 40867–40876. [DOI] [PubMed] [Google Scholar]
- [29].Hong M, Critical domains within the sequence of human organic anion transporting polypeptides, Current drug metabolism, 15 (2014) 265–270. [DOI] [PubMed] [Google Scholar]
- [30].Hagenbuch B, Stieger B, The SLCO (former SLC21) superfamily of transporters, Molecular aspects of medicine, 34 (2013) 396–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tsigelny IF, Kovalskyy D, Kouznetsova VL, Balinskyi O, Sharikov Y, Bhatnagar V, Nigam SK, Conformational changes of the multispecific transporter organic anion transporter 1 (OAT1/SLC22A6) suggests a molecular mechanism for initial stages of drug and metabolite transport, Cell biochemistry and biophysics, 61 (2011) 251–259. [DOI] [PubMed] [Google Scholar]
- [32].Zhu C, Nigam KB, Date RC, Bush KT, Springer SA, Saier MH Jr., Wu W, Nigam SK, Evolutionary Analysis and Classification of OATs, OCTs, OCTNs, and Other SLC22 Transporters: Structure-Function Implications and Analysis of Sequence Motifs, PloS one, 10 (2015) e0140569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tazoe Y, Hayashi H, Tsuboi S, Shioura T, Matsuyama T, Yamada H, Hirai K, Tsuji D, Inoue K, Sugiyama T, Itoh K, Reduced folate carrier 1 gene expression levels are correlated with methotrexate efficacy in Japanese patients with rheumatoid arthritis, Drug metabolism and pharmacokinetics, 30 (2015) 227–230. [DOI] [PubMed] [Google Scholar]
- [34].Eudes A, Kunji ER, Noiriel A, Klaus SM, Vickers TJ, Beverley SM, Gregory JF 3rd, Hanson AD, Identification of transport-critical residues in a folate transporter from the folate-biopterin transporter (FBT) family, The Journal of biological chemistry, 285 (2010) 2867–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Vastermark A, Driker A, Li J, Saier MH Jr., Conserved movement of TMS11 between occluded conformations of LacY and XylE of the major facilitator superfamily suggests a similar hinge-like mechanism, Proteins, 83 (2015) 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wong WC, Maurer-Stroh S, Eisenhaber F, Not all transmembrane helices are born equal: Towards the extension of the sequence homology concept to membrane proteins, Biol Direct, 6 (2011) 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wong WC, Maurer-Stroh S, Schneider G, Eisenhaber F, Transmembrane helix: simple or complex, Nucleic acids research, 40 (2012) W370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE, Finn RD, The Pfam protein families database in 2019, Nucleic acids research, 47 (2019) D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhai Y, Saier MH Jr., A web-based program for the prediction of average hydropathy, average amphipathicity and average similarity of multiply aligned homologous proteins, Journal of molecular microbiology and biotechnology, 3 (2001) 285–286. [PubMed] [Google Scholar]
- [40].Medrano-Soto A, Moreno-Hagelsieb G, McLaughlin D, Ye ZS, Hendargo KJ, Saier MH Jr., Bioinformatic characterization of the Anoctamin Superfamily of Ca2+-activated ion channels and lipid scramblases, PloS one, 13 (2018) e0192851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic acids research, 25 (1997) 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fu L, Niu B, Zhu Z, Wu S, Li W, CD-HIT: accelerated for clustering the next-generation sequencing data, Bioinformatics (Oxford, England), 28 (2012) 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pearson WR, Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms, Genomics, 11 (1991) 635–650. [DOI] [PubMed] [Google Scholar]
- [44].Wong WC, Maurer-Stroh S, Eisenhaber F, More than 1,001 problems with protein domain databases: transmembrane regions, signal peptides and the issue of sequence homology, PLoS Comput Biol, 6 (2010) e1000867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Katoh K, Standley DM, MAFFT multiple sequence alignment software version 7: improvements in performance and usability, Mol Biol Evol, 30 (2013) 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T, trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses, Bioinformatics (Oxford, England), 25 (2009) 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhai Y, Saier MH Jr., A web-based program (WHAT) for the simultaneous prediction of hydropathy, amphipathicity, secondary structure and transmembrane topology for a single protein sequence, Journal of molecular microbiology and biotechnology, 3 (2001) 501–502. [PubMed] [Google Scholar]
- [48].Tusnady GE, Simon I, The HMMTOP transmembrane topology prediction server, Bioinformatics (Oxford, England), 17 (2001) 849–850. [DOI] [PubMed] [Google Scholar]
- [49].Bernsel A, Viklund H, Hennerdal A, Elofsson A, TOPCONS: consensus prediction of membrane protein topology, Nucleic acids research, 37 (2009) W465–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Reddy A, Cho J, Ling S, Reddy V, Shlykov M, Saier MH, Reliability of nine programs of topological predictions and their application to integral membrane channel and carrier proteins, Journal of molecular microbiology and biotechnology, 24 (2014) 161–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL, OPM database and PPM web server: resources for positioning of proteins in membranes, Nucleic acids research, 40 (2012) D370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tusnady GE, Dosztanyi Z, Simon I, PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank, Nucleic acids research, 33 (2005) D275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Frishman D, Argos P, Knowledge-based protein secondary structure assignment, Proteins, 23 (1995) 566–579. [DOI] [PubMed] [Google Scholar]
- [54].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Overview of the CCP4 suite and current developments, Acta Crystallogr D Biol Crystallogr, 67 (2011) 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Krissinel E, Henrick K, Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions, Acta Crystallogr D Biol Crystallogr, 60 (2004) 2256–2268. [DOI] [PubMed] [Google Scholar]
- [56].Zhang Y, Skolnick J, TM-align: a protein structure alignment algorithm based on the TM-score, Nucleic acids research, 33 (2005) 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yen MR, Choi J, Saier MH Jr, Bioinformatic Analyses of Transmembrane Transport: Novel Software for Deducing Protein Phylogeny, Topology, and Evolution, Journal of molecular microbiology and biotechnology, 17 (2009) 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Eddy SR, Accelerated Profile HMM Searches, PLoS Comput Biol, 7 (2011) e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Valdes R, Elferich J, Shinde U, Landfear SM, Identification of the intracellular gate for a member of the equilibrative nucleoside transporter (ENT) family, The Journal of biological chemistry, 289 (2014) 8799–8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD, The equilibrative nucleoside transporter family, SLC29, Pflugers Archiv : European journal of physiology, 447 (2004) 735–743. [DOI] [PubMed] [Google Scholar]
- [61].Harley KT, Djordjevic GM, Tseng TT, Saier MH, Membrane-fusion protein homologues in gram-positive bacteria, Mol Microbiol, 36 (2000) 516–517. [DOI] [PubMed] [Google Scholar]
- [62].Harley KT, Saier MH Jr., A novel ubiquitous family of putative efflux transporters, Journal of molecular microbiology and biotechnology, 2 (2000) 195–198. [PubMed] [Google Scholar]
- [63].Van Dyk TK, Templeton LJ, Cantera KA, Sharpe PL, Sariaslani FS, Characterization of the Escherichia coli AaeAB efflux pump: a metabolic relief valve?, Journal of bacteriology, 186 (2004) 7196–7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yeh KY, Yeh M, Glass J, Interactions between ferroportin and hephaestin in rat enterocytes are reduced after iron ingestion, Gastroenterology, 141 (2011) 292–299, 299.e291. [DOI] [PubMed] [Google Scholar]
- [65].Madejczyk MS, Ballatori N, The iron transporter ferroportin can also function as a manganese exporter, Biochimica et biophysica acta, 1818 (2012) 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Delaby C, Pilard N, Puy H, Canonne-Hergaux F, Sequential regulation of ferroportin expression after erythrophagocytosis in murine macrophages: early mRNA induction by haem, followed by iron-dependent protein expression, The Biochemical journal, 411 (2008) 123–131. [DOI] [PubMed] [Google Scholar]
- [67].Mitchell CJ, Shawki A, Ganz T, Nemeth E, Mackenzie B, Functional properties of human ferroportin, a cellular iron exporter reactive also with cobalt and zinc, American journal of physiology. Cell physiology, 306 (2014) C450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Taniguchi R, Kato HE, Font J, Deshpande CN, Wada M, Ito K, Ishitani R, Jormakka M, Nureki O, Outward- and inward-facing structures of a putative bacterial transition-metal transporter with homology to ferroportin, Nat Commun, 6 (2015) 8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Nomura N, Verdon G, Kang HJ, Shimamura T, Nomura Y, Sonoda Y, Hussien SA, Qureshi AA, Coincon M, Sato Y, Abe H, Nakada-Nakura Y, Hino T, Arakawa T, Kusano-Arai O, Iwanari H, Murata T, Kobayashi T, Hamakubo T, Kasahara M, Iwata S, Drew D, Structure and mechanism of the mammalian fructose transporter GLUT5, Nature, 526 (2015) 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yonezawa A, Masuda S, Katsura T, Inui K, Identification and functional characterization of a novel human and rat riboflavin transporter, RFT1, American journal of physiology. Cell physiology, 295 (2008) C632–641. [DOI] [PubMed] [Google Scholar]
- [71].Powers HJ, Riboflavin (vitamin B-2) and health, The American Journal of Clinical Nutrition, 77 (2003) 1352–1360. [DOI] [PubMed] [Google Scholar]
- [72].Yamamoto S, Inoue K, Ohta KY, Fukatsu R, Maeda JY, Yoshida Y, Yuasa H, Identification and functional characterization of rat riboflavin transporter 2, Journal of biochemistry, 145 (2009) 437–443. [DOI] [PubMed] [Google Scholar]
- [73].Ernst CM, Peschel A, Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids, Mol Microbiol, 80 (2011) 290–299. [DOI] [PubMed] [Google Scholar]
- [74].Ernst CM, Staubitz P, Mishra NN, Yang SJ, Hornig G, Kalbacher H, Bayer AS, Kraus D, Peschel A, The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion, PLoS Pathog, 5 (2009) e1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ernst CM, Kuhn S, Slavetinsky CJ, Krismer B, Heilbronner S, Gekeler C, Kraus D, Wagner S, Peschel A, The lipid-modifying multiple peptide resistance factor is an oligomer consisting of distinct interacting synthase and flippase subunits, MBio, 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Motzik A, Nechushtan H, Foo SY, Razin E, Non-canonical roles of lysyl-tRNA synthetase in health and disease, Trends in molecular medicine, 19 (2013) 726–731. [DOI] [PubMed] [Google Scholar]
- [77].Kamensky Y, Liu W, Tsai AL, Kulmacz RJ, Palmer G, Axial ligation and stoichiometry of heme centers in adrenal cytochrome b561, Biochemistry, 46 (2007) 8647–8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Nakanishi N, Takeuchi F, Tsubaki M, Histidine cycle mechanism for the concerted proton/electron transfer from ascorbate to the cytosolic haem b centre of cytochrome b561: a unique machinery for the biological transmembrane electron transfer, Journal of biochemistry, 142 (2007) 553–560. [DOI] [PubMed] [Google Scholar]
- [79].Liu W, Rogge CE, da Silva GF, Shinkarev VP, Tsai AL, Kamensky Y, Palmer G, Kulmacz RJ, His92 and His110 selectively affect different heme centers of adrenal cytochrome b(561), Biochimica et biophysica acta, 1777 (2008) 1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lu P, Ma D, Yan C, Gong X, Du M, Shi Y, Structure and mechanism of a eukaryotic transmembrane ascorbate-dependent oxidoreductase, Proceedings of the National Academy of Sciences of the United States of America, 111 (2014) 1813–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lu Q, Lu JP, Li XD, Liu XH, Min H, Lin FC, Magnaporthe oryzae MTP1 gene encodes a type III transmembrane protein involved in conidiation and conidial germination, J Zhejiang Univ Sci B, 9 (2008) 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Pao GM, Wu LF, Johnson KD, Hofte H, Chrispeels MJ, Sweet G, Sandal NN, Saier MH Jr., Evolution of the MIP family of integral membrane transport proteins, Mol Microbiol, 5 (1991) 33–37. [DOI] [PubMed] [Google Scholar]
- [83].Abascal F, Irisarri I, Zardoya R, Diversity and evolution of membrane intrinsic proteins, Biochimica et biophysica acta, 1840 (2014) 1468–1481. [DOI] [PubMed] [Google Scholar]
- [84].Palmieri F, The mitochondrial transporter family SLC25: identification, properties and physiopathology, Molecular aspects of medicine, 34 (2013) 465–484. [DOI] [PubMed] [Google Scholar]
- [85].Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G, Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside, Nature, 426 (2003) 39–44. [DOI] [PubMed] [Google Scholar]
- [86].Staubitz P, Neumann H, Schneider T, Wiedemann I, Peschel A, MprF-mediated biosynthesis of lysylphosphatidylglycerol, an important determinant in staphylococcal defensin resistance, FEMS Microbiol Lett, 231 (2004) 67–71. [DOI] [PubMed] [Google Scholar]
- [87].Andra J, Goldmann T, Ernst CM, Peschel A, Gutsmann T, Multiple peptide resistance factor (MprF)-mediated Resistance of Staphylococcus aureus against antimicrobial peptides coincides with a modulated peptide interaction with artificial membranes comprising lysyl-phosphatidylglycerol, The Journal of biological chemistry, 286 (2011) 18692–18700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Slavetinsky CJ, Peschel A, Ernst CM, Alanyl-phosphatidylglycerol and lysyl-phosphatidylglycerol are translocated by the same MprF flippases and have similar capacities to protect against the antibiotic daptomycin in Staphylococcus aureus, Antimicrob Agents Chemother, 56 (2012) 3492–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Hebecker S, Krausze J, Hasenkampf T, Schneider J, Groenewold M, Reichelt J, Jahn D, Heinz DW, Moser J, Structures of two bacterial resistance factors mediating tRNA-dependent aminoacylation of phosphatidylglycerol with lysine or alanine, Proceedings of the National Academy of Sciences of the United States of America, 112 (2015) 10691–10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Asard H, Barbaro R, Trost P, Berczi A, Cytochromes b561: ascorbate-mediated trans-membrane electron transport, Antioxid Redox Signal, 19 (2013) 1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Berczi A, Zimanyi L, The trans-membrane cytochrome b561 proteins: structural information and biological function, Curr Protein Pept Sci, 15 (2014) 745–760. [DOI] [PubMed] [Google Scholar]
- [92].Lane DJ, Bae DH, Merlot AM, Sahni S, Richardson DR, Duodenal cytochrome b (DCYTB) in iron metabolism: an update on function and regulation, Nutrients, 7 (2015) 2274–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The program TMSOC was used to classify TMSs into three types: complex, twilight and simple. A) Frequency of the type of TMS per TMS position as reported by TMSOC across 69 MFS members with 14 TMSs. B) Frequency of simple TMSs per TMS position. Note that 1) all TMSs are predominantly complex in the 2 repeat units (TMSs 1–6 and 9–12); 2) the two central TMSs 7–8, which are not part of the 6-TMS repeat units, have higher frequencies of simple TMSs; 3) the first repeat unit has more simple TMSs in the first TMS compared to the third TMS, but the third TMS still has more simple TMSs compared to TMSs in positions 2, 4 and 5; and 4) the frequencies of simple TMSs in the second repeat unit follows the same pattern as the 12-TMS MFS members in Fig 2.
Sequences were renamed to facilitate visualization of the tree. The family name is followed by the 2 last digits of the TC numbers, separated by underscores. Families with names larger than 4 characters were shortened following the convension: Cytb561 (Cytb), DUF1275 (DUF), POT/PTR (POT), and E-RFT (RFT).
Table S1. Comparison of 9 new MFS families with families in the negative control set.
Hydropathy plots are presented across the homology transitivity path between family ArAE and the MFS. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of ArAE member S7PTY2 (TC# 2.A.85.3.5). B. Hydropathy plot of the alignment (E-value: 1.9×10−184) between ArAE member S7PTY2 and its homolog KII87451. C. Hydropathy plot of ArAE homolog KII87451. D. Hydropathy plot of MFS member A0ZXK6 (TC# 2.A.1.1.43). E. Hydropathy plot of the alignment (E-value: 2.6×10−79) between MFS member A0ZXK6 and its homolog EIE83441. F. Hydropathy plot of MFS homolog EIE83441. G. Hydropathy plot of the 6-TMS alignment (E-value: 9.5×10−11) between ArAE homolog KII87451 and MFS homolog EIE83441. According to the TMSOC classification, only TMS 6 in KII87451and TMS 12 in EIE83441 are simple. This is compatible with the MFS distribution of simple TMSs observed in Fig 2. The Pfam domain characteristic of family MFS (PF00083) can be projected to ArAE homolog KII87451 (E-value: 10−6), further supporting the relationship between these two families.
The leaves are labeled following the same format as for File S1.
Table S2. Relative confidence scores of homology inferences.
Hydropathy plots are presented across the homology transitivity path between families Fpn and GPH. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of Fpn member Q6MLJ0 (TC# 2.A.100.2.1). B. Hydropathy plot of the alignment (E-value: 5.0×10−110) between Fpn member Q6MLJ0 and its homolog WP_015091118. C. Hydropathy plot of Fpn homolog WP_015091118. D. Hydropathy plot of GPH member P94488 (TC# 2.A.2.3.2). E. Hydropathy plot of the alignment (E-value: 2.5×10−54) between GPH member P94488 and its homolog KLE28886. F. Hydropathy plot of GPH homolog KLE28886. G. Hydropathy plot of the 10-TMS alignment (E-value: 2.8×10−10) between Fpn homolog WP_015091118 and GPH homolog KLE28886. According to TMSOC, and compatible with MFS (Fig 2), only the twelfth TMS in both proteins is simple and it is not part of the alignment. The Pfam domains characteristic of families Fpn (PF06963) and GPH (PF03083) are included in the alignment and belong to the same clan (CL0015), further supporting the relationship between these two families.
Ribbon representation of the structural superposition between TMSs 1–6 of Fpn member Q6MLJ0 (TC# 2.A.100.2.1; PDB ID: 5AYN; green) and TMSs 1–6 of the GPH homolog P30878 (PDB ID: 4M64; cyan). The alignment has an RMSD value of 2.93 Å with 93% coverage of the smallest 6-helix bundle. A) Top view and B) side view.
Ribbon representation of the structural superposition between TMSs 1–12 of Fpn member Q6MLJ0 (TC# 2.A.100.2.1; PDB ID: 5AYN; cyan) and TMSs 1–12 of the MFS member P11551 (TC# 2.A.1.7.1; PDB ID: 3O7P; green). The alignment has an RMSD value of 3.0 Å with 98% coverage of the smallest structure. A) Top view and B) side view.
Ribbon representation of the structural superposition between TMSs 1–6 of Fpn member Q6MLJ0 (TC# 2.A.100.2.1; PDB ID: 5AYN; cyan) and TMSs 1–6 of the MFS member P11551 (TC# 2.A.1.7.1; PDB ID: 3O7P; green). The alignment has an RMSD value of 2.27 Å with 97% coverage of the smallest 6-helix bundle. A) Top view and B) side view.
Hydropathy plots are presented across the homology transitivity path between families E-RFT and ENT. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of E-RFT member D2VY17 (TC# 2.A.125.1.5). B. Hydropathy plot of the alignment (E-value: 1.2×10−20) between E-RFT member D2VY17 and its homolog XP_004334153. C. Hydropathy plot of E-RFT homolog XP_004334153. D. Hydropathy plot of ENT member Q99808 (TC# 2.A.57.1.1). E. Hydropathy plot of the alignment (E-value: 1.5×10−135) between ENT member Q99808 and its homolog XP_005604017. F. Hydropathy plot of ENT homolog XP_005604017. G. Hydropathy plot of the 10-TMS alignment (E-value: 9.3×10−12) between E-RTF homolog XP_004334153 and ENT homolog XP_005604017. According to the TMSOC classification, only protein XP_005604017 has simple TMSs (TMSs 3 and 9) in agreement with the MFS distribution observed in Fig 2. The Pfam domain characteristic of family ENT (PF01733) can be projected to E-RFT homolog XP_004334153 (E-value: 2.2×10−7), further supporting the relationship between these two families.
Hydropathy plots are presented comparing families MprF and LysS. Vertical black lines with wedges denote the regions of the proteins in panels A and B involved in the alignment shown in panel C. A. Hydropathy plot of MprF member Q3M879 (TC# 4.H.1.1.6). Hydrophobic peaks (inferred TMSs) are highlighted in tan colored bars. B. Hydropathy plot of LysS member WP_047224658 (TC# 9.B.111.1.4). Inferred TMSs are highlighted with cyan bars. C. Hydropathy plot of the highly significant 6-TMS alignment (E-value: 1.3×10−48) between MprF member Q3M879 and LysS member WP_047224658. Overlapping TMSs are highlighted with green bars. According to the TMSOC classification, only TMS 3 in MprF member Q3M879 is simple, and in LysS member WP_047224658 TMSs 4–5 are simple. See Section 1.3.2.5 for the discussion explaining how these differences are compatible with MFS. Notwithstanding the two simple TMS in WP_047224658, the high significance of the pairwise alignment fully covering both proteins (E-value: 1.3×10−48; Fig S8C) leaves little doubt that they are related. The relationship is further supported by the highly significant projection of the Pfam domain of family LysS (PF16995) to MprF member Q3M879 (E-value: 6.9×10−16). The C-terminal TMS in both proteins is not highlighted because it is not part of the MFS-like repeat unit.
The program TMSOC was used to classify TMSs into three types: complex, twilight and simple. A) Frequency of the type of TMS per TMS position across 124 LysS members with 6 TMSs. B) Frequency of simple TMSs per TMS position. Note that 1) TMSs 3 and 6 have the highest frequencies of simple TMSs in agreement the reference distribution of the 12-TMS MFS members in Fig 2, and 2) TMSs 4 and 5 show higher frequencies of simple TMSs compared to positions 1 and 2.
Hydropathy plots are presented across the homology transitivity path between families LysS and MFS. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of LysS member O69916 (TC: 9.B.111.1.2). B. Hydropathy plot of the alignment (E-value: 5.3×10−209) between LysS member O69916 and its homolog WP_030772239. C. Hydropathy plot of LysS homolog WP_030772239. D. Hydropathy plot of MFS member Q8NHS3 (TC: 2.A.1.2.56). E. Hydropathy plot of the alignment (E-value: 3.7×10−32) between MFS member Q8NHS3 and its homolog EYC21101. F. Hydropathy plot of MFS homolog EYC21101. G. Hydropathy plot of the 6-TMS alignment (E-value: 4.2×10−8) between LysS homolog WP_030772239 and MFS homolog EYC21101. According to the TMSOC classification, TMSs 3 and 6 of both proteins are simple in agreement with the patterns observed for MFS (Fig 2) and LysS (Fig S9). The Pfam domain characteristic of family LysS (PF16995) can be projected to MFS homolog EYC21101 (E-value: 2×10−5), further supporting the relationship between these two families.
Hydropathy plots are presented across the homology transitivity path between families Cytb561 and POT. To save space, Cytb561 is displayed as Cytb in the plots. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of Cytb561 member Q53TN4 (TC: 5.B.2.1.3). B. Hydropathy plot of the alignment (E-value: 2.3×10−77) between Cytb561 member Q53TN4 and its homolog XP_005393290. C. Hydropathy plot of Cytb561 homolog XP_005393290. D. Hydropathy plot of POT member Q9LQL2 (TC: 2.A.17.3.19). E. Hydropathy plot of the alignment (E-value: 2.8×10−181) between POT member Q9LQL2 and its homolog XP_002320578. F. Hydropathy plot of POT homolog XP_002320578. G. Hydropathy plot of the 6-TMS alignment (E-value: 2.5×10−9) between Cytb561 homolog XP_005393290 and POT homolog XP_002320578. According to the TMSOC classification, only the first TMS in Cytb561 homolog XP_005393290 is simple. This is compatible with MFS because there are established MFS families that show a tendency to have the first TMS simple (Fig S1), and because the global distribution of simple/complex TMSs in Cytb561 members with 6 TMSs also shows that TMSs 3 and 6 have higher frequencies of simple TMSs (Fig S12), which is compatible with MFS (Fig 2). The Pfam domain characteristic of family POT (PF00854) can be projected to Cytb561 homolog XP_005393290 (E-value: 9.6×10−6), further supporting the relationship between these two families.
The program TMSOC was used to classify TMSs into three types: complex, twilight or simple. A) Frequency of the type of TMS per TMS position across 693 Cytb561 members with 6 TMSs. B) Frequency of simple TMSs per TMS position. Note that although the first TMS shows high frequency of simple TMSs, TMSs 3 and 6 have considerably higher frequencies of simple TMSs relative positions 2, 4 and 5, which is in agreement with the reference distribution of the 12- and 14-TMS MFS members in Figures 2, and S1.
Hydropathy plots are presented across the homology transitivity path between families CCPG and Cytb561. To save space, family Cytb561 is abbreviated as Cytb in the titles of panels D and F. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of CCPG member G4MKH1 (TC: 9.B.57.1.1). B. Hydropathy plot of the alignment (E-value: 9.6×10−103) between CCGP member G4MKH1 and its homolog KUL88187. C. Hydropathy plot of CCGP homolog KUL88187. D. Hydropathy plot of Cytb561 member R0K6W2 (TC: 5.B.2.3.1). E. Hydropathy plot of the alignment (E-value: 2.4×10−63) between Cytb561 member R0K6W2 and its homolog OAK96959. F. Hydropathy plot of Cytb561 homolog OAK96959. G. Hydropathy plot of the 5-TMS alignment (E-value: 7.1×10−11) between CCGP homolog KUL88187 and Cytb561 homolog OAK96959. According to the TMSOC classification, only the third TMS of KUL88187 (TMS 4 of the ancestral 12-TMS precursor) and the fifth TMS of OAK96959 (TMS 6 of the ancestral 6-TMS precursor) are simple. See Section 1.3.2.7 for the discussion explaining how these differences are compatible with MFS. More importantly, the Pfam domain PF10348 is present (hmmscan E-value: 3.2×10−7) in both families, and the domain characteristic of family Cytb561 (PF03188) belongs to the same clan as domain PF10348 (CL0328), which further supports the relationship between these two families.
Hydropathy plots are presented across the homology transitivity path between families DUF1275 and the MFS. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of DUF1275 member Q8PUP5 (TC: 9.B.143.2.3). B. Hydropathy plot of the alignment (E-value: 4.8×10−35) between DUF1275 member Q8PUP5 and its homolog WP_003499261. C. Hydropathy plot of DUF1275 homolog WP_003499261. D. Hydropathy plot of MFS member Q07282 (TC: 2.A.1.2.75). E. Hydropathy plot of the alignment (E-value: 3.2×10−18) between MFS member Q07282 and its homolog WP_015325006. F. Hydropathy plot of MFS homolog WP_015325006. G. Hydropathy plot of the 5-TMS alignment (E-value: 1.3×10−9) between DUF1275 homolog WP_003499261 and MFS homolog WP_015325006. According to the TMSOC classification, none of the TMSs in both proteins is simple, which is compatible with the distribution observed for MFS (Fig 2A) The Pfam domain characteristic of family DUF1275 (PF06912) can be projected to MFS homolog WP_015325006 (E-value: 7.7×10−7), further supporting the relationship between these two families.
Hydropathy plots are presented across the homology transitivity path between families MIP and POT. Refer to the legend of Fig 3 for a detailed description of the format. A. Hydropathy plot of MIP member P55087 (TC: 1.A.8.8.5). B. Hydropathy plot of the alignment (E-value: 7.6×10−40) between MIP member P55087and its homolog CAX48992. C. Hydropathy plot of MIP homolog CAX48992. D. Hydropathy plot of POT member Q59ST2 (TC: 2.A.17.2.5). E. Hydropathy plot of the alignment (E-value: 9.9×10−104) between POT member Q59ST2 and its homolog EEH20305. F. Hydropathy plot of POT homolog EEH20305. G. Hydropathy plot of the alignment (E-value: 5.0×10−8) between MIP homolog CAX48992 and POT homolog EEH20305. Notice in panel G that the hydropathies of TMSs 1, 3 and 5 are not properly aligned. In addition, the Pfam domain of family MIP (PF00230) is projected to POT homolog EEH20305 with marginal significance (E-value: 1.4×10−4), thus decreasing our confidence in this relationship.