

Abstract
There is a significant need for new antibacterial agents as pathogenic bacteria continue to threaten human health through the acquisition of resistance and tolerance towards existing antibiotics. Over the last several years, our group has been developing a novel series of halogenated phenazines that demonstrate potent antibacterial and biofilm eradication activities against critical Gram-positive pathogens, including: Staphylococcus aureus, Staphylococcus epidermidis and Enterococcus faecium. Here, we report the design, chemical synthesis and initial biological assessment of a halogenated phenazine-erythromycin conjugate prodrug 5 aimed at enhancing the translational potential for halogenated phenazines as a treatment of bacterial infections.
Keywords: drug discovery, drug design, antibacterial prodrug conjugate, halogenated phenazine, antibiotic resistance
Graphical Abstract
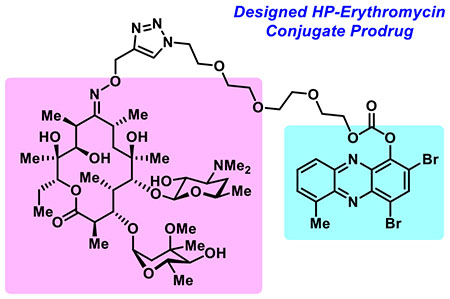
We report the synthesis and initial biological assessment of a halogenated phenazine-erythromycin conjugate prodrug 5 aimed to treat bacterial infections.
Despite significant efforts to treat bacterial infections over the last century, pathogenic bacteria continue to devastate human life.1–5 Bacteria are capable of demonstrating incredible resilience to conventional antibiotics during infection as a result of two distinct biological phenomena, including: (1) antibiotic resistance, and (2) tolerance.5 Bacteria can acquire, or develop, resistance towards antibiotic threat through well-defined mechanisms (e.g., mutation of antibacterial target, efflux pump promoted removal of antibiotics, enzymatic inactivation of antibiotics).5,6 Currently in the United States, there are ~2.8 million new cases and >35,000 deaths each year as a result of antibiotic-resistant bacterial infections.7 In contrast to acquired resistance, metabolically-dormant persister cells demonstrate innate tolerance towards all classes of conventional antibiotics.5,8–10 Surface-attached bacterial biofilms have enriched persister cell populations and are the underlying cause of chronic and recurring infections.5,10
Our group has discovered a synthetically tunable series of halogenated phenazines (HPs) that demonstrates potent antibacterial and biofilm-killing activities against Gram-positive pathogens, including: Staphylococcus aureus, S. epidermidis and Enterococcus faecium.11–15 Several halogenated phenazines have also shown good to excellent antibacterial activities against Mycobacterium tuberculosis12–15, a slow-growing pathogen responsible for killing 1.4 million humans worldwide in 2019.16 We have produced several HPs that demonstrate antibacterial activities with minimum inhibitory concentrations (MICs) ≤ 0.5 μM against Gram-positive pathogens, which is near the potency of vancomycin. HPs have demonstrated outstanding biofilm-killing activities with minimum biofilm eradication concentrations (MBECs) ≤ 10 μM against S. aureus, S. epidermidis and E. faecium, whereas front-running methicillin-resistant S. aureus (MRSA) therapies (vancomycin, daptomycin, linezolid) are unable to eradicate Staphylococcal biofilms at 2,000 μM when tested alongside HPs.12–15
Using RNA-seq technology for transcript profiling, we showed that halogenated phenazine analogue HP-14 eradicates MRSA biofilms through a rapid induction of iron starvation.17 Halogenated phenazines have a metal-coordinating structural moiety that forms a stable 5-membered chelate following binding from the 1-hydroxyl oxygen atom and the adjacent nitrogen embedded in the phenazine heterocycle, which allows iron binding and starvation in bacteria. In addition, we have shown that several HP analogues bind iron(II) using UV-vis spectroscopy.13–15 Despite this mechanism, halogenated phenazines have demonstrated excellent cytotoxicity profiles during our investigations.12–15
In addition to structure-activity relationship (SAR) exploration and mode of action studies, we are working to translate our HP molecules as therapies against antibiotic-resistant and -tolerant bacterial infections. Our current aim is to develop phenol-based prodrug strategies that mitigate the metal-chelation properties of our lead HPs while enhancing physicochemical properties (e.g., water solubility).14,15,18 HP-17 is an analogue we previously reported to demonstrate potent antibacterial (MRSA isolates, MIC = 0.1 – 0.39 μM; Figure 1) and biofilm killing (MRSA BAA-1707, MBEC = 6.25 μM).14 We also reported a PEG-carbonate version of HP-17 (structure not shown) that is unable to bind iron, yet has enhanced antibacterial activities (MRSA isolates, MIC = 0.003 – 0.1 μM) compared to HP-17. We believe the PEG-carbonate version of HP-17 acts as a prodrug with enhanced bacterial-penetrating abilities as the phenolic hydroxyl group of HP-17 has a pKa ~ 7 and exists largely as an anion.
Figure 1.
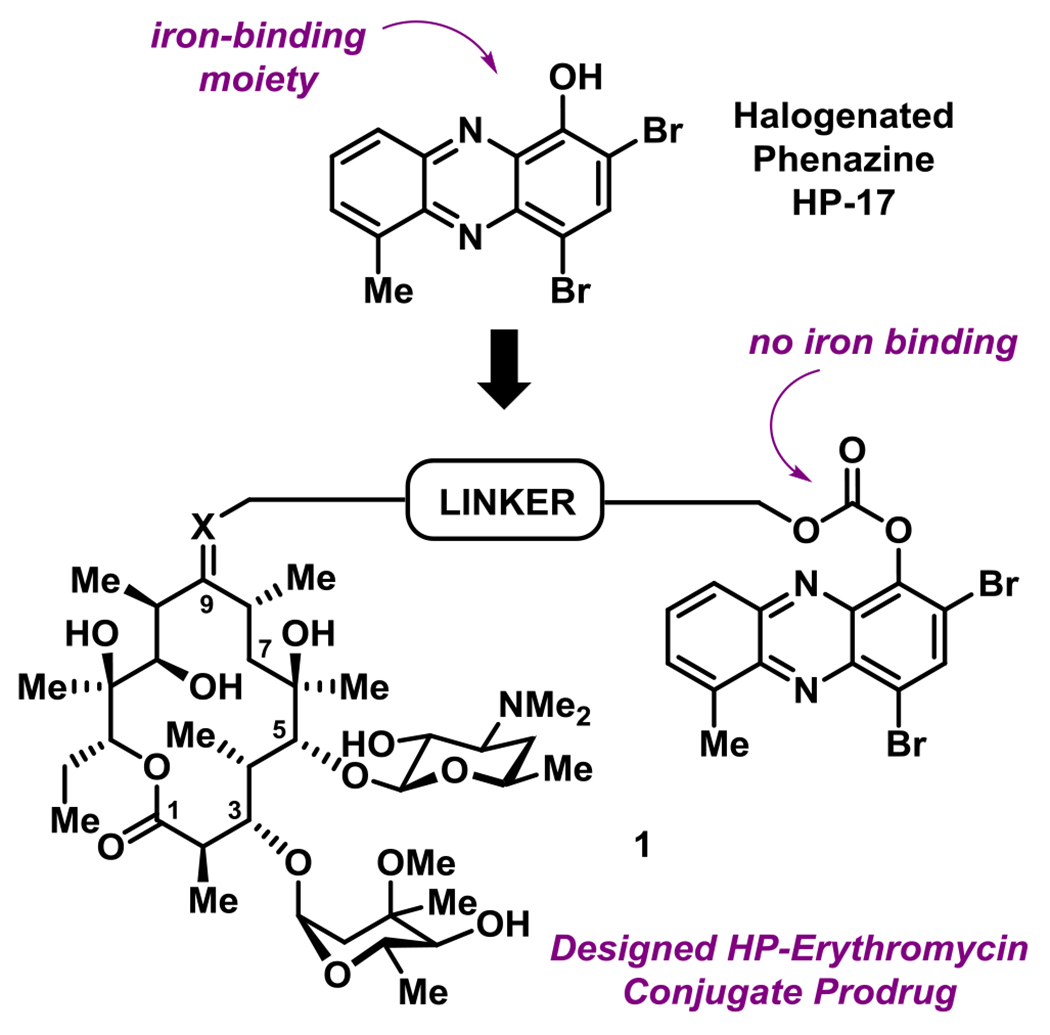
General design strategy for an HP-erythromycin conjugate prodrug 1 that mitigates iron-binding, alters physicochemical properties and aims to enhance translational potential of HP antibacterial agents.
As we continue efforts involving the design and synthesis of new HP prodrugs, we consider antibiotic conjugates to be valuable options for translational purposes (Figure 1).19 We believe that clinically used antibiotics have multiple attributes for conjugate applications, including: (1) antibiotic moieties that dramatically alter physicochemical properties of an investigational antibacterial agent, (2) antibiotics used in humans typically have good pharmacokinetics (PK)/toxicology profiles, thus a great starting point for conjugation, and (3) well-designed conjugates can lead to dual action antibiotics.
Erythromycin is a conventional antibiotic that binds bacterial ribosomes and inhibits protein synthesis.20 This bacteriostatic mode of action inhibits the replication (growth) of bacteria during infection. The desosamine sugar at C-5 of erythromycin’s macrolide architecture serves to anchor this antibiotic to the bacterial ribosome; however, erythromycin’s C-9 ketone is solvent exposed during this binding event and provides an ideal handle for conjugation.20 These features inspired us to design and synthesize an erythromycin-HP conjugate. Our strategy was to build on previous work related to PEG-carbonate analogues of potent HP agents, which operate through the hydrolysis and release of free (“active”) HPs.
The conjugate was designed to be a carbonate prodrug of HP-17 linked to erythromycin at the C-9 ketone to maximize the potential for dual ribosome binding with subsequent HP liberation and iron starvation (Figure 2). Although the linker can be diversified extensively for optimization, we decided to use a PEG linker with four polyethylene glycol units, similar to our previous HP-17 PEG-carbonate prodrug.14 To link the HP and erythromycin, we utilized oxime-based chemistry that others have reported.21–23
Figure 2.
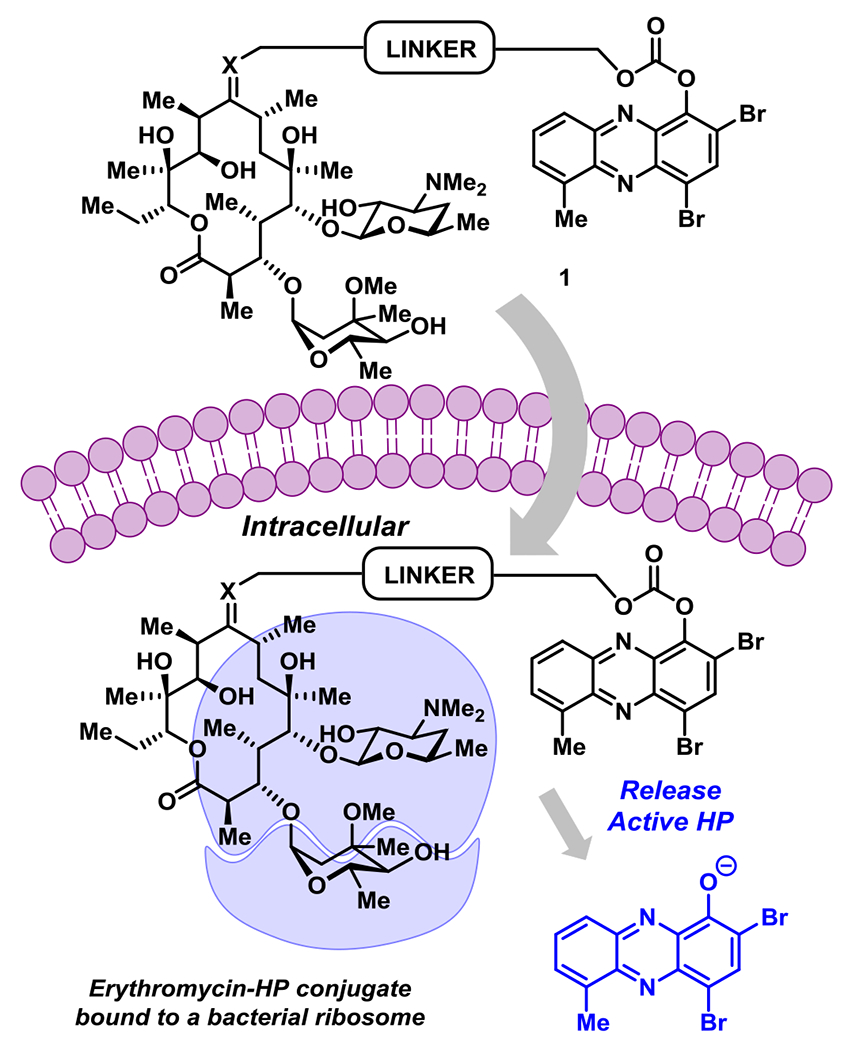
HP-erythromycin conjugate prodrug 1 has the potential to act as a dual ribosome inhibitor and iron-starving agent.
We designed a convergent synthetic route to HP-erythromycin conjugate 5 that hinged on a click reaction to establish the linker between the HP carbonate and erythromycin (Scheme 1). This synthesis began with HP-17 reacting with chloroformate 2 (generated in situ) to give carbonate 3 in 25% yield. In parallel, erythromycin was initially condensed with hydroxylamine to give the corresponding oxime at C-9 (77% yield) followed by alkylation with propargyl bromide in the presence of potassium hydroxide to give 4 (29% yield). Erythromycin’s oxime and 4 have been previously reported21–23; however, we were met with challenges in the purification of these compounds and unable to locate spectra to compare, so we characterized both intermediates by 1H/13C NMR and HRMS. In addition, we purchased 9(E)-Erythromycin A oxime (Cayman Chemical) for direct comparison to our synthetic sample for confirmation in our analytical methods. With key synthetic intermediates 3 and 4 in hand, a click reaction was performed using copper(II) sulfate and ascorbic acid to yield the target erythromycin-HP conjugate 5 in 39% yield.
Scheme 1.
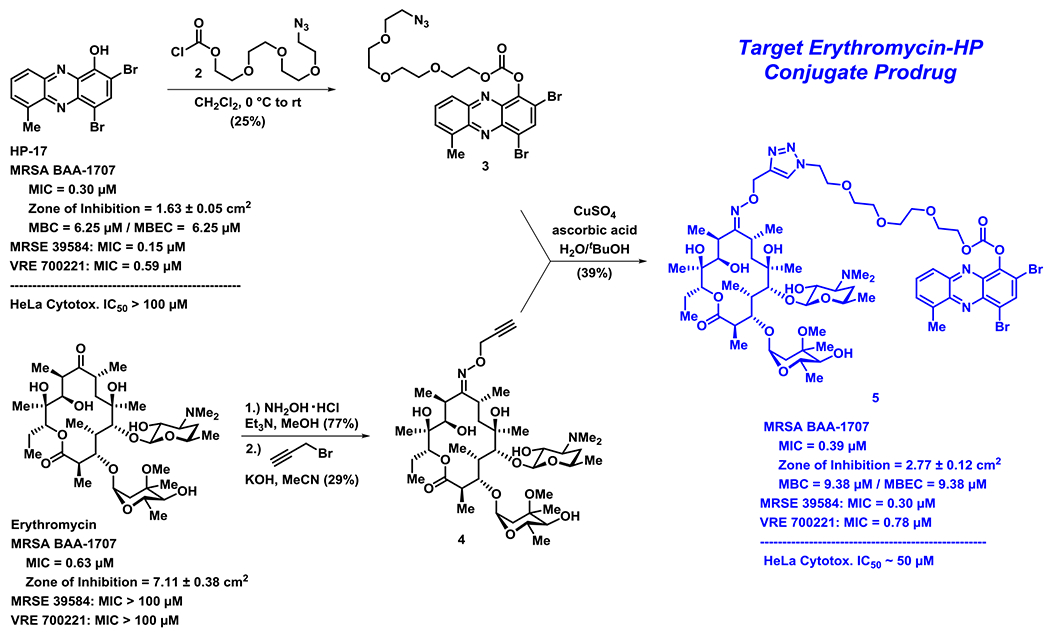
The convergent chemical synthesis and biological data for erythromycin-HP conjugate prodrug 5. Note: MBC = minimum bactericidal concentration.
Following the chemical synthesis of erythromycin-HP 5, we demonstrated that this conjugate does not bind iron(II), as designed (Figure 3A, HP-17 rapidly binds iron(II) as indicated by a new peak at HP-17 has a ~570 nm, conjugate 5 does not bind iron(II) based on UV-vis spectroscopy). We then evaluated conjugate 5 in MIC, zone of inhibition and MBEC assays to determine antibacterial and biofilm-killing activity. We were delighted to see 5 demonstrated potent antibacterial activity against MRSA BAA-1707 (MIC = 0.39 μM), which is near equipotent to parent HP-17 (MIC = 0.30 μM) and erythromycin (MIC = 0.63 μM). Interestingly, 5 demonstrated a similar activity profile against methicillin-resistant S. epidermidis 35984 (5, MIC = 0.30 μM; HP-17, MIC = 0.15 μM) and vancomycin-resistant E. faecium 700221 (5, MIC = 0.78 μM; HP-17, MIC = 0.59 μM); however, both strains were resistant to erythromycin (MIC > 100 μM). From initial MIC results, it appears 5 is operating as a prodrug and not through a dual mechanism (ribosomal binding and iron starvation); however, additional work is needed to investigate dual activity potential against erythromycin-sensitive strains.
Figure 3.
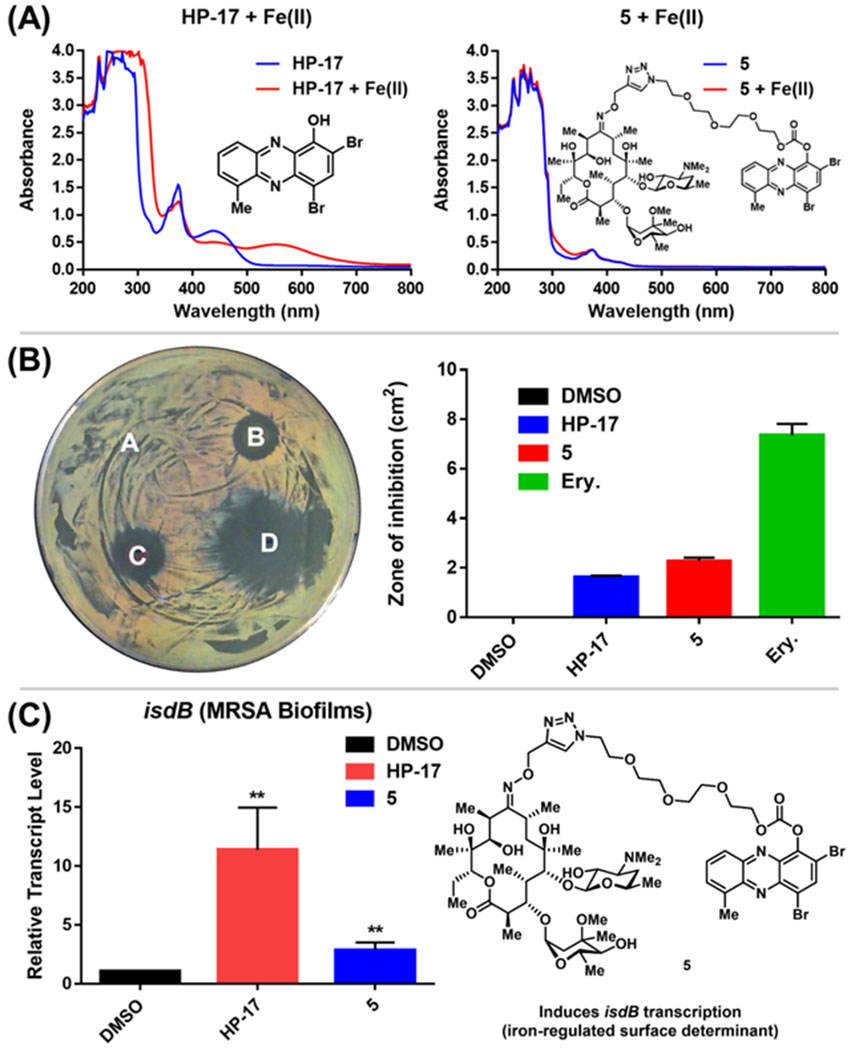
(A) UV-vis spectroscopy with HP-17 and HP-erythromycin conjugate 5. (B) Agar diffusion assay to determine zone of inhibition against MRSA BAA-1707 regarding A.) DMSO (control, zone of inhibition: 0 cm2), B.) HP-17 (zone: 1.63 ± 0.05 cm2), C.) HP-erythromycin conjugate 5 (zone: 2.77 ± 0.12 cm2), and D.) Erythromycin (zone: 7.11 ± 0.38 cm2). Note: Ery. = erythromycin. (C) Relative transcript levels of isdB from MRSA BAA-1707 biofilms treated with 1 μM of HP-17 and 5 for 4 h (RT-qPCR; **p-values < 0.01, Student’s t test). All biological data was obtained from three independent experiments.
In zone of inhibition experiments against MRSA BAA-1707 (Figure 3B), HP-erythromycin conjugate 5 produced a significantly larger zone of inhibition (clearance zone: 2.77 ± 0.12 cm2) than parent HP-17 (clearance zone: 1.63 ± 0.05 cm2) and a reduced clearance zone compared to erythromycin (7.11 ± 0.38 cm2). In addition, HP-erythromycin 5 demonstrated near equipotent activity to HP-17 in biofilm eradication experiments against MRSA BAA-1707 (MBEC = 9.38 μM, Calgary Biofilm Device assays). In separate experiments, established MRSA BAA-1707 biofilms were treated with HP-17 or HP-erythromycin conjugate 5 at 1 μM for 4 h to evaluate transcript levels of isdB (iron-surface determinant gene, involved in iron uptake from heme)17 as a biomarker for iron starvation. As expected, HP-17 induced isdB levels in MRSA biofilms 11.4-fold higher while HP-erythromycin conjugate 5 up-regulated this transcript 2.9-fold compared to vehicle control (Figure 3C, RT-qPCR). These results are indicative of (1) HP-17 and 5 inducing iron starvation in MRSA biofilms, and (2) HP-erythromycin 5 operating through a prodrug mode as the parent conjugate does not bind iron(II). The lower levels of isdB up-regulation induced by 5 compared to HP-17 can be attributed to time required for prodrug processing (carbonate cleavage of 5 to liberate free HP-17, which then induces iron starvation).
When tested against HeLa cells, conjugate 5 (IC50 ~ 50 μM) displayed more cytotoxicity than HP-17 at high concentrations (100 μM). However, the bacterial targeting of 5 is good with a selectivity index of ~128 (HeLa cytotoxicity IC50 value / MRSA MIC value).
In conclusion, we have designed an erythromycin-HP conjugate prodrug aimed at enhancing the translational potential of HP molecules to treat pathogenic bacteria. We developed a convergent synthesis of erythromycin-HP 5 that required only four steps to synthesize from erythromycin (commercially available) and HP-17. Initial biological assessment of 5 demonstrates prodrug activities as HPs are active upon carbonate release of their free phenol that enables iron binding and subsequent induction of iron starvation. Additional studies related to this work are needed; however, our findings showcase the potential to rapidly access HP-erythromycin conjugate prodrugs and can serve as a platform to improve treatment options against human pathogens in the clinic.
Supplementary Material
Acknowledgments
We would like to acknowledge the National Institute of General Medical Sciences of the National Institutes of Health for providing generous financial support for this work (R35GM128621 to R.W.H.). High-resolution mass spectra for synthesized analogues were obtained from the University of Florida’s Mass Spectrometry Research and Education Center and supported by NIH S10 OD021758-01A1.
Footnotes
Conflicts of Interest
There are no conflicts to declare.
References and Notes
- 1.Lewis K, Cell, 2020, 181, 29. [DOI] [PubMed] [Google Scholar]
- 2.Lewis K, Nat. Rev. Drug Discov, 2013, 12, 371. [DOI] [PubMed] [Google Scholar]
- 3.Brown ED and Wright GD, Nature, 2016, 529, 336. [DOI] [PubMed] [Google Scholar]
- 4.Fisher JF and Mobashery S, MedChemComm, 2016, 7, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abouelhassan Y, Garrison AT, Yang H, Chávez-Riveros A, Burch GM and Huigens RW III, J. Med. Chem, 2019, 62, 7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munita JM and Arias CA, Microbiol. Spectr, 2016, 4, VMBF-0016–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzpatrick MA, Lancet Infect. Dis, 2020, 20, 1108.32505229 [Google Scholar]
- 8.Wood TK, Biotechnol. Bioeng, 2016, 113, 476. [DOI] [PubMed] [Google Scholar]
- 9.Yan J and Bassler BL, Cell Host Microbe, 2019, 26, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis K, Annu. Rev. Microbiol, 2010, 64, 357. [DOI] [PubMed] [Google Scholar]
- 11.Borrero NV, Bai F, Perez C, Duong BQ, Rocca JR, Jin S and Huigens RW III, Org. Biomol. Chem, 2014, 12, 881. [DOI] [PubMed] [Google Scholar]
- 12.Garrison AT, Abouelhassan Y, Kallifidas D, Bai F, Ukhanova M, Mai V, Jin S, Luesch H and Huigens RW III, Angew. Chem. Int. Ed, 2015, 54, 14819. [DOI] [PubMed] [Google Scholar]
- 13.Garrison AT, Abouelhassan Y, Norwood VM, Kallifidas D, Bai F, Nguyen MT, Rolfe M, Burch GM, Jin S, Luesch H and Huigens RW III, J. Med. Chem, 2016, 59, 3808. [DOI] [PubMed] [Google Scholar]
- 14.Yang H, Abouelhassan Y, Burch GM, Kallifdas D, Huang G, Yousaf H, Jin S, Luesch H and Huigens RW III, Sci. Rep, 2017, 7, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrison AT, Abouelhassan Y, Kallifidas D, Tan H, Kim YS, Jin S, Luesch H and Huigens RW III, J. Med. Chem, 2018, 61, 3962. [DOI] [PubMed] [Google Scholar]
- 16.Global Tuberculosis Report. World Health Organization. 2020. https://www.who.int/tb/publications/global_report/en/ (accessed 11-3-2020)
- 17.Abouelhassan Y, Zhang Y, Jin S and Huigens RW III, Angew. Chem. Int. Ed, 2018, 57, 15523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huigens RW III, Abouelhassan Y and Yang H, ChemBioChem, 2019, 20, 2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao T, Liu K and Huigens RW III, Bioorg. Med. Chem. Lett, 2020, 30, 127515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katz L and Ashley GW, Chem. Rev, 2005, 105, 499. [DOI] [PubMed] [Google Scholar]
- 21.Pereira DE, PCT Int. Appl. 2012034058, 2012. [Google Scholar]
- 22.Cochrane SA, Li X, He S, Yu M, Wu M and Vederas JC, J. Med. Chem, 2015, 58, 9779. [DOI] [PubMed] [Google Scholar]
- 23.Tian JC, Han X, Lv W, Li YX, Wang H, Fan BZ, Cushman M and Liang JH, Bioorg. Med. Chem. Lett, 2017, 27, 1513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.