

Abstract
Cachexia presents in 80% of advanced cancer patients; however, cardiac atrophy in cachectic patients receives little attention. This cardiomyopathy contributes to increased occurrence of adverse cardiac events compared to age-matched population norms. Research on cardiac atrophy has focused on remodeling; however, alterations in metabolic properties may be a primary contributor.
Purpose:
Determine how cancer-induced cardiac atrophy alters mitochondrial turnover, mitochondrial mRNA translation machinery and in-vitro oxidative characteristics.
Methods:
Lewis lung carcinoma (LLC) tumors were implanted in C57BL6/J mice and grown for 28days to induce cardiac atrophy. Endogenous metabolic species, and markers of mitochondrial function were assessed. H9c2 cardiomyocytes were cultured in LLC-conditioned media with(out) the antioxidant MitoTempo. Cells were analyzed for ROS, oxidative capacity, and hypoxic resistance.
Results:
LLC heart weights were ~10% lower than controls. LLC hearts demonstrated ~15% lower optical redox ratio (FAD/FAD+NADH) compared to PBS controls. When compared to PBS, LLC hearts showed ~50% greater COX-IV and VDAC, attributed to ~50% lower mitophagy markers. mt-mRNA translation machinery was elevated similarly to markers of mitochondrial content. mitochondrial DNA-encoded Cytb was ~30% lower in LLC hearts. ROS scavengers GPx-3 and GPx-7 were ~50% lower in LLC hearts. Treatment of cardiomyocytes with LLC-conditioned media resulted in higher ROS (25%), lower oxygen consumption rates (10% at basal, 75% at maximal), and greater susceptibility to hypoxia (~25%) -- which was reversed by MitoTempo.
Conclusion:
These results substantiate metabolic cardiotoxic effects attributable to tumor-associated factors and provide insight into interactions between mitochondrial mRNA translation, ROS mitigation, oxidative capacity and hypoxia resistance.
Keywords: Cardiac cachexia, optical redox imaging, mitochondrial translation, cardiac wasting, cardio-oncology
1. Introduction
Cancer cachexia is a progressive deterioration of functional capacity characterized predominantly by a loss of skeletal muscle mass with impacts on multiple other organs[1]. While cachexia is present in as many as 80% of advanced cancer patients[2], the comorbidity of cardiac atrophy in cachectic patients has received little attention[3]. The clinical focus in targeting the underlying malignancy often dismisses cardiotoxic effects of radiation and chemotherapeutic treatment approaches[4],[5]. However, recent data demonstrates that advanced cancer contributes to detrimental cardiac alterations including reduced left ventricular systolic function and decreased cardiac musculature resulting from tumor-derived factors rather than from chemo/radiotherapies[6],[7]. While the compounding effects of tumor-related cardiac alterations, treatment modalities, and potential of underlying heart disease make it difficult to determine the etiology of cardiac atrophy seen in cancer patients[5], further research is desperately needed to understand how cancer contributes to changes in the cardiac metabolic state.
Heart failure has been tightly linked to alterations in metabolic substrate utilization and detriments to mitochondrial oxidative capacity[8],[9]. Specifically, patients with heart disease exhibit a shift from fatty acid oxidation towards greater reliance on glucose as a source for ATP[10]. Mitochondria have therefore become a key target in combatting this metabolic reprogramming in heart disease as these organelles contribute ~90% of ATP generated in the healthy myocardium[11]–[13]. Previous research supports these efforts to target mitochondria because electron transport through oxidative phosphorylation (OXPHOS) complexes I and III limits oxygen flux during left ventricular systolic dysfunction[14]. When electron transport at these specific OXPHOS sites slows, they can become primary producers of reactive oxygen species (ROS)[15]. These free radicals, when left unmitigated[16], can contribute to DNA mutations and damage, protein modifications, and membrane lipid oxidation[17]–[23]. Because of the proximity to mitochondrial DNA (mtDNA), ROS produced at OXPHOS complexes has the potential to mutate mtDNA and further contribute to detriments in OXPHOS electron transport seen in heart failure[24]. This interconnected relationship between mtDNA mutations, OXPHOS deficits, and ROS production provides the basis for Harman’s free radical theory of aging[25] and more specifically, the mitochondrial theory of heart disease[26]. While progressive loss of mtDNA copy number, increased mtDNA mutations[27],[28], OXPHOS deterioration[14], and excess ROS[29] are well established contributors to traditional cardiomyopathies, the mitochondrial alterations during cancer-induced cardiac atrophy remain largely unexplored.
Key to elucidating mitochondrial bioenergetics during cancer-induced cardiac atrophy is understanding the processes of mitochondrial turnover and maintenance of OXPHOS complex activity. The balance of mitochondrial biogenesis and mitophagy contribute to fluctuations in organelle volume within the myocardium, however, mitochondrial mRNA (mt-mRNA) translation machinery likewise contributes to OXPHOS activity and proper electron transport[30]–[35]. A balance between mt-mRNA translation and cytoplasmic translation is required for proper assembly of OXPHOS complexes[36] and during situations of impaired mt-mRNA translation, metabolic side-effects are evident[37]–[39]. Improper translational insertion[40] by mt-mRNA translation machinery also contributes to ROS production and alters antioxidant defenses through the mitochondrial unfolded protein response[41],[42]. The connection between mt-mRNA translation and myocyte oxidative capacity has been established and may contribute to changes in this form of cardiac dysfunction[43],[44].
In an effort to better understand the impact of cancer-induced cardiac atrophy on mitochondrial oxidative characteristics, the purpose of this investigation was to examine mitochondrial turnover and mt-mRNA translation alterations during this unique form of heart disease. Furthermore, we investigated tumor-associated changes in antioxidant defense as a contributor to excess ROS burden and how this may contribute to the deleterious effects on cardiac oxidative reserve capacity and resistance to hypoxia. By using an established, in-vivo model of cancer-induced cardiac atrophy we present label-free metabolic oxidation-reduction (redox) characteristics indicative of heart disease, alterations in mitochondrial content, mt-mRNA translation, and ROS scavengers. We further demonstrate the potential efficacy of mitochondrially-targeted antioxidants to mitigate excessive ROS production and how this approach protects against hypoxic insult to cardiomyocytes in-vitro.
2. Materials and Methods
2.1. Animal model of cancer-induced cardiac atrophy
All methods were approved by the Institutional Animal Care and Use Committee at the University of Arkansas. Eight week old, male C57BL6/J mice were injected with 1×106 Lewis Lung Carcinoma cells (LLC, n = 8) or equal volume sterile phosphate-buffered saline (PBS, n = 8) to the hind flank as previously described[45],[46]. LLC cells were purchased from American Type Culture Collection (ATCC, CRL-1642) and grown in DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (pen/strep) with media changed every second day. Cells were trypsinized, centrifuged, and counted prior to implantation. Tumors were allowed to grow for 28 days before mice were euthanized and hearts excised. Multiple small sections of the left ventricle were separated to allow protein and histological assessment of the same sample. Heart samples were snap frozen and stored at −80°C before further analysis. Mice were injected with 99.9% 2H2O in the peritoneum (20μL/g body weight, Sigma-Aldrich, St. Louis, MO, USA) 24 hours prior to euthanasia. 4% 2H2O in H2O(v/v) was provided ad libitum for 24 hours as a precursor for the intracellular creation of 2H-Alanine to measure protein synthesis[47].
2.2. Two-photon excitation fluorescence of endogenous NADH and FAD
Two-photon excitation fluorescence (TPEF) was used to measure endogenous fluorescence of NADH and FAD as previously described[48]. The oxidized forms (NADH, FAD) are referenced because the reduced forms (NAD+ and FADH2) and are not fluorescent. A ratio of FAD/(NADH+FAD) fluorescence intensity has been shown to be highly correlated with the ratio of intracellular cofactor concentrations NAD+/(NADH+NAD+) as determined with liquid chromatography/mass spectrometry[58]. Briefly, laser excitation by MaiTai Ti:Sapphire laser source (Spectra-Physics, Santa Clara CA) was tuned to 755nm or 860nm. Images were acquired using a resonant-galvo scanner and GaAsP photomultipler tubes (H7422–40, Hamamatsu) with 460/40nm (NADH), 525/45nm (FAD), and 600/70nm (Rhodamine) filters. 10μm section of the left ventricle were cut along the short axis. Slides were imaged (512×512 pixels, 16bit depth, 260μm2) and the pixel-wise FAD/(NADH+FAD) ratio normalized to rhodamine was calculated. Multiple fields were imaged for each sample and averaged to represent an individual biological sample. NADH and FAD image analysis was completed using MATLAB (MathWorks, Natick, MA).
2.3. Immunoblot analysis
Protein was extracted from small fractions of muscle taken directly from the left ventricle, gently boiled at 50C for 30 minutes, separated by 8% or 12% SDS-PAGE and transferred to polyvinylidene fluoride membrane as described[49],[50] alongside molecular weight ladder. Membranes were blocked using 5% milk in Tris-buffered saline (w/v) with 0.2% tween and incubated with specific primary antibodies at 4°C overnight. Primary antibodies were specific to HIF-1α (NB100–105, Novus Biologicals), COX-IV (Cell Signaling 4855S) VDAC (Cell Signaling, 4866S), PGC-1α (Santa Cruz sc-13067), PPARα (Santa Cruz sc-9000), PPARδ (Santa Cruz sc-7197), BNIP3 (Cell Signaling 3769), PINK-1 (Santa Cruz sc-33796), Parkin (Cell Signaling 42115), pSer65-Parkin (Abcam ab154995), mtIF2 (Santa Cruz sc-365477), mtIF3 (Origene TA800421), TACO1 (Abcam ab121688), Cytb (Santa Cruz 11436), Total OXPHOS antibody cocktail (Abcam ab110413, Complex I – NDUFB8, Complex II – SDHB, Complex III – UQCRC2, Complex IV – MTCO1, Complex V – ATP5A), SOD1 (Genetex GTX100554), SOD2 (Cell Signaling 131945), SOD3 (R and D Systems AF4817), GPx7 (Genetex GTX117516), GPx3 (Genetex GTX89142), and Catalase (Cell Signaling 140975). Using appropriate HRP-conjugated or fluorescent conjugated secondary antibodies, protein antigens were visualized within a linear range by either ECL on Protein Simple FluorChemM (Minneapolis, MN) or near-infrared fluorescence on Odyssey Fc (LI-COR, Lincoln, NE) and analyzed using ImageStudio Software (LI-COR). Bands were normalized to the 45kDa band of PonceauS as loading control. For each experiment, all groups were represented on each membrane and normalized to control.
2.4. Mitochondrial Isolation and 24-hour Protein Synthesis
Isolation of mitochondrial subpopulation and fractional synthesis rates were determined from samples as previously described and using 2H2O administration as outlined in Section 2.1[47]. Briefly, 30 mg of left ventricle was homogenized isolation buffer 1 (10 mM HEPES, 20 mM sucrose, 50 mM mannitol, 2mM EDTA, 0.25% v/v protease inhibitor cocktail, pH 7.4) and centrifuged at 650g. The supernatant was then further centrifuged at 10,000g for 10 minutes and pellet collected for analysis of subsarcolemmal (SS) mitochondria. SS mitochondria were washed in isolation buffer 2 (50 mM HEPES, 5mM EGTA, 1mM ATP, 100mMKCl, 5 mM MgSO4, 0.25% v/v protease inhibitor cocktail, pH 7.4) The intermyofibrillar (IMF) mitochondria were separated from the myofibrils in the pellet created in the first centrifugation first by dounce homogenization in isolation buffer 3 (100 mM KCl, 50 mM Tris, 5 mM MgCl2, 1 mM EDTA, 10 mM Beta-glycerophosphate, 1.5% w/v BSA, 0.25% v/v protease inhibitor cocktail, pH 7.5). The resulting homogenate was centrifuged at 650g and the resulting supernatant was centrifuged at 10,000g for 10 minutes and mitochondrial-rich pellet collected. SS and IMF proteins were hydrolyzed into individual amino acids by heating for 24 hours at 100°C in 6N HCl. An aliquot of the hydrolysate was dried down and mixed with a 3:2:1 solution of methyl-8, methanol, and acetonitrile to determine 2H incorporation into alanine on its methyl-8 derivative. The solution was then placed in a GCMS and analyzed for the ratio of labeled alanine to unlabeled alanine which was used to calculate protein synthesis. In order to normalize results based on the precursory pool of 2H2O, plasma was reacted with 10 M NaOH and a 5% solution of acetone in acetonitrile for 24 h in order to conjugate the free 2H2O to acetone. The solution was extracted by adding Na2SO4 and chloroform, and analyzed on the GCMS. Fractional synthetic rate (FSR) of proteins was calculated using the equation EA × [EBW × 3.7 × t (h)]−1 × 100, where EA represents amount of protein-bound [2H]alanine (mole% excess), EBW is the quantity of 2H2O in body water (mole% excess), 3.7 represents the exchange of 2H between body water and alanine (3.7 of 4 carbon-bound hydrogens of alanine exchange with water) and t(h) represents the time the label was present in hours.
2.5. H9c2 Culture experiments
H9c2 ventricular cardiomyocytes were purchased from ATCC (CRL-1446) and grown at 37°C, 5%CO2, and 20% O2 in DMEM containing 10% FBS and 1% pen/strep (GM) and changed every second day. When cells were ~75% confluent, 2×104 cells/well were sub-cultured in 96-well plates for 24 hours before media was replaced with control media (CM; DMEM containing 2.5% FBS, 1% pen/strep) or LLC-conditioned media (LCM) as previously described[51],[52]. To generate LCM, LLC growth media was collected after two days of incubation in 162cm2 flask with LLC density ending ~75% confluence. Media was centrifuged and filtered to remove cells and cell debris and diluted 1:4 (v/v) with serum-free DMEM. MitoTEMPO (MitoT; SML0737, Sigma-Aldrich, St. Louis, MO) diluted in PBS was added to CM or LCM at a concentration of 2 μM. After 2 hours incubation in respective media, 5μM MitoSOX Red (M36008, Invitrogen) in PBS was added to cells for 10 minutes, rinsed, and cells were visualized at 510/580nm (ex/em) on Nikon TiS epifluorescent microscope (Melville, NY) to assess mitochondrial superoxide production.
2.6. Bioenergetic flux analysis
Oxygen consumption rates (OCR) were analyzed using Seahorse XFp extracellular flux analyzer (Agilent, Santa Clara, CA) according to manufacturer instructions and as previously described[53]. Briefly, 2×104 cells were seeded per well in GM. After 24 hours, media was removed, cells were rinsed with sterile PBS, and replaced with CM or LCM with or without 2 μM MitoT. After 24 hours incubation, media was removed and replaced with Seahorse Assay media containing 7 mM glucose, 2 mM pyruvate, and 2 mM glutamine. OCR was measured prior to and following sequential addition of 1 μM oligomycin, 1 μM FCCP, and 1 μM rotenone/Antimycin A. This allowed assessment of cellular OCR related to basal respiration and maximal uncoupled respiration. Reserve respiration was determined as the difference between maximal and basal after normalizing to non-mitochondrial oxygen consumption.
2.7. Hypoxic exposure and MTT viability assessment
In order to assess the resistance of H9c2 cells to hypoxic challenge, 2×104 cells/well were plated in 96-well plate and incubated in GM for 24 hours. Media was replaced with CM or LCM with or without 2 μM MitoT. After 24 hours incubation at 5% CO2 and 20% O2, media was replaced with serum-free DMEM and cells were placed in a dual gas controlled (Oxycycler C42, Biospherix, Parish, NY) incubator sub-chamber. Oxygen was flushed by nitrogen and maintained at 1% O2 and 5% CO2 for 6 hours. The combination of serum-free media with a hypoxic environment was used to simulate ischemic conditions[54]. Following hypoxic exposure, 1 mM MTT (M6494, Invitrogen) was added to cells and incubated ~21% O2 for 2 hours. The resulting formazan crystals were solubilized by addition of 100 μL of 350 mM SDS in 0.01% HCl and absorbance was read at 570 nm. A separate plate was maintained at ~21% O2 to serve as 100% viability control.
2.8. Statistical Analysis
Statistics were calculated and visualized using GraphPad Prism v6.0. To compare PBS to LLC groups, Student’s t-test was used with α set at 0.05. For cell culture experiments, a 2×2 ANOVA was used to compare main effects or interactions between groups (CM × LCM vs. Control × MitoT). Where significant omnibus differences occurred, Tukey’s post-hoc analysis was used to investigate changes within groups. Cell culture experiments were analyzed using 3 biological replicates and at least 3 technical replicates. Data were analyzed for Gaussian (normal) distribution using Kolmogorov-Shmirnov test of normality. Data presented represent mean ± SEM.
3. Results and Discussion
3.1. Cancer cachexia contributes to cardiac atrophy and altered optical metabolic properties
Four weeks of LLC tumor implantation resulted in severe skeletal muscle and fat mass loss including a ~30% reduction in tibialis anterior muscle cross-sectional area as we have previously reported in the same animal cohort demonstrating cancer cachexia in this model[45],[46]. In the current study, we used the same animal cohort to analyze cardiac alterations associated with tumor implantation[45],[46]. Total wet weight of the heart was 10% lower in LLC compared to PBS demonstrating atrophy of the myocardium (p < 0.01, Table 1). Tibia lengths were not different between experimental conditions suggesting body size was similar between groups so we have presented raw heart mass. Additionally, heart mass relative to tumor free body weight was 6% less in LLC compared to PBS (p = 0.013, Table 1). To further characterize the metabolic alterations associated with this form of cardiac atrophy, we performed TPEF of endogenous FAD and NADH. This approach has been demonstrated as a powerful, label-free assessment of metabolic characteristics that has been validated in a variety of cell and tissue types[53],[55]–[58]. TPEF allows the identification of metabolic intermediates in a label-free assessment of cellular metabolic state[59]. In calculating the optical redox ratio, we identified a significantly lower redox ratio in LLC heart samples compared to PBS (Figure 1A, B; mean of 0.75 in PBS vs. 0.68 in LLC, p < 0.01). A lower optical redox ratio has been used as a marker of lower utilization of mitochondrial oxidative metabolism and greater reliance on glucose as a substrate[56]. Additionally, immunoblot analysis of HIF-1α indicates ~100% greater content in LLC compared to PBS (Figure 1C), suggestive of the induction of a hypoxic condition in hearts of LLC tumor-bearing mice. HIF-1α is a key sensor of oxygen levels and can downregulate mitochondrial oxidative metabolism while promoting glycolytic enzymes; a key characteristic of metabolic changes in cardiomyopathy[60],[61]. While further morphological data of the myocardium would aid in our interpretation of these findings, these results are supported by and extend upon previous evidence demonstrating decreased heart size in colorectal cancer-induced cardiac atrophy[62] and the shift away from β-oxidation towards glycolysis seen in other forms of heart failure[8].
Table 1.
Descriptive statistics of heart weight.
| Heart wet weight (mg) | Heart weight / (Body weight – Tumor weight) (mg/g) | |
|---|---|---|
| PBS (n = 21) | 120.08±3.15 | 4.90±0.13* |
| LLC (n = 14) | 108.21±3.36* | 4.60±0.09 |
Data represent M±SEM.
p < 0.05 compared to PBS.
Figure 1.
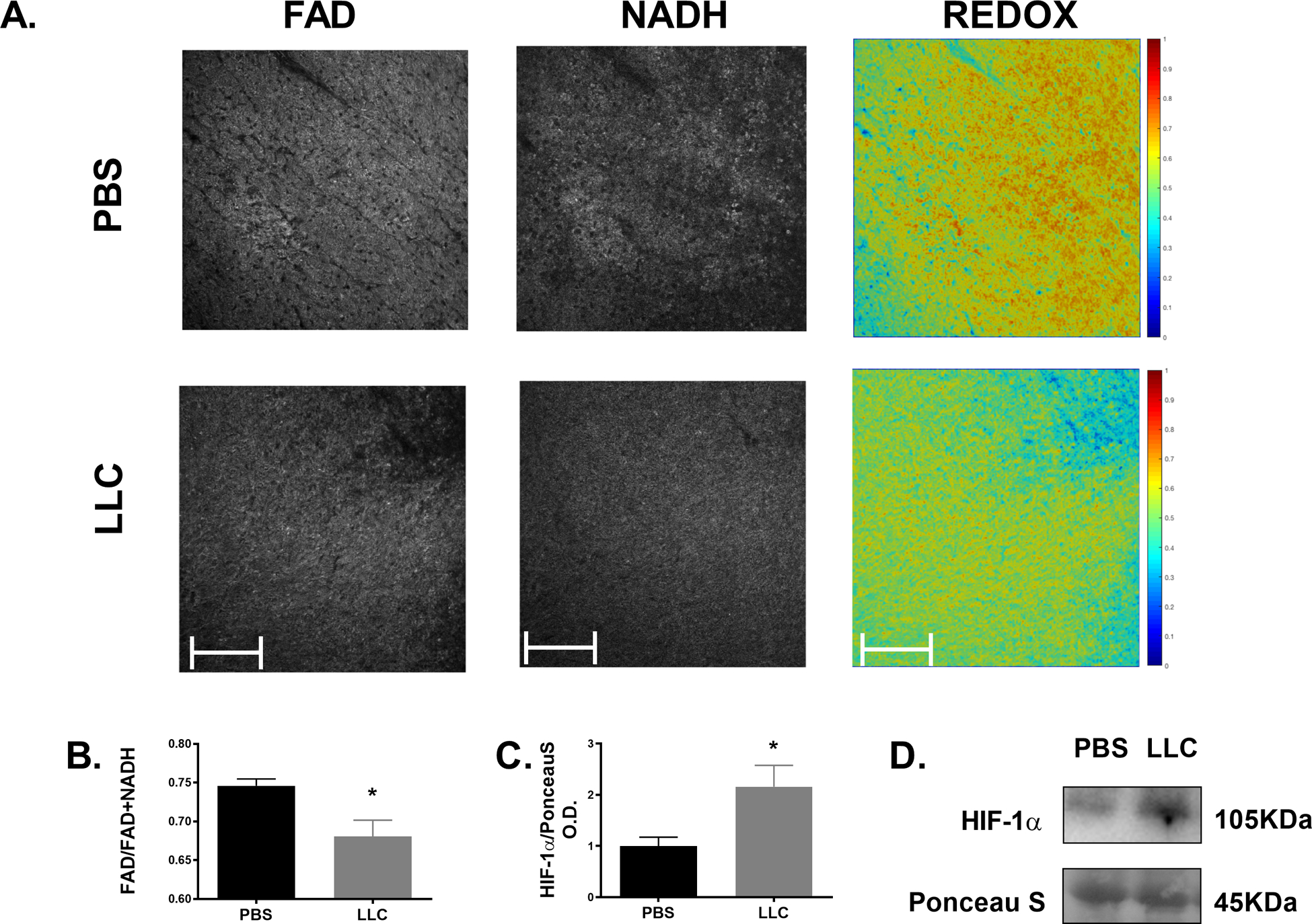
Cancer-induced cardiac atrophy demonstrates metabolic characteristics of heart disease. (A) Representative images of FAD, NADH, and redox ratio of TPEF of endogenous redox species within the myocardium of PBS and LLC left ventricle sections. (B) Calculated optical redox ratio and (C) HIF-1α protein content of PBS and LLC hearts. * indicates p < 0.05.
3.2. Cardiac atrophy disrupts mitochondrial clearance
Mitochondrial function has been a target for heart failure treatments to combat against the metabolic shift that occurs[13]; therefore, in lieu of directly assessing mitochondrial respiratory function, we analyzed proteins associated with mitochondrial content and biogenesis. Both COX-IV and VDAC levels were significantly greater in LLC compared to PBS by ~40% and ~75%, respectively (Figure 2A, p < 0.05) indicating elevated mitochondrial content in LLC hearts. There were no differences measured between experimental groups in either subsarcolemmal or intermyofibrillar mitochondria protein fractional synthetic rates (FSR) nor in protein content of regulators of mitochondrial biogenesis including PGC-1α, PPARα, or PPARδ (Figure 2B,C; p > 0.05). These data combine content measures of markers of biogenesis with direct assessment of FSR of mitochondrial protein subfractions to suggest that elevated mitochondrial content in this model is not due to increased mitochondrial biogenesis. In other models of heart failure, downregulation of proteins associated with lipid metabolism including PPARα and PGC-1α as well as respiratory-chain complex activity have been reported[63]. However, electron microscopy reveals that the amount of mitochondria are increased in many forms of cardiomyopathy despite other structural malformations[64]. Greater mitochondrial content (COX-IV, VDAC) absent upregulation of mitochondrial biogenesis (i.e. mitochondrial FSR, PPARs, PGC-1α) suggests alternate processes contributing to the overall mitochondria pool. Total mitochondrial content is a highly regulated balance between biogenesis and mitochondrial-specific autophagy (mitophagy)[30], thus an imbalance in mitochondrial content between PBS and LLC with no alteration in biogenesis predicates mitophagy as a contributing factor. This led us to examine markers of two common mitophagic pathways, BNIP3 and PINK1/Parkin. Protein content of BNIP3 was unchanged between PBS and LLC hearts (p > 0.05) while the total content of PINK1 and the ratio of phosphorylated to total Parkin were both decreased by ~50% (Figure 3; p < 0.05). These markers indicate PINK1/Parkin mediated mitophagy detriments may contribute to a build-up of (presumably) defective mitochondria which has, as yet, remained unclear during chronic heart disease[31]. Parkin-deficient mouse models present with accumulation of depolarized mitochondria following myocardial infarction indicating the importance of this pathway in the clearance of these damaged organelles in the myocardium[65]. These results suggest an important role for Parkin-dependent mitochondrial clearance in cancer-induced cardiomyopathies which could result in accumulation of depolarized mitochondria. The consequences of depolarized mitochondria that are not efficiently broken down in the myocardium remains uncertain.
Figure 2.
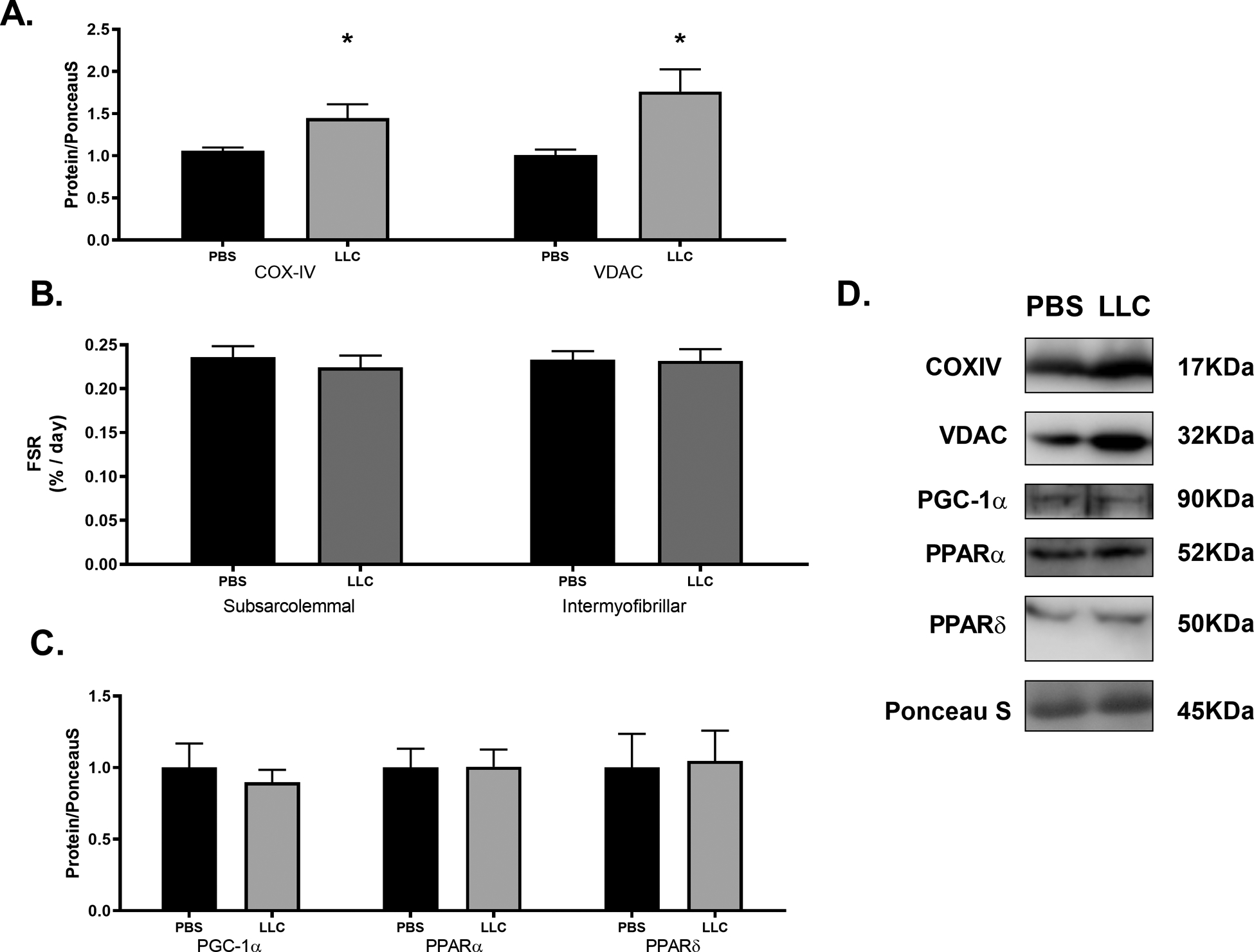
Cancer-induced cardiac atrophy alters mitochondrial content markers but not mitochondrial biogenesis protein content. (A) Markers of mitochondrial content, (B) subsarcolemmal and intermyofibrillar mitochondral FSRs and (C) markers of mitochondrial biogenesis in PBS and LLC hearts. (D) Sample immunoblot images of protein target indicated. Bands were cropped at indicated molecular weight and to show each group side-by-side. * indicates p < 0.05.
Figure 3.
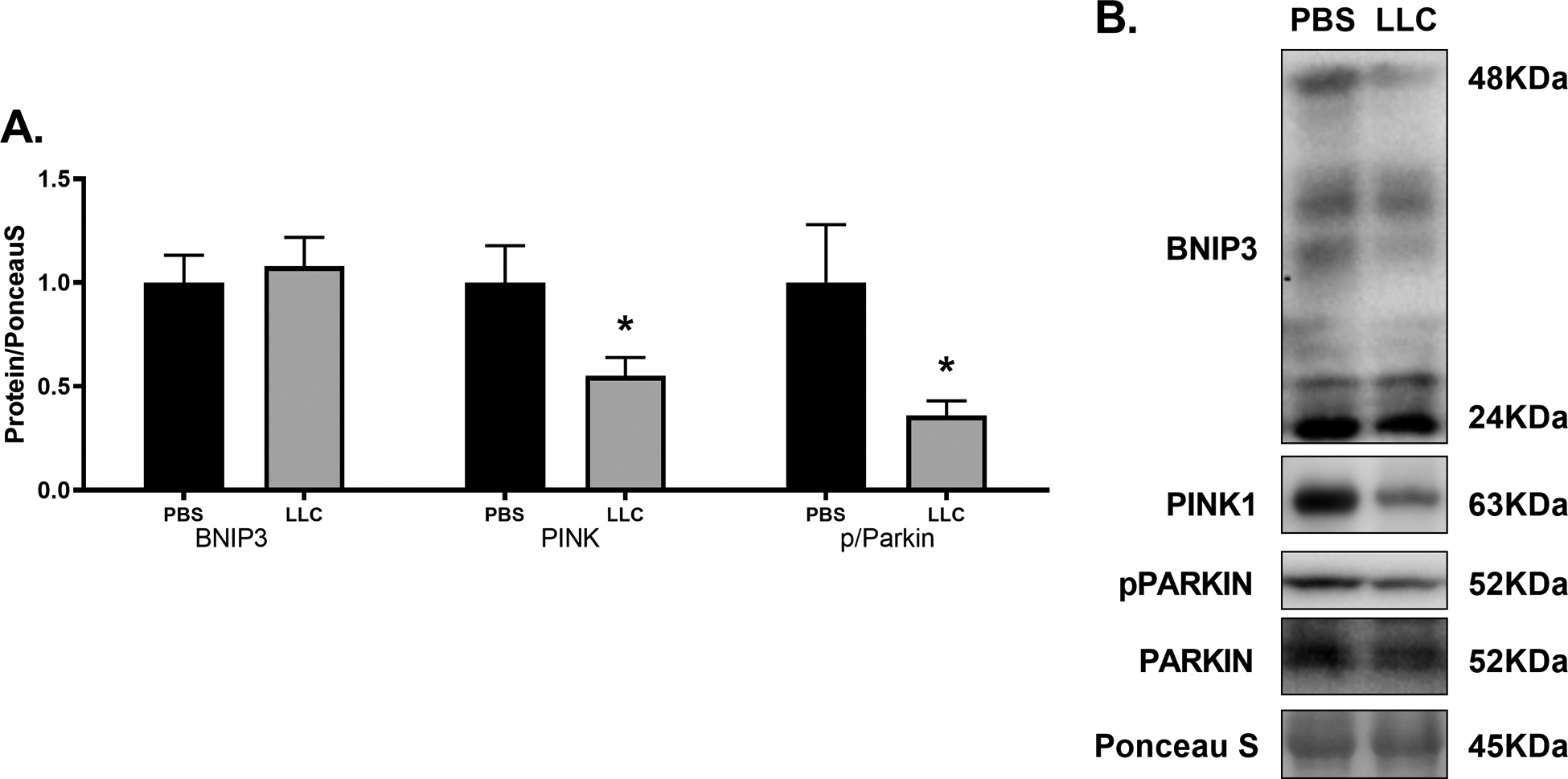
(A) Cancer-induced cardiac atrophy disrupts mitochondrial clearance. Markers of mitochondrial autophagy BNIP3, PINK, and phosphorylated to total Parkin in PBS and LLC hearts. (B) Sample immunoblot images of protein target indicated. Bands were cropped at indicated molecular weight and to show each group side-by-side. * indicates p < 0.05.
3.3. LLC myocardial mt-mRNA translation machinery is unable to maintain mtDNA-encoded OXPHOS subunits
mt-mRNA translation machinery allows proper translation of the 13 canonical protein subunits encoded by mt-DNA which are required for proper production of core portions of OXPHOS complexes[34]. TFAM is a key factor involved in mtDNA transcription[66] and the primary controllers of mt-mRNA translation are two mammalian mitochondrial initiation factors mtIF2 and mtIF3[67]. Additionally, co-translational insertion using translational coactivators – such as TACO1 in mammals – is required for proper assembly of OXPHOS complexes[40]. These factors work to create a balance between mt-mRNA translation and cytoplasmic translation to produce and assemble OXPHOS complexes in a manner that optimizes electron transport and ATP production[36]. TFAM, mtIF2, mtIF3, and TACO1 protein contents were all significantly higher in LLC compared to PBS by ~50–125% (p < 0.05). However, the mt-mRNA translation product Cytb was not significantly altered by the LLC tumor (Figure 4A; p > 0.05). To assess content of these proteins relative to the total mitochondrial pool, we normalized mt-mRNA translation proteins to VDAC protein. We observed no differences in contents of TFAM, mtIF2, mtIF3, or TACO1 between experimental groups (p > 0.05); however, Cytb, when normalized to VDAC as a marker of mitochondrial content, was ~45% lower in LLC compared to PBS (Figure 4B; p < 0.05). These data would suggest that mt-mRNA translation is unable to maintain expression of mtDNA-encoded proteins to match the level of mitochondrial content. When complex content was assessed using immunoblot analysis of individual core subunits, we found no significant difference between groups for Complex I, IV, or V (p > 0.05) but found significantly elevated content of Complexes II and III (Figure 4C ,p < 0.05). The proteins encoded by mtDNA are integral, core subunits in OXPHOS complexes I, III, IV, and V[34]. In each complex, the mtDNA-encoded channels are vital for appropriate electron transport or proton translocation across the membrane[68]. Where mutations or alterations in expression of mtDNA-encoded proteins arise, oxidative complications associated with cardiac illness are evident[19],[24]. The results presented here suggest that during this form of cardiomyopathy, the machinery responsible for expression of mtDNA-encoded transcripts is maintained relative to the mitochondrial content as measured by VDAC. However, resulting mtDNA-encoded protein subunits of OXPHOS may not be equally maintained suggesting other possible issues responsible for deficient mitochondria-encoded protein expression. By assessing amount of complex formation through immunoblot of subunits that are labile when not assembled, we found that total content of complexes with a large proportion of mtDNA-encoded proteins were no different between groups (Complex I, IV, V) while those with primarily nDNA-encoded portions (Complex II, III) were elevated concomitant with mitochondrial content markers. This reduced content of mtDNA-encoded proteins can result in misincorporation of OXPHOS subunits and prevent efficient electron flow through complexes I and III and result in greater production of superoxide, presumably through reverse electron transport[69]. Mitigation of free radicals by antioxidant enzymes may present as a potential mechanism to protect against excessive ROS produced during heart disease[29].
Figure 4.
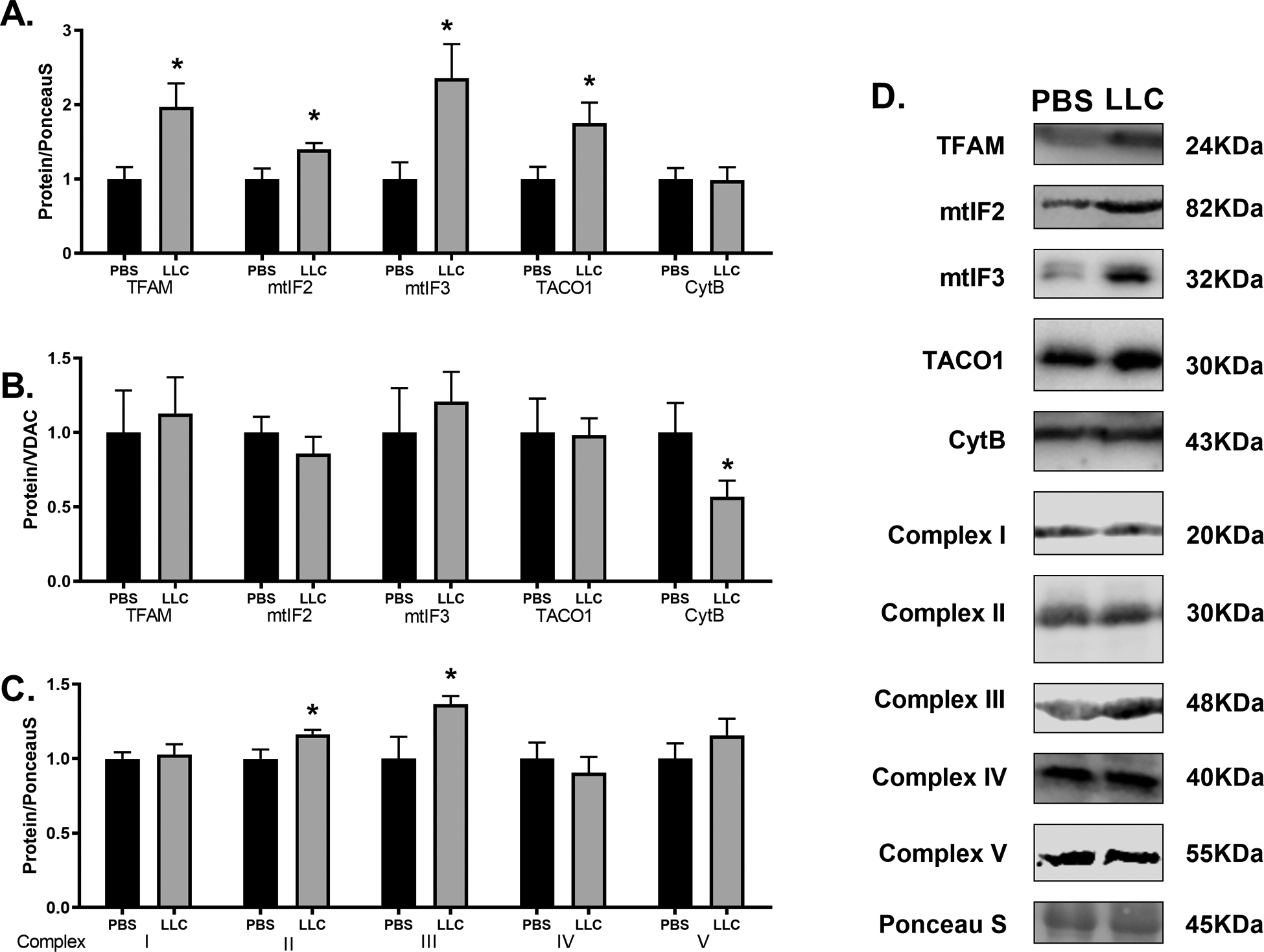
Cancer-induced cardiac atrophy disrupts mt-mRNA translation products but not mt-mRNA translation machinery. (A) Mitochondrial transcription and translation machinery (TFAM, mtIF2, mtIF3, TACO1) and a mt-mRNA translation product (Cytb) normalized to total protein content by PonceauS and (B) normalized to mitochondrial content marker VDAC in PBS and LLC hearts. (C) Immunoblot analysis of OXPHOS complex content as determined through protein subunits required for complex formation. Specific target for each subunit is indicated in the materials and methods section. (D) Sample immunoblot images of protein target indicated. Bands were cropped at indicated molecular weight and to show each group side-by-side. * indicates p < 0.05.
3.4. LLC contributes to altered ROS scavenger protein levels
In an effort to better understand handling of ROS, we assessed various proteins involved in superoxide (superoxide dismutases: SOD1, 2, 3) and hydrogen peroxide dissipation (Catalase, Glutathione Peroxidases: GPx-3,7). No differences in protein content of SOD-1, 2, or 3 were found between PBS and LLC hearts (Figure 5A; p > 0.05). Protein content of Catalase was 80% more abundant in LLC animals compared to PBS (p < 0.05), while GPx-3 and GPx-7 were both significantly lower in LLC compared to PBS by ~60% (p < 0.01) and ~50% (Figure 5B; p < 0.05), respectively. Taken together, these results suggest no alterations in the control of superoxide radicals through dismutase proteins. However, alterations appear in proteins involved in hydrogen peroxide (H2O2) clearance. Specifically, the elevation seen in Catalase content was unexpected because, previously, elevated Catalase has been linked to mitigation of age-dependent heart disease in mice[70]. The levels of GPx-3 and −7 were both reduced by the tumor-associated cardiomyopathy. Taken together, this could signify an overall decrease in ability to handle H2O2 because GPx’s maintain greater affinity for[71] and reactivity with H2O2 compared to catalase at physiologically relevant concentrations[72]. Excessive H2O2 has been seen in aging-induced heart disease despite greater activity of Catalase[73], which may contribute to cardiac cell death[74] and reduced mitochondrial oxygen consumption[75]. It is worth noting that these markers would relate to the whole cell’s capacity to process H2O2 which we believe is principally generated by the mitochondria. A limitation of these results is a lack of direct measures of redox enzyme activity or ROS content in vivo because the presence/absence of these protein markers may be disconnected from ROS production and their mitigation activity. While direct measurements of ROS production and OCR were not assessed in PBS and LLC hearts, we next sought to determine how tumor-associated factors could alter mitochondrial ROS production and OCR in cardiac cells in-vitro.
Figure 5.
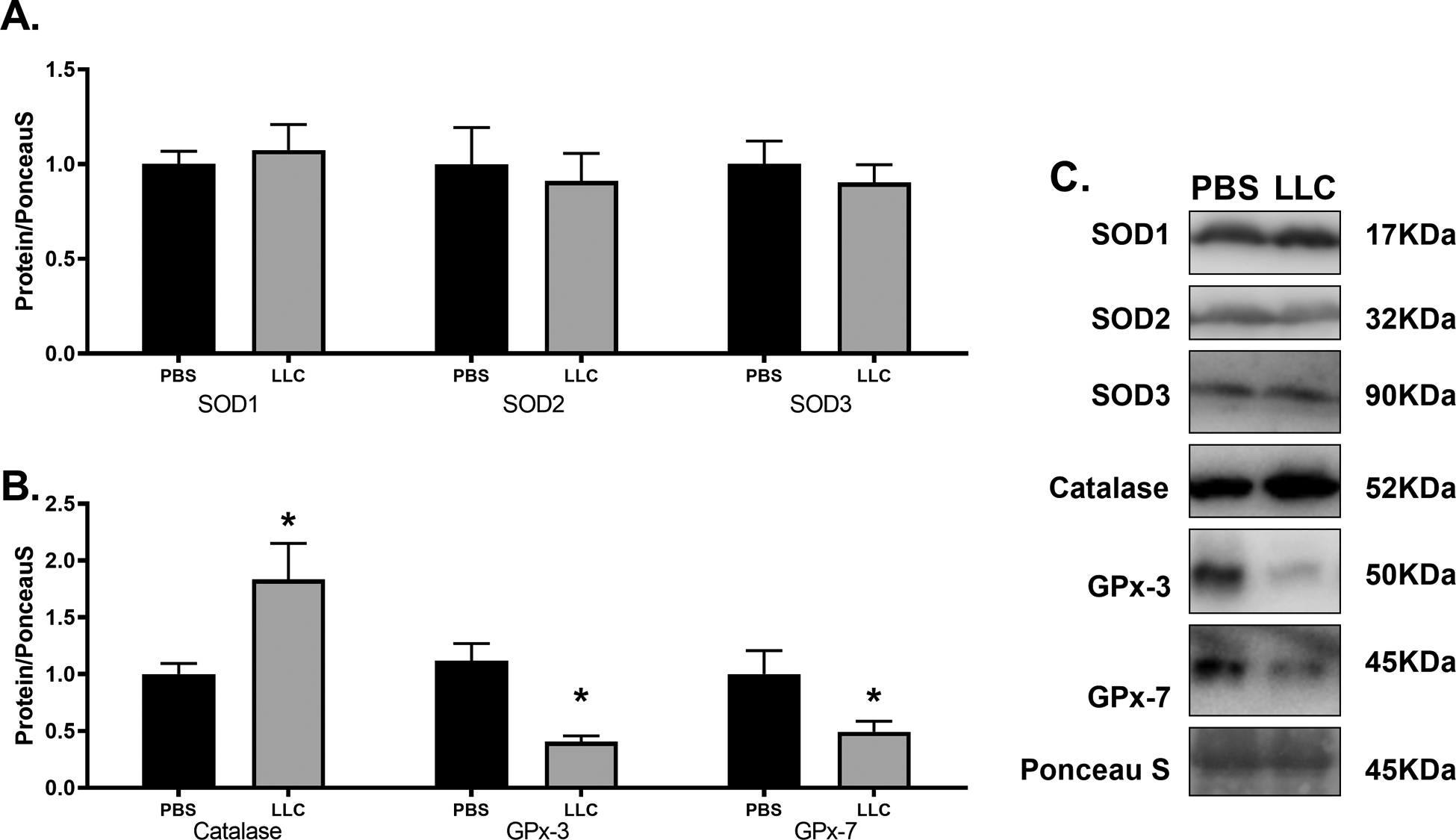
Cancer-induced cardiac atrophy disrupts hydrogen peroxide clearance protein content. (A) Superoxide dismutase protein isoforms and (B) hydrogen peroxide mitigation proteins catalase, and glutathione peroxidases −3 and −7 in PBS and LLC hearts. (C) Sample immunoblot images of protein target indicated. Bands were cropped at indicated molecular weight and to show each group side-by-side. * indicates p < 0.05
3.5. Mitochondrial antioxidants mitigate effects of tumor-associated factors on cardiomyocyte ROS production and oxidative capacity
To directly assess impacts of tumor-associated factors on the myocardium, independent of other circulating factors (immune cells, compliment), cardiomyocytes were cultured in CM or LCM alone or in combination with MitoT, a mitochondria-targeted antioxidant. Two hours following treatment, ROS accumulation was assessed using MitoSOX fluorescence. H9c2 cardiomyocytes showed ~25% greater MitoSOX fluorescence when incubated in LCM alone (p < 0.001). The combination with MitoT had no effect on MitoSOX in CM-treated cells (p > 0.05), however, combined LCM and MitoT treatment showed significantly lower MitoSOX fluorescence compared to LCM alone (p < 0.001) to the point that LCM + MitoT was not different compared to CM (Figure 6A, B; p > 0.05). To further test the detrimental effects of LCM, cardiomyocytes were treated for 24 hours and oxygen flux analysis was assessed (Figure 6C). Basal OCR was ~15% lower following 24 hours of LCM treatment compared to CM (p < 0.01) with no effects of MitoT on LCM treated cells (Figure 6D; p > 0.05). When cells were treated with FCCP to simulate maximal electron flux through the ETC, LCM control cells had a FCCP-stimulated OCR that was ~1/3 that of the CM (p < 0.01). When combining LCM with MitoT, FCCP-OCR was higher than LCM control (p < 0.01) and not different from either CM treatment (Figure 6E; p > 0.05). Using these values, we were able to calculate the reserve OCR which followed a similar pattern of severe reduction (~75% capacity) relative to CM following 24 hours LCM (p < 0.01) but not when combined with MitoT (Figure 6F; p > 0.05). These results demonstrate a clear connection between tumor-associated factors and alterations in mitochondrial metabolic characteristics in cardiac muscle cells. The initial increases in ROS production following LCM treatment can be mitigated using a mitochondrial antioxidant and overtime can result in functional rescue of mitochondrial oxidative reserve. One way myocardial oxidative capacity contributes to cardioprotection is through resistance to hypoxic-insult[76]–[78]. In order to test if our results in OCR reflected susceptibility to hypoxia-induced cell death, we exposed cells to 1% O2 for 6 hours and assessed cell viability. All groups demonstrated significantly lower viability following hypoxia compared to normoxic control (dashed line) with LCM control being significantly lower compared to CM and LCM + MitoT cells by ~30% (Figure 7A; p < 0.001). These data suggest that cancer-induced changes to electron flux capacity may be rescued through mitigation of mitochondrial ROS. Because these oxidative measures were assessed on immortalized cardiomyocytes, there remains considerable need to better characterize the oxidative metabolic properties of the myocardium undergoing cancer-induced structural alterations in vivo to understand the translational value of these data. These results substantiate cardiotoxic effects attributable to tumor-associated factors and provide new insight into interactions between ROS mitigation, cardiac oxidation and hypoxia resistance.
Figure 6.
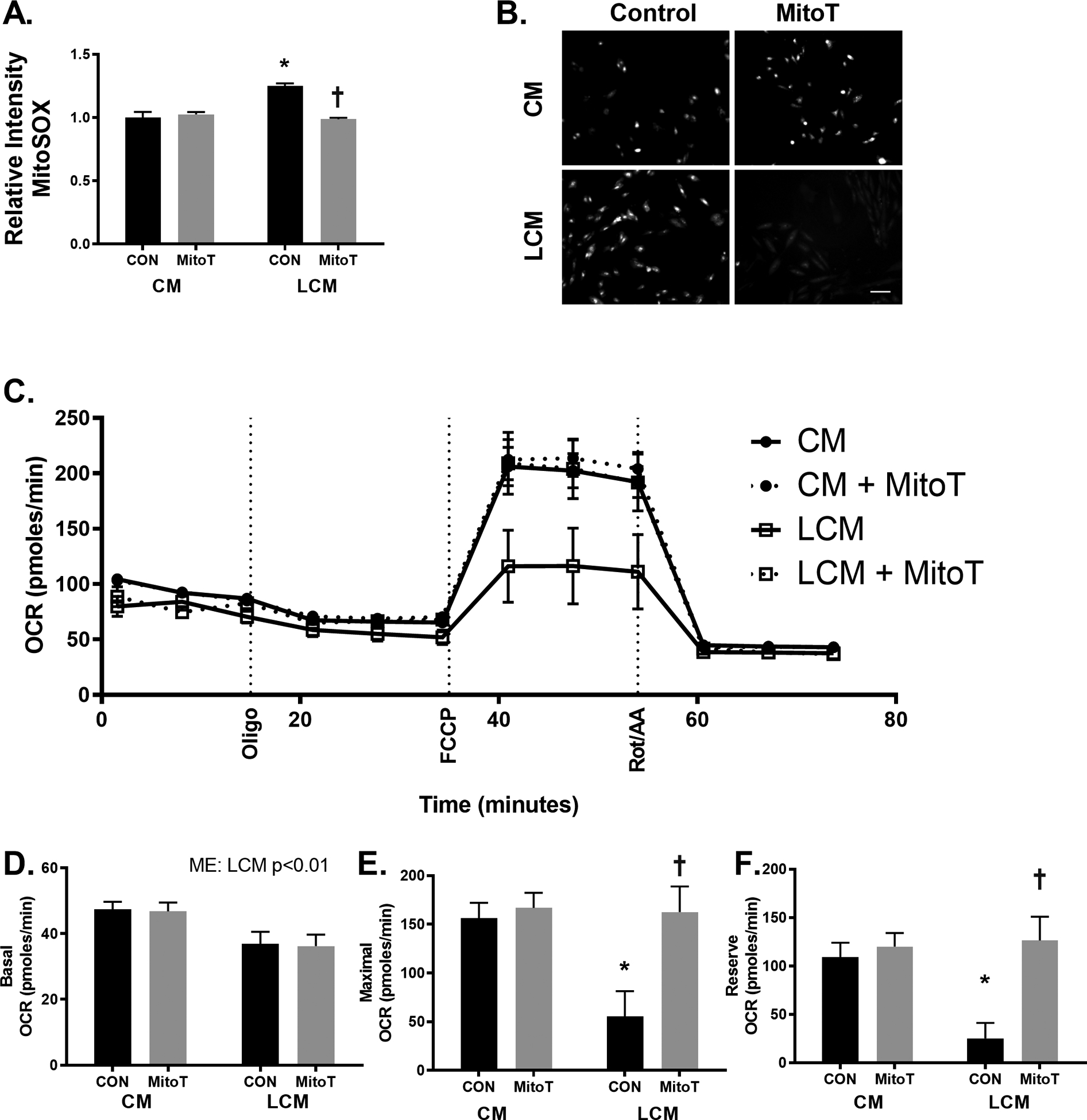
Media from LLC cancer cells contribute to greater ROS and reduced oxidative capacity. H9c2 cells were treated with a combination of LLC media (LCM) and 2μM of the mitochondrial targeted antioxidant (MitoT) for 2 hours and ROS assessed using MitoSOX (A,B) or similar treatment for 24 hours analyzing cellular bioenergetic flux analysis (C) to analyze oxygen consumption rates at (D) basal and (E) maximal (that is FCCP-stimulated) rates, or calculated reserve rates (F). Scale bar = 100μm. * indicated p < 0.05 vs. control media (CM) with no MitoT (con); † indicated p < 0.05 vs. con cells receiving same media condition.
Figure 7.
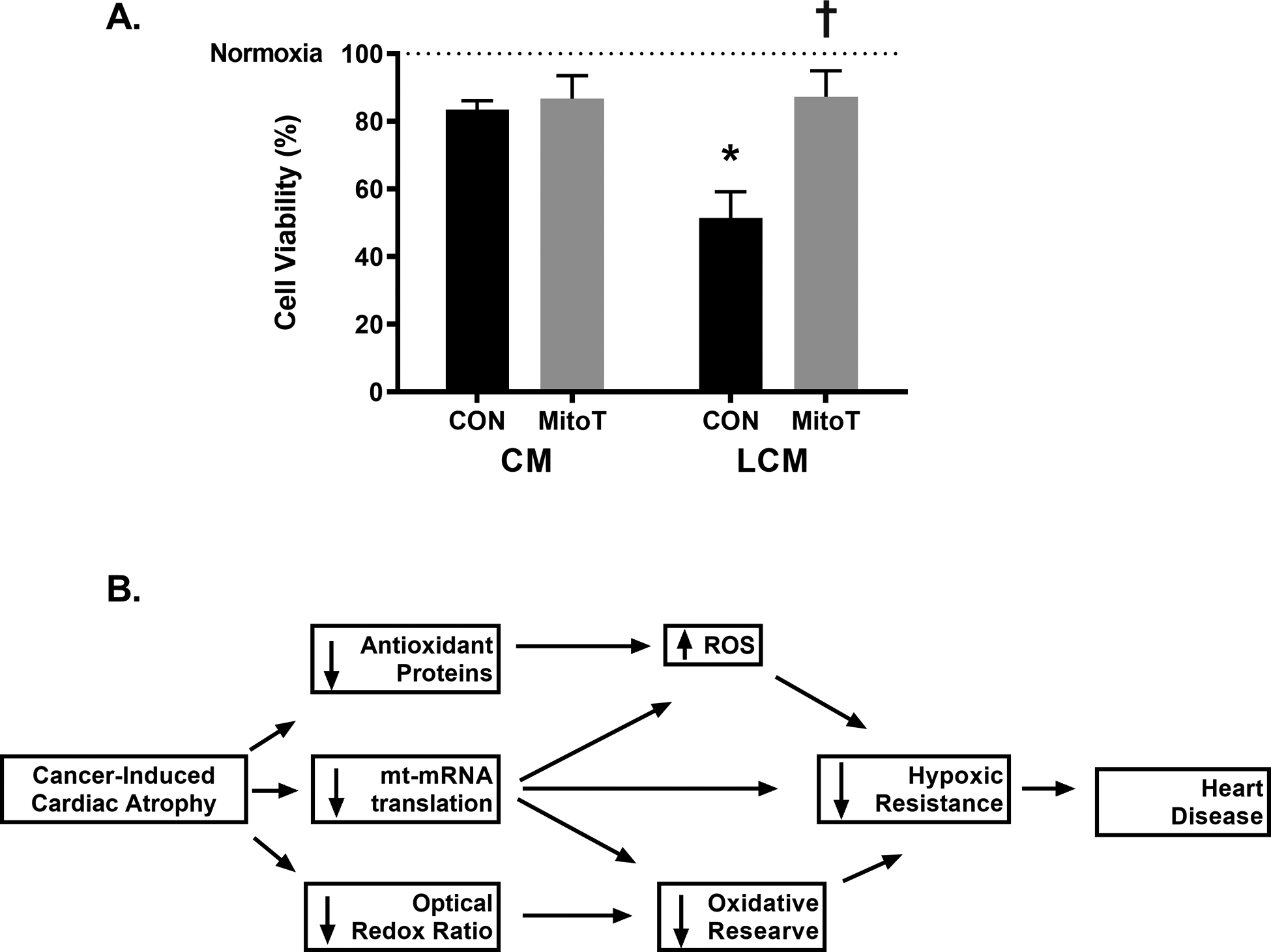
Media from LLC cancer cells reduces resistance to hypoxic insult in cardiomyocytes. (A) H9c2 cells were treated with a combination of LLC media (LCM) and 2μM of the mitochondrial targeted antioxidant (MitoT) for 24 hours and exposed to 1% oxygen and serum-free media for 6 hours followed by MTT viability assessment. * indicates p < 0.05 vs. control media (CM) with no MitoT (con); † indicates p < 0.05 vs. con cells receiving same media condition; dashed line at 100% indicates control cells that were maintained ~21% O2 in standard growth media. (B) Graphical representation of suggested relationship between cancer-induced cardiac atrophy and oxidative characteristics.
Our findings suggest cancer-induced cardiac atrophy presents with altered metabolic properties associated with heart disease including greater reliance on glycolysis assessed using a label free measurement of endogenous redox species and elevation in HIF-1α. We additionally present impaired mitochondrial clearance, disruptions in mt-encoded protein expression, and lowered content of proteins involved in ROS mitigation, specifically peroxides. Finally, in-vitro experiments show that tumor-specific factors exacerbate cardiac ROS production, lead to detriments in oxidative reserve, and enhanced susceptibility to hypoxic challenge – all of which can be reversed using mitochondria targeted antioxidants. The results presented here extend previous research (focused specifically on cardiac remodeling) by demonstrating metabolic and mitochondrial protein markers are altered in this unique form of heart disease in vivo and mitochondrial flux analysis in vitro. Maintenance of mitochondrial oxidative function is key in tailoring therapeutic approaches to limit cardiotoxic effects in treating the primary malignancy as well as limiting tumor-specific cardiomyopathy (Figure 7B). This is paramount in a clinical setting because cancer patients have increased occurrence of both myocardial ischemia[79] and adverse cardiac events[80] compared to age-matched population norms. We now provide evidence for the utility of a mitochondrial antioxidant to combat cancer-induced oxidative impairments in the myocardium. Others have attempted to use pharmaceutical approaches to treat excessive ROS production in heart disease[81]–[83] but present with adverse effects on physiological ROS signaling in multiple other systems[84]. Our results suggest two pathways involved in pathological accumulation of free radicals through 1) loss of functional expression of mt-mRNA translation products for OXPHOS and 2) downregulation of GPx proteins for H2O2 clearance. An alternative approach to combat excess ROS may be utilizing alternative pharmaceuticals which could indirectly reduce myocardial ROS[85].
4. Conclusion
In summary, we present evidence of mitochondrial alterations contributing to ROS generation during cancer-induced cardiac atrophy using a combination of protein markers from in vivo experiments and in vitro oxidative characterizations. We build on these results by demonstrating factors excreted by the tumor cells contribute to reduction in cardiomyocyte oxidative reserve in-vitro. With heart disease and cancer competing for the leading cause of mortality worldwide, the contribution of malignancy to cardiomyopathy must be made clear and approaches to mitigate it researched. Further research should focus on the compounding effects of cancer and chemotherapeutics on cardiac function and how other cancers less associated with cachexia may still contribute to heart disease.
New and Noteworthy.
Cancer leads to altered metabolism and atrophy of the heart. Mitochondrial turnover, mRNA translation, and ROS scavenging are all affected in the myocardium during advanced cancer in mice. Mitochondrial ROS scavenging may protect aerobic capacity and resistance to hypoxia in vitro. Antioxidant drugs might mitigate the effects of tumor-associated changes to the heart.
Acknowledgements:
We would like to acknowledge Dr. Timothy Muldoon for use of his two-photon microscope and support from the University of Arkansas Exercise Science Research Center. Funding for this project was provided by the Arkansas Biosciences Institute to NPG, by National Institute of Arthritis and Musculoskeletal and Skin Diseases and National Institute of General Medical Sciences of the NIH Award Number R15AR069913 to NPG and ACSM Foundation Doctoral Student Research Grant to DEL.
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest associated with this publication.
Disclosures: None
References
- 1.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 2.Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer 2002;2:862–871. [DOI] [PubMed] [Google Scholar]
- 3.Wilens SL, Dische MR, Henderson D. The low incidence of terminal myocardial infarction and the reversibility of cardiac hypertrophy in cachexia. Am J Med Sci 1967;253:651–60. [DOI] [PubMed] [Google Scholar]
- 4.Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol 2015;12:620–620. [DOI] [PubMed] [Google Scholar]
- 5.Murphy KT. The pathogenesis and treatment of cardiac atrophy in cancer cachexia. Am J Physiol Hear Circ Physiol 2016;310:H466–77. [DOI] [PubMed] [Google Scholar]
- 6.Tian M, Nishijima Y, Asp ML, Stout MB, Reiser PJ, Belury MA. Cardiac alterations in cancer-induced cachexia in mice. Int J Oncol 2010;37:347–53. [DOI] [PubMed] [Google Scholar]
- 7.Manne ND, Lima M, Enos RT, Wehner P, Carson JA, Blough E. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int J Oncol 2013;42:2134–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol Rev 2005;85:1093–1129. [DOI] [PubMed] [Google Scholar]
- 9.Fukushima A, Milner K, Gupta A, Lopaschuk GD. Myocardial Energy Substrate Metabolism in Heart Failure : from Pathways to Therapeutic Targets. Curr Pharm Des 2015;21:3654–64. [DOI] [PubMed] [Google Scholar]
- 10.Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail 2014;7:1022–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mootha VK, Arai AE, Balaban RS. Maximum oxidative phosphorylation capacity of the mammalian heart. Am J Physiol 1997;272:H769–75. [DOI] [PubMed] [Google Scholar]
- 12.Balaban RS. Domestication of the cardiac mitochondrion for energy conversion. J Mol Cell Cardiol 2009;46:832–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol 2017;14:238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stride N, Larsen S, Hey-Mogensen M, Sander K, Lund JT, Gustafsson F et al. Decreased mitochondrial oxidative phosphorylation capacity in the human heart with left ventricular systolic dysfunction. Eur J Heart Fail 2013;15:150–157. [DOI] [PubMed] [Google Scholar]
- 15.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015;163:560–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies MJ. The oxidative environment and protein damage. Biochim Biophys Acta - Proteins Proteomics 2005;1703:93–109. [DOI] [PubMed] [Google Scholar]
- 18.Kehrer JP, Klotz L-O. Free radicals and related reactive species as mediators of tissue injury and disease: implications for Health. Crit Rev Toxicol 2015;45:765–798. [DOI] [PubMed] [Google Scholar]
- 19.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 2003;17:1195–1214. [DOI] [PubMed] [Google Scholar]
- 20.Bürkle A, Virág L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol Aspects Med 2013;34:1046–1065. [DOI] [PubMed] [Google Scholar]
- 21.Poulsen HE, Specht E, Broedbaek K, Henriksen T, Ellervik C, Mandrup-Poulsen T et al. RNA modifications by oxidation: a novel disease mechanism? Free Radic Biol Med 2012;52:1353–61. [DOI] [PubMed] [Google Scholar]
- 22.Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol 2014;2:411–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baraibar MA, Liu L, Ahmed EK, Friguet B. Protein Oxidative Damage at the Crossroads of Cellular Senescence, Aging, and Age-Related Diseases. Oxid Med Cell Longev 2012;2012:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casademont J, Miró O. Electron transport chain defects in heart failure. Heart Fail Rev 2002;7:131–9. [DOI] [PubMed] [Google Scholar]
- 25.Harman D Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956;11:298–300. [DOI] [PubMed] [Google Scholar]
- 26.Harman D The biologic clock: the mitochondria? J Am Geriatr Soc 1972;20:145–7. [DOI] [PubMed] [Google Scholar]
- 27.Ozawa T, Sugiyama S, Tanaka M, Hattori K. Mitochondrial DNA mutations and disturbances of energy metabolism in myocardium. Jpn Circ J 1991;55:1158–64. [DOI] [PubMed] [Google Scholar]
- 28.Ozawa T, Tanaka M, Sugiyama S, Hattori K, Ito T, Ohno K et al. Multiple mitochondrial DNA deletions exist in cardiomyocytes of patients with hypertrophic or dilated cardiomyopathy. Biochem Biophys Res Commun 1990;170:830–6. [DOI] [PubMed] [Google Scholar]
- 29.Zhang P-Y, Xu X, Li X. Cardiovascular diseases : oxidative damage and antioxidant protection. Eur Rev Med Pharmacol Sci 2014;18:3091–3096. [PubMed] [Google Scholar]
- 30.Palmeira CM, Rolo AP. Mitophagy and Mitochondrial Balance. Mitochondrial Regul Methods Protoc 2014;1241:1–194. [Google Scholar]
- 31.Saito T, Sadoshima J. The Molecular Mechanisms of Mitochondrial Autophagy/Mitophagy in the Heart. Cancer Control 2015;116:1477–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dorn GW, Vega RB, Kelly DP, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 2015;29:1981–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gustafsson CM, Falkenberg M, Larsson N-G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu Rev Biochem 2016;85:133–160. [DOI] [PubMed] [Google Scholar]
- 34.Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet 2012;13:878–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ott M, Amunts A, Brown A. Organization and Regulation of Mitochondrial Protein Synthesis. Annu Rev Biochem 2016;85:77–101. [DOI] [PubMed] [Google Scholar]
- 36.Suhm T, Ott M. Mitochondrial translation and cellular stress response. Cell Tissue Res 2017;367:21–31. [DOI] [PubMed] [Google Scholar]
- 37.Lee DE, Brown JL, Rosa ME, Brown LA, Perry RA, Washington TA et al. Translational machinery of mitochondrial mRNA is promoted by physical activity in Western diet-induced obese mice. Acta Physiol 2016;218:167–177. [DOI] [PubMed] [Google Scholar]
- 38.Greene NP, Nilsson MI, Washington TA, Lee DE, Brown LA, Papineau AM et al. Impaired Exercise-Induced Mitochondrial Biogenesis in the Obese Zucker Rat, Despite PGC-1α Induction, is Due to Compromised Mitochondrial Translation Elongation. Am J Physiol Endocrinol Metab 2014;306:E503–11. [DOI] [PubMed] [Google Scholar]
- 39.Christian BE, Spremulli LL. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta 2012;1819:1035–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ott M, Herrmann JM. Co-translational membrane insertion of mitochondrially encoded proteins. Biochim Biophys Acta - Mol Cell Res 2010;1803:767–775. [DOI] [PubMed] [Google Scholar]
- 41.Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 2004;117:4055–66. [DOI] [PubMed] [Google Scholar]
- 42.Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell 2015;58:123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boczonadi V, Horvath R. Mitochondria: impaired mitochondrial translation in human disease. Int J Biochem Cell Biol 2014;48:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol 2010;2010:737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee DE, Brown JL, Rosa-Caldwell ME, Blackwell TA, Perry RAJ, Brown LA et al. Cancer cachexia-induced muscle atrophy: evidence for alterations in microRNAs important for muscle size. Physiol Genomics 2017;49:253–260. [DOI] [PubMed] [Google Scholar]
- 46.Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J Cachexia Sarcopenia Muscle 2017;8:926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nilsson MI, Greene NP, Dobson JP, Wiggs MP, Gasier HG, Macias BR et al. Insulin resistance syndrome blunts the mitochondrial anabolic response following resistance exercise. Am J Physiol Endocrinol Metab 2010;299:E466–E474. [DOI] [PubMed] [Google Scholar]
- 48.Alhallak K, Jenkins SV., Lee DE, Greene NP, Quinn KP, Griffin RJ et al. Optical imaging of radiation-induced metabolic changes in radiation-sensitive and resistant cancer cells. J Biomed Opt 2017;22:060502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greene NP, Lee DE, Brown JL, Rosa ME, Brown LA, Perry RA et al. Mitochondrial quality control, promoted by PGC-1α, is dysregulated by Western diet-induced obesity and partially restored by moderate physical activity in mice. Physiol Rep 2015;3:e12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown JL, Rosa-Caldwell ME, Lee DE, Brown LA, Perry RA, Shimkus KL et al. PGC-1 α 4 gene expression is suppressed by the IL-6-MEK-ERK 1/2 MAPK signalling axis and altered by resistance exercise, obesity and muscle injury. Acta Physiol 2017;220:275–288. [DOI] [PubMed] [Google Scholar]
- 51.Gao S, Carson JA. Lewis Lung Carcinoma Regulation of Mechanical Stretch-Induced Protein Synthesis in Cultured Myotubes. Am J Physiol - Cell Physiol 2015;ajpcell.00052.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang G, Jin B, Li Y-P. C/EBPβ mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J 2011;30:4323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alhallak K, Rebello LG, Muldoon TJ, Quinn KP, Rajaram N. Optical redox ratio identifies metastatic potential-dependent changes in breast cancer cell metabolism. Biomed Opt Express 2016;7:4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuznetsov AV, Javadov S, Sickinger S, Frotschnig S, Grimm M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim Biophys Acta 2015;1853:276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu HN, Nioka S, Glickson JD, Chance B, Li LZ. Quantitative mitochondrial redox imaging of breast cancer metastatic potential. J Biomed Opt 2010;15:036010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walsh AJ, Cook RS, Manning HC, Hicks DJ, Lafontant A, Arteaga CL et al. Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer. Cancer Res 2013;73:6164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varone A, Xylas J, Quinn KP, Pouli D, Sridharan G, McLaughlin-Drubin ME et al. Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues. Cancer Res 2014;74:3067–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quinn KP, Sridharan GV, Hayden RS, Kaplan DL, Lee K, Georgakoudi I. Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci Rep 2013;3:3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li LZ, Zhou R, Xu HN, Moon L, Zhong T, Kim EJ et al. Quantitative magnetic resonance and optical imaging biomarkers of melanoma metastatic potential. Proc Natl Acad Sci U S A 2009;106:6608–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177–185. [DOI] [PubMed] [Google Scholar]
- 61.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006;3:187–97. [DOI] [PubMed] [Google Scholar]
- 62.Cosper PF, Leinwand LA. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res 2011;71:1710–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neubauer S The Failing Heart ? An Engine Out of Fuel. N Engl J Med 2007;356:1140–1151. [DOI] [PubMed] [Google Scholar]
- 64.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 2001;88:529–35. [DOI] [PubMed] [Google Scholar]
- 65.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 2013;288:915–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi Y, Dierckx A, Wanrooij PH, Wanrooij S, Larsson N-G, Wilhelmsson LM et al. Mammalian transcription factor A is a core component of the mitochondrial transcription machinery. Proc Natl Acad Sci U S A 2012;109:16510–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaur R, Grasso D, Datta PP, Krishna PD V, Das G, Spencer A et al. A single mammalian mitochondrial translation initiation factor functionally replaces two bacterial factors. Mol Cell 2008;29:180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burger G, Gray MW, Lang BF. Mitochondrial genomes: anything goes. Trends Genet 2003;19:709–16. [DOI] [PubMed] [Google Scholar]
- 69.Mailloux RJ. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol 2015;4:381–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dai D-F, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010;9:536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 2008;88:1243–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Türker FS, Doğan A, Ozan G, Kıbar K, Erışır M. Change in Free Radical and Antioxidant Enzyme Levels in the Patients Undergoing Open Heart Surgery with Cardiopulmonary Bypass. Oxid Med Cell Longev 2016;2016:1783728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Judge S Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J 2004;19:419–421. [DOI] [PubMed] [Google Scholar]
- 74.Ku HJ, Park J-W. Downregulation of IDH2 exacerbates H 2 O 2 -mediated cell death and hypertrophy. Redox Rep 2017;22:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jang S, Javadov S. Association between ROS production, swelling and the respirasome integrity in cardiac mitochondria. Arch Biochem Biophys 2017;630:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Powers SK, Sollanek KJ, Wiggs MP, Demirel HA, Smuder AJ. Exercise-induced improvements in myocardial antioxidant capacity: the antioxidant players and cardioprotection. Free Radic Res 2014;48:43–51. [DOI] [PubMed] [Google Scholar]
- 77.Lee Y, Min K, Talbert EE, Kavazis AN, Smuder AJ, Willis WT et al. Exercise protects cardiac mitochondria against ischemia-reperfusion injury. Med Sci Sports Exerc 2012;44:397–405. [DOI] [PubMed] [Google Scholar]
- 78.Kavazis AN, McClung JM, Hood DA, Powers SK. Exercise induces a cardiac mitochondrial phenotype that resists apoptotic stimuli. Am J Physiol Hear Circ Physiol 2008;294:H928–35. [DOI] [PubMed] [Google Scholar]
- 79.Yusuf SW, Razeghi P, Yeh ETH. The Diagnosis and Management of Cardiovascular Disease in Cancer Patients. Curr Probl Cardiol 2008;33:163–196. [DOI] [PubMed] [Google Scholar]
- 80.Mamidanna R, Nachiappan S, Bottle A, Aylin P, Faiz O. Defining the timing and causes of death amongst patients undergoing colorectal resection in England. Color Dis 2015;n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 81.Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics 2003;16:29–37. [DOI] [PubMed] [Google Scholar]
- 82.Batinić-Haberle I, Rebouças JS, Spasojević I. Superoxide dismutase mimics: chemistry, pharmacology, and therapeutic potential. Antioxid Redox Signal 2010;13:877–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Z-W, Xu X-C, Liu T, Yuan S. Mitochondrion-Permeable Antioxidants to Treat ROS-Burst-Mediated Acute Diseases. Oxid Med Cell Longev 2016;2016:6859523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alleman RJ, Katunga LA, Nelson MA, Brown DA, Anderson EJ. The ‘Goldilocks Zone’ from a redox perspective-Adaptive vs. deleterious responses to oxidative stress in striated muscle. Front Physiol 2014;5:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Du Y, Zhang J, Fang F, Wei X, Zhang H, Tan H et al. Metformin ameliorates hypoxia/reoxygenation-induced cardiomyocyte apoptosis based on the SIRT3 signaling pathway. Gene 2017;626:182–188. [DOI] [PubMed] [Google Scholar]
- 86.von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017; 8: 1081–1083) [DOI] [PMC free article] [PubMed] [Google Scholar]